
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diesel production via standalone and co-hydrotreating of catalytic fast pyrolysis oil†
Xiaolin
Chen
 a,
Kellene A.
Orton
a,
Calvin
Mukarakate
a,
Luke
Tuxworth
b,
Michael B.
Griffin
a and
Kristiina
Iisa
*a
a,
Kellene A.
Orton
a,
Calvin
Mukarakate
a,
Luke
Tuxworth
b,
Michael B.
Griffin
a and
Kristiina
Iisa
*a
aNational Renewable Energy Laboratory, Golden, CO, USA. E-mail: kristiina.iisa@nrel.gov
bJohnson Matthey Technology Center, Billingham, Cleveland, UK
First published on 1st May 2024
Abstract
Hydrotreating catalytic fast pyrolysis (CFP) oil is a promising technology for producing diesel fuel from lignocellulosic biomass to reduce greenhouse gas emissions. Compared to fast pyrolysis oil, CFP oils exhibit a low oxygen content and high stability and can be processed in a single hydrotreater. Two strategies were evaluated for biogenic carbon-incorporation into the diesel production: co-hydrotreating of CFP oil with straight run diesel (SRD) and standalone hydrotreating of CFP oil. Co-hydrotreating 80 vol% SRD and 20 vol% CFP oil at typical conditions of SRD hydrotreating employed at petroleum refineries (325 °C, 55 bar) over sulfided NiMo and CoMo catalysts led to products with desirable fuel properties, including indicated cetane numbers (ICNs) of 42–45. While oxygen was efficiently removed from products by co-hydrotreating over both catalysts, NiMo resulted in a higher formation of cycloalkanes and a higher ICN. Over 90% of the carbon in CFP oil was incorporated into the hydrotreated product as determined by C-14 analysis. Standalone hydrotreating of CFP oil was performed at 385 °C, 125 bar over NiMo in a single hydrotreating reactor with a two-zone configuration consisting of an initial zone from 150 °C to the hydrotreating temperature followed by an isothermal zone. Compared to previously reported isothermal hydrotreating, the new hydrotreating configuration resulted in a similar carbon efficiency but a higher fraction (95 vs. 84 wt%) boiling in the fuel range and significantly improved fuel properties (cetane number of 45 vs. 24). Both co-hydrotreating and standalone hydrotreating of CFP oil strategies showed great potential to produce sustainable diesel fuel with properties meeting standard diesel specifications in an existing petroleum refinery and a customized biorefinery, respectively.
1. Introduction
Approximately 47 billion gallons of petroleum-derived diesel were consumed by the U.S. transportation sector in 2021, which resulted in 472 million metric tons of CO2 emissions.1 This amount was equal to 26% of total U.S. transportation sector CO2 emissions and equal to 10% of total U.S. energy-related CO2 emissions in 2021. While electrification has become a promising replacement for gasoline utilization in the light-duty vehicle sector, petroleum-derived diesel is still dominant in heavy-duty transportation applications (e.g., rail, trucking, and marine).2–4 It is urgent to develop solutions to reduce petroleum diesel-derived CO2 emissions. Petroleum diesel substitutes (e.g., traditional biodiesel and green diesel) are produced from organic lipid feedstocks, namely fats, oils, and greases (FOG).5 However, there is a limited supply of FOG and using it as a feedstock for fuel products contributes to the competition between food and energy. In comparison, lignocellulosic biomass is a better feedstock for diesel production due to its higher abundance and domestic availability and no risks to food security.6,7Catalytic fast pyrolysis (CFP) of lignocellulosic biomass is a promising approach to produce a stabilized low-oxygen bio-oil, which can be used as a fuel precursor.8–10 A variety of catalysts have been applied to CFP. While zeolites (e.g., HZSM-5) are effective in producing aromatic hydrocarbons from biomass pyrolysis vapors, bifunctional metal-acid catalysts (e.g., Pt/TiO2) with co-fed H2 can improve carbon efficiencies and reduce coke formation.11–17 The Pt sites in Pt/TiO2 activate hydrogen for hydrogenation or hydrodeoxygenation (HDO) reactions, and the Lewis acid sites catalyze dehydration or C–C coupling reactions.15 Pt/TiO2 catalyst enables the hydrogenation of coke precursors to inhibit coke formation and thus achieve high carbon yields in the CFP oil.13,18 Advances in the CFP technology for making sustainable transportation fuels are presented in a recent perspective.19
CFP oils need to be further deoxygenated and upgraded to fuels compatible with current infrastructure, and this can be accomplished in a process similar to petroleum hydroprocessing.20 Compared to non-catalytic fast pyrolysis, CFP can produce higher quality bio-oil with lower contents of oxygenates, including acids and reactive components such as aldehydes, and thus enable single-stage hydroprocessing to further remove the residual oxygen. Sulfided NiMo and CoMo on alumina are proven bifunctional catalysts for heavy oil upgrading, where the metals (i.e., Ni and Co) are responsible for hydrogenation and heteroatom removal, and the alumina support contributes to hydrocracking.21,22 Sulfided CoMo and NiMo have both demonstrated successful hydrotreating of CFP oils to a product with less than 1 wt% of oxygen in single-stage continuous fixed-bed reactors operated under industrially relevant conditions.13,23 CFP oil could be processed either in dedicated biorefineries via standalone hydrotreating or by co-hydrotreating with petroleum streams in existing petroleum refineries. Co-hydrotreating CFP oil with petroleum streams has potential for incorporating biogenic carbon into the existing petroleum refinery infrastructure and capital cost savings compared to standalone hydrotreating at a biorefinery. However, co-hydrotreating operating conditions are constrained to avoid interfering with the original petroleum production at the refinery. Otherwise, it could bring technical risks to the refinery, such as inferior fuel properties, plugging and fouling, and corrosion.24,25 Despite standalone CFP oil hydrotreating requiring a higher capital investment, it offers a greater flexibility for optimizing fuel production through customization of process design, catalyst, and operating conditions. It also reduces the risk of interfering with petroleum refinery operations. A recent technoeconomic and lifecycle analysis compared conceptual processes of co-hydrotreating CFP oil at a petroleum refinery and standalone hydrotreating in a biorefinery based on experimental bench-scale results.26 Co-hydrotreating CFP oil showed an economic advantage; however, greenhouse gas emissions were significantly lower for standalone hydrotreating, mainly because co-hydrotreating utilizes fossil-derived hydrogen, whereas standalone hydrotreating uses hydrogen derived from reforming of the CFP off-gases.
Diesel in petroleum refineries is processed via hydrotreating of straight-run diesel (SRD), which is directly produced from the petroleum distillation column.27 Typically, the commercial process of SRD hydrotreating is conducted at a temperature range of 330–350 °C, a pressure range of 41–62 bar, and liquid hourly space velocities of 1–2.5 h−1 to produce diesel with a good cetane number.28 These conditions are much less severe than those used for standalone hydrotreating of CFP oil to produce low-oxygen products (∼400 °C, >100 bar, LHSV ∼0.2 h−1).13,23,29 Less severe conditions have led to higher and increasing oxygen contents as a function of time on stream for standalone hydrotreating, and the lower severity conditions for co-hydrotreating may pose a challenge in particular with high fractions of CFP.30,31 Previously, bench-scale co-hydrotreating 90 vol% SRD and 10 vol% CFP oil was conducted at 340 °C, 83 bar over sulfided NiMo and achieved 95% biogenic carbon incorporation into the fuel product, product oxygen content below detection limit (<0.5 wt%), and an ICN of 50 for the diesel fraction.18
Standalone hydrotreating of CFP oil can produce a highly oxygenated diesel-range product, but the reported cetane numbers are low, 13–26 vs. the minimum U.S. requirement for on-road vehicles of 40.13,20 Aromatic hydrocarbons constituted over 30% of the identified compounds in the hydrotreated products. Aromatics have lower cetane numbers than the corresponding cycloalkanes, e.g., 22 for naphthalene vs. 42–48 for decahydronaphthalene and 16 for propylbenzene vs. 52 for propylcyclohexane.32 We hypothesize that the cetane number can be improved by hydrogenation where alkenes and aromatics are saturated into alkanes and cycloalkanes. In addition, deoxygenation can also contribute to cetane number improvement where oxygen is eliminated with the formation of the corresponding hydrocarbons.33,34 Therefore, it is necessary to enhance hydrogenation and deoxygenation during hydrotreating CFP oil to obtain an acceptable cetane number for the diesel fuel product.
The objective of this contribution is to evaluate the potential for high-quality diesel products with acceptable cetane numbers from lignocellulosic biomass using both co-hydrotreating CFP oil with SRD and standalone hydrotreating of CFP oil. The CFP oil was produced over a bifunctional metal-acid catalyst (Pt/TiO2), and we investigated hydrotreating over industrially relevant sulfided hydrotreating catalysts: NiMo and CoMo on alumina support. To the best of our knowledge, this contribution is the first to show diesel-range product with an acceptable cetane number from standalone hydrotreating of CFP oil. Compared to prior literature, co-hydrotreating was expanded to cover a higher co-hydrotreating level of 20%, which enables higher biogenic carbon incorporation in the product.18 In addition, we compared the performance of sulfided NiMo and CoMo catalysts for co-hydrotreating and compared co-hydrotreating to standalone hydrotreating of CFP oil at the same temperature and pressure. This work will bring insights to producing diesel with acceptable cetane numbers through either standalone hydrotreating of lignocellulosic biomass-derived CFP oil in a customized biorefinery or co-hydrotreating with SRD in an existing petroleum refinery. These experiments of short duration did not allow us to evaluate catalyst deactivation.
2. Experimental
2.1 Materials
The CFP oil was produced from pine and pine residues over 0.5 wt% Pt/TiO2 in an ex situ fluidized bed-fixed bed combination with co-fed H2 at atmospheric pressure in the 2-inch Fluidized Bed Reactor (2FBR) system at the National Renewable Energy Laboratory (NREL).13,16,18 The biomass feedstocks were supplied by Idaho National Laboratory (INL) and the ultimate analysis is given in Table S1 (ESI†). In the 2FBR, biomass was fed into the fluidized bed pyrolysis reactor after which the pyrolysis vapors were deoxygenated over Pt/TiO2 in the fixed bed upgrading reactor prior to condensing into CFP oil. The pyrolysis temperature was 500–525 °C, and the upgrading temperature was 400 °C. The Pt/TiO2 catalyst was prepared via strong electrostatic adsorption technique on a TiO2 support provided by Johnson Matthey.13 The SRD was provided by ExxonMobil.The catalysts used for hydrotreating were NiMo on Al2O3 and CoMo on Al2O3 provided by Johnson Matthey. SiC from Panadyne (Green Silicon Carbide, 24 grit, average particle size 686 μm) was used to dilute the catalyst in some experiments.
2.2 Hydrotreating experiments
Hydrotreating experiments were conducted in a continuous trickle-bed hydrotreating system, which consisted of five sections: gas feeding and compression, liquid feeding, catalytic hydrotreating, liquid product collection, and gas product analysis (Fig. 1). H2 with 150 ppm H2S from a custom gas cylinder was compressed and stored in a gas accumulator, from which it was fed to the reactor via a mass flow controller. The liquids were fed to the reactor by two 500-mL ISCO syringe pumps. The combination of the liquid feed(s) and the gas feed were fed to the top of the reactor via a syringe tip and flowed through the catalyst bed. The reactor was a tube-in-tube heat exchanger using compressed air as a working fluid heated by a four-zone electric 2000-W furnace. The reactor was 122 cm tall with an inner diameter of 8.4 mm, and it was equipped with six thermocouples along the centerline. The outside diameter of the thermocouple well was 3.2 mm, and the catalyst was placed in the 2.7-mm wide annulus surrounding the thermocouple well (see insert in Fig. 1). The purpose of the narrow width of the catalyst layer and the enhanced heat transfer provided by the heat transfer fluid was to ensure isothermal operation. The highest exotherms measured were ∼1 °C. The working fluid could also ensure cooling in case of a run-away reaction.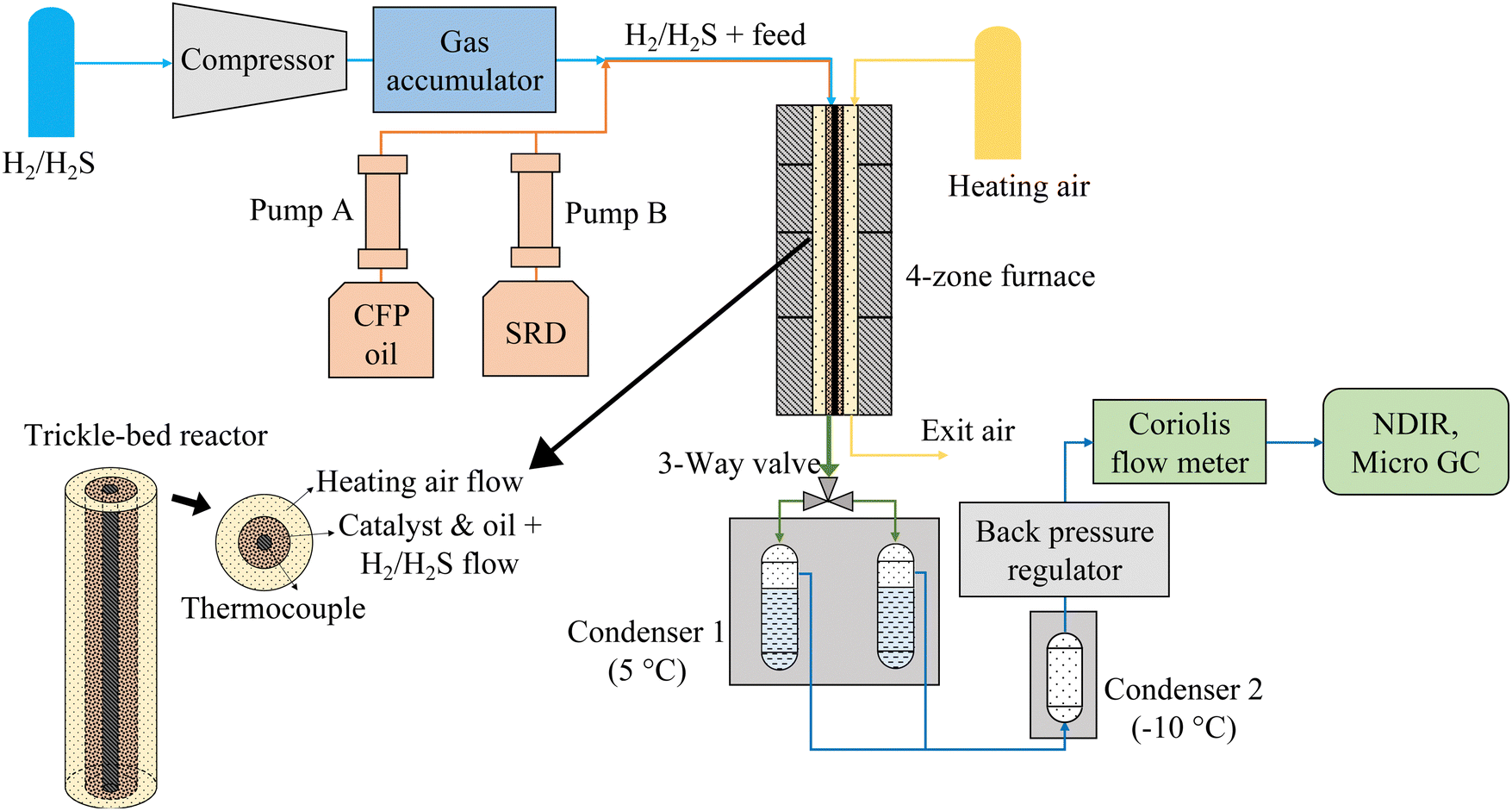 | ||
| Fig. 1 Continuous hydrotreater system with co-current heating air flow. For counter-current heating air flow, the air flow direction was reversed. | ||
The liquid product was collected in two alternating 150-mL collection vessels chilled in a 5 °C bath followed by a secondary −10 °C condenser used to fully condense any residual liquids in the gas flow (<10 mL was collected in the second condenser). The exit gas flow passing through a back pressure regulator was measured by a Coriolis flow meter and the exit gas composition was measured by a micro-GC for C1–C5 hydrocarbons, CO, CO2, and H2. The gases were additionally monitored for CO, CO2, and CH4 by NDIR analyzers. The experiments involve flammable and toxic gases and high pressures and temperatures. Safety precautions are included in the ESI.†
The catalyst was loaded into the reactor in its oxide form. At the beginning of each experiment, the catalyst was presulfided in situ in the reactor with a sulfiding agent (i.e., 35 wt% di-tert-butyl disulfide in decane). The catalyst was initially heated to 150 °C at a ramp rate of 2.5 °C min−1 in a flow of 150 standard mL min−1 (smL min−1) of the H2/H2S gas, after which the sulfiding liquid flow was started at 0.05 mL min−1 by an HPLC pump. The catalyst temperature was held at 150 °C for 2 h, then heated to the desired temperature (325–385 °C) at a ramp rate of 1.5 °C min−1 and held at the set temperature for another 4 h, all at the reaction pressure and with 100–125 smL min−1 of H2/H2S gas. The 150 ppm of H2S in H2 during hydrotreating process was used to maintain the activation of the catalyst. After catalyst presulfidation, CFP oil and SRD were fed from pump A and pump B, respectively, which allowed either standalone hydrotreating of one feed or co-hydrotreating of both feeds.
For co-hydrotreating experiments, the reactor was heated by a counter-current air flow to obtain an isothermal temperature profile at the top of the reactor (Fig. 2(a)). The temperature was 325 °C, pressure 55 bar with a WHSV of 1 h−1 and an H2:liquid ratio of 600 sL L−1. Standalone SRD hydrotreating was conducted first, followed by co-hydrotreating of 80 vol% SRD and 20 vol% CFP oil over the same catalyst bed for two catalysts: sulfided NiMo and CoMo. Standalone CFP oil hydrotreating was conducted over sulfided NiMo at the same temperature and pressure (325 °C, 55 bar) as a comparison with a fresh catalyst bed. However, the WHSV was lower at 0.4 h−1 because a higher flow of CFP oil would have increased CFP oil polymerization and led to plugging above the catalyst bed in this configuration.
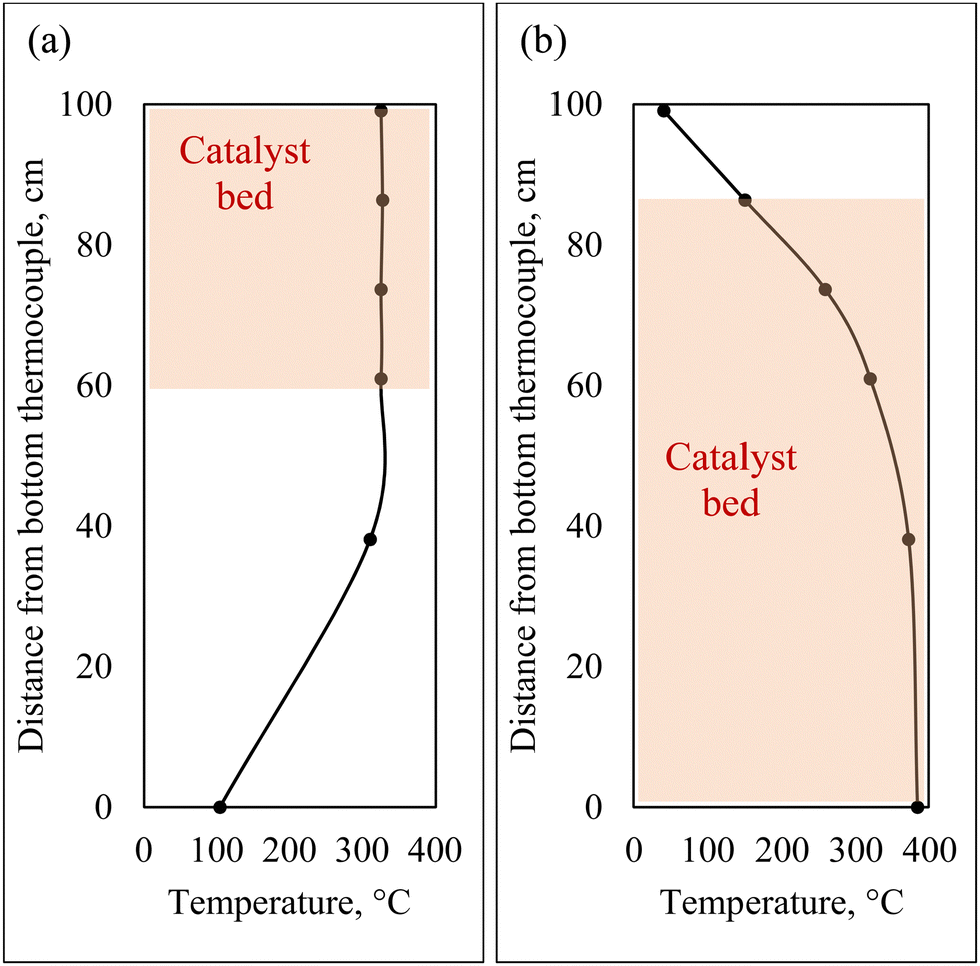 | ||
| Fig. 2 Hydrotreater reactor temperature profiles and catalyst bed locations: (a) counter-current air flow for co-hydrotreating, (b) co-current air flow for standalone hydrotreating. | ||
For standalone CFP oil hydrotreating in two zones, co-current air heating was utilized to obtain a low-temperature transition zone prior to an isothermal zone (Fig. 2(b)). CFP oil was hydrotreated at 385 °C in the isothermal zone, 125 bar with a total WHSV of 0.16 h−1 and an H2:liquid of 3750 sL L−1, over sulfided NiMo with a fresh catalyst bed. Standalone CFP oil hydrotreating to a fully deoxygenated product requires severe operating conditions: high temperature, high pressure, and low WHSV. The temperature and pressure of the standalone hydrotreating of CFP oil in this study were set to the maximum limits of the hydrotreater in this configuration, and the WHSV was selected based on prior literature results.13
The liquid products were collected approximately every 12 h and weighed. The liquid products collected from standalone CFP oil hydrotreating and co-hydrotreating of SRD and CFP oil consisted of an organic phase and an aqueous phase while the liquid product from standalone SRD hydrotreating only consisted of an organic phase. The liquid products with two phases were separated and both phases were weighed. For each 12-h product, mass yields of liquids (organic and aqueous phases) and each gas component were calculated. H2 consumption (g H2 g−1 CFP oil) was calculated based on the difference between inlet and outlet H2 mass flow divided by the CFP oil flow. Mass balance closures were evaluated based on oil feed and hydrogen consumption. More experimental details are given in the ESI.†
2.3 Liquid characterization
The hydrotreated oil product (organic phase), was analyzed for CHNS, direct oxygen and water by Karl–Fisher titration at Huffman-Hazen Laboratory (Golden, CO).35 The aqueous phase and the feed CFP oil and SRD were analyzed for CHN using a LECO Analyzer and direct sulfur by inductively coupled plasma-optical emission spectroscopy (ICP-OES) spectroscopy and water by Karl–Fischer titration (NREL/TP-5100-80968, ASTM D7544) at NREL. The total acid number (TAN) of the SRD was determined by ASTM D664 and the carboxylic acid number (CAN) of the CFP oil by modified ASTM D664.36 The molar concentration of carbonyls (aldehydes and ketones) was determined using ASTM E3146-20. The density was determined at 15 °C using ASTM D4052 on Mettler Toledo D4.Simulated distillation by ASTM D2887 was performed to estimate distribution of fuel fractions based on boiling point. The CFP oil was analyzed by GC–MS-FID, the hydrotreated products from co-hydrotreating by GC-VUV, and the diesel product from two-zone hydrotreating by GC × GC TOFMS-FID. See the ESI† for analytical details.
The product from standalone hydrotreating with two temperature zones was fractionated into gasoline and diesel fractions using a B/R 800 micro spinning band distillation system equipped with a metal band with fourteen theoretical plates. The hydrotreated oil and approximately 5 g of alumina boiling chips were loaded with into a 250 mL boiling flask with a thermocouple well. The low-temperature fraction was collected from 30 °C to 100 °C under atmospheric conditions. The distillation was then stopped and allowed to cool. 14 g of alumina boiling chips were added to the pot before starting the distillation again. The distillation column and pot were then brought to 30 Torr and heated. Fractions were collected from different temperature ranges of 100–115 °C, 115–130 °C, 130–145 °C, 145–245 °C, 245–260 °C, 260–300 °C, and 300–330 °C. All collected fractions were prepared and analyzed according to ASTM D2887. Using an in-house blending model, fractions were then blended together to obtain an ideal gasoline and diesel sample, in this case <145 °C for gasoline and 145–330 °C for diesel.
The fractions boiling in the diesel range were evaluated for fuel properties. ICN was measured by ASTM D8183 using the Advanced Fuel Ignition Delay Analyzer (AFIDA). The ICN is comparable to derived cetane number (DCN) measured by ASTM D6890 using a constant volume combustion chamber.37
The biogenic carbon content of the fuel products from co-hydrotreating was determined by Carbon-14 Analysis at Beta Analytic Inc. (Miami, FL).
2.4 Hydrotreating catalyst characterization
The catalysts were characterized for elemental composition by XRF, for metal areas by H2 chemisorption, for surface area and pore structure by N2 physisorption, and Hg porosimetry.For XRD, the finely ground samples were top loaded and pressed into an X-ray transparent sample holder and loaded into a Bruker D8 Advance powder diffractometer. The instrument was operated in a Bragg–Brentano (Reflection) mode using a copper X-ray tube operating at 40 KV and 40 mA with Göbel mirror optics and a 0.2 mm divergence slit. A diffraction pattern was collected over a 10–130° 2Theta range with a 0.02° step size and 1 second per step.
X-ray florescence (XRF) was carried out using Panalytical Axios WD XRF instrument. The samples were prepared by bead fusion of a powdered sample and analyzed on predefined, calibrated programs.
The surface areas were measured using a Micromeritics 2420 ASAP physisorption analyzer by application of the BET method in accordance with ASTM Method D 3663-03; Standard Test for Surface Area. Nitrogen was used as the adsorbate and the measurements carried out at liquid nitrogen temperature. The pore size distributions were derived using the BJH model from the adsorption branch of the isotherm.
Mercury intrusion/extrusion data was measured on a Micromeritics AutoPore 9600 mercury porosimeter in accordance with ASTM Method D4284-03; Test Method for Determining Pore Volume Distributions of Catalysts by Mercury Intrusion Porosimetry. Intrusion curves were measured over the pressure range of 0.5 to 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 psia followed by extrusion down to atmospheric pressure.
000 psia followed by extrusion down to atmospheric pressure.
Hydrogen chemisorption was measured on a Micromeritics HTP 6 Station Chemisorption Analyzer. The samples were reduced with 100% hydrogen at 650 °C for 120 minutes. After purging with helium, the sample is cooled under vacuum to 35 °C. At the analysis temperature the sample is dosed with 100% hydrogen over a range of pressures between 100 and 760 mmHg. At each pressure the chemisorbing hydrogen is allowed to equilibrate, and the volume of hydrogen uptake is measured.
3. Results and discussion
3.1 Hydrotreating catalyst characterization
The chosen hydrotreating catalysts, NiMo and CoMo on Al2O3, were characterized for physicochemical properties. All characterizations were performed on the oxidized forms of the catalysts before presulfidation. XRF analysis was conducted to determine the elemental composition of the catalysts. The catalysts contained 9–12 wt% of Mo and 3–4 wt% of Ni or Co (Table 1), giving similar Ni/Mo and Co/Mo mass ratios. CoMo/Al2O3 exhibited a higher BET area, a higher pore volume but a lower median pore diameter compared to NiMo/Al2O3 measured by N2-physisorption. The metal areas measured by H2-chemisoprtion were similar between both catalysts.| Catalyst | NiMo/Al2O3 | CoMo/Al2O3 |
|---|---|---|
| Elemental composition (wt%) by XRF | ||
| Co | <0.01 | 3.03 |
| Ni | 3.65 | <0.01 |
| Mo | 11.48 | 9.29 |
| Ni/Mo or Co/Mo, g g−1 | 0.32 | 0.33 |
| N2 physisorption | ||
| BET area, m2 g−1 | 170 | 207 |
| Pore volume, cm3 g−1 | 0.41 | 0.45 |
| Median pore diameter, Å | 97 | 86 |
| H2 chemisorption | ||
| Metal area [st 0] (m2 gcat−1) | 0.5 | 0.5 |
| Metal area [tot 0] (m2 gcat−1) | 1.1 | 1.0 |
| Mercury intrusion porosimetry | ||
| Corrected intrusion volume (cm3 g−1) | 0.43 | 0.47 |
| Entrapment (% v/v) | 100 | 100 |
| Median pore diameter (Å) | 113 | 100 |
The structural characterization of the catalysts was conducted by XRD, and the patterns indicate the catalyst samples are poorly crystalline but exhibit features which can be attributed to γ-Al2O3 with peaks around 2θ of 37°, 46°, and 67° as demonstrated in Fig. 3. There were no distinct peaks associated with crystalline Ni, Co or Mo oxides, which suggests no crystalline phases large enough (<3 nm) to be detected in the catalysts.
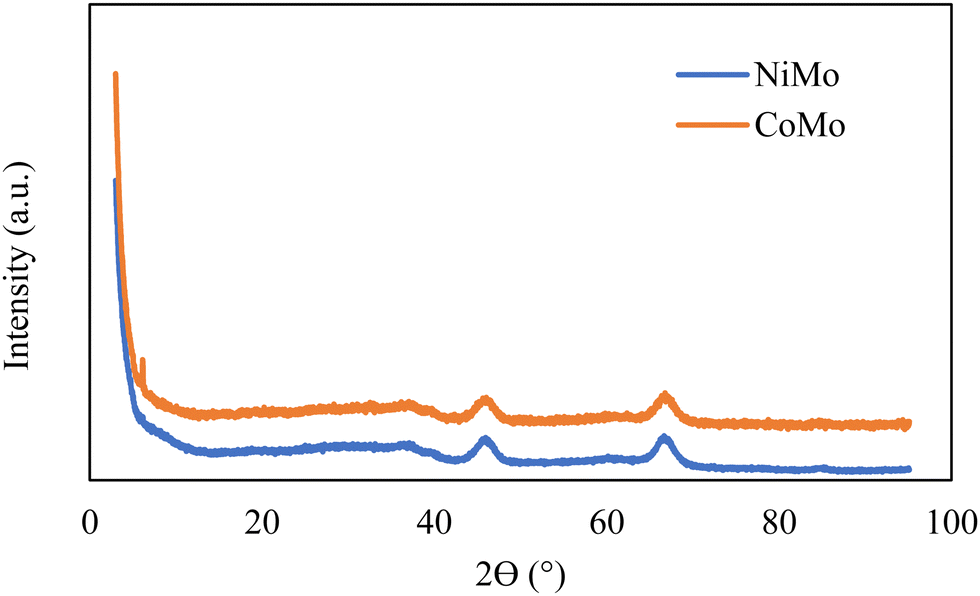 | ||
| Fig. 3 XRD results of hydrotreating catalysts. | ||
3.2 Co-hydrotreating SRD and CFP oil
The compositions of the CFP oil and the SRD are summarized in Table 2. The SRD mainly consisted of carbon and hydrogen, whereas the CFP oil contained 19.4 wt% of oxygen due to the presence of water and oxygenated organic compounds. In addition, the SRD contained 0.03 wt% of nitrogen and 0.2 wt% of sulfur, while the CFP oil contained 0.2 wt% of nitrogen and sulfur was below detection level. By the GC–MS analysis of the CFP oil, the oxygenated compounds included phenols, carbonyls, furans, carboxylic acids, and anhydrosugars, in accordance with previous results for this type of CFP oil.13,16 Carbonyls (i.e., ketones and aldehydes) have been indicated as potential factors causing catalyst bed plugging.38,39 The carbonyl concentration was 1.7 mol kg−1, which is lower than the limit of 2.5 mol kg−1 suggested for successful hydrotreating of pyrolysis-based oils in the literature.40 Carboxylic acids accounted for 1.4 wt% of the CFP oil, and the CAN was 39, which is significantly higher than the TAN for the SRD. More details on the CFP oil GC–MS analysis are given in the ESI.†| Elemental analysis (wet basis, wt%) | ||
|---|---|---|
| CFP oil | SRD | |
| a TAN for SRD, CAN for CFP oil. | ||
| C | 72.6 | 86.6 |
| H | 7.3 | 13.2 |
| O | 19.4 | ≤0.3 |
| N | 0.2 | 0.03 |
| S | <0.01 | 0.2 |
| H:C, mol mol−1 |
1.21 | 1.83 |
| H:Ceff = (H-2O)/C, mol mol−1 |
0.81 | 1.83 |
| H2O | 2.8 | <0.01 |
| Acid numbera, mg KOH g−1 | 39 | 0.05 |
| Carbonyls, mol kg−1 | 1.7 | <0.01 |
| % Modern carbon | 104.1 ± 0.3 | <0.44 |
| CFP oil GC–MS analysis (wt%) | |
|---|---|
| Phenols without methoxy groups | 11.1 |
| Methoxyphenols | 1.8 |
| Naphthols/indenols | 0.8 |
| Ketones | 6.1 |
| Aldehydes | 0.5 |
| Furans | 1.5 |
| Acids | 1.4 |
| Aromatic hydrocarbons | 0.1 |
| Sugars | 0.7 |
SRD hydrotreating and co-hydrotreating 80 vol% SRD and 20 vol% CFP oil were conducted at 325 °C, 55 bar over sulfided NiMo and CoMo. Due to the vastly different sizes of oil refineries and anticipated sizes of plants producing CFP oil, the blend ratio for co-hydroprocessing is expected to remain initially low and, therefore, we selected this blend ratio, which, however, is higher than in the previous study (i.e., 90 vol% SRD and 10 vol% CFP oil).18Table 3 summarizes the results from SRD hydrotreating and co-hydrotreating studies in this work. While SRD produced only an oil phase, the co-hydrotreated liquid products consisted of oil and aqueous phases, due to water formation as a result of hydrodeoxygenation reactions. Hydrodeoxygenation was efficient, and the co-hydrotreated products had oxygen contents below the reliable detection limit of the method. Co-hydrotreating increased gas formation and led to higher hydrogen consumptions compared to standalone SRD hydrotreating. This is expected since CFP oils are deficient in hydrogen as shown by a low effective hydrogen index of 0.81 compared to 1.83 for SRD (Table 2). Co-hydrotreating slightly increased the product sulfur and nitrogen contents, which suggests that removing oxygen from CFP oil may have inhibited the ability of the catalysts to remove nitrogen and sulfur. For co-hydrotreating, NiMo resulted in a higher product H:C ratio (1.82 vs. 1.79) and a higher hydrogen consumption (0.014 g H2 g−1 feed vs. 0.011 g H2 g−1 feed) than CoMo did. The higher H:C could imply a higher heating value and a higher cetane number. The density of the hydrotreated product was not influenced by co-hydrotreating or using different catalysts.
| Feed | SRD | SRD + CFP | CFP | SRD | SRD + CFP |
|---|---|---|---|---|---|
| Catalyst type | NiMo | NiMo | NiMo | CoMo | CoMo |
| Oil, g g−1 oil | 100% | 96% | 85% | 101% | 91% |
| Aqueous, g g−1 oil | — | 5.5% | 16% | — | 6.1% |
| Gas, g g−1 oil | 0.27% | 1.4% | 5.8% | 0.03% | 1.5% |
| H2 consumption, g g−1 oil | 0.24% | 1.4% | 3.6% | 0.23% | 1.1% |
| Mass balance, g g−1 (oil + H2 consumed) | 101% | 101% | 102% | 101% | 98% |
| Oil, g C per g C in oil | 100% | 100% | 95% | 101% | 95% |
| Gas, g C per g C in oil | 0.30% | 1.3% | 5.8% | 0.03% | 1.4% |
| Carbon balance, g C per g C in oil | 100% | 101% | 101% | 101% | 96% |
| C, wt% | 86.34 | 86.98 | 81.44 | 87.01 | 86.77 |
| H, wt% | 13.39 | 13.22 | 10.32 | 13.48 | 12.95 |
| O, wt% | ≤0.3 | ≤0.3 | 8.24 | ≤0.3 | ≤0.3 |
| N, wt% | 0.03 | 0.04 | <0.01 | 0.02 | 0.04 |
| S, wt% | 0.01 | 0.03 | <0.01 | 0.02 | 0.04 |
| H:C, mol:mol |
1.86 | 1.82 | 1.52 | 1.86 | 1.79 |
| Density, g mL−1 | 0.83 | 0.83 | 0.94 | 0.83 | 0.83 |
| Modern carbon, % | <0.44 | 19.6 ± 0.1 | 85 | <0.44 | 19.0 ± 0.1 |
| ICN | 50 | 45 | 16 | 48 | 42 |
The chemical composition of the hydrotreated products by GC-VUV analysis are given in Fig. 4(a). Here, aromatics include partially hydrogenated aromatics, i.e., cycloalkenes. Compared to standalone SRD hydrotreating, co-hydrotreating enhanced the formation of aromatic hydrocarbons and cycloalkanes. This is consistent with the highly aromatic nature of the CFP oil and the results for standalone hydrotreating of CFP oil, which produced mainly cycloalkanes, aromatic hydrocarbons, and phenols. For both SRD hydrotreating and co-hydrotreating, NiMo resulted in higher cycloalkane and lower aromatics contents, compared to CoMo. This indicates that NiMo can enhance hydrogenation in a more significant way where aromatics are converted to cycloalkanes. The enhanced hydrogenation over NiMo was also indicated by the aforementioned higher hydrogen consumption and higher product H:C over NiMo than over CoMo during co-hydrotreating (Table 3). The differences between the catalysts are as expected based on literature results and catalyst properties. The higher hydrogenation activity of NiMo, compared to CoMo, is well known,21,41 and for example in a recent comparison of NiMo and CoMo on alumina support for aromatics hydrogenation, the Ni metal promoter enhanced the hydrogenating activity of Mo more than Co did.22 In addition, co-hydrotreating over NiMo resulted in a lower phenolics content than co-hydrotreating over CoMo did, in accordance with previous studies showing that NiMo exhibited a higher deoxygenation functionality than CoMo did.42
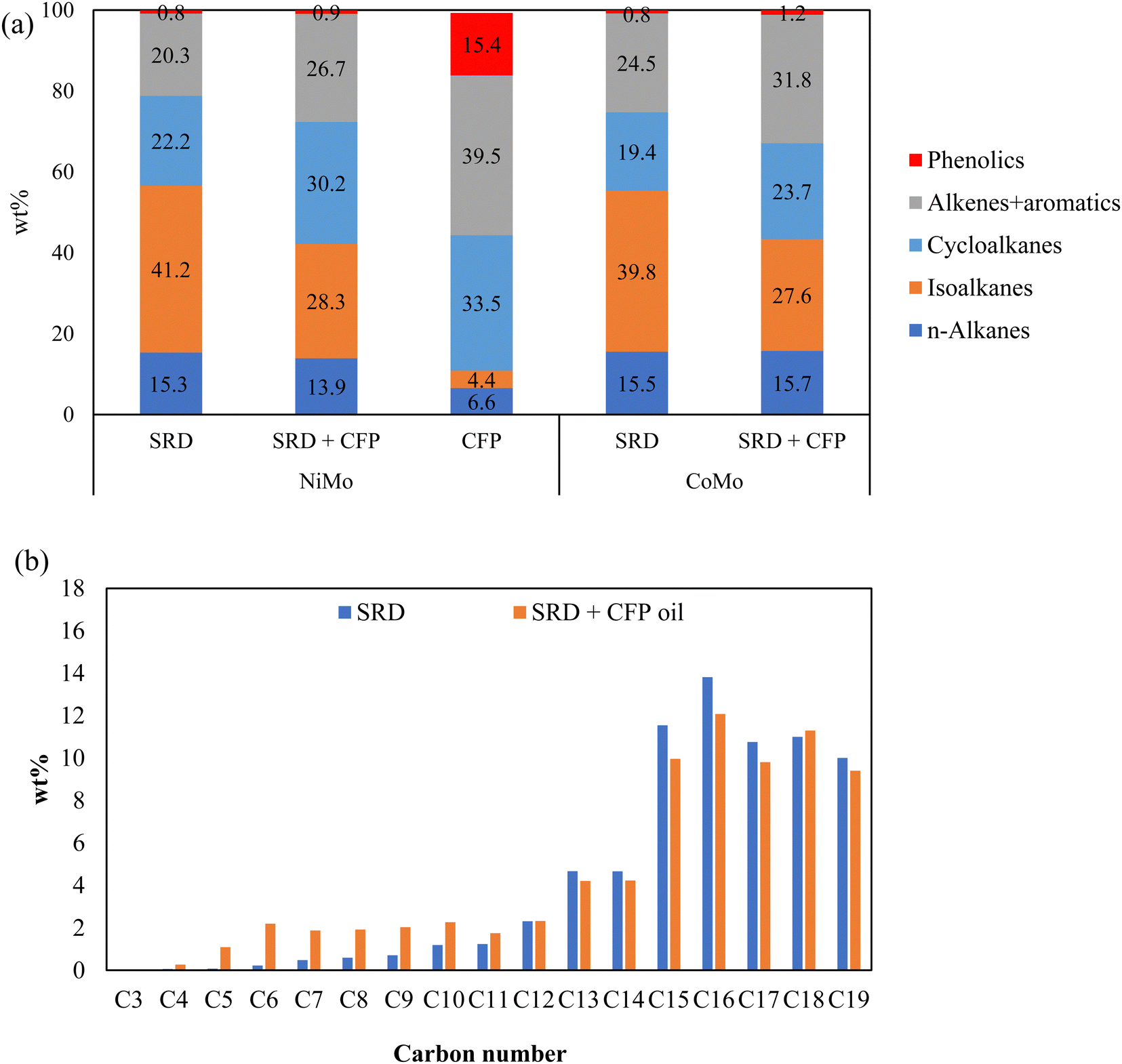 | ||
| Fig. 4 (a) GC-VUV analysis of oil products from hydrotreating at 325 °C, 55 bar over sulfided NiMo/sulfided CoMo; (b) carbon number distribution of oil products from SRD hydrotreating and co-hydrotreating of SRD and CFP oil over sulfided NiMo. | ||
The carbon number distributions of the hydrotreated products over NiMo by GC-VUV analysis are given in Fig. 4(b). Compared to SRD hydrotreating, co-hydrotreating significantly increased the formation of C4–C11 molecules. This is consistent with the simulated distillation results in Fig. 5. Co-hydrotreating increased the fraction of low-boiling compounds (<200 °C) and also slightly enhanced the formation of residual compounds (>400 °C) vs. SRD hydrotreating. The low-boiling components are attributed to sugar-derived molecules (e.g., acetone, furans, cyclopentenones, see Table S3, ESI†) in the CFP oil and the residue to oligomers formed from polymerization of oxygenates at high temperatures. The volatility of the products was not impacted by the choice of the catalyst.
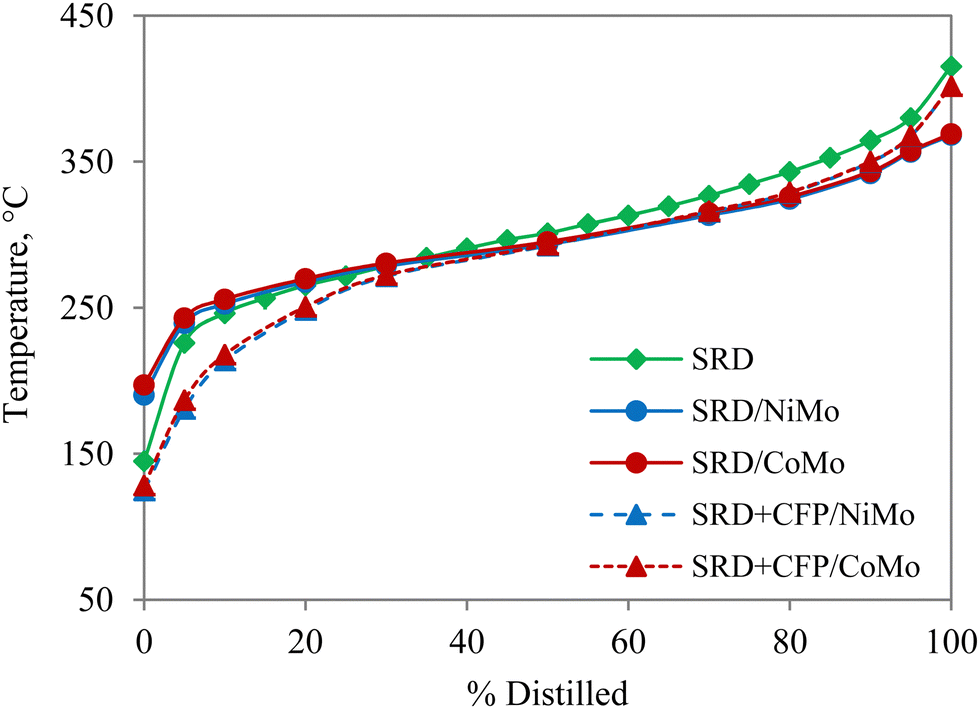 | ||
| Fig. 5 Simulated distillation of unreacted SRD, and hydrotreated products from SRD hydrotreating and co-hydrotreating of SRD and CFP oil in a 4:1 volumetric ratio over sulfided NiMo and CoMo. | ||
CFP oil was hydrotreated separately at the co-hydrotreating temperature and pressure of 325 °C and 55 bar (Table 3), which resulted in a product with a high oxygen content of 8 wt% and poor product quality, including an ICN of 16. The poorer performance, compared to hydrotreating of a similar CFP oil, which gave 0.4% oxygen and a cetane number of 24 is due to the lower hydrotreating temperature and pressure and a higher WHSV in the current study.13 The carbon yield, in contrast, was higher than in the previous study (95% vs. 89%) due to the milder operating conditions. The elemental composition (Table 3) and GC-VUV (Fig. 4) suggest better deoxygenation and hydrogenation of the CFP oil during co-hydrotreating than during standalone CFP oil hydrotreating. The better performance during co-hydrotreating may be due to a lower flow of oxygen heteroatoms per catalyst active site, compared to the experiment with CFP oil only. In addition, a possible synergistic effect between the SRD and the CFP oil, such as hydrogen transfer from the SRD to the CFP oil, could enhance hydrogenation and deoxygenation. The reason for the better performance should be ascertained by further studies.
We measured trace amounts of phenolics in the products from standalone hydrotreating of SRD, at levels comparable to those produced during co-hydrotreating of SRD and CFP oil (0.8 wt% vs. 0.9–1.2 wt%). This may be a misidentification by the analysis method although some phenols have been reported in petroleum diesel fuel by other researchers.43
Co-hydrotreating decreased ICN compared to SRD hydrotreating (Table 3); however, the ICNs of co-hydrotreated products were 42–45, which meet the US on-road vehicle specification with respect to cetane number (>40). Compared to CoMo, NiMo led to higher ICNs for both SRD hydrotreating and co-hydrotreating with CFP oil. Overall, between the two hydrotreating catalysts, NiMo was better at promoting hydrogenation and thus increasing the cetane number, which resulted in higher-quality diesel. NiMo has been similarly reported as a more suitable catalyst for co-hydroprocessing petroleum fractions and lipids.41
None of the products met the sulfur limit of 15 ppm for ultra-low sulfur diesel (ULSD) mandated for onroad vehicles by EPA since 2010.44 Co-hydrotreating increased product sulfur contents, compared to SRD hydrotreating (Table 3), which suggests that the presence of CFP oil may negatively impact the desulfurization ability of the catalyst employed. In order to meet the sulfur content requirement, a more effective catalyst and/or an optimized reactor configuration, e.g. deoxygenation followed by desulfurization, is required.
C-14 analysis was applied here to differentiate carbon atoms originating from SRD (millions of years old) and carbon atoms originating from biomass (less than 100 years old) and thus determine the biogenic carbon content of the biofuel (Table 3). The fraction of modern carbon exceeded 100% in the CFP oil and was below the detection limit for the SRD. Modern carbon values over 100% are possible in woody biomass because of higher 14C values in the atmosphere due to residues from nuclear bomb experiments.45 The co-hydrotreated products contained 19–20% of modern carbon, which suggests approximately 95% incorporation of the carbon in the CFP oil into the final product. This percentage (95%) is the same as previously reported for co-hydrotreating 10 vol% CFP oil with SRD.18 CFP oil carbon can, therefore, be incorporated at similar efficiencies at higher blend ratios, leading to a higher fraction of biogenic carbon in the product (19–20% vs. 9.5%) due to the higher blend ratio.
The biogenic carbon incorporation via hydroprocessing is higher than the values reported for co-processing via fluid catalytic cracking (FCC).46,47 For example, co-processing of catalytic fast pyrolysis oil with VGO in a Davidson circulating reactor suggested over 50% of biogenic carbon lost to light gases and coke.47 The high biogenic carbon incorporation is a significant advantage for hydroprocessing.
3.3 Two-zone standalone hydrotreating of CFP oil
Standalone CFP oil hydrotreating was conducted at 385 °C, 125 bar in a single hydrotreater with a two-zone hydrotreating process: a non-isothermal entrance zone where the temperature gradually increased from 150 °C to the final hydrotreating temperature followed by an isothermal zone at the final hydrotreating temperature. The temperature profile in the entrance zone was tailored to enhance hydrogenation and thus improve the diesel product cetane number. NiMo was chosen as the standalone hydrotreating catalyst due to its better hydrogenation performance. The hydrotreating performance and product properties are summarized in Table 4. The two-zone standalone CFP oil hydrotreating resulted in an oil carbon yield of 89%, which is the same as the reported value for isothermal hydrotreating.13 However, the hydrogen consumption in the two-zone process was higher (13 wt% vs. 4 wt%), as was the product H:C ratio (2.01 vs. 1.71).13 This indicates the two-zone configuration applied here resulted in a more complete hydrogenation compared to the reported isothermal hydrotreating process without additional loss of carbon.| Condition | 385 °C, 125 bar |
|---|---|
| Oil, g g−1 CFP oil | 75% ± 1% |
| Aqueous, g g−1 CFP oil | 25% ± 1% |
| Gas, g g−1 CFP oil | 9% ± 0.4% |
| H2 consumption, g g−1 CFP oil | 13% ± 1% |
| Mass balance, g g−1 (CFP oil + H2 consumed) | 97% ± 2% |
| Oil, g C per g C in CFP oil | 89% ± 1% |
| Gas, g C per g C in CFP oil | 10% ± 0.5% |
| Carbon balance, g C per g C in CFP oil | 99% ± 2% |
| C, wt% | 84.9 |
| H, wt% | 14.2 |
| O, wt% | ≤0.3 |
| N, wt% | <0.01 |
| S, wt% | <0.01 |
| H:C, mol:mol |
2.01 |
| Density, g mL−1 | 0.74 |
The CFP process produces a lower oxygen content, more stable bio-oil, compared to non-catalytic fast pyrolysis. Hydrotreating CFP oil can be performed at very high carbon efficiencies in both standalone and co-hydrotreating scenarios, 89–95% shown here (Tables 3 and 4). These values are significantly higher than those reported for hydrotreating of non-catalytic fast pyrolysis oils (53–77%).40,48 Because of the higher carbon efficiency of fast pyrolysis, compared to CFP, the overall carbon efficiency from biomass to hydrotreated product could be higher for fast pyrolysis than for CFP. However, due to the complex composition of oxygenates, hydrotreating of non-catalytic fast pyrolysis oil is challenging and requires multiple stages of hydrotreating to overcome the technical challenges, e.g., plugging and catalyst deactivation issues.
The product fractionation results, diesel fraction composition by GC × GC-TOFMS-FID, and diesel fraction cetane number are given in Table 5. 95 wt% of the hydrotreated product boiled in the fuel range, including 51 wt% in the gasoline range and 45 wt% in the diesel range. This represents an improvement over the reported total fuel range value of 84 wt% (45 wt% in the gasoline range, 39 wt% in the diesel range) for isothermal hydrotreating.13
| Distillation fractions, wt% | |
| Gasoline (<145 °C) | 50.9% |
| Diesel (145–330 °C) | 44.5% |
| Residue | 3.6% |
| Losses | 1.0% |
| Diesel GC × GC analysis, wt% | |
| n-Alkane | 4.9% |
| Isoalkane | 4.0% |
| Cycloalkane | 77.7% |
| Aromatic hydrocarbons | 0.8% |
| Unidentified | 0.1% |
| Diesel cetane number | |
| ICN | 45 |
Nearly 87 wt% of the compounds in the diesel fraction were identified by GC × GC-TOFMS-FID analysis. Cycloalkanes, which were the desired product, were the most abundant product group (78 wt%), aromatic hydrocarbons were low (0.8 wt%), and no oxygenates were detected. The high fraction of cycloalkanes is consistent with the high H:C ratio and the high hydrogen consumption. The GC × GC analysis showed a high abundance of C10 compounds (Fig. S2, ESI†), which consisted of cyclohexane derivatives, such as methyl-propyl- and butyl-cyclohexane, but also of decahydronaphthalene (Table S4, ESI†). The cyclohexanes are likely deoxygenated lignin monomer derivatives in which the aromatic ring has been hydrogenated.
The ICN of the diesel fraction was 45, which meets the US on-road vehicle specification with respect to cetane number (>40) and is significantly higher than the reported derived cetane number (DCN) of 24 from isothermal hydrotreating.13 Therefore, standalone hydrotreating of CFP oil in this two-zone configuration significantly enhanced hydrogenation and produced a diesel product with cycloalkanes as the main compound group and an ICN meeting the US on-road standard.
4. Conclusion
This contribution investigated diesel production through co-hydrotreating 80 vol% SRD and 20 vol% CFP oil and standalone hydrotreating of CFP oil. Co-hydrotreating was performed at a mild condition (325 °C, 55 bar) typical for industrial SRD hydrotreating over two different catalysts: sulfided NiMo and CoMo. NiMo was better at hydrogenating aromatics to cycloalkanes and thus resulted in a higher-quality co-hydrotreated product with a higher ICN of 45. Over 90% of biogenic carbon was incorporated from CFP oil into the co-hydrotreated product as determined by C-14 analysis. Possible synergy between SRD and CFP oil during co-hydrotreating enhanced hydrogenation and deoxygenation.Standalone CFP oil hydrotreating was conducted at 385 °C, 125 bar over sulfided NiMo in a two-zone configuration aimed at a more complete hydrogenation. Compared to isothermal hydrotreating in previous studies, standalone CFP oil hydrotreating in the two-zone process gave a similar carbon efficiency but significantly increased the formation of cycloalkanes through enhanced hydrogenation of aromatics in the initial low-temperature zone. The standalone hydrotreated product was fractionated into 49% of diesel and 45% of gasoline. The diesel fraction mainly consisted of cycloalkanes (78 wt%) with an ICN of 45, which exceeds the US on-road vehicle specification.
Co-hydrotreating and standalone hydrotreating strategies demonstrated for CFP oil in this contribution have brought insight into producing sustainable diesel fuel from lignocellulosic biomass-derived CFP oils either in existing refinery infrastructures or in customized biorefineries. To make the processes viable, research is required into the long-term performance of the hydrotreating process, including catalyst deactivation and the impact of impurities in the CFP oil, and optimization of hydrotreating conditions and catalysts. In the future, more types of sustainable heavy-duty transportation fuels such as marine and aviation fuel, with satisfactory fuel properties will be produced from lignocellulosic biomass using this integrated thermochemical converting technology, namely CFP and hydrotreating, to help reduce overall greenhouse gas emissions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was authored in part by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under contract no. DE-AC36-08GO28308. Funding provided by U.S. Department of Energy Office of Energy Efficiency and Renewable Energy Bioenergy Technologies Office. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes. The authors gratefully acknowledge the experimental contributions by Sean West, Andy Young, Alex Rein, Earl Christensen, Jon Luecke, Scott Palmer, and Dr Rick French.References
- U.S. Energy Information Administration (EIA), 2022, Diesel fuel explained, https://www.eia.gov/energyexplained/diesel-fuel/diesel-and-the-environment.php, (accessed April 25, 2023).
- X. Sun, Z. Li, X. Wang and C. Li, Energies, 2019, 13(1), 90 CrossRef.
- K. L. Fleming, A. L. Brown, L. Fulton and M. Miller, Curr. Sustainable/Renewable Energy Rep., 2021, 8(3), 180–188 CrossRef CAS.
- S. Paltsev, A. Ghandi, J. Morris and H. Chen, Econ. Energy Environ. Policy, 2022, 11(1), 165–191 CrossRef.
- P. Vignesh, A. R. Pradeep Kumar, N. S. Ganesh, V. Jayaseelan and K. Sudhakar, Oil Gas Sci. Technol., 2021, 76, 6 CrossRef.
- N. N. Deshavath, V. D. Veeranki and V. V. Goud, J. Sustainable Bioenergy Syst., 2019, 1–19 Search PubMed.
- S. L. Douvartzides, N. D. Charisiou, K. N. Papageridis and M. A. Goula, Energies, 2019, 12(5), 809 CrossRef CAS.
- K. Iisa, R. J. French, K. A. Orton, A. Dutta and J. A. Schaidle, Fuel, 2017, 207, 413–422 CrossRef CAS.
- R. J. French, K. A. Orton and K. Iisa, Energy Fuels, 2018, 32(12), 12577–12586 CrossRef CAS.
- C. A. Mullen and A. A. Boateng, Fuel, 2019, 245, 360–367 CrossRef CAS.
- T. Dickerson and J. Soria, Energies, 2013, 6(1), 514–538 CrossRef CAS.
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell III, J. Wang, L. Moens and J. E. Hensley, Green Chem., 2014, 16(2), 454–490 RSC.
- M. B. Griffin, K. Iisa, H. Wang, A. Dutta, K. A. Orton, R. J. French, D. M. Santosa, N. Wilson, E. Christensen, C. Nash, K. M. Van Allsburg, F. G. Baddour, D. A. Ruddy, E. C. Tan, H. Cai, C. Mukarakate and J. A. Schaidle, Energy Environ. Sci., 2018, 11(10), 2904–2918 RSC.
- J. Liang, G. Shan and Y. Sun, Renewable Sustainable Energy Rev., 2021, 139, 110707 CrossRef CAS.
- F. Lin, Y. Lu, K. A. Unocic, S. E. Habas, M. B. Griffin, J. A. Schaidle, H. M. Meyer, Y. Wang and H. Wang, ACS Catal., 2021, 12(1), 465–480 CrossRef.
- R. J. French, K. Iisa, K. A. Orton, M. B. Griffin, E. Christensen, S. Black, K. Brown, S. E. Palmer, J. A. Schaidle, C. Mukarakate and T. D. Foust, ACS Sustainable Chem. Eng., 2021, 9(3), 1235–1245 CrossRef CAS.
- C. Mukarakate, K. Iisa, S. E. Habas, K. A. Orton, M. Xu, C. Nash, Q. Wu, R. M. Happs, R. J. French, A. Kumar, E. M. Miller, M. R. Nimlos and J. A. Schaidle, Chem. Catal., 2022, 2(7), 1819–1831 CrossRef CAS.
- M. M. Yung, C. Mukarakate, K. Iisa, N. Wilson, M. R. Nimlos, S. E. Habas, A. Dutta, K. A. Unocic, J. A. Schaidle and M. B. Griffin, Green Chem., 2023, 25, 6809–6822 RSC.
- C. J. Wrasman, A. N. Wilson, O. D. Mante, K. Iisa, A. Dutta, M. S. Talmadge, D. C. Dayton, S. Uppili, M. J. Watson, X. Xu, M. B. Griffin, C. Mukarakate, J. A. Schaidle and M. R. Nimlos, Nat. Catal., 2023, 6(7), 563–573 CrossRef.
- K. Iisa, C. Mukarakate, R. J. French, F. A. Agblevor, D. M. Santosa, H. Wang, N. Wilson, E. Christensen, M. B. Griffin and J. A. Schaidle, Energy Fuels, 2023, 37, 19653–19663 CrossRef CAS PubMed.
- K. G. Knudsen, B. H. Cooper and H. Topsøe, Appl. Catal., A, 1999, 189, 205–215 CrossRef CAS.
- A. Hart, M. Adam, J. P. Robinson, S. P. Rigby and J. Wood, Catalysts, 2020, 10(5), 497 CrossRef CAS.
- F. A. Agblevor, D. C. Elliott, D. M. Santosa, M. V. Olarte, S. D. Burton, M. Swita, S. H. Beis, K. Christian and B. Sargent, Energy Fuels, 2016, 30(10), 7947–7958 CrossRef CAS.
- M. Al-Sabawi and J. Chen, Energy Fuels, 2012, 26(9), 5373–5399 CrossRef CAS.
- X. Han, H. Wang, Y. Zeng and J. Liu, Energy Convers. Manage.: X, 2021, 10, 100069 CAS.
- A. Dutta, H. Cai, M. S. Talmadge, C. Mukarakate, K. Iisa, H. Wang, D. M. Santosa, L. Ou, D. S. Hartley, A. Nolan Wilson, J. A. Schaidle and M. B. Griffin, Chem. Eng. J., 2023, 451, 138485 CrossRef CAS.
- C. Kochaphum, S. H. Gheewala and S. Vinitnantharat, J. Cleaner Prod., 2012, 37, 142–146 CrossRef.
- F. X. Zhu, R. Hoehn, V. Thakkar and E. Yuh, Hydroprocessing for Clean Energy: Design, Operation, and Optimization, American Institute of Chemical Engineers, New York, 2017 Search PubMed.
- D. M. Santosa, C. Zhu, F. A. Agblevor, B. Maddi, B. Q. Roberts, I. V. Kutnyakov, S.-J. Lee and H. Wang, ACS Sustainable Chem. Eng., 2020, 8(13), 5156–5164 CrossRef CAS.
- O. D. Mante, D. C. Dayton, J. Gabrielsen, N. L. Ammitzboll, D. Barbee, S. Verdier and K. Wang, Green Chem., 2016, 18(22), 6123–6135 RSC.
- S. Verdier, O. D. Mante, A. B. Hansen, K. G. Poulsen, J. H. Christensen, N. Ammtizboll, J. Gabrielsen and D. C. Dayton, Sustainable Energy Fuels, 2021, 5(18), 4668–4679 RSC.
- J. Yanowitz, M. A. Ratcliff, R. L. McCormick, J. D. Taylor and M. J. Murphy, 2017, Compendium of Experimental Cetane Numbers DOI:10.2172/1345058, (accessed April 8, 2024).
- J. A. Tilton, W. M. Smith and W. G. Hockberger, Ind. Eng. Chem., 1948, 40(7), 1269–1273 CrossRef CAS.
- R. Santana, P. Do, M. Santikunaporn, W. Alvarez, J. Taylor, E. Sughrue and D. Resasco, Fuel, 2006, 85(5–6), 643–656 CrossRef CAS.
- K. Iisa, R. J. French, K. A. Orton, M. M. Yung, D. K. Johnson, J. ten Dam, M. J. Watson and M. R. Nimlos, Energy Fuels, 2016, 30(3), 2144–2157 CrossRef CAS.
- E. Christensen, J. Ferrell, M. V. Olarte, A. B. Padmaperuma and T. Lemmon, 2016, Acid number determination of pyrolysis bio-oils using potentiometric titration, NREL/TP-5100-65890, March 2016, https://www.nrel.gov/docs/fy16osti/65890.pdf.
- R. L. McCormick, G. Fioroni, J. Luecke and S. Yellapantula, 2022, Measuring critical fuel properties for simulations to accelerate fuel qualification, https://www.osti.gov/servlets/purl/1887881, (accessed January 25, 2023).
- E. D. Christensen, G. M. Chupka, J. Luecke, T. Smurthwaite, T. L. Alleman, K. Iisa, J. A. Franz, D. C. Elliott and R. L. McCormick, Energy Fuels, 2011, 25(11), 5462–5471 CrossRef CAS.
- A. H. Zacher, M. V. Olarte, D. M. Santosa, D. C. Elliott and S. B. Jones, Green Chem., 2014, 16(2), 491–515 RSC.
- A. H. Zacher, D. C. Elliott, M. V. Olarte, D. M. Santosa, F. Preto and K. Iisa, Energy Fuels, 2014, 28, 7510–7516 CrossRef CAS.
- S. Bezergianni, A. Dimitriadis and G. Meletidis, Fuel, 2014, 125, 129–136 CrossRef CAS.
- N. Priharto, F. Ronsse, W. Prins, I. Hita, P. J. Deuss and H. J. Heeres, Biomass Bioenergy, 2019, 126, 84–93 CrossRef CAS.
- N. Kolbe, O. V. Rheinberg and J. T. Andersson, Energy Fuels, 2009, 23(6), 3024–3031 CrossRef CAS.
- U. S. Environmental Protection Agency (EPA), 2023, Overview of Diesel Standards, https://www.epa.gov/diesel-fuel-standards/diesel-fuel-standards-and-rulemakings, (accessed April 25, 2023).
- P. J. Reimer, T. A. Brown and R. W. Reimer, Radiocarbon, 2004, 46(3), 1299–1304 CrossRef CAS.
- S. Dell’Orco, E. D. Christensen, K. Iisa, A. K. Starace, A. Dutta, M. S. Talmadge, K. A. Magrini and C. Mukarakate, Anal. Chem., 2021, 93, 4351–4360 CrossRef PubMed.
- R. Seiser, J. L. Olstad, K. A. Magrini, R. D. Jackson, B. H. Peterson, E. D. Christensen and M. S. Talmadge, Biomass Bioenergy, 2022, 163, 106484 CrossRef CAS.
- D. C. Elliott, H. Wang, R. French, S. Deutch and K. Iisa, Energy Fuels, 2014, 28, 5909–5917 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00098f |
| This journal is © The Royal Society of Chemistry 2024 |