
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transboundary transport of air pollution in eastern Canada†
Robin
Stevens
 a,
Charles
Poterlot
a,
Nicole
Trieu
ab,
Henry Alejandro
Rodriguez
a and
Patrick L.
Hayes
*a
a,
Charles
Poterlot
a,
Nicole
Trieu
ab,
Henry Alejandro
Rodriguez
a and
Patrick L.
Hayes
*a
aDepartment of Chemistry, Faculty of Arts and Sciences, Université de Montréal, Montréal, Québec, Canada. E-mail: patrick.hayes@umontreal.ca
bDepartment of Chemical Engineering & Applied Chemistry, Faculty of Applied Science & Engineering, University of Toronto, Toronto, Ontario, Canada
First published on 31st January 2024
Abstract
We assess the GEOS-Chem chemical transport model against observations from the Quebec Air Quality Monitoring Network (RSQAQ) for carbon monoxide (CO), nitrogen oxides (NOX; nitrogen oxide (NO) and nitrogen dioxide (NO2)), fine particles having a diameter of less than 2.5 μm (PM2.5), ozone (O3), sulphur dioxide (SO2), and elemental carbon (EC), a component of PM2.5 known to have effects on both human health and climate. These pollutants have lifetimes that span from hours to weeks, allowing them to cross national borders and affect air quality far from their emission sites. We then investigate the sources of air pollution in Quebec through two complementary methods: back trajectory analysis using potential source contribution function (PSCF) and chemical transport modelling using GEOS-Chem. We perform three sensitivity studies with GEOS-Chem to determine the contributions from three source regions (Quebec, the rest of Canada, and the United States) to the concentrations of each of the investigated pollutants in Quebec. The PSCF calculations show that southern Quebec (local sources), the east coast of the United States, and southeastern Ontario are associated with days of high concentrations of several pollutants. Depending on the season, southern Quebec is associated with high concentrations of NOX, SO2, and CO; the east coast of the United States with high concentrations of PM2.5, NOX, O3, and CO; and southeastern Ontario with high concentrations of PM2.5 and EC. The GEOS-Chem results reveal that anthropogenic emissions from Quebec contribute the greatest amount (53%, 58%, 30%, and 44%) to concentrations of NOX, SO2, PM2.5, and EC in Quebec. Anthropogenic emissions from the US were the greatest contributor to CO concentrations (11%) and summertime O3 concentrations (17%). We find that removing all anthropogenic emissions from Quebec would reduce the fraction of the population of Quebec living in regions that exceed the recommended annual mean WHO PM2.5 concentration threshold of 5.0 μg m−3 from 87.7% to about 0.0%. While an absolute cessation of anthropogenic emissions is neither feasible nor desirable, our results suggest that substantial improvements in air quality in Quebec would be possible through reductions in local emissions alone despite the strong influence of transboundary transport.
Environmental significanceIt is well-known that ozone, fine aerosol particles (PM2.5), sulphur dioxide, nitrogen oxides, and carbon monoxide are harmful to human health. From 2016–2022, Montreal experienced between 21 and 43 poor air quality days per year, and air quality monitoring stations in Quebec reported from 2 to 278 “poor” air quality days in 2021. Therefore, it continues to be important to understand the sources of air pollution in Montreal and in the province of Quebec more generally. Specifically, we investigate in this study the contributions from local sources which can be addressed through local action and transboundary transport which required cooperation with other jurisdictions to address. |
1. Introduction
Air pollution is well known to have harmful effects on human health.1–4 However, the lifetimes of many common air pollutants span from days to weeks, allowing these air pollutants to cross national and sub-national borders, negatively affecting human health in other countries and even other continents than those where they were emitted.5–10 Addressing the problem of air pollution therefore requires cooperation across jurisdictions, which should be informed by knowledge of the relationships between source emissions and atmospheric concentrations in receptor regions.From 2016–2022, Montreal experienced between 21 and 43 poor air quality days per year, defined as days with concentrations of fine particles having a diameter of less than 2.5 μm (PM2.5) greater 35 μg m−3. Annual mean concentrations of PM2.5 were 7.1 μg m3 in Montreal in 2022, beyond the WHO air quality guideline of 5 μg m−3.11 The province of Quebec uses an Air Quality Index calculated based on the concentrations of ozone (O3), PM2.5, sulphur dioxide (SO2), nitrogen oxides (NOX; nitrogen oxide (NO) and nitrogen dioxide (NO2)), and carbon monoxide (CO). Based on this Air Quality Index, the stations in Quebec reported from 2 to 278 “poor” air quality days in 2021.12 Therefore, it continues to be important to understand the sources of air pollution in Montreal and in the province of Quebec more generally. Specifically, we wish to investigate in this study the contributions from local sources which can be addressed through local action and transboundary transport which required cooperation with other jurisdictions to address.
It is well known that emissions from anthropogenic sources within the US contribute to concentrations at the surface within Quebec and that air quality in Quebec is episodically influenced by biomass burning emissions in northwestern Canada.13,14 Summertime surface-level ozone in eastern Canada has been shown to be greatest during periods of southwesterly flow.15,16 A previous study has examined the contributions to PM2.5 in Canada by source sector, and highlighted the importance of wildfires, transportation, and residential combustion.17 They additionally found that sources within the US were responsible for 33% of population-weighted PM2.5 in Central Canada, a region which includes Manitoba, Ontario, and Quebec.
Montreal constitutes a large city (metropolitan population of four million) in an otherwise rural region, located in the outflow region of the industrialized northeastern US, including the megacity of New York City. This is therefore a good region for studying the transport and transformation of urban and industrial emissions into a populated boreal region. Historically, transport of US emissions into the Quebec region has been examined closely due to concerns of acid rain,13,18,19 but continued examination of this transboundary transport is important because of possible changes in transport with climate change,20 historical changes in emissions,21,22 an expanded set of pollutants of interest, expansions and improvements in monitoring,11,23 and improvements in techniques for attributing transboundary transport, including chemical transport modelling24–28 and back-trajectory modelling.29–32
We investigate several pollutants monitored by the Quebec Air Quality Monitoring Network (RSQAQ, Réseau de Surveillance de la Qualité de l'Air du Québec).23 The data collected at the RSQAQ sites is included in the Canadian National Air Pollution Monitoring Network (NAPS) Program,33 and those stations on the island of Montreal are operated by the city of Montreal Air Quality Monitoring Network (RSQA, Réseau de surveillance de la qualité de l'air).11 We will describe the observational data in more detail in Section 2.1. Specifically, we investigate CO, NOX, PM2.5, O3, SO2, and elemental carbon (EC). It is well known that these pollutants can be harmful to human health.1–4 We include EC in our analysis because it is a particularly interesting component of PM2.5: it absorbs solar radiation and can contribute to global warming. Thus, efforts to reduce the concentration of EC will improve air quality while reducing radiative forcing leading to climate change. The distribution by sector of PM2.5 in Canada has previously been investigated by Meng et al. (2019)17 for the year 2013. In this study, we focus on a breakdown by region in order to specify the concentrations of pollutants from Quebec or outside Quebec. The years analyzed are 2014, 2015 and 2016. Quebec is Canada's largest province by size and second-largest by population and is located to the north and east of major population centers in Canada and the USA, and thus it is susceptible to receiving transboundary pollution transported by prevailing winds.
We present a general back-trajectory analysis for the province of Quebec. Calculating back trajectories is a well-established method for identifying geographic regions that are potential sources of air pollution.7,30–32 To better explain this method, consider the calculation of a normal trajectory that advances in time. The simulation begins at a given point in space and time that would correspond to the release of a pollutant from a source (e.g. a factory or a smelter). From this point, the movements of the pollutant can be calculated using meteorological data, including wind direction and speed. This simulation operates over time to generate a trajectory. If it is repeated several times starting from different times, for example on subsequent days, the set of trajectories can be superimposed to make a frequency map of the path of the trajectory. To calculate a back trajectory, the simulation is built in the same way, but the calculations proceed in the opposite direction in time, compared to the normal trajectory. Thus, if the simulation begins at an observation site, the regions upwind of the site are identified.
In order to take into account the observed concentrations of pollutants, maps of the Potential Source Contribution Function (PSCF) were calculated. PSCF is a method of weighting a set of back trajectories using data from a monitoring station so that source regions that are associated with high concentrations of a pollutant at a station can be identified.29,34,35 PSCF maps are calculated for all the pollutants specified above, i.e. CO, NOX, PM2.5, O3, SO2, and EC at each station. To facilitate the interpretation of the results, the PSCF maps for several stations in Quebec are averaged for each pollutant.
A complementary approach to assess transboundary transport is the use of chemical transport models. In these models, the atmosphere is divided into a three-dimensional grid based on latitude, longitude and altitude. Atmospheric processes are comprehensively simulated in three dimensions, including emissions, transport and chemistry. Such models are used to make air quality forecasts, to help develop environmental policies and for research. Since these models include emission inventories, it is possible to modify these emissions to study the impact of different sectors or regions on air pollutant concentrations. At the same time, emissions inventories may contain errors or may not accurately represent actual emissions. This is a limitation of this approach, and it is therefore desirable to combine this approach with other methods such as back trajectory calculations.
Therefore, to complement the PSCF analysis, we use the GEOS-Chem chemical transport model to simulate pollutant concentrations in Quebec for four scenarios. The first scenario includes the emission inventories without modification. In the other scenarios, emissions were removed in Quebec, the United States or the other Canadian provinces and territories. This series of scenarios allows us to estimate the quantities of a pollutant from each of these regions. We begin with the results of the GEOS-Chem simulations to assess the transboundary input of air pollutants, and more specifically, the quantities coming from the United States or the rest of Canada (RoC). Then, back-trajectory analysis and PSCF maps are used to identify more precisely the most important source regions in the United States and Canada.
The article is organized as follows: Section 2 explains in detail the methodologies used, including the observational data (Section 2.1), the calculations to produce the back trajectories and PSCF maps (Section 2.2) and the chemical transport modelling by GEOS-Chem (Section 2.3). Section 3 presents the validation of GEOS-Chem using the observational data (Section 3.1), the assessment of transboundary input based on GEOS-Chem results (Section 3.2) and the results of the PSCF maps (Section 3.3). We compare the PSCF and chemical transport modelling results in Section 4. Finally, the conclusions of the study are summarized in Section 5.
2. Experimental methods
2.1 Observational data
The observational data used in this study was collected at stations operated by either the RSQAQ or the city of Montreal air quality monitoring network, and all of the stations participate in the NAPS program. The RSQAQ and the City of Montreal's air quality monitoring network use similar instrumentation and the same data collection and quality assurance protocols. For a complete description of the data collection methods, we refer the reader to the Ambient Air Monitoring and Quality Assurance/Quality Control Guidelines, National Air Pollution Surveillance Program.33 For simplicity, we will refer to all observation stations as RSQAQ stations in the rest of this article.We use the data from the RSQAQ stations for two purposes: First, to evaluate the results of the base-case GEOS-Chem simulation, and secondly, to perform the PSCF analysis. To evaluate GEOS-Chem, we use data from 2012 to 2016 inclusive. For the PSCF analysis, we focus on data for the years 2014 to 2016 inclusive, in order to avoid periods of missing data and to have a more continuous dataset. EC was measured at two stations (Tables A1 and A2†) with a frequency of one day in three or one day in six and an interval of 24 hours. All other pollutants were monitored continuously year-round and reported by the government as hourly averages. We aggregated the pollutant concentration data into monthly averages for comparison with the GEOS-Chem results, and into daily averages for the PSCF calculations. The stations used in this report to obtain data for the PSCF calculations are summarized in Table A1 and Fig. A1.† We list the stations used to evaluate GEOS-Chem in Table A2.†
Since the RSQAQ stations are positioned to monitor population exposure, the majority of the stations are in urban centers or on their outskirts. As the population of Quebec is concentrated on the shores of the St. Lawrence River which is in the south of the province, the stations are also geographically concentrated in the south of the province. However, the purpose of this study is to investigate transboundary sources of pollution. The location of the stations could therefore present some challenges for the PSCF analysis: the first is that the stations located in urban centers measure ambient pollution which is more likely to be strongly influenced by local emissions at a scale finer than that which can be resolved by the PSCF analysis, and the cross-border contribution is added to this local pollution. If the monitoring station were in a rural setting, the transboundary input would become a larger part of the total concentration, and it would therefore be easier to quantify the transboundary input and its sources. We also do not want the PSCF analysis to be dominated by sources that only affect Montreal or Québec City, where the greatest number of stations are located. To limit the influence of local emissions for stations located in urban areas and to ensure that the PSCF analysis is regionally representative, we only used data from one station in each of Montreal and Québec City in the PSCF analysis. The second problem is the sampling bias towards the south of the province due to the large number of stations concentrated in this part of the province. As the vast majority of the population of Quebec lives in the south of the province, we expect that our results will nonetheless be representative of the pollution sources that affect the majority of Quebec's population. In addition, the largest sources outside of Quebec are to the south and southwest of the province, and they are not expected to exert as large an influence on air pollutant concentrations in northern Quebec. In fact, this assumption is supported by the GEOS-Chem results presented below. Thus, as the purpose of this study is to study transboundary contributions, it is useful to have a greater number of datasets from monitoring stations near the south and southwest borders of the province.
For the evaluation of GEOS-Chem, we exclude observations from seven stations that are known to be directly downwind of large point sources (Table A3†). The model results yield pollutant concentrations averaged over the resolution of our simulations (0.5° latitude by 0.625° longitude), and thus it is impossible for the model to capture highly concentrated plumes, such as those measured at these stations. For the model evaluation, we use data from a total of 34 stations, of which 32 measure PM2.5, 32 measure O3, 12 measure NOX, 8 measure SO2, 6 measure CO, and 2 measure EC.
2.2 Back-trajectory and PSCF analysis
Several trajectory models have been developed to study atmospheric transport.30–32 In this study, we used the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model developed by the National Oceanic and Atmospheric Administration (NOAA) in the United States.30 The backward trajectories of the air masses observed at the RSQAQ stations were calculated for the period from 2014–2016 inclusive. Meteorological data from the Global Data Assimilation System (GDAS)36 with a resolution of 1° were obtained from the NOAA website and used to carry out the simulations.To estimate the potential locations of sources of air pollutants transported over long distances, we used the ZeFir software package37 to calculate PSCF maps using the back-trajectories calculated with HYSPLIT. From the HYSPLIT trajectory modelling results, the air mass movement can be represented as segments that create a trajectory describing the air movements upstream of the site. If a segment of a trajectory is in a geographical cell defined on a map, this cell will then be considered as a potential contributor of pollutants transported to the receiving site. PSCF is used to generate a probability map of cells around a monitoring site where the probability that a cell is associated with a high concentration of a pollutant is indicated. The probabilities are calculated according to eqn (1) below where nij is the total number of segments entering cell ij and mij is the number of segments entering the same cell for which the concentration of a pollutant when the trajectory arrives at the site is within the 90th percentile.
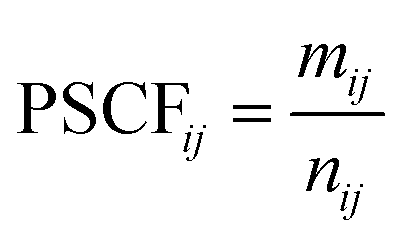 | (1) |
The daily concentrations of pollutants at the RSQAQ sites were used in the calculations of the PSCF maps. The trajectories were calculated 5 days backwards in time and were started at a height of 500 m above ground level; this altitude was chosen to limit the effects of local topography. The domain of interest displayed in the PSCF graphs extends from 20° N to 80° N and from 40° W to 150° W. In a PSCF analysis, small values of nij can generate high PSCF probabilities with large uncertainties due to lack of data.29,35 Therefore, to improve the visual representation of the results, a sigmoidal weighting function, w(nij), was applied to the PSCFij values.38–40
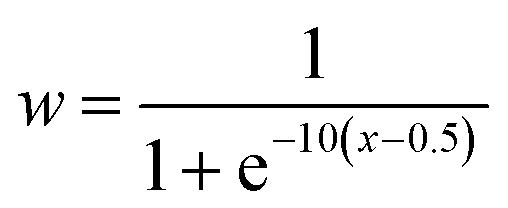 | (2) |
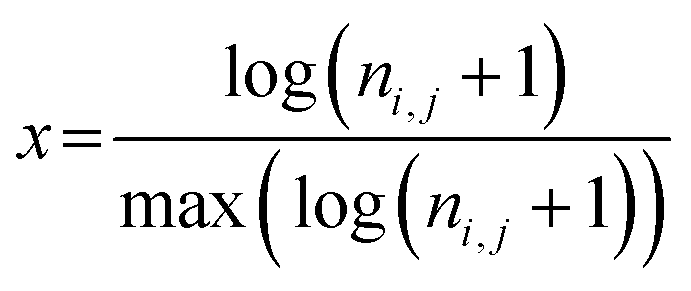 | (3) |
Finally, there were a number of days when it was not possible to perform the PSCF analysis due to a lack of meteorological data or a lack of measurements of a pollutant. The percentages of days missing from the PSCF analysis are summarized in Tables A3–A8.†
2.3 GEOS-Chem simulations
We perform simulations using the GEOS-Chem chemical transport model (version 14.0.2, https://doi.org/10.5281/zenodo.7383132).24,25 GEOS-Chem is driven by assimilated meteorology from the Modern-Era Retrospective analysis for Research and Applications, Version 2 (MERRA-2) dataset produced by the NASA Global Modeling and Assimilation Office (GMAO). We generate boundary conditions using a global simulation at 2° latitude x 2.5° longitude resolution. We then use a nested grid with 0.5° latitude × 0.625° longitude resolution, spanning 35° N to 65° N, 90° W to 50° W in order to include the full province of Quebec as well as the strong source regions of the Great Lakes region and the northeastern US. We performed simulations from 2012–2016, but we report results in Section 3.2 only from 2014–2016 for consistency with the PSCF analysis. Our simulations therefore have a spin-up period of two years for the analysis, and a spin-up period of one month for the evaluation against the RSQAQ observations. The atmosphere is resolved using 47 vertical layers from the surface to 0.01 hPa. The vertical resolution of the model is about 100 m near the surface, but it becomes coarser at higher altitudes. Biomass-burning emissions are simulated from the Global Fire Emissions Database version 4 (GFED4).41 Anthropogenic emissions from Canada are provided by the Air Pollutant Emissions Inventory (APEI), recently processed for use in chemical transport models as described in Crippa et al. (2023).21 Anthropogenic emissions from the US are provided by the National Emissions Inventory for 2016,42 with annual scaling factors to account for changes in emissions relative to 2016. Shipping emissions are provided by the Community Emissions Data System version 2 (CEDSv2),22 and aircraft emissions are provided by the Aviation Emissions Inventory Code (AEIC).43,44 We use the recently developed wet particle entrainment treatment described by Luo et al. (2019, 2020),27,28 which has been shown to provide better agreement with nitrate and ammonium concentrations over eastern North America.In order to examine the sensitivity of air pollutant concentrations to transboundary transport, we perform a base-case simulation with no changes in emissions and three sensitivity simulations, each with anthropogenic emissions from a geographic region turned off. In the first sensitivity simulation (noQC), we do not allow any anthropogenic emissions within the borders of the province of Quebec. In the second simulation (noRoC), we do not allow any anthropogenic emissions within the borders of Canada, except for emissions from the province of Quebec (Rest of Canada, RoC). In the third simulation (noUS), we do not allow any anthropogenic emissions within the borders of the contiguous United States. We include emissions from shipping and aircraft within the boundaries of the specified region as anthropogenic emissions, therefore we reduce these to zero as well. We have not changed emissions from wildland fire or natural sources. We note that for the purposes of masking emissions, each model grid cell is considered to be entirely within one province or country; the resolution of the provincial or national masks is the same as the model resolution. By calculating the differences in the concentrations of air pollutants in the sensitivity simulations compared to the base-case simulation, we estimate the proportions of air pollutants in Quebec due to sources within Quebec, sources in the rest of Canada, and sources in the contiguous United States.
Despite the two complementary investigative techniques, there are still limits to our methodology. The chemical transport model results shown here rely on high-quality, spatially and temporally resolved emissions inventories. Reporting and monitoring of emissions is crucial to the creation and updating of these emissions inventories. In addition, the chemical transport model and the back trajectory analysis are dependent on the accuracy of the reanalysis meteorological data, which in turn depends in part on local meteorological monitoring.
3. Results and discussion
3.1 Evaluation of GEOS-Chem against observations for Quebec, Canada
We evaluate the results of our GEOS-Chem simulations against the observations made at the RSQAQ stations. This evaluation is summarized in Tables 1 and 2 and shown in Fig. 1–3. We use as our metrics of evaluation the Pearson's correlation coefficient (R), the mean error (ME), the normalized mean error (NME), the mean bias (MB), and the normalised mean bias (NMB), where ME, NME, MB, and NMB are defined as follows. The xi variables indicate the model predictions and the yi variables indicate the observed data for a given station, both as monthly averages, and N is the number of model-observation pairs: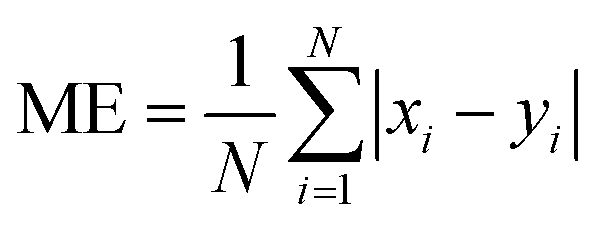 | (4) |
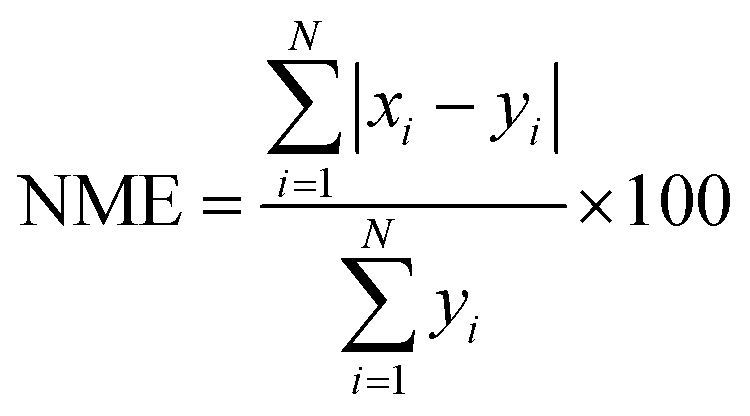 | (5) |
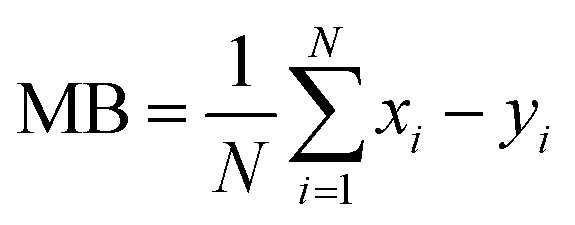 | (6) |
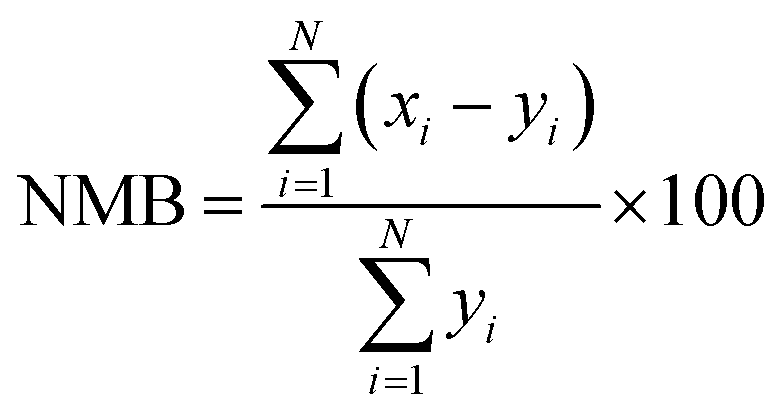 | (7) |
| Pollutant | R | Temporal R | Spatial R | ME | NME [%] | MB | NMB [%] |
|---|---|---|---|---|---|---|---|
| PM2.5 [μg m−3] | 0.55 | 0.73 | 0.65 | 2.03 | 30 | −0.229 | −3 |
| CO [ppm] | 0.29 | 0.74 | 0.11 | 0.051 | 24 | −0.042 | −20 |
| NOX [ppb] | 0.61 | 0.91 | 0.42 | 4.79 | 50 | −4.70 | −49 |
| SO2 [ppb] | 0.41 | 0.37 | 0.57 | 0.469 | 60 | 0.105 | 13 |
| O3 [ppb] | 0.75 | 0.79 | 0.61 | 7.32 | 28 | 7.17 | 27 |
| EC [μg m−3] | 0.76 | 0.62 | — | 0.137 | 31 | −0.084 | −19 |
| Pollutant | Season | ME | NME [%] | MB | NMB [%] |
|---|---|---|---|---|---|
| PM2.5 [μg m−3] | Winter | 2.70 | 34 | −0.871 | −11 |
| Summer | 1.73 | 23 | −0.421 | −6 | |
| CO [ppm] | Winter | 0.058 | 24 | −0.045 | 19 |
| Summer | 0.043 | 21 | −0.029 | −14 | |
| NOX [ppb] | Winter | 7.10 | 52 | −6.93 | −50 |
| Summer | 2.95 | 48 | −2.91 | −47 | |
| SO2 [ppb] | Winter | 0.468 | 46 | 0.052 | 5 |
| Summer | 0.438 | 82 | 0.223 | 42 | |
| O3 [ppb] | Winter | 4.49 | 17 | 4.12 | 15 |
| Summer | 8.24 | 33 | 8.20 | 33 | |
| EC [μg m−3] | Winter | 0.160 | 30 | −0.079 | −15 |
| Summer | 0.160 | 35 | −0.154 | −34 |
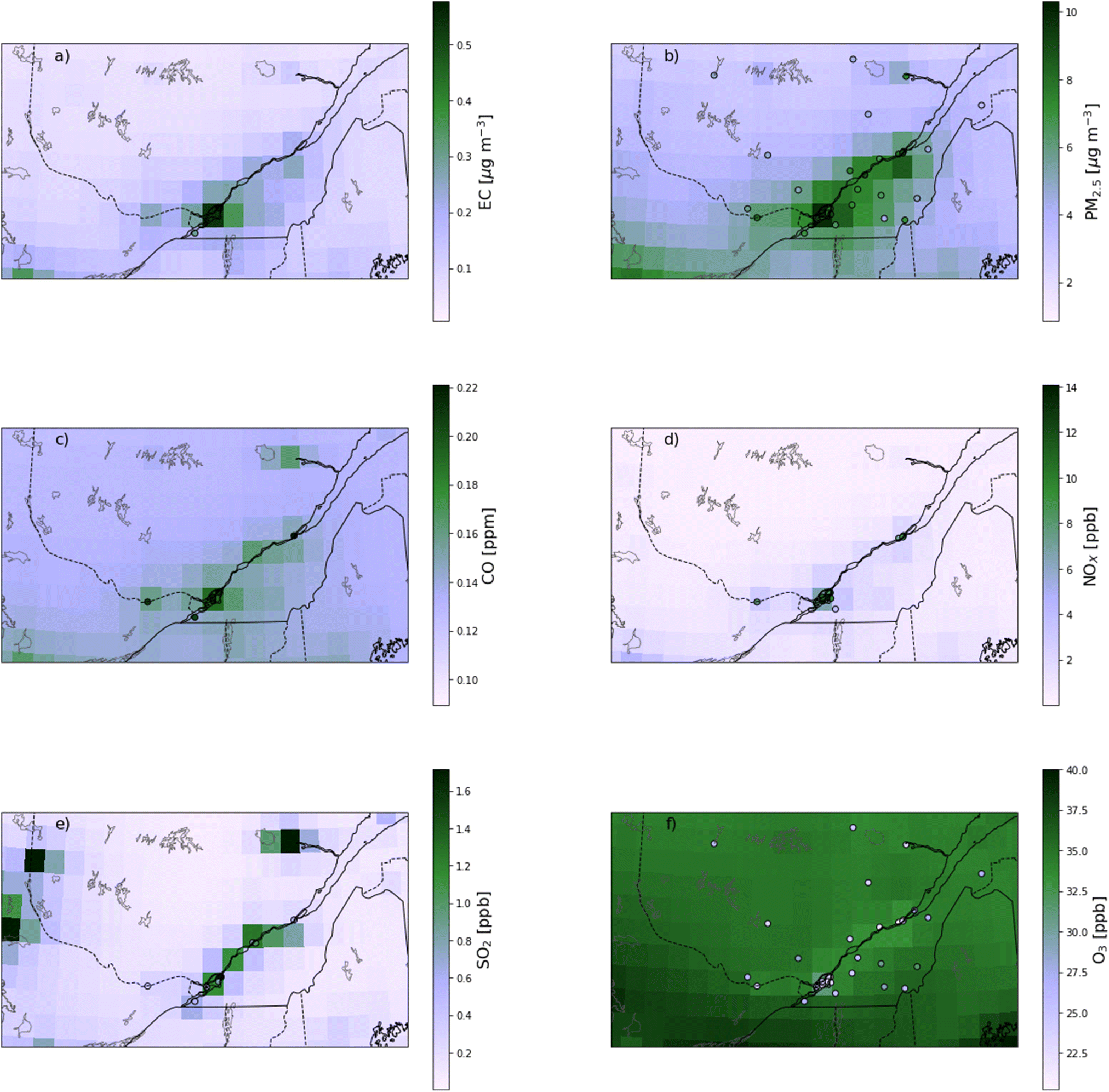 | ||
| Fig. 1 Map of mean concentrations simulated by the base case GEOS-Chem simulation, with the mean concentrations at the RSQAQ stations superimposed. Values are shown for (a) EC, (b) PM2.5, (c) CO, (d) NOX, (e) SO2, and (f) O3. | ||
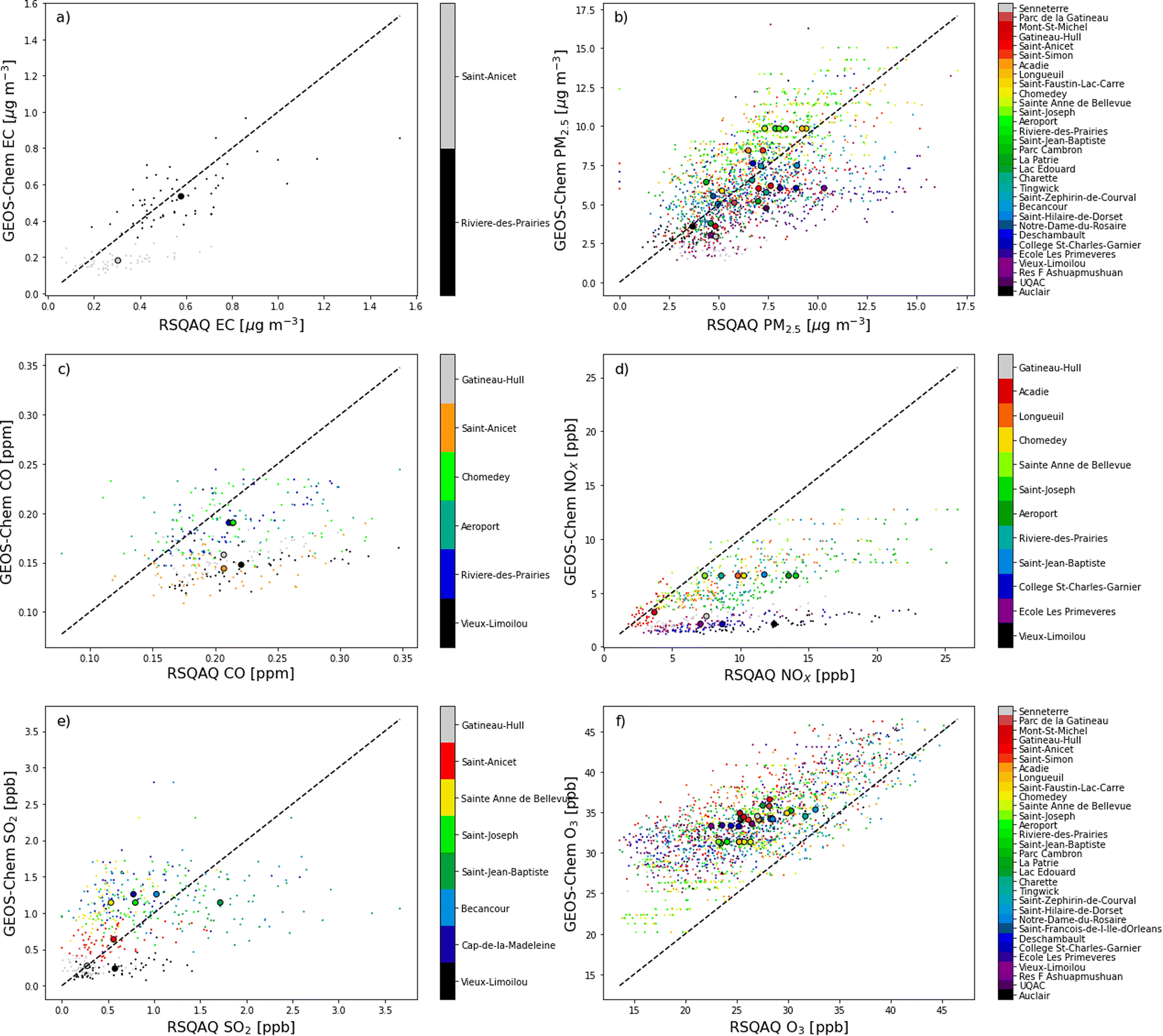 | ||
| Fig. 2 Scatterplot of GEOS-Chem simulated concentrations vs. concentrations observed at the RSQAQ stations. Temporal averages at each station are displayed as large dots. Values are shown for (a) EC, (b) PM2.5, (c) CO, (d) NOX, (e) SO2, and (f) O3. | ||
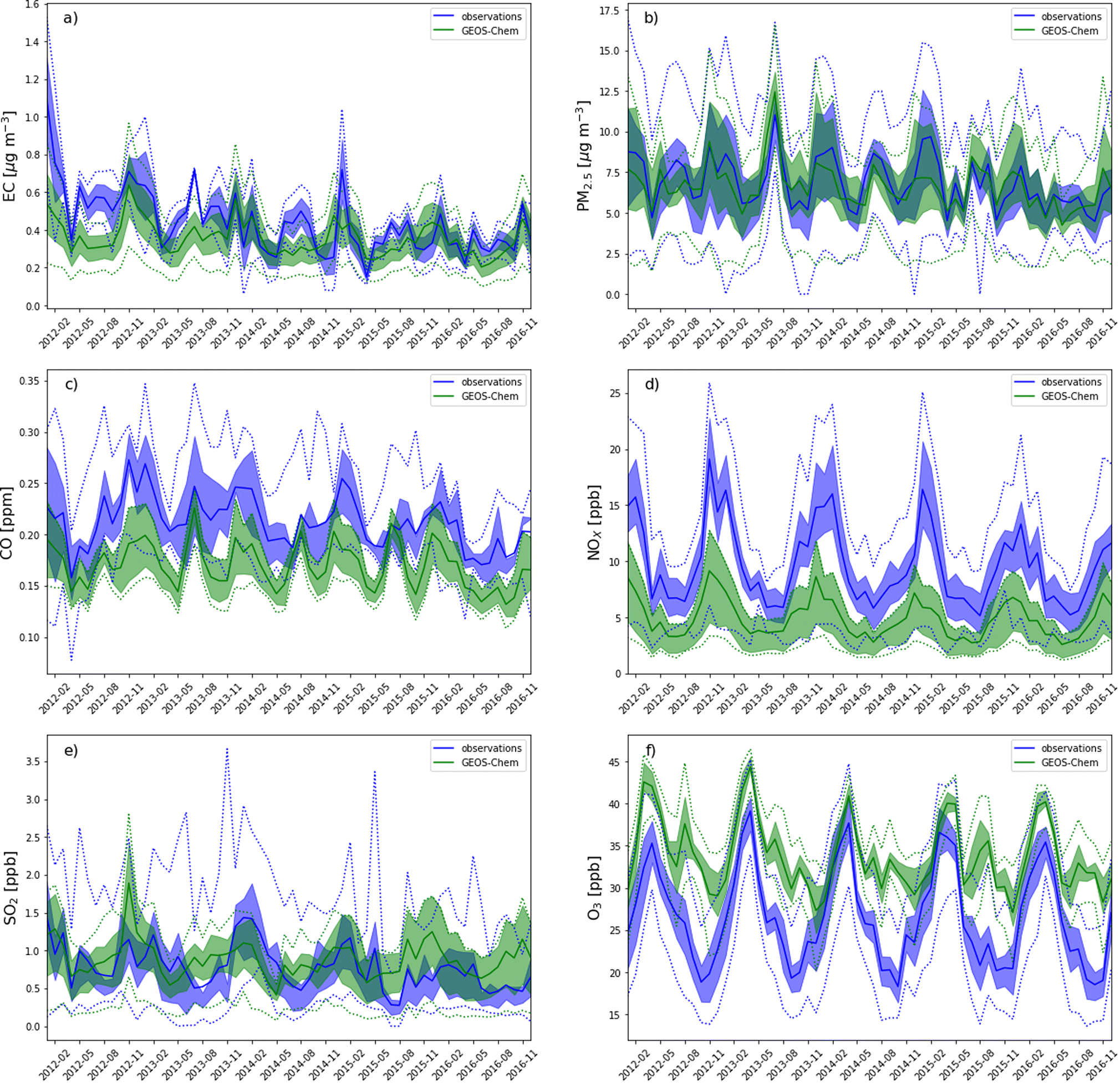 | ||
| Fig. 3 Time series of pollutant concentrations averaged at RSQAQ station locations. Values simulated by GEOS-Chem are in green, while RSQAQ observations are in blue. The shaded region gives the interval between the 25th and 75th percentiles. Dotted lines indicate minimums and maximums. Values are shown for (a) EC, (b) PM2.5, (c) CO, (d) NOX, (e) SO2, and (f) O3. | ||
In order to provide a more in-depth assessment of the accuracy of GEOS-Chem regarding the spatial and temporal variations for each pollutant, we also recalculate R after averaging the model and observation data for all stations (temporal R) or after averaging these data over time for each station individually (spatial R). We do not average multiple stations within a single GEOS-Chem grid cell in agreement with previous studies that state that this artificially improves model-observations agreement.6
When all seasons are considered, the correlation coefficients range from 0.29 for CO to 0.76 for EC. Temporal correlation coefficients vary from 0.37 for SO2 to 0.91 for NOX, and spatial correlation coefficients vary from 0.11 for CO to 0.65 for PM2.5. We note that with only two stations where EC was measured, it is not useful to calculate a spatial correlation coefficient for EC. The higher temporal correlations show that GEOS-Chem is able to reproduce the seasonal cycles of each pollutant, which is also visible in the time series comparisons presented in Fig. 3. The low spatial correlation between the GEOS-Chem results and the CO observations are partly due to the low spatial variability of the observed concentrations. Mean CO concentrations differ by less than 0.014 ppm between stations (∼7%, Fig. 1c and 2c), and this small variability is less likely to be due to regional-scale differences in CO concentrations than to other factors, especially smaller scale variability that cannot be reproduced by GEOS-Chem. Furthermore, in our analysis, 3 of the 6 stations measuring CO are located in the same GEOS-Chem grid cell (Montréal – Rivière Des Prairies, Montréal – Aéroport de Montréal, and Laval – Chomedey), and the modelling results will therefore give exactly the same CO concentrations for these three stations. The same challenge exists for simulating NOX concentrations: 5 of the 12 stations used in our analysis are in the Montreal area, and one station is in the city of Laval (Fig. 1d and 2d). These six stations are all within a single GEOS-Chem grid cell, and therefore GEOS-Chem predicts the same NOX concentrations for all 6 stations. Similarly, 3 of the 12 stations are in the Québec City region, also covered by a single GEOS-Chem grid cell.
There are small biases for CO and EC and a moderate bias for NOX. There may be a contribution to the bias in NOX concentrations from an underestimate in NOX emissions or an overestimate of O3 concentrations, which would lead to an overestimation of NOX oxidation. As we will show in Section 3.2, NOX concentrations have one of the greatest relative contributions from local sources (53.2% from sources within Quebec) of the pollutants we examine. As all but one of the stations measuring NOX were in urban locations (Fig. 1d), the grid-cell means from GEOS-Chem would be expected to be less than the point measurements from the RSQAQ stations. Silvern et al. (2019)45 previously found that GEOS-Chem underestimated NO2 concentrations in a similar manner at the United States Environmental Protection Agency Air Quality System sites, which are mainly in urban locations. The same study found a greatly reduced low bias for GEOS-Chem NO2 concentrations compared against observations at rural locations. Moreover, the relative interannual trend at both rural and urban locations from 2005–2017 was well reproduced by GEOS-Chem in that study. The effect of coarse resolution leading to a low bias with respect to surface observations of NO2 has also been previously shown to affect satellite observations in a similar manner.46–48 This model resolution effect likely also contributes to the low biases in CO and EC concentrations.
Conversely, the stations measuring PM2.5 were almost evenly split between urban and rural locations (15 and 17, respectively), so the effect of model resolution on the bias would be less uniformly negative. Similar to this work, Kim et al. (2015)49 found a good correlation (R = 0.65) for PM2.5 concentrations when comparing against observations from US measurement networks for September and August of 2013, and that study also found a slightly negative bias of −1.4%, which is similar to our NMB of −3%, despite the finer resolution of the model (0.25° × 0.3125°) used in this previous study. The model error and model bias in PM2.5 concentrations is greater in winter than in summer (Table 2). We will show in Section 3.2.2 (Tables 3 and 4, Fig. A10 and A11†) that GEOS-Chem predicts both a larger fraction of PM2.5 from Quebec anthropogenic sources and a larger fraction of PM2.5 from anthropogenic sources in general in winter vs. summer. It is therefore likely that there is an increased contribution from local primary sources in winter such as emissions from residential home heating. These sources are more difficult to resolve in 0.5° latitude × 0.625° longitude grid cells than secondary formation of PM2.5, which would be more prominent in summer, and this would lead to a low model bias as discussed above for NOX concentrations.
| 2014–2016 | DJF | JJA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| noQC | noRoC | noUS | noQC | noRoC | noUS | noQC | noRoC | noUS | |
| O3 | 0.9% | 2.4% | 11.1% | −2.2% | −0.4% | 4.3% | 4.5% | 5.2% | 16.6% |
| NOX | 53.2% | 14.1% | 25.4% | 48.3% | 15.9% | 35.2% | 56.6% | 11.5% | 13.2% |
| SO2 | 57.6% | 26.7% | 18.3% | 55.5% | 28.6% | 25.1% | 59.7% | 25.9% | 9.7% |
| CO | 5.6% | 2.5% | 11.2% | 7.1% | 3.5% | 15.1% | 4.3% | 1.9% | 7.9% |
| PM2.5 | 30.2% | 13.5% | 27.9% | 38.7% | 15.1% | 34.1% | 18.3% | 10.7% | 19.6% |
| EC | 43.8% | 14.9% | 32.8% | 45.3% | 14.9% | 36.8% | 37.4% | 14.6% | 25.6% |
| 2014–2016 | DJF | JJA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| noQC | noRoC | noUS | noQC | noRoC | noUS | noQC | noRoC | noUS | |
| O3 | 0.296 | 0.832 | 3.789 | −0.733 | −0.138 | 1.441 | 1.368 | 1.601 | 5.099 |
| NOX | 0.395 | 0.104 | 0.188 | 0.577 | 0.190 | 0.420 | 0.242 | 0.049 | 0.057 |
| SO2 | 0.118 | 0.055 | 0.038 | 0.147 | 0.076 | 0.067 | 0.102 | 0.044 | 0.016 |
| CO | 7.441 | 3.364 | 14.982 | 10.219 | 4.952 | 21.557 | 6.070 | 2.695 | 11.063 |
| PM2.5 | 1.166 | 0.523 | 1.080 | 1.457 | 0.569 | 1.285 | 0.841 | 0.492 | 0.897 |
| EC | 0.043 | 0.015 | 0.032 | 0.055 | 0.018 | 0.045 | 0.035 | 0.014 | 0.024 |
GEOS-Chem exhibits a positive bias in O3 concentrations which is greater in summer and fall and smaller in winter and spring (Tables 2, A10,†Fig. 3f). Several atmospheric models (including GEOS-Chem) have been previously shown by Reidmiller et al. (2009) to have a summer peak in O3 concentrations that is too late when compared to observations in Maine and Vermont, which resulted in a multi-model mean JJA mean bias of 10 ppb.5 However, we find that the seasonal cycle in O3 concentrations is reproduced well for Quebec, albeit with a consistently high bias that is similar to the previous work of Reidmiller et al. (2009). One possible source of this bias is the difference in vertical sampling between the model and the observations: the lowest model vertical level is centered at 65 m above ground level, while the observations were typically sampled at about 2 m above ground level. Previous studies have attempted to correct for this bias, and have calculated that this effect is responsible for a 3 ppb overestimation in southeastern US maximum daily 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h average (MDA8) surface ozone50 and approximately 0.5 μg m−3 in continental US PM2.5.51 The method that was used to correct for this bias assumes no surface emissions of the species being corrected, and thus is only applicable to O3 and the secondary components of PM2.5, not to any of the other pollutants discussed in this study. Additionally, the low model bias in NOX concentrations may contribute to the high O3 bias where NOX titration occurs, specifically in urban locations in wintertime. We performed a correlation analysis between the model biases in O3 concentrations and the model biases in NOX concentrations at stations that measure both pollutants, and we found spatial correlation coefficient of −0.76: The stations with higher mean NOX concentrations have greater underestimations in NOX concentrations, and also greater overestimations of mean O3 concentrations (Fig. A2,† also compare Fig. 1d and f, 2d and f). This spatial correlation was strongest in winter (spatial R = −0.89) when NOX titration is expected to occur, and weakest in summer (spatial R = −0.15).
h average (MDA8) surface ozone50 and approximately 0.5 μg m−3 in continental US PM2.5.51 The method that was used to correct for this bias assumes no surface emissions of the species being corrected, and thus is only applicable to O3 and the secondary components of PM2.5, not to any of the other pollutants discussed in this study. Additionally, the low model bias in NOX concentrations may contribute to the high O3 bias where NOX titration occurs, specifically in urban locations in wintertime. We performed a correlation analysis between the model biases in O3 concentrations and the model biases in NOX concentrations at stations that measure both pollutants, and we found spatial correlation coefficient of −0.76: The stations with higher mean NOX concentrations have greater underestimations in NOX concentrations, and also greater overestimations of mean O3 concentrations (Fig. A2,† also compare Fig. 1d and f, 2d and f). This spatial correlation was strongest in winter (spatial R = −0.89) when NOX titration is expected to occur, and weakest in summer (spatial R = −0.15).
In summary, GEOS-Chem can reproduce the observed concentrations and variability of PM2.5, O3 and EC. The observed variability of NOX is well reproduced, but the concentrations simulated by GEOS-Chem have a negative bias. This bias is likely due in part to small-scale variations in NOX concentrations that are not relevant to the regional-scale analysis that we will present in Section 3.2. Small-scale variations also likely impact CO spatial correlation and bias. The model performs somewhat less well in predicting SO2 and CO concentrations, but it is still useful in assessing transboundary transport, albeit with larger uncertainties. Further work is needed to improve simulations of these species with GEOS-Chem for Quebec, possibly by using higher resolution model runs that can be performed with the high performance version of GEOS-Chem.26
Finally, we note that in our analysis, we will be examining the differences between two sets of simulations. Therefore, many potential sources of error, including uncertainties in natural emissions, long-range transport from outside of North America, and model meteorology would affect the results of all simulations in the same way. We therefore expect that results based on the differences between simulations will be more robust than the predictive power of the base-case simulation.
3.2 Apportionment of pollutants to emissions in Quebec, the RoC, and the US
To evaluate the contributions from emissions in Quebec, the RoC, and the US to O3, NOX, SO2, CO, PM2.5, and EC, the results from the GEOS-Chem simulations are plotted as differences between the sensitivity simulations and the base-case simulation. Specifically, for each pollutant, we average the surface concentrations annually, and over either winter (DJF) or summer (JJA) of the 2014–2016 period. Fig. 4–9 show the absolute differences (base case – sensitivity case) and the relative differences (base case – sensitivity case)/(base case) in the mean concentrations. We show the summer mean for O3 and annual means for all other pollutants. Figures for winter and summer for all pollutants are given in the ESI (Fig. A2–A13†). To facilitate comparison of the GEOS-Chem modelling results with the PSCF results shown in Sect. 3.3, we also show in Tables 3 and 4 the relative and absolute differences averaged over 45° N to 49° N, 78.75° W to 68.75° W, a region that encompasses all of the RSQAQ sampling stations used in the PSCF analysis. We note that as many of the RSQAQ sampling stations have been placed with the objective of determining the exposure of Quebec's population to atmospheric pollution, this region also encompasses the area where the vast majority of Quebec's population is located, including the cities of Montreal and Québec City.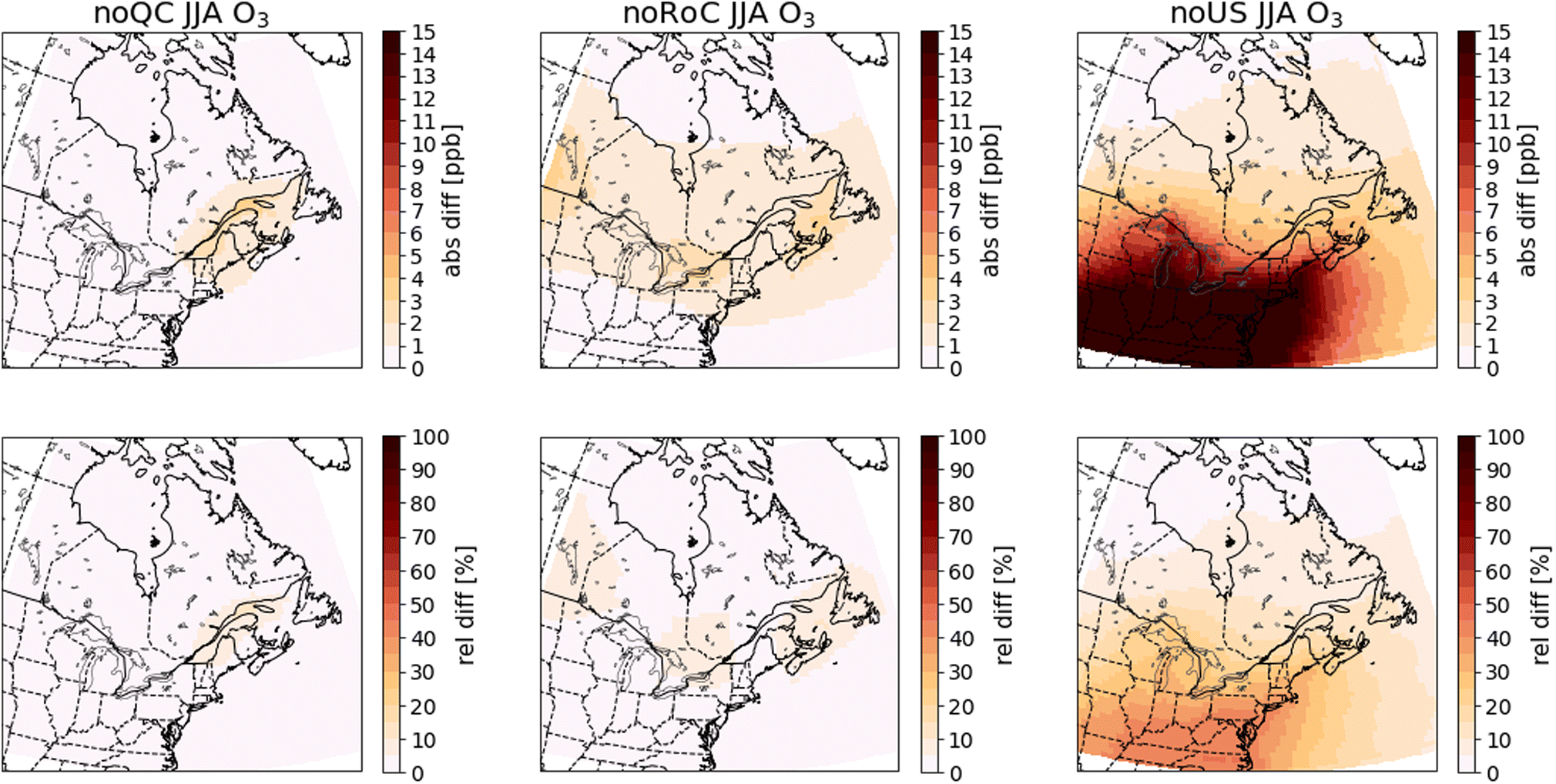 | ||
| Fig. 4 Differences in summer (JJA) surface O3 concentrations between the sensitivity simulations and the base-case simulation. Absolute differences are shown in the top row and relative differences in the bottom row. The three columns show the results of the noQC (left), noRoC (center) and noUS (right) sensitivity simulations. | ||
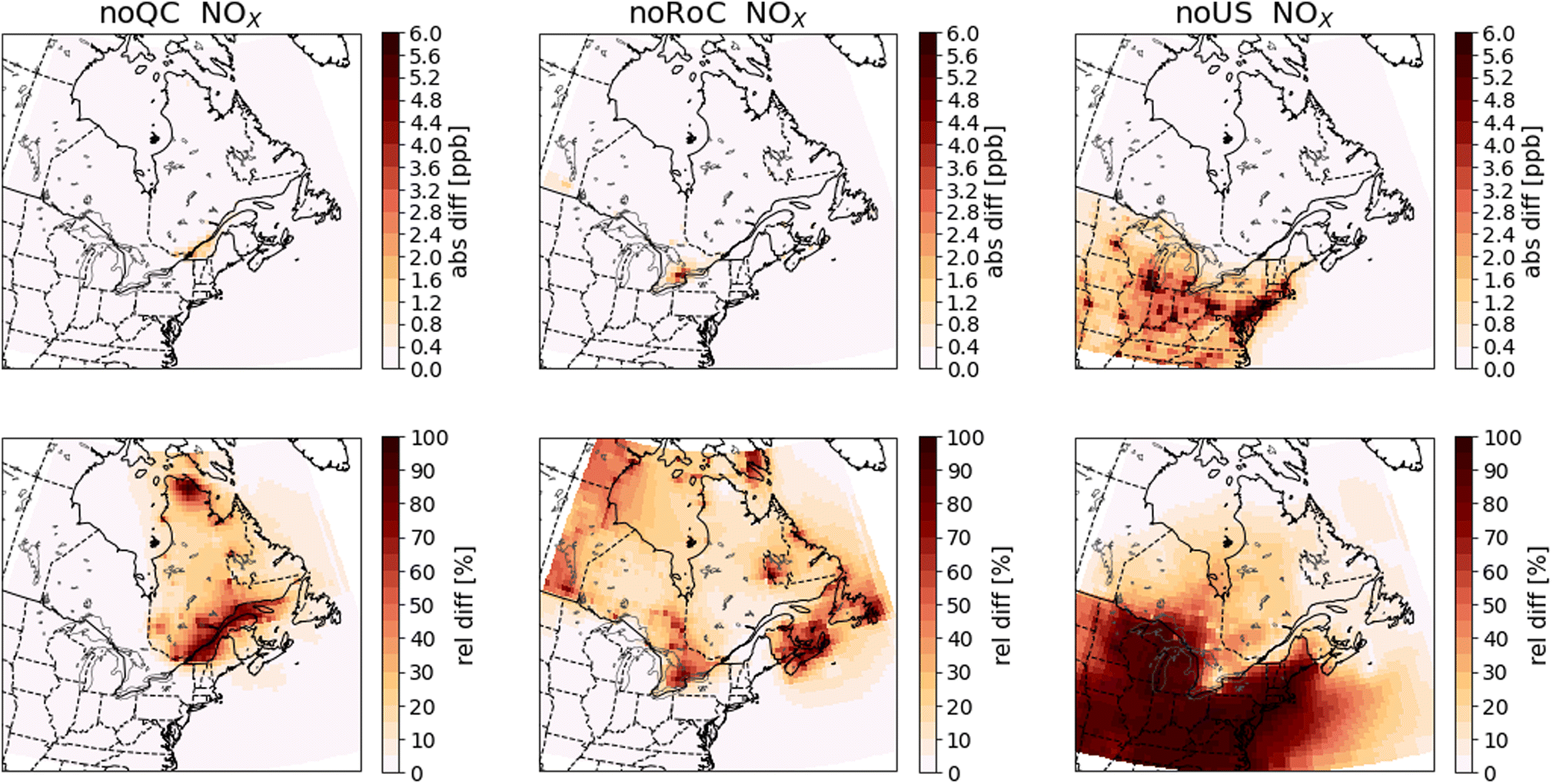 | ||
| Fig. 5 Differences in annual mean surface NOX concentrations between the sensitivity simulations and the base case simulation. Absolute differences are shown in the top row and relative differences in the bottom row. The three columns show the results of the noQC (left), noRoC (center) and noUS (right) sensitivity simulations. | ||
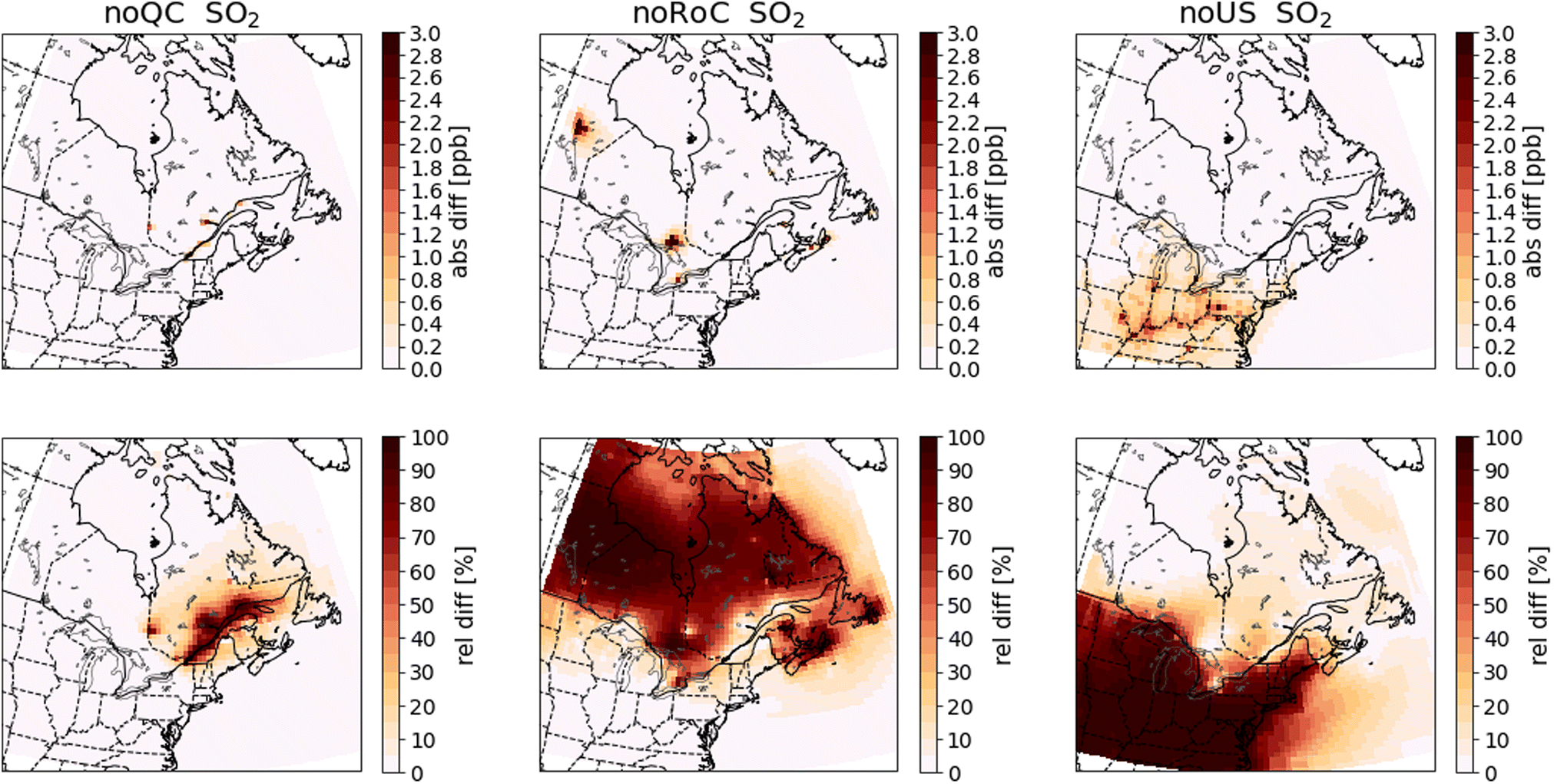 | ||
| Fig. 6 Differences in annual mean surface SO2 concentrations between the sensitivity simulations and the base-case simulation. Absolute differences are shown in the top row and relative differences in the bottom row. The three columns show the results of the noQC (left), noRoC (center) and noUS (right) sensitivity simulations. | ||
 | ||
| Fig. 7 Differences in annual mean surface CO concentrations between the sensitivity simulations and the base case simulation. Absolute differences are shown in the top row and relative differences in the bottom row. The three columns show the results of the noQC (left), noRoC (center) and noUS (right) sensitivity simulations. | ||
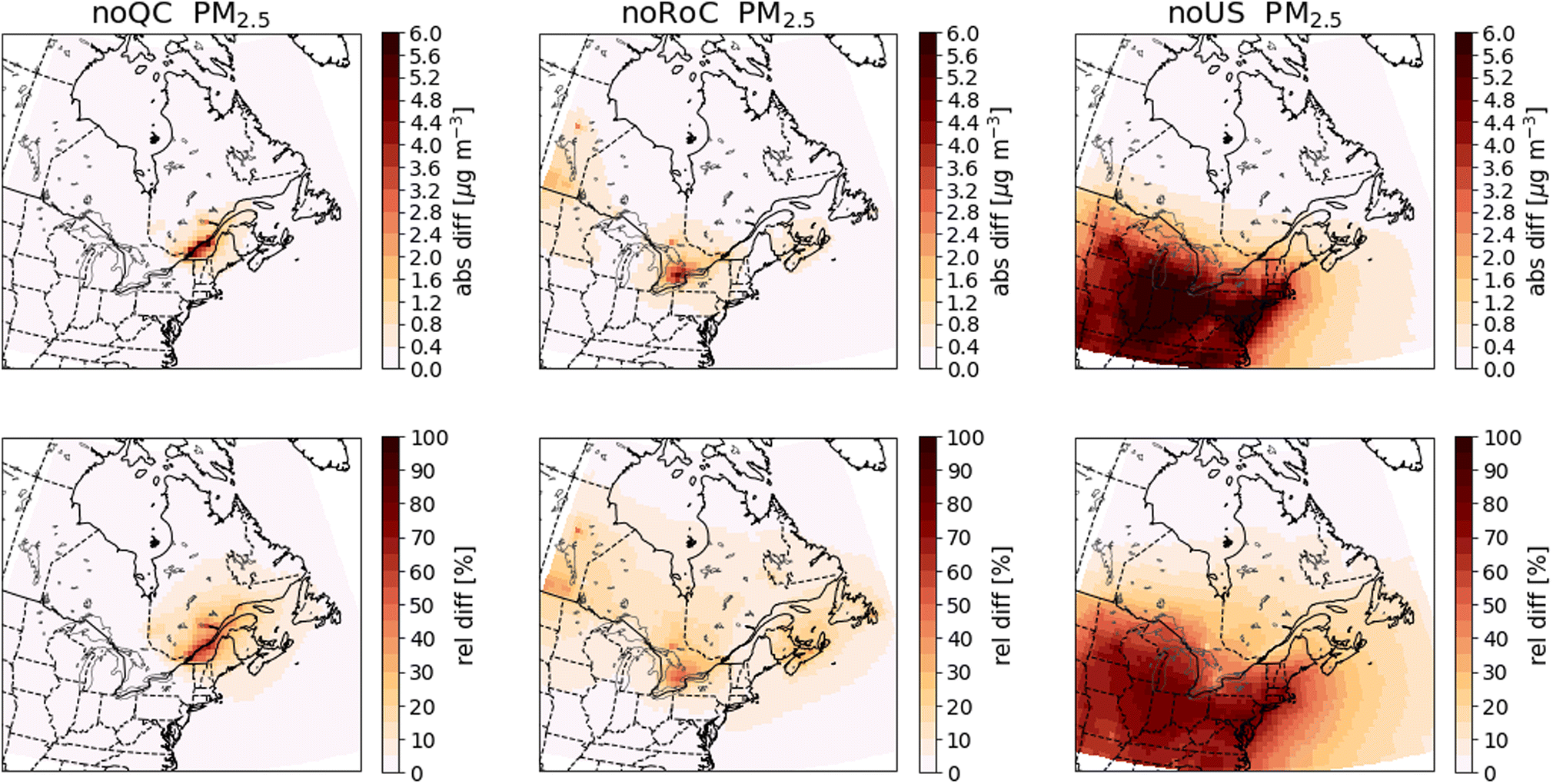 | ||
| Fig. 8 Differences in annual mean surface PM2.5 concentrations between the sensitivity simulations and the base-case simulation. Absolute differences are shown in the top row and relative differences in the bottom row. The three columns show the results of the noQC (left), noRoC (center) and noUS (right) sensitivity simulations. | ||
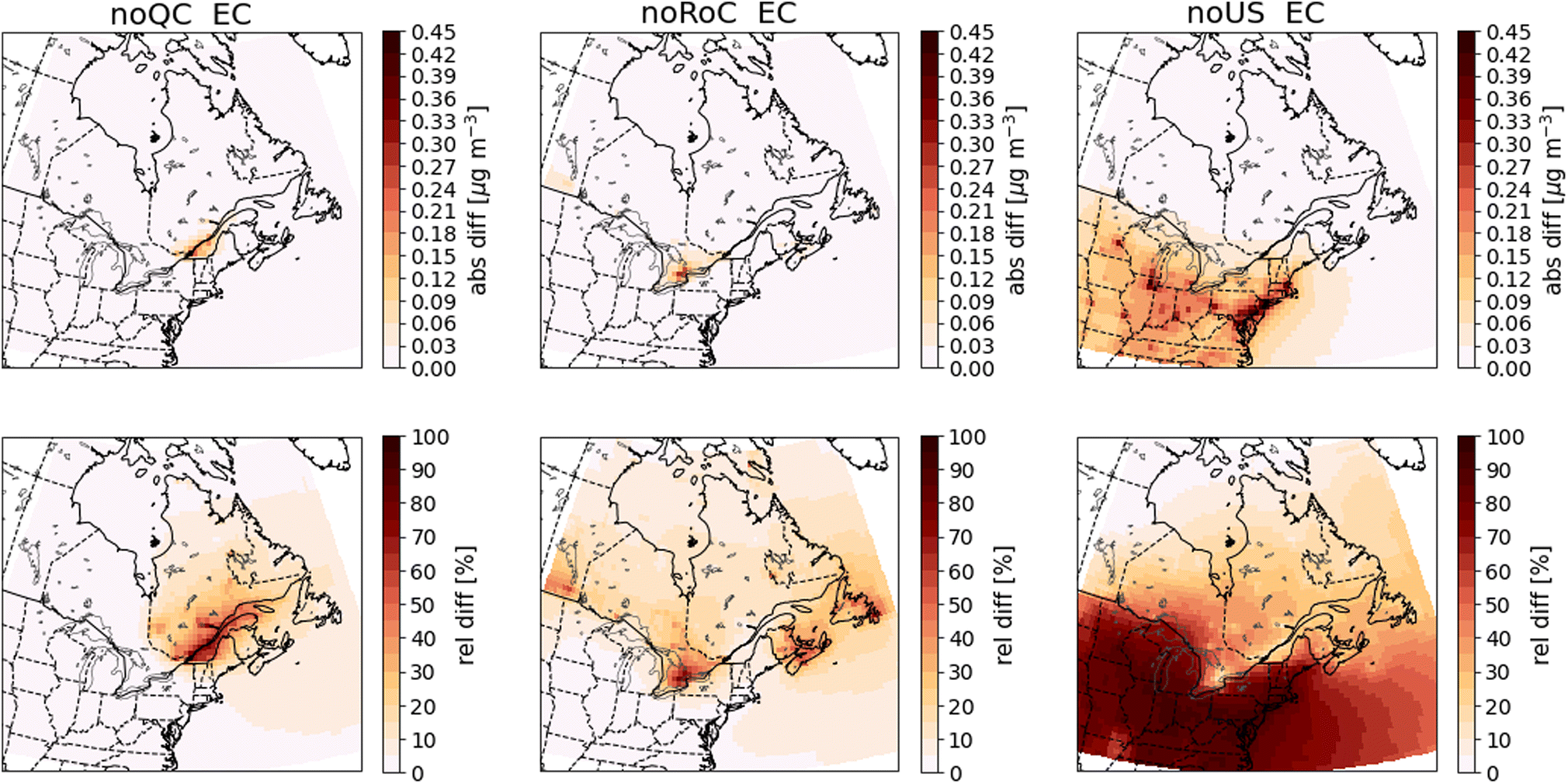 | ||
| Fig. 9 Differences in annual mean surface EC concentrations between the sensitivity simulations and the base-case simulation. Absolute differences are shown in the top row and relative differences in the bottom row. The three columns show the results of the noQC (left), noRoC (center) and noUS (right) sensitivity simulations. | ||
As expected, most pollutants show the greatest differences in concentrations over the emissions regions that are set to zero in the sensitivity simulations, and absolute differences in concentrations are larger in the southern part of Quebec where the RSQAQ stations are located than in the northern parts of the province. Additionally, when emissions from one of the three regions are removed, differences in concentrations are typically larger during winter than summer. One contributing factor to this seasonal cycle is the seasonal cycle in boundary layer heights: due to warmer surface temperatures, boundary layer heights are greater in summer than in winter. The same mass of pollution is therefore diluted in a larger vertical layer in summer than winter. Another factor is the concentration of oxidants which will react with certain pollutants such as SO2. Concentrations of oxidants are generally higher in summer, which decreases the lifespan and therefore the accumulation of these pollutants. Therefore, in the absence of seasonal differences in emissions or horizontal transport, we would expect higher concentrations of any primary pollutant in winter than in summer.
We note that while the maximum O3 concentrations occur in spring, the effect of anthropogenic emissions on O3 concentrations is most positive in summer and most negative in winter, suggesting that the spring maximum and fall minimum are features of the background O3 in this region, due to either long-range transport from outside of North America, biogenic emissions, or stratospheric–tropospheric exchange. This also suggests that anthropogenic emissions in this region have the effect of shifting the annual O3 maximum later in the year, as has been discussed in other studies.52,53
NOX and SO2 are reactive gases with a relatively short lifetime in the atmosphere. Therefore, the absolute differences in NOX and SO2 concentrations between the simulations are largely constrained to the source regions (Fig. 5 and 6). Within Quebec, only the noQC simulation shows differences in NOx greater than 0.8 ppb, and these are observed in the region surrounding the St. Lawrence River. In this simulation, the annual mean difference in NOX concentrations in Montreal reaches almost 5.4 ppb. Similarly, the greatest changes to SO2 concentrations in Quebec are seen in the noQC simulation, with a small number of grid cells showing increases due to Quebec emissions of more than 1 ppb, likely due to strong point sources. The impacts of emissions removal extends over a larger region in winter than summer (compare Fig. A4 with Fig. A5 and Fig. A6 with Fig. A7†), because oxidation of NOX and SO2 is slower due to lower oxidant concentrations during this season. For example, emissions from the United States increase NOX concentrations by 0.4 ppb up to a latitude of 47° N in winter, but they do not yield increases greater than 0.4 ppb beyond the Quebec-US border in summer. In winter, emissions from outside of Quebec do not increase concentrations of SO2 within Quebec by more than 0.2 ppb, except emissions from the RoC close to the Ontario-Quebec border.
Carbon monoxide has a longer lifetime than either NOX or SO2, and therefore increases in CO persist further from the source regions than for NOX or SO2 (Fig. 7). This allows emissions from the US to enhance CO concentrations by more than 4 ppb throughout all of Quebec. Of the three emissions regions investigated in this study, emissions from the US have the greatest effect on CO concentrations in Quebec, except close to large emissions sources in Quebec. In these locations, the effect of Quebec emissions exceeds the effect of US emissions. However, we note that unlike NOx and SO2, the GEOS-Chem simulations indicate that CO in Eastern Canada, including Quebec at the scale resolved by GEOS-Chem is not primarily due to North American anthropogenic emissions. This is in agreement with a previous source apportionment study, which showed that Asian anthropogenic CO emissions contributed more to CO concentrations within Quebec than North American anthropogenic CO emissions in April of 2008.54
An important consideration in this study is that summertime EC concentrations can be influenced by outdoor biomass combustion emissions, especially from forest fires. As we have not included these biomass combustion emissions in the anthropogenic emissions that we modify in the sensitivity simulations, the EC concentration derived from outdoor biomass combustion emissions will be identical in all simulations. In regions where outdoor biomass combustion is the main source of EC, the differences in its concentration after removal of anthropogenic emissions will be small. This effect is responsible for the small relative differences in EC concentrations in northern Quebec, especially during summer (Fig. 9, A12†). This consideration applies to all species emitted from biomass combustion, including CO (Fig. 7), if their emissions from biomass combustion are predominant. As shown in Fig. 9, the differences in EC between the base case simulation and the sensitivity simulations has a spatial pattern similar to that observed for PM2.5. Emissions from Quebec cause increases in EC that are largest along the St. Lawrence River, while emissions from the RoC do not produce increases within Quebec greater than 0.03 μg m−3. Emissions from the United States produce increases greater than 0.03 μg m−3 as far north as 47° N.
We further examine the importance of emissions from the three regions to PM2.5 concentrations in Quebec by calculating the area of Quebec that exceeds the annual average concentration thresholds recommended for 2020 by the Canadian Ambient Air Quality Standards (CAAQS) (8.8 μg m−3)55 or the World Health Organization (WHO) air quality guidelines (5 μg m−3)56 in each simulation. The results are summarized in Table 5, and the values for individual years are in Tables A12 and A13.†
| Threshold | Base case | noQC | noRoC | noUS |
|---|---|---|---|---|
| Area exceeding 8.8 μg m−3 | 1.5% | 1.3% | 1.3% | 1.3% |
| Area exceeding 5.0 μg m−3 | 7.3% | 3.5% | 6.4% | 4.7% |
| Population exceeding 8.8 μg m−3 | 39.0% | 0.0% | 0.0% | 0.0% |
| Population exceeding 5.0 μg m−3 | 87.7% | 0.0% | 85.8% | 66.8% |
We supplement this analysis by using 10 km data for the 2016 Canadian population57 to calculate the Quebec population living in geographic cells where PM2.5 concentrations exceed thresholds of 8.8 μg m−3 or of 5.0 μg m−3. The percentages of the population of Quebec living in the geographic cells that exceed these thresholds are presented in Table 5, and the values for each year are given in Tables A14 and A15.† In the base-case simulation, the areas covering 39.0% and 87.7% of the total population of Quebec exceed respectively 8.8 or 5 μg m−3. By removing the emissions from either Quebec, the RoC or the United States, the population of Quebec where the mean concentration exceeds the CAAQS standard of 8.8 μg m−3 is reduced to approximately 0.0%. Similarly, removing emissions from Quebec, the RoC or the United States reduces the relative population of Quebec where the average concentration exceeds the WHO guideline of 5 μg m−3 to 0.0%, 85.8% or 66.8%, respectively.
These results suggest significant improvements in air quality in Quebec would be possible by controlling emissions only in Quebec. It is important to note that the model cell that contains the island of Montreal constitutes 0.2% of the area of Quebec, but contains 39.0% of the population of Quebec, and it is here that we find the greatest concentrations of several pollutants, including PM2.5. In addition, there were forest fires in 2013 that caused a large area of Quebec to have elevated PM2.5 concentrations, but affected principally regions with small populations.
We also performed the same analysis for SO2 and NO2, but the annual average concentrations were, in all cases, below the 2020 CAAQS thresholds (5.0 ppb for SO2 and 17.0 ppb for NO2), and even below the 2025 thresholds (4.0 ppb for SO2 and 12.0 ppb for NO2) everywhere. Among all the grid cells in Quebec, the maximum annual mean concentration of SO2 in the base-case simulation was 2.48 ppb in 2012, and that of NO2 was 6.73 ppb in 2012. Within Quebec, average NO2 concentrations from 2012 to 2016 exceed the WHO threshold (10 μg m−3 NO2, ≅5.2 ppb at normal temperature and pressure) in the cell covering the island of Montreal, and only in this grid cell, for all simulations except the noQC simulation. There is no WHO guideline for annual SO2 concentrations. As our model evaluation showed a consistent low bias of about 50% in NOX concentrations, we also compared against a NO2 threshold of 2.6 ppb to account for this low bias. In the base-case simulation and the noRoC simulation, the grid cell just east of Montreal had a mean NO2 concentration in excess of 2.6 ppb, bringing the population exposed to more than 2.6 ppb to 46.4% of Quebec's population. Otherwise, the results were identical to the comparison against a threshold of 5.2 ppb.
3.3 PSCF results
In the next section, we will discuss the results of the PSCF analysis. As there is frequently more variability between seasons in the spatial patterns of the PSCF maps than for the GEOS-Chem results, we present both summer and winter maps for each species in Fig. 10–15.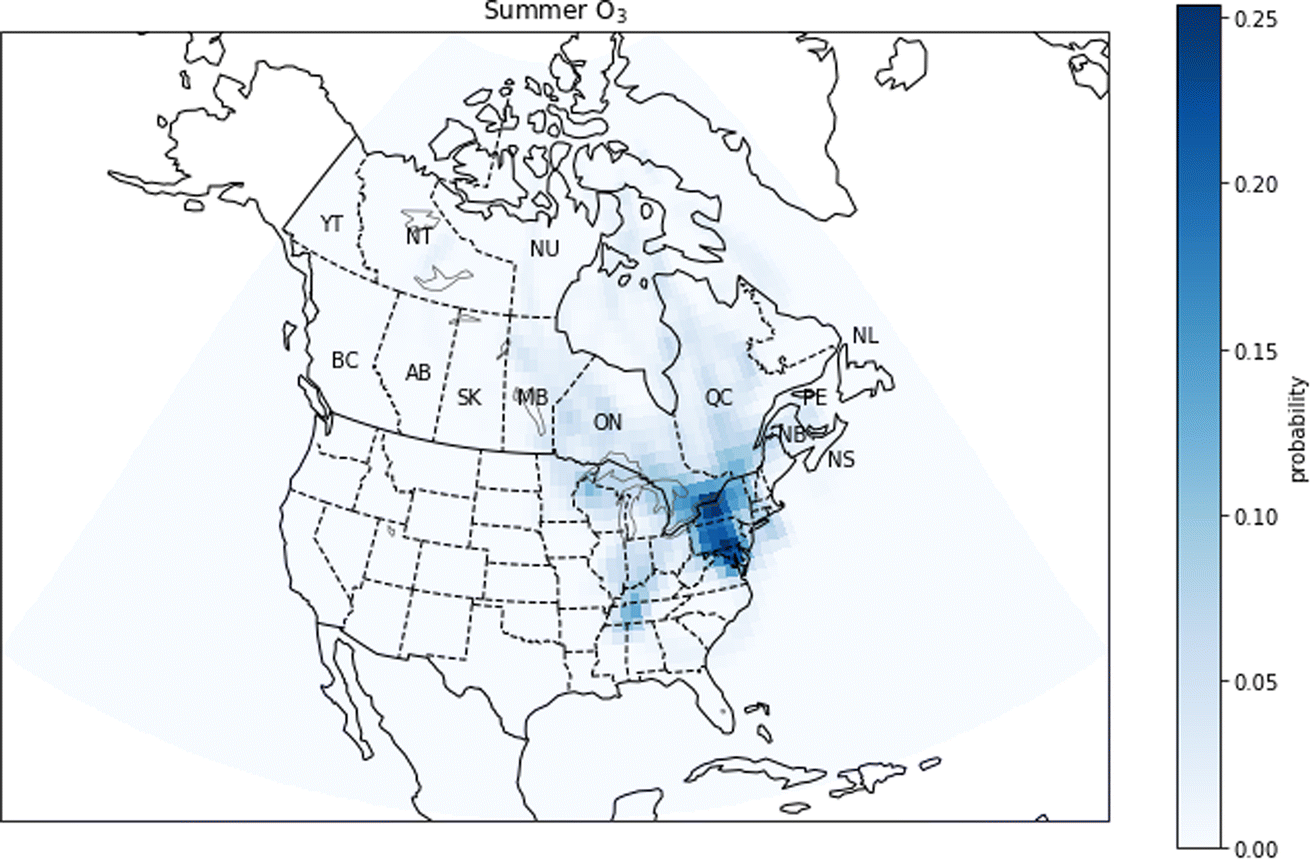 | ||
| Fig. 10 Summer probability source contribution function map calculated using the O3 measurements. Canadian provinces and territories are labeled by postal abbreviation. NL: Newfoundland and Labrador, PE: Prince Edward Island, NS: Nova Scotia, NB: New Brunswick, QC: Quebec, ON: Ontario, MB: Manitoba, SK: Saskatchewan, AB: Alberta, BC: British Columbia, YT: Yukon, NT: Northwest Territories, NU: Nunavut. | ||
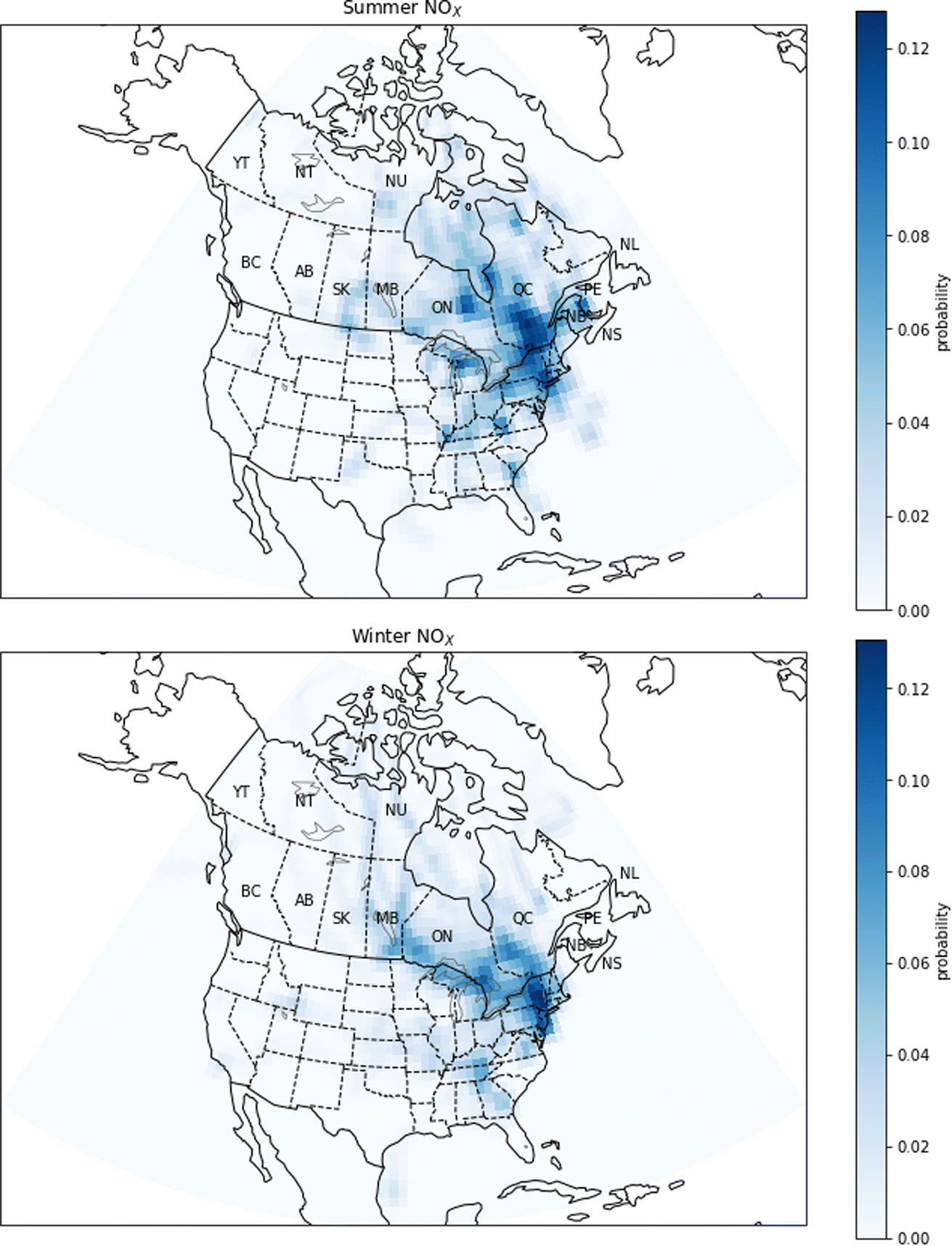 | ||
| Fig. 11 Probability source contribution function maps calculated using the NOX measurements. Top figure: summer probabilities. Bottom figure: winter probabilities. Canadian provinces and territories are labeled by postal abbreviation. NL: Newfoundland and Labrador, PE: Prince Edward Island, NS: Nova Scotia, NB: New Brunswick, QC: Quebec, ON: Ontario, MB: Manitoba, SK: Saskatchewan, AB: Alberta, BC: British Columbia, YT: Yukon, NT: Northwest Territories, NU: Nunavut. | ||
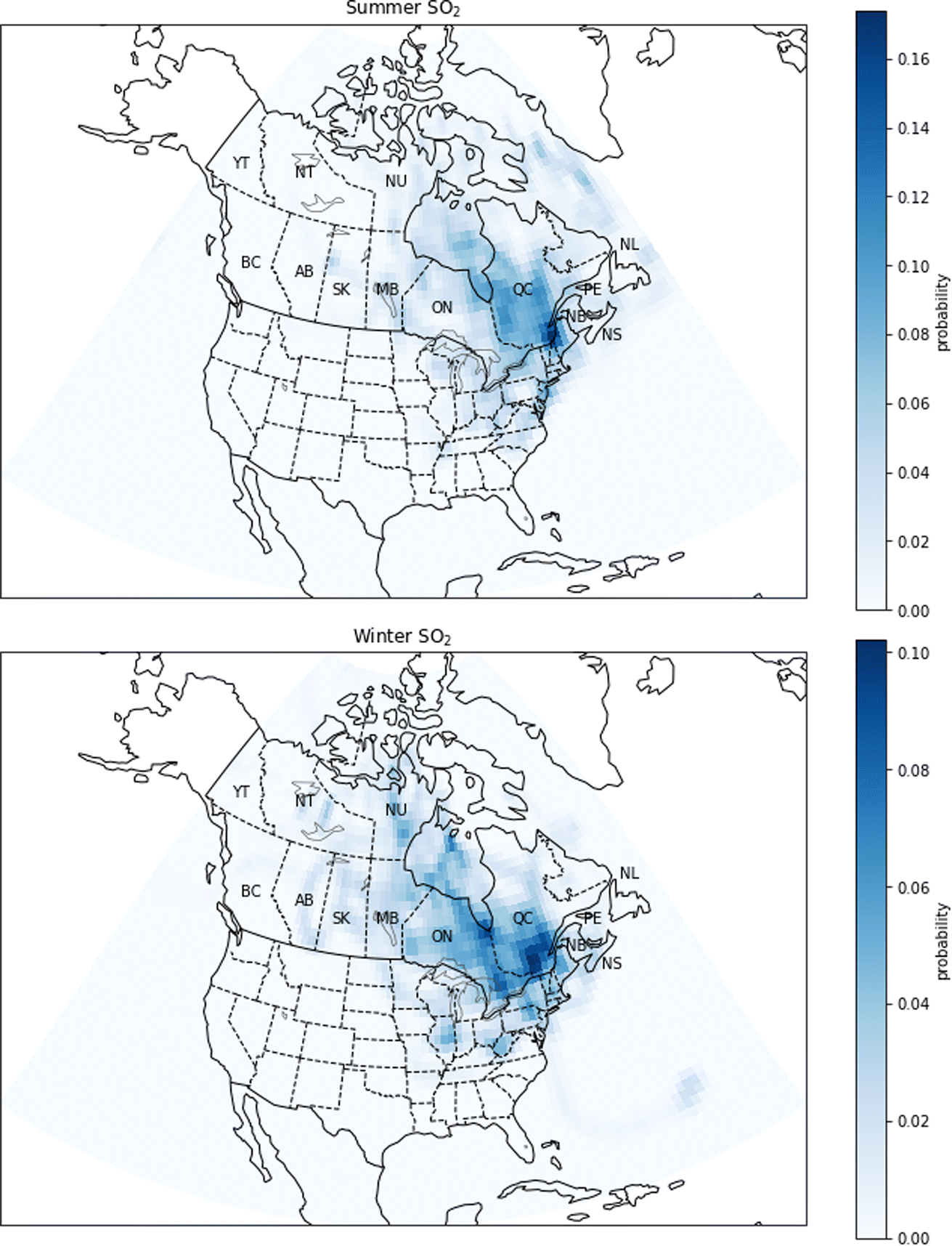 | ||
| Fig. 12 Probability source contribution function maps calculated using the SO2 measurements. Top figure: summer probabilities. Bottom figure: winter probabilities. Canadian provinces and territories are labeled by postal abbreviation. NL: Newfoundland and Labrador, PE: Prince Edward Island, NS: Nova Scotia, NB: New Brunswick, QC: Quebec, ON: Ontario, MB: Manitoba, SK: Saskatchewan, AB: Alberta, BC: British Columbia, YT: Yukon, NT: Northwest Territories, NU: Nunavut. | ||
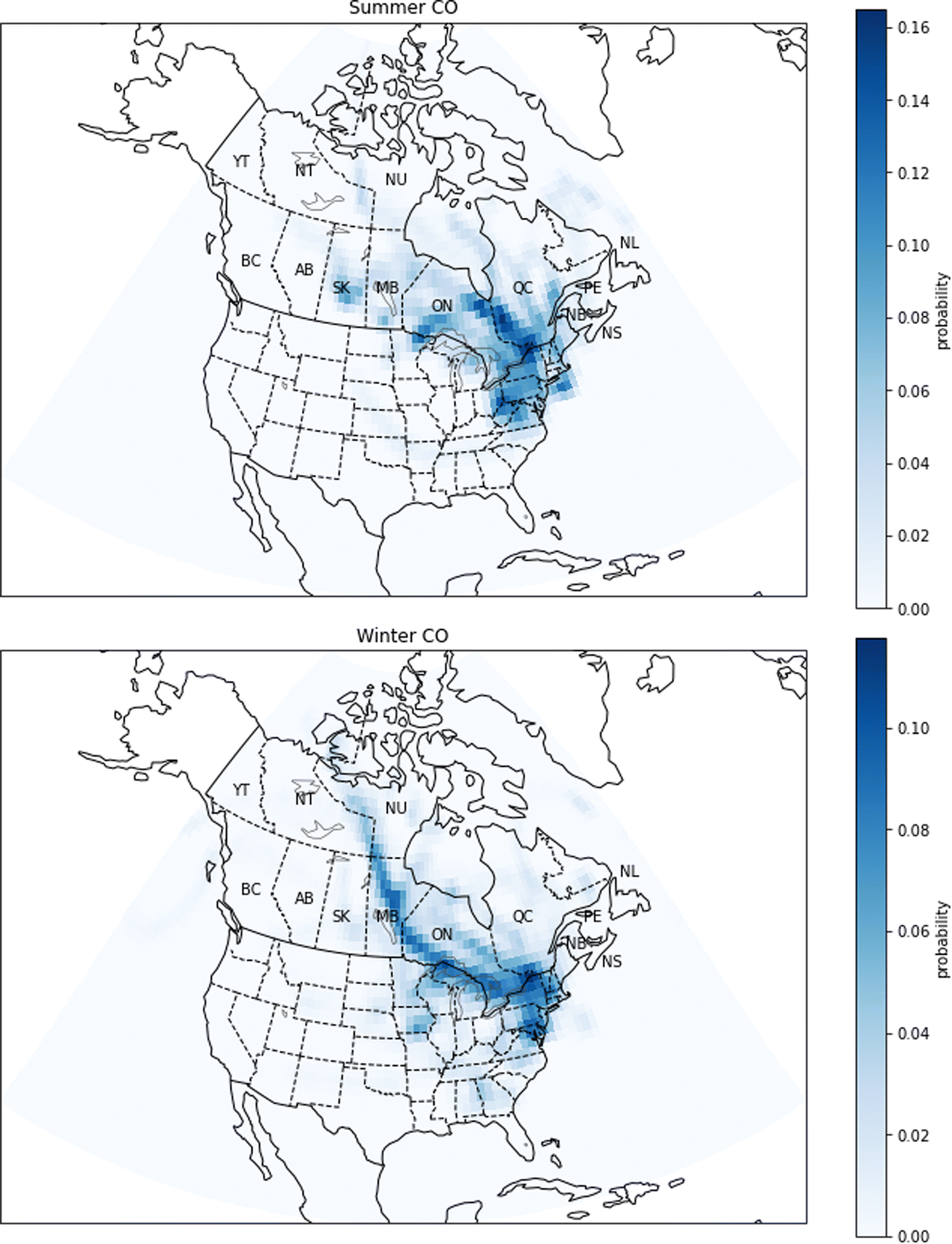 | ||
| Fig. 13 Probability source contribution function maps calculated using the CO measurements. Top figure: summer probabilities. Bottom figure: winter probabilities. Canadian provinces and territories are labeled by postal abbreviation. NL: Newfoundland and Labrador, PE: Prince Edward Island, NS: Nova Scotia, NB: New Brunswick, QC: Quebec, ON: Ontario, MB: Manitoba, SK: Saskatchewan, AB: Alberta, BC: British Columbia, YT: Yukon, NT: Northwest Territories, NU: Nunavut. | ||
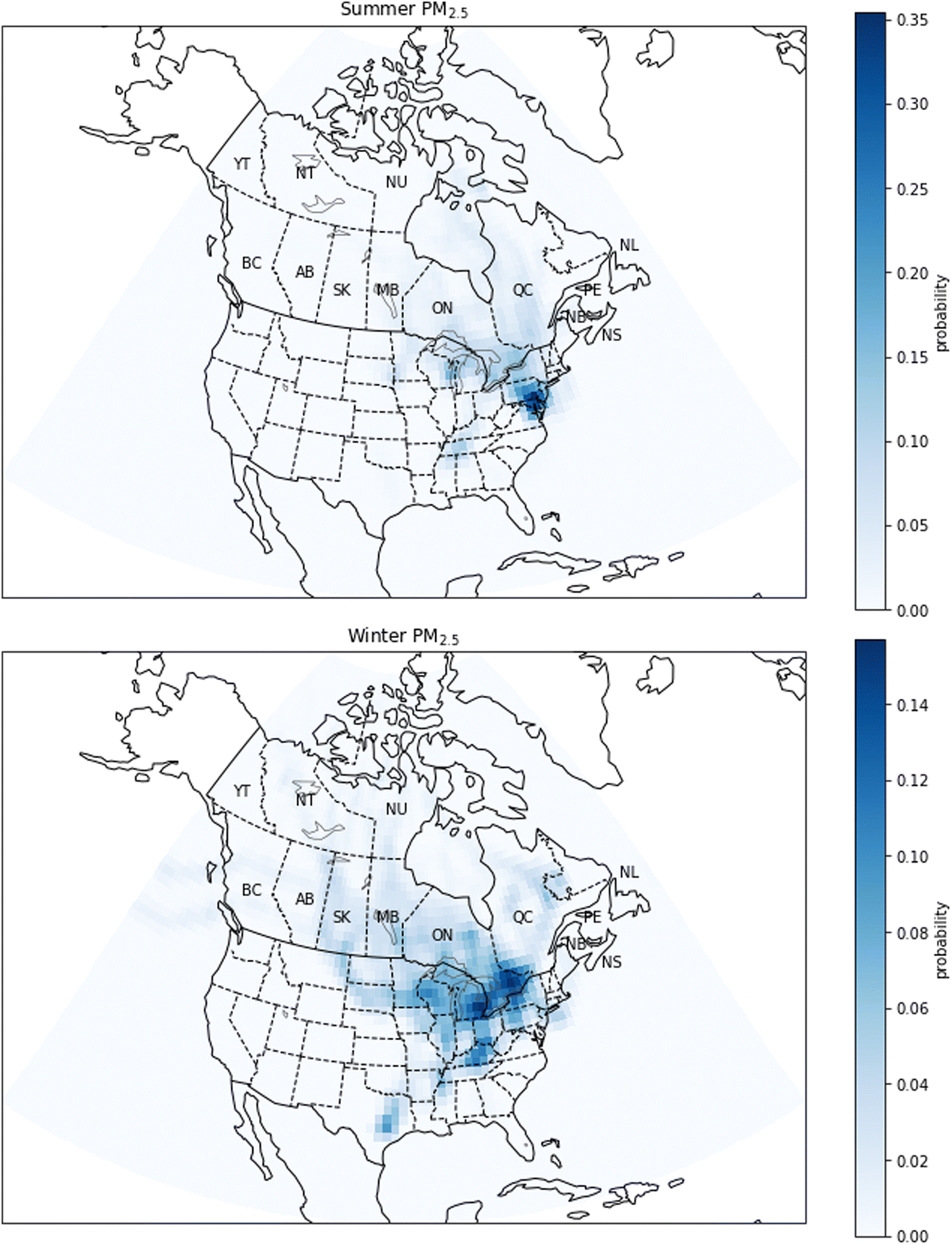 | ||
| Fig. 14 Probability source contribution function maps calculated using the PM2.5 measurements. Top figure: summer probabilities. Bottom figure: winter probabilities. Canadian provinces and territories are labeled by postal abbreviation. NL: Newfoundland and Labrador, PE: Prince Edward Island, NS: Nova Scotia, NB: New Brunswick, QC: Quebec, ON: Ontario, MB: Manitoba, SK: Saskatchewan, AB: Alberta, BC: British Columbia, YT: Yukon, NT: Northwest Territories, NU: Nunavut. | ||
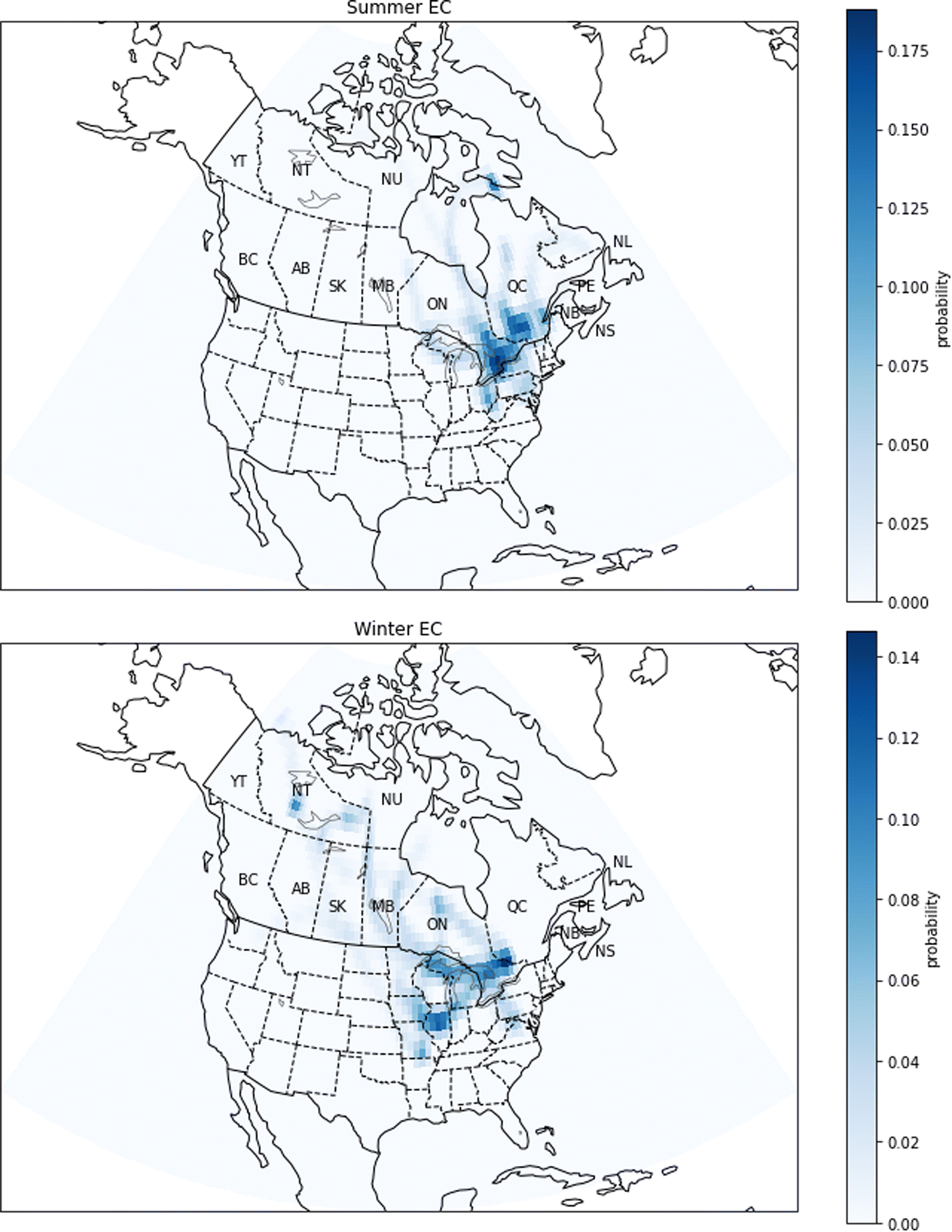 | ||
| Fig. 15 Probability source contribution function maps calculated using the EC measurements. Top figure: summer probabilities. Bottom figure: winter probabilities. Canadian provinces and territories are labeled by postal abbreviation. NL: Newfoundland and Labrador, PE: Prince Edward Island, NS: Nova Scotia, NB: New Brunswick, QC: Quebec, ON: Ontario, MB: Manitoba, SK: Saskatchewan, AB: Alberta, BC: British Columbia, YT: Yukon, NT: Northwest Territories, NU: Nunavut. | ||
For NOX, the PSCF calculations show a large difference between the potential sources in winter and in summer (Fig. 11). In summer, pollution comes from southern Quebec and areas south of Quebec such as the New York City metropolitan area. In winter, PSCF probabilities are highest in regions south of Quebec, specifically the east coast of the United States and major urban centers such as New York, Philadelphia and Washington. Although there are natural sources of NOX such as biogenic emissions from soils, the GEOS-Chem simulations suggest most NOX in urban centres is due to anthropogenic sources (Fig. 5). These urban anthropogenic sources would include vehicle exhaust. The increase in relative probability during winter in the United States compared to Quebec may be explained by the use of gas and oil heating in the United States, while in Quebec this type of heating is less important.58–60
In Quebec, SO2 emissions come from aluminum production, industrial processes related to metallurgy and other minor sources.61 PSCF calculations show differences, but also similarities in the regions of potential sources between winter and in summer (Fig. 12). In summer and winter, the high probabilities are mainly concentrated in southern Quebec, near the cities of Quebec and Montreal. At the same time, in winter, the intermediate probabilities extend over a larger region, including parts of the United States and Ontario, which is explained by the lifetime of SO2 being longer in winter. This seasonal change is in agreement with GEOS-Chem simulations which showed that the relative differences between the base-case and the noCA or noUS simulations are greater in winter (Fig. A6 and A7†).
The largest CO emissions in Quebec come from the aluminum industry, including aluminum smelters in Bécancour (between Montreal and Québec City), Saguenay (north of Québec City) and Sept-Îles (on the north bank of the Gulf of St. Lawrence). Another important source of CO is vehicular combustion of fossil fuels. This means that major urban centers like New York and Toronto have high CO emissions. The PSCF calculations show a similarity between the regions of potential sources in winter and summer (Fig. 13). In summer, high probabilities are found in areas of southern Quebec, such as Montreal and Québec City. Regions south of Quebec, such as New York and southeastern Ontario, also have intermediate probabilities. In winter, PSCF probabilities are high in the same regions, but another area of high probabilities has emerged, which passes through southern Ontario and Manitoba. We have examined wildfire emissions for the winters of 2014–2016 according to the GFED4 inventory, and we did not find any large emissions that would explain this trajectory. We also note that large wildfires are not expected during the Canadian winter. We also examined possible sources in western Ontario, Manitoba, and the Northwest Territories from the National Pollutant Release Inventory (NPRI), and we did not find a clear and obvious source that explains this track. Due to the long atmospheric lifetime of CO, it is possible that this trajectory pathway is associated with long-range transport across the Arctic, but it may instead be associated with a synoptic weather pattern that transports CO from the industrial region of southern Ontario or another more local source.
There are also seasonal differences in PSCF maps for EC (Fig. 15). The high probabilities are generally observed near large cities where there are sources of combustion emissions, for example vehicles. In summer, EC has high probabilities especially in the Greater Toronto Area, but there are also high probabilities in southwestern Quebec. Notably, in summer, the probabilities are close to zero in western and northern Canada indicating that there is not a strong contribution of forest fires to the EC concentration. At the same time, our confidence in this conclusion is limited by the sampling frequency which was 1 day in 3 or 1 day in 6, and it is therefore possible that there are smoke events which have not been captured by the measurements. On the other hand, in winter, the PSCF map shows a medium probability in the province of Ontario in an area close to the Quebec border, near Ottawa and Gatineau. Also, the map shows that the EC can come from the United States, more precisely from the Chicago area and the state of Illinois. The high-probability regions indicated by the PSCF analysis for PM2.5 and EC show some consistency, particularly in winter when the probability is high in Ontario. This is expected as EC is a component of PM2.5.
4. Discussion
Before comparing the results of the PSCF and GEOS-Chem analyses, it is important to note the different information provided by the two methods: our PSCF analysis isolates the periods with the highest concentrations of each pollutant, and determines the source regions of air masses during those periods, regardless of whether the source was natural or anthropogenic. On the other hand, the GEOS-Chem modelling results give contributions from anthropogenic sources from broad regions averaged over long periods. Natural sources may be highlighted by the PSCF analysis but not by the GEOS-Chem results. Also, a source with a significant but temporally-invariant contribution to pollutant concentrations would be highlighted by the GEOS-Chem analysis, but may be missed by the PSCF analysis.Both GEOS-Chem and the PSCF results agree on an important role for emissions from the US on PM2.5 concentrations, and in particular that the US is a relatively more important source region during the summer months. However, in winter, the PSCF results suggest a possible strong contribution from southern Ontario, while the GEOS-Chem results suggest that the US and Quebec are more important source regions than the rest of Canada (including southern Ontario). It is clear that PM2.5 in Quebec is due to a variety of sources, but our analysis highlights the role of urban emissions from outside of Quebec. Both methods also show that local emissions contribute strongly to NOX and SO2 concentrations, with additional contributions from Ontario and the US, particularly in winter. These results suggest that local traffic emissions are likely the most important contributor to local NOX concentrations, and local industry, including aluminum production and industrial processes related to metallurgy, is likely to be the most important contributor to local SO2 concentrations.
Due to the predicted titration of O3 in winter by anthropogenic emissions in the GEOS-Chem simulations, it is only meaningful to compare the GEOS-Chem and the PSCF results in summer. Both the PSCF and GEOS-Chem results show an important contribution of US emissions to O3 concentrations. The PSCF analysis in particular highlights urban regions. This suggests that urban emissions of O3 precursors from outside of Quebec, including vehicular NOX and anthropogenic VOCs, are an important contributor to O3 concentrations in the Quebec region.
Regarding CO, the GEOS-Chem and PSCF results agree about the importance of local and US emissions. We note that contributions to CO from forest fires and anthropogenic sources outside of North America would be included in the PSCF analysis but were not perturbed in the GEOS-Chem sensitivity simulations. However, the generally low percentage of CO attributable to anthropogenic emissions from any of the three regions in the GEOS-Chem results would be in agreement with a either a significant natural source of CO or long-range transport from outside of North America.
There is an apparent contrast in the GEOS-Chem and PSCF results for EC, with GEOS-Chem giving a greater importance to US emissions and the PSCF analysis showing a greater importance for sources in southern Ontario. However, we note that EC presents some special challenges when comparing the two analyses: EC was sampled less frequently and at fewer stations than the other pollutants. Also, forest fires are an important natural source of EC that would be highlighted by the PSCF analysis but not the GEOS-Chem analysis. We do not have reason to believe that there were large forest fires in southern Ontario during the study period, but air masses originating from forest fires may have traveled over southern Ontario before arriving at the measurement sites, leading to higher probabilities over southern Ontario. It is also possible that the emissions inventories used in GEOS-Chem underestimate EC from Canadian sources and/or overestimate EC from US sources.
5. Conclusions
We have investigated the sources of air pollution in Quebec through two complementary methods: back trajectory analysis using PSCF and chemical transport modelling. The PSCF analysis provides information on which source regions are best correlated with high-concentration events in Quebec, while the chemical transport modelling gives estimates of the contributions from three different source regions (Quebec, the rest of Canada (RoC), and the United States) to the concentrations of each of the investigated pollutants in Quebec.The GEOS-Chem chemical transport modelling results reveal that all three regions considered have significant contributions to air pollutant concentrations in Quebec. For most pollutants, emissions from the RoC had a smaller contribution to concentrations in Quebec than local (Quebec) emissions or US emissions. Our results suggest that O3 in summertime, when exceedances are expected to occur, is in a NOX-limited regime in Quebec. We find that removing all anthropogenic emissions from any one of the three regions would reduce the fraction of the Quebec population living above the annual mean CAAQS PM2.5 concentration threshold of 8.8 μg m−3 from 39.0% to about 0.0%. In contrast, only removal of emissions from Quebec decreases the fraction of the Quebec population living above the more stringent WHO PM2.5 concentration threshold of 5.0 μg m−3 (from 87.7% to about 0.0%). While an absolute cessation of anthropogenic emissions is neither feasible nor desirable, our results suggest that significant improvements in air quality in Quebec would be possible through reductions in local emissions alone. At the same time, the achievable improvements will be limited by the strong transboundary contribution of certain pollutants, in particular for summertime O3 for which the contribution from the United States exceeds that of Quebec.
The PSCF calculations show that there are a few regions associated with days of high concentrations of several pollutants. These regions include southern Quebec (local sources), the east coast of the United States, including the cities of New York, Washington and Philadelphia, and southeastern Ontario, including Toronto. Depending on the season, southern Quebec is associated with high concentrations of NOX, SO2, and CO; the east coast of the United States is associated with high concentrations of PM2.5, NOX, O3, and CO; and southeastern Ontario is associated with high concentrations of PM2.5 and EC.
The PSCF analysis relies on high-quality air pollutant measurements that are not strongly influenced by local sources. In contrast, several RSQAQ sites were specifically located with the intent of measuring local sources, making them less useful for this type of analysis. In addition, the RSQAQ stations are currently located in the southern portions of the province, predominantly along the St. Lawrence valley. This choice of placement allows for better understanding of the pollutant exposure in the most populous region of Quebec, but limits our ability to evaluate pollutant transport to the northern regions of the province.
The two methods provided some contrasting results regarding the sources of CO and EC in Quebec. This is potentially due to the fact that we did not perturb natural emissions in the GEOS-Chem simulations, including forest fires, which would influence the PSCF analysis. The smaller number of stations and the less frequent sampling of EC is also a source of greater uncertainty in the PSCF analysis. However, both methods highlight the importance of local emissions for NOX and SO2 concentrations, non-Quebec sources for summertime O3 concentrations, and an important contribution from US emissions to PM2.5 concentrations in Quebec.
Author contributions
PH provided conceptualization and funding acquisition. RS, NT, and HR performed the chemical transport modelling and analysis, and CP performed the PSCF modelling and analysis. RS wrote the original draft with input from PH and CP, and all authors reviewed the text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank James King of the Université de Montréal and Hélène Côté and Biljana Music or Ouranos for thoughtful discussions about this work. This work was supported by the Ministère de l'Environnement, de la Lutte contre les changements climatiques, de la Faune et des Parcs du Québec. This research was enabled in part by support provided by Cacul Québec (https://calculquebec.ca) and the Digital Research Alliance of Canada (https://alliancecan.ca). NT acknowledges financial support from the Natural Sciences and Engineering Research Council of Canada through the Undergraduate Student Research Awards program.Notes and references
- C. A. Pope and D. W. Dockery, J. Air Waste Manage. Assoc., 2006, 56, 709–742 CrossRef CAS PubMed.
- C. A. Pope, M. Ezzati and D. W. Dockery, N. Engl. J. Med., 2009, 360, 376–386 CrossRef CAS PubMed.
- F. Laden, J. Schwartz, F. E. Speizer and D. W. Dockery, Am. J. Respir. Crit. Care Med., 2006, 173, 667–672 CrossRef CAS PubMed.
- D. M. Stieb, S. Judek and R. T. Burnett, J. Air Waste Manag. Assoc., 2002, 52, 470–484 CrossRef PubMed.
- D. R. Reidmiller, A. M. Fiore, D. A. Jaffe, D. Bergmann, C. Cuvelier, F. J. Dentener, B. N. Duncan, G. Folberth, M. Gauss, S. Gong, P. Hess, J. E. Jonson, T. Keating, A. Lupu, E. Marmer, R. Park, M. G. Schultz, D. T. Shindell, S. Szopa, M. G. Vivanco and A. Zuber, Atmos. Chem. Phys., 2009, 9, 5027–5042 CrossRef CAS.
- J. M. Kelly, E. A. Marais, G. Lu, J. Obszynska, M. Mace, J. White and R. J. Leigh, City Environ. Interact., 2023, 18, 100100 CrossRef.
- U. Jeong, J. Kim, H. Lee and Y. G. Lee, Atmos. Environ., 2017, 150, 33–44 CrossRef CAS.
- S. C. Anenberg, J. J. West, H. Yu, M. Chin, M. Schulz, D. Bergmann, I. Bey, H. Bian, T. Diehl, A. Fiore, P. Hess, E. Marmer, V. Montanaro, R. Park, D. Shindell, T. Takemura and F. Dentener, Air Qual. Atmos. Health, 2014, 7, 369–379 CrossRef CAS.
- Q. Liang, R. S. Stolarski, S. R. Kawa, J. E. Nielsen, A. R. Douglass, J. M. Rodriguez, D. R. Blake, E. L. Atlas and L. E. Ott, Atmos. Chem. Phys., 2010, 10, 2269–2286 CrossRef CAS.
- F. Dentener, T. Keating, H. Akimoto, N. Pirrone, S. Dutchak and A. Zuber, Convention on Long-range Transboundary Air Pollution. Task Force on Hemispheric Transport of Air Pollution, United Nations Economic Commission for Europe and UNECE Task Force on Emission Inventories and Projections, Hemispheric Transport of Air Pollution 2010: Part A – Ozone and Particulate Matter (Air Pollution Studies), United Nations, New York, Illustrated, 2011 Search PubMed.
- Ville de Montréal Service de l'environnement, Environmental Assessment Report (EAR) 2022 Air Quality in Montréal, Division de la planification et du suivi environnemental, Montréal, Québec, Canada, 2023 Search PubMed.
- Gouvernement de Québec, Ministère de l'Environnement, de la Lutte contre les changements climatiques and de la Faune et des Parcs, Statistiques annuelles de l'IQA, 2021, https://www.iqa.environnement.gouv.qc.ca/contenu/grille_stat_jour.asp?annee=2021, accessed July 20, 2023 Search PubMed.
- R. Vet and C.-U. Ro, Atmos. Environ., 2008, 42, 2518–2529 CrossRef CAS.
- Q. Li, D. J. Jacob, R. Park, Y. Wang, C. L. Heald, R. Hudman, R. M. Yantosca, R. V. Martin and M. Evans, J. Geophys. Res., 2005, 110, D10301 Search PubMed.
- E. Chan and R. J. Vet, Atmos. Chem. Phys., 2010, 10, 8629–8647 CrossRef CAS.
- T. Zhang, Y. Chen, R. Tan and X. Xu, Atmosphere, 2021, 12, 1300 CrossRef CAS.
- J. Meng, R. V. Martin, C. Li, A. van Donkelaar, Z. A. Tzompa-Sosa, X. Yue, J.-W. Xu, C. L. Weagle and R. T. Burnett, Environ. Sci. Technol., 2019, 53, 10269–10278 CrossRef CAS PubMed.
- L. G. Chestnut and D. M. Mills, J. Environ. Manage., 2005, 77, 252–266 CrossRef CAS PubMed.
- J. Dupont, La problématique des lacs acides au Québec, Direction du suivi de l'état de l'environnement, Ministère de l'Environnement, 2004 Search PubMed.
- L. J. Mickley, D. J. Jacob, B. D. Field and D. Rind, Geophys. Res. Lett., 2004, 31, L24103 CrossRef.
- M. Crippa, D. Guizzardi, T. Butler, T. Keating, R. Wu, J. Kaminski, J. Kuenen, J. Kurokawa, S. Chatani, T. Morikawa, G. Pouliot, J. Racine, M. D. Moran, Z. Klimont, P. M. Manseau, R. Mashayekhi, B. H. Henderson, S. J. Smith, H. Suchyta, M. Muntean and K. Foley, Earth Syst. Sci. Data, 2023, 15, 2667–2694 CrossRef.
- R. M. Hoesly, S. J. Smith, L. Feng, Z. Klimont, G. Janssens-Maenhout, T. Pitkanen, J. J. Seibert, L. Vu, R. J. Andres, R. M. Bolt, T. C. Bond, L. Dawidowski, N. Kholod, J. Kurokawa, M. Li, L. Liu, Z. Lu, M. C. P. Moura, P. R. O'Rourke and Q. Zhang, Geosci. Model Dev., 2018, 11, 369–408 CrossRef CAS.
- Gouvernement du Québec – Ministère de l'Environnement et de la Lutte contre les changements climatiques, Réseau de surveillance de la qualité de l'air du Québec, https://www.environnement.gouv.qc.ca/air/reseau-surveillance/Carte.asp, accessed August 18, 2022 Search PubMed.
- I. Bey, D. J. Jacob, R. M. Yantosca, J. A. Logan, B. D. Field, A. M. Fiore, Q. Li, H. Y. Liu, L. J. Mickley and M. G. Schultz, J. Geophys. Res., 2001, 106, 23073–23095 CrossRef CAS.
- R. J. Park, D. J. Jacob, B. D. Field, R. M. Yantosca and M. Chin, J. Geophys. Res., 2004, 109, D15204 Search PubMed.
- S. D. Eastham, M. S. Long, C. A. Keller, E. Lundgren, R. M. Yantosca, J. Zhuang, C. Li, C. J. Lee, M. Yannetti, B. M. Auer, T. L. Clune, J. Kouatchou, W. M. Putman, M. A. Thompson, A. L. Trayanov, A. M. Molod, R. V. Martin and D. J. Jacob, Geosci. Model Dev., 2018, 11, 2941–2953 CrossRef CAS.
- G. Luo, F. Yu and J. M. Moch, Geosci. Model Dev., 2020, 13, 2879–2903 CrossRef CAS.
- G. Luo, F. Yu and J. Schwab, Geosci. Model Dev., 2019, 12, 3439–3447 CrossRef CAS.
- P. K. Hopke, J. Air Waste Manag. Assoc., 2016, 66, 237–259 CrossRef PubMed.
- A. F. Stein, R. R. Draxler, G. D. Rolph, B. J. B. Stunder, M. D. Cohen and F. Ngan, Bull. Amer. Meteor. Soc., 2015, 96, 2059–2077 CrossRef.
- I. Pisso, E. Sollum, H. Grythe, N. I. Kristiansen, M. Cassiani, S. Eckhardt, D. Arnold, D. Morton, R. L. Thompson, C. D. Groot Zwaaftink, N. Evangeliou, H. Sodemann, L. Haimberger, S. Henne, D. Brunner, J. F. Burkhart, A. Fouilloux, J. Brioude, A. Philipp, P. Seibert and A. Stohl, Geosci. Model Dev., 2019, 12, 4955–4997 CrossRef.
- J. C. Lin, J. Geophys. Res., 2003, 108, ACH2 Search PubMed.
- Canadian Council of Ministers of the Environment, Ambient Air Monitoring and Quality Assurance/Quality Control Guidelines, National Air Pollution Surveillance Program, https://ccme.ca/en/res/ambientairmonitoringandqa-qcguidelines_ensecure.pdf, accessed August 24, 2022 Search PubMed.
- L. L. Ashbaugh, W. C. Malm and W. Z. Sadeh, Atmos. Environ., 1985, 19, 1263–1270 CrossRef CAS.
- Y. Zeng and P. K. Hopke, Atmos. Environ., 1989, 23, 1499–1509 CrossRef CAS.
- M. Kanamitsu, Weather Forecasting, 1989, 4, 335–342 CrossRef.
- J. E. Petit, O. Favez, A. Albinet and F. Canonaco, Environ. Model. Software, 2017, 88, 183–187 CrossRef.
- F. Shen, L. Zhang, L. Jiang, M. Tang, X. Gai, M. Chen and X. Ge, Environ. Int., 2020, 137, 105556 CrossRef CAS PubMed.
- Y. Yuan, X. Zhang, J. Zhao, F. Shen, D. Nie, B. Wang, L. Wang, M. Xing and M. I. Hegglin, Front. Public Health, 2023, 11, 1075262 CrossRef PubMed.
- S. S. Raj, O. O. Krüger, A. Sharma, U. Panda, C. Pöhlker, D. Walter, J. Förster, R. P. Singh, T. Klimach, E. Darbyshire, S. T. Martin, G. McFiggans, H. Coe, J. Allan, V. K. Soni, H. Su, M. O. Andreae, U. Pöschl and S. S. Gunthe, J. Geophys. Res., 2021, 126, e2021JD035681 CrossRef.
- L. Giglio, J. T. Randerson and G. R. van der Werf, J. Geophys. Res. Biogeosci., 2013, 118, 317–328 CrossRef.
- US EPA, 2016v1 Air Emissions Modeling Platform, https://www.epa.gov/air-emissions-modeling/2016v1-platform, accessed May 2, 2023 Search PubMed.
- M. E. J. Stettler, S. Eastham and S. R. H. Barrett, Atmos. Environ., 2011, 45, 5415–5424 CrossRef CAS.
- N. W. Simone, M. E. J. Stettler and S. R. H. Barrett, Transport. Res. Transport Environ., 2013, 25, 33–41 CrossRef.
- R. F. Silvern, D. J. Jacob, L. J. Mickley, M. P. Sulprizio, K. R. Travis, E. A. Marais, R. C. Cohen, J. L. Laughner, S. Choi, J. Joiner and L. N. Lamsal, Atmos. Chem. Phys., 2019, 19, 8863–8878 CrossRef CAS.
- M. J. Cooper, R. V. Martin, C. A. McLinden and J. R. Brook, Environ. Res. Lett., 2020, 15, 104013 CrossRef CAS.
- L. M. Judd, J. A. Al-Saadi, S. J. Janz, M. G. Kowalewski, R. B. Pierce, J. J. Szykman, L. C. Valin, R. Swap, A. Cede, M. Mueller, M. Tiefengraber, N. Abuhassan and D. Williams, Atmos. Meas. Tech., 2019, 12, 6091–6111 CrossRef CAS PubMed.
- S. K. Kharol, R. V. Martin, S. Philip, B. Boys, L. N. Lamsal, M. Jerrett, M. Brauer, D. L. Crouse, C. McLinden and R. T. Burnett, Atmos. Environ., 2015, 118, 236–245 CrossRef CAS.
- P. S. Kim, D. J. Jacob, J. A. Fisher, K. Travis, K. Yu, L. Zhu, R. M. Yantosca, M. P. Sulprizio, J. L. Jimenez, P. Campuzano-Jost, K. D. Froyd, J. Liao, J. W. Hair, M. A. Fenn, C. F. Butler, N. L. Wagner, T. D. Gordon, A. Welti, P. O. Wennberg, J. D. Crounse and A. E. Perring, Atmos. Chem. Phys., 2015, 15, 10411–10433 CrossRef CAS.
- K. R. Travis and D. J. Jacob, Geosci. Model Dev., 2019, 12, 3641–3648 CrossRef CAS.
- Y. Li, R. V. Martin, C. Li, B. L. Boys, A. van Donkelaar, J. Meng and J. R. Pierce, Atmos. Chem. Phys., 2023, 23, 12525–12543 CrossRef CAS.
- D. D. Parrish, K. S. Law, J. Staehelin, R. Derwent, O. R. Cooper, H. Tanimoto, A. Volz-Thomas, S. Gilge, H. E. Scheel, M. Steinbacher and E. Chan, Geophys. Res. Lett., 2013, 40, 1631–1636 CrossRef CAS.
- H. Bowman, S. Turnock, S. E. Bauer, K. Tsigaridis, M. Deushi, N. Oshima, F. M. O'Connor, L. Horowitz, T. Wu, J. Zhang, D. Kubistin and D. D. Parrish, Atmos. Chem. Phys., 2022, 22, 3507–3524 CrossRef CAS.
- J. A. Fisher, D. J. Jacob, M. T. Purdy, M. Kopacz, P. Le Sager, C. Carouge, C. D. Holmes, R. M. Yantosca, R. L. Batchelor, K. Strong, G. S. Diskin, H. E. Fuelberg, J. S. Holloway, E. J. Hyer, W. W. McMillan, J. Warner, D. G. Streets, Q. Zhang, Y. Wang and S. Wu, Atmos. Chem. Phys., 2010, 10, 977–996 CrossRef CAS.
- Le conseil canadien des ministres de l'environnement, Qualité de l'air, https://www.ccme.ca/fr/qualite-de-lair#slide-7, accessed May 2, 2023 Search PubMed.
- World Health Organization, WHO Global Air Quality Guidelines: Particulate Matter (PM2.5 and PM10), Ozone, Nitrogen Dioxide, Sulfur Dioxide and Carbon Monoxide, Geneva, 2021 Search PubMed.
- Agriculture and Agri-Food Canada, Population of Canada, 10km Gridded - Open Government Portal, https://open.canada.ca/data/en/dataset/c6c48391-fd2f-4d8a-93c8-eb74f58a859b, accessed May 9, 2023 Search PubMed.
- Gouvernement du Québec - Ministère de l’Énergie et des Ressources naturelles, Gaz naturel, https://mern.gouv.qc.ca/energie/hydrocarbures/gaz-naturel/, accessed June 27, 2022 Search PubMed.
- U.S. Energy Information Administration, Use of energy explained - Energy use in homes, https://www.eia.gov/energyexplained/use-of-energy/homes.php, accessed June 27, 2022 Search PubMed.
- J. Whitmore and P.-O. Pineau, État de l'énergie au Québec 2019, Chaire de gestion du secteur de l'énergie, HEC Montréal, préparé pour Transition énergétique Québec, Montréal, 2018 Search PubMed.
- Government of Canada, National Pollutant Release Inventory Dashboard, https://www.canada.ca/en/environment-climate-change/services/national-pollutant-release-inventory/tools-resources-data/all-year-dashboard.html, accessed June 27, 2022 Search PubMed.
- L. Liu, W. Xu, X. Lu, B. Zhong, Y. Guo, X. Lu, Y. Zhao, W. He, S. Wang, X. Zhang, X. Liu and P. Vitousek, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2121998119 CrossRef CAS PubMed.
- M. Mozurkewich, Atmos. Environ. Part A Gen. Top., 1993, 27, 261–270 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00307h |
| This journal is © The Royal Society of Chemistry 2024 |