
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-driven hydrogen evolution via a novel pincer/no pincer mechanism including a possible concerted proton electron transfer†‡
Wolfgang
Viertl-Strasser
a,
Johann
Pann
a,
Richard
Pehn
a,
Helena
Roithmeyer
 a,
Holger
Kopacka
a,
Thomas S.
Hofer
a,
Mark E.
Thompson
b,
Austin
Mencke
b and
Peter
Brüggeller
*a
a,
Holger
Kopacka
a,
Thomas S.
Hofer
a,
Mark E.
Thompson
b,
Austin
Mencke
b and
Peter
Brüggeller
*a
aUniversity of Innsbruck, Centrum for Chemistry and Biomedicine, Institute of General, Inorganic and Theoretical Chemistry, Innrain 82, 6020 Innsbruck, Austria. E-mail: Peter.Brueggeller@uibk.ac.at
bUniversity of Southern California, Irani Chair of Chemistry Department of Chemistry, Chemical Engineering and Materials Science, Seeley G. Mudd Bldg. 3620 McClintock Ave, Los Angeles, CA 90089-1062, USA
First published on 13th December 2023
Abstract
The road to efficient molecular catalysts for artificial photosynthesis requires a great deal of basic research to address the challenges of anthropogenic global warming. An unexpected result of such research is a novel pincer/no pincer mechanism discovered for the DFT-calculated catalytic cycle of PNP-C2 complexes. Structural parameters from theoretical modeling are supported by single crystal X-ray data for the corresponding complexes. Using the examples of [M(II)Cl(PNP-C2-R)]+ (M = Pd2+, Ni2+; R = CH3, H; PNP-C2 = N,N-bis{(di(2-methoxy-phenyl)phosphanyl)ethyl}-N-alkylamine) this pincer/no pincer mechanism is supported by differential pulse voltammetric, cyclic voltammetric, UV-Vis spectroscopic (combined with voltammetry) and 31P nuclear magnetic resonance spectroscopic studies. The efficiency of hydrogen evolution during artificial photosynthesis depends significantly on the ratio of photosensitiser and water reduction catalysts. At first sight, the results from irradiation experiments (gas chromatography and mass spectrometry) seem obvious, but show a considerable impact of secondary and tertiary amines within the pincer structure. The best result obtained for the palladium pincer complex was with a secondary amine as a proton relay, delivering a turnover number of 2237 after only 2.5 hours. In contrast the analogous water reduction catalyst with a methylated (tertiary) amine instead of a secondary one gives only 972 turnovers under the same experimental conditions, over the same time. For all photocatalytic measurements the amount of hydrogen is approximately 2.5-times higher when a secondary amine is used as the proton relay. The influences of these different amines on the activity of water reduction catalysts during irradiation experiments in H2O or D2O lead to evidence of a kinetic isotope effect, supporting a possible concerted proton electron transfer. DFT calculations of reaction enthalpies, activation energies and intrinsic reaction coordinates of the catalytic cycle support the high efficiency of the presented catalysts. In particular, results from secondary amines suggest new candidates as examples of the rarely known concerted proton electron transfer.
Introduction
Nowadays, great effort can be recognised in the research and development of artificial photosynthesis. Due to climate change and energy economy, the motivation to find competitive alternatives for fossil fuels is becoming bigger every year. Today, there are three main approaches in the context of artificial photosynthesis: solid state systems,1–4 quantum dots plus nanoparticles5–10 and molecular photosensitisers (PS) plus water reduction catalysts (WRCs). Heterogeneous photosynthesis of solid state compounds, like artificial leaves,2 is quite exciting, because of high efficiencies and recent usage of earth abundant metals, but there is an immense drawback. This approach produces exclusively oxyhydrogen gas (combination of oxidative and reductive water splitting) and has already led to dangerous accidents, e.g. when upscaling artificial leaves.2 The intermediate level between solids and homogeneous, molecular compounds is photoreactive quantum dots and nanoparticles, which are quite efficient as well. Unfortunately, they are expensive, and are often toxic and harmful to the environment. Molecular systems too incorporate low-cost materials, where the efficiencies of various systems fluctuate between turnover numbers from 0 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and the catalytic lifetimes can be a few seconds up to a number of days. One can see that the competition for useable artificial photosynthesis is still running and there are very promising candidates ranging from heterogeneous to homogeneous catalysis. Nature has also been an excellent advisor to develop applicable approaches for all kinds of scientific questions, with biomimicry playing a key role in artificial photosynthesis.
000 and the catalytic lifetimes can be a few seconds up to a number of days. One can see that the competition for useable artificial photosynthesis is still running and there are very promising candidates ranging from heterogeneous to homogeneous catalysis. Nature has also been an excellent advisor to develop applicable approaches for all kinds of scientific questions, with biomimicry playing a key role in artificial photosynthesis.
For billions of years nature has been using the principle of photosynthesis. In primeval times, the first photosynthetic organisms used hydrogen and hydrogen sulfide as electron sources.11 An excellent example of dihydrogen production in nature is in the form of hydrogenases. These enzymes are believed to be part of primeval microorganisms, which were already generating/storing energy as dihydrogen.12 Although there are three main types of these hydrogenases, only one shows an amine base as a proton relay, the properties of which are of high interest for research, especially in the field of artificial photosynthesis. Nature employs three main types of hydrogenase to produce dihydrogen; of special note is the [FeFe] hydrogenase. X-ray diffraction studies of this enzyme reveal a bimetallic iron compound bridged by an S–N–S linkage, with each sulfur binding to both irons. The amine base of this linkage is then found sharing a bridge hydrogen with one of these metal centers.13–19
One can see in Fig. 1 that the enzymatic machinery consists of three separate channels for protons, dihydrogen and electrons. The pendant base, the nitrogen connecting the sulphur atoms in Fig. 1, plays a key role during the formation and cleavage of dihydrogen, functioning as a proton relay. The electron channel works as an electron transport chain with the help of [Fe4S4] clusters, which react with specific redox partners on the outer face of the enzyme to allow the regulation of the overall process.13,20 It seems quite reasonable that the way protons and electrons are provided to the catalytic metal of the active site is essential for the catalysis. Therefore, special attention should be paid to this aspect in the design of synthetic catalysts for H2 production and oxidation.20 Homogeneous hydrogen production, in general and in artificial photosynthesis, is closely connected with the working groups of M. Beller,21–23 M. Fontecave24–26 and R. Eisenberg.27–30 Furthermore, the importance of the pendant base acting as a proton relay has been intensively studied by D. DuBois20,31–34 in particular. PCET (proton coupled electron transfer)/CPET (concerted proton electron transfer) is a part of the investigated areas in the context of artificial photosynthesis, where activities in this community have been ongoing already for several years.
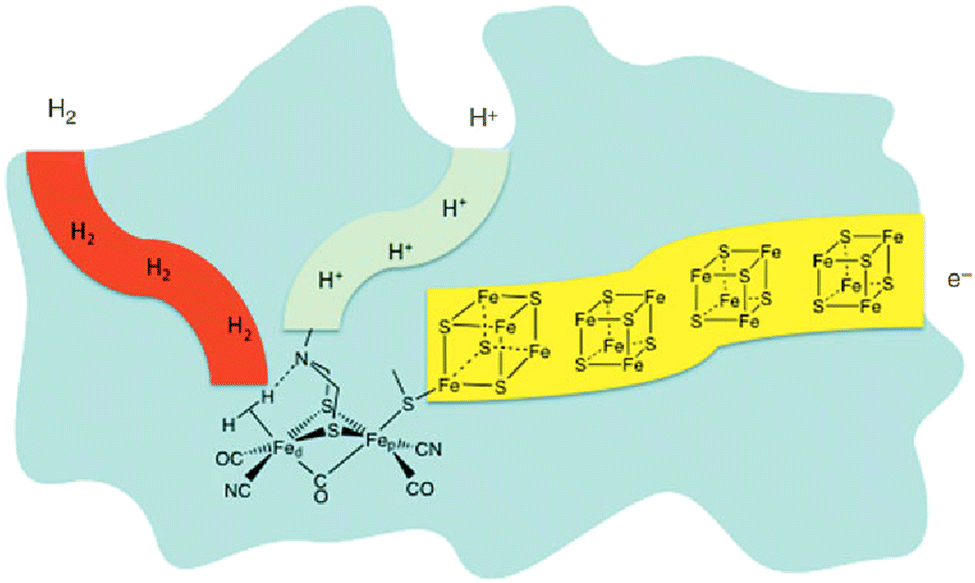 | ||
| Fig. 1 Depiction of hydrogenase enzyme emphasizing channels for proton, electron and H2 transport. Fed and Fep represent distal and proximal Fe atoms, respectively.17–20 | ||
The roles of Ni2+ and Pd2+ centers during catalysis have been calculated by DFT (see Fig. 2 and 4). As a whole, both Ni2+ and Pd2+ accept two electrons and two protons. This first leads to bonded H2 and then to the release of hydrogen, thus closing the catalytic cycle.
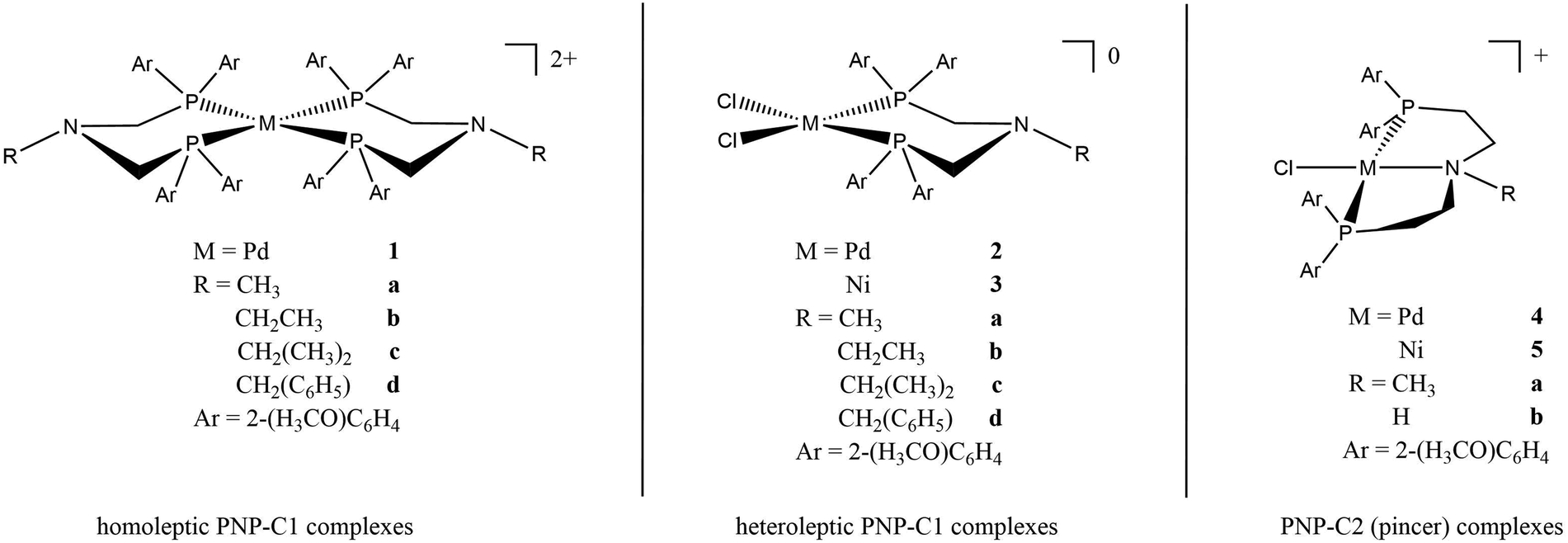 | ||
| Fig. 2 Prepared complexes 1-5 with different ligands (PNP-C1-R, PNP-C2-R; R = H, Me, Et, iPr, Bz) and metal centres (Pd, Ni). By enlarging the PNP backbone, the coordination chemistry and the mechanism of the catalytic cycle change dramatically. As is known for pincer ligands with the structure PNP-C2, a tridentate coordination is observed in contrast to the no pincer bidentate ligands of the structure PNP-C1. Thus, pincer ligands seem to stabilise complexes more, due to the five-membered ring chelate effect. In the pincer/no pincer mechanism a tridentate coordination changes to a bidentate during the catalytic cycle. | ||
Concerted proton electron transfer (CPET)
In contrast to traditional electron and proton transfers of a reaction that occur in a consecutive manner, concerted transfers move both particles simultaneously. In doing so, high energy intermediates are avoided and both the reaction rate and the turnover number of a reaction can increase dramatically.35–40 When describing the general process of transferring electrons and protons, T. J. Meyer uses the term proton-coupled-electron-transfer, but electron-proton-transfer for the specific cases of simultaneous, i.e. concerted, transfers.40 Other scientists prefer terms for concerted transfers like electron-transfer-proton-transfer,41 electron-proton-transfer42 or concerted-proton-electron-transfer.43For clarity reasons, we use in this work the terms as described in the following. This nomenclature is in accordance with Bonin and Routier36 or Saveant:44–47
EPT – electron transfer followed by a proton transfer.
PET – proton transfer followed by an electron transfer.
CPET – concerted proton electron transfer, i.e. both transfers occurring simultaneously.
As mentioned earlier the phenomenon of CPET is a young research field. Though preliminary work dates back to the 1950's, it took nearly 30 years to gain essential insights.48–50 The first published notation of the concerted transfer came from T. J. Meyer in 1981.51 After that he continued research40 and others joined in to investigate this phenomenon. Some of them, highlighted by name, are Cukier,41,52–55 Hammes–Schiffer,56,57 Nocera,41 Babcock58–60 and Saveant.61 Despite preparative setups to prove and describe the existence as well as the mechanism of CPET, theoretical chemistry provided a very useful approach too. In reviews, examinations by DFT are considered when exploring the effect of CPET on redox enzymes.62,63
Proof for the existence of CPET and data about the concrete mechanism result from several techniques like spectroscopy, electrochemical measurements and DFT calculations. The key parameters influencing CPET and its reaction constant according to T. J. Meyer are temperature, free enthalpies, activation energies, reorganisation energies, general solvation, H-bonding including distance dependence and quantum effects. Besides the obvious contributions of energetics, the distance dependence is crucial for CPET and dominated by proton transfer because of its short-ranged nature.40 This aspect is key when investigating CPET for proton relays. Interestingly, the free enthalpy and the reaction constant are independent of the pH of the external solution, since the elementary step of (de)protonation stays unaffected for CPET.40 For the general class of proton and electron transfers, a pH dependence occurs if more than one form of a pH-dependent couple is involved.40
More couples lead to an effect of irreversible thermodynamics. An inorganic example in the literature for CPET is the comproportionation reaction of cis-[RuIV(bpy)2(py)(O)]2+ and cis-[RuII(bpy)2(py)(H2O)]2+ to cis-[RuIII(bpy)2(py)(OH)]2+. This is a comparable model with regard to the presented complexes in this work (especially compounds 4b and 5b in Fig. 2). Hence, for the Ru complexes and eventually the pincer complexes with (secondary) amines as proton relays, the observed rate enhancements may derive from an existing CPET. In the case of cis-[RuIII(bpy)2(py)(OH)]2+, high energy intermediates for the sequential transfer of a proton and electron (ΔG ≥ +0.55 eV for the first reaction step, no matter if either proton or electron transfer) are eliminated by CPET, resulting in significantly lower Gibbs free energy (−0.11 eV), a lower activation barrier (0.44 eV) and a rate enhancement of approximately 100.51,64–66 Moreover, a kinetic isotope effect is observed for the Ru comproportionation with k(H2O)/(D2O) = 16.1, which is larger than the O–H/O–D classical limit of 7.9.40 For the presented pincer complexes with secondary amines, a kinetic isotope effect and a rate enhancement can be observed as well, although to a lesser extent, as reported for cis-[RuIII(bpy)2(py)(OH)]2+. Other respectable examples of PCET that involve H2-evolution comprise work published by I. Siewert67–70 or C. Streb.71,72
Results and discussion
Preliminary insights and catalytic cycles of pincer complexes
Earlier studies of complexes with PNP-C1 ligands as in 1-3 (Fig. 2) proved the efficiency increasing effect of pendant amines as proton relays on the photochemical production of H2.73 Two parameters play key roles in determining the catalytic performance. Firstly, the steric hindrance of amine substitutions plays a major role in photocatalytic hydrogen production. Small and flexible substitutions on the proton relay site enhance the efficiency, whereas bulkier, aliphatic ones decrease the turnover numbers. Secondly, one has to consider that the proton transfer from the surrounding solution to the catalytically active site of the WRC via a proton relay is essentially an acid/base reaction and therefore depends on the pKa values of the used amines.74–80 This influence is believed to be of less importance than the steric pressure. Nevertheless, less basic proton relays with benzyl-substituted amines provide higher hydrogen outputs than more basic ones like aliphatic-substituted amines. These results from earlier studies73 are well in compliance with DuBois et al.81Surprisingly, complexes with PNP-C2 ligands 4-5 (Fig. 2) show even higher turnover numbers than complexes containing PNP-C1 ligands 1-3, although PNP-C2 ligands act as pincer ligands. These well-known pincer ligands82–84 coordinate in a threefold manner to the metal. So, the amine seems to be deactivated in its function as a proton relay while coordinating together with the phosphine groups to the metal centre. X-ray diffraction studies verify this deactivation of the proton relay via coordination in the coordination environment of the pre-catalyst (4b presented in Fig. 3; 4b can be found in the ESI‡). Despite this, the pincer complexes (4-5) produce more than twice the amount of hydrogen in contrast to diphosphine complexes (1-3).73 This contradiction can be explained with a new coordination behaviour of pincer ligands and a novel catalytic cycle of their complexes (Fig. 4). Depending on the oxidation state of the metal ion in the catalyst, the pendant amine acts as either a coordination partner or a proton relay. DFT studies of the pincer complex 5a revealed this possible mechanism of water reduction catalysts with PNP-C2 ligands. In the following, evidence of a catalytic cycle for compound 5b to promote the HER is shown, based on modelling studies and experimental evidence for the catalysis.
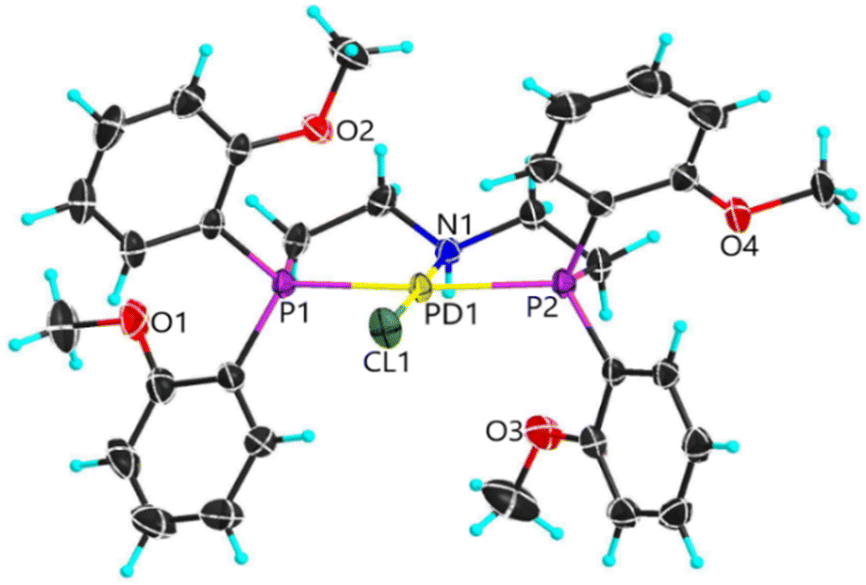 | ||
| Fig. 3 Molecular structure of [Pd(II)Cl(PNP-C2-H)](PF6) 4b (SXRD). | ||
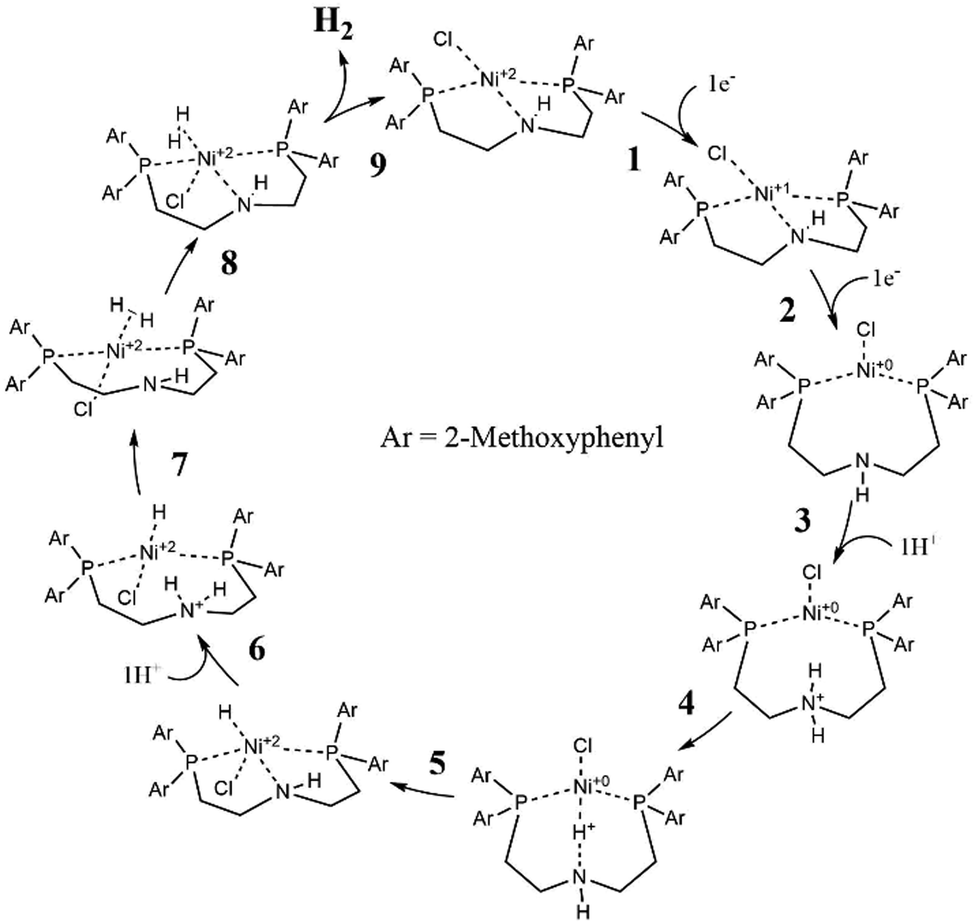 | ||
| Fig. 4 Catalytic cycle of [Ni(II)(PNP-C2-H)]+5b (DFT-PCM). | ||
Catalytic cycle of pincer catalysts by DFT
In Fig. 4, the catalytic cycle of the Ni-WRC 5b, calculated by DFT is presented. As previously reported for pincer ligands with the structure PNP-C2,85–87 a tridentate coordination is observed in contrast to the bidentate ligands of the structure PNP-C1. Thus, pincer ligands appear to have a stabilising effect on the associated complexes due to the five-membered ring chelate effect.When receiving electrons from photosensitisers (steps 1 and 2), the metal centre, in this case nickel, changes its oxidation state (and its Ni–N distance) from +II (2.0 Å) to +I (2.2 Å) finally to 0 (3.9 Å). These distances are derived from DFT calculations based on the polarisable continuum model (PCM), which are quite comparable to the calculated distances in the gas phase. More information is provided in the ESI.‡ During the reduction steps 1 and 2, a change in the coordination sphere can be observed. The nitrogen, formerly coordinating in a pincer mode, shifts to a bidentate coordination mode to the metal centre, recovering the proton relay function. Next, step 3 shows the first protonation of the amine. Alternatively, if protonation takes place prior to reduction of the pre-catalyst, the amine becomes an ammonium and its coordination ability is lost as well (for more information on these two reaction pathways see the CPET results and the ESI‡). In each case, the pendant amine can either coordinate or provide protons for reduction depending on the oxidation state of the metal or its own protonation state. After the protonation (step 3) the proton relay can transfer hydrogen to the metal centre (step 4), which reduces the proton, forming a metal hydride (step 5). After a second protonation (step 6) and proton transfer (step 7), dihydrogen is formed (step 8) leading to a square pyramidal structure (sp). Finally, molecular hydrogen is released (step 9). The deprotonated proton relay is free to coordinate again to the Ni(II) metal centre with a coordinative bond length of 2.0 Å. The sp conformation returns to the square planar structure of the pre-catalyst after H2 release (step 9).
The catalytic cycle with PNP-C2-H (5b) is almost identical to that with PNP-C2-Me (5a, see the ESI‡). A significant difference occurs due to the sterically very flexible secondary amine, when the hydride formation (step 5) takes place. The deprotonated amine and the formally oxidised nickel centre in the +II oxidation state seem to bind again (Ni–N distance 2.0 Å) by forming an sp geometry (with the chlorine as pyramidal top) before the second protonation (step 6) resolves this coordination again (Ni–N distance 2.8 Å). This behaviour is only observed in the PCM calculation, whereas during gas phase calculations no coordination bond between the pincer nitrogen and metal centre was detected. This leads to the assumption that depending on either the availability of protons and/or a corresponding solvent environment, there may be an additional stabilising state, which may affect the catalytic efficiency. Apart from that, the catalytic cycle (steps 1–9 in Fig. 4) is the same in the gas phase as for PCM and identical in respect to PNP-C2 ligands with tertiary amines (compounds 4a and 5a).
Pincer/no pincer by DPV, CV, UV-Vis and NMR spectroscopy
This new coordination behaviour of a pincer complex was investigated by spectroscopic, electrochemical and spectroelectrochemical methods to support the proposed pincer/no pincer mechanism, especially steps 1–3 during the catalytic cycle (Fig. 4). As described earlier,73 Pd(PNP-C2-Me) (compound 4a) was examined with cyclic voltammetry (CV) and differential pulse voltammetry (DPV), measured relative to an internal ferrocene reference in anhydrous acetonitrile and with addition of water during the CV experiment (Fig. 5).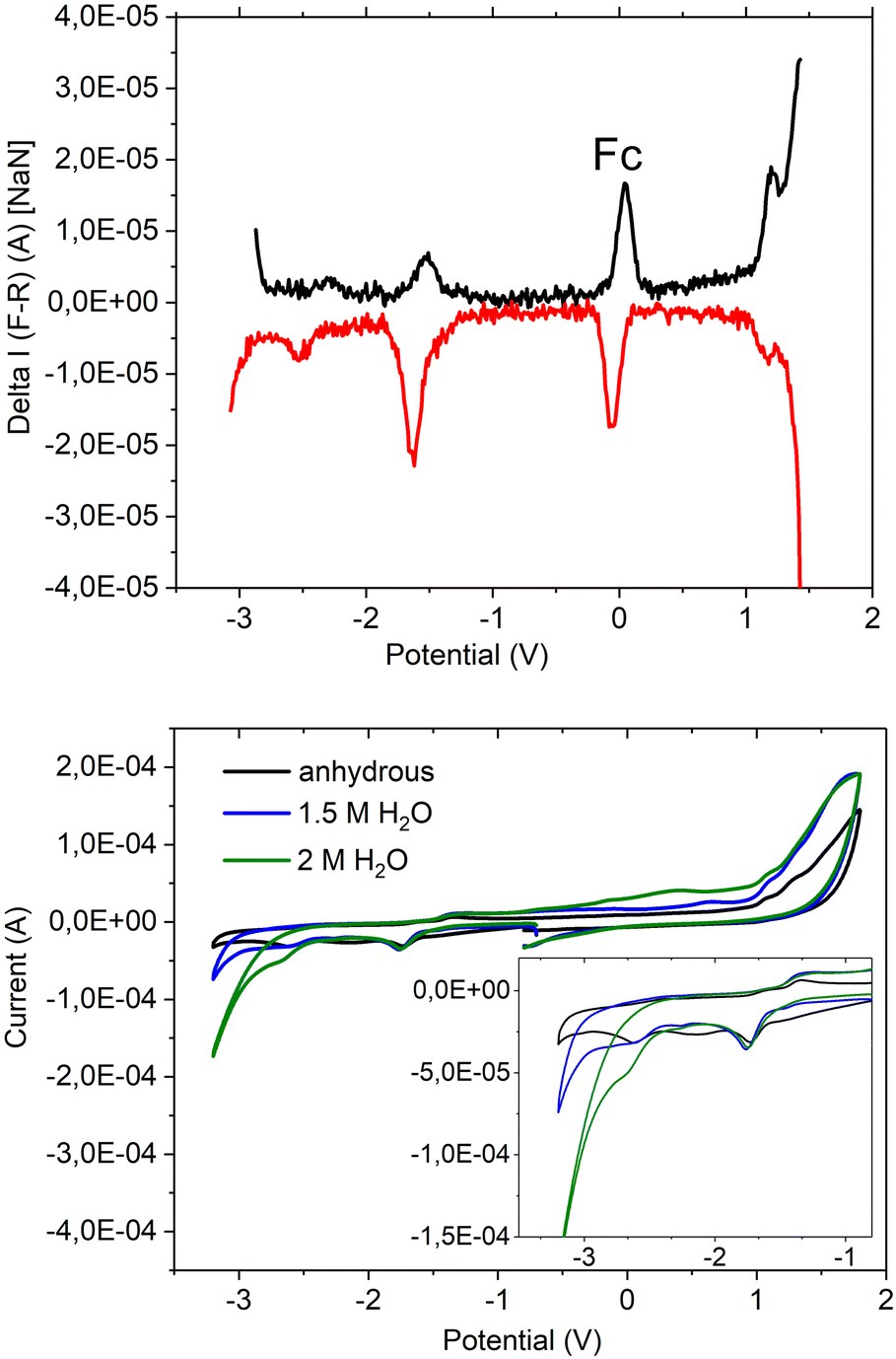 | ||
| Fig. 5 Electrochemical properties of compound 4a [Pd(PNP-C2-Me)] in anhydrous acetonitrile. Differential pulse voltammetry (voltages referred to ferrocene/ferrocenium, the y-axis shows ΔCurrent (A). NaN stands for “not a number”, F–R for “forward minus reverse” indicating how the DPV measures its signal) in anhydrous acetonitrile is shown above and cyclic voltammetry including electrochemical traces in the presence of water below: 4a in anhydrous acetonitrile (black), 1.5 M H2O in acetonitrile (blue), and 2 M H2O in acetonitrile (green). During the study of different amounts of water added, the electrodes were completely stable. | ||
In the DPV traces (Fig. 5, top), one can see two quasireversible electron transfers at potentials of −1.55 V and −2.4 V, corresponding to M2+/1+ and M1+/0 transitions, respectively. The second reduction wave and its reversible oxidation wave are less significant than the first ones, indicating a less stable compound and less reversibility in the absence of a redox agent like hydrogen or water. So, one can conclude that the reduction from Pd2+ develops stepwise via Pd1+ to Pd0 in the activated catalyst, which forms colloidal palladium and therefore leads to the loss of the pincer complex. Moreover, the reduction seems more likely to happen before the first protonation step. Nevertheless, both pathways are possible. Oxidation of the compound takes place at a potential of >1 V and is irreversible.
In the CV (Fig. 5 below), the two reduction waves are clearly visible as well and show a minor shift, like the catalytic wave itself, when water was added (1.5 and 2 M). Despite that, the CV shows a shift of the onset of the catalytic wave from −3.0 V to −2.5 V, which results in an overlap with the second reduction potential at −2.4 V. Hence, the pincer catalyst is capable of lowering the overpotential for water reduction even as an electrochemical catalyst.
UV-Vis absorbance can be detected during a chronoamperometry experiment to observe the spectroscopic properties of Pd(PNP-C2-Me) (compound 4a) while being reduced under anhydrous conditions. The applied voltage of −2.0 V vs. QRE was determined to be the potential of the second reduction versus CV. Under this applied bias in a 0.1 M n-Bu4NPF6 anhydrous acetonitrile solution the first ligand centred transition band of 4a grows over time, while the d–d transition is bleached out. These results imply, as mentioned earlier, that Pd(II) is converted into colloidal Pd, ruling out first electron transfer followed by proton transfer as a possible next step (Fig. 6(a)). In addition, it is important to mention that during CV experiments the supply of electrons is enormous in contrast to ordinary irradiation experiments, in which an electron transfer occurs relatively rarely, since these transfers are dependent on the natural frequency of collisions in solution of the photosensitisers in excited states and the pre-catalysts. As a consequence of the increased electron supply, the irreversible reduction of Pd(II) to Pd(0) in the CV experiment is expected. Attempts to directly reduce H+ were performed in a 0.1 M HCl 2:1 acetonitrile/water (v/v) solution, using a Pt working electrode. However, hydrogen evolution at the Pt surface was favourable over 4a, which is inferred by the fact that the UV-Vis spectra are invariant over time.
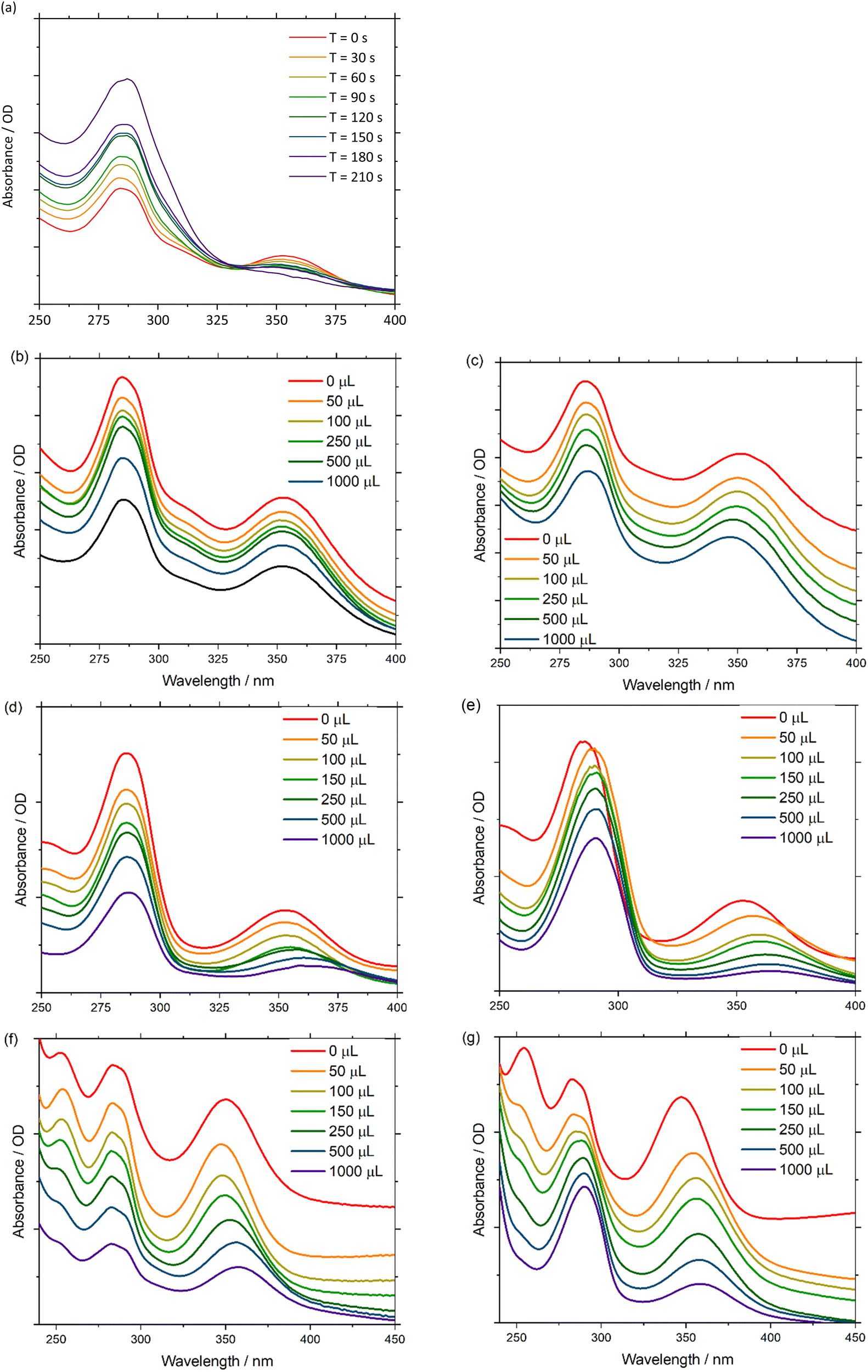 | ||
| Fig. 6 UV-Vis absorbance spectra of: (a) Pd(PNP-C2-Me) 4a overtime at an applied potential of −2 V vs. Ag QRE in a 0.1 M n-Bu4NPF6 anhydrous acetonitrile solution, (b) Pd(PNP-C2-Me) 4a in an anhydrous acetonitrile solution with various amounts of water added, (c) Pd(PNP-C2-Me) 4a in an anhydrous acetonitrile solution with various amounts of 65% HBF4 added, (d) Ni(PNP-C2-Me) 5a in an anhydrous acetonitrile solution with various amounts of water added, (e) Ni(PNP-C2-Me) 5a in an anhydrous acetonitrile solution with various amounts of 65% HBF4 added, (f) Ni(PNP-C2-H) 5b in an anhydrous acetonitrile solution with various amounts of water added, (g) Ni(PNP-C2-H) 5b in an anhydrous acetonitrile solution with various amounts of 65% HBF4 added. | ||
Next, the spectroscopic properties of compounds 4a, 5a and 5b (Pd(PNP-C2-Me), Ni(PNP-C2-Me), Ni(PNP-C2-H)) were examined in more detail. By adding water as a proton source to an anhydrous acetonitrile solution of the Pd compound 4a, no change of the first ligand centred transition band (π–π*) or of the d–d transition band in the UV-Vis absorbance spectra was observed. In contrast, the d–d transition band of the two nickel compounds 5a and 5b (Fig. 6(d) and (f)) showed a redshift due to the hydration of the metal centre. This may be explained with the higher affinity of nickel complexes to form octahedral structures with the solvent molecules, especially with water.88 It is confirmed by the red shift of the d–d transition zone of [Ni(en)3]2+, as soon as en is replaced by H2O forming [Ni(H2O)6]2+. These six water molecules shift the absorption from 333 to 400 nm (see page 839 of ref. 88).
Under anhydrous conditions with the addition of fluoroboric acid to protonate the investigated compounds 4a, 5a and 5b an additional band shift arises. Upon dropwise addition of 65% HBF4 to a solution of acetonitrile, the peak intensities in the spectra remain constant. However, the first ligand centred transition redshifts, while the d–d transition blueshifts for 4a and redshifts for 5a and 5b (Fig. 6(c), (e) and (g)). The π–π* band shift, caused by the acid, seems consistent with a rapid protonation of the amine site, which is thereby forced to lose coordination. Interestingly, the d–d transition shift of palladium does not appear in a hydrous environment alone but only in the presence of acid (Fig. 6(b) and (c)). Acid-induced solvation of palladium is due to the loss of amine coordination, resulting in a square planar geometry.
The d–d bands of the nickel compounds show stronger shifts in the acidified solvent than in the hydrous environment (Fig. 6(d)–(g)), which corresponds to a significant coordination change as well, since the protonated amine cannot coordinate. Due to all of these shift changes and the calculated protonation of the pincer complex, one can conclude that a new complex species is established by either reduction and/or protonation of pincer complexes, which correspond to a diphosphine complex with an activated (and loaded) proton relay like in the catalytic cycle (Fig. 4).
To further explore the pincer/no pincer mechanism, a platinum analogue Pt(PNP-C2-Me), corresponding to compound 4a, was protonated with drops of hydrochloric acid (2 M) and investigated by 31P nuclear magnetic resonance spectroscopy (NMR). The complex shows a clear signal at 31.0 ppm in dichloromethane, when the pincer ligand is coordinated in a conventionally threefold manner. As expected, satellites of the platinum nucleus can be noticed in the spectrum with a coupling constant of 1JPtP = 2757 Hz, derived from the trans-positioned phosphorus atoms. After the addition of three drops of hydrochloric acid, a second species arises with a high-field shift difference of approximately 50 ppm. The second signal with a chemical shift of −18.6 ppm may refer to an active proton relay, since the formation of an 8-membered ring out of two 5-membered rings forces a high-field shift in this range (see Chart S1, ESI‡).
This platinum analogue with the diphosphine ligand PNP-C1-Me (see Fig. 2) was synthesised as previously reported.73 Unfortunately, the former pincer complex with an activated proton really is not stable for long periods and with greater amounts of acid. By adding 1 mL acid in total, an additional protonation of phosphorus atoms is observed, resulting in a total loss of catalytic activity. This observed behaviour during NMR spectroscopy shows the earlier mentioned colloid formation during CV and that an excess of acidic protons, as well as multiple reductions, destroy the highly active but less stable catalyst. After examining the data, the proposed pincer/no pincer mechanism is supported.
Comparison of secondary and tertiary amines as proton relays in hydrogen production
In the following, the compounds [M(II)Cl(PNP-C2-Me)]+ (4a, 5a) and [M(II)Cl(PNP-C2-H)]+ (4b, 5b) are compared. The difference here is the exchange of the methyl group with a proton, i.e. exchanging a tertiary pendant amine with a secondary one as the proton relay. The mechanisms during the catalysis can be supposed to be the same for all four water reduction catalysts. Hence, both pendant amines can act either as coordinating ligands or as proton relay.As described elsewhere,73 it became obvious that water reduction catalysts with PNP-C2 (pincer) ligands produce approximately twice the amount of hydrogen as PNP-C1 (diphosphine) ligands. This led to the first investigations of the mechanism of the catalytic cycle. Moreover, it became also obvious that secondary amines produce significantly more hydrogen than comparable catalysts with tertiary amines. Experiments include irradiation of a water/acetonitrile solution (1:1) of different water reduction catalysts, the well-known iridium photosensitiser [Ir(bpy)(ppy)2](PF6) and triethylamine as a sacrificial donor with a 150 W Hg medium pressure lamp or 2 W LED lamps (470 nm) and measurements of the evolved hydrogen by gas chromatography (and mass spectrometry).
The results in Table 1 show for palladium and nickel catalysts that complexes with secondary amines deliver much more hydrogen gas and a largely enhanced turnover number (TON). The reason for this efficiency increase may be the sterically less hindered amine. Basicity is likely not a factor, since the secondary amine is expected to perform worse due to its higher pKa and preliminary studies.73 Another possible reason may be a concerted proton electron transfer mechanism (CPET), which is discussed below.
| Compound | TON | Ratio |
|---|---|---|
| Pd(PNP-C2-Me) 4a | 515 (972) | 2.6 (for Hg lamp) |
| Pd(PNP-C2-H) 4b | 1328 (2237) | |
| Ni(PNP-C2-Me) 5a | 68 (6) | 2.7 (for Hg lamp) |
| Ni(PNP-C2-H) 5b | 183(26) |
Several further experiments were carried out to ensure that the HER efficiency of pincer catalysts is tied to the nature of the amine, i.e. the secondary amine-based pincer is more efficient than a tertiary amine-based analogue, and not due to an interaction with the photosensitizer (Fig. 7(a)–(d), detailed information about all irradiation experiments see ESI‡). Excess photosensitiser, with ratios between the photosensitiser and water reduction catalyst (PS/WRC) of 1, 2, 5 or 25 for each experiment were used. After each hydrogen measurement, a corresponding equivalent of PS was added. For the excess/ratio of 1 (stars in Fig. 7(a) and (b)) the life time of the photosensitiser is clearly determined to be 45 min, since no further hydrogen evolvement takes place. When fresh PS is added, hydrogen production resumes. The photosensitiser decomposes due to the irradiation, forming metallic iridium. The Ir-complex and the metallic iridium are not catalytically active.73 For all of the four different PS excesses, the pincer catalyst with the secondary amine (Fig. 7(b)) outperforms its analogue with a tertiary amine (Fig. 7(a)).
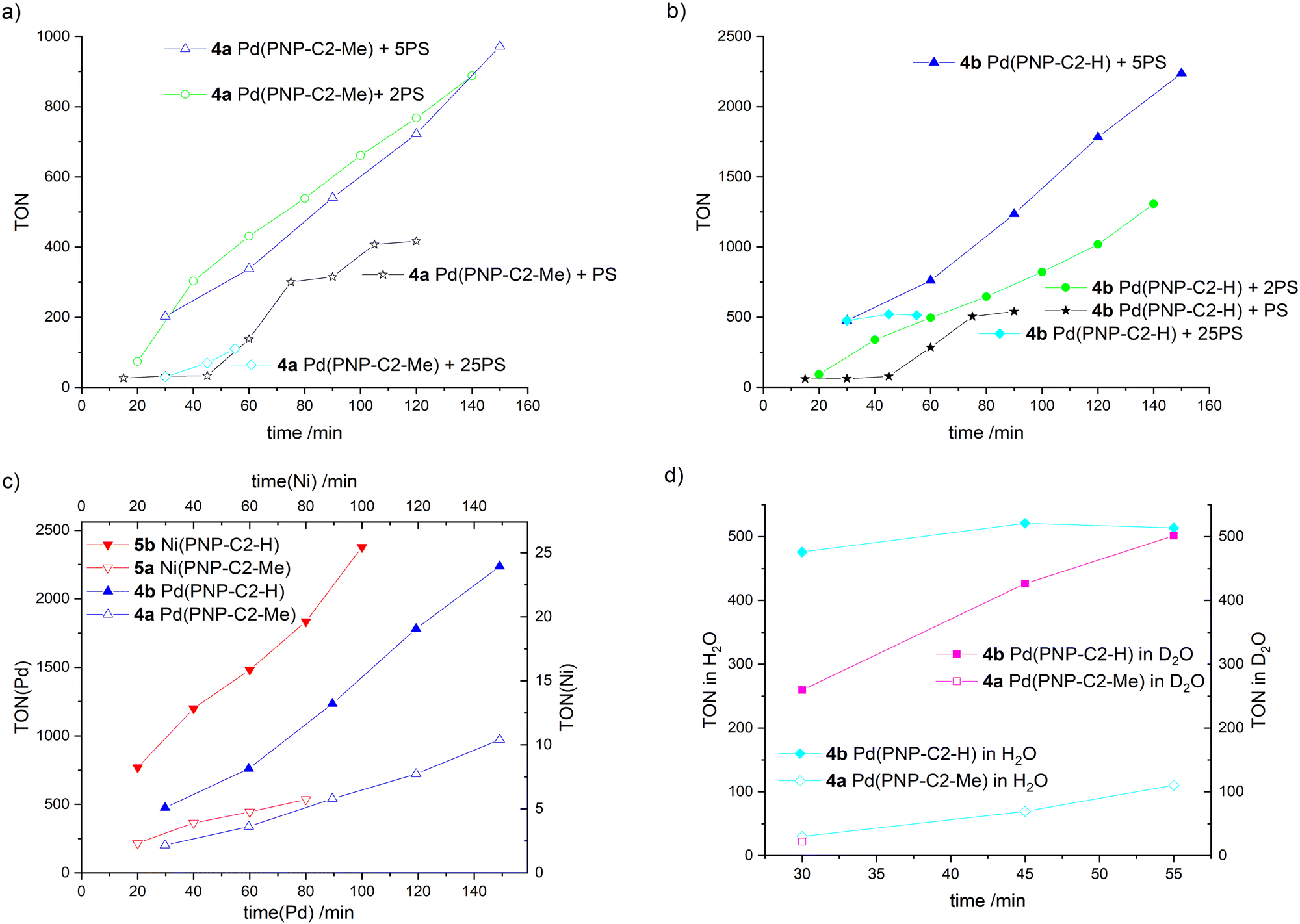 | ||
| Fig. 7 Time-dependent hydrogen production of water reduction catalysts (WRC) 4a Pd(PNP-C2-Me), 4b Pd(PNP-C2-H), 5a Ni(PNP-C2-Me) and 5b Ni(PNP-C2-H), addition of an extra equivalent of photosensitiser (PS) after each hydrogen measurement except for experiments with 25-fold excess of PS from the beginning (diamonds), irradiation with 470 nm and 100 mW (except for nickel catalysts – 160 mW), measurement of hydrogen concentration with a maximal relative standard deviation of 1.5%. This standard deviation originates from an average over triplicate measurements. The solvent is MeCN/H2O (v/v = 1:1). The sacrificial electron donor is triethylamine (TEA). In all cases TEA was always constantly concentrated in the irradiation solution with 10% (v/v). The pH/pD is always kept at 10. (a) and (b) Comparison of compounds 4a (empty symbols) and 4b (filled symbols) with starting PS/WRC ratio of 1 (stars), 2 (circles), 5 (triangles) and 25 (diamonds). (c) Comparison of compounds 4a, 4b, 5a and 5b with starting PS/WRC ratio of 5. (d) Comparison of compounds 4a and 4b in H2O and D2O with a starting PS/WRC ratio of 25, measured with gas chromatography as well as mass spectrometry (sensitive to hydrogen isotopes). Further determining an increased kinetic isotope effect (KIE) is a good indication for a reaction that involves proton tunnelling and where a proton transfer is the rate determining step, such as a CPET. By contrast, for EPT or PET the electron transfer is assumed to be the rate determining step, where none or a small KIE is measurable.89 | ||
Contrary to our expectations, higher excess of photosensitiser does not correlate to higher turnover numbers. When the ratio of 25 is used (diamonds), hydrogen evolution is as low as or even lower than for the ratio of 1 (stars). Higher PS concentrations can lead to shadowing of the solution and hence less performance. Furthermore, a large amount of photosensitiser decomposes due to long times in an excited (reduced) state, subsequently forming metallic iridium. In addition, a large excess of PS may be disadvantageous for the catalyst as well. The high availability of excited electrons may correspond with multiple reductions of the catalyst, as discussed above. In addition to photosensitiser decomposition over time, the catalyst itself decomposes due to over reduction. Despite that, excesses of 2 (empty and filled circles in Fig. 7(a) and (b)) and 5 (empty and filled triangles in Fig. 7(a) and (b)) show better results than a ratio PS/WRC of 1, which is quite reasonable and well in accordance with expectations.
The best conditions for hydrogen evolution are when the photosensitiser is in a five-fold excess. After 150 min, compound 4a Pd(PNP-C2-Me) reaches a maximum turnover of 972 and compound 4b Pd(PNP-C2-H) a TON of 2237.
The pincer catalyst with the methylated, tertiary amine M(II)(PNP-C2-Me) (4a and 5a empty triangles) and the secondary amine M(II)(PNP-C2-H) (4b and 5b filled triangles) are compared in more detail in Fig. 7(c). For clarity reasons and a better comparison, the hydrogen evolvements of compounds 4a-5b are overlayed in a single plot in Fig. 7(c). More efficient HER is observed for the secondary over tertiary amine based pincer complexes irrespective of the metal used to form the complex: the same trend is seen for both Ni and Pd based catalysts. Independently of the metal centre (palladium or nickel) the progression of the hydrogen evolution matches perfectly for the species with tertiary and secondary amines, respectively. One can see that the palladium catalysts show approximately a hundred fold higher hydrogen production than their nickel counterparts. Additionally, the turnover frequencies (TOF) of the catalysts with the secondary amine are remarkable.
They perform 2.3–3.3 times faster and therefore more efficiently, which is very consistent with previous results from Table 1. The distinct higher turnovers for palladium relative to nickel catalysts is in contrast to previous results, which likely arise from different irradiation conditions like the duration (5.5 h vs. 60/120 min), the light source (150 W Hg lamp vs. LED) and the WRC/PS ratio (10 vs. 5). Again, there is the question, if this efficiency enhancement derives only from the sterically less hindered secondary amine. A concerted proton electron transfer may be a possible explanation as well.
Possibility of a concerted proton electron transfer
A concerted proton electron transfer (CPET) is observed when the transfer of an electron and a proton occurs simultaneously. This recently reported phenomenon is not yet fully understood. Even examples in the literature are very rare.36,40 One piece of strong evidence for CPET is a reaction rate enhancement of several orders of magnitude by eliminating the high energy intermediates derived from sequential electron and proton transfer. The increase in the rate of catalysis when a secondary amine is used as the proton relay in this study is not large. Nevertheless, there is still a possibility of a CPET, since this effect responds quite delicately to temperature, free enthalpies, activation energies, reorganisation energies, general solvation, H-bonding including distance dependence and quantum effects.40Besides the specific efficiency increase of secondary amines as proton relays and the general enhancement of pincer catalysts (4-5) in contrast to diphosphine complexes (1-3), signs for CPET come from irradiation experiments in H2O and D2O, as presented in Fig. 7(d). Here, the results correspond to irradiations with a PS excess of 25 after 55 min. Compound 4a Pd(PNP-C2-Me) delivers a lower hydrogen outcome due to possessing a tertiary amine as discussed above. Despite that, the first measurement after 30 min results in 30 turnovers for the irradiation in H2O (empty diamonds) and 22 in D2O (empty square). These low and comparable turnover numbers were detected by gas chromatography and additionally with mass spectrometry when D2O was used. The irradiation solution consists of acetonitrile suitable as a proton source, and only water is deuterated. However, it is known that under photocatalytic conditions H/D exchange is common. For the catalyst with the secondary amine the turnovers are very different. After 30 min the efficacy of D2O is low with a TON of 260 in contrast to 476 for irradiation in H2O. Interestingly, the hydrogen evolution in D2O recovers within a total irradiation time of 55 min and reaches nearly the same outcome as in H2O (TON 502 in D2O and 514 in H2O) due to photochemical H/D exchange. This behaviour can be explained with a reduced activity and therefore a reduced reaction rate forced by the deuterated water. The reduced access to protons leads to significant deceleration of the reaction rate but in the end the same hydrogen outcome. Fig. 7(d) clearly demonstrates that there is an equilibrium between the protons of acetonitrile, triethylamine and water. However, this equilibrium is present for both secondary and tertiary amine catalysts. Hence, this is a kinetic isotope effect, which is sometimes observed for concerted proton electron transfers.40
Subsequently, the thermodynamics and activation barriers of compounds 5a Ni(PNP-C2-Me) and 5b Ni(PNP-C2-H) are of special interest to support the possibility of a CPET. The different geometries of the catalytic cycle (Fig. 4) and their corresponding energy levels were calculated by density functional theory. To take solvent influences into account, the calculations were undertaken in an acetonitrile/water environment (1:1) with the help of the polarisable continuum model (PCM). The reaction enthalpies and activation energies for the first (steps 4 and 5) and second proton transfer (steps 7 and 8) from the pendant amine to the metal centre as well as the second protonation step of the proton relay (step 6) are presented in Table 2 and Fig. 8.
| Compound | ΔH (PT1) | E a (PT1) | ΔH (PT2) | E a (PT2) |
|---|---|---|---|---|
| Ni(PNP-C2-Me) 5a | −8.1 | 1.0 | 46.0 | 128.9 |
| Ni(PNP-C2-H) 5b | −47.8 | 0.2 | 19.4 | 82.8 |
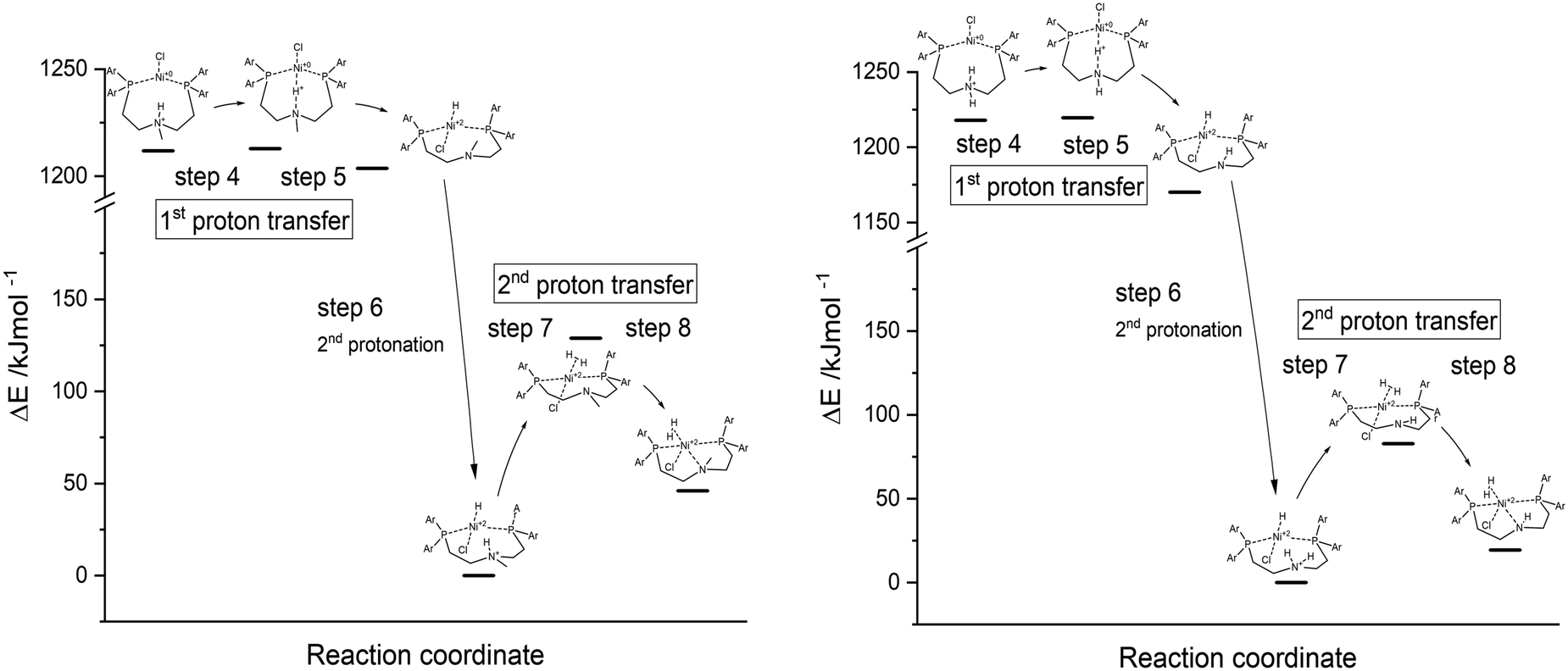 | ||
| Fig. 8 Energy diagrams of first and second proton transfer (steps 4–8) of Ni(PNP-C2-Me) 5a (left) and Ni(PNP-C2-H) 5b (right), calculated as DFT-PCM. | ||
For both compounds 5a Ni(PNP-C2-Me) and 5b Ni(PNP-C2-H), the first proton transfers are exothermic and the activation energies are in the range of 1 kJ mol−1. The second proton transfers are again both endothermic and show a large energy barrier corresponding to the speed-determining step and hence the resting state. Therefore, the energetics of catalysts with secondary and tertiary amines appear similar. Although, the rate enhancement and higher TON of the secondary amine could derive from the significant more favourable second proton transfer, since the reaction enthalpy decreases from 46.0 to 19.4 kJ mol−1 and the activation energy drops from 128.9 to 82.8 kJ mol−1. Despite this, the pincer/no pincer mechanism favours especially the first proton transfer in contrast to endothermic proton transfers and large activation energies for catalysts with diphosphine ligands (1-3).73 Pincer catalysts seem to promote strongly the formation of a metal hydride. This behaviour looks alike for secondary and tertiary amines.
In this case, the assumption is that the electron transfers to the catalysts take place prior to the first protonation of the proton relay. As discussed above, the sequence can switch. When the amine within the pincer ligand gets protonated, it loses the coordination to the metal as well, becomes an activated proton relay and waits for the reduction potential from the excited photosensitiser. These two mechanisms are classical proton electron transfers in a sequential manner. Otherwise, if the first protonation of the proton relay happens, there may be also the chance for CPET, a simultaneous transfer of electrons. Hence, the reaction path of the first proton transfer was calculated. This intrinsic reaction coordinate (IRC) is depicted in Fig. 9 (red points) and displays an activated catalyst (reduced Ni0 and protonated amine), which passes a proton from the amine to the metal.
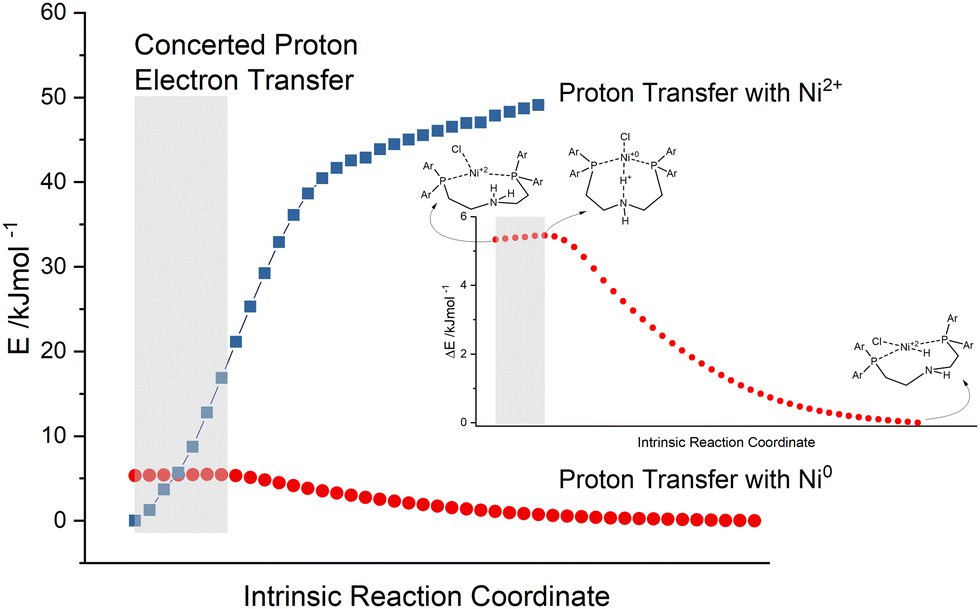 | ||
| Fig. 9 Intrinsic reaction coordinate (IRC) calculation (red points with Ni0) and single point (SP) energies of corresponding geometries minus two electrons (blue squares with Ni2+) of the first proton transfer of Ni(PNP-C2-H) 5b (PCM). | ||
The single point (SP) energies of these intermediate geometries from the first proton transfer were recalculated in the absence of two electrons, simulating a proton transfer of an inactive Ni2+ catalyst (blue squares in Fig. 9). By comparing these reaction paths, one can see that the energy of the protonated but inactive catalyst rises very rapidly (zero point energy chosen arbitrarily). This general behaviour is naturally self-evident, since a positively charged proton gets closer to the Ni2+ centre of the complex. Nevertheless, this progression increases to such an extent that the intersection must be located somewhere before the transition state of the protonated and reduced catalyst. As described earlier, the concerted transfer possesses a large distance dependency, dominated by the proton transfer. In the grey highlighted area the N–H bond length ranges from 118 pm to 127 pm. This extremely small difference of 9 pm suggests together with the intersection of the IRC (red points) and the SP energy progression (blue squares) that in this area a concerted proton electron transfer may be quite likely. The vibrational mode of the N–H bond can already favour the acceptance of reduction potential from the photosensitiser. Moreover, the movements of the two protons of the secondary amine may force a kind of high energy intermediate, which is skipped by a concerted proton electron transfer and results in the rate enhancement measured in previous experiments.
Conclusions
After investigation of pincer complexes with the structural motif of [M(II)Cl(PNP-C2-R)]+ (M = Pd2+, Ni2+; R = CH3, H; PNP-C2 = N,N-bis{(di(2-methoxyphenyl)phosphanyl)ethyl}-N-alkylamine), new insights arise. Though the proton transfer may be complicated by shuttling effects, induced through the described pincer/no pincer behaviour of PNP ligands, several features suggest a CPET mechanism.89 With the excellent structural data from single crystal diffractometry, theoretical calculations deliver the catalytic cycle of a pincer complex with a secondary amine as a proton relay. Electrochemical and/or spectroscopic results support the postulation of a catalytic cycle with a novel pincer/no pincer mechanism. Moreover, pincer catalysts with secondary amines instead of tertiary ones are certainly more efficient and lead to 2.3–3.3 times higher turnover numbers. E.g. the best hydrogen evolvement of a palladium pincer catalyst produces 2237 turnovers, when featuring the secondary amine, whereas the tertiary analogue reaches only a TON of 972. This efficiency enhancement in artificial photosynthesis points together with calculations of the energetics during catalysis to the possibility of a concerted proton electron transfer. In irradiation experiments with H2O or D2O an observed kinetic isotope effect supports this assumption. With calculated intrinsic reaction coordinates, the possible concerted proton electron transfer seems even more likely. To validate the novel pincer/no pincer mechanism and a possible CPET for the presented pincer complexes, further experiments must be continued. This could also shed some light on the investigation of multinuclear WRCs and their mutual dependence, where it is shown that multiple metal centres support the proton reduction due to the presence of more catalytically active centres.90The single crystal X-ray structures of complexes containing the PNP-C1 ligand73 clearly indicate that a chemical bond between PNP-C1 and the metals is not possible and hence also during the catalytic cycle only a no pincer mechanism occurs. The methoxy O atoms of PNP-C1 additionally endow the framework of PNP-C1 with good stability. The single crystal structure of [PdCl2(PNP-C1)] is also known (see ESI‡ of ref. 73). Therefore, the synthesis of highly crystalline PNP-C1 coordinated to Pd2+ is fully controllable. This material stability is enhanced in pincer complexes containing PNP-C2 due to the presence of thermodynamically favoured five-membered rings.
The mineralization rate of water reduction catalysts containing PNP-C1 and PNP-C2 has been investigated. After successful photochemical hydrogen evolution, the mineralization is complete and no further hydrogen evolution occurs. This means that the mineralization rate corresponds to the endpoint of the hydrogen evolution diagrams in this work and in ref. 73. At this point the catalytic mixture shows the appearance of palladium black being no more catalytically active. The PNP-C1 and PNP-C2 ligands are irreversibly oxidized to the corresponding phosphine dioxides also indicating the end of catalytic activity, where this has been proved by 31P{1H} NMR spectroscopy.
Experimental
Materials and methods
All preparative work was carried out using standard Schlenk techniques at ambient temperature and pressure. For inert gas, Argon 5.0 (Messer) was used. Solvents (Acros Organics/Fisher Scientific, extra dry, with molecular sieve and crown cap) were either degassed by ‘freeze–pump–thaw’ or by usage of a vacuum pump for at least 15 minutes, dependent on individual boiling point. Further dehydration was not performed, unless otherwise indicated. Deuterated solvents were purchased from euro-isotope and used as received. The educt for all PNP syntheses, di-(2-methoxyphenyl)phosphine, was prepared by the strategy of working group Brüggeller.91 Other starting materials as amines, metal salts and bases (Sigma Aldrich, ABCR) were purchased. For filtration, a Durapore Membrane Filter (Merck Millipore) was used.Synthesis
The syntheses of the PNP-C2 ligands and their metal complexes (4a, 4b, 5a, 5b) are presented in the ESI.‡ Preliminary results for 4a [Pd(II)Cl(PNP-C2-Me)](PF6), PNP-C1 ligands and their metal complexes are published elsewhere.73Structural analysis
1H, 13C and 31P NMR spectra were recorded on a Bruker Avance DPX 300 spectrometer with internal 2D-lock. Measurements were undertaken at 121.497 MHz for 31P{1H}-NMR (phosphoric acid, 85%, as external standard), at 75.476 MHz for 13C{1H}-NMR (calibration based on solvent signal) and at 300.13 MHz for 1H-NMR (calibration based on solvent signal).Mass spectra of the synthesised compound were received on a Finnigan MAT-95 apparatus using MALDI for ionisation and a DAN (1,5-diaminonaphthalene) matrix.
Prior to the XRD measurements, the single crystals were handpicked using a Leica Wild M10 microscope with vertical illumination and a polarising filter. X-ray diffraction analysis was performed on a Bruker D8 diffractometer with an incoatex-microfocus-channel, a multi-layer-monochromator and the new CMOS-technology from Bruker (D8 Quest). Cell refinement, data reduction and the empirical absorption correction were performed using the Apex III and Saint-V 8.34 A programmes. All structure determination calculations were realised using SHELXTL-NT V6.1 and SHELXL-2014/7. Final refinements on F2 were done with anisotropic thermal parameters for all non-hydrogen atoms.
Electrochemical measurements
Cyclic voltammetry and differential pulsed voltammetry were performed using a VersaSTAT 3 potentiostat. Anhydrous acetonitrile (DriSolv) was further dried on 3 Å molecular sieves and was used as the solvent under an inert atmosphere, with 0.1 M tetra(n-butyl)ammonium hexafluorophosphate (TBAF) used as the supporting electrolyte. A glassy carbon rod was used as the working electrode, a platinum wire was used as the counter electrode, and a silver wire was used as a pseudo reference electrode. The redox potentials are based on values measured from differential pulsed voltammetry and are reported relative to a ferrocene/ferrocenium (Cp2Fe/Cp2Fe+) redox couple used as an internal reference, while electrochemical reversibility was determined using cyclic voltammetry. A water splitting experiment was carried out by adding deionized water to the solution via a micropipette.Absorptivity spectra were measured using a UV-VIS Agilent 4853 diode array spectrometer.
Spectroelectrochemical experiments were performed using a VersaSTAT 3 potentiostat and a UV-VIS Agilent 4853 diode array spectrometer. Samples were prepared at an optical density of 0.6–0.8 in anhydrous acetonitrile (Drisolv), which had been further dried over 3 Å molecular sieves. 0.1 M tetra(n-butyl)-ammoniumhexafluorophosphate (TBAF) was used as the supporting electrolyte. Samples were measured in a 2 mm path length cuvette using a platinum wire mesh electrode as the working electrode placed in the UV-vis beam path, a platinum wire as the counter electrode, and a silver wire as the pseudo reference electrode. A one sweep cyclic voltammogram was measured before each experiment to measure the voltage drift of the QRE. Absorbance spectra were measured every 30 seconds while the working electrode was held in a chronoamperometry setup at a 100 mV overpotential relative to the redox process of interest.
Irradiation and hydrogen measurement
First attempts were performed in a self-made chamber containing a water-cooled mercury medium pressure lamp with 150 W (TQ 150, λ = 280–700 nm, λmax = 360 nm, Heraeus). Later irradiation experiments were carried out with a “solar hydrogen lab-luminaire”, planned, designed and produced by Bartenbach GmbH in cooperation with Johann Pann, MSc Dr. This luminaire was equipped with NCSCE17A LEDs from Nichia (1.5 W per LED), a non-in situ photon-flux control with an optical sensor and temperature control of the irradiation solution with a thermostat Grande Fleur from Peter Huber Kältemaschinen AG. The maximum radiative power is 200 mW, focused on the irradiated Schlenk flask, which corresponds to 2820 W m−2. All irradiations were undertaken by 100 mW and 20 °C (except for nickel compounds with 160 mW).Hydrogen measurements and chromatograms were received on a gas chromatograph (Fusion-GC, Inficon) with Argon 5.0 as a carrier gas and with a thermal conductivity detector. The calibration window ranged from 1000 ppm to 10000 ppm concentration of hydrogen. Experimental errors were measured for calibration gases with hydrogen contents of 1000 ppm and 100000 ppm. Here, a flask was filled with calibration gas and measured seven times with gas chromatography. The relative standard deviations were 0.4 and 1.5%. Analyses were carried out by usage of EZ IQ programmes. Mass spectrometric analyses were performed on an HSense from V&F Analyse- und Messtechnik GmbH (EI-MS, detection limit < 1 ppm), sensitive to H2 and D2 gas.
In all cases the electron donor triethylamine (TEA) was always constantly concentrated in the irradiation solution with 10% (v/v). The standard deviation of 1.5% originates from an average over triplicate measurements. The Hg lamps show a spectrum (see ESI,‡ Chart S2) that has similarities to unfiltered sunlight. However, LED light consists of a specific wavelength. The light sources have been changed to explain, if a wavelength matching the absorption of the photosensitiser is enough. Additional wavelengths as in the case of Hg lamps could be beneficial or detrimental depending on the kind of chromophore and WRC used. In this work it is shown that LED light outperforms the light from a Hg lamp producing the largest TON values in the case of Pd (see Table 1).
DFT calculations
DFT calculations were performed by using the Gaussian16 program. Therefore, geometry optimisations (energy minima and transition states) in the gas phase and in a solvent environment were executed. The minima and TS structures were verified using harmonic frequency calculations. For calculations in the gas phase the B3LYP hybrid functional and the basis set 6-31G(d,p) were used. For calculations in a solvent environment, the polarisable continuum model (PCM) was used and the hybrid functional was changed to BP86, while the basis set was the same. The solvent was chosen as in the experimental setup during irradiation. It was a solvent mixture of water and acetonitrile in the ratio 1:1. Hence, the relative permittivity was averaged and set to 57.02 as well as the square of the index of refraction with 1.792.
Basis structural data derived from single crystal X-ray data of compounds Pd(PNP-C2-Me), 4a and Pd(PNP-C2-H), 4b. To calculate the catalytic cycle the metal centre palladium was exchanged with nickel. For these changes as well as any visualisations the programs Gausview09 and Mercury 3.9 were used.
Computing was performed on LEO3 and LEO4, high performance compute clusters of the Research Area Scientific Computing at the University of Innsbruck in operation since September 2011 and November 2018, respectively. LEO3 consists of 1944 Intel Xeon (Gulftown) computer cores and is equipped with 24 GB RAM per node, i.e., about 4 TB of main memory altogether. The nodes and GPFS storage system are joined by a 40 GB s−1 Infiniband high speed interconnect. LEO4 consists of 1452 Intel Xeion (Broadwell or Skylake) computer cores and is equipped with at least 65 GB RAM per node, i.e., 8.4 TB of main memory altogether. The system has a high-performance low-latency Infiniband interconnect for MPI communications between nodes and GPFS file system traffic.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from University Innsbruck, Hypo Tirol Bank, Tyrolean Research Fonds (TWF), the Tyrolean government (Land Tirol) and European Union (ERDF, Project Solar Hydrogen) is gratefully acknowledged. Cooperation with Bartenbach GmbH and V&F Analyse- und Messtechnik Absam was essential. This research was executed as a part of the center ‘materials & nanoscience’. Mencke and Thompson were supported by the Department of Energy, Office of Basic Energy Science (Award: DE-SC0016450).Notes and references
- Y. Surendranath, D. K. Bediako and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15617 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767 CrossRef CAS PubMed.
- D. Nocera, M. T. Huynh, D. Bediako, N. Li and C. Liu, Abstracts of Papers, 251st ACS National Meeting & Exposition, San Diego, CA, United States, 2016, March 13-17, 772.
- J. Gurudayal, D. Sabba, M. H. Kumar, L. H. Wong, J. Barber, M. Grätzel and N. Mathews, Nano Lett., 2015, 15, 3833 CrossRef PubMed.
- C. Wadia, A. P. Alivisatos and D. M. Kammen, Environ. Sci. Technol., 2009, 43, 2072 CrossRef CAS PubMed.
- S. M. Reimann and M. Manninen, Rev. Mod. Phys., 2002, 74, 1283 CrossRef CAS.
- M. Berr, A. Vaneski, A. S. Susha, J. Rodríguez-Fernández, M. Döblinger, F. Jäckel, A. L. Rogach and J. Feldmann, Appl. Phys. Lett., 2010, 97, 93108 CrossRef.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321 CrossRef CAS PubMed.
- T.-L. Li, C.-D. Cai, T.-F. Yeh and H. Teng, J. Alloys Compd., 2013, 550, 326 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851 CrossRef CAS.
- J. M. Olson, Photosynth. Res., 2006, 88, 109 CrossRef CAS.
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 5411 CrossRef CAS.
- M. Frey, ChemBioChem, 2002, 3, 153 CrossRef CAS.
- J. W. Peters, Curr. Opin. Struct. Biol., 1999, 9, 670 CrossRef CAS.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853 CrossRef CAS.
- Y. Nicolet, A. L. de Lacey, X. Vernède, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596 CrossRef CAS.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592 RSC.
- W. Lubitz, E. Reijerse and M. van Gastel, Chem. Rev., 2007, 107, 4331 CrossRef CAS.
- J. Y. Yang, R. M. Bullock, M. R. DuBois and D. L. DuBois, MRS Bull., 2011, 36, 39 CrossRef CAS.
- S.-P. Luo, E. Mejía, A. Friedrich, A. Pazidis, H. Junge, A.-E. Surkus, R. Jackstell, S. Denurra, S. Gladiali, S. Lochbrunner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 419 CrossRef CAS.
- E. Mejía, S.-P. Luo, M. Karnahl, A. Friedrich, S. Tschierlei, A.-E. Surkus, H. Junge, S. Gladiali, S. Lochbrunner and M. Beller, Chemistry, 2013, 19, 15972 CrossRef.
- S. Fischer, D. Hollmann, S. Tschierlei, M. Karnahl, N. Rockstroh, E. Barsch, P. Schwarzbach, S.-P. Luo, H. Junge, M. Beller, S. Lochbrunner, R. Ludwig and A. Brückner, ACS Catal., 2014, 4, 1845 CrossRef CAS.
- P.-A. Jacques, V. Artero, J. Pécaut and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20627 CrossRef CAS PubMed.
- E. S. Andreiadis, P.-A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pécaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2013, 5, 48 CrossRef CAS PubMed.
- P. Zhang, P.-A. Jacques, M. Chavarot-Kerlidou, M. Wang, L. Sun, M. Fontecave and V. Artero, Inorg. Chem., 2012, 51, 2115 CrossRef CAS PubMed.
- P. Du, J. Schneider, G. Luo, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 4952 CrossRef CAS PubMed.
- Z. Han, W. R. McNamara, M.-S. Eum, P. L. Holland and R. Eisenberg, Angew. Chem., Int. Ed., 2012, 51, 1667 CrossRef CAS.
- Z. Han, L. Shen, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2013, 135, 14659 CrossRef CAS.
- Z. Han and R. Eisenberg, Acc. Chem. Res., 2014, 47, 2537 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863 CrossRef CAS.
- M. Rakowski DuBois and D. L. DuBois, Acc. Chem. Res., 2009, 42, 1974 CrossRef CAS.
- J. Y. Yang, R. M. Bullock, W. J. Shaw, B. Twamley, K. Fraze, M. R. DuBois and D. L. DuBois, J. Am. Chem. Soc., 2009, 131, 5935 CrossRef CAS PubMed.
- K. Redin, A. D. Wilson, R. Newell, M. R. DuBois and D. L. DuBois, Inorg. Chem., 2007, 46, 1268 CrossRef CAS.
- G. A. Parada, Z. K. Goldsmith, S. Kolmar, B. Pettersson Rimgard, B. Q. Mercado, L. Hammarström, S. Hammes-Schiffer and J. M. Mayer, Science, 2019, 364, 471 CrossRef CAS PubMed.
- J. Bonin and M. Routier, Artif. Photosynth., 2013, 1, 6 Search PubMed.
- A. Aster, S. Wang, M. Mirmohades, C. Esmieu, G. Berggren, L. Hammarström and R. Lomoth, Chem. Sci., 2019, 10, 5582 RSC.
- M. Natali, S. Campagna and F. Scandola, Chem. Soc. Rev., 2014, 43, 4005 RSC.
- H.-J. Wolff, D. Burssher and U. E. Steiner, Pure Appl. Chem., 1995, 67, 167 CrossRef CAS.
- M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004 CrossRef CAS PubMed.
- R. I. Cukier and D. G. Nocera, Annu. Rev. Phys. Chem., 1998, 49, 337 CrossRef CAS.
- H. Decornez and S. Hammes-Schiffer, J. Phys. Chem. A, 2000, 104, 9370 CrossRef CAS.
- L. Pause, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2001, 123, 4886 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2007, 129, 5870 CrossRef CAS PubMed.
- C. Costentin, Chem. Rev., 2008, 108, 2145 CrossRef CAS.
- C. Costentin, M. Robert, J.-M. Savéant and A.-L. Teillout, ChemPhysChem, 2009, 10, 191 CrossRef CAS.
- C. Costentin, V. Hajj, C. Louault, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2011, 133, 19160 CrossRef CAS PubMed.
- J. Hudis and R. W. Dodson, J. Am. Chem. Soc., 1956, 78, 911 CrossRef CAS.
- H. L. Friedman and M. D. Newton, J. Electroanal. Chem. Interfacial Electrochem., 1986, 204, 21 CrossRef CAS.
- W. L. Reynolds and R. W. Lumry, J. Chem. Phys., 1955, 23, 2460 CrossRef CAS.
- R. A. Binstead, B. A. Moyer, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1981, 103, 2897 CrossRef CAS.
- R. I. Cukier, J. Phys. Chem., 1994, 98, 2377 CrossRef CAS.
- R. I. Cukier, J. Phys. Chem., 1995, 99, 16101 CrossRef CAS.
- X. G. Zhao and R. I. Cukier, J. Phys. Chem., 1995, 99, 945 CrossRef CAS.
- R. I. Cukier, J. Phys. Chem., 1996, 100, 15428 CrossRef CAS.
- J.-Y. Fang and S. Hammes-Schiffer, J. Chem. Phys., 1997, 107, 8933 CrossRef CAS.
- J.-Y. Fang and S. Hammes-Schiffer, J. Chem. Phys., 1997, 106, 8442 CrossRef CAS.
- C. Tommos and G. T. Babcock, Acc. Chem. Res., 1998, 31, 18 CrossRef CAS.
- C. W. Hoganson and G. T. Babcock, Science, 1997, 277, 1953 CrossRef CAS PubMed.
- G. T. Babcock, M. Espe, C. Hoganson, N. Lydakis-Simantiris, J. McCracken, W. Shi, S. Styring, C. Tommos, K. Warncke and B. R. Wood, Acta Chem. Scand., 1997, 51, 533 CrossRef CAS PubMed.
- C. Costentin, D. H. Evans, M. Robert, J.-M. Savéant and P. S. Singh, J. Am. Chem. Soc., 2005, 127, 12490 CrossRef CAS PubMed.
- F. Himo and P. E. M. Siegbahn, Chem. Rev., 2003, 103, 2421 CrossRef CAS PubMed.
- T. Lovell, Coord. Chem. Rev., 2003, 238–239, 211 CrossRef CAS.
- R. A. Binstead and T. J. Meyer, J. Am. Chem. Soc., 1987, 109, 3287 CrossRef CAS.
- R. A. Binstead, L. K. Stultz and T. J. Meyer, Inorg. Chem., 1995, 34, 546 CrossRef CAS.
- A. E. Pond, M. Sono, E. A. Elenkova, D. B. Goodin, A. M. English and J. H. Dawson, Biospectroscopy, 1999, 5(5), 42 CrossRef.
- M. van der Meer, E. Glais, I. Siewert and B. Sarkar, Angew. Chem., Int. Ed., 2015, 54, 13792 CrossRef CAS PubMed.
- M. Kügler, J. Scholz, A. Kronz and I. Siewert, Dalton Trans., 2016, 45, 6974 RSC.
- S. Nestke, E. Ronge and I. Siewert, Dalton Trans., 2018, 47, 10737 RSC.
- I. Siewert, Chemistry, 2015, 21, 15078 CrossRef CAS PubMed.
- M. Schulz, N. Hagmeyer, F. Wehmeyer, G. Lowe, M. Rosenkranz, B. Seidler, A. Popov, C. Streb, J. G. Vos and B. Dietzek, J. Am. Chem. Soc., 2020, 142, 15722 CrossRef CAS PubMed.
- D. Gao, R. Liu, J. Biskupek, U. Kaiser, Y.-F. Song and C. Streb, Angew. Chem., Int. Ed., 2019, 58, 4644 CrossRef CAS PubMed.
- W. Viertl, J. Pann, R. Pehn, H. Roithmeyer, M. Bendig, A. Rodríguez-Villalón, R. Bereiter, M. Heiderscheid, T. Müller, X. Zhao, T. S. Hofer, M. E. Thompson, S. Shi and P. Brueggeller, Faraday Discuss., 2019, 215, 141 RSC.
- CRC handbook of chemistry and physics, A ready-reference book of chemical and physical data, ed. R. C. Weast, CRC Pr, Cleveland, Ohio, 56th edn, 1975 Search PubMed.
- B. M. Fernandez, C. B. Schapira and S. Lamdan, J. Heterocycl. Chem., 1980, 17, 667 CrossRef CAS.
- H. P. Marshall and E. Grunwald, J. Am. Chem. Soc., 1954, 76, 2000 CrossRef CAS.
- V. Frenna, N. Vivona, G. Consiglio and D. Spinelli, J. Chem. Soc., Perkin Trans. 2, 1985, 1865 RSC.
- J. Hansson, Sven. Kem. Tidskr., 1955, 256, 67 Search PubMed.
- J. Hine and Y. Chou, J. Org. Chem., 1981, 46, 649 CrossRef CAS.
- J. Hine and M. N. Khan, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1992, 427, 31 Search PubMed.
- P. Zhang, M. Wang, J. Dong, X. Li, F. Wang, L. Wu and L. Sun, J. Phys. Chem. C, 2010, 114, 15868 CrossRef CAS.
- D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201 CrossRef CAS PubMed.
- J. Serrano-Becerra and D. Morales-Morales, Curr. Org. Synth., 2009, 6, 169 CrossRef CAS.
- P. Hermosilla, P. López, P. García-Orduña, F. J. Lahoz, V. Polo and M. A. Casado, Organometallics, 2018, 37, 2618 CrossRef CAS.
- (a) D. Rabinovich, J. Chem. Educ., 2000, 77, 311 CrossRef CAS; (b) S. Wiese, U. J. Kilgore, M.-H. Ho, S. Raugei, D. L. DuBois, R. M. Bullock and M. L. Helm, ACS Catal., 2013, 3, 2527 CrossRef CAS.
- (a) J. M. Darmon, N. Kumar, E. B. Hulley, C. J. Weiss, S. Raugei, R. M. Bullock and M. L. Helm, Chem. Sci., 2015, 6, 2737 RSC; (b) T. A. Tronic, W. Kaminsky, M. K. Coggins and J. M. Mayer, Inorg. Chem., 2012, 51, 10916 CrossRef CAS PubMed; (c) M. O’Hagan, W. J. Shaw, S. Raugei, S. Chen, J. Y. Yang, U. J. Kilgore, D. L. DuBois and R. M. Bullock, J. Am. Chem. Soc., 2011, 133, 14301 CrossRef PubMed.
- P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726 CrossRef CAS PubMed.
- F. A. Cotton, Advanced inorganic chemistry, Wiley, New York, Chichester, 1999 Search PubMed.
- J. Pann, W. Viertl, H. Roithmeyer, R. Pehn, T. S. Hofer and P. Brüggeller, Isr. J. Chem., 2022, 62, e202100035 CrossRef CAS.
- H. Roithmeyer, R. Pehn, J. Pann, W. Viertl, B. Trübenbacher, J. Dutzler, H. Kopacka, T. Müller and P. Brüggeller, Energy Adv., 2023, 2, 513 RSC.
- G. Czermak, PhD Thesis, University of Innsbruck, 2006.
Footnotes |
| † Dedicated by W. V.-S. to Prof. Dr Peter Brüggeller on the occasion of his retirement. |
| ‡ Electronic supplementary information (ESI) available: Syntheses for 4a-5b, molecule structure of a Pd(II)-pincer-complex, 4b, the catalytic cycles and energy diagrams of 5a and 5b, reaction pathways of reduction steps versus first protonation and hydrogen measurements for 4a-5b. CCDC 2282395. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ya00354j |
| This journal is © The Royal Society of Chemistry 2024 |