
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNaked eye detection of arsenite, arsenate, and H2S by a Schiff base naphthaldehyde conjugate using a single paper strip, based on a deprotonation mechanism†
Diptiman
De
ac,
Priyotosh
Ghosh
a,
Sriman
De
 b and
Prithidipa
Sahoo
*a
b and
Prithidipa
Sahoo
*a
aDepartment of Chemistry, Visva-Bharati University, Santiniketan-731235, India. E-mail: prithidipa.sahoo@visva-bharati.ac.in
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur-741246, India
cDepartment of Chemistry, Gushkara Mahavidyalaya, Guskara-713128, West Bengal, India
First published on 27th August 2024
Abstract
Considering the significant toxicity of arsenite (AsO2−), arsenate (AsO43−), and hydrogen sulphide (H2S), the early detection of these ions and gas using simple methods like naked-eye chemosensing could have substantial implications for environmental and industrial applications. With these factors in mind, we have developed a novel and straightforward colorimetric chemosensor called NADNP (2-hydroxy naphthaldehyde conjugated 2,4-dinitrophenyl hydrazine) for swift paper-based colorimetric detection of arsenite, arsenate, and H2S, based on a deprotonation mechanism. NADNP exhibits strong binding affinity towards sulfide, arsenite, and arsenate, with very lower detection limits (LOD) of 0.17 μM, 0.15 μM and 0.15 μM respectively, and the binding stoichiometry between these detected ions and NADNP is determined to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 through Job's plot analysis. Structural elucidation and electronic properties calculation have been conducted via DFT (Density Functional Theory) studies for correlation with the spectroscopic analyses. The ‘three-in-one’ paper strip-based chemosensor could be considered a promising colorimetric tool for rapid, cost-effective, selective, and sensitive “on-spot” sensing and monitoring of arsenite, arsenate, and sulfide in environmental samples.
:1 through Job's plot analysis. Structural elucidation and electronic properties calculation have been conducted via DFT (Density Functional Theory) studies for correlation with the spectroscopic analyses. The ‘three-in-one’ paper strip-based chemosensor could be considered a promising colorimetric tool for rapid, cost-effective, selective, and sensitive “on-spot” sensing and monitoring of arsenite, arsenate, and sulfide in environmental samples.
Environmental significanceArsenic (As) pollution in the groundwater of India poses significant health hazards, encompassing a spectrum of adverse effects on human health. Conversely, hydrogen sulfide (H2S), traditionally known as a toxic gas with a foul odor resembling rotten eggs, has emerged as a gasotransmitter. Arsenic and H2S contamination has significant ambivalent effects on society, primarily impacting human health, the environment, and economic development. Therefore, addressing these untoward effects is crucial for maintaining the environment and human health. To tackle this challenge, we have developed a novel and straightforward colorimetric chemosensor for rapid paper-based detection of arsenite, arsenate, and H2S. This user-friendly and cost-effective paper strip-based kit is an easy-to-use tool for common people and hence plays a vital role in protecting human health by enabling early detection, empowering communities, improving accessibility to testing, facilitating targeted interventions, preventing health risks, and promoting community engagement and awareness. |
Introduction
The term “environmental hazards” or “toxicants” describes compounds or pollutants found in the environment that can be detrimental to ecosystems, human health, or both. These substances can stem from natural sources or human activities and can cause harmful effects on living organisms and their surroundings. Heavy metals, volatile toxic gases, reactive oxygen species (ROS), persistent organic pollutants (POPs), particulate matter (PM), endocrine disrupting chemicals (EDCs), radioactive contaminants, etc. are a few examples of environmental hazards or toxicants. The development of selective and sensitive approaches for detecting toxic species in biological and environmental samples has grown in importance in recent years. In this research work, we have chosen arsenic (As3+/As5+) and hydrogen sulfide (H2S) as their toxic effects lead to adverse health effects through inhibition of essential enzymes, which ultimately leads to death from multi-system organ failure.Arsenic (As) stands as a highly toxic water pollutant with grave implications for human health and is designated as a Group-1 human carcinogen by the International Agency for Research on Cancer.1,2 The predominant forms of inorganic arsenic found in aquatic organisms are arsenite (As3+) and arsenate (As5+). Additionally, various organo-arsenics, such as arsenobetaine, arsenosugars, and methylated arsenic acids, exist, each possessing distinct chemical properties and toxicity levels.3,4 Arsenite is known to be more toxic and mobile in groundwater, while arsenate, though less toxic, is more commonly found in surface water and soils. Arsenic compounds have diverse applications in agriculture, serving as pesticides and alloying agents, and in the manufacture of semiconductors, lasers, transistors, metal adhesives, explosives, and pharmaceutical products. Arsenic interferes with the chemical and biochemical activities of P(V), a Group-VB element, within living systems. Human exposure to arsenic primarily occurs through drinking contaminated water or consuming plants grown in polluted areas.5–7 In India, regions such as West Bengal, Bihar, and Uttar Pradesh commonly experience groundwater contamination with arsenic levels exceeding the World Health Organization (WHO) guidelines for safe drinking water (10 μg L−1).8 Arsenite can induce DNA strand breakage, leading to skin lesions, increased cellular levels of nitric oxide and superoxide, disruption of cardiovascular and nervous system functions, heightened cancer risk for skin and other organs, and interference with protein phosphorylation through binding to vicinal thiols.9–14 Arsenate can disrupt cellular processes by substituting phosphate in biochemical reactions, thereby disrupting essential metabolic pathways such as ATP synthesis.15–18
Traditionally recognized as a toxic gas with a foul odor resembling rotten eggs, hydrogen sulfide (H2S) has emerged as the third gasotransmitter, following carbon monoxide (CO) and nitric oxide (NO).19,20 Even at low concentrations, H2S poses a wide array of adverse health effects due to its reducibility and high lipid solubility.21–23 Reportedly, the typical concentration of H2S in blood falls within the range of 10 to 600 μM.24–27 H2S primarily originates from the decomposition of organic compounds or as a by-product of various industries such as petroleum refining, farming, waste management, and natural gas production.28–30 Anaerobic bacteria can convert sulfate to sulfide through microbial reduction, and sulfur-containing amino acids present in biosystems also contribute to its production.31 In hydrolysis, sulfide ions produce H2S, which is even more toxic than S2−.32 However, elevated levels of H2S in living systems can lead to human diseases such as Alzheimer's disease, Down's syndrome, hypertension, and liver cirrhosis.33,34
Continuing exposure to these hazards can result in various health issues and consequently, detecting elements poses a formidable challenge for researchers. Numerous methods have been documented for determining arsenic in environmental samples like water, soil, and sediment.35–42 These sophisticated analytical techniques require extensive sample preparation procedures before instrumental measurement, making them laborious, time-consuming, and demand substantial reagent quantities. Moreover, their application at pollution sources is cumbersome. Alternatively, colorimetric methods employing organic reagents such as molybdenum blue,43 silver diethyldithiocarbamate,44 methylene blue,45 and rhodamine-B46 offer simplicity and cost-effectiveness for arsenic determination, allowing application directly at the sample source. However, these reagents have many shortcomings and drawbacks that remain a challenge. Thus, an alternative colorimetric method that is quite simple, inexpensive, accessible, sensitive, selective, and addresses the drawbacks of conventional techniques is desirable. In recent years, interest has surged in detection methods employing chemosensors, as they possess key practical sensing properties including high sensitivity, selectivity, portability, specificity, cost-effectiveness, and real-time sensing capability.47–54 To date, numerous chemosensors have been developed for the detection of arsenite/arsenate or sulfide ions34 (see the comparison in ESI Tables S1 and S2†). These probes typically operate via three crucial mechanisms: displacement of metal ions by sulfide ions, coordination of sulfide ions to the metal center, and sulfide-specific chemical conversion. Few Schiff base sulfide ion sensors have been reported, primarily based on the deprotonation of acidic hydrogen atoms. However, in many cases, the synthetic procedures are overly complex, or the detection limits are not sufficiently low.55,56 Consequently, there remains a scarcity of studies focusing on sulfide ion sensing via anion-induced deprotonation with high sensitivity.
Given the high toxicity of arsenic and H2S, early detection of these ions and gases through simple methods like naked-eye detection could significantly impact environmental and industrial applications. The attractive advantages of colorimetric analysis over traditional analytical procedures include cost-effectiveness, relatively simple synthetic procedures, high selectivity and sensitivity, and ease of operation. Therefore, our primary focus was on developing a simple Schiff's base colorimetric chemosensor that could enable naked-eye detection of arsenic (As3+/As5+) and hydrogen sulfide (H2S) together. Taking all these factors into consideration, we have developed a novel and straightforward colorimetric chemosensor named NADNP (2-hydroxy naphthaldehyde conjugated 2,4-dinitrophenyl hydrazine) with a good yield for the swift detection of arsenic (both As3+ and As5+) and hydrogen sulfide (H2S) (Scheme 1) following some literature reports.57,58 The uniqueness of this probe is that it can detect arsenic (both As3+ and As5+) and hydrogen sulfide (H2S) simultaneously with strong binding affinity as well as very low detection limits compared to many other reported probes (Tables S1 and S2†). To the best of the authors' knowledge, there are no reports in the literature on the detection of both As3+/As5+ and H2S colorimetrically using a single chemosensor like NADNP. The NADNP probe has been thoroughly characterized using techniques such as UV-vis, 1H NMR (Fig. S1†), 13C NMR (Fig. S2†), HRMS, and DFT studies.
 | ||
| Scheme 1 Colorimetric change of NADNP upon interaction with AsO2−, AsO43−, and H2S (S2−). | ||
The significance of this work lies in the fact that a single chemosensor can rapidly detect colorimetrically both arsenite (As3+) and arsenate (As5+) along with H2S (S2−). Moreover, the colorimetric chemosensor has been incorporated into paper strips for the rapid and selective visualization of these analytes without the need for sophisticated equipment, making it applicable to various types of water samples such as ground-level water and water from unused wells.
Results and discussion
UV-visible titration studies
Utilizing absorbance titration in DMSO:H2O (4:1 v/v, pH 7.0, 10 mM Tris–HCl buffer), the interactions between the NADNP probe and AsO2−/AsO43− were examined. A titration of a 10 μM solution of NADNP was performed against different known concentrations of AsO2− (0 to 0.52 μM) and AsO43− (0 to 2.35 μM). At 424 nm, NADNP showed a distinctive absorption band. Bathochromic shifts of 95 nm and 91 nm were seen in the absorption spectra for titration with AsO2− and AsO43−, signifying the transition of the intra-molecular charge transfer (ICT) due to the deprotonation of the NADNP sensor by AsO2−/AsO43−. After adding 0.52 μM AsO2− and 3.35 μM AsO43−, the optimum absorbance was measured at 519 nm and 515 nm, respectively. The newly developed bands at 519 and 515 nm showed a linear rise in intensity, while the band at 424 nm declined in both cases (Fig. 1). In both cases, there were distinct isosbestic points at 458 nm and 462 nm, suggesting the formation of deprotonated NADNP. This resulted in a robust interaction between NADNP and AsO2−/AsO43−, exhibiting a deep purple color.
 | ||
| Fig. 1 (a) Comparative absorbance spectra of NADNP (10 μM) after gradual addition of NaAsO2 (0 to 0.52 μM) to DMSO:H2O (4:1 v/v, pH 7.0, 10 mM Tris–HCl buffer). (b) Comparative absorbance spectra of NADNP (10 μM) after gradual addition of Na3AsO4 (0 to 3.35 μM) to DMSO:H2O (4:1 v/v, pH 7.0, 10 mM Tris–HCl buffer). | ||
A 1:1 stoichiometric ratio of NADNP with AsO2− and AsO43− was determined to be established by binding interactions from Job's plot (Fig. S3 and S4†). Absorption spectra revealed association constants of 3.7 × 103 M−1 and 4.9 × 102 M−1, respectively (Fig. S5 and S6†). The detection limit of NADNP for AsO2− and AsO43− was determined to be 0.15 μM in both the instances (Fig. S7 and S8†).
In DMSO:H2O (4:1 v/v, pH 7.0, 10 mM Tris–HCl buffer), a UV-visible titration experiment was carried out to determine the sensitivity of NADNP to H2S. NADNP showed an absorption band at 424 nm and the intensity at 424 nm gradually dropped with increasing concentration of S2− (which ranges from 0 to 0.169 μM) and simultaneously a new absorption peak appeared at 519 nm (Fig. 2). The definite isosbestic point at 460 nm implies the emergence of a stable complex with a specific stoichiometric ratio (1:1) while interacting with sulphide ions, which was further supported by Job's plot (Fig. S9†). The association constant of NADNP with H2S was found to be 1.2 × 104 M−1 with a very low detection limit of 0.17 μM (Fig. S10 and S11†). The bathochromic shift of the absorption band led to the proposal of an intramolecular charge transfer transition (ICT) that also occurred through the deprotonation of NADNP in the presence of S2− showing a distinct purple color.59
 | ||
| Fig. 2 Absorbance spectra of NADNP (10 μM) after gradual addition of H2S (0 to 0.169 μM) to DMSO:H2O (4:1 v/v, pH 7.0, 10 mM Tris–HCl buffer). | ||
Selectivity studies
With UV-visible spectroscopy, a comparative selectivity test was carried out in the presence of several metal ions and some relevant anions to evaluate the sensitivity of NADNP towards AsO2−, AsO43−, and H2S in DMSO:H2O (4:1 v/v, pH 7.0, 10 mM Tris–HCl buffer) (Fig. 3). NADNP changes its color from yellow to purple only in the presence of AsO2− and AsO43− (Fig. 4 and 5) while its colour remains the same in the other cases.
 | ||
| Fig. 3 (a) Color change observed for NADNP (10 μM) in DMSO:H2O (4:1 v/v, pH 7.0, 10 mM Tris–HCl buffer) with addition of 3 equiv. of different cations. (b) Colour change observed for NADNP (10 μM) in DMSO:H2O (4:1 v/v, pH 7.0, 10 mM Tris–HCl buffer) with addition of 3 equiv. of different anions. | ||
 | ||
| Fig. 4 (a) UV-vis spectral changes of NADNP (10 μM) with addition of 3 equiv. of AsO2−, AsO43− and other cations. (b) Histogram plot of NADNP with different cations. | ||
 | ||
| Fig. 5 (a) UV-vis spectral changes of NADNP (10 μM) with addition of 3 equiv. of H2S and other anions. (b) Histogram plot of NADNP with different anions. | ||
Alternatively, amongst all tested anions H2S and hypochlorite ions (ClO−) both turned NADNP to deep purple or pale pink, respectively (Fig. 3). However, ClO− displayed a significantly larger LOD value and a very low association constant, and we did not select it as a candidate for our experiment. Furthermore, when the other investigated cations and pertinent analytes are added, NADNP does not exhibit any discernible spectrum or color changes (Fig. 3).
Effect of pH
pH has a great impact on the binding mechanism. To examine the influence of pH on the binding of arsenite, arsenate, and sulphide with NADNP, the detection was shown in a range of solutions with pH values ranging from 1 to 14. Usually, deprotonation is less likely to happen in acidic pH, but above pH 9, a significant HO− was produced and interfered with NADNP. From the comprehensive pH study, it was determined that the effectiveness of NADNP shows its best at neutral pH while binding with analytes (Fig. S12†).Determination of a plausible mechanism through 1H NMR titration and mass spectroscopy
Depending on the results obtained from UV-visible spectroscopy, we carried out additional studies using 1H NMR titration and mass spectroscopy to validate the binding mechanism of cations with chemosensors. The 1H-NMR spectra of the complexes (DMSO-d6 and D2O) showed notable shifts while titrating NADNP with specific ions, namely AsO2− and AsO43−. The imine proton peak at 9.58 ppm showed a slight drop in intensity upon the sequential addition of both AsO2− and AsO43− and shifted upfield to 9.38 ppm (Fig. 6).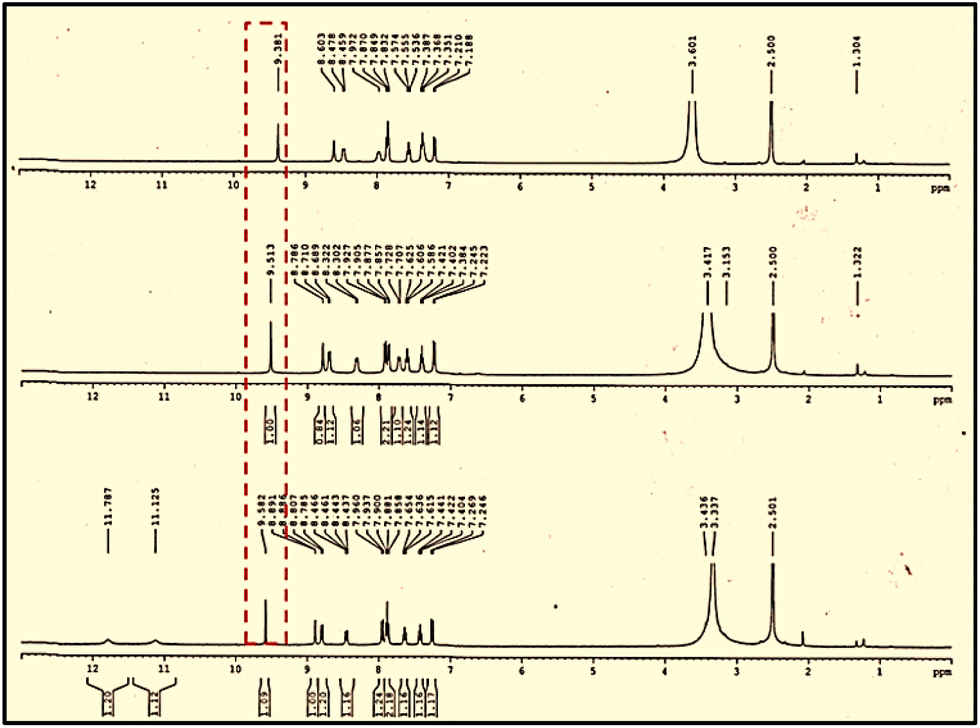 | ||
| Fig. 6 1H NMR titration spectrum [400 MHz] of NADNP in DMSO-d6 with the addition of different number of equiv. of NaAsO2/Na3AsO4 to D2O. | ||
This illustrated the rapid interaction of NADNP with AsO2−/AsO43− followed by the deprotonation of phenolic –OH in NADNP. Furthermore, there was an upfield shift in the aromatic region of both the DNP and naphthaldehyde moieties, indicating that electron delocalization was facilitated by the naphthalene imine moiety.60 The results obtained from NMR were validated by the mass spectra analysis of the aforementioned conjugates (Fig. S13 and S14†).
1H NMR titrations were carried out to understand the binding mechanism of NADNP with H2S. After the addition of 1.0 equivalent of H2S to the probe, the imine proton at 9.58 ppm moved upfield to 9.29 ppm (Fig. 7) along with the upfield movement of the aromatic protons. This implies the quick deprotonation of the phenolic –OH of NADNP by S2−, produced from H2S, and the electron was delocalized throughout the NADNP molecule. The HRMS study corroborated the fact supporting the genesis of NADNP–Na+ as a result of the deprotonation (Fig. S15†).
 | ||
| Fig. 7 1H NMR titration spectrum [400 MHz] of NADNP in d6-DMSO with the addition of different equiv. of H2S to D2O. | ||
The pH analysis of NADNP in the presence of strong bases like NaOH also provided evidence in favor of the deprotonation mechanism. The spectrum behavior and color resemble those of arsenite, arsenate, sulphide and ions. When potassium sulphide (K2S) and ammonium sulphide ((NH4)2S) were present in an aqueous solution, an identical outcome was once more attained. The basicity order of anions can be utilized to clarify the deprotonation of the receptor NADNP caused by sulphide ions or arsenite/arsenate. The anions in an aqueous solution have the following basicity order: S2− > AsO2−/AsO43− > PO42− > CO32− > CN− > OAc− > F−. As a result, the basic sulphide ion deprotonates the hydroxyl group of the naphthalene moiety and the negative charge is delocalized to the naphthalene imine moiety. Since a significant shift in absorbance and color was seen in an ideal aqueous medium, the impact of hydrogen bonding on the sensing mechanism is minimal. Because water itself regenerates the free NADNP from the anion-bonded receptor NADNP–S2− and competes with hydrogen bonding interactions. Additionally, the strong base sulphide ion (S2−) may be able to take up a proton from water to produce a relatively weak base ion (HS−), increasing the possibility of deprotonation by HS− to the receptor NADNP. The cations As3+/As5+ are present in aqueous medium as AsO2−/AsO43−; therefore, the acidic hydrogen of the hydroxyl group is deprotonated as S2− by the negative portion of arsenite/arsenate. Sulphide ions respond faster than the others while being more basic than the others. Consequently, other anions and cations are unable to participate in the deprotonation process.
Corroboration with computational studies
Density functional theory (DFT), which is included in the Gaussian 09 programme package, was used to assist with all computational calculations.61 Without any symmetry constraints, the B3LYP exchange–correlation functional62 and the 6-31+G(d) basis set63 were used to optimize the geometries of all the complexes (Table S3†). The optimized geometries were subjected to single-point calculations at the B3LYP/6-31+G(d,p) level, accounting for the solvation effects caused by DMSO (dielectric constant ε = 46.826). The SMD continuum solvation model64 developed in Gaussian09 was used to simulate the influence of solvent (Table S4†). We have carried out the TDDFT calculations at the B3LYP/6-31+G(d,p)/SMD//B3LYP/6-31+G(d) level of theory under the SMD model (solvent = DMSO) to interpret the UV-vis spectra. The recognizable band was observed at λ = 482.9 nm and 570.82 nm in the theoretical UV absorption of NADNP and NADNP–Na+, with oscillator strengths of 0.6222 and 0.4573, respectively.This indicates that HOMO → LUMO+1 excitations were classified in both cases (Fig. 8 and Table S5†), and this is consistent with the band of NADNP and NADNP–Na+ that is observed experimentally at λ = 424 nm and 519 nm, respectively. HOMO and LUMO+1 energy distributions for NADNP and NADNP–Na+ complexes have been calculated and highlighted in Fig. 8 (Table S6†). The observed redshift in the UV-vis spectra of NADNP–Na+ complexation is consistent with the significant lowering of the energy gap on NADNP–Na+ compared to the NADNP. For NADNP, the HOMO–LUMO+1 energy gap was determined to be 2.899 eV, but for NADNP–Na+, the energy gap was decreased to 2.499 eV. The UV-vis absorption shift (Δλmax) of 95 nm validates this transition in the HOMO–LUMO+1 energy gap. The naphthalene ring has the distribution of HOMOs for both NADNP and NADNP–Na+. The LUMOs of NADNP and NADNP–Na+, on the other hand, were localized in the 2,4-DNP moiety.
 | ||
| Fig. 8 Frontier molecular orbital comparison between NADNP and NADNP–Na+ (isosurface = 0.050 a.u.). | ||
Application
To assess the sensing efficacy of NADNP towards H2S, arsenite, and arsenate, a variety of water samples were taken from the villages in West Bengal where groundwater is severely polluted with these hazards. The residents of these communities suffer from serious chronic illnesses due to the regular usage of such water for irrigation, drinking, and domestic purposes. To raise awareness among common people, we have developed an economical, user-friendly sensing strip that can easily detect the contaminants and hence the risks of consuming groundwater.To validate the above-mentioned sensing strategy, the detection and estimation of those toxicants were performed in some polluted water collected from an abandoned well and a tube well. The experiment was conducted three times for each sample, and the swift color change of the paper strip demonstrated convincing results that have been shown in Fig. 9. Overall, the technique has great potential for on-site and real-time colorimetric monitoring of arsenite, arsenate, and sulfide in environmental and biological samples (Fig. S16–S18†). Also, statistical evaluation of the paper strip and a spiked recovery experiment were performed (Tables S3–S5†). We also checked the differences in sensor sensitivity between solutions and paper strips (Table S6†).
 | ||
| Fig. 9 Photographs of two different water samples and portable paper strips as well as solutions after being in contact with water samples spiked with different volumes of unknown concentrations of arsenic and H2S. | ||
Experimental section
Materials and methods
We purchased sodium arsenite (NaAsO2), sodium arsenate (Na3AsO4), sodium hypochlorite (NaOCl), sodium sulphide (Na2S), 2,4-dinitrophenyl hydrazine (2,4-DNP), and 2-hydroxy naphthaldehyde from Sigma-Aldrich Pvt. Ltd. All materials were obtained from commercial suppliers, unless otherwise noted, and were utilized without further purification. Standard techniques were used to dry the solvents. All studies used water from an Elix Millipore system. NMR spectra at 400 MHz were acquired with Bruker Ascend 400 apparatus (Switzerland). For NMR spectra, DMSO-d6 was utilized as the solvent; for NMR titration, DMSO-d6 and D2O were utilized, with TMS (tetramethyl silane) acting as the internal standard. The units used to indicate chemical shifts are δ ppm, while the coupling constants (1H–1H and 1H–C) are specified in Hz. A Hitachi U-2910 spectrophotometer (Japan) was used to record the UV-vis spectra. HRMS spectra were recorded using a Waters Mass spectrometer (Xevo G2-Xs QTof, Massachusetts). The abbreviations listed below indicating spin multiplicities in 1H NMR spectra: s = singlet; d = doublet; t = triplet; m = multiplet.Synthetic procedure
In a single process, 2-hydroxy naphthaldehyde and 2,4-dinitrophenyl hydrazine were used for production of NADNP. 30 ml of methanol and 1.40 g (8.13 mmol) of 2-hydroxy naphthaldehyde were added to a 100 ml round-bottom flask. To this combination, 2,4-DNP (1.93 g, 9.75 mmol) was added. Following that, the mixture was refluxed for 16 hours at 70 °C (Scheme 2). Cold brine was added to the reaction mixture after the reaction finished (as seen by thin layer chromatography) to precipitate the product once the methanol evaporated. The red precipitate was filtered and repeatedly cleaned with water. To obtain the product, it was first dried in a hot air oven and then purified using column chromatography utilizing the eluent CHCl3:PET (3:1 v/v).
 | ||
| Scheme 2 Synthesis of NADNP. | ||
1H NMR (400 MHz, DMSO-d6): δ (ppm) = 7.26–7.24 (d, 1H, J = 8 Hz), 7.44–7.40 (t, 1H, J = 8 Hz), 7.65–7.61 (t, 1H, J = 8 Hz), 7.90–7.85 (t, 2H, 8 Hz), 7.96–7.93 (d, 1H, J = 12 Hz), 8.46–8.44 (d, 1H, J = 12 Hz), 8.80–8.78 (d, 1H, J = 8 Hz), 8.89 (s, 1H), 9.58 (s, 1H), 11.12 (s, 1H), 11.78 (s, 1H) (Fig. S1†). 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 109.93, 116.03, 118.24, 123.21, 123.67, 128.13, 128.87, 130.20, 131.39, 133.44, 143.75, 149.12, 157.68 (Fig. S2†). Analytical data for C17H12N4O5, HRMS (ESI-MS) (m/z) 353.1587 [M + H]+ (353.0880 calc.).
UV-vis titration studies
In DMSO, a NADNP solution with a concentration of 1 × 10−5 M was created. Millipore water was used to create solutions of NaAsO2, Na3AsO4, and Na2S at concentrations of 5 × 10−3 M and 1 × 10−3 M, respectively. All experiments were conducted in DMSO:H2O (4:1 v/v, pH 7.0, 10 mM Tris–HCl buffer). The 1 × 10−5 M NADNP solution was continuously added to a quartz optical cell that had a 1 cm optical path length during the titration procedure. Several analytes' stock solutions were gradually added to the quartz optical cell using a micropipette.
Conclusions
In summary, we have successfully established a unique colorimetric sensor (NADNP) for the quick and straightforward detection of arsenite, arsenate, and sulfide in an aqueous solution. With a very low detection limit, NADNP exhibits a rapid color change from yellow to purple, demonstrating selective detection even in the presence of significant metal ions and anions. Additionally, theoretical calculations have been performed to clarify the binding mechanism of NADNP with the analytes. More intriguingly, for the use of NADNP in practical application, a sensor-coated paper strip has been made for common people to verify whether the groundwater is contaminated with these hazards. Overall, the technique introduced in this study has great potential for on-site and real-time colorimetric monitoring of arsenite, arsenate, and sulfide in environmental and biological samples.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† Raw data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
DD: writing – original draft, visualization, validation, methodology, investigation, formal analysis, editing, and performing all the spectroscopic experiments as well as interpreting the data. PG: helping throughout the spectroscopic analysis, data interpretation, theoretical study, and ESI† preparation. SD: performed the theoretical calculation, wrote the theoretical portion, and drafted the review. PS: conceptualization, designing the experiment, funding acquisition, data correction, writing – review & editing, visualization, investigation, supervision.Conflicts of interest
There are no conflicts of interest.Acknowledgements
P. S. acknowledges SERB, India, for awarding her a SERB Power grant [Project file no. Ref. No. SPG/2020/000713]. Authors sincerely acknowledge the Department of Chemistry, Visva-Bharati, Santiniketan, India for providing infrastructural facilities. The authors also acknowledge Jiko Raut and Shrodha Mondal for helping with the application part.Notes and references
- Priority list of hazardous substances, http://www.atsdr.cdc.gov/spl/index.html.
- International Agency for Research on Cancer, Some Metals and Metallic Compounds, IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, World Health Organisation, 1980, vol. 23 Search PubMed.
- B. K. Mandal and K. T. Suzuki, Talanta, 2002, 58, 201–235 CrossRef CAS.
- P. L. Smedley and D. G. Kinniburgh, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- S. J. Hill, Chem. Soc. Rev., 1997, 26, 291–298 RSC.
- K. Shrivas and K. S. Patel, Anal. Lett., 2004, 37, 333–344 CrossRef CAS.
- H. K. Das, A. K. Mitra, P. K. Sengupta, A. Hossain, F. Islam and G. H. Rabbani, Environ. Int., 2004, 30, 381–385 CrossRef PubMed.
- World Health Organization, Guidelines for Drinking Water Quality, 1993, p. , p. 41 Search PubMed.
- M. M. Rahman, K. Paul, T. R. Chowdhury, C. R. Chanda and D. Lodh, Arsenic Exposure and Health Effects, Amsterdam: Elsevier, 2001, p. 27 Search PubMed.
- K. S. Patel, K. Shrivas, R. Brandt, N. Jokubowski and W. Corns, Environ. Geochem. Health, 2005, 27, 131–145 CrossRef CAS.
- C. J. Chen, Y. M. Hsueh, M. S. Lai, M. P. Shyu, S. Y. Chen, M. M. Wu, T. L. Kuo and T. Y. Tai, Hypertension, 1995, 25, 53–60 CrossRef CAS PubMed.
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713–764 CrossRef CAS.
- M. Morita and J. S. Edmonds, Pure Appl. Chem., 1992, 64, 575–590 CrossRef.
- A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS.
- M. F. Hughes, Toxicol. Lett., 2002, 133(1), 1–16 CrossRef CAS.
- (a) Z. Drobna, M. Styblo and D. J. Thomas, Curr. Protoc. Toxicol., 2009, 42(431), 4 Search PubMed; (b) L. M. Tam, N. E. Price and Y. Wang, Chem. Res. Toxicol., 2020, 33(3), 709–726 Search PubMed.
- R. K. Kwok, R. B. Kaufmann and M. Jakariya, J. Health Popul. Nutr., 2006, 24(2), 190–205 Search PubMed.
- J. C. States, S. Srivastava, Y. Chen and A. Barchowsky, Toxicol. Sci., 2009, 107(2), 312–323 CrossRef CAS PubMed.
- K. R. Olson, J. A. Donald, R. A. Dombkowski and S. F. Perry, Respir. Physiol. Neurobiol., 2012, 184, 117 CrossRef CAS PubMed.
- L. Ling, R. Peter and P. K. Moore, Annu. Rev. Pharmacol. Toxicol., 2011, 51, 169 CrossRef.
- X. Lin, X. Lu, J. Zhou, H. Ren, X. Dong, W. Zhao and Z. Chen, Spectrochim. Acta, Part A, 2019, 213, 416–422 CrossRef CAS PubMed.
- A. I. Ayesh, A. F. S. Abu-Hani, S. T. Mahmoud and Y. Haik, Sens. Actuators, B, 2016, 231, 593–600 CrossRef CAS.
- D. Fu, W. Zhi, L. Lv, Y. Luo, X. Xiong, X. Kang, w. Hou, J. Yan, H. Zhao and L. Zheng, Spectrochim. Acta, Part A, 2020, 224, 117391 CrossRef CAS.
- K. Abe and H. Kimura, J. Neurosci., 1996, 16, 1066 CrossRef CAS PubMed.
- A. Papapetropoulos, A. Pyriochou, Z. Altaany, G. Yang, A. Marazioti, Z. Zhou, M. G. Jeschke, L. K. Branski, D. N. Herndon and R. Wang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21972 CrossRef CAS.
- L. Li, M. Bhatia, Y. Z. Zhu, Y. C. Zhu, R. D. Ramnath, Z. J. Wang, F. B. M. Anuar, M. Whiteman, M. Salto-Tellez and P. K. Moore, FASEB J., 2005, 19, 1196 CrossRef CAS.
- World Health Organization, Hydrogen Sulfide, Environmental Health Criteria No. 19, Geneva, 1981 Search PubMed.
- Y. Hu, X. Li, Z. Zhang, G. Chen, H. Liang, D. Zhang and C. Liu, Sens. Actuators, B, 2018, 272, 308–313 CrossRef CAS.
- S. Tang, T. Qi, D. Xia, M. Xu, M. Xu, A. Zhu, W. Shen and H. K. Lee, Anal. Chem., 2019, 91, 5888–5895 CrossRef CAS PubMed.
- R. K. Jha, J. V. D'Costa, N. Sakhuja and N. Bhat, Sens. Actuators, B, 2019, 297, 126687 CrossRef CAS.
- C. Szabó, Nat. Rev. Drug Discovery, 2007, 6, 917–935 CrossRef.
- B. Peng, W. Chen, C. Liu, E. W. Rosser, A. Pacheco, Y. Zhao, H. C. Aguilar and M. Xian, Chem.–Eur. J., 2014, 20, 1010–1016 CrossRef CAS.
- A. H. Sharif, M. Iqbal and B. Manhoosh, et al. , Neurochem. Res., 2023, 48, 1981–1996 CrossRef CAS.
- R. McRae, P. Bagchi, S. Sumalekshmy and C. J. Fahrni, Chem. Rev., 2009, 109, 4780–4827 CrossRef CAS.
- E. M. Becker, M. B. Dessuy, W. Boschetti, M. G. R. Vale, S. L. C. Ferreira and B. Welz, Spectrochim. Acta, Part B, 2012, 71–72, 102–106 CrossRef CAS.
- H. Abdolmohammad-Zadeh, A. Jouyban and R. Amini, Talanta, 2013, 16, 604–610 CrossRef PubMed.
- P. Cava-Montesinos, M. L. Cervera, A. Pastor and M. de la Guardia, Talanta, 2003, 60, 787–799 CrossRef CAS.
- J. Entwisle and R. Hearn, Spectrochim. Acta, Part B, 2006, 61, 438–443 CrossRef.
- J. F. R. Paula, R. E. S. Froes-Silva and V. S. T. Ciminelli, Microchem. J., 2012, 104, 12–16 CrossRef CAS.
- H. Barros, L. M. M. Parra, L. Bennun and E. D. Greaves, Spectrochim. Acta, Part B, 2010, 65, 489–492 CrossRef.
- Z. L. Gang and X. J. Huang, Trends Anal. Chem., 2014, 60, 25–35 CrossRef.
- K. Deepa and Y. Lingappa, Spectrochim. Acta, Part A, 2014, 124, 102–107 CrossRef CAS PubMed.
- V. S. S. Rao, S. C. S. Rajan and N. V. Rao, Talanta, 1993, 40, 653–656 CrossRef CAS PubMed.
- G. Stratton and H. C. Whitehead, J.–Am. Water Works Assoc., 1962, 54, 861–864 CrossRef CAS.
- S. Kundu, S. K. Ghosh, M. Mandal, T. Pal and A. Pal, Talanta, 2002, 58, 935–942 CrossRef CAS PubMed.
- A. Pillai, G. Sunita and V. K. Gupta, Anal. Chim. Acta, 2000, 408, 111–115 CrossRef CAS.
- S. Liu, L. Zhang, T. Yang, H. Yang, K. Y. Zhang, X. Zhao, W. Lv, Q. Yu, X. Zhang, Q. Zhao, X. Liu and W. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 11013–11017 CrossRef CAS.
- H. S. Sarkar, A. Ghosh, S. Das, P. K. Maiti, S. Maitra, S. Mandal and P. Sahoo, Sci. Rep., 2018, 8, 3402–3409 CrossRef PubMed.
- L. Qin, G. Zeng, C. Lai, D. Huang, C. Zhang, P. Xu, T. Hu, X. Liu, M. Cheng, Y. Liu, L. Hu and Y. Zhou, Sens. Actuators, B, 2017, 24, 3946–3954 Search PubMed.
- S. Das and P. Sahoo, Sens. Actuators, B, 2019, 291, 287–292 CrossRef CAS.
- R. M. Mohamed, S. M. El-Sheikh, M. W. Kadi, A. A. Labib and S. M. Sheta, RSC Adv., 2023, 13, 11751–11761 RSC.
- R. R. Sheta, S. M. Sheta, M. A. Hamouda, S. M. El-Sheikh, A. T. Kandil and O. I. Ali, J. Environ. Radioact., 2023, 270, 107287 CrossRef.
- S. M. Sheta, A. S. Abdelelmoaty, H. M. Abu Hashish, A. M. Kamel, M. M. Abd-Elzaher and S. M. El-Sheikh, Anal. Bioanal. Chem., 2022, 414, 8379–8388 CrossRef CAS.
- S. M. Sheta, M. A. Hamouda, O. I. Ali, A. T. Kandil, R. R. Sheha and S. M. El-Sheikh, RSC Adv., 2023, 13, 25182–25208 RSC.
- (a) S. Y. Lee and C. Kim, RSC Adv., 2016, 6, 85091 RSC; (b) D. Y. Park, K. Y. Ryu, J. A. Kim, S. Y. Kim and C. Kim, Tetrahedron, 2016, 72, 3930–3938 CrossRef CAS.
- K. Y. Ryu, S. Y. Lee, D. Y. Park, S. Y. Kim and C. Kim, Sens. Actuators, B, 2017, 242, 792–800 CrossRef CAS.
- J. Chen, Q. Lin, Q. Li, W. T. Li, Y.-M. Zhang and T. B. Wei, RSC Adv., 2016, 6(89), 86627–86631 RSC.
- R. Behura, P. P. Dash, P. Mohanty, S. Behera, M. Mohanty, R. Dinda, S. K. Behera, A. K. Barick and B. R. Jali, J. Mol. Struct., 2022, 1264, 133310 CrossRef CAS.
- S. Y. Lee and C. Kim, RSC Adv., 2016, 6, 85091–85099 RSC.
- N. Yadava and A. K. Singh, RSC Adv., 2016, 6, 100136–100144 RSC.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- W. J. Hehre, L. Radom, L. V. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, NY, 1986 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4va00213j |
| This journal is © The Royal Society of Chemistry 2024 |