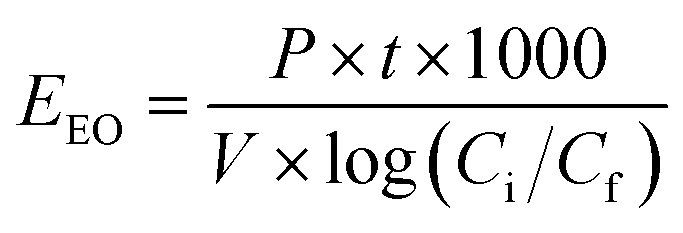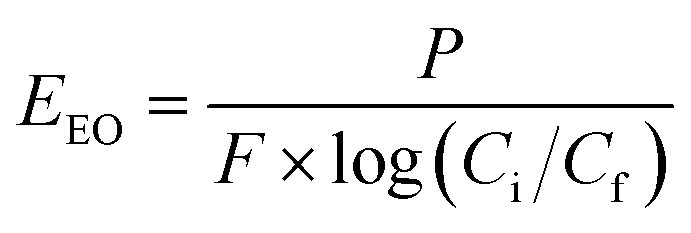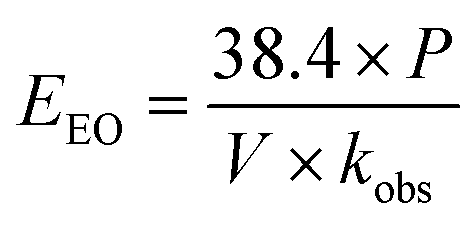From batch system toward continuous UV/PS based AOP reactor: the case of tramadol effluent degradation†
Received
3rd April 2024
, Accepted 25th June 2024
First published on 26th June 2024
Abstract
The stability of pharmaceutical active ingredients (APIs) and their resistance to conventional treatment methods necessitates the development of degradation methods as point-source treatment before mixing with municipal wastewater. Advanced oxidation processes utilize oxidants such as H2O2 or persulfate (PS) to treat organic contaminants and have shown promising results for eliminating APIs from wastewater. This research investigated the degradation of tramadol (TRA), a fully synthetic opioid, in a UVC/PS system, which was selected after evaluating thermal and simulated solar activation techniques. Different concentrations of PS were tested, and the UVC/PS system with [PS]0 = 0.4 mM achieved complete degradation of 10 mg L−1 [TRA]0 in 6 min with kobs of 0.90 min−1 and was chosen for this study. The system was evaluated under different conditions and showed a decrease in reaction rate under acidic conditions and in the presence of bicarbonates or competing natural organic matter. Additionally, high levels of chlorides and nitrates inhibited the degradation. Building on insights from batch treatment experiments, a pilot-scale treatment plant was developed utilizing elements from commercially available UV water-disinfection kits for continuous-flow treatment of pharmaceutical industry effluent. After optimization, the system achieved full degradation of 360 L per day of 10 mg L−1 [TRA]0 at a cost of $0.296 per m3.
Environmental significance
Tramadol is an opioid that causes damage when improperly disposed of in the environment, disrupting aquatic ecosystems and particularly affecting crustaceans. One major way pharmaceuticals such as tramadol infiltrate the environment is through pharmaceutical industry effluents. This research evaluated UVC/PS as an advanced oxidation process for the elimination of tramadol from pharmaceutical effluent, hence tackling the issue at the source. The UVC/PS system was applied in both batch treatment and continuous-flow treatment. A new pilot-scale setup for continuous treatment was developed for this purpose and showed promising results regarding scalability and adaptability to other pollutants.
|
1. Introduction
Pharmaceuticals and personal care products (PPCPs) are organic and inorganic contaminants that have gained global interest in recent years owing to their occurrence in various water bodies and the risks they pose to both human and environmental health. PPCPs encompass a wide range of chemical compounds, including medications, cosmetics, fragrances, and personal hygiene products. The widespread use and improper disposal of these compounds contribute to their continuous release into surface, ground, and drinking water. The ubiquity of these contaminants in the environment1–4 can be linked to two main factors: the increases in human population and global livestock production.5 In 2007, the European Union (EU) added PPCPs to its list of priority pollutants, aiming to remove them from the environment within 20 years to protect the ecological systems to which they are harmful.6 PPCPs infiltrate the environment through various pathways. Understanding these pathways is crucial for developing effective strategies to mitigate the impact of PPCPs. Major sources of PPCPs include wastewater treatment plants (WWTPs), agricultural activities, landfill leachate, healthcare centers, and direct discharge from industrial facilities.7
An important class of compounds contributing to PPCP pollution are active pharmaceutical ingredients (APIs). These are biologically active ingredients that are often resistant to conventional/biological wastewater treatment methods,4,8–11 which can be attributed to the stability of these compounds, rendering them less susceptible to full degradation by natural photolysis and biodegradation, the latter being one of the main methods used in wastewater treatment plants (WWTPs).12,13 Many WWTPs employ activated sludge as a primary treatment process, which is a process combining adsorption and biodegradation to remove contaminants. However, this introduces another challenge for conventional WWTPs since the low pKa of many APIs means that they occur as ions in neutral pH conditions and are less likely to be removed by adsorption onto the sludge.14 Furthermore, the stability of APIs necessitates long sludge retention times for effective degradation, contributing to the cost of treatment.15 The removal of pharmaceuticals from wastewater by adsorption is a widely studied strategy.16 Activated carbon (AC) is a common adsorbent that can be functionalized to treat different kinds of pollutants owing to its high surface area, stability, and low cost. Balarak et al. studied the removal of ciprofloxacin from water using AC magnetized using iron(III) oxide nanoparticles17 and ZnO nanoparticles18 to improve its adsorptivity. Loading such photocatalysts onto AC or graphene oxide can lead to hybrid adsorption/photodegradation strategies where reactive oxygen species are released under the right activation conditions to further degrade contaminants. Photocatalysts such as TiO2 are often employed in these strategies.19
The transformation products (TPs) produced during the treatment of APIs present an additional challenge since they may possess equivalent or higher toxicity than the original pollutant.8,20
Among these APIs is tramadol (TRA), a fully synthetic opioid that is widely prescribed for relieving moderate-to-severe pain and as a serotonin-norepinephrine reuptake inhibitor (SNRI).21 A recent study showed that TRA is one of the most widely consumed opioids, linking that to its high risk of dependency, and reported an increase in consumption of TRA globally between 2009–2019, particularly in high-income countries.22 Multiple studies reported the occurrence of this drug in multiple wastewater samples at relatively high concentrations (up to 6 μg L−1).8,23–25 The effects of TRA on the environment are particularly apparent in terms of the disruption of aquatic ecosystems,26–28 particularly regarding crustaceans. One study found that exposure to environmentally relevant concentrations of TRA causes stress-related heart rate increase in crayfish.29 Another study found that the occurrence of TRA in water at concentrations as low as 1 μg L−1 can affect the behavior of native European fish, causing them to become inactive and less exploratory.30
TRA has proven difficult to treat using conventional methods.31 Studies of multiple WWTPs reported difficulties in effectively removing TRA.2,32–36 Loganathan et al. emphasized the challenging nature of the wastewater matrix as one of the main factors causing difficulty in eliminating PPCPs in WWTPs using conventional treatment methods.37 Alternatively, treatment of PPCPs can be employed at the point source. Pharmaceutical industry facilities and hospitals are the leading sources of PPCP leaks, including TRA, into the environment. Unlike municipal wastewater, the effluent makeup from these facilities and their specific pollutants are often known, creating an opportunity to use specifically tailored remediation strategies to ensure environmental compliance before discharge.
Research on the on-site treatment of TRA from industrial effluent is limited. Treatment of industrial effluents containing other pharmaceuticals using activated sludge,38 chemical and electrocoagulation,39 and oxidative treatment methods including electro-Fenton, photo-Fenton, photocatalytic oxidation, and hydrodynamic cavitation have been reported.39,40Table 1 presents the results of several studies on the degradation of TRA in aqueous solutions, specifically in DI water or ultrapure water. These results are comparable to those obtained in our study. Hybrid processes were also explored. Eniola et al. recommended the use of hybrid technologies owing to their ability to achieve higher removal efficiencies of pollutants with different properties.41 The nature of the treated effluent is a crucial parameter for choosing the appropriate treatment strategy. For example, photo-induced oxidation strategies are not suitable for effluents with high levels of organic contamination, particularly if the contamination results in the coloration of the effluent.39 Combining such techniques with a pre-treatment process would be ideal.
Table 1 Comparison table showing documented outcomes of using batch/continuous AOPs for the degradation of tramadol in aqueous solutions
| Method |
Operating conditions |
Mode |
Degradation efficiency |
k
obs
|
Ref. |
| Gamma irradiation, nanofiltration |
[TRA]0 = 20 mg L−1 |
Batch |
100% |
— |
108
|
| Dose rate = 46 Gy min−1 |
| Absorbed dose = 5 kGy |
| Electro-Fenton |
[TRA]0 = 26.3 mg L−1, [Fe2+] = 0.2 mM |
Batch |
100% (in 8 min) |
0.70 min−1 |
103
|
| Constant current = 500 mA |
| [TRA]0 = 39.51 mg L−1, [Fe2+] = 0.1 mM |
Batch |
100% (in 12 min) |
0.498 min−1 |
109
|
| Constant current = 300 mA |
| Solar/TiO2 |
[TRA]0 = 10 mg L−1, [TiO2] = 100 mg L−1 |
Batch |
100% (in 30 min) |
0.153 min−1 |
68
|
| Light source: solar simulator |
| UV/H2O2 |
[TRA]0 = 0.005 mg L−1, [H2O2] = 1 mg L−1 |
Batch |
100% (in 15 min) |
0.2005 min−1 |
110
|
| Incident photon flux = 1.04 × 10−6 Einstein per L per s−1 |
| UVC/Cl2 |
[TRA]0 = 0.005 mg L−1, [Cl2] = 1 mg L−1 |
Batch |
100% (in 10 min) |
0.3785 min−1 |
110
|
| Incident photon flux = 1.04 × 10−6 Einstein per L per s−1 |
| UVC/PS |
[TRA]0 = 8.8 mg L−1, [PS] = 0.380 mM |
Batch |
99% (in 10 min) |
7.85 m3 kW−1 h−1 |
59
|
| Incident photon flux = 2.84 × 10−7 Einstein per s |
| UVC/PS |
[TRA]0 = 10 mg L−1, [PS] = 0.4 mM |
Batch |
100% (in 6 min) |
0.90 min−1 |
This work |
| Incident photon flux = 8.07 × 10−8 Einstein per s |
| Pulsed-corona discharge/PS |
[TRA]0 = 8.8 mg L−1, [PS] = 0.380 mM |
Continuous flow |
94% (in 28 min) |
3.10 m3 kW−1 h−1 |
59
|
| Flow rate = 1 m3 h−1 (circulated) 50 pps with input 9W |
| Energy consumed = 0.42 kW h m−3 |
| Dielectric barrier discharge plasma treatment |
[TRA]0 = 5 mg L−1 |
Continuous flow |
93% (in 60 min) |
0.056 min−1 |
111
|
| Input voltage = 8 kV |
| UVC/PS |
[TRA]0 = 10 mg L−1, [PS] = 0.4 mM |
Continuous flow |
100% |
0.090 min−1 |
This work |
| Flow rate = 0.014 m3 h−1 |
Persulfate-based advanced oxidation processes (PS-AOPs) have become a popular choice among AOPs owing to multiple factors. First, sulfate radicals have more effective redox potential than hydroxyl radicals, with E° = 2.60–3.10 V compared to 1.90–2.70 V, respectively, at neutral to high pH, which is often the relevant pH in developing water treatment strategies.42 Second, sulfate radicals are better mineralizing agents than hydroxyl radicals owing to faster reaction rates and higher selectivity for attacking certain functional groups.42 Third, PS-AOPs systems have lower operational costs owing to the widespread availability of persulfate salts and lower transportation costs compared to those of hydrogen peroxide, which is liquid and corrosive.43
The activation of persulfate can be achieved chemically, e.g., using Fe2+ (eqn (1))
| | | S2O82− + Fe2+ → SO42− + SO4˙− + Fe3+ | (1) |
Other possible methods include thermal,44,45 ultrasound,46 and/or UV-activation (eqn (2))
Various heterogeneous catalysts have also been used.42,47,48
Most research on PPCP treatment using oxidation methods was experimental using batch reactors and focused on the theoretical aspects rather than the real-world applications. Some techniques may show promising laboratory results; however, studies often emphasize the importance of further investigating their viability in real-world applications since they may be difficult to scale up without exacerbating the treatment cost. Scaling up a technology requires initial evaluation using a pilot-scale reactor; however, published research is limited. Continuous treatment systems have the advantage of lower reactor volume requirements. Therefore, such systems are more adaptable for pilot and industrial-scale reactors. Grilla et al. developed a pilot-scale continuous flow reactor to evaluate UV/TiO2/PS in a real-life scenario. The study was conducted on a solar-powered pilot plant, and different matrix effects were tested for the degradation of trimethoprim.49 Plakas et al. developed a fully automated photocatalytic membrane reactor for the degradation of diclofenac using a UVC/TiO2/ultrafiltration system, achieving up to 100% degradation; however, total organic carbon (TOC) removal was limited to 52%.50
In this work, the degradation of TRA by a UVC/PS system was evaluated under different matrix conditions. TRA degradation was evaluated in solutions with varied pH, salinity, hardness, and nitrate levels, in addition to the presence of natural organic matter and dissolved oxygen. The performance of the UVC/PS system was subsequently compared to that of UVC/H2O2 and UVC/PS/H2O2 systems, and the system was evaluated for real-life application by testing it on a simulated effluent. Furthermore, the transformation products were identified to elucidate the degradation pathway. The findings were used to optimize a newly developed, mobile, pilot-scale continuous flow treatment plant. To the best of our knowledge, this is the first study that investigated the degradation of tramadol by a UVC/PS and its application in a continuous-flow system.
2. Chemicals
Tramadol hydrochloride was obtained by dissolving TRAMAL® 50 mg capsules (STADA, Italy) in deionized (DI) water and filtering twice using 0.45 μm S-Pak® membrane filters (Merck, Germany). Sodium persulfate (PS) (Na2S2O8, ≥99.0%) was purchased from Sigma-Aldrich, China. To study matrix effects on the degradation, sodium phosphate monobasic (NaH2PO4, ≥99.0%), sodium phosphate dibasic (Na2HPO4, 98–100.5%), sodium bicarbonate (NaHCO3, ≥99.7%), humic acid sodium salt (technical grade), and potassium iodide (KI, 99.0–100.5%) for the persulfate quantification method were purchased from Sigma-Aldrich, Germany. Sodium chloride (NaCl, ≥99.5%), mild steel (MS)-grade solvents (water, methanol, and acetonitrile), and MS-grade formic acid were purchased from Fisher Scientific, UK. High-performance liquid chromatography (HPLC)-grade acetonitrile and methanol were purchased from Honeywell, Germany. Hydrogen peroxide (PERDROGEN™ ≥30% w/w) for use as a comparative oxidant to PS was obtained from Sigma Aldrich, Germany. DI water was used to prepare all solutions used in this research.
3. Reaction setups
3.1. Batch reactor
A bench scale experimental setup was constructed using elements of a commercial UVC water disinfection apparatus: a low-pressure mercury lamp (LPHgL) (Philips TUV 11W G11 T5, Poland) and its corresponding quartz insert tube (Fig. 1). This setup was used in previous studies by our research group.51–53 A full description of the reactor and the methods of operation are provided in Text S1 of the ESI.† The absorbed photon flux in the reactor under our experimental conditions was determined to be 8.07 × 10−8 Einstein per s using iodine/iodate actinometry, according to the standard IUPAC method.54,55 More detailed calculations are presented in Text S2.† An initial concentration [TRA]0 = 10 mg L−1 was used in all degradation experiments, except where otherwise mentioned. The reaction was initiated by inserting the preheated UV lamp into the quartz insert. Two control samples were obtained before and after PS addition (labeled t = −2 and t = −1, respectively). This concentration falls within the expected range for wastewater effluent from a pharmaceutical production facility after dilution with the factory discharge. Our lab has partnered with local pharmaceutical manufacturing industries to examine the effluent resulting from cleaning powder mixers used for capsule manufacturing. The concentrations of pharmaceuticals in the effluent were found to range between 10 mg L−1 and 200 mg L−1, depending on the quantity of water used for washing. A reasonable volume for washing results in 10 ppm of TRA wastewater. This was reported in our previous study on theophylline degradation by UV/PS.56 Therefore, a simulated effluent of [TRA]0 = 10 ppm was used for this study.
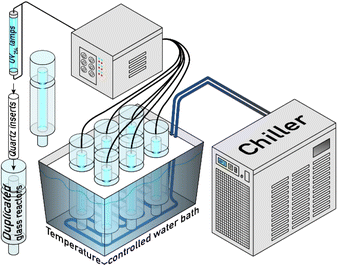 |
| | Fig. 1 Experimental setup for the batch experiment using 11 W LPHgLs irradiating solutions in custom reactors with Vtotal = 350 mL. | |
3.2. Continuous-flow reactor
A continuous-flow system (CFS) was built using the same elements of the commercial UVC water disinfection apparatus used for the batch experiments, in addition to its corresponding stainless-steel casing. Five stainless steel reactors were connected in series to two peristaltic pumps: one for pumping the solution to be treated (KHM-24B3S40, Kamoer) and the second for pumping PS (KAS-SE-B083, Kamoer). The solutions were pumped into the reactors through a Tee connector, and mixing occurred before the solutions passed through the reactors. After passing through the reactors, a portion was diverted to a flow cell in a Thermo-Scientific GENESIS 10S UV-Vis spectrophotometer for online monitoring of the absorbance at 254 nm as an indicator for organic contamination.57
4. Chemical analyses
4.1. TRA quantification
[TRA] was determined using an HPLC instrument (HPLC, ThermoFisher Scientific UltiMate 3000 RS series), equipped with a quaternary pump, a vacuum degasser, an autosampler unit with cooling maintained at 4 °C, and a thermally controlled column compartment set at 30 °C. The HPLC was coupled to a fluorescence detector (FLD) set up for 2D acquisition, with λex = 200 nm, λem = 300 nm, and temperature maintained at 43 °C. The column was a Discovery® HS C-18 reverse phase column (5 μm; 4.6 mm internal diameter × 250 mm length), equipped with a guard column HS C-18 (5 μm; 4.0 mm internal diameter × 20 mm length) (Pennsylvania, USA). The mobile phase constituted a phosphate buffer solution (0.01 M, pH = 3) and acetonitrile (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25) with 1% triethylamine under an isocratic flow rate of 1 mL min−1. The injection volume was 10 μL. The TRA peak had a retention time at t = 6 min. The calibration curve for TRA is shown in Fig. S1.† Four replicates were produced for each point, and error bars were calculated based on the formula
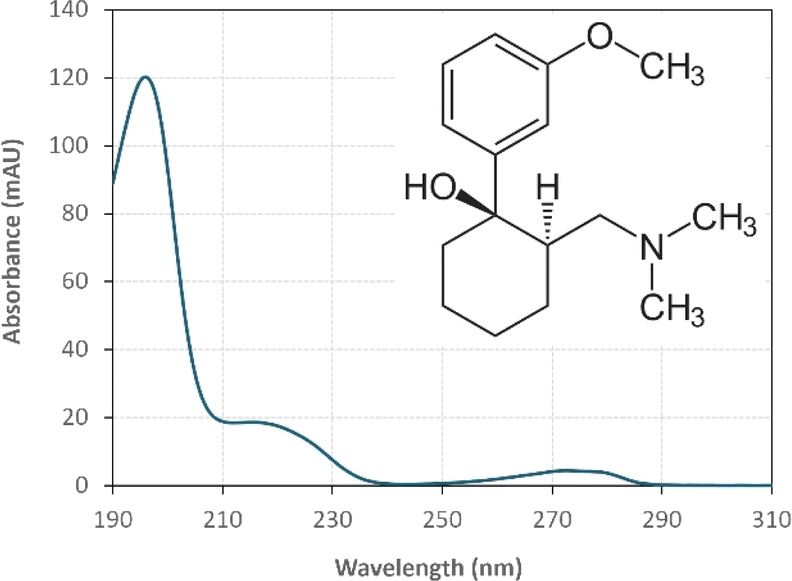
:25) with 1% triethylamine under an isocratic flow rate of 1 mL min−1. The injection volume was 10 μL. The TRA peak had a retention time at t = 6 min. The calibration curve for TRA is shown in Fig. S1.† Four replicates were produced for each point, and error bars were calculated based on the formula 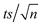 , where t is the student's t with degrees of freedom = 3, s is the standard deviation, and n is the number of replicates = 4. The absorbance spectrum of TRA is shown in Fig. 2. TRA shows three maxima of absorbance at 200, 215, and 272 nm. All three wavelengths were evaluated as excitation wavelengths for fluorescence analysis, and emission scans were conducted to determine the maximum emission value. While λex = 215 nm and λem = 320 nm would yield a more specific chromatographic peak, λex = 200 nm and λem = 300 nm showed better sensitivity, without sacrificing specificity; therefore, it was chosen for the FLD instrumental method.
, where t is the student's t with degrees of freedom = 3, s is the standard deviation, and n is the number of replicates = 4. The absorbance spectrum of TRA is shown in Fig. 2. TRA shows three maxima of absorbance at 200, 215, and 272 nm. All three wavelengths were evaluated as excitation wavelengths for fluorescence analysis, and emission scans were conducted to determine the maximum emission value. While λex = 215 nm and λem = 320 nm would yield a more specific chromatographic peak, λex = 200 nm and λem = 300 nm showed better sensitivity, without sacrificing specificity; therefore, it was chosen for the FLD instrumental method.
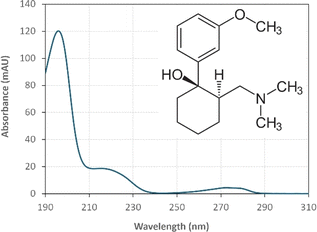 |
| | Fig. 2 UV absorbance spectrum of [TRA] = 10 mg L−1 (inset: tramadol molecule structure). | |
4.2. Identification of transformation products
To propose a degradation pathway for TRA, we conducted a high-resolution mass spectrometry analysis to determine the TPs. Sample preparation was achieved by diluting samples taken at t = 2 min by a factor of 10 and later samples by a factor of 5 and filtering using 0.45 μm PTFE syringe filters. Additional details regarding the employed LC and MS methods are presented in Text S3 in the ESI.† The mass spectra of TRA transformation products were acquired using HPLC coupled to quadrupole time-of-flight tandem mass spectrometry (Sciex X500R HPLC-QTOF-MS/MS, Sciex Applied Biosystems, Framingham, MA, USA).
4.3. Persulfate quantification
[PS] was determined by an in-house validated analytical method developed by Baalbaki et al.58 The method employs a modified HPLC unit coupled to bypass capillary loop columns and a diode array detector (DAD). A mobile phase of concentrated potassium iodide (KI) solution is used to reduce PS inside the capillary loop columns (eqn (3) and (4)). Triiodide anion (I3−) absorbs at 352 nm, minimizing interferences from organic contaminants (OCs) present in the samples.| | | S2O82− + 2I− → 2SO42− + I2 | (3) |
5. Results and discussion
5.1. PS activation method
An initial assessment was conducted to determine the applicability of different activation techniques for TRA removal. Three common activation techniques were investigated for this purpose: UVA (simulating solar radiation), UVC, and thermal. The results are presented in Fig. S3.† UVA activation with T5-UVA lamps (F8T5/BL, Haichao) was ineffective for the treatment of [TRA]0 by direct photolysis (DP) or by a UVA/PS system. Less than 10% degradation of TRA was achieved after 60 min in the UVA/PS system ([PS]0 = 1 mM), and no significant degradation was observed under DP. The emission spectrum of the UVA lamps used is presented in Fig. S2.† Thermal activation was inefficient since only temperatures >60 °C showed positive results. Full degradation of [TRA]0 with [PS]0 = 1 mM at T = 70 °C required more than 60 min. No significant degradation was achieved by heating the solution without PS. The high energy required for the thermally activated PS system is inapplicable for treating industrial effluents.
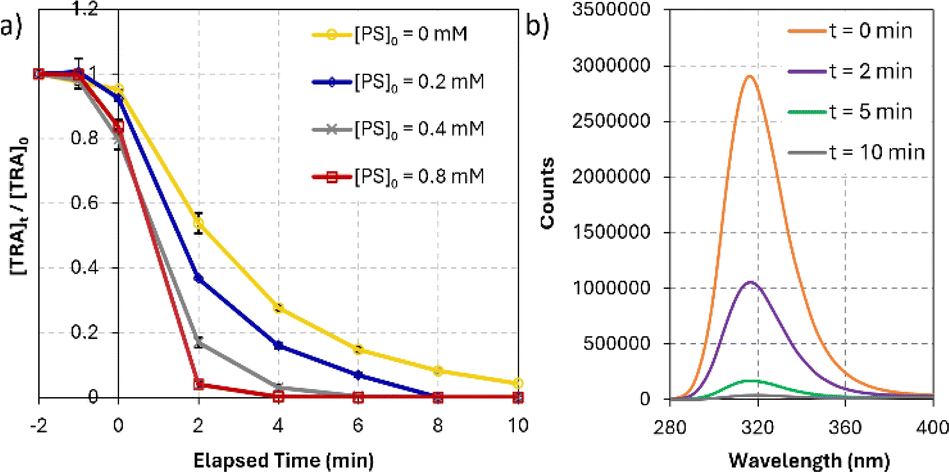
On the other hand, TRA was degraded much faster under DP by UVC and UVC/PS systems (Fig. 3a). DP by UVC achieved 99.7% degradation of TRA in 30 min. The main advantage of UVC/PS was the elimination of a recalcitrant transformation product (TP-X) that persisted in the UVC-only system. The FLD-generated chromatogram showed TP-X after 10 min of reaction at a retention time of 13 min using [PS]0 = 0.4 mM. The UVC/PS system was successful in fully degrading TRA in only 6 min with [PS]0 = 0.4 mM. The ratio of [PS]:[TRA] in our study was 11:1. The required stoichiometrically amount could be theoretical or empirical/experimental. However, the theoretical value is seldom used since the required reaction coefficients are difficult to obtain in such reactions. The experimental ratio was only once in the literature and was found to be similar to our ratio of 11:1.59 Notably, the amount of sulfates added to the solution in this process was calculated to be 76.85 mg L−1, which is below the limit of 250 mg L−1 set by the WHO60 and the EPA61 for drinking water. The decline in the absorbance and fluorescence spectra of TRA and its TP was recorded using 3D acquisition setting on the DAD and FLD detectors, ensuring the complete elimination of any light absorbing/fluorescent compounds in detectable concentrations (Fig. 3b). Therefore, UVC activation was selected for this study.60,61
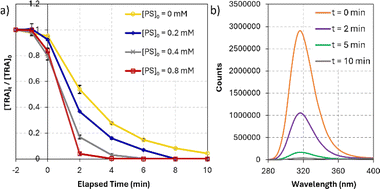 |
| | Fig. 3 (a) Degradation of TRA in UVC/PS system. Experimental conditions: [PS] = 0.0–0.8 mM, [TRA]0 = 10 mg L−1, (b) the fluorescence spectrum of TRA at different reaction times. Experimental conditions: [PS] = 0.4 mM; [TRA]0 = 10 mg L−1. Error bars were calculated using 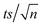 , where absent bars fall behind the point marker. , where absent bars fall behind the point marker. | |
5.2. Kinetic order determination
The degradation of OCs by PS-AOPs typically follows pseudo-first-order kinetics.62,63 In this study, the rate of degradation of TRA was evaluated at four different [PS]0 (Fig. 3a). The results were successfully fitted into a pseudo-first-order kinetic model, as proven by the regression coefficients when 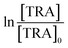 for different [PS]0 was plotted against time (Fig. S4†). kobs was then calculated using eqn (5) and proved to be proportional to [PS]0 within the studied range. The degradation followed pseudo-first-order kinetics under all experimental conditions, regardless of the tested matrix/pH conditions.
for different [PS]0 was plotted against time (Fig. S4†). kobs was then calculated using eqn (5) and proved to be proportional to [PS]0 within the studied range. The degradation followed pseudo-first-order kinetics under all experimental conditions, regardless of the tested matrix/pH conditions.| | 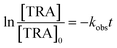 | (5) |
where kobs is the observed pseudo-first-order rate constant (min−1) and t is time (min).
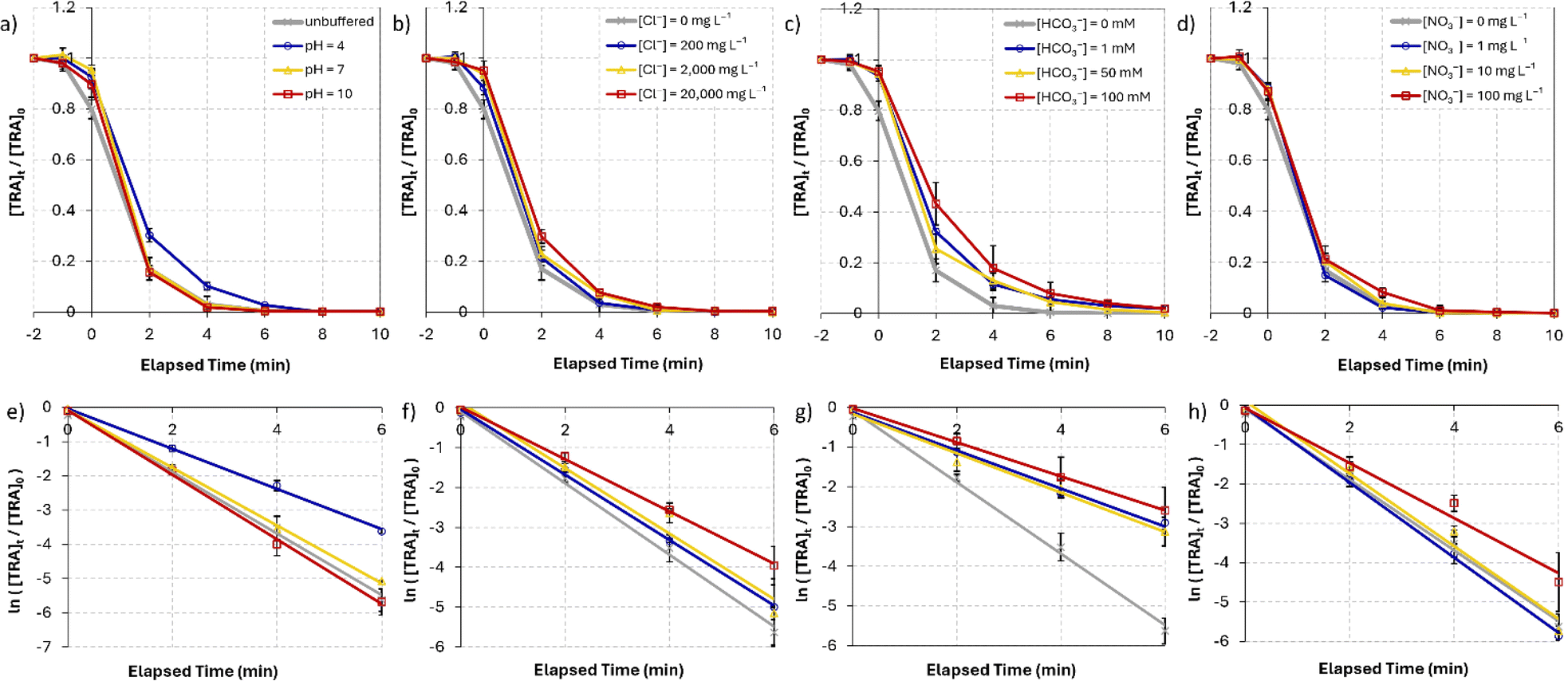
5.3. pH effects
The performance of the UVC/PS system was evaluated in 20 mM phosphate buffer solutions under acidic (pH = 4), neutral (pH = 7), and basic (pH = 10) conditions. The degradation curves under these conditions are shown in Fig. 4a. The results show complete degradation of TRA at t = 10 min in neutral and basic buffered solutions. Acidic pH hindered the degradation of TRA, and full degradation required 15 min, with a significant decrease in kobs from 0.90 to 0.59 min−1 (Table 2). Studies in the literature have reported a more efficient degradation of pharmaceuticals under acidic conditions;51,62 however, other studies showed agreement with our findings of improved degradation efficiency under basic conditions.64–67 This variation in results suggests that the degradation efficiency of the UVC/PS system under different pH conditions is dependent on the nature of the analyte/contaminant, which dictates its degradation pathway.62 The changes in the rate constant Δkobs for pH = 7 and pH = 10 were minimal at +6% and −5%, respectively. The inhibition observed under acidic conditions could be attributed to the abundance of the protonated tertiary amine under acidic conditions, causing this site to be less susceptible to oxidation by electrophilic radical species.68 Interestingly, no detectable TPs were observed after 10 min in solutions buffered at pH = 10, whereas acidic and neutral conditions showed lower degradation efficiency of the TPs. Similar results were reported by Gao et al. who investigated the degradation of sulfamethazine in the UV/PS system.67 In general, PS activation is enhanced under basic conditions, partly due to the formation of superoxide radicals (O2˙−) (eqn (6))51,69–71 and further formation of highly oxidative (E° = 1.8–2.7 V)66 and less specific ˙OH through eqn (7), leading to the degradation of a wider range of contaminants in solution.| | 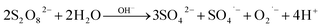 | (6) |
| | | SO4˙− + OH− → SO42− + ˙OH | (7) |
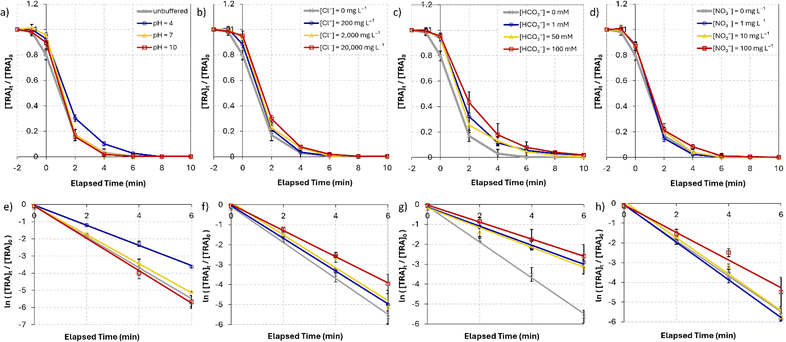 |
| | Fig. 4 (a) Degradation of [TRA]0 = 10 mg L−1 using [PS]0 = 0.4 mM (a) in solutions buffered at pH = 4, 7 and 10, (b) under different salinity conditions, [NaCl] = 0–20000 mg L−1, (c) [NaHCO3] = 0–100 mM, (d) [NO3−] = 0–100 mg L−1 (e)–(h) show the plots of 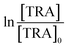 vs. elapsed reaction time corresponding to the experiments shown in (a)–(d), respectively. Error bars were calculated using vs. elapsed reaction time corresponding to the experiments shown in (a)–(d), respectively. Error bars were calculated using 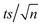 where absent bars fall behind the point marker. where absent bars fall behind the point marker. | |
Table 2
k
obs calculated at different degradation conditions, Δkobs showing the difference in kobs compared to the control, and initial and final pH values
| Matrix elements |
k
obs (min−1) |
Δkobs |
pHi |
pHf |
|
None (control)
|
0.90 ± 0.04 |
0% |
5.04 |
3.22 |
| pH = 4 |
0.59 ± 0.02 |
−35% |
4.04 |
3.60 |
| pH = 7 |
0.84 ± 0.02 |
−6% |
7.19 |
7.13 |
| pH = 10 |
0.94 ± 0.03 |
5% |
9.85 |
8.95 |
|
|
Chloride
|
| [Cl−] = 200 mg L−1 |
0.82 ± 0.03 |
−9% |
5.02 |
3.47 |
| [Cl−] = 2000 mg L−1 |
0.82 ± 0.10 |
−9% |
5.05 |
3.60 |
| [Cl−] = 20000 mg L−1 |
0.65 ± 0.02 |
−27% |
5.45 |
3.59 |
|
|
Bicarbonate
|
| [HCO3−] = 1 mM |
0.48 ± 0.03 |
−47% |
8.38 |
7.95 |
| [HCO3−] = 50 mM |
0.49 ± 0.04 |
−45% |
8.55 |
8.41 |
| [HCO3−] = 100 mM |
0.43 ± 0.01 |
−52% |
8.53 |
8.47 |
|
|
Nitrate
|
| [NO3−] = 1 mg L−1 |
0.95 ± 0.02 |
6% |
6.82 |
3.41 |
| [NO3−] = 10 mg L−1 |
0.92 ± 0.09 |
3% |
6.53 |
3.41 |
| [NO3−] = 100 mg L−1 |
0.70 ± 0.07 |
−22% |
5.81 |
3.34 |
|
|
Humic acids
|
| [HA] = 0.5 mg L−1 |
0.73 ± 0.05 |
−19% |
6.14 |
3.40 |
| [HA] = 5 mg L−1 |
0.39 ± 0.01 |
−57% |
6.09 |
3.20 |
| [HA] = 20 mg L−1 |
0.16 ± (0.03 × 10−1) |
−82% |
6.34 |
3.43 |
|
|
Anoxic
|
0.56 ± 0.03 |
−37% |
— |
— |
|
|
Chloroform
|
0.51 ± 0.01 |
−43% |
— |
— |
|
|
UVC/H
2
O
2
|
0.86 ± 0.09 |
−7% |
— |
— |
|
|
UVC/PS/H
2
O
2
|
0.95 ± 0.06 |
3% |
— |
— |
|
|
Simulated effluent
|
0.77 ± 0.06 |
−14% |
— |
— |
5.4. Matrix effects
5.4.1. Chloride effect.
Industrial pharmaceutical effluent may contain significant concentrations of chlorides, particularly in coastal locations where brackish water can be mixed with the pharmaceutical effluents. We tested the effect of chlorides on the degradation efficiency of the UVC/PS system using three concentrations, 200, 2,000, and 20000 mg L−1 (Fig. 4b), simulating fresh, brackish, and saline water, respectively. In fresh and brackish conditions, chlorides in solution resulted in a 9% decrease in kobs from 0.90 for the control to 0.82 min−1 in both. A more significant decrease of 27% reaching 0.65 min−1 was observed under saline conditions (Table 2).
Some studies have found that the degradation efficiency of UV/PS systems is enhanced in freshwater compared to DI water conditions owing to the formation of reactive chlorine radicals (Cl˙);51,72–75 however, other studies found that even low concentrations of chloride ions in solution can inhibit the degradation efficiency of the studied contaminant.76–79 Different target molecules undergo different mechanisms during their degradation process, and Cl˙ (E° = 2.432 V),80 while having a lower redox potential than ˙OH and SO4˙−, is more reactive towards electron-rich OCs.81 Therefore, TRA, which has an amine group and an aromatic ring connected to an electron-donating group (−OCH3), would be susceptible to oxidation by Cl˙. This could explain how only minor inhibition was observed under freshwater and brackish conditions. Furthermore, ˙OH forms in solutions containing chlorides (eqn (10)) and contributes to the degradation of TRA and its TPs. The results obtained under brackish and saline conditions are in agreement with previous reports.51,74,76–79 As the concentration of chloride ions increases, the scavenging of ˙OH and SO4˙− in solution (eqn (8) and (10))78,82 results in a more significant decrease in kobs. Additionally, self-quenching reactions of chlorine radicals that convert into less-reactive form Cl2˙− (E° = 2.126 V), according to eqn (11),74 may lead to further inhibition. Similar results were observed by Ghauch et al. regarding the degradation of o-toluidine and chloramphenicol.52,83 The detection of two unidentified TPs at retention times of 3.69 and 4.80 min under saline conditions indicates that the presence of an excess of chlorides in solution has an inhibitory effect on the mineralization. Regardless, full degradation of TRA was successful in all studied systems.
| | | SO4˙− + Cl− → SO42− + Cl˙, k = 3.1 × 108 M−1 s−1 | (8) |
| | | Cl˙ + H2O ⇌ ClOH˙− + H+, kfor = 1.3 × 103 M−1 s−1, | (9) |
| | | ClOH˙− ⇌ ˙˙OH + Cl−, kfor = 6.1 × 109 M−1 s−1, krev = 4.3 × 109 M−1 s−1 | (10) |
| | | Cl˙ + Cl− → Cl2˙ (k = 2.1 × 1010 M−1 s−1) | (11) |
5.4.2. Bicarbonate effect.
To study the effect of bicarbonate and ionic strength on the degradation of TRA, experiments were performed in solutions containing [HCO3−]0 = 1, 50, and 100 mM (Fig. 4c). The addition of sodium bicarbonate inhibited the degradation of TRA at all studied concentrations. [HCO3−]0 = 1 and 50 mM conditions caused kobs to decrease by 47% and 45%, respectively. A slightly more pronounced decrease of 52% was observed when [HCO3−]0 was increased to 100 mM (Table 2).
The added bicarbonate leads to an equilibrium of the H2CO3/HCO3−/CO32− due to the hydrolysis of HCO3− (eqn (12) and (13)).74 These ions can scavenge reactive radicals and generate HCO3˙ and CO3˙− with lower redox potentials.81 HCO3− and CO32− react with SO4˙− and ˙OH (eqn (14)–(17))65 to generate HCO3˙ and CO3˙− with lower redox potentials.84 The addition of bicarbonates to the reaction medium had a similarly significant inhibitory effect in studies using UVC/PS systems.66,79,83,85–87
| | | HCO3− + H2O ⇌ H2CO3 + OH−, pKa = 6.35 | (12) |
| | | HCO3− ⇌ H+ + CO32−, pKa = 10.3 | (13) |
| | | SO4˙− + HCO3− → HCO3˙ + SO42−, k = 2.8 × 106 M−1 s−1 | (14) |
| | | SO4˙− + CO32− → CO3˙− + SO42−, k = 6.1 × 106 M−1 s−1 | (15) |
| | | ˙OH + HCO3− → HCO3˙ + OH−, k = 8.5 × 106 M−1 s−1 | (16) |
| | | ˙OH + CO32− → CO3˙− + OH−, k = 3.9 × 108 M−1 s−1 | (17) |
An increase in the degradation efficiency of target molecules in the presence of carbonate/bicarbonate anions was observed in previous studies using UV/PS-activated systems.53,64,74,75,78 Notably, the observed increase in kobs in the aforementioned studies reaches a plateau or reverses, and the degradation efficiency starts decreasing when the concentration of bicarbonate exceeds a certain threshold.53,64,65,74,76,78 This may be attributed to the dominance of the scavenging reactions over the reactions with the OC and its TPs. In real-life applications, the presence of bicarbonate in the matrix negatively affects the degradation efficiency of the system; therefore, longer treatment times or higher [PS]0 counteracting the quenching should be considered.
5.4.3. Nitrate effect.
The influence of nitrates on the degradation of TRA was investigated by adding sodium nitrate at three increasing concentrations: 1, 10, and 100 mg L−1 (Fig. 4d). The results showed that the addition of [NO3−]0 = 1 and 10 mg L−1 had an insignificant positive effect on the degradation rate of TRA, increasing kobs by 6% and 3%, respectively. The addition of [NO3−] = 100 mg L−1 led to a 22% decrease in kobs (Table 2).
The negative effect of nitrates on the degradation of organic contaminants in water has been reported by multiple studies.79,82,83,85,86 The photolysis of NO3− can generate ˙OH (eqn (18) and (19)),88 which contributes to the degradation of TRA. However, the molar absorption coefficient and quantum yield of NO3− at 254 nm is low; therefore, this effect is predicted to be insignificant at low concentrations.89 Additionally, multiple studies reported that NO3− does not play a role in the quenching of ˙OH and emphasized that the rate of SR scavenging by NO3− (eqn (20)) is too low for this reaction to have a significant effect, particularly at low [NO3−]0.82,86 This can explain why low [NO3−]0 does not significantly impact the degradation of TRA. Meanwhile, higher concentrations exacerbate the “inner filter effect” caused by the nitrate ions and significantly inhibit the degradation. To further understand this inner filter effect, Sörensen et al. investigated the effect of nitrates on the degradation of EDTA in a UV/H2O2 system.90 Interestingly, the authors found that the DP of EDTA benefits from increasing the concentration of nitrates (up to 100 mg L−1), and the opposite effect occurred in the UV/H2O2 systems. This could indicate that the inner filter effect reported by some studies may inhibit the photolysis of H2O2 and reduce the concentration of generated oxidative radicals, rather than hinder the DP of the organic contaminants. Studies on the thermal activation of PS for the degradation of OCs show less dependence on [NO3−], with minimal to no inhibition observed.91–94 These results further indicate that the inner filter effect is the main factor inhibiting the degradation of TRA in the UVC/PS system.
| | 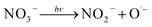 | (18) |
| | | O˙− + H2O → ˙OH + OH− | (19) |
| | | SO4˙− + NO3− → SO42− + NO3˙, k = 5.0 × 104 M−1 s−1 | (20) |
5.4.4. Humic acids.
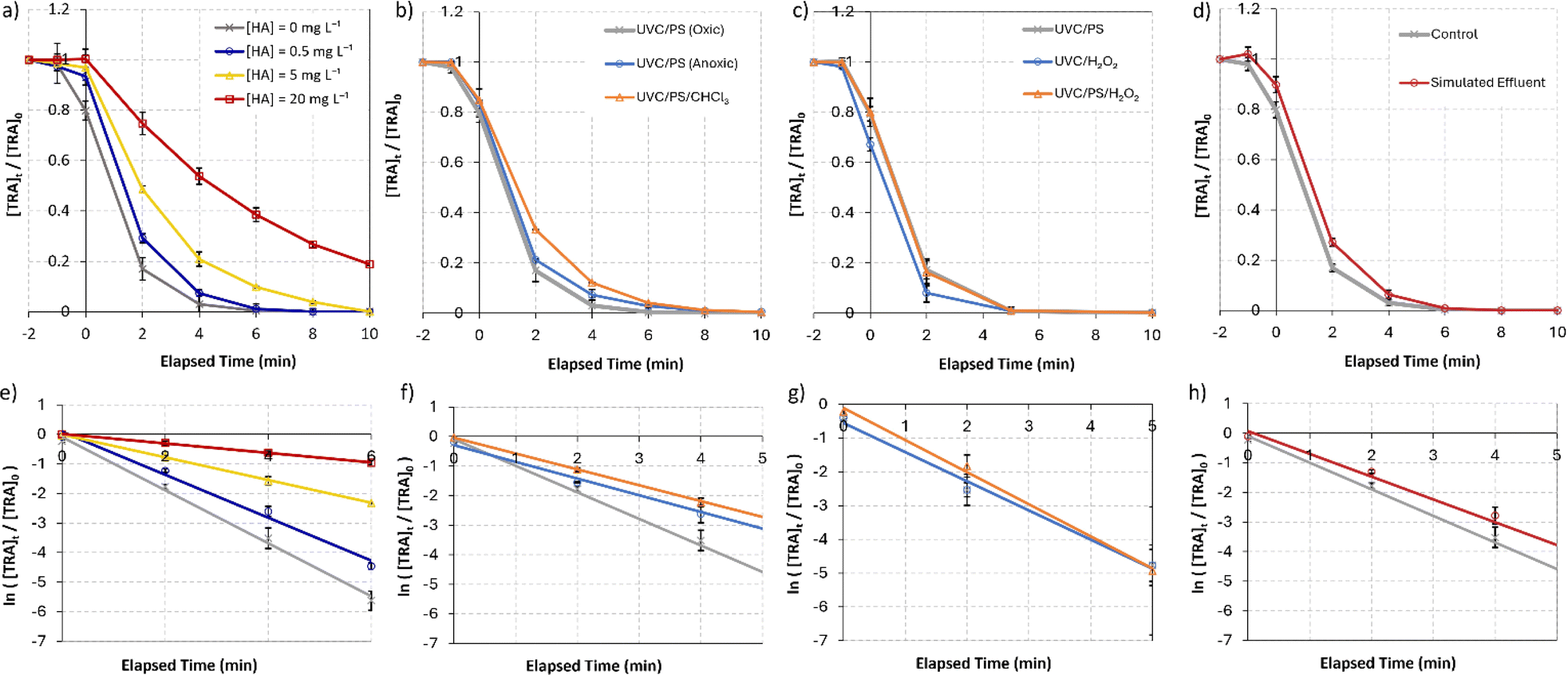
To simulate the effect of natural organic matter (NOM), three different concentrations of humic acids (HA) were tested: [HA]0 = 0.5, 5, and 20 mg L−1. An inhibitory effect on the degradation was observed, with a significant decrease in kobs with each increased concentration (Fig. 5a). When [HA]0 was increased from 0 to 0.5 mg L−1, kobs decreased by 19% from 0.90 to 0.73 min−1. When [HA]0 was increased to 5 and 20 mg L−1, kobs decreased to reach values as low as 0.39 and 0.16 min−1, respectively (Table 2).
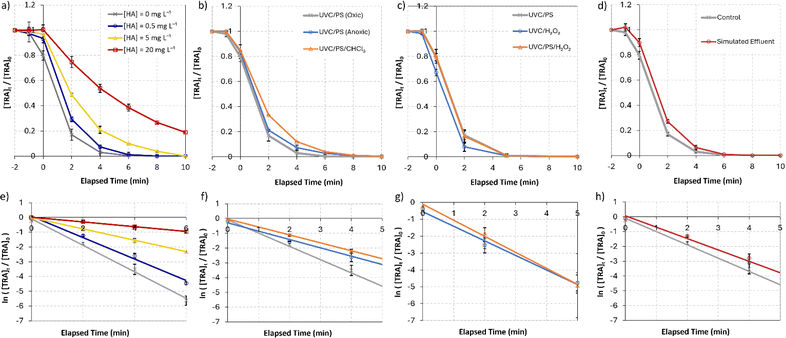 |
| | Fig. 5 Degradation of [TRA]0 = 10 mg L−1 using [PS]0 = 0.4 mM, (a) in systems containing different concentrations of humic acids, [HA] = 0–20 mg L−1, (b) in oxic UVC/PS (control), anoxic UVC/PS (purged with N2) and the UVC/PS/CHCl3 (CHCl3 quenches O2˙−), (c) in UVC/PS ([PS]0 = 0.4 mM), UVC/H2O2 ([H2O2]0 = 0.4 mM), and UVC/PS/H2O2 ([PS]0 = 0.2 mM, [H2O2]0 = 0.4 mM), (d) under simulated effluent conditions, (e)–(h) show the plots of 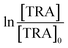 vs. elapsed reaction time corresponding to (a)–(d), respectively. Error bars were calculated using vs. elapsed reaction time corresponding to (a)–(d), respectively. Error bars were calculated using 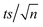 where absent bars fall behind the point marker. where absent bars fall behind the point marker. | |
These results are consistent with the outcomes reported in the literature.64,74,76,87,95–98 The observed inhibition can be linked to two main factors. First, the addition of HA to the solutions caused a visible color change from clear to light brown. The inner filter effect caused by the added light-absorbing HA inhibits both the DP of TRA and the UVC-activation of persulfate. Second, the added HA competes with TRA, and the oxidation of HA consumes the reactive species in solution, significantly reducing the degradation efficiency of TRA.97
5.5. Effect of dissolved oxygen
Dissolved oxygen (DO) is an influential factor in the UVC/PS system. To evaluate the effect of dissolved oxygen on the degradation of TRA, the reactor was purged with nitrogen gas for 1 h before spiking with persulfate to ensure the elimination of most of the O2 in solution.53 The results show a decrease in kobs by 37% in the anoxic system. This may be linked to the generation of superoxide radicals (O2˙−) under oxic conditions, which in turn promote the generation of more H2O2,99 leading to more ˙OH. Additionally, studies have reported that the quantum yield of persulfate activation is lower under anoxic conditions (1.4 mol Einsteins s−1) compared to oxic (1.8 mol Einsteins s−1).100,101 This could be linked to the ability of O2˙− generated in solution to activate PS and further generate SO4˙− (eqn (21)).| | | O2˙− + S2O82− → SO4˙− + SO42− + O2 | (21) |
To further evaluate the role of O2˙− on the degradation of [TRA], chloroform was used as a superoxide quencher (Fig. 5b). A 43% decrease in kobs was observed after the addition of CHCl3 to the UVC/PS system (Table 2). This decrease is comparable to that observed under anoxic conditions, possibly indicating that the generation of O2˙− under oxic conditions is the main cause for the enhanced degradation of TRA.
5.6. Comparative study: UVC/H2O2vs. UVC/PS vs. UVC/H2O2/PS
PS was compared to H2O2, a common oxidative reagent in UVC/AOPs systems, to assess its performance and evaluate the viability of systems with different oxidants for the continuous flow system (CFS) in development. The degradation of TRA was evaluated in UVC/PS, UVC/H2O2, and UVC/PS/H2O2 systems (Fig. 5c). One mole of each oxidant generates two reactive radicals; therefore, the total concentration of the oxidant(s) added was kept the same at 0.4 mM to achieve comparison. In the UVC/PS/H2O2 system, [PS]0 = 0.2 mM and [H2O2]0 = 0.2 mM were adopted. The results showed no significant difference in the performance of the three systems. A 4% decrease in kobs was obtained in the UVC/H2O2 system compared to a 6% increase in kobs in the UVC/PS/H2O2 system. The insignificant difference between the three systems can be attributed to the relatively low contribution of SO4˙− in the UVC/PS system when compared to DP. This would lead to similar results when PS is replaced with H2O2 since both contribute to the degradation of TRA primarily by generating ˙OH.
Considering the insignificant difference between the three systems, PS presents valuable advantages for real-world applications. The ease of transport and storage of PS salts compared to the liquid, corrosive H2O2, its longer shelf life, and lower cost allow it to be a more desirable choice for AOPs treating industrial wastewater.
5.7. Simulated effluent
The degradation of TRA by UVC/PS was attempted under simulated effluent conditions to evaluate the performance of the system in real-life applications. The simulated effluent constituted a non-filtered stock solution of TRA prepared using TRAMAL® 50 mg capsules (STADA, Italy). To obtain comparable data to the control conditions, the initial TRA concentration was maintained at 10 mg L−1. The results showed a 14% decrease in kobs (Fig. 5d).
This decrease in degradation efficiency can be primarily linked to the inner filter effect of the excipients present in the drug formulation: microcrystalline cellulose, sodium starch glycolate, colloidal anhydrous silica, and magnesium stearate. Notably, the aforementioned excipients are all insoluble; therefore, they do not affect the degradation by competing for reactions with reactive species in solution.
5.8. Identification of transformation products
Samples from the UVC/PS system with [TRA]0 = 10 mg L−1 and [PS]0 = 0.4 mM were screened for common TRA oxidation TPs obtained from the literature. These possible TPs are compiled in Table S3.† Eight species with [M + H]+ of m/z 280.1914–280.1919 Da were observed in the extracted ion chromatogram (XIC). These peaks belong to TPs with m/z equal to an added oxygen onto TRAH+; therefore, they might correspond to either TP280a (N-OXID) or it's a single hydroxylation product TP280b.68,102 The MS Spectra obtained for each of the eight peaks are shown in Fig. S7 and S8.† The highlighted red portion shown in the XIC (Fig. S6†) is the background selected to be subtracted from the spectral data. Additional hydroxylation products were possibly formed, with two TPs having m/z = 296.1865 Da (Fig. S12†), indicating double-hydroxylated TP296, and m/z = 312.1816 Da, indicating triple-hydroxylated TP312 (Fig. S13†).
The XIC for formula C15H24NO2 showed two major peaks with m/z = 250.1810 ± 0.01 Da (Fig. S9†). These peaks may belong to the dealkylation TPs N-desmethyltramadol (TP250a) and O-desmethyltramadol (TP250b). TP236 possibly corresponds to further dealkylation at the nitrogen atom (Fig. S10†). The XIC for C14H21NO2 shows a clear peak at 2.778 min, with the MS spectrum showing a peak at 236.1653 Da.
Finally, TP278 with formula C16H24NO4 and m/z = 278.1759–278.1762 could be formed by the addition of oxygen to one of the carbons around the nitrogen, resulting in either an aldehyde or a ketone.102 Fig. S11† shows all the MS spectra for TP278.
Based on these findings and previous research on the oxidation of TRA by various AOPs,68,102,103 three degradation pathways, A, B and C, were proposed, as shown in Fig. 6. Pathway A begins with the oxidation of the methyl group on either the tertiary amine or the methoxy group, followed by demethylation. Pathway B is the oxidation of the amine by oxygen transfer. Pathway C is a multi-hydroxylation pathway that begins with the hydroxylation of the aromatic or cyclohexane ring.
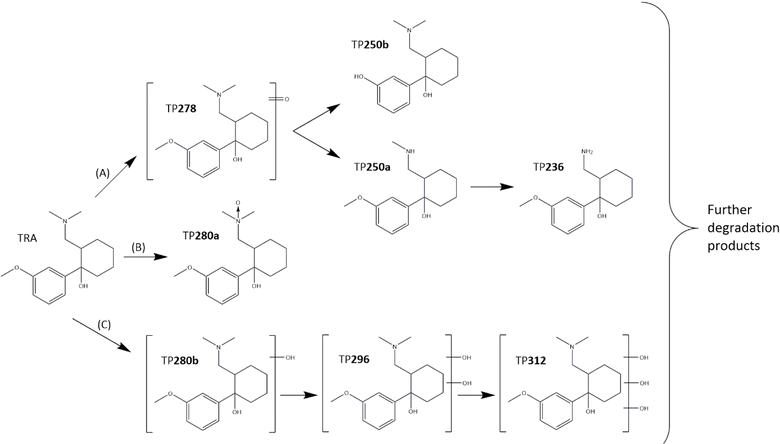 |
| | Fig. 6 Proposed TRA degradation mechanism. Experimental conditions: [TRA]0 = 10 mg L−1 and [PS]0 = 0.4 mM. | |
5.9. Continuous-flow system
Our research group has been conducting studies on AOPs, primarily PS-based technology. The studies first tested chemical and thermal activation of PS, followed by UVC activation. The first few projects using UVC activation were conducted via research-grade tools, i.e., reactors and lamps, which do not resemble the working conditions within an industrial environment. The research evolved to incorporate commercially available, common, low-cost elements, such as UVC water disinfection elements, as a replacement for the reactors; however, the elements were not used as per their original design. Instead, they were used in a batch process to study the degradation of pharmaceuticals under different conditions. This step is important for optimizing the system's performance under different conditions.
After conducting several successful studies on the elimination of PPCPs using UVC/PS batch treatment, which requires the collection and storage of the effluent, increasing process complexity and cost, it became necessary to develop a skid-mounted continuous-flow system (CFS) at a pilot-scale. The system had to be portable, easy to operate, and ultimately a stand-alone plug-and-play unit capable of being installed in industries with hazardous wastewater treating separate streams in-line with production. For this purpose, the unit shown in Fig. 7a was developed by our research group and was tested for the treatment of TRA at a flow rate of 360 L per day of [TRA]0 = 10 mg L−1.
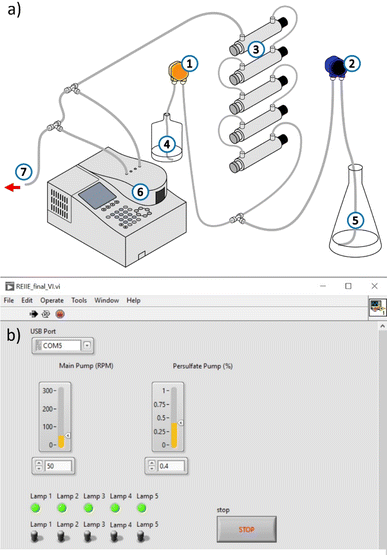 |
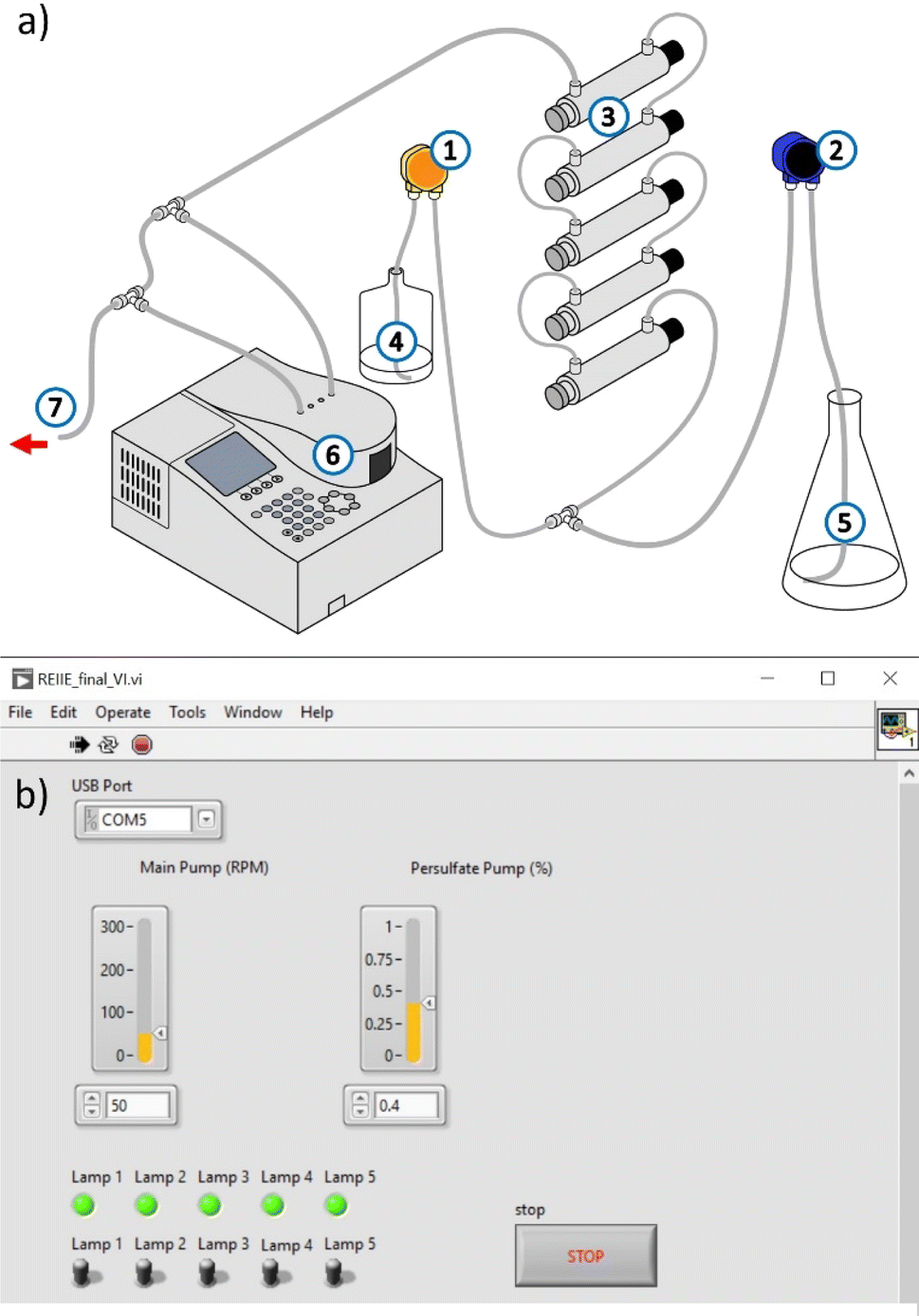
| | Fig. 7 (a) Schematic diagram of the continuous-flow system developed for the treatment of industrial effluent ((1) PS pump, (2) influent pump, (3) reactor containing UVC lamp, (4) PS solution, (5) influent, (6) Spectrophotometer containing a flow cell for online monitoring, (7) system output/treated effluent). (b) User interface used to control the flow rates of the pumps and the number of UVC lamps in use. | |
The operation parameters of the CFS were adjusted to mimic the batch conditions required for the full degradation of [TRA]0 = 10 mg L−1. These parameters include the volume of the reactor, the UVC dosage, and the temperature of the solution. The batch reactor had a total volume of 350 mL and required 6 min for the total elimination of TRA. To achieve an equivalent UVC irradiation time in the CFS with 5 lamps turned on and a total reactor volume of 1434 mL, a flow rate of 239 mL min−1 should be set.
A volume of 2.5 L of TRA solution was pumped into the CFS via the main pump, with the flow rate set to 235 mL min−1. To achieve [PS]delivered = 0.04 mM, the PS flow rate was set to 4 mL min−1. At this flow rate, 24 mM of PS solution was required.
To ensure that the set flow rates were delivering the correct concentrations, the system was operated at the selected parameters with the lamps kept off and only one solution being pumped at a time. The temperature of the solution was monitored before and after passing through the CFS. The increase in temperature was <5 °C. The parameters of the system, i.e., the flow rate of fluid exiting each pump and the UV dosage are controlled using the user interface shown in Fig. 7b. The system was modeled to mimic the batch reactor and maintain the same experimental conditions. [PS]final was below the detection limit in both the batch and continuous flow experiments. Control experiments conducted without PS showed a similar degradation profile to that of the batch experiments: TRA was almost completely degraded, whereas TPs persisted.
The results of the degradation experiment are shown in Fig. 8. Full degradation of [TRA]0 = 10 mg L−1 was achieved under the aforementioned CFS parameters. Samples were obtained at every minute from the start of the run and [TRA] was determined using HPLC/FLD. TRA was not detected in any of the samples. The system was subsequently operated with 4, 3, 2, and 1 lamp(s), reducing the irradiation time in the UVC reactors. The results showed almost full degradation of TRA, with a minimum percentage reached, e.g., 75.57% for the configuration using one operational lamp. However, the degradation improved to 84.79, 93.19, 99.86, and 100% for the configurations using 2, 3, 4, and 5 lamps, respectively (Fig. 8b). These results showed the high potential of a similar system to fully degrade industrial effluents charged with OCs, i.e., pharmaceuticals, while performing effluent characterization and optimizing parameters such as the concentration of PS used, the retention time of the effluent in the in-series reactors, and the related flow rate of the effluent entering the CFS.
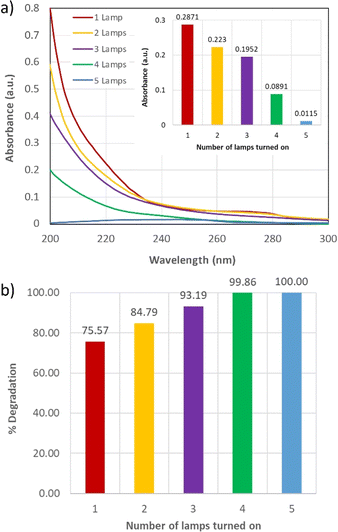 |
| | Fig. 8 (a) Absorbance spectra of the effluent from the CFS, recorded by the online spectrophotometer during experiments testing the effect of UV dosage on the degradation of TRA (inset: absorbance at 215 nm under different UV dosage conditions), (b) % degradation of TRA at different UV dosages. The samples taken for this experiment were analyzed using HPLC/FLD. Experimental conditions: [TRA]0 = 10 mg L−1 and [PS]0 = 0.4 mM. | |
5.10. Economic feasibility
The economic feasibility was assessed based on a method by Bolton et al. published in an IUPAC technical report.104 The required energy is represented by the electric energy per order (EEO). EEO (kW h per m3 per order) is defined as the electric energy required to degrade contaminants by one order of magnitude, e.g., from 10 mg L−1 to 1 mg L−1, in 1 m3 of contaminated water, and is calculated using eqn (22) for a batch system and eqn (23) for a continuous flow system.| | 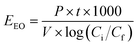 | (22) |
| | 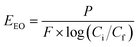 | (23) |
where P is the power supplied to the system in kW, t is the duration of treatment in hour, V is the volume treated in L, Ci and Cf are the influent and effluent concentrations, respectively, and F is the influent flow rate in m3 h−1. The conversion factor from L to m3 is 1000.
This equation assumes first-order kinetics; therefore, log(Ci/Cf) is equivalent to 0.4343 × kobs × t, where kobs is the first-order reaction rate constant (min−1) and t is the reaction time (min). By substitution, eqn (22) can be written in the form of eqn (24), simplified to yield eqn (25) for a batch reactor.
| | 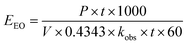 | (24) |
| | 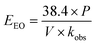 | (25) |
In
eqn (24), 60 is the conversion factor from min
−1 to h
−1.
The electrical energy cost for each system is calculated using the price of $0.079 per kW h, based on the average cost for industries in the US.105,106 The cost of reagent, i.e., persulfate, is based on the price of $2 per kg of sodium persulfate (price obtained from Jinan Shijitongda Chemical Co., Ltd). The calculation steps used to obtain the mass and price of sodium persulfate required per m3 in this system and other systems are elucidated in Table S4.†
For the batch system, EEO was evaluated for the UVC/PS control system using [PS]0 = 0.4 mM and [TRA]0 = 10 mg L−1, with total reactor volume V = 0.35 L, power of the UVC lamp used P = 11 W, and kobs = 0.90 min−1. The electric energy per order was calculated accordingly using eqn (25) to be EEO (control, batch) = 1.341 kW h per m3 per order. The total cost was $0.296 per m3. The detailed cost for each considered system is presented in Table 3. According to the survey done by Miklos et al., the median value 0.67 kW h per m3 per order for UV/PS systems.107 Our EEO value of 1.341 kW h per m3 per order is above the median considered in their study. It falls within the data set and is lower than studies where the EEO reaches an order of 101.
Table 3 Total system cost based on electricity price rates in the US
| System |
E
EO (kW h per m3 per order) |
Electrical cost ($ per m3) |
Reagent cost ($ per m3) |
Total cost ($ per m3) |
| Control (batch) |
1.341 |
0.106 |
0.190 |
0.296 |
| Control (CFS) |
5.177 |
0.409 |
0.113 |
0.522 |
| Simulated effluent (batch) |
1.567 |
0.124 |
0.190 |
0.314 |
| Simulated effluent (CFS) |
— |
— |
— |
0.554 (projected) |
For the continuous flow system, the total power is equivalent to the sum of power used by the 3 lamps (since 3 lamps are needed to achieve a degradation of one order of magnitude), the pumps used, and miscellaneous power use (mainly computing power). Each lamp uses 11 W of power, the influent pump uses 20 W, the PS pump uses 10 W, and the general required computing power was rounded to 10 W. The total power used was equal to 73 W. The flow rate of influent treated was equal to 235 mL min−1. Using eqn (23), EEO (control, CFS) = 5.177 kW h per m3 per order, which translates to an electrical cost of $0.409 per m3 and a total cost of $0.522 per m3.
The batch UVC/PS system treating simulated effluent uses the same amount of PS; therefore, it has the same cost of reagent. On the other hand, EEO (simulated effluent, batch) = 1.567 kW h per m3 per order, leading to an electric energy cost of $0.124 per m3 and a total cost of $0.314 per m3.
The total cost for control conditions moving from batch to continuous increased by 77%. Therefore, the cost for treatment of simulated effluent using the developed CFS is projected to be $0.554 per m3.
6. Conclusions
The degradation of TRA by UVC/PS was investigated in this research. Experiments in a lab-scale batch treatment setup showed that the system achieved full degradation of TRA (10 mg L−1) using low [PS]0 = 0.4 mM, at a cost of $0.296 per m3. The performance of the system under the effect of various factors including the pH, the presence of inorganic ions such as chloride, bicarbonates, and nitrates, the presence of NOM, and the level of dissolved oxygen was evaluated. The results showed that the degradation was significantly inhibited by low pH, the presence of bicarbonates, and elevated concentrations of chlorides and nitrates. The degradation also benefits from the dissolved oxygen in solution, as shown by the decrease in degradation efficiency under anoxic conditions. NOM present in solution significantly slowed down degradation and scavenged reactive species that oxidize TRA. The system managed to fully degrade TRA under simulated effluent conditions. The UVC/PS system was employed in a pilot-scale continuous flow treatment system that was able to replicate the results obtained in the batch treatment. Full degradation of [TRA]0 = 10 mg L−1 at a rate of 360 L per day was achieved at a cost of $0.296 per m3. Notably, this system is modular/scalable and can be modified to accommodate effluents from industries of greater capacity.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This research was funded in part by the University Research Board (Award Number 103603) of the American University of Beirut and USAID-Lebanon through The National Academy of Sciences under PEER project 5-18 (Award number 103262) and the MEPI TLP program (Award number 103992). The authors are thankful to Eng. Joan Younes, senior technician Simon Al-Ghawi, digital media artist Dany Khoury and the glass blower at the chemistry department Boutros Sawaya for their technical assistance, and Rita Al Kady for copyediting the final draft. They are also very thankful to the personnel of the K. Shair CRSL for their kind help.
References
- L. Cizmas, V. K. Sharma, C. M. Gray and T. J. McDonald, Environ. Chem. Lett., 2015, 13(4), 381–394 CrossRef CAS PubMed.
- M. Patel, R. Kumar, K. Kishor, T. Mlsna, C. U. Pittman and D. Mohan, Chem. Rev., 2019, 119, 3510–3673 CrossRef CAS PubMed.
- N. J. D. G. Reyes, F. K. F. Geronimo, K. A. V. Yano, H. B. Guerra and L. H. Kim, Water, 2021, 13, 1159 CrossRef CAS.
- Y. Yang, Y. S. Ok, K. H. Kim, E. E. Kwon and Y. F. Tsang, Sci. Total Environ., 2017, 596–597, 303–320 CrossRef CAS PubMed.
- K. E. Arnold, A. R. Brown, A. R. Brown, G. T. Ankley and J. P. Sumpter, Philos. Trans. R. Soc., B, 2014, 369(1956) DOI:10.1098/RSTB.2013.0569.
- M. Kumar, S. Sridharan, A. D. Sawarkar, A. Shakeel, P. Anerao, G. Mannina, P. Sharma and A. Pandey, Sci. Total Environ., 2023, 859, 160031 CrossRef CAS PubMed.
- Z. Li, X. Yu, F. Yu and X. Huang, Environ. Sci. Pollut. Res., 2021, 28(17), 20903–20920 CrossRef CAS PubMed.
- M. Ibáñez, L. Bijlsma, E. Pitarch, F. J. López and F. Hernández, Trends Environ. Anal. Chem., 2021, 29, e00118 CrossRef.
- W. C. Li, Environ. Pollut., 2014, 187, 193–201 CrossRef CAS PubMed.
- M. Čelić, M. Gros, M. Farré, D. Barceló and M. Petrović, Sci. Total Environ., 2019, 652, 952–963 CrossRef PubMed.
- N. Collado, S. Rodriguez-Mozaz, M. Gros, A. Rubirola, D. Barceló, J. Comas, I. Rodriguez-Roda and G. Buttiglieri, Environ. Pollut., 2014, 185, 202–212 CrossRef CAS PubMed.
- K. M. Onesios, J. T. Yu and E. J. Bouwer, Biodegradation, 2009, 20, 441–466 CrossRef CAS PubMed.
- A. K. Priya, L. Gnanasekaran, S. Rajendran, J. Qin and Y. Vasseghian, Environ. Res., 2022, 204, 112298 CrossRef CAS PubMed.
- J. L. Tambosi, L. Y. Yamanaka, H. J. José, R. De Fátima Peralta Muniz Moreira and H. F. Schröder, Quim. Nova, 2010, 33, 411–420 CrossRef CAS.
- S. Zorita, L. Mårtensson and L. Mathiasson, Sci. Total Environ., 2009, 407, 2760–2770 CrossRef CAS PubMed.
- D. Balarak, A. D. Khatibi and K. Chandrika, Int. J. Pharm. Invest., 2020, 10, 106–111 CrossRef CAS.
- M. Yilmaz, T. J. Al-Musawi, M. khodadadi Saloot, A. D. Khatibi, M. Baniasadi and D. Balarak, Biomass Convers. Biorefin., 2024, 14, 649–662 CrossRef CAS.
- A. A. Alameri, R. H. C. Alfilh, S. A. Awad, G. S. Zaman, T. J. Al-Musawi, M. M. Joybari, D. Balarak and G. McKay, Biomass Convers. Biorefin., 2022, 1, 1–14 Search PubMed.
- D. Balarak, N. Mengelizadeh, P. Rajiv and K. Chandrika, Environ. Sci. Pollut. Res., 2021, 28, 49743–49754 CrossRef CAS PubMed.
- J. Maculewicz, D. Kowalska, K. Świacka, M. Toński, P. Stepnowski, A. Białk-Bielińska and J. Dołżonek, Sci. Total Environ., 2022, 802, 149916 CrossRef CAS PubMed.
-
Tramadol: MedlinePlus Drug Information, https://medlineplus.gov/druginfo/meds/a695011.html, accessed 25 June 2023 Search PubMed.
- S. Jayawardana, R. Forman, C. Johnston-Webber, A. Campbell, S. Berterame, C. de Joncheere, M. Aitken and E. Mossialos, eClinicalMedicine, 2021, 42, 101198 CrossRef PubMed.
- G. D. Foster and A. Leahigh, ACS ES&T Water, 2021, 1, 1447–1455 Search PubMed.
- D. Sadutto, V. Andreu, T. Ilo, J. Akkanen and Y. Picó, Environ. Pollut., 2021, 271, 116353 CrossRef CAS PubMed.
- M. C. Campos-Mañas, I. Ferrer, E. M. Thurman and A. Agüera, Trends Environ. Anal. Chem., 2018, 20, e00059 CrossRef.
- R. L. Bachour, O. Golovko, M. Kellner and J. Pohl, Chemosphere, 2020, 238, 124587 CrossRef CAS PubMed.
- P. Sehonova, L. Plhalova, J. Blahova, P. Berankova, V. Doubkova, M. Prokes, F. Tichy, V. Vecerek and Z. Svobodova, Environ. Toxicol. Pharmacol., 2016, 44, 151–157 CrossRef CAS PubMed.
- L. Plhalova, P. Sehonova, J. Blahova, V. Doubkova, F. Tichy, C. Faggio, P. Berankova and Z. Svobodova, Appl. Sci., 2020, 10, 2349 CrossRef CAS.
- F. Ložek, I. Kuklina, K. Grabicová, J. Kubec, M. Buřič, R. Grabic, T. Randák, P. Císař and P. Kozák, Aquat. Toxicol., 2019, 213, 105217 CrossRef PubMed.
- M. E. S. Santos, P. Horký, K. Grabicová, P. Hubená, O. Slavík, R. Grabic, K. Douda and T. Randák, Ecotoxicol. Environ. Saf., 2021, 212, 111999 CrossRef CAS PubMed.
- T. Mackuľak, L. Birošová, I. Bodík, R. Grabic, A. Takáčová, M. Smolinská, A. Hanusová, J. Híveš and M. Gál, Sci. Total Environ., 2016, 539, 420–426 CrossRef PubMed.
- V. G. Beretsou, M. C. Nika, K. Manoli, C. Michael, Q. Sui, L. Lundy, D. M. Revitt, N. S. Thomaidis and D. Fatta-Kassinos, Sci. Total Environ., 2022, 852, 158391 CrossRef CAS PubMed.
- E. Archer, B. Petrie, B. Kasprzyk-Hordern and G. M. Wolfaardt, Chemosphere, 2017, 174, 437–446 CrossRef CAS PubMed.
- O. Golovko, S. Örn, M. Sörengård, K. Frieberg, W. Nassazzi, F. Y. Lai and L. Ahrens, Sci. Total Environ., 2021, 754, 142122 CrossRef CAS PubMed.
- R. Loos, R. Carvalho, D. C. António, S. Comero, G. Locoro, S. Tavazzi, B. Paracchini, M. Ghiani, T. Lettieri, L. Blaha, B. Jarosova, S. Voorspoels, K. Servaes, P. Haglund, J. Fick, R. H. Lindberg, D. Schwesig and B. M. Gawlik, Water Res., 2013, 47, 6475–6487 CrossRef CAS PubMed.
- D. Sadutto, V. Andreu, T. Ilo, J. Akkanen and Y. Picó, Environ. Pollut., 2021, 271, 116353 CrossRef CAS PubMed.
- P. Loganathan, S. Vigneswaran, J. Kandasamy, A. K. Cuprys, Z. Maletskyi and H. Ratnaweera, Membranes, 2023, 13, 158 CrossRef CAS PubMed.
- P. Verlicchi, E. Zambello and M. Al Aukidy, Compr. Anal. Chem., 2013, 62, 231–286 CAS.
- J. Lalwani, A. Gupta, S. Thatikonda and C. Subrahmanyam, J. Environ. Chem. Eng., 2020, 8, 104190 CrossRef CAS.
- J. Lalwani, A. Gupta, S. Thatikonda and C. Subrahmanyam, J. Environ. Chem. Eng., 2020, 8, 104281 CrossRef CAS.
- J. O. Eniola, R. Kumar, M. A. Barakat and J. Rashid, J. Cleaner Prod., 2022, 356, 131826 CrossRef CAS.
- J. Lee, U. Von Gunten and J. H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed.
- T. Pottage, C. Richardson, S. Parks, J. T. Walker and A. M. Bennett, J. Hosp. Infect., 2010, 74, 55–61 CrossRef CAS PubMed.
- A. Ghauch, A. M. Tuqan and N. Kibbi, Chem. Eng. J., 2015, 279, 861–873 CrossRef CAS.
- S. Al Hakim, A. Baalbaki, O. Tantawi and A. Ghauch, RSC Adv., 2019, 9, 33472–33485 RSC.
- G. Z. Kyzas, N. Mengelizadeh, M. khodadadi Saloot, S. Mohebi and D. Balarak, Colloids Surf., A, 2022, 642, 128627 CrossRef CAS.
- R. El Asmar, A. Baalbaki, Z. Abou Khalil, S. Naim, A. Bejjani and A. Ghauch, Chem. Eng. J., 2021, 405, 126701 CrossRef CAS.
- S. Ghazali, A. Baalbaki, W. Bou Karroum, A. Bejjani and A. Ghauch, Environ. Sci.: Adv., 2024, 3(1), 119–131 CAS.
- E. Grilla, V. Matthaiou, Z. Frontistis, I. Oller, I. Polo, S. Malato and D. Mantzavinos, Catal. Today, 2019, 328, 216–222 CrossRef CAS.
- K. V. Plakas, V. C. Sarasidis, S. I. Patsios, D. A. Lambropoulou and A. J. Karabelas, Chem. Eng. J., 2016, 304, 335–343 CrossRef CAS.
- M. Amasha, A. Baalbaki and A. Ghauch, Chem. Eng. J., 2018, 350, 395–410 CrossRef CAS.
- M. Amasha, A. Baalbaki, S. Al Hakim and R. El Asmar, J. Adv. Oxid. Technol., 2018, 21(1), 1–13 CrossRef.
- S. Al Hakim, S. Jaber, N. Zein Eddine, A. Baalbaki and A. Ghauch, Chem. Eng. J., 2020, 380, 122478 CrossRef CAS.
- H. J. Kuhn, S. E. Braslavsky and R. Schmidt, Pure Appl. Chem., 2004, 76, 2105–2146 CAS.
- R. O. Rahn, Photochem. Photobiol., 1997, 66, 450–455 CrossRef CAS.
- S. Al Hakim, S. Jaber, N. Zein Eddine, A. Baalbaki and A. Ghauch, Chem. Eng. J., 2020, 380, 122478 CrossRef CAS.
- J. L. Weishaar, G. R. Aiken, B. A. Bergamaschi, M. S. Fram, R. Fujii and K. Mopper, Environ. Sci. Technol., 2003, 37, 4702–4708 CrossRef CAS PubMed.
- A. Baalbaki, N. Zein Eddine, S. Jaber, M. Amasha and A. Ghauch, Talanta, 2018, 178, 237–245 CrossRef CAS PubMed.
- D. Nikitin, B. Kaur, S. Preis and N. Dulova, Process Saf. Environ. Prot., 2022, 165, 22–30 CrossRef CAS.
-
World Health Organization, Sulfate in Drinking-water: Background document for development of WHO Guidelines for Drinking-water Quality, 2024 Search PubMed.
-
Drinking Water Regulations and Contaminants, US EPA, https://www.epa.gov/sdwa/drinking-water-regulations-and-contaminants, accessed 31 May 2024 Search PubMed.
- Z. Honarmandrad, X. Sun, Z. Wang, M. Naushad and G. Boczkaj, Water Resour. Ind., 2023, 29, 100194 CrossRef CAS.
- M. Sayed, M. Ismail, S. Khan, S. Tabassum and H. M. Khan, Environ. Technol., 2016, 37, 590–602 CrossRef CAS PubMed.
- Y. qiong Gao, N. yun Gao, D. qiang Yin, F. xiang Tian and Q. feng Zheng, Chemosphere, 2018, 201, 50–58 CrossRef PubMed.
- X. Ao, W. Sun, S. Li, C. Yang, C. Li and Z. Lu, Chem. Eng. J., 2019, 361, 1053–1062 CrossRef CAS.
- X. Ao and W. Liu, Chem. Eng. J., 2017, 313, 629–637 CrossRef CAS.
- Y. Gao, N. Gao, Y. Deng, Y. Yang and Y. Ma, Chem. Eng. J., 2012, 195–196, 248–253 CrossRef CAS.
- M. Antonopoulou and I. Konstantinou, Appl. Catal., A, 2016, 515, 136–143 CrossRef CAS.
- O. S. Furman, A. L. Teel and R. J. Watts, Environ. Sci. Technol., 2010, 44, 6423–6428 CrossRef CAS PubMed.
- A. Tsitonaki, B. Petri, M. Crimi, H. Mosbk, R. L. Siegrist and P. L. Bjerg, Crit. Rev. Environ. Sci. Technol., 2010, 40, 55–91 CrossRef CAS.
- J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- A. Ghauch and A. M. Tuqan, Chem. Eng. J., 2012, 183, 162–171 CrossRef CAS.
- C. Tan, N. Gao, Y. Deng, W. Rong, S. Zhou and N. Lu, Sep. Purif. Technol., 2013, 109, 122–128 CrossRef CAS.
- X. Lu, Y. Shao, N. Gao, J. Chen, Y. Zhang, H. Xiang and Y. Guo, Ecotoxicol. Environ. Saf., 2017, 141, 139–147 CrossRef CAS PubMed.
- C. Tan, N. Gao, S. Zhou, Y. Xiao and Z. Zhuang, Chem. Eng. J., 2014, 253, 229–236 CrossRef CAS.
- A. Angkaew, C. Sakulthaew, T. Satapanajaru, A. Poapolathep and C. Chokejaroenrat, J. Environ. Chem. Eng., 2019, 7, 102858 CrossRef CAS.
- Z. Fang, P. Chelme-Ayala, Q. Shi, C. Xu and M. Gamal El-Din, Chemosphere, 2018, 211, 271–277 CrossRef CAS PubMed.
- L. Zhou, C. Ferronato, J. M. Chovelon, M. Sleiman and C. Richard, Chem. Eng. J., 2017, 311, 28–36 CrossRef CAS.
- Y. qiong Gao, N. yun Gao, Y. Deng, D. qiang Yin and Y. sen Zhang, Environ. Sci. Pollut. Res., 2015, 22, 8693–8701 CrossRef PubMed.
- Z. Liu, H. Lan, Y. Wang, J. Zhang, J. Qin, R. Zhang and N. Dong, Chem. Eng. J., 2022, 429, 132485 CrossRef CAS.
- J. Wang and S. Wang, Chem. Eng. J., 2021, 411, 128392 CrossRef CAS.
- E. Can-Güven, Y. Daniser, S. Yazici Guvenc, F. Ghanbari and G. Varank, J. Photochem. Photobiol., A, 2022, 433, 114139 CrossRef.
- A. Ghauch, A. Baalbaki, M. Amasha, R. El Asmar and O. Tantawi, Chem. Eng. J., 2017, 317, 1012–1025 CrossRef CAS.
- J. Guo, Q. Gao, S. Yang, F. Zheng, B. Du, S. Wen and D. Wang, Process Saf. Environ. Prot., 2021, 146, 686–693 CrossRef CAS.
- J. Dan, Q. Wang, K. Mu, P. Rao, L. Dong, X. Zhang, Z. He, N. Gao and J. Wang, Environ. Sci., 2020, 6, 2510–2520 CAS.
- L. Chen, T. Cai, C. Cheng, Z. Xiong and D. Ding, Chem. Eng. J., 2018, 351, 1137–1146 CrossRef CAS.
- Y. Fu, X. Gao, J. Geng, S. Li, G. Wu and H. Ren, Chem. Eng. J., 2019, 356, 1032–1041 CrossRef CAS.
- Y. Gao, J. Zhou, J. Zhang, C. Li, N. Gao and D. Yin, Sep. Purif. Technol., 2021, 256, 117819 CrossRef CAS.
- O. K. Türk, G. Adalar, S. Yazici Guvenc, E. Can-Güven, G. Varank and A. Demir, Environ. Sci. Pollut. Res., 2023, 30, 869–883 CrossRef PubMed.
- M. Sörensen and F. H. Frimmel, Water Res., 1997, 31, 2885–2891 CrossRef.
- J. Cai, M. Zhou, W. Yang, Y. Pan, X. Lu and K. G. Serrano, Chemosphere, 2018, 212, 784–793 CrossRef CAS PubMed.
- A. Ghauch, A. M. Tuqan and N. Kibbi, Chem. Eng. J., 2015, 279, 861–873 CrossRef CAS.
- H. Shi, G. Zhou, Y. Liu, Y. Fu, H. Wang and P. Wu, RSC Adv., 2019, 9, 31370–31377 RSC.
- Q. Wang, X. Lu, Y. Cao, J. Ma, J. Jiang, X. Bai and T. Hu, Chem. Eng. J., 2017, 328, 236–245 CrossRef CAS.
- Y.-M. Lee, G. Lee and K.-D. Zoh, J. Hazard. Mater., 2021, 403, 123591 CrossRef CAS PubMed.
- H. Liu, Y. Meng, J. Li, X. Wang and T. Zhang, Chemosphere, 2022, 308, 136418 CrossRef CAS PubMed.
- J. L. Acero, F. J. Benítez, F. J. Real and E. Rodríguez, Sep. Purif. Technol., 2018, 201, 41–50 CrossRef CAS.
- Z. Frontistis, J. Photochem. Photobiol., A, 2019, 378, 17–23 CrossRef CAS.
- J. Du, C. Wang, Z. Zhao, F. Cui, Q. Ou and J. Liu, Chem. Eng. Sci., 2021, 241, 116683 CrossRef CAS.
- Y. Xu, Z. Lin and H. Zhang, Chem. Eng. J., 2016, 285, 392–401 CrossRef CAS.
- G. Mark, M. N. Schuchmann, H. P. Schuchmann and C. von Sonntag, J. Photochem. Photobiol., A, 1990, 55, 157–168 CrossRef CAS.
- S. G. Zimmermann, A. Schmukat, M. Schulz, J. Benner, U. von Gunten and T. A. Ternes, Environ. Sci. Technol., 2012, 46, 876–884 CrossRef CAS PubMed.
- H. Monteil, N. Oturan, Y. Péchaud and M. A. Oturan, Chemosphere, 2020, 247, 125939 CrossRef CAS PubMed.
- J. R. Bolton, K. G. Bircher, W. Tumas and C. A. Tolman, Pure Appl. Chem., 2001, 73, 627–637 CrossRef CAS.
-
U.S.: Industrial Electricity Prices Monthly 2023, Statista, https://www.statista.com/statistics/1395805/monthly-electricity-price-industrial-sector-united-states/#statisticContainer, accessed 2 February 2024 Search PubMed.
-
Electric Power Monthly, U.S. Energy Information Administration (EIA), https://www.eia.gov/electricity/monthly/epm_table_grapher.php?t=epmt_5_6_a, accessed 2 February 2024 Search PubMed.
- D. B. Miklos, C. Remy, M. Jekel, K. G. Linden, J. E. Drewes and U. Hübner, Water Res., 2018, 139, 118–131 CrossRef CAS PubMed.
- S. Ghazouani, F. Boujelbane, D. J. Ennigrou, B. Van der Bruggen and N. Mzoughi, Process Saf. Environ. Prot., 2022, 159, 442–451 CrossRef CAS.
- W. Benkayba, M. El Karbane, K. El Kacemi, M. R. Arhoutane, K. Karrouchi and A. Guessous, Int. J. Environ. Anal. Chem., 2023, 1, 1–15 CrossRef.
- M. Cobo-Golpe, V. Fernández-Fernández, T. Arias, M. Ramil, R. Cela and I. Rodríguez, J. Environ. Chem. Eng., 2022, 10, 107854 CrossRef CAS.
- S. O. Babalola, M. O. Daramola and S. A. Iwarere, J. Water Process Eng., 2023, 56, 104294 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Amir
Nasreddine
,
Amir
Nasreddine

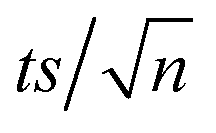 , where t is the student's t with degrees of freedom = 3, s is the standard deviation, and n is the number of replicates = 4. The absorbance spectrum of TRA is shown in Fig. 2. TRA shows three maxima of absorbance at 200, 215, and 272 nm. All three wavelengths were evaluated as excitation wavelengths for fluorescence analysis, and emission scans were conducted to determine the maximum emission value. While λex = 215 nm and λem = 320 nm would yield a more specific chromatographic peak, λex = 200 nm and λem = 300 nm showed better sensitivity, without sacrificing specificity; therefore, it was chosen for the FLD instrumental method.
, where t is the student's t with degrees of freedom = 3, s is the standard deviation, and n is the number of replicates = 4. The absorbance spectrum of TRA is shown in Fig. 2. TRA shows three maxima of absorbance at 200, 215, and 272 nm. All three wavelengths were evaluated as excitation wavelengths for fluorescence analysis, and emission scans were conducted to determine the maximum emission value. While λex = 215 nm and λem = 320 nm would yield a more specific chromatographic peak, λex = 200 nm and λem = 300 nm showed better sensitivity, without sacrificing specificity; therefore, it was chosen for the FLD instrumental method.
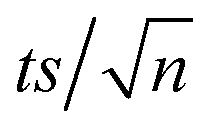 , where absent bars fall behind the point marker.
, where absent bars fall behind the point marker. for different [PS]0 was plotted against time (Fig. S4†). kobs was then calculated using eqn (5) and proved to be proportional to [PS]0 within the studied range. The degradation followed pseudo-first-order kinetics under all experimental conditions, regardless of the tested matrix/pH conditions.
for different [PS]0 was plotted against time (Fig. S4†). kobs was then calculated using eqn (5) and proved to be proportional to [PS]0 within the studied range. The degradation followed pseudo-first-order kinetics under all experimental conditions, regardless of the tested matrix/pH conditions.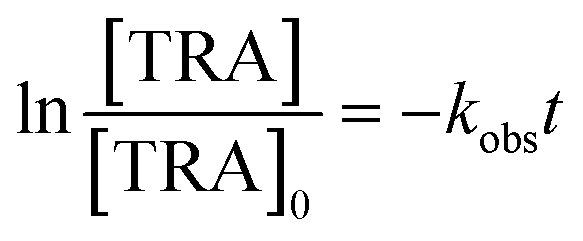
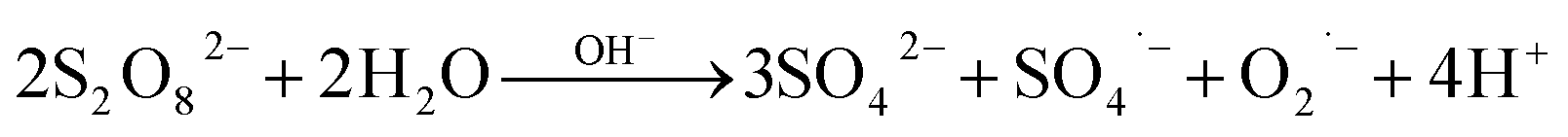
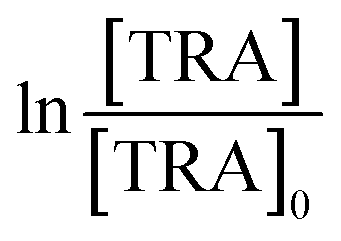 vs. elapsed reaction time corresponding to the experiments shown in (a)–(d), respectively. Error bars were calculated using
vs. elapsed reaction time corresponding to the experiments shown in (a)–(d), respectively. Error bars were calculated using 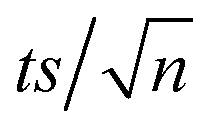 where absent bars fall behind the point marker.
where absent bars fall behind the point marker.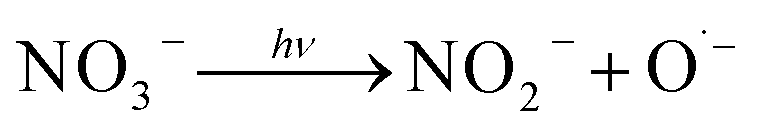
 vs. elapsed reaction time corresponding to (a)–(d), respectively. Error bars were calculated using
vs. elapsed reaction time corresponding to (a)–(d), respectively. Error bars were calculated using 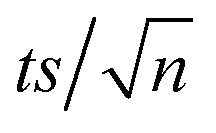 where absent bars fall behind the point marker.
where absent bars fall behind the point marker.