
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNoninvasive and in situ identification of the phenotypes and differentiation stages of individual living cells entrapped within hydrogels†
Isamar
Pastrana-Otero
 a,
Apurva R.
Godbole
a and
Mary L.
Kraft
*abc
a,
Apurva R.
Godbole
a and
Mary L.
Kraft
*abc
aDepartment of Chemical and Biomolecular Engineering, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, USA. E-mail: mlkraft@illinois.edu
bDepartment of Chemistry, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, USA
cCenter for Biophysics and Quantitative Biology, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, USA
First published on 31st March 2025
Abstract
Microscale screening platforms that allow cells to interact in three dimensions (3D) with their microenviroment have been developed as a tool for identifying the extrinsic cues that might stimulate stem cells to replicate without differentiating within artificial cultures. Though these platforms reduce the number of valuable stem cells that must be used for screening, analyzing the fate decisions of cells in these platforms can be challenging. New noninvasive approaches for identifying the lineage-specific differentiation stages of cells while they are entrapped in the hydrogels used for these 3D cultures are especially needed. Here we used Raman spectra acquired from individual, living cells entrapped within a hydrogel matrix and multivariate analysis to identify cell phenotype noninvasively and in situ. We collected a single Raman spectrum from each cell of interest while it was entrapped within a hydrogel matrix and used partial least-squares discriminant analysis (PLS-DA) of the spectra for cell phenotype identification. We first demonstrate that this approach enables identifying the lineages of individual, living cells from different laboratory lines entrapped within two different hydrogels that are used for 3D culture, collagen and gelatin methacrylate (gelMA). Then we use a hematopoietic progenitor cell line that differentiates into different types of macrophages to show that the lineage-specific differentiation stages of individual, living hematopoietic cells entrapped inside of gelMA scaffolds may be identified by PLS-DA of Raman spectra. This ability to noninvasively identify the lineage-specific differentiation stages of cells without removing them from a 3D culture could enable tracking the differentiation of the same cell over time.
1. Introduction
Microscale culture platforms have been developed to reduce the number of valuable cells, such as rare stem cells, that must be used to screen the fate decisions elicited by extracellular matrix (ECM) proteins, substrate stiffness, and other potential extrinsic cues.1–4 The number of valuable cells required for screening might be further reduced if the fate decisions of individual stem and progenitor cells in a microscale screening platform could be noninvasively identified in situ with a fast and objective approach. Noninvasive fate identification in situ would permit the same cell to be analyzed in the platform at multiple time points during differentiation, enabling multiple fate decisions to be observed for each cell employed. Rapid identification would allow the responses of numerous cells and cues to be screened efficiently, and objective methods could increase their reproducibility. A method that enables such cell fate identification is likely to be adopted by others, especially if it is easy to implement. For this reason, much effort has been made to combine single-cell Raman microspectroscopy with multivariate analysis to permit the in situ identification of the phenotypes of cells on microscale culture platforms.5–20Numerous reports by ourselves and others have demonstrated that the differentiation stages of stem and progenitor cells may be identified by multivariate analysis of Raman spectra acquired from individual, living cells.7–14,17,19 We recently demonstrated that partial least-squares discriminant analysis (PLS-DA) of single-cell Raman spectra enables noninvasively identifying the lineage-specific differentiation stages of individual, living hematopoietic stem and progenitor cells cultured on top of hydrogels functionalized with ECM proteins.17 The cells cultured on these functionalized hydrogels could only interact with the ECM proteins on the substrate beneath them. This largely confines the cell–ECM interactions to two dimensions (2D), so we refer to this as 2D culture. In contrast, cells in the body may interact with their surroundings in three dimensions (3D), and 3D interactions may be required to stimulate stem and progenitor cells to make certain fate decisions in vitro.21–23 Consequently, microscale screening platforms that allow cells to interact with their environment in 3D have been created.23
The in situ identification of the differentiation stages of living stem or progenitor cells in 3D cultures using multivariate analysis of single-cell Raman spectra acquired with a non-imaging approach has not previously been reported. However, a recent publication has established the feasibility of performing Raman imaging on living cells embedded within hydrogels24,25 and using the imaging spectra to identify cell phenotype.25 Motivated by this development, here we explore the feasibility of acquiring a single Raman spectrum from each individual, living cell of interest entrapped within a 3D culture and applying PLS-DA to the spectra to identify each cell's phenotype. For this study, we used collagen and gelatin methacrylate (gelMA) hydrogels as the 3D matrix. Collagen-based 3D scaffolds have been used to culture stem and progenitor cells because collagen contains sequences for cell adhesion and signaling that promote cell growth, proliferation, and differentiation.26 GelMA, which consists of collagen conjugated to a methacrylate monomer, has also been used to synthesize 3D culture platforms because it has the biocompatibility of collagen and the controllable mechanical properties of synthetic polymers.27–30 We first demonstrate that the phenotypes of cells from three different lines in 3D culture could be accurately identified by PLS-DA of single-cell Raman spectra acquired from living cells within collagen and gelMA hydrogels. Then we use the THP-1 progenitor cell line that differentiates into different types of macrophages31–33 to show that the lineage-specific differentiation stages of living hematopoietic progenitor cells within gelMA hydrogels may be identified by PLS-DA of single-cell Raman spectra.
2. Experimental
2.1 Cell culture
Chinese Hamster Ovary cells (CHO-K1, ATCC®, CCL-61™) were cultured in 6 cm culture dishes (Falcon) with Roswell Park Memorial Institute (RPMI) medium, 10% fetal bovine serum (FBS, ThermoFisher Scientific), and 1% penicillin–streptomycin (P/S, ThermoFisher Scientific) in a 37 °C incubator under 5% CO2. CHO-K1 cells that were transfected so they stably expressed HaloTag and SNAP-tag® proteins fused to two different organelle-specific proteins,16 referred to as CHO-T cells, were cultured under the same conditions as CHO cells but with the addition of hygromycin (30 μL, 0.3 mg mL−1) and geneticin (30 μL of 0.3 mg mL−1 geneticin) to the medium. Madin–Darby Canine Kidney (MDCK, ATCC®, CCL-34™) cells were cultured in 6 cm culture dishes with Dulbecco's Modified Eagle Medium (DMEM), 10% v/v FBS, and 1% v/v PS in a 37 °C incubator at under 5% CO2. Human monocyte cells (THP-1, ATCC® TIB-202) were cultured in T-25 flasks (Sarstedt) with RPMI medium, 10% v/v heat-inactivated FBS, and 1% v/v P/S. THP-1 cells in 6 cm culture dishes were differentiated by culturing them in medium that contained 200 nM phorbol 12-myristate 13-acetate (PMA) for 3 d and then in fresh medium without PMA for another 5 days.2.2 Synthesis of collagen hydrogels with entrapped cells
Raman-compatible substrates, which are subsequently referred to as Raman substrates, were fabricated as previously reported16 and immobilized in polydimethylsiloxane (PDMS) wells as described.17 A separate 2 mg mL−1 collagen hydrogel was prepared for each cell lineage according to published methods.34 Briefly, 100 μL of 10× phosphate buffered saline (PBS) and 12 μL of 1 N NaOH were mixed in a sterile tube and chilled on ice. Collagen type 1 stock solution from rat tail (Corning®, 522.2 μL, 3.83 mg mL−1) and culture medium containing cells from a single line (365 μL, cell concentration > 0.5 × 106 cells per mL) were added to the tube on ice. The solution was pipetted up and down to mix. 18 μL of collagen solution was deposited onto a Raman substrate surrounded by a PDMS well in a culture dish and incubated at 37 °C for 40 min. Once the collagen had gelled around the cells, fresh culture medium was added to each sample.2.3 Synthesis of gelMA hydrogels with entrapped cells
Clean Raman substrates were immersed in 0.05 N NaOH and placed on an orbital shaker for 1 h. After removing the NaOH solution, the substrates were rinsed 5 times with distilled water (dH2O) and dried with filtered compressed air. The substrates were immersed in a freshly prepared solution of 2% v/v 3-(trimethoxysilyl)propyl methacrylate (3-TPM) in ethanol and placed on an orbital shaker for 30 min. The 3-TPM solution was removed, and the substrates were immersed in ethanol and placed on an orbital shaker for 5 min. The substrates were removed from the ethanol, dried with filtered compressed air, and baked at 110 °C on a hot plate until dry (5 min).Soft and stiff gelatin methacrylate (gelMA) hydrogels were synthesized from gelMA precursor solutions that contained 4% w/v and 7% w/v gelMA (Allevi, Inc.), respectively.27,28 All vials used to prepare or store the gelMA precursor solutions were covered with aluminum foil to prevent ambient light from degrading the photoinitiators or initiating photopolymerization. For each gelMA precursor solution, a 0.5% w/v lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) solution was prepared by vortexing 0.005 g LAP in 200 μL PBS. In a separate vial, 800 μL of culture medium and the appropriate amount (0.04 g or 0.07 g) of gelMA were combined in a vial and the vial was placed in a 70 °C water bath for approximately 40 min to dissolve the gelMA. While the gelMA was dissolving, cells were detached from the culture vessel and counted. When the gelMA had dissolved, the gelMA solution was removed from the water bath and mixed with the LAP solution, producing 1 mL of gelMA precursor solution without interleukin 4 (IL-4). A gelMA precursor solution containing 10 ng mL−1 interleukin 4 (IL-4) was prepared by combining 10 μL of 1 μg mL−1 IL-4 in PBS with 990 μL gelMA precursor solution. A volume of culture medium containing 0.5–1.0–1.0 × 106 cells was centrifuged and the resulting cell pellet was resuspended in 1 mL of gelMA precursor solution (with or without IL4). A 30 μL or 100 μL aliquot of gelMA precursor solution containing cells was deposited onto a silanized custom Raman substrate or coverslip, respectively. The deposited gelMA precursor solution was covered with a chlorosilanized coverslip and exposed to UV light (365 nm wavelength, 15 mW cm−2, Daigger Scientific Inc.) for 2 min to polymerize it. Then the chlorosilanized coverslip was removed and the gelMA hydrogel containing the cells was incubated in a culture dish containing fresh culture medium at 37 °C and 5% CO2.
2.4 Differentiation of THP-1 cells to inactive macrophages
THP-1 cells were differentiated into inactive macrophages (M0 cells) via phorbol 12-myristate-13-acetate (PMA) treatment. Briefly, THP-1 cells were cultured in medium containing 200 nM PMA at 37 °C for 72 h. Then the PMA-containing medium was removed and fresh PMA-free medium was added. At this point, most cells had attached to and spread on the surface of the culture dish, which indicated they had differentiated. The cells were incubated in PMA-free medium for 5 more days (120 h), detached from the culture dish using CTS™ TrypLE™ Select Enzyme (Gibco), and encapsuled within a soft gelMA hydrogel as described above.2.5 Differentiation of THP-1 cells entrapped in hydrogels to pro- or anti-inflammatory macrophages
To differentiate THP-1 cells into pro-inflammatory M1-like or anti-inflammatory M2-like macrophages, THP-1 cells were first encapsulated within a soft gelMA hydrogel in the absence or presence of IL-4, respectively. Then the gelMA hydrogels with entrapped cells were placed in fresh medium and incubated in a 37 °C incubator under 5% CO2. The spent medium was replaced with fresh medium every 3 d, and Raman spectra were acquired from cells after 8 d.2.6 Labeling of CHO cells with Hoechst nuclear stain
Hoechst staining solution was prepared by diluting a stock solution of 10 mg mL−1 Hoechst 33342 (16.2 mM, ThermoFisher Scientific) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 with 2% w/v BSA in PBS and mixed by pipetting up and down. CHO cells were detached from the culture dish, counted, and solution was adjusted to obtain a cell concentration of at least 0.5 × 106 cells per mL. The cell solution was centrifuged at 1500 rpm for 7 min, the medium was removed, and the cell pellet was resuspended in 500 μL of Hoechst staining solution at rt for 10 min. Then the cells were centrifuged at 1500 rpm for 7 min, the staining solution was removed, and the cell pellet was resuspended in culture medium. Collagen hydrogels containing these stained cells were synthesized as described above.
:1000 with 2% w/v BSA in PBS and mixed by pipetting up and down. CHO cells were detached from the culture dish, counted, and solution was adjusted to obtain a cell concentration of at least 0.5 × 106 cells per mL. The cell solution was centrifuged at 1500 rpm for 7 min, the medium was removed, and the cell pellet was resuspended in 500 μL of Hoechst staining solution at rt for 10 min. Then the cells were centrifuged at 1500 rpm for 7 min, the staining solution was removed, and the cell pellet was resuspended in culture medium. Collagen hydrogels containing these stained cells were synthesized as described above.
2.7 Labeling collagen hydrogels with 5(6)-carboxy fluorescein N-succinimidyl ester
A solution of 5(6)-carboxy fluorescein N-succinimidyl ester (CFSE, 20 mM, ThermoFisher Scientific) was prepared by adding 10.92 μL of CFSE stock solution (10 mg mL−1 CFSE in DMSO) to a conical tube, bringing the volume up to 2 mL with 50 mM borate buffer, and vortexing it. To label the collagen fibrils in the hydrogels, the collagen hydrogel was incubated at rt with 4 mL of 50 mM borate buffer (pH 9.0) for 6 min, the borate buffer was carefully aspirated from the culture dish, and the CFSE solution was added to the collagen hydrogel. The culture dish was wrapped with aluminum foil to protect it from light and the sample was placed on a rocking platform for 1 h at rt to allow the dye to conjugate to the collagen hydrogel. Next, the CFSE solution was removed by aspiration, 10 mL of 50 mM Tris buffer (pH 7.5) was added to quench the unreacted dye and the sample was incubated while rocking for 10 min. Finally, the CFSE-stained collagen hydrogel was rinsed three times by immersion in 4 mL of PBS for 10 min.2.8 Labeling of THP-1 cells with Hoechst stain and LysoTracker™ Red DND-99
THP-1 cells encapsulated within gelMA hydrogels were labeled with Hoechst nuclear staining and LysoTracker™ Red DND-99 (Invitrogen™). Cells were incubated in phenol red-free medium overnight before fixing with 4% v/v paraformaldehyde in PBS for 15 min at rt. Then the cells were permeabilized with 0.25% v/v Triton X-100 for 10 min at rt, washed with PBS three times for 5 min each, and stained with Hoechst nuclear stain as described above for CHO cells. Finally, cells were labeled with LysoTracker™ Red by incubation for 2 h at rt with 75 nM LysoTracker™ in phenol red-free RPMI.2.9 Fluorescence confocal microscopy
A Leica SP8 fluorescence confocal microscope equipped with UV/visible laser and a 40x oil immersion objective was used to acquire fluorescence images from living CHO cells within and outside of collagen hydrogels, and fixed THP-1 monocytes and differentiated THP-1 macrophages within gelMA hydrogels. The Hoechst nuclear stain was excited using the 405 nm laser and its fluorescence emission was detected at 460 nm. The CFSE-labeled fibrils in the collagen hydrogels were imaged using ex/em of 492 nm/517 nm. The fluorescence from the LysoTracker™ used for labeling the lysosomes was performed with ex/em of 561 nm/590 nm. Images were processed using Fiji software (previously known as ImageJ).2.10 Acquisition of Raman spectra
Each hydrogel containing cells was incubated at 37 °C in 5% CO2 for at least 3 h before Raman spectra were acquired. Then the culture medium bathing each sample was replaced with 12 mL of fresh DMEM medium containing 1% P/S for CHO, CHO-T, and MDCK cells and 12 mL RPMI medium containing 1% P/S for THP-1 cells. A single Raman spectrum was acquired from each individual living cell within the hydrogel matrix with a Horiba LabRam HR 3D confocal Raman imaging system with a pinhole size of 500 μm, a slit size of 100 μm, and a grating of 300 groves per mm using a water-dipping objective with a working distance of 2 mm (Olympus 60×, NA 1.0) that was immersed directly into the culture medium. A diffraction-limited spot of 1 μm diameter was focused on each cell with a 785 nm laser (200 mW) such that neighboring cells were excluded from the focal area. Raman scattering data was acquired from 600 to 1750 cm−1 for 30 s per cell for at least 20 cells for each sample. A custom heat stage was used to maintain the hydrogel samples containing the live cells at 37 °C during Raman spectra acquisition.2.11 Spectral analysis
Spectra were first preprocessed using LabSpec 5 (Horiba Scientific) to manually remove any cosmic spikes. Spectra were aligned to the peak of phenylalanine at 1004 cm−1 using an in-house MATLAB (R2016b, MathWorks Inc.) code. Hydrogel-related spectral features that were not produced by the cells were partially removed from the spectra with a reported background subtraction algorithm35 that we modified in-house. Cell spectra were uploaded into the PLS Toolbox (v.8.9, Eigenvector Research) run in MATLAB. For each spectrum, the second derivative was taken using a Savitzky–Golay algorithm (2nd order polynomial, window = 25 points) to smooth each spectrum and emphasize the lower intensity peaks, and normalized to all peaks over the 600–1750 cm−1 range. The data set was mean-centered. Half the spectra acquired for each cell type were used to construct a PLS-DA model that related the combination of Raman peaks most distinctive of each cell lineage to a matrix of variables indicative of that lineage. The minimum number of latent variables (LVs), which are the linear combinations of Raman spectral features that are the most useful for identifying cell type, required to capture at least 80% of the spectral variance were retained in each model. This PLS-DA model was applied to the remaining half of the cell spectra to calculate a prediction value that was used to identify cell lineage. Cells with prediction values that exceed the Bayesian threshold for a specific cell population, which is represented by a red dashed line in the identification plot, were identified as members of that population.3. Results and discussion
3.1 Fluorescence imaging of CHO cells within collagen hydrogels
We began by confirming our hydrogel synthesis procedure suspended the cells within the hydrogel. We used our standard procedure to encapsulate cells within hydrogels, and for comparison, we also made a sample consisting of a collagen hydrogel polymerized on top of cells attached to a substrate, and a sample in which the cells were seeded on top of a collagen hydrogel. The collagen fibrils and encapsulated CHO cells within each sample were labeled with CFSE and Hoechst nuclear stain, respectively. Then fluorescence microscopy was used to visualize the CHO cells with respect to the collagen fibrils in the sample consisting of cells on top of the collagen gel (Fig. 1a–d), the sample made with our standard encapsulation procedure (Fig. 1e–h) and the sample consisting of cells underneath the collagen hydrogel (Fig. 1i–l). Green fluorescence locates the CFSE-labeled collagen fibrils and overlap of this green fluorescence with the blue fluorescence from the Hoechst nuclear stain makes the cells appear cyan (white arrows, Fig. 1).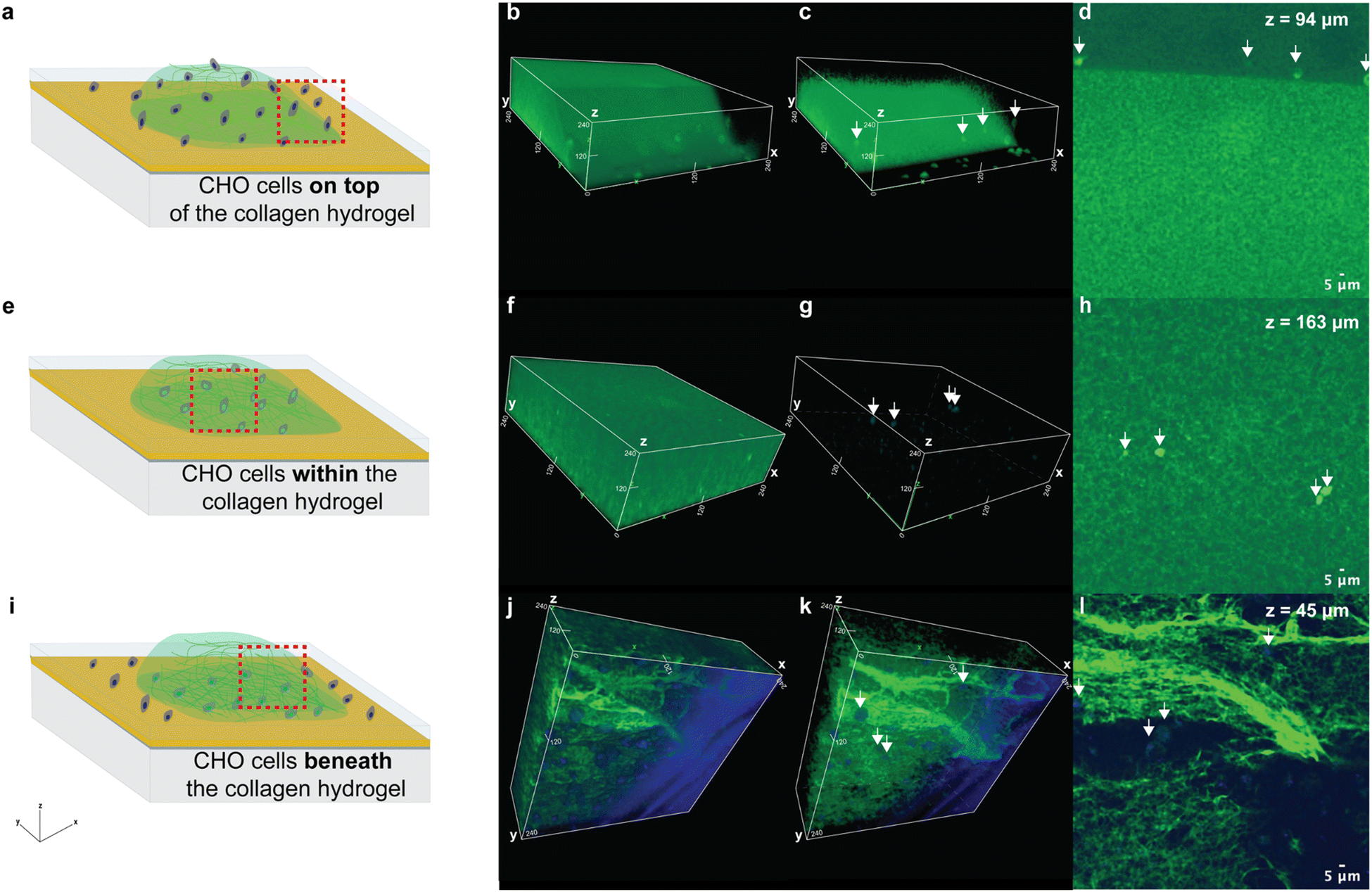 | ||
| Fig. 1 Schematics show the CHO cells positioned (a) on top of, (e) within, or (i) on the substrate beneath the collagen hydrogel (green). The region outlined with a red-dashed line approximates the location where fluorescence imaging was performed. Volumetric confocal microscopy images of CHO cells that were labeled with Hoechst nuclear stain (blue) (b and c) on top of, (f and g) within, or (j and k) beneath a collagen hydrogel, which was labeled with CFSE (green). White arrows indicate the positions of the cells. Cross-sectional confocal microscopy images acquired of the sample containing cells (white arrows) positioned (d) on top of, (h) within, or (l) beneath the collagen hydrogel. The z-coordinate where the image was taken is shown on the figure. Low values are closer to the substrate beneath the hydrogel and high values are closer to the top surface of the hydrogel. | ||
For the sample consisting of cells seeded on top of the hydrogel, the volumetric fluorescence microscopy image (Fig. 1b and c) shows the hydrogel's top surface slopes downward, and the cells are resting on top of it. The cross-sectional confocal microscopy image of this sample shows the cells are positioned above the bright green region that denotes the CFSE-labeled collagen hydrogel (Fig. 1d). Note that the z-position where this cross section was acquired has an intermediate value (Fig. 1d) because the surface of the hydrogel that the cells are resting on slopes downward. In contrast, for the sample created with our standard cell encapsulation procedure, the 3D volumetric image shows the cells are surrounded by the hydrogel (Fig. 1e–h). The cross-sectional confocal microscopy image acquired at an intermediate z-position within this sample (Fig. 1h) shows the CFSE-labeled collagen hydrogel above and below the cells, confirming they were suspended within the hydrogel matrix. In comparison, the volumetric confocal microscopy images of the sample consisting of cells below the hydrogel are rotated to show the bottom portion of the gel so that the cells would be visible (Fig. 1j–l). The z-position where the confocal microscopy cross-sectional image of this sample (Fig. 1l) was acquired confirms these cells were positioned very close to the substrate. Overall, these findings confirm that the general procedure used for hydrogel synthesis entraps cells within a hydrogel matrix.
3.2 Raman microspectroscopy for 3D cultures
The features in a Raman spectrum are specific to the chemical bonds in the molecules within the sample. If the Raman features associated with the collagen fibrils in the hydrogel overlap with those from the biomolecules in the cells, the spectral signatures of the cell populations might be obscured enough to prevent identifying the cell's phenotype with PLS-DA. As the first step in exploring this possibility, we identified the Raman peaks associated with collagen hydrogels. Fig. 2 shows the average baseline-corrected spectra collected from regions of collagen, soft gelMA, and stiff gelMA hydrogels that did not contain cells. We refer to the Raman spectra acquired from cell-free regions of the hydrogels as background spectra. The features at 1400 cm−1 and 1650 cm−1 in the collagen hydrogel spectra (Fig. 2a) were similar to those in the spectra of the polyacrylamide hydrogels that we have used for 2D cell cultures in prior work.16,28 The background spectra from the soft and stiff gelMA hydrogels also contained peaks associated with denatured and partially hydrolyzed collagen (highlighted in gray in Fig. 2a) that occur at approximately the same wavelengths as typical peaks in cell spectra. However, few peaks from the collagen are visible in the spectra of the CHO-T cells entrapped in the same hydrogel (Fig. 2b). The few features that are common to both the hydrogel spectrum and the spectrum of cells embedded in that hydrogel include the peak at 1000 cm−1 (phenylalanine), the broad peak at 1446 cm−1 (CH2 bending and scissoring modes), and the peak at 850 cm−1 (nonspecific C–C stretching). The average preprocessed spectra acquired from individual, living cells from each line in each hydrogel are presented in Fig. S1.† Altogether, these spectra demonstrate that the hydrogel-related spectral features had negligible overlap with features in the cell spectra. | ||
| Fig. 2 (a) Average preprocessed Raman spectra collected from collagen (n = 8, orange), soft gelMA (n = 5, pink) and stiff gelMA (n = 5, magenta) hydrogels at regions devoid of CHO-T cells. (b) Average preprocessed spectra from CHO-T cells within collagen (n = 45), soft gelMA (n = 62), and stiff gelMA (n = 61). In (a) and (b), black lines represent the standard deviation from the average spectra and features highlighted in gray are associated with denatured and partially hydrolyzed collagen in the hydrogel. | ||
3.3 Identifying the lineage of individual living cells from laboratory lines within hydrogels
Next, we used cells from three laboratory lines (CHO, CHO-T, and MDCK cells) to assess whether PLS-DA models of single-cell Raman spectra could be used to identify the phenotypes of living cells within collagen and gelMA hydrogels. The main difference between the CHO-T line and the parent CHO line is CHO-T cells stably express two proteins that are not expressed by CHO cells, HaloTag and SNAP-tag fused to organelle-specific proteins. Thus CHO and CHO-T lines are very similar to one another, but they each differ more substantially from the MDCK line, which is from a different species and tissue. Therefore, the CHO-T and CHO lines are likely as similar to one another as stem cells are to the progeny cell populations they differentiate into, but more different from MDCK cells than stem cells and their progeny are from the cells in the niche where they reside. Consequently, we expected the CHO, CHO-T and MDCK lines would roughly approximate the challenges of identifying the phenotypes of closely related stem and progenitor cells in the presence of niche cells.A single Raman spectrum was acquired from each living cell entrapped within the collagen hydrogels. A total of 45, 45, and 39 single-cell spectra were acquired from CHO, CHO-T and MDCK cells. Roughly half of these spectra (23, 23, and 20 spectra from individual, living CHO, CHO-T and MDCK cells, respectively) were used to construct a PLS-DA model that contained 4 latent variables, which are linear combinations of spectral features. We selected this number of LVs because it was the minimum number that captured at least 80% (86.7%) of the total spectral variation in the calibration spectra. We previously demonstrated that this criterion best minimizes the error of cell phenotype prediction, whereas the optimal number of LVs varies depending on the number of cell phenotypes included in the model.18 This criterion is validated by the higher errors that were obtained when PLS-DA models containing other numbers of LVs that captured different amounts of spectral variance were employed for cell phenotype identification (Table S1†).
The PLS-DA model containing four LVs was applied to a separate test set that contained 22, 22 and 19 spectra, each acquired from a single, living CHO, CHO-T or MDCK cell, respectively, that was entrapped within a collagen hydrogel. Cells that exceed the classification threshold (red dashed line), which is based on Bayes theorem, in the identification plots for each cell lineage were identified as belonging to that lineage (Fig. 3a–c). The identification accuracy was remarkably high, 98.8%, 97.6%, and 97.4% for the test CHO, CHO-T, and MDCK cells, respectively. Inspection of the variable importance for prediction (VIP) scores revealed that the 685 cm−1 peak, which is associated with citric acid,36 and the 1438 cm−1 peak that is attributed to CH2 bending in lipids have high (>1) VIP scores, indicating they contributed to identifying the CHO cells and MDCK cells. Because citric acid is a primary metabolite in the tricarboxylic acid (TCA) cycle and lipid synthesis increases when cells divide, the high importance of the peaks at 685 cm−1 and 1438 cm−1 suggest that the CHO and MDCK cells were actively participating in the cell cycle. The protein-related peaks at 883 cm−1 (CH2 rocking) and 1061 cm−1 (C–C in-plane bending and C–N stretching) had high VIP scores that indicate they were important for identifying the CHO-T cells. The high importance of these protein-related peaks towards identifying the CHO-T cells is consistent with the overexpression of two organelle-specific proteins fused to HaloTag and SNAP-tag by cells from this line. Thus, the lineages of individual, living cells within collagen hydrogels may be identified with high accuracy by PLS-DA of single-cell Raman spectra.
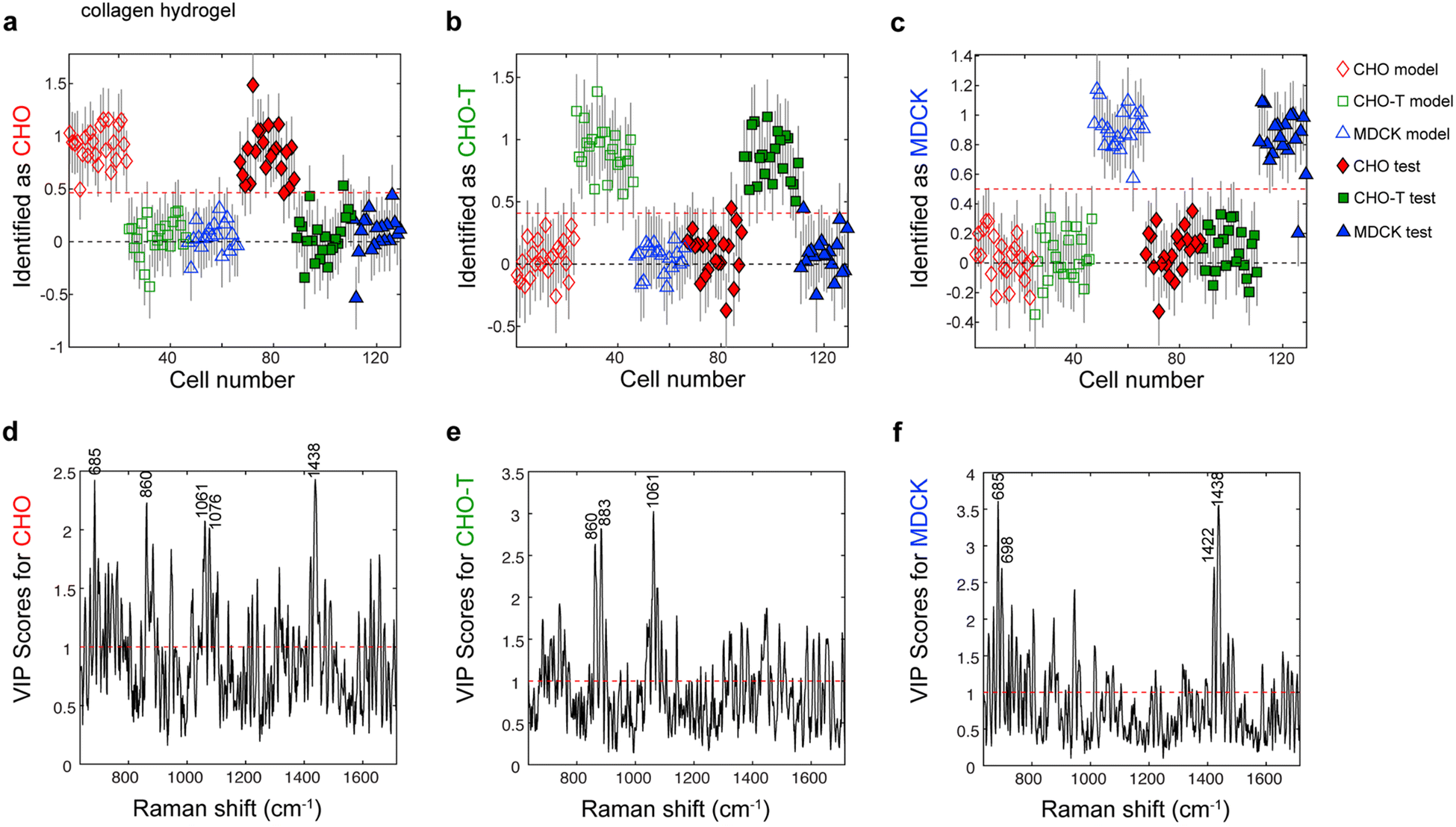 | ||
| Fig. 3 Identification plots for the PLS-DA model constructed using Raman spectra of acquired from individual, living (a) CHO, (b) CHO-T, and (c) MDCK cells encapsulated in collagen hydrogels identify the lineage of each cell. Markers located above the classification threshold (red dashed line) indicate that cell was identified as a (a) CHO cell (red diamonds, nmodel = 23, ntest = 22), (b) CHO-T cell (green squares, nmodel = 23, ntest = 22), or (c) MDCK cell (blue triangles, nmodel = 20, ntest = 19). Variable importance for prediction (VIP) scores plots for (d) CHO cells, (e) CHO-T cells, and (f) MDCK cells encapsulated in collagen hydrogels show the VIP scores for various Raman peaks. Peaks with high (>1) scores are important for identifying the indicated cell type. Assignments of labeled peaks: 685 cm−1 (citric acid), 698 cm−1 (methionine), 860 cm−1 (tyrosine, collagen, and phosphates), 883 cm−1 (proteins), 1061 cm−1 (proteins), 1076 cm−1 (lipids), 1422 cm−1 (adenine, guanine, and deoxyribose), and 1438 cm−1 (lipids). | ||
Next, we assessed whether the lineages of cells entrapped within the gelMA hydrogels could be accurately identified by using PLS-DA of single-cell Raman spectra. For this task, we used CHO, CHO-T, and MDCK cells encapsulated within both soft and stiff gelMA hydrogels that approximated the stiffness of two regions in the bone marrow niche where HSCs reside in the body.37 We used both soft and stiff gelMA hydrogels for PLS-DA model construction due to their relevance to screens of HSC fate decisions. However, the use of spectra from cells within gels with different stiffness to train the same PLS-DA model values presents new challenges because the PLS-DA model must be trained to exclude any spectral variation related to hydrogel stiffness while capturing the variation that is important for identifying cell phenotype.6
A PLS-DA model was constructed using a calibration set consisting of 62, 62, and 60 spectra of individual, living CHO, CHO-T and MDCK cells, respectively, entrapped in soft and stiff gelMA hydrogels. The resulting PLS-DA model contained 6 latent variables that captured approximately 80% of the total variation in the calibration spectra (Table S2†). This PLS-DA model was applied to a separate set of test spectra that were acquired from 61, 61 and 59 individual, living CHO, CHO-T and MDCK cells, respectively, that were entrapped in soft and stiff gelMA hydrogels. The accuracy for identifying the CHO, CHO-T, and MDCK cells in the test set was 98.8%, 95.0%, and 97.5%, respectively (Fig. 4). Examination of the VIP scores plots for this PLS-DA model (Fig. 4d–f) indicated that the biomolecules that were important for identifying each cell phenotype in the PLS-DA model of the cells entrapped within collagen were similar to those that were important for identification by this model. Specifically, peaks associated with lipids and energy sources (i.e., 716 cm−1, 1015 cm−1, and 1022 cm−1 from lipids, carbohydrates and glycogen, respectively) were important for identifying the CHO and MDCK cells. Peaks associated with proteins (1442 cm−1 from fatty acids and collagen and 1585 cm−1 from phenylalanine and hydroxyproline) were among those important for identifying the CHO-T cells. Altogether, these findings demonstrate that the lineages of individual, living cells within gelMA hydrogels of varying stiffness may be accurately identified by PLS-DA of single-cell Raman spectra that encode for cell biochemistry.
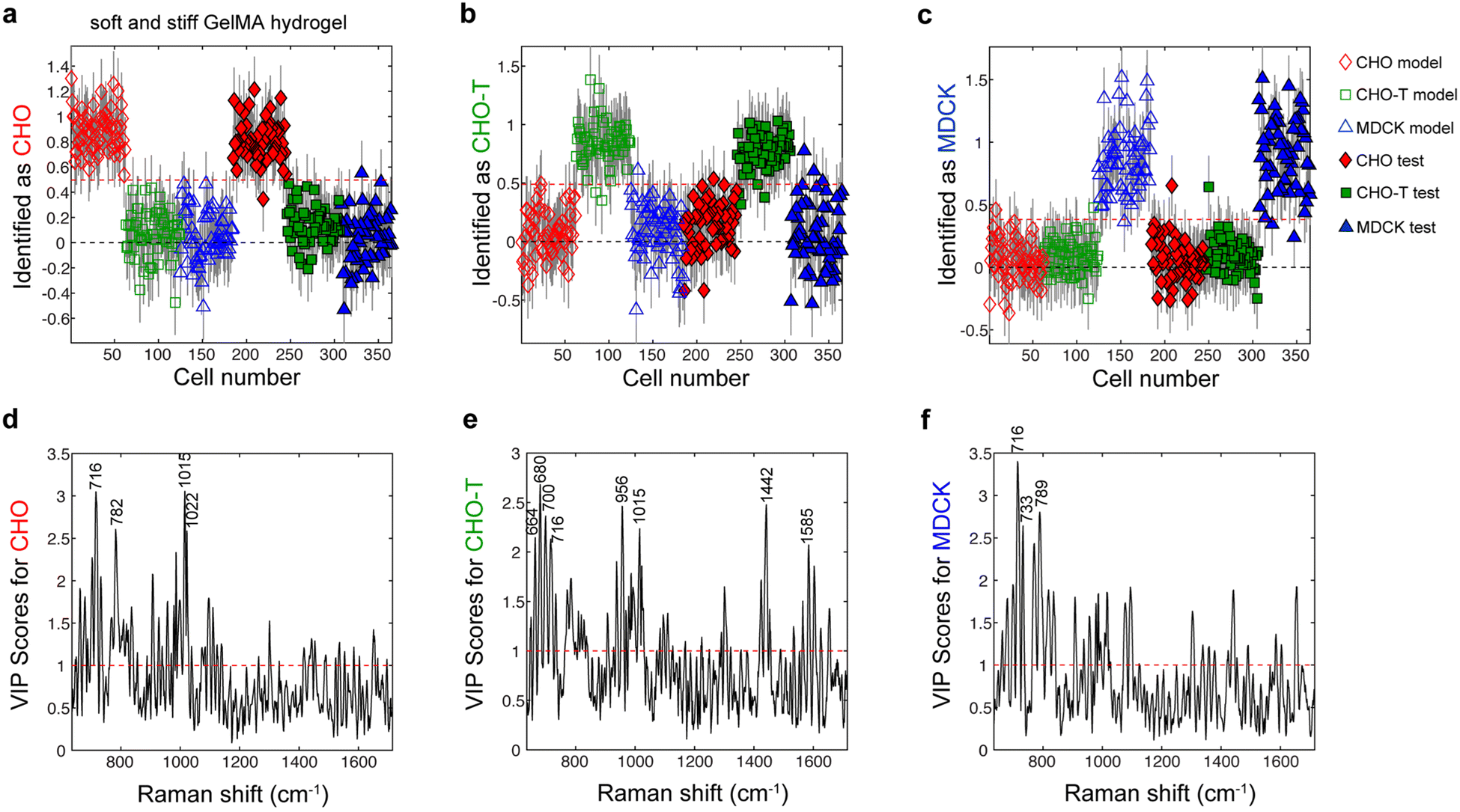 | ||
Fig. 4 Identification plots for the PLS-DA model constructed using Raman spectra acquired from individual, living (a) CHO, (b) CHO-T, and (c) MDCK cells encapsulated in gelMA hydrogels. Cells located above the classification threshold denoted by the red dashed line were identified as (a) CHO cells ( and and  , nmodel = 62, ntest = 61), (b) CHO-T cells ( , nmodel = 62, ntest = 61), (b) CHO-T cells ( and and  , nmodel = 62, ntest = 61), and (c) MDCK cells ( , nmodel = 62, ntest = 61), and (c) MDCK cells ( and and  , nmodel = 60, ntest = 59). Variable importance for prediction (VIP) plots for (d) CHO cells, (e) CHO-T cells, and (f) MDCK cells show which peaks have high (>1) scores that denote importance for identifying that cell type. Assignment of labeled peaks: 664 cm−1 (DNA bases), 680 cm−1 (DNA bases), 700 cm−1 (methionine), 716 cm−1 (lipids), 733 cm−1 (phosphatidylserine), 782 cm−1 (DNA/RNA), 789 cm−1 (phosphodiester bands), 956 cm−1 (cholesterol), 1015 cm−1 (carbohydrates), 1022 cm−1 (glycogen), 1442 cm−1 (fatty acids and collagen), and 1585 cm−1 (phenylalanine and hydroxyproline). , nmodel = 60, ntest = 59). Variable importance for prediction (VIP) plots for (d) CHO cells, (e) CHO-T cells, and (f) MDCK cells show which peaks have high (>1) scores that denote importance for identifying that cell type. Assignment of labeled peaks: 664 cm−1 (DNA bases), 680 cm−1 (DNA bases), 700 cm−1 (methionine), 716 cm−1 (lipids), 733 cm−1 (phosphatidylserine), 782 cm−1 (DNA/RNA), 789 cm−1 (phosphodiester bands), 956 cm−1 (cholesterol), 1015 cm−1 (carbohydrates), 1022 cm−1 (glycogen), 1442 cm−1 (fatty acids and collagen), and 1585 cm−1 (phenylalanine and hydroxyproline). | ||
3.4 Identifying the differentiation stages of THP-1 cells and differentiated macrophages entrapped in hydrogels
We next probed whether the differentiation stages of individual, living immune cells entrapped within hydrogels could be noninvasively identified by PLS-DA of single-cell Raman spectra. For this task, we employed the spontaneously immortalized human monocyte-like cells from the THP-1 cell line that may be differentiated into three different types of macrophage-like cells, M0, M1–M2, and M2. Treatment of THP-1 cells in 2D cultures with phorbol-12-myristate-13-acetate (PMA), interferon-gamma, or IL-4 induces differentiation to inactive M0 macrophage-like cells, active pro-inflammatory M1 macrophage-like cells, or active anti-inflammatory M2 macrophage-like cells, respectively.31,33,38 A previous report has established that by the eighth day in culture, THP-1 cells embedded in soft gelMA hydrogels containing IL-4 differentiate into M2-like macrophages,39 which we refer to as M2 cells. In contrast, by the eighth day in culture, THP-1 cells embedded in soft gelMA hydrogels lacking IL-4 differentiate into cells that exhibit characteristics common to both M1 and M2 macrophage-like cells,39 which we refer to as M1-M2 cells. Therefore, we embedded THP-1 cells in soft gelMA hydrogels and cultured them without IL-4 for 8 d to induce THP-1 cell differentiation to M1-M2 cells. We also cultured THP-1 cells within soft gelMA hydrogels that contained IL-4 for 8 d to induce differentiation to M2 cells. Raman spectra were acquired from individual living cells within the gelMA hydrogels on day 1 when the cells still exhibited the THP-1 phenotype, and on day 8 when the cells in the gelMA hydrogel with or without IL-4 were at the M2 or M1–M2 phenotype, respectively.39 Because the effects of hydrogel encapsulation on THP-1 cell differentiation to M0 cells following PMA treatment have not been reported, we first used PMA to induce THP-1 cell differentiation to M0 macrophage-like cells, which we refer to as M0 cells, in 2D culture following an established protocol.38 Then we entrapped these M0 cells in soft gelMA hydrogels and acquired Raman spectra from individual, living M0 cells in the hydrogel 8 d following PMA treatment.We confirmed the phenotypes of the cells when the Raman spectra were acquired by using fluorescence microscopy assessment of the cells’ lysosomal content. Fluorescence microscopy imaging of cell lysosomes stained with LysoTracker™ in cells prepared in parallel with those analyzed with Raman spectroscopy indicated the THP-1 cells (cells on the first day in the hydrogel) had relatively low lysosomal content (Fig. S2†). In comparison, higher lysosomal staining was observed in the M0 cells (Fig. S2†), which is consistent with the higher number of lysosomes in M0 cells relative to THP-1 cells. Likewise, the fluorescence from the LysoTracker™ was higher in the M1–M2 cells and M2 cells than the THP-1 cells (Fig. S2†), which is also consistent with the high abundance of lysosomes in M1 and M2 macrophages.40
The average single-cell spectra acquired from the living THP-1 cells and differentiated progeny M0, M1–M2, and M2 cells encapsulated in soft gelMA hydrogels are shown in Fig. 5. The most obvious difference in the averaged spectra is the height of the 960 cm−1 peak, which is greatest in the averaged M2 cell spectrum, intermediate in the averaged M1–M2 cell spectrum, and nearly absent in the averaged M0 and THP-1 cell spectra. The identity of this peak is unclear because it has been assigned to β-carotene, cholesterol, and hydroxyapatite elsewhere,8,41,42 but we were unable to confirm any of these assignments. Compared to the other averaged cell spectra, the peak at 785 cm−1 that is associated with DNA/RNA bases was largest in the averaged THP-1 cell spectrum, and the 1302 cm−1 peak from collagen and lipids was largest in the averaged M0 cell spectrum. However, the intensities of the peaks at 785 cm−1, 960 cm−1, and 1302 cm−1 varied substantially between each spectrum acquired from a single cell from the same population, so none could be used alone to identify a cell's lineage and differentiation stage.
 | ||
| Fig. 5 Averages spectra acquired from individual, living THP-1 cells (red, n = 121), M0 cells (green, n = 79), M1–M2 cells (blue, n = 42), and M2 cells (cyan, n = 42) within soft gelMA hydrogels. Spectra were background subtracted, baseline corrected using a weighted-least-squares method, normalized, and offset for clarity. Black lines represent 1 standard deviation from the averaged spectra. Assignment of labeled peaks: 702 cm−1 (cholesterol), 718 cm−1 (choline), 785 cm−1 (DNA/RNA bases), 832 cm−1 (tyrosine and asymmetric O–P–O stretching), 854 cm−1 (C–C stretching), 960 cm−1 (unidentified), 1004 cm−1 (phenylalanine), 1093 cm−1 (phosphodiester bonds in DNA), 1127 cm−1 (proteins), 1175 cm−1 (cytosine and guanine), 1209 cm−1 (tryptophan and phenylalanine), 1265 cm−1 (lipids and collagen), 1302 cm−1 (lipids and collagen), 1342 cm−1 (guanine), 1445 cm−1 (lipids and collagen), 1460 cm−1 (lipids and collagen), 1582 cm−1 (phenylalanine), 1612 cm−1 (cytosine and tyrosine), and 1658 cm−1 (amide I). | ||
We next assessed whether each cell's lineage and differentiation stage could be determined by PLS-DA of the spectra acquired from individual, living cells while they were entrapped within a gelMA hydrogel. A PLS-DA model was constructed using 61, 40, 21, and 21 spectra from individual, living THP-1, M0, M1–M2 and M2 cells, respectively, entrapped within gelMA hydrogels. The PLS-DA model, which contained 5 LVs that captured 83% of the variation in the calibration set (Table S3†), was applied to a test set consisting of 60, 39, 21 and 21 spectra of individual, living THP-1, M0, M1–M2 and M2 cells, respectively, within hydrogels. The cells that exceeded the red dashed line that denotes the classification threshold for the lineage-specific differentiation stage specified on the y-axis of the identification plots shown in Fig. 6a–d were identified as belonging to that population. The accuracies of identifying the THP-1, M0, M1–M2 and M2 cells in the test set were 99.2%, 97.7%, 99.2%, and 95.2%, respectively. The VIP scores from this PLS-DA model (Fig. 6e–h) showed that the peaks at 788 cm−1 from DNA, 1441 cm−1 from lipids, 960 cm−1 from an unknown source and 981 cm−1 from proteins were among those that were important for identifying the THP-1 cells, M0 cells, M1–M2 cells, and M2 cells, respectively. Altogether, these results demonstrate that the differentiation stages of individual, living hematopoietic cells within hydrogels can be identified with high accuracy by using PLS-DA models of single-cell Raman spectra.
 | ||
Fig. 6 Identification plots for the PLS-DA model of Raman spectra from (a) THP-1, (b) M0, (c) M1–M2, and (d) M2 cells within soft gelMA hydrogels. Cells located above the red dashed line that represents the classification threshold for each plot were identified as (a) THP-1 cells ( and and  , nmodel = 61, ntest = 60), (b) M0 cells ( , nmodel = 61, ntest = 60), (b) M0 cells ( and and  , nmodel = 40, ntest = 39), (c) M1–M2 cells ( , nmodel = 40, ntest = 39), (c) M1–M2 cells ( and and  , nmodel = 21, ntest = 21), and (d) M2 cells ( , nmodel = 21, ntest = 21), and (d) M2 cells ( and and  , nmodel = 21, ntest = 21). VIP score plots for (e) THP-1 cells, (f) M0 cells, (g) M1–M2 cells, and (h) M2 cells. Assignments of labeled peaks: 743 cm−1 (DNA and tryptophan), 788 cm−1 (phosphodiester bonds in DNA), 960 cm−1 (unidentified), 981 cm−1 (proteins), 1013 cm−1 (tryptophan), 1302 cm−1 (collagen and lipids), 1309 cm−1 (collagen and lipids), 1422 cm−1 (DNA/RNA), 1441 cm−1 (lipids), and 1657 cm−1 (lipids, protein, and DNA/RNA). , nmodel = 21, ntest = 21). VIP score plots for (e) THP-1 cells, (f) M0 cells, (g) M1–M2 cells, and (h) M2 cells. Assignments of labeled peaks: 743 cm−1 (DNA and tryptophan), 788 cm−1 (phosphodiester bonds in DNA), 960 cm−1 (unidentified), 981 cm−1 (proteins), 1013 cm−1 (tryptophan), 1302 cm−1 (collagen and lipids), 1309 cm−1 (collagen and lipids), 1422 cm−1 (DNA/RNA), 1441 cm−1 (lipids), and 1657 cm−1 (lipids, protein, and DNA/RNA). | ||
4. Conclusions
We have demonstrated that a single Raman spectrum may be noninvasively acquired from each living cell of interest that is entrapped within a collagen and gelMA hydrogel. Though spectral features related to the hydrogel matrix were present in each single-cell spectrum, they had low intensities and did not obscure the spectral features that are collectively characteristic of cell phenotype. By applying PLS-DA to the spectra acquired from individual cells belonging to four different closely related immune cell populations, THP-1, and their progeny M0, M1–M2, and M2 populations, the lineage-specific differentiation stage of each cell entrapped within a hydrogel was noninvasively identified in situ with high accuracy. This method is advantageous because it could allow the differentiation stages of individual, living cells in 3D cultures to be identified in situ, which could enable the noninvasive tracking of cell differentiation over time. This capability could facilitate efforts to identify the cues that elicit desired stem cell fate decisions in vitro.In this feasibility study, we employed the THP-1 cell line and its three progeny populations, M0, M1–M2, and M2. The THP-1 cells were embedded in gels during their differentiation to M1–M2 and M2 cells because prior work has established the effects of hydrogel encapsulation on THP-1 cell differentiation to these phenotypes. Specifically, hydrogel encapsulation induces THP-1 cell differentiation to the M1–M2 phenotype, which is distinct from the THP-1 and M2 phenotypes, whereas hydrogel encapsulation does not affect IL-4-induced differentiation of THP-1 cells to the M2 phenotype. In contrast, whether the PMA-induced differentiation of THP-1 cells to M0 macrophage-like cells is affected by THP-1 cell encapsulation within a hydrogel is not known. To ensure that the third population of differentiated cells had the well-characterized M0 macrophage-like phenotype, and thus, could be identified by lysosomal staining, we exposed THP-1 cells on nonporous substrates to PMA and encapsulated the resulting M0 cells within hydrogels. Although cell encapsulation in gels during differentiation might produce higher cell-to-cell heterogeneity, we expect that a similarly low error of classification would have been obtained if the THP-1 cells were encapsulated in gels during PMA-induced differentiation. This expectation is based on the observation that the error of classification for the M0 cells (97.7%) was between the classification errors for the M1–M2 (99.2%) and M2 (95.2%) cells that were encapsulated within the gel during differentiation. We attribute this low error of classification despite the potential presence of within-population spectral variation to our use of PLS-DA to model the spectra. PLS-DA emphasizes the spectral variation between the cell populations and deemphasizes the within-population spectral variance.14 Therefore, to obtain low errors of identification, the cell spectra that are used for PLS-DA model construction must contain as much within-population spectral variation as the cell populations that they are used to identify.14
We selected immune cells at different differention stages to test this PLS-DA and Raman spectroscopy approach for noninvasively identifying the phenotypes of cells within hydrogels due to the clinical importance of another hematopoietic cell population, hematopoietic stem cells (HSCs). HSC transplants are an effective treatment for leukemia and other blood and immune disorders due to their ability to produce all the mature blood and immune cells in the body throughout life. Because the availability of this lifesaving treatment might increase if HSCs would divide without differentiating in vitro as they do in the body, much effort has been made to identify the cues within the HSC niche that might induce HSC self-renewal in vitro.37,43,44 Noninvasive, location-specific methods that enable routinely identifying the fate decisions made by HSCs and early hematopoietic cell progenitors within microscale screening platforms are needed. With further development, we anticipate that the Raman spectroscopy and PLS-DA approach for noninvasive cell phenotype identification described herein might address this need.
The feasibility of using PLS-DA of Raman spectra to noninvasively screen the effects of 3D interactions on HSC fate decisions in vitro is suggested by the work reported herein and our previously reported use of this approach to noninvasively identify the differentiation stages of individual, living cells from the six most immature hematopoietic cell populations in 2D cultures.17 However, subsequent studies are needed to confirm the robustness of this approach. Future work must establish that the subtle spectral differences characteristic of the most immature hematopoietic cell populations are not masked by spectral features produced by the hydrogel matrix that they are embedded in. Traditional approaches such as immunolabeling and fluorescence microscopy or analysis of gene expression should also be used to confirm the accuracy of the differentiation stage assignments made by PLS-DA of Raman spectra. The development of a platform that is compatible with single-cell Raman spectroscopy and permits locating the same cell multiple times would facilitate such correlated analyses with complementary techniques, and could also enable tracking the fate decisions made by the same cell over time. Ultimately, PLS-DA of Raman spectra for noninvasive phenotype identification could reduce the cost of screening the effects of 3D interactions on stem cell fate decisions in microscale screening platforms and accelerate identification of the cues that elicit desired stem cell fate decisions in vitro.
Data availability
Data for this article in the form of the raw spectra are available in the Illinois Data Bank at DOI: 10.13012/B2IDB-5669414_V1.45Conflicts of interest
The authors declare no competing interests.Acknowledgements
Raman microspectroscopy and fluorescence microscopy was performed in the Microscopy Suite at the Beckman Institute for Advanced Science and Technology at UIUC. This work was partially supported by the National Institutes of Health under R21 HL132642 and funding from the Department of Chemical & Biomolecular Engineering at UIUC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors also acknowledge support from a 3M Corporate Fellowship (I. P.-O.) and an AT Widiger Fellowship (A. G.) from the Department of Chemical & Biomolecular Engineering at UIUC.References
- C. J. Flaim, S. Chien and S. N. Bhatia, Nat. Methods, 2005, 2, 119–125 CrossRef CAS PubMed.
- S. Gobaa, S. Hoehnel, M. Roccio, A. Negro, S. Kobel and M. P. Lutolf, Nat. Methods, 2011, 8, 949–955 CAS.
- G. H. Underhill and S. N. Bhatia, Curr. Opin. Chem. Biol., 2007, 11, 357–366 CAS.
- A. Roch, S. Giger, M. Girotra, V. Campos, N. Vannini, O. Naveiras, S. Gobaa and M. P. Lutolf, Nat. Commun., 2017, 8, 221 Search PubMed.
- S. Rangan, H. G. Schulze, M. Z. Vardaki, M. W. Blades, J. M. Piret and R. F. B. Turner, Analyst, 2020, 145, 2070–2105 CAS.
- Y. Ilin and M. L. Kraft, Curr. Opin. Biotechnol., 2015, 31, 108–116 CrossRef CAS PubMed.
- A. Germond, Y. Panina, M. Shiga, H. Niioka and T. M. Watanabe, Anal. Chem., 2020, 92, 14915–14923 CrossRef CAS PubMed.
- C. De Bleye, M. Fontaine, E. Dumont, P.-Y. Sacré, P. Hubert, N. Theys and E. Ziemons, J. Pharm. Biomed. Anal., 2020, 186, 113319 CrossRef CAS PubMed.
- T. Ichimura, L.-D. Chiu, K. Fujita, S. Kawata, T. M. Watanabe, T. Yanagida and H. Fujita, PLoS One, 2014, 9, e84478 CrossRef PubMed.
- C.-C. Hsu, J. Xu, B. Brinkhof, H. Wang, Z. Cui, W. E. Huang and H. Ye, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 18412–18423 CrossRef CAS PubMed.
- S. O. Konorov, H. G. Schulze, B. K. Gage, T. J. Kieffer, J. M. Piret, M. W. Blades and R. F. B. Turner, Anal. Chem., 2015, 87, 10762–10769 CrossRef CAS PubMed.
- N. Pavillon, A. J. Hobro, S. Akira and N. I. Smith, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E2676–E2685 CrossRef PubMed.
- H. G. Schulze, S. O. Konorov, N. J. Caron, J. M. Piret, M. W. Blades and R. F. B. Turner, Anal. Chem., 2010, 82, 5020–5027 CrossRef CAS PubMed.
- Y. Ilin, J. S. Choi, B. A. C. Harley and M. L. Kraft, Anal. Chem., 2015, 87, 11317–11324 CrossRef CAS PubMed.
- S. Majumdar and M. L. Kraft, Biointerphases, 2020, 15, 041010 CrossRef CAS PubMed.
- I. Pastrana-Otero, S. Majumdar, A. E. Gilchrist, B. L. Gorman, B. A. C. Harley and M. L. Kraft, Analyst, 2020, 145, 7030–7039 RSC.
- I. Pastrana-Otero, S. Majumdar, A. E. Gilchrist, B. A. C. Harley and M. L. Kraft, Anal. Chem., 2022, 94, 11999–12007 CrossRef CAS PubMed.
- Y. Ilin and M. L. Kraft, Analyst, 2014, 139, 2177–2185 RSC.
- J. S. Choi, Y. Ilin, M. L. Kraft and B. A. C. Harley, Bioconjugate Chem., 2018, 29, 3121–3128 CrossRef CAS PubMed.
- V. Charwat, K. Schütze, W. Holnthoner, A. Lavrentieva, R. Gangnus, P. Hofbauer, C. Hoffmann, B. Angres and C. Kasper, J. Biotechnol., 2015, 205, 70–81 CrossRef CAS PubMed.
- N. S. Hwang, S. Varghese and J. Elisseeff, Adv. Drug Delivery Rev., 2008, 60, 199–214 CrossRef CAS PubMed.
- D. R. Albrecht, G. H. Underhill, T. B. Wassermann, R. L. Sah and S. N. Bhatia, Nat. Methods, 2006, 3, 369–375 CrossRef CAS PubMed.
- A. A. Chen, G. H. Underhill and S. N. Bhatia, Integr. Biol., 2010, 2, 517–527 CrossRef CAS PubMed.
- C. Kallepitis, M. S. Bergholt, M. M. Mazo, V. Leonardo, S. C. Skaalure, S. A. Maynard and M. M. Stevens, Nat. Commun., 2017, 8, 14843 CrossRef CAS PubMed.
- J. Marzi, E. Fuhrmann, E. Brauchle, V. Singer, J. Pfannstiel, I. Schmidt and H. Hartmann, ACS Appl. Mater. Interfaces, 2022, 14, 30455–30465 CrossRef CAS PubMed.
- V. Korzhikov-Vlakh and I. Pepelanova, in Basic Concepts on 3D Cell Culture, ed. C. Kasper, D. Egger and A. Lavrentieva, Springer International Publishing, Cham, 2021, ch 4, pp. 79–104, DOI:10.1007/978-3-030-66749-8_4.
- A. E. Gilchrist, S. Lee, Y. Hu and B. A. C. Harley, Adv. Healthcare Mater., 2019, 8, 1900751 CrossRef CAS PubMed.
- J.-W. E. Chen, S. Pedron and B. A. C. Harley, Macromol. Biosci., 2017, 17, 1700018 CrossRef PubMed.
- B. P. Mahadik, S. Pedron Haba, L. J. Skertich and B. A. C. Harley, Biomaterials, 2015, 67, 297–307 CrossRef CAS PubMed.
- M. Sun, X. Sun, Z. Wang, S. Guo, G. Yu and H. Yang, Polymers, 2018, 10, 1290 CrossRef PubMed.
- E. W. Baxter, A. E. Graham, N. A. Re, I. M. Carr, J. I. Robinson, S. L. Mackie and A. W. Morgan, J. Immunol. Methods, 2020, 478, 112721 CrossRef CAS PubMed.
- H. Bosshart and M. Heinzelmann, Ann. Transl. Med., 2016, 4, 438 CrossRef PubMed.
- P. Italiani and D. Boraschi, Front. Immunol., 2014, 5, 514 Search PubMed.
- B. P. Mahadik, N. A. K. Bharadwaj, R. H. Ewoldt and B. A. C. Harley, Biomaterials, 2017, 125, 54–64 CrossRef CAS PubMed.
- B. D. Beier and A. J. Berger, Analyst, 2009, 134, 1198–1202 RSC.
- J. De Gelder, K. De Gussem, P. Vandenabeele and L. Moens, J. Raman Spectrosc., 2007, 38, 1133–1147 CrossRef CAS.
- J. S. Choi and B. A. C. Harley, Sci. Adv., 2017, 3, e1600455 CrossRef PubMed.
- M. Daigneault, J. A. Preston, H. M. Marriott, M. K. B. Whyte and D. H. Dockrell, PLoS One, 2010, 5, e8668 CrossRef PubMed.
- B.-H. Cha, S. R. Shin, J. Leijten, Y.-C. Li, S. Singh, J. C. Liu, N. Annabi, R. Abdi, M. R. Dokmeci, N. E. Vrana, A. M. Ghaemmaghami and A. Khademhosseini, Adv. Healthcare Mater., 2017, 6, 1700289 CrossRef PubMed.
- P. J. Murray and T. A. Wynn, Nat. Rev. Immunol., 2011, 11, 723–737 CrossRef CAS PubMed.
- P. Bouzy, S. O'Grady, H. Madupalli, M. Tecklenburg, K. Rogers, F. Palombo, M. P. Morgan and N. Stone, Lab. Invest., 2021, 101, 1267–1280 CrossRef CAS PubMed.
- A. Bonetti, A. Bonifacio, A. Della Mora, U. Livi, M. Marchini and F. Ortolani, Eur. J. Histochem., 2015, 59, 93–97 CAS.
- A. C. Wilkinson, R. Ishida, H. Nakauchi and S. Yamazaki, Nat. Protoc., 2020, 15, 628–648 CrossRef CAS PubMed.
- B. S. Monika, S. L. Lalita and P. K. Vaijayanti, Haematologica, 2012, 97, 651–660 CrossRef PubMed.
- I. Pastrana-Otero, A. R. Godbole and M. L. Kraft, Data for Noninvasive and In Situ Identification of the Phenotypes and Differentiation Stages of Individual Living Cells Entrapped Within Hydrogels, University of Illinois Urbana-Champaign, 2025, DOI:10.13012/B2IDB-5669414_V1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00800f |
| This journal is © The Royal Society of Chemistry 2025 |