
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModelling and predicting liquid chromatography retention time for PFAS with no-code machine learning†
Yunwu
Fan
a,
Yu
Deng
a,
Yi
Yang
a,
Xin
Deng
a,
Qianhui
Li
a,
Boqi
Xu
a,
Jianyu
Pan
a,
Sisi
Liu
 ab,
Yan
Kong
c and
Chang-Er
Chen
*ab
ab,
Yan
Kong
c and
Chang-Er
Chen
*ab
aSchool of Environment, MOE Key Laboratory of Theoretical Chemistry of Environment, South China Normal University, Guangzhou 510006, China. E-mail: changer.chen@m.scnu.edu.cn; Tel: +86 20 39311529
bEnvironmental Research Institute, Guangdong Provincial Key Laboratory of Chemical Pollution and Environmental Safety, South China Normal University, Guangzhou 510006, China
cWomen and Children's Hospital, Qingdao University, Qingdao 266000, China
First published on 1st December 2023
Abstract
Machine learning is increasingly popular and promising in environmental science due to its potential in solving various environmental problems. One such worldwide issue is the pollution caused by the persistent chemicals – per- and polyfluoroalkyl substances (PFAS), threatening the environment and human beings. Here, we introduce a no-code machine learning approach for modelling the quantitative structure–retention relationship (QSRR) of liquid chromatographic retention time (LC-RT) for PFAS. This approach aims to streamline the modelling process, particularly for environmental professionals who may find intensive coding cumbersome. The QSRR models were developed using the no-code machine learning tool, Orange, employing simple 2D molecular descriptors as input features. Through a systematic analysis, 12 descriptors were identified as pivotal properties essential for developing optimal models (including multiple linear regression – MLR and support vector machine – SVM). These selected models demonstrate great internal validation metrics (R2 > 0.98, MAE < 6.5 s) and reasonable external robustness (R2 > 0.80, MAE ∼ 40 s). Furthermore, a concise model interpretation was conducted to elucidate the molecular factors influencing LC-RT. It is anticipated that our models, capable of predicting the LC-RT for over 2000 PFAS within the Norman Network, will be instrumental in addressing this environmental challenge. This study not only contributes valuable insights into PFAS LC behaviour but also serves as a catalyst for future endeavours in the development and applications of no-code machine learning models.
Environmental significanceMachine learning is popular and promising for environmental science and engineering and should be accessible to any professional, even without coding experience. Here we demonstrated a no-code machine learning methodology with Orange to develop quantitative structure–property relationship (QSPR) models for the liquid chromatographic retention time (LC-RT) of PFAS with simple 2D molecular descriptors as input. Twelve features/descriptors were identified as the key properties that can be employed to develop the best models (including multiple linear regression-MLR and support vector machine-SVM). The selected models have great internal validation metrics (R2 > 0.98, MAE < 6.5 s) and reasonable external robustness (R2 > 0.80, MAE ∼ 40 s). A brief model interpretation was also performed to reveal the molecular factors affecting LC-RT. We anticipate that our models can effectively predict the LC-RT of >2000 PFAS in the Norman Network, and this study serves as inspiration for the development of future models based on no-code machine learning. |
1. Introduction
In recent years, the swift progress of high-performance computing and the continual advancement of modelling software have propelled the rapid evolution and widespread application of machine learning.1 This surge includes a growing presence in environmental science and engineering, fueled by the abundance of extensive datasets.2,3 Machine learning proves invaluable in uncovering intricate and concealed patterns, discerning correlations, and predicting outcomes across diverse domains within the environmental sciences. Its utility extends to forecasting particulate matter concentrations in the air, assessing water variables, pinpointing crucial features (environmental parameters or chemicals), detecting anomalies, and even unearthing novel materials or chemicals with potential environmental implications.3Of the applications in the field of environment, quantitative structure–property/activity relationship (QSPR/QSAR) models are commonly used in environmental chemistry and toxicology. These models are tailored to predict or assess specific properties or toxicities of organic chemicals based on their chemical structures or readily available properties.2–5 Notably, machine learning has demonstrated its capacity to enhance QSPR/QSAR models by mitigating errors and augmenting explanatory power.6–8 Despite the increasing advantages and applications of machine learning in this context, a notable barrier exists – many applications demand proficiency in computer programming languages such as Python or R. The requisite intensive learning and practice associated with coding can be daunting for environmental professionals, impeding the broader utilisation of these advanced techniques.9 Addressing this challenge, simple machine learning tools with little to no code requirements (often referred to as ‘coding-free’ solutions)9 aim to provide accessibility for practitioners in environmental science and engineering without necessitating extensive coding expertise, thereby fostering the broader adoption of versatile and modern artificial intelligence (AI) techniques.10 One such notable machine learning tool fitting this description is Orange. Of course, a fundamental understanding of the underlying algorithmic principles of each algorithm is still necessary. It should be noted that users can only tune the models with the pre-defined parameters within the tools.
Orange is an open-source interactive data analysis tool,11 offering many popular machine learning algorithms and data visualisation capabilities for learners and experts. Notably, it serves as an excellent modelling software for those new to the field, enabling users to analyse data without extensive coding. This approach streamlines the often complex data analysis pipeline, making it more accessible and comprehensible to users without a coding background (further details can be found on the official website at https://orangedatamining.com/). The user-friendly nature of Orange makes it particularly well-suited for individuals in the environmental field who lack coding experience. Consequently, we employed Orange to develop a quantitative structure–retention relationship (QSRR) model for predicting the liquid chromatographic retention time (LC-RT) for per- and poly-fluoroalkyl substances (PFAS). Through leveraging Orange's capabilities, we seek to demonstrate the efficacy of a no-code machine learning approach in addressing environmental challenges related to PFAS.
PFAS are a class of synthetic fluorinated organic compounds widely used in everyday products and across various industrial and civil sectors.12 PFAS have been detected worldwide in all environmental media and organisms. These compounds exhibit persistence, bioaccumulation, and toxicity to organisms,13 prompting significant attention and research efforts on a global scale in recent decades.14 Despite the substantial focus on PFAS, the sheer diversity of over 8000 PFAS variants in the market15 poses challenges for their identification and quantification through traditional target methods, often proving costly or impractical. The start-of-art technique for PFAS identification in the environment relies on non-target screening approaches utilising high-resolution mass spectrometry.16 However, even with this advanced methodology, challenges persist, especially in discerning PFAS with similar chemical structures in chromatographs.17 According to QSRR, the PFAS structural information may determine their RT on LC. Leveraging machine learning to assist QSRR models specifically tailored for LC-RT can furnish orthogonal information crucial for identifying organic chemicals.7,18–22 However, no dedicated model is available for understanding and predicting the RT of PFAS on LC. Recognising the existing gap, there is a compelling interest in developing QSRR models that draw from simple chemical structure information, such as 2D molecular descriptors calculated from open-source tools like PaDEL.
Therefore, in this study, we employed Orange as the machine learning platform to develop models for predicting the LC-RT of PFAS with simple 2D molecular descriptors. We aimed to (1) demonstrate that cutting-edge AI techniques, such as machine learning, can be effectively employed by leveraging freely available tools like Orange by anyone in the field of environmental science and engineering even without coding experiences, and (2) develop a QSRR model capable of predicting the LC-RT of PFAS. By utilizing a simple machine learning tool like Orange, we aim to significantly streamline the application of machine learning methods, thereby facilitating the identification of PFAS in the environment. This approach aligns with the broader objective of making advanced AI techniques more accessible to practitioners in the environmental sciences, ultimately fostering advancements in the field.
2. Materials and method
2.1 Data collection
The RT data was obtained on ultra-high-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) with commonly used reverse-phase C18 LC column (BEH C18, 2.1 × 50 mm, 1.7 μm), mobile phases (ammonium acetate in ultrapure water and pure acetonitrile) at a flow rate of 0.4 mL min−1 (total runtime 10.5 min), and multi-reaction monitoring (MRM) mode (detailed elsewhere in our previous study23), which included 58 PFAS, covering a wide range of physiochemical properties (detailed in Table S1 and Fig. S3 of ESI†).Molecular descriptors are a set of numerical values that quantify different properties of molecules (such as physicochemical, topological and structural) to facilitate observation and comparison of properties of different compounds. Previous research has demonstrated that even simple molecular descriptors can be effectively used in machine learning approaches to establish accurate QSPR models24,25 and, particularly, QSRR models.20–22 To quantify the properties of 58 PFAS compounds, we computed the 1D and 2D molecular descriptors with the open-source tool – PaDEL-descriptor (v2.21 for Windows with Java 1.8.0_301, https://yapcwsoft.com/dd/padeldescriptor/) by inputting 1D and 2D Structure Data Format (SDF) files obtained from PubChem. In total, 1444 molecular descriptors were obtained for each compound and used as the input/features for model development (detailed in Table S2†).
2.2 Data preprocessing
Not all features are necessary, particularly for this small dataset. The following steps were conducted to preprocess the data: (1) variance filtering. If a feature has a variance that is too small, it contributes little to the model and should be considered for removal.20,26 The “Aggregate Columns” widget in Orange was utilised to calculate the variances of features, and the “Transpose” widget was connected to assist in the calculation (Fig. 1). The “Data Table” widget was connected to the output of the “Aggregate Columns” widget to sort the variances. A threshold of 0.1 for the variances was implemented to eliminate features,26 resulting in 651 features (detailed in Table S3†); (2) normalisation. The “Continuize” widget in Orange was utilised to normalise the data into the range of [−1,1] to mitigate the impact of scale on the models. | ||
| Fig. 1 Widget connections in the Orange machine learning canvas. | ||
2.3 Model development
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, in this case, feature number ≤ 12), the selected models were evaluated using top 2, 3, 5, 10, 12, 15, 20, 25 and 30 features as inputs respectively to determine the optimal number of features that yielded the best performance. This was done by connecting the preprocessed data and the “Lasso” model widget to the “Feature Importance” widget from the explain group in the Orange (Fig. 1). This analysis aimed to balance data informativeness and feature dimensionality.
:5, in this case, feature number ≤ 12), the selected models were evaluated using top 2, 3, 5, 10, 12, 15, 20, 25 and 30 features as inputs respectively to determine the optimal number of features that yielded the best performance. This was done by connecting the preprocessed data and the “Lasso” model widget to the “Feature Importance” widget from the explain group in the Orange (Fig. 1). This analysis aimed to balance data informativeness and feature dimensionality.
2.4 Application domain
The application domain (AD) of a QSAR model refers to the specific area in the response and chemical structure space where the model can make reliable predictions, and descriptors generally represent the chemical structure space.32 The descriptor space covered by chemicals in the training sets, also known as the descriptor domain, was used as AD in this study. The distance between any two compounds in the descriptor space can represent the molecular similarity and be used to determine the boundary of the descriptor space. The most commonly used is the Euclidean distance, which was calculated as follows: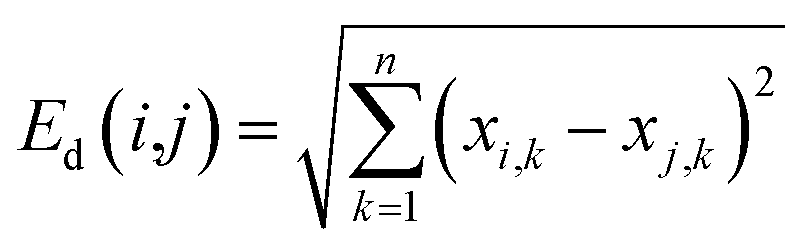 | (1) |
2.5 Norman PFAS list RT prediction
In the Norman Network (https://www.norman-network.com/), more than 4000 PFAS chemicals have been registered, while their RT values on LC are mostly unknown. Therefore, the developed models will be applied to predict the RT values for these PFAS chemicals. The molecular descriptors (selected features) were calculated by PaDEL and used as input for the prediction models. Then, the “Prediction” widget was employed to predict the RT values for the Norman PFAS list (including 4777 PFAS) by connecting the established models and the selected features of the 4777 PFAS to its input. Whether the predicted RTs are within the AD will also be discussed.3. Results and discussion
3.1 Data preprocessing
By setting the variance threshold to 0.1, the number of features was reduced from 1444 to 651. However, considering the small dataset (only 58 observations), it is still not small enough that further feature selection was performed later and tested with a supervised method. The dataset underwent normalization to the range of [−1,1]. This normalization step could avoid the loss function containing regular terms ignoring the features with increasing scale. This precaution was particularly pertinent for the subsequent application of a supervised method, specifically Lasso with regularization. Moreover, a noteworthy positive outcome of this normalization was the observed significant enhancement in the running speed of the “Feature Importance” widget.3.2 Feature selection
The result of the top features calculated by the “Feature Importance” widget under the supervision of Lasso is shown in Fig. S2.† The changes in the pre-selected model evaluation metrics (R2 and MAE from different validation methods: CV10, RS and LOO) against different numbers of top features are shown in Fig. 2. For the LR and SVM models, there was a significant difference among the results of selecting the top 10, 12 and 15 features (p < 0.05), while no significant difference was found between the top 15 and 20 (p > 0.05). Adhering to the feature-case ratio principle, we opted for top 12 features, namely nAcid, AATSC1v, ATS4s, MDEC-44, MWC10, AATS5v, ATSC3m, SpMax6_Bhm, maxsOH, mindssC, ATSC2i and minssCH2 (refer to Tables S4 and S5† for detailed explanations). Commonly recognized is the positive correlation between retention time (RT) and hydrophobicity, often indicated by logP or carbon chain length. Indeed, correlation analysis revealed strong correlations (r > 0.80) between these features and RTs. Intriguingly, features such as XlogP, AlogP, nX, nF, MW, etc., typically associated with hydrophobicity, were not identified during the supervised feature selection step with Lasso. This omission can be attributed to their high collinearities (r > 0.90) with at least one of the shortlisted features, specifically MWC10, as elucidated in the ‘Model Interpretation’ section. Utilising these top 12 features, the models demonstrated exceptional performance (R2 > 0.97 and MAE < 6.5 s). However, for tree-based models (AdaBoost, GBoost, and RF), the optimal performance was achieved with only the top 3 features. Despite this, their R2 remained below 0.95 (some below 0.90), and MAE exceeded 10 s, highlighting the nuanced dynamics in feature importance across different machine learning algorithms.
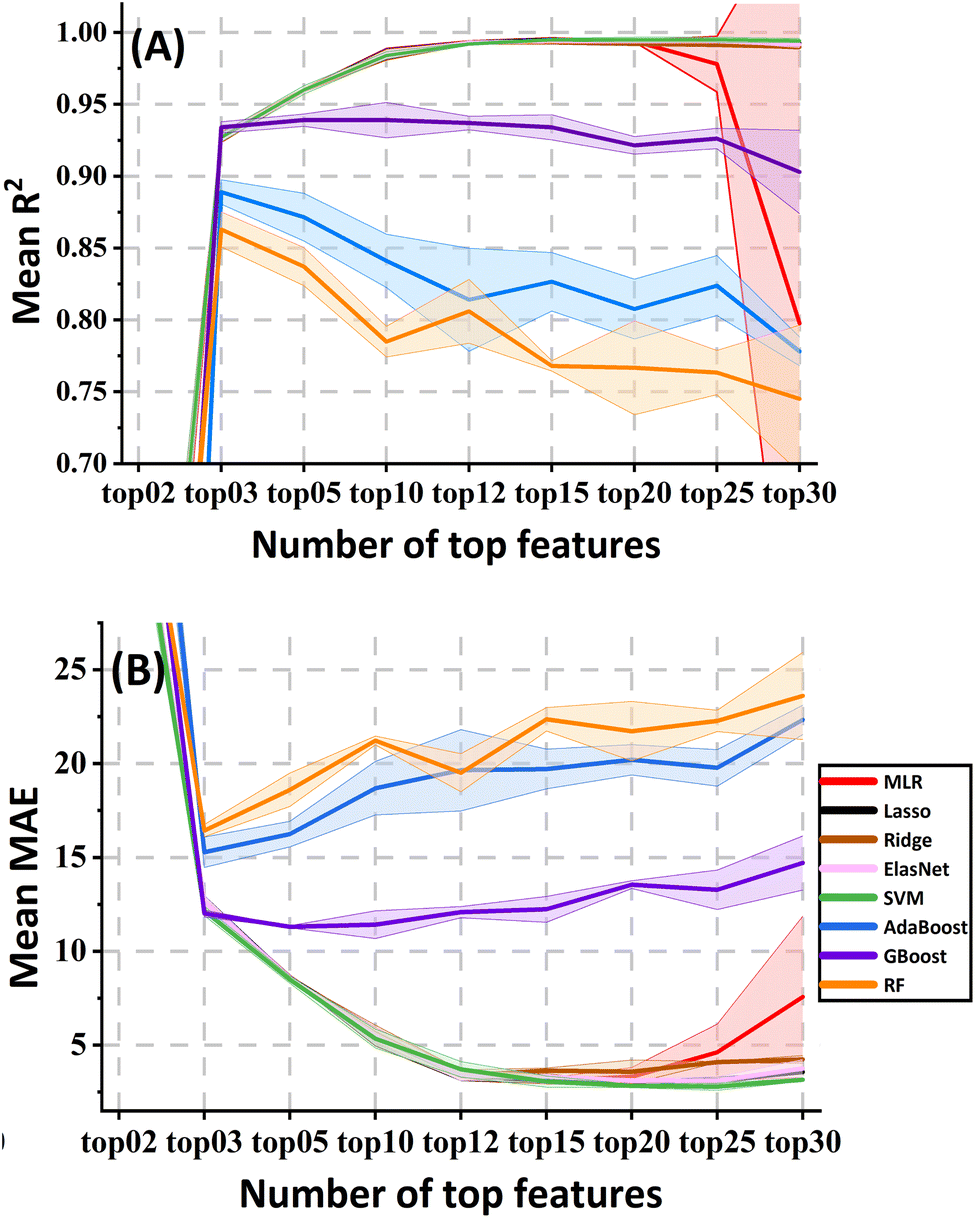 | ||
| Fig. 2 The metrics changes of the pre-selected models with the different top features selected in the “Feature Importance” widget. (A) R2. (B) MAE. There is a large overlap between LRs and SVM. | ||
3.3 Model selection and evaluation
The chosen models underwent fine-tuning by adjusting their hyperparameters in each learner widget to optimise their performance. As depicted in Fig. 2, the tree-based models did not undergo further consideration due to their comparatively lower performance. The detailed metrics results and optimized parameters for the remaining models are presented in Table 1. Evaluation based on R2 and mean absolute error (MAE) values derived from 10-fold cross-validation with the entire dataset revealed exceptional performance for all LR and SVM models, with R2 consistently surpassing 0.97. Moreover, no significant differences were identified among these models (p > 0.05). Considering the simplicity and interpretability outlined by the OECD guideline for QSAR,34 MLR (without regularisation) emerged as the preferred linear model over other LR variants. The rationale for this choice stems from the observation that regularization does not appear necessary for this dataset and its associated problem. The robustness of both MLR and SVM models was further confirmed through rigorous validation methods, including RS, LOO cross-validation, and manual data splitting (train/test = 8:2). In all instances, the resulting R2 values for MLR and SVM consistently exceeded 0.98, while MAE values remained below 5.5 s. These findings attested to the high goodness of fit and a minimal predicted error for both models. Notably, MLR and SVM models were also proposed as the best QSRR models to predict the RTs for other organic chemicals in previous studies.20,21
| Model | Number of features | Parameters | R 2 (mean/sd) | MAE (s) (mean/sd) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CV10a | RSb | LOOc | Testd | CV10 | RS | LOO | Test | |||
|
a 10-fold cross-validation.
b Random sampling, repeat train/test 10 times and training set size = 80%.
c Leave one out.
d Manually split the dataset (8:2) 5 times and tested on the test set (20%).
|
||||||||||
| MLR | 12 | Fit intercept | 0.992/0.085 | 0.989/0.110 | 0.992/0.093 | 0.983/0.008 | 3.38/3.96 | 4.10/3.95 | 3.44/4.48 | 5.15/1.35 |
| Lasso | 12 | Fit intercept, α = 0.01 | 0.992/0.067 | 0.988/0.098 | 0.993/0.089 | 0.983/0.008 | 3.37/3.13 | 4.38/3.68 | 3.46/4.48 | 5.18/1.26 |
| Ridge | 12 | Fit intercept, α = 0.01 | 0.992/0.074 | 0.989/0.121 | 0.991/0.105 | 0.983/0.007 | 3.34/3.91 | 4.54/4.11 | 3.43/4.46 | 5.12/1.29 |
| ElasNet | 12 | Fit intercept, α = 0.001, L1:L2 = 3:1 |
0.989/0.092 | 0.985/0.135 | 0.987/0.129 | 0.974/0.025 | 4.22/3.95 | 6.45/5.37 | 4.48/5.51 | 6.32/1.73 |
| SVM | 12 | C = 10.00, ε = 1.00, linear, Nt = 0.5, iter = 150 | 0.995/0.071 | 0.994/0.087 | 0.995/0.088 | 0.988/0.007 | 2.75/2.38 | 3.62/2.86 | 2.82/3.52 | 4.30/1.51 |
External validation of the models was conducted by introducing 17 new PFAS into the LC-MS system, and their corresponding RTs were measured (see Table S7† for data details). The predictive capabilities of both the MLR and SVM models were scrutinized against these new PFAS, revealing commendable performance. As depicted in Fig. 3, both models exhibited robust predictions, yielding R2 values exceeding 0.80 and MAE hovering around 40 s. Remarkably, this performance is better than or comparable to previous models based on deep learning or graph-neutral networks.7,35 In contrast, the alternative models, particularly the tree-based ones, displayed relatively inferior metrics with R2 values falling below 0.75. Furthermore, statistical analysis confirmed significant differences between the tree-based models and the MLR and SVM models (p < 0.05). This conclusive evidence supports the assertion that MLR and SVM are the two best-performing models in this study. Their consistent and reliable predictions across both internal and external validations reinforce their utility in accurately predicting the retention times of PFAS.
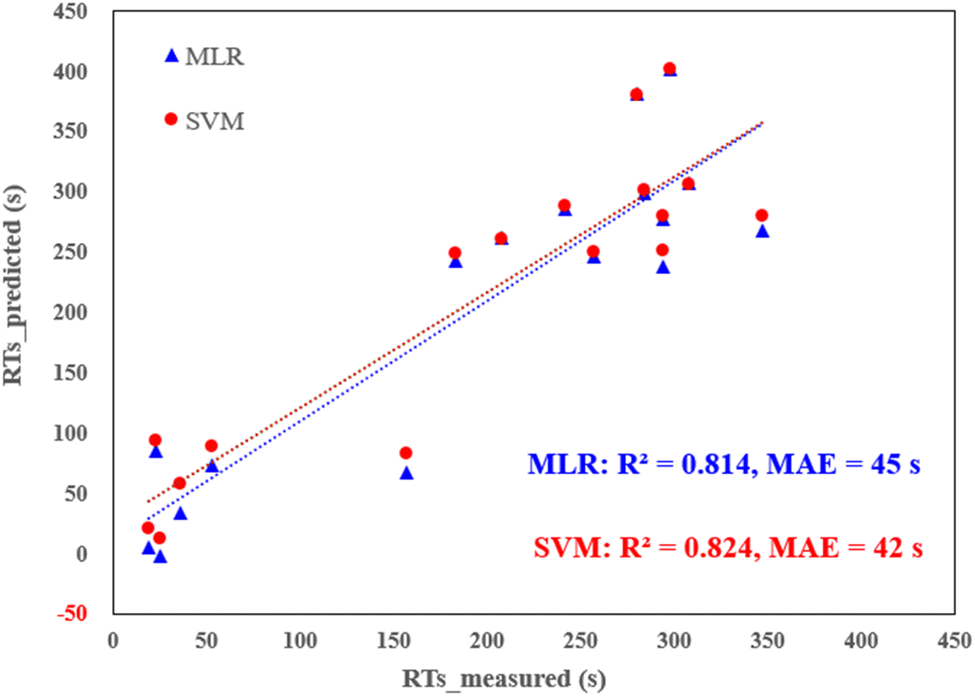 | ||
| Fig. 3 Predicted RTs (via MLR and SVM) vs. measured RTs for external validation dataset (17 new PFAS, detailed in Table S7†). | ||
3.4 Model interpretation
Model interpretation plays a crucial role post-model development, ensuring the congruence between model predictions and the underlying principles of the relevant domain science.3 In this study, the 12 ultimately selected features, each described in detail and categorized based on the six classes outlined by PaDEL, are enumerated in Table S5† as 2D molecular descriptors. To delve into the contribution of each feature to the predictions, we employed the SHapley Additive exPlanations (SHAP) analysis.36 This approach facilitated the elucidation of feature importance and their effects on predictions, presenting a comprehensive overview in the SHAP summary plot (Fig. 4). The plot effectively ranks the features from the most to the least important, providing valuable insights into the variables driving the model's predictive performance. This interpretative step enhances the transparency of the model's decision-making process, establishing a vital link between the identified features and their impact on the LC-RT prediction for PFAS.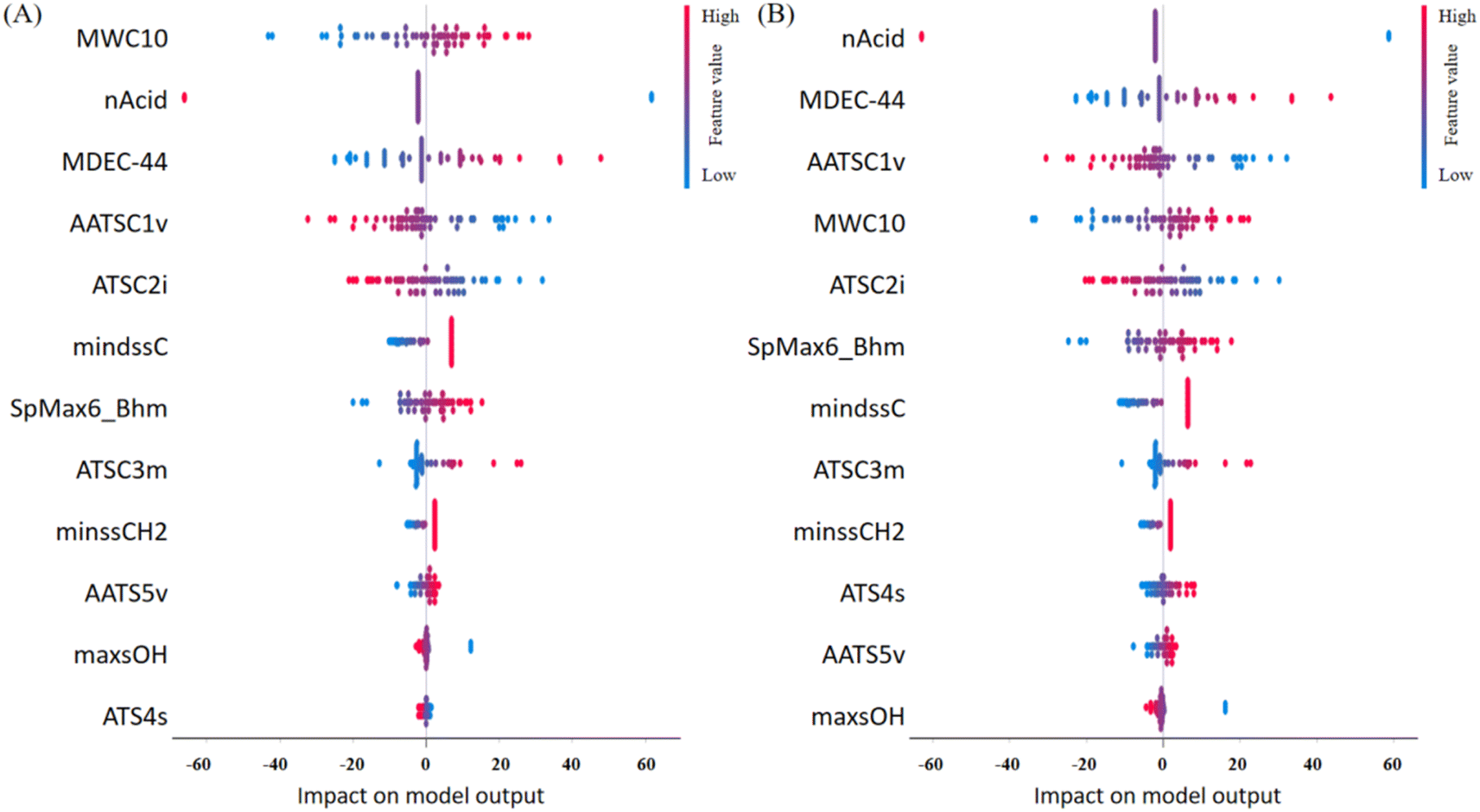 | ||
| Fig. 4 The SHAP value plots of the best models obtained from the “Explain Model” widget. (A) MLR. (B) SVM. The vertical axis ranks feature importance from the most important (top) to the least important (bottom). The horizontal axis indicates the SHAP values. Red indicates a positive association with RTs, and blue indicates a negative association with RTs. | ||
Among the 12 selected features, the variable nAcid, denoting the number of acidic groups within a molecule, emerged as particularly influential. Its significance was underscored by its ranking as the top 2 feature in both the MLR and SVM models (Fig. 4). A closer examination of the SHAP values in the same figure reveals a noteworthy trend: larger values of nAcid correspond to shorter retention times (RTs), as indicated by the blue or negative values in the plot.This observation is intuitive and expected. A higher count of acidic groups within the chemical structure typically translates to increased polarity,37 reducing the affinity of the compound to the stationary phase (the reverse phase LC column). Consequently, chemicals with larger values of nAcid exhibit shorter RTs.
A notable subset of the selected features, comprising over one-third of the total, consisted of autocorrelation descriptors (AATSC1v, ATS4s, ATSC2i, ATSC3m, and AATS5v). These descriptors are widely employed to characterize the distribution of specific physicochemical properties along molecular topology, providing crucial insights into essential molecular structural information.38 It has been documented that these autocorrelation descriptors are extremely useful in QSAR studies.39 These autocorrelation descriptors play a key role in characterising the distribution of van der Waals volumes, first ionisation potential, mass and intrinsic state on a PFAS molecule. Based on their ranking of importance in MLR and SVM models, it is evident that the distributions of van der Waals volumes and the first ionisation potential distribution contributed more significantly to the models. Following them, the mass distribution exhibited a certain degree of correlation. The intrinsic state distribution, while contributing, had a relatively minor impact on the models.
Three features (mindssC, maxsOH and minssCH2) are the electrotopological state atom type descriptors, which are topological indexes at the atomic level that characterise the electronic state of the bonded atom and its topological properties within the molecular skeleton.40 They can recognise atoms or molecular fragments that effectively influence molecular properties.41 They are each associated with the intrinsic electronic properties of the bonded atoms in different structures, in this case, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
C![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) , –OH, and –CH2–, respectively. Moreover, these features are interconnected with the electrotopological environment shaped by surrounding atoms. In the SHAP analysis (Fig. 4), their importance generally ranks lower, indicating a relatively lower contribution to the models.
, –OH, and –CH2–, respectively. Moreover, these features are interconnected with the electrotopological environment shaped by surrounding atoms. In the SHAP analysis (Fig. 4), their importance generally ranks lower, indicating a relatively lower contribution to the models.
The SpMax6_Bhm is a derived index obtained from the Burden modified matrix with relative mass weighting, is often recommended for use in conjunction with other indices (e.g. the e-state descriptors) to enhance models for predicting molecular properties.42 It provides information on the molecular mass of PFAS. As the molecular mass increases, the mobility of PFAS molecules decreases in the liquid phase, resulting in a higher tendency to adsorb onto stationary phases. Consequently, this leads to bigger RTs in LC analysis. The positive correlation observed between SpMax6_Bhm and RTs in Fig. 4 substantiates this phenomenon, reinforcing the notion that molecular mass plays a pivotal role in dictating the behavior of PFAS.
Two additional features, MWC10 and MDEC-44, offer valuable insights into the structural characteristics of PFAS and their impact on LC-RTs. MWC10 represents the total walk count of the tenth order in the molecular graph, where a walk count signifies the number of edges in a sequence of pairwise adjacent edges leading from one vertex to another.43 This descriptor is closely associated with the length of PFAS molecules and the presence of branched chains, providing information on the molecular complexity. On the other hand, MDEC-44 signifies the count of C–C bonds between all quaternary carbons in the molecule. This descriptor is intricately linked to the number of carbon atoms and indirectly characterises the size of a PFAS molecule. Both MWC10 and MDEC-44 play crucial roles in influencing the adsorption of PFAS molecules onto the stationary phase and their mobility within the liquid phase. These features impact the complexity and degree of spatial crimping of PFAS molecules, thereby influencing their behavior in LC. For both the MLR and SVM models, their importance is ranked at the top (Fig. 4). Particularly noteworthy is the top ranking of MWC10 in the MLR model, emphasising its significant influence on RTs during LC analysis. These insights deepen our understanding of the structural determinants affecting PFAS behavior in LC and further validate the importance of these features in predicting LC-RTs using machine learning models.
The hierarchical clustering for the 12 features was employed to better understand the underlying mechanisms and the key factors influencing RTs in the models (Fig. 5). The 12 features were clustered into three distinct clusters (C1, C2 and C3). Notably, ATSC2i stands alone in C1, suggesting it acts as an independent factor with an inverse correlation with RTs. The first ionisation potential is considered a valuable indicator of a compound's stability, which, in turn, can influence its behaviour in LC and subsequently impact its RTs. The C2 can be interpreted as a composite descriptor that considers the factors of molecular length, mass and complexity. All of the features within C2 exhibit a positive correlation with RTs, indicating a direct relationship between the size of a PFAS molecule and its RTs in LC. In practical terms, larger and more complex molecules tend to have longer RTs, signifying a slower elution from the stationary phase compared to smaller molecules.
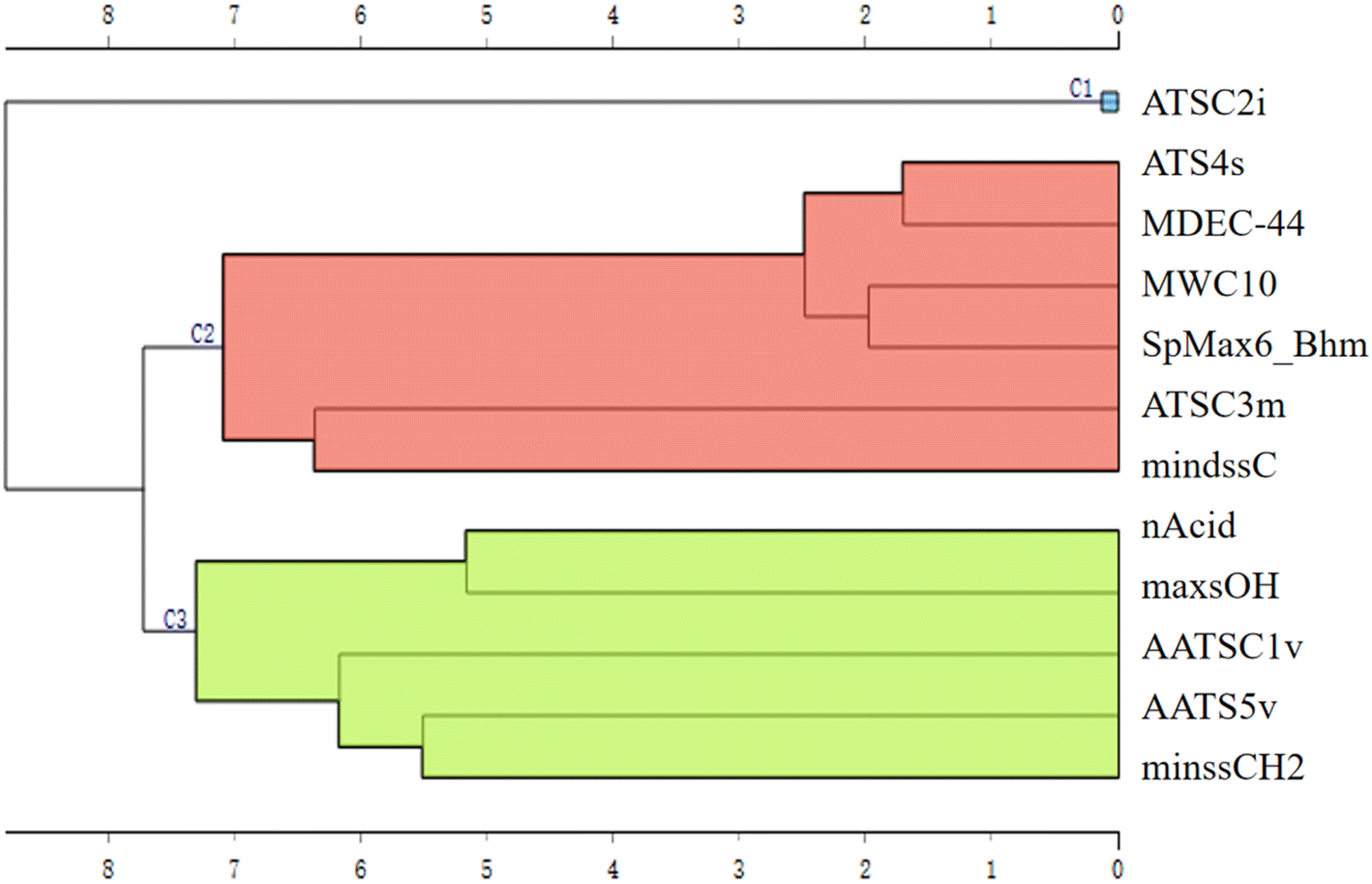 | ||
| Fig. 5 Cluster plot for 12 features obtained in the “Hierarchical Clustering” widget. | ||
Cluster C3 represents a dimension that integrates specific functional groups and intermolecular forces. Among the specific functional groups (including carboxyl, acidic group and methylene) considered in this cluster, it is noteworthy that only minssCH2 shows a positive correlation with RTs. This positive correlation could be attributed to the presence of methylene as a component of the carbon chain structure. In contrast, others tend to enhance the affinity with the liquid phase by increasing molecular polarity, resulting in shorter RTs. AATS5v and the AATSC1v are related to van der Waals volumes. These intermolecular forces can impact the charge distribution within the molecule and, consequently, its polarity. Therefore, these features characterised the influence of molecular polarity on RTs. This further supports the notion that three key factors affecting RTs are molecular stability, size, and polarity.
3.5 Application domain and prediction
The 12 features from the training set were used to construct the AD, with the method set to “Euclidean distance” in Ambit Discovery. Analysis of the training set demonstrated that all 58 PFAS within the set fell within boundaries of the AD. Subsequently, the 12 key features for the extensive list of 4777 PFAS in the Norman PFAS list were calculated and fitted into the AD. It was found that 2101 PFAS from the Norman PFAS list (44%) reside within the established AD. The visual representation of the chemical space based on principal component analysis (PCA) is illustrated in Fig. 6, highlighting the AD (depicted by red dots/area) and the training dataset (represented by cross shapes). Notably, some chemicals from the training dataset overlap with others within the AD. These 58 PFAS in the training set exhibit a broad range of PFAS in terms of molecular weight (MW, 214–1204 g mol−1), acid group number (0, 1, 2), MWC10 (11.4–13.7), ATSC2i (−216 to 3.9) as shown in Fig. S3.† Notably, there is significant overlap between the training dataset and the Norman PFAS list. For the Norman PFAS list, approximately 56% of PFAS were found outside the AD (blue dots in Fig. 6). Whiel acknowledging that the AD obtained from the 58 PFAS may not encompass the entire range of the targeted > 4000 PFAS, it does encompass a considerable number (>2000). This coverage surpasses many target and non-target analyses of PFAS in the environment contexts. Therefore, it will be helpful for the non-target screening of PFAS by offering a comprehensive foundation for future investigations in the environmental domain in the future.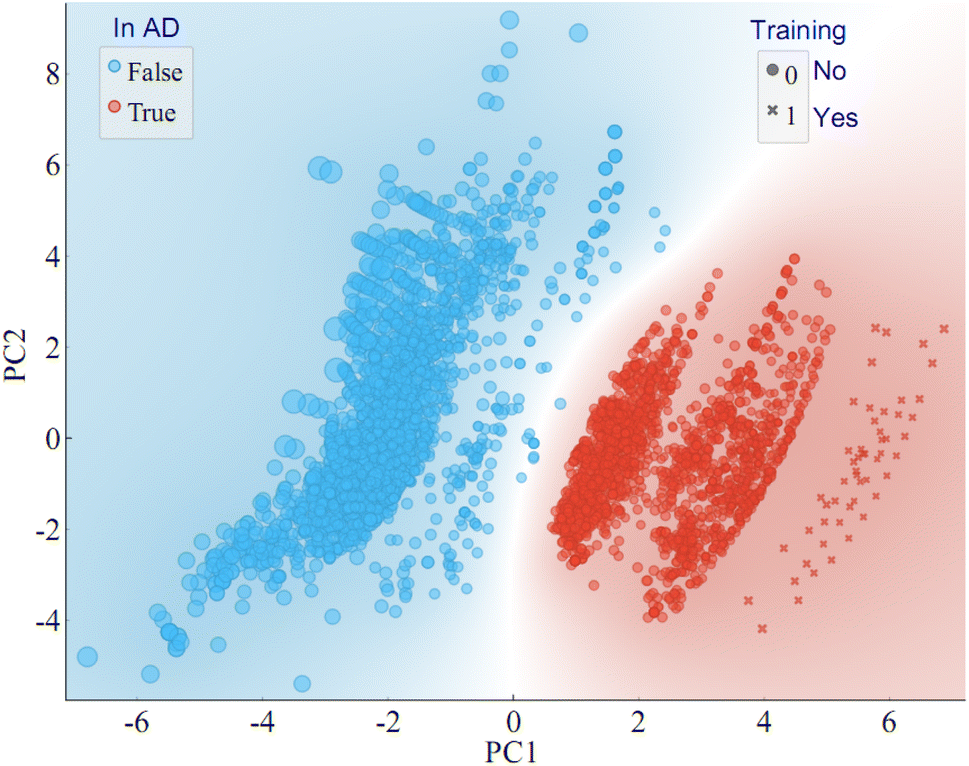 | ||
| Fig. 6 The application domain of the MLR model is characterised by a PCA-based approach for the LC-RT of PFAS. Chemicals in AD are shown in red, otherwise in blue; chemicals in the training dataset are in ‘x’ shape, otherwise in dot shape; the size of the symbols represents the Euclidean distance. | ||
The predicted RTs of the Norman PFAS list are listed in Table S7.† RT predictions for PFAS within the AD can be considered relatively reliable, aiding in the identification of novel PFAS in the environment. External validation of the predictions using an external dataset reveals that all PFAS within the AD exhibit mean absolute errors (MAE) less than 60 s, affirming the accuracy of the predictions within the established AD. However, caution is warranted when considering the reliability of predicted RTs for PFAS outside the AD. External validation indicates that many PFAS outside the AD display higher MAE values, exceeding 60 seconds and, in some instances, reaching up to 100 seconds. An important consideration is that our training dataset did not include chemicals with more than two acid groups, which predisposes these chemicals to fall outside the AD. Notably, some intriguing observations arise when comparing chemicals within and outside the AD. For instance, chemicals lacking an acid group (nAcid = 0), are likely to fall outside the AD if their ionisation potential (indicated by ATSC2i) is either too low (<−200) or too high (>200). This observation aligns with the understanding that chemicals unable to ionize cannot be detected by LC-MS. To improve the prediction of more reliable RTs and enlarge the AD for PFAS, more PFAS with distinct chemical structures can be incorporated into our models, which is subject to our future work.
It is crucial to highlight that the LC method employed to acquire the RTs follows a widely used reversed phase liquid chromatography (RPLC) approach. This method involves a C18 column and mobile phases comprising pure acetonitrile and ammonium acetate in pure water. This RPLC method is standard for a broad spectrum of organic chemicals, including PFAS. Therefore, the models developed based on this method are expected to be applicable to other PFAS under the same or similar RPLC conditions. Nevertheless, it is important to acknowledge that absolute RTs are inherently LC system-dependent. Even with similar LC conditions, such as the use of the same LC column, mobile phases, and gradient, absolute RTs may vary between different LC systems. To extend the applicability of the models to other RPLC systems, potential approaches include RT mapping with tools like PredRet44 or using the RTI system,20 albeit these considerations fall beyond the scope of the present study. Ongoing research efforts are actively exploring these possibilities. In the future, as additional PFAS standards become accessible, the models can be easily reconstructed or validated using newly measured RTs. This continuous refinement ensures that the models remain robust and adaptable, contributing to their reliability and efficacy in diverse LC systems.
4. Conclusions
In summary, this study successfully developed QSRR models utilising simple 2D molecular descriptors obtained from open-source software PaDEL. The no-code machine learning tool, Orange, was instrumental in constructing models aimed understanding and predicting the LC-RTs of PFAS. From a pool of over 1000 features, 12 key descriptors were identified and employed as input for the model development. The resultant models exhibited impressive internal validation metrics and demonstrated reasonable robustness when applied to external chemicals. Notably, the investigation elucidated that the molecular stability, size and polarity are the pivotal factors influencing the LC-RT of PFAS. This study demonstrated the efficacy of no-code machine learning tools, exemplified by Orange, as valuable resources for environmental professionals, particularly those lacking coding experience. The accessibility of such tools can empower practitioners to harness the capabilities of machine learning for problem-solving and pattern identification in environmental science.3 However, it is important to acknowledge the limitation of this study, primarily related to the scale of the data. The potential enhancement of model performance through the enlargement of the PFAS RT dataset is recognized and constitutes an avenue for future work. Continued efforts in expanding the dataset aim to further refine the models, ensuring their applicability and accuracy in predicting LC-RTs for a broader spectrum of PFAS compounds in environmental contexts.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2022YFC3902102), the National Natural Science Foundation of China (No. 42277457), the Guangdong Basic and Applied Basic Research Foundation (2023A1515011515), the Young Talent Support Project of Guangzhou Association for Science and Technology (Si-si Liu), South China Normal University Extracurricular Research Gold Seed Cultivation Project (23HJGB03) and Guangdong Provincial Key Laboratory of Chemical Pollution and Environmental Safety (2019B030301008).References
- N. Artrith, K. T. Butler, F. X. Coudert, S. Han, O. Isayev, A. Jain and A. Walsh, Best practices in machine learning for chemistry, Nat. Chem., 2021, 13, 505–508 CrossRef CAS PubMed.
- S. Gupta, D. Aga, A. Pruden, L. Zhang and P. Vikesland, Data Analytics for Environmental Science and Engineering Research, Environ. Sci. Technol., 2021, 55, 10895–10907 CrossRef CAS PubMed.
- S. Zhong, K. Zhang, M. Bagheri, J. G. Burken, A. Gu, B. Li, X. Ma, B. L. Marrone, Z. J. Ren, J. Schrier, W. Shi, H. Tan, T. Wang, X. Wang, B. M. Wong, X. Xiao, X. Yu, J. J. Zhu and H. Zhang, Machine Learning: New Ideas and Tools in Environmental Science and Engineering, Environ. Sci. Technol., 2021, 55, 12741–12754 CAS.
- M. W. H. Wang, J. M. Goodman and T. E. H. Allen, Machine Learning in Predictive Toxicology: Recent Applications and Future Directions for Classification Models, Chem. Res. Toxicol., 2021, 34, 217–239 Search PubMed.
- D. Xia, J. Chen, Z. Fu, T. Xu, Z. Wang, W. Liu, H. B. Xie and W. Peijnenburg, Potential Application of Machine-Learning-Based Quantum Chemical Methods in Environmental Chemistry, Environ. Sci. Technol., 2022, 56, 2115–2123 CrossRef CAS PubMed.
- G. Gini and F. Zanoli, Ecotoxicological QSARs, ed. K. Roy, 2020, pp. 111–149 Search PubMed.
- Q. Yang, H. Ji, H. Lu and Z. Zhang, Prediction of Liquid Chromatographic Retention Time with Graph Neural Networks to Assist in Small Molecule Identification, Anal. Chem., 2021, 93, 2200–2206 CrossRef CAS PubMed.
- S. Zhong, Y. Zhang and H. Zhang, Machine Learning-Assisted QSAR Models on Contaminant Reactivity Toward Four Oxidants: Combining Small Data Sets and Knowledge Transfer, Environ. Sci. Technol., 2021, 56, 681–692 CrossRef PubMed.
- M. Z. Naser, Machine learning for all! Benchmarking automated, explainable, and coding-free platforms on civil and environmental engineering problems, Journal of Infrastructure Intelligence and Resilience, 2023, 2, 100028 CrossRef.
- L. Lei, R. Pang, Z. Han, D. Wu, B. Xie and Y. Su, Current applications and future impact of machine learning in emerging contaminants: a review, Crit. Rev. Environ. Sci. Technol., 2023, 1–19 Search PubMed.
- J. Demšar, T. Curk, A. Erjavec, Č. Gorup, T. Hočevar, M. Milutinovič, M. Možina, M. Polajnar, M. Toplak, A. Starič, M. Štajdohar, L. Umek, L. Žagar, J. Žbontar, M. Žitnik and B. Zupan, Orange: data mining toolbox in python, J. Mach. Learn. Res., 2013, 14, 2349–2353 Search PubMed.
- J. Glüge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, An overview of the uses of per- and polyfluoroalkyl substances (PFAS), Environ. Sci.: Processes Impacts, 2020, 22, 2345–2373 RSC.
- X. Jiao, Q. Shi and J. Gan, Uptake, accumulation and metabolism of PFASs in plants and health perspectives: a critical review, Crit. Rev. Environ. Sci. Technol., 2020, 51, 2745–2776 CrossRef.
- R. Naidu, P. Nadebaum, C. Fang, I. Cousins, K. Pennell, J. Conder, C. J. Newell, D. Longpré, S. Warner, N. D. Crosbie, A. Surapaneni, D. Bekele, R. Spiese, T. Bradshaw, D. Slee, Y. Liu, F. Qi, M. Mallavarapu, L. Duan, L. McLeod, M. Bowman, B. Richmond, P. Srivastava, S. Chadalavada, A. Umeh, B. Biswas, A. Barclay, J. Simon and P. Nathanail, Per- and poly-fluoroalkyl substances (PFAS): current status and research needs, Environ. Technol. Innovation, 2020, 19, 100915 CrossRef.
- Z. Wang, A. M. Buser, I. T. Cousins, S. Demattio, W. Drost, O. Johansson, K. Ohno, G. Patlewicz, A. M. Richard, G. W. Walker, G. S. White and E. Leinala, A New OECD Definition for Per- and Polyfluoroalkyl Substances, Environ. Sci. Technol., 2021, 55, 15575–15578 CrossRef CAS PubMed.
- K. Ng, N. Alygizakis, A. Androulakakis, A. Galani, R. Aalizadeh, N. S. Thomaidis and J. Slobodnik, Target and suspect screening of 4777 per- and polyfluoroalkyl substances (PFAS) in river water, wastewater, groundwater and biota samples in the Danube River Basin, J. Hazard. Mater., 2022, 436, 129276 CrossRef CAS PubMed.
- H. Ryu, B. Li, S. De Guise, J. McCutcheon and Y. Lei, Recent progress in the detection of emerging contaminants PFASs, J. Hazard. Mater., 2021, 408, 124437 CrossRef CAS PubMed.
- X. A.-O. Domingo-Almenara, C. Guijas, E. A.-O. Billings, J. A.-O. Montenegro-Burke, W. Uritboonthai, A. E. Aisporna, E. Chen, H. A.-O. Benton and G. A.-O. Siuzdak, The METLIN small molecule dataset for machine learning-based retention time prediction, Nat. Commun., 2019, 10, 5811 CrossRef CAS PubMed.
- C. Feng, Q. Xu, X. Qiu, Y. Jin, J. Ji, Y. Lin, S. Le, J. She, D. Lu and G. Wang, Evaluation and application of machine learning-based retention time prediction for suspect screening of pesticides and pesticide transformation products in LC-HRMS, Chemosphere, 2021, 271, 129447 CrossRef CAS PubMed.
- R. Aalizadeh, N. A. Alygizakis, E. L. Schymanski, M. Krauss, T. Schulze, M. Ibáñez, A. D. McEachran, A. Chao, A. J. Williams, P. Gago-Ferrero, A. Covaci, C. Moschet, T. M. Young, J. Hollender, J. Slobodnik and N. S. Thomaidis, Development and Application of Liquid Chromatographic Retention Time Indices in HRMS-Based Suspect and Nontarget Screening, Anal. Chem., 2021, 93, 11601–11611 CrossRef CAS PubMed.
- R. Aalizadeh, M.-C. Nika and N. S. Thomaidis, Development and application of retention time prediction models in the suspect and non-target screening of emerging contaminants, J. Hazard. Mater., 2019, 363, 277–285 CrossRef CAS PubMed.
- F. Gritti, Perspective on the Future Approaches to Predict Retention in Liquid Chromatography, Anal. Chem., 2021, 93, 5653–5664 CrossRef CAS PubMed.
- C. E. Chen, Y. Y. Yang, J. L. Zhao, Y. S. Liu, L. X. Hu, B. B. Li, C. L. Li and G. G. Ying, Legacy and alternative per- and polyfluoroalkyl substances (PFASs) in the West River and North River, south China: occurrence, fate, spatio-temporal variations and potential sources, Chemosphere, 2021, 283, 131301 CrossRef CAS PubMed.
- W. Cheng and C. A. Ng, Using Machine Learning to Classify Bioactivity for 3486 Per- and Polyfluoroalkyl Substances (PFASs) from the OECD List, Environ. Sci. Technol., 2019, 53, 13970–13980 CrossRef CAS PubMed.
- A. Raza, S. Bardhan, L. Xu, S. S. R. K. C. Yamijala, C. Lian, H. Kwon and B. M. Wong, A Machine Learning Approach for Predicting Defluorination of Per- and Polyfluoroalkyl Substances (PFAS) for Their Efficient Treatment and Removal, Environ. Sci. Technol. Lett., 2019, 6, 624–629 CrossRef CAS.
- Y. Wang, J. fan, S. Wang, G. Huang and Z. Yan, Predict Toxicity Effects of Endocrine Disruptor Chemicals on Aquatic Organisms Using Machine Learning, Asian J. Ecotoxicol., 2022, 17, 148–163 Search PubMed.
- Z. Li, Z. Long, S. Lei, L. Yang, W. Zhang and T. Zhang, Explicit expressions of the saturation flux density and thermal stability in Fe-based metallic glasses based on Lasso regression, Intermetallics, 2021, 139, 107361 CrossRef CAS.
- J. Cervantes, F. Garcia-Lamont, L. Rodríguez-Mazahua and A. Lopez, A comprehensive survey on support vector machine classification: applications, challenges and trends, Neurocomputing, 2020, 408, 189–215 CrossRef.
- S. Wu and H. Nagahashi, Analysis of Generalization Ability for Different AdaBoost Variants Based on Classification and Regression Trees, J. Electr. Comput. Eng., 2015, 2015, 835357 Search PubMed.
- Z. Yan and H. Wen, Comparative Study of Electricity-Theft Detection Based on Gradient Boosting Machine, 2021 IEEE International Instrumentation and Measurement Technology Conference (I2MTC), 2021, pp. 1–6 Search PubMed.
- H. Jiang, J. Li, R. Sun, C. Tian, J. Tang, B. Jiang, Y. Liao, C.-E. Chen and G. Zhang, Molecular Dynamics and Light Absorption Properties of Atmospheric Dissolved Organic Matter, Environ. Sci. Technol., 2021, 55, 10268–10279 CrossRef CAS PubMed.
- Z. Wang, J. Chen, Z. Fu and X. Li, Characterization of applicability domains for QSAR models, Chin. Sci. Bull., 2022, 67, 255–266 CrossRef.
- W. Ou, H. Liu, J. He and X. Yang, Development of chicken and fish muscle protein - water partition coefficients predictive models for ionogenic and neutral organic chemicals, Ecotoxicol. Environ. Saf., 2018, 157, 128–133 CrossRef CAS PubMed.
- OECD, Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models, 2014 Search PubMed.
- X. Domingo-Almenara, C. Guijas, E. Billings, J. R. Montenegro-Burke, W. Uritboonthai, A. E. Aisporna, E. Chen, H. P. Benton and G. Siuzdak, The METLIN small molecule dataset for machine learning-based retention time prediction, Nat. Commun., 2019, 10, 5811 CrossRef CAS PubMed.
- S. Akbar, F. Ali, M. Hayat, A. Ahmad, S. Khan and S. Gul, Prediction of antiviral peptides using transform evolutionary & SHAP analysis based descriptors by incorporation with ensemble learning strategy, Chemom. Intell. Lab. Syst., 2022, 230, 104682 CrossRef CAS.
- S. Joudan, R. Z. Liu, J. C. D’Eon and S. A. Mabury, Unique analytical considerations for laboratory studies identifying metabolic products of per- and polyfluoroalkyl substances (PFASs), Trends Anal. Chem., 2020, 124, 115431 CrossRef CAS.
- G. Moreau and P. Broto, The autocorrelation of a topological structure: a new molecular descriptor, Nouv. J. Chim., 1980, 4, 359–360 CAS.
- J. L. Velázquez-Libera, J. Caballero, A. P. Toropova and A. A. Toropov, Estimation of 2D autocorrelation descriptors and 2D Monte Carlo descriptors as a tool to build up predictive models for acetylcholinesterase (AChE) inhibitory activity, Chemom. Intell. Lab. Syst., 2019, 184, 14–21 CrossRef.
- L. B. Kier and L. H. Hall, An Atom-Centered Index for Drug QSAR Models, in Advances in Drug Design, ed. B. Testa, Academic Press, 1992, vol. 22 Search PubMed.
- L. Jiao, H. Liu, L. Qu, Z. Xue, Y. Wang, Y. Wang, B. Lei, Y. Zang, R. Xu, Z. Zhang, H. Li and O. A. A. Alyemeni, QSPR Studies on the Octane Number of Toluene Primary Reference Fuel Based on the Electrotopological State Index, ACS Omega, 2020, 5, 3878–3888 CrossRef CAS PubMed.
- F. R. Burden, A chemically intuitive molecular index based on the eigenvalues of a modified adjacency matrix, Quant. Struct.-Act. Relat., 1997, 16, 309–314 CrossRef CAS.
- G. Rüecker and C. Rüecker, Counts of all walks as atomic and molecular descriptors, J. Chem. Inf. Comput. Sci., 1993, 33, 683–695 CrossRef.
- J. Stanstrup, S. Neumann and U. Vrhovšek, PredRet: Prediction of Retention Time by Direct Mapping between Multiple Chromatographic Systems, Anal. Chem., 2015, 87, 9421–9428 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00242j |
| This journal is © The Royal Society of Chemistry 2024 |