
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deciphering colour mechanisms in co-crystals and salts containing violuric acid and chosen L-amino acids†
Agnieszka
Rydz
,
Marlena
Gryl
 *,
Katarzyna
Ostrowska
and
Katarzyna Marta
Stadnicka
*,
Katarzyna
Ostrowska
and
Katarzyna Marta
Stadnicka
Faculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Cracow, Poland. E-mail: gryl@chemia.uj.edu.pl
First published on 5th September 2024
Abstract
The understanding of colour origins in single-component materials is well established, however comprehending the factors that influence colour in multi-component crystalline phases remains a complex endeavor. This study addresses the challenge of predicting absorption properties in solid-state materials, crucial for designing systems for specific applications. We focused on six multi-component crystals based on violuric acid (VA) and selected L-amino acids, chosen for their chemical and structural diversity. This approach allowed us to explore various factors affecting absorption properties, including co-crystal and salt formation, hydrogen bonding, π–π interactions, amino acid side chain conformations, and solvent inclusion. Techniques such as X-ray diffraction, 1H NMR spectroscopy, and UV-Vis spectroscopy were employed for detailed analysis of the structure and optical properties of the studied systems. The intermolecular interactions were scrutinized through the topology of electron density, Hirshfeld surface analysis, and the non-covalent interaction (NCI) index. UV-Vis titrations of crystallization solutions were also conducted to determine the binding constants of salts. This comprehensive study aims to enhance our understanding of absorption properties in multi-component materials.
Introduction
Colour chemistry attracts much attention because of the widespread applications of chromic materials as sensors,1–3 molecular optical devices,4,5 thermochromic inks6,7 or dyes.8 Chromic effects arise from the absorption or emission processes in a material due to applied external stimuli which can be, e.g. light, temperature, pressure, electric field or the presence of another substance.7 Light absorption processes in the solid state seem to be well understood in many single-component systems, in which molecule under the influence of external stimulus undergoes, e.g., cis/trans conformational transformation,9 keto–enol tautomerism,10,11 cyclization reactions,12 or exhibit changes in the geometry of intermolecular interactions.13,14 However, inducing absorption or emission at a precisely selected wavelength using specified chemical compounds remains a formidable challenge. Engineering such properties necessitates a profound understanding of molecular aggregation processes, which lead to distinct molecular arrangements found in the solid state.Moreover, many acknowledged chromogens and chromophores may not be environmentally benign and sometimes are even considered toxic. Familiar instances include carcinogenic azo compounds15 and the notorious pollution from textile dyes.16 These cases pose substantial challenges to industrial applications. Finding new colorants, less harmful, is of crucial importance to the textile industry, environmental sustainability, and public health, as it addresses the pressing need for safer production practices, reduces the ecological footprint, and minimizes exposure to toxic chemicals for workers and consumers alike.
An alternative to typical single-component chromic materials might be multicomponent systems, where one chromogen can be used to generate a set of compounds with absorption depending upon the co-former incorporated in the crystal structure. The colour modifiers could be environmentally friendly molecules (here L-amino acids).
Growing interest in co-crystals and organic salts in fields such as pharmaceutical sciences17 or optoelectronics is due to the possibility of tuning properties like stability,18 bioavailability,19,20 permeability21 and SHG efficiency.22–24 A comparable strategy involving cocrystallization techniques can be employed with chromic materials to fine-tune their absorption or emission properties.25,26 In principle, if we understand the origin of colour in such a multicomponent system, we can tune the absorption at will. Using a co-crystal or an organic salt limits the usage of heavy metal ions and selection of a chromogen capable of forming vibrantly coloured products restricts the concentrations employed. Recently, we have demonstrated that designing intermolecular interactions between a chromogen and a co-former leads to distinctly coloured compounds.27 The core component, violuric acid (VA)—an isonitroso derivative of barbituric acid (Fig. 2a)—is a colorless solid under ambient conditions. Deprotonation of VA during synthesis or cocrystallization results in the generation of intensely coloured products, with the hue attributable to n–π* transitions.28 For instance, cocrystallization with tyramine, another colourless compound, produced two differently coloured salts: red and violet pseudopolymorphs. Additionally, we obtained a red-brown polymorph through a single crystal-to-single crystal phase transition. We have elucidated the colour phenomena in tyraminium violurates, and attributed it to a two-step process: (1) proton transfer between VA and tyramine and (2) redistribution of electron density within the oxime group of VA, influenced by intermolecular interactions.
This research27 inspired us to explore the chromic phenomena of multicomponent materials containing VA, broadening our scope, which constitutes the focus of this study. Consequently, as co-formers for VA, we opted for representatives of amino acids (AA), namely: L-serine (L-Ser), L-tyrosine (L-Tyr), L-tryptophan (L-Trp), L-lysine (L-Lys), L-histidine (L-His), and L-arginine (L-Arg), as depicted in Fig. 1. This class of compounds carries a broad chemical and structural diversity, depending on the structure of the AA side chain. The presence of identical functional groups (carboxylate and ammonium in the zwitterionic form) in each amino acid, along with the fixed donor and acceptor sites in VA, simplifies the elucidation of the molecular recognition mechanism in the studied phases.
In this manuscript, we showcase six multicomponent crystalline phases, each comprising VA and a selected AA, namely: VASer (L-serine violuric acid water 〈1/1/2〉), VATyr (L-tyrosine violuric acid water 〈1/1/3〉), VATrp (L-tryptophan violuric acid 〈1/1〉), VATrp* (L-tryptophan violuric acid water 〈1/1/1〉) VAHis (L-histidinium violurate water 〈2+/2−/2〉) and VALys (L-lysinium violurate water 〈2+/2−/2〉). The names for the multicomponent solids were devised based on a nomenclature system proposed by two members of our team.29 All, except VASer, exhibit absorption in the range of 428–586 nm. The origin of colour in these materials was extensively examined with the help of X-ray diffraction, UV-Vis spectroscopy and 1H NMR studies of parent and product solutions. Binding constants for salts were determined through UV-Vis titration experiments. Analysis of weak interactions (hydrogen bonds, π–π interactions) was performed using theoretical calculations of non-covalent interaction (NCI) index30 and QTAIM topological analysis of electron density in the solid state (Crystal1731,32 and Topond33) to gain insight into the color phenomena of obtained crystalline materials.
Results and discussion
Crystal engineering and structure analysis
To develop multicomponent chromic materials that incorporate VA, we have strategically selected AA that exhibit diverse characteristics. L-Lysine and L-arginine were chosen as co-formers for salt formation due to their inherent basic nature. Additionally, the presence of an imidazole ring in L-His facilitates π–π stacking interactions. We also aimed to explore how weak dispersive interactions affect colour phenomena. For this purpose, we considered L-tyrosine (L-Tyr) and L-tryptophan (L-Trp), each of which has an aromatic ring in its side chain. The possibility of creating coloured crystals using VA and a co-former without aromatic rings and resistance to protonation was uncertain. L-Serine (L-Ser), a simple amino acid with a hydroxyl group in the side chain, emerged as a viable option. Its classification as a polar, uncharged amino acid, due to its hydroxyl group, contrasts with L-tryptophan's (L-Trp) non-polar characteristics. This difference could offer insights into their distinct hydrogen-bonding interactions with water molecules.Therefore, the final selection of co-formers for cocrystallization experiments with VA includes L-Lys, L-His, L-Arg, L-Ser, L-Tyr, and L-Trp (Fig. 1). The specifics of the employed crystal engineering procedure, which includes a search for potential synthons in the CSD database, are comprehensively detailed in the ESI† (Fig. S1, S2 and Table S1).
 | ||
| Fig. 1 violuric acid (1), violurate ion (2), L-Trp (3), L-Tyr (4), L-Lys (5), L-Arg (6), L-His (7), L-Ser (8). | ||
As previously stated, a primary objective of our study was to produce either co-crystals or salts to establish whether the final colour of a product is predominantly determined by the proton transfer between an acid and a base. Typically in such a case, a pKa rule helps to predict the possible product of cocrystallisation. This rule is based on the difference between the pKa of a base and an acid, here ΔpKa = pKAAa2 − pKAa. If ΔpKa is less than 0, then a co-crystal formation is favoured. If the ΔpKa is larger than 2–3 a salt formation is expected. This rule, however, does not correctly predict the formation of co-crystal or salt, if ΔpKa is in the range of 0–2/3. The rule has been validated only for scenarios involving acid–base pairs where either a single proton is transferred, or specifically, where the transfer involves the most acidic proton of the acid to the most basic atom of the base, considering only interactions associated with the first ionization constant.34
For the sake of comparison Table 1 presents the pKa values for VA, L-Ser, L-Tyr, L-Trp, L-Lys, L-His and L-Arg as well as obtained values of isoelectric point (PI) for a particular amino acid. We have included the ΔpKa values calculated as a difference between the pKa1 of VA and PI of the amino acid.
| VA | L-Trp | L-Tyr | L-Lys | L-Arg | L-His | L-Ser | |
|---|---|---|---|---|---|---|---|
| pKa1 | 4.35 | 2.38 | 2.24 | 2.15 | 2.03 | 1.70 | 2.13 |
| pKa2 | 9.64 | 9.34 | 9.04 | 9.16 | 9.00 | 9.09 | 9.05 |
| pKa3 | 14.2 | — | 10.10 | 10.67 | 12.10 | 6.04 | — |
| PI | — | 5.89 | 5.66 | 9.74 | 10.76 | 7.59 | 5.68 |
| ΔpKa | 1.54 | 1.31 | 5.39 | 6.41 | 3.24 | 1.33 | |
| VATrp | VATyr | VALys | VAArg | VAHis | VASer | ||
| Co-crystal | Co-crystal | Salt | Salt | Salt | Co-crystal |
All the above considerations helped us to obtain four co-crystals: VASer, VATyr, VATrp, and VATrp* and two salts: VALys and VAHis. Despite multiple attempts to crystallize VAArg for X-ray diffraction analysis using various techniques, the resulting dark purple product was amorphous, not crystalline. All of the obtained phases crystallise in noncentrosymmetric space groups with amino acids as zwitterions in their L-form (crystallographic data and refinement details are given in Table S2 and asymmetric units are shown in Fig. S3, ESI†).
In the VASer co-crystal, the molecule of VA forms intermolecular interactions with four water molecules, which in turn reduces the potential for hydrogen bonding with L-Ser. Three distinct hydrogen-bond motifs between VA and L-Ser can be recognized: R22(9) and R21(6) rings, and D11(2) discrete motif (Fig. 2a) at the second and first graph-set level,36 respectively. All three motifs are formed through N–H⋯O interactions, with nitrogen atoms from VA's NH group or AA's NH3+ as donors and oxygen atoms from carboxylate or carbonyl groups as acceptors. The hydroxyl group on the L-Ser's side-chain binds with two water molecules, a result of the polar O–H group's strong affinity for water. In the three remaining co-crystal structures, the arrangement of water molecules around VA varies: four water molecules interact with VA in VATyr, two water molecules associate with VA in VATrp*, and no solvent molecules are incorporated in VATrp structure.
 | ||
| Fig. 2 Recognized synthons (shown in colour) in the (a) VASer, (b) VATyr, (c) VATrp, (d) VATrp*, (e) VAHis and (f) VALys crystal structures. Hydrogen atoms which do not participate in hydrogen bonds were omitted for clarity. | ||
In these three co-crystals, two specific interactions/motifs between VA and amino acid can be recognized. The first one is a hydrogen bond of the O–H⋯O type (marked as a pink ellipse in Fig. 2b–d) involving the O–H oxime group and the carboxylate oxygen atom of L-Tyr or L-Trp. The second motif is an R33(12) ring (shown in yellow in Fig. 2b–d) incorporating the first motif and additional two hydrogen bonds: N8B–H8B3⋯O4Aiv and N8B–H8B1⋯O91Biv (in VATyr) or N11B–H11D⋯O4Aiv and N11B–H11A⋯O13Biv (in VATrp). Moreover, in both VATyr and VATrp structures, zig-zag shaped hydrogen bonds along [100] and [010] form the C11(5) motif via N8B–H8B1⋯O91Biv and N11B–H11A⋯O13Biv hydrogen bonds (Fig. 2b and c), respectively. Such an architecture of hydrogen bonds leads to a parallel arrangement of phenol (L-Tyr) and indole (L-Trp) groups with VA molecules allowing π–π interactions to be present (what will be discussed later). Analysing the crystal structures of salts: VALys and VAHis, it can be seen that the number of water molecules engaged in hydrogen bonds with VA is reduced to two per anion. Water molecules function as connectors, offering steric complementarity for the densest possible packing arrangement (1.526 and 1.661 g cm−3 VALys and VAHis, respectively). Salt bridges between lysinium cations in VALys and histidinium ones in VAHis lead to the crystal structure built of alternate and interconnected by hydrogen bond layers of violurate anions and lysinium/histidinium cations (Fig. S4, ESI†). Detailed geometrical parameters of all hydrogen bonds can be found in Table S3 (ESI†) and hydrogen bonds around the VA oxime are depicted in Fig. S5 (ESI†). As we expected from the synthon formation analysis based on the CSD search, in salt crystal structures, i.e. VALys and VAHis, the R22(8) ring motifs are observed. However, in co-crystals (VASer, VATyr and VATrp), these synthons were replaced by interactions with AA or water molecules.
Conformation and π–π interactions
Structural analysis revealed that in all co-crystals, both the ammonium group and the side chain group—hydroxyl in L-Ser, phenyl in L-Tyr, and indole in L-Trp—adopt a synclinal conformation along the Cα–Cβ bond (χ1). Torsion angles are equal to 57.77° in VASer, −65.83° in VATyr, −49.72° in VATrp and 51.28° in VATrp*. In salts, there are two independent cations of L-Lys and L-His in the asymmetric unit, namely C- and D-cations. The C-cations of L-Lys and L-His have a synclinal conformation (torsion angles χ1 are 56.93° and −50.74°, respectively), and cations adopt antiperiplanar conformation with a torsion angle −170.30° (L-Lys) and −164.66° (L-His) – see Fig. S6 (ESI†). As was mentioned earlier, in VATyr, VATrp* and VATrp crystal structures, several π–π interactions were found. According to the literature,37 the usual π interaction is characterized by an offset or slipped stacking arrangement. The ring normal and the vector between the ring centroids (Cg) form an angle of about 20° up to centroid–centroid distances of 3.8 Å.The carbonyl group of VA is in the close vicinity to the phenyl ring of L-Tyr and with the 5- and 6-membered indole ring of L-Trp (Cg⋯Cg distances in the range of 3.24–3.56 Å). Also, the double –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of the oxime group participates in π–π interaction with L-Trp (3.40 Å). Similar interactions are observed in VAHis, where distances of Cg(C5AN5A)⋯Cg(3) and Cg(C5BN5B)⋯Cg(4) are 3.37 Å and 3.45 Å, respectively. Moreover, in VAHis, the O1W oxygen atom is located in close vicinity of L-His 5-membered ring with a Lp(O1W)⋯Cg(4) distance of 3.386 Å at an angle of 89.95°. Detailed geometrical description of all mentioned above interactions is given in Table S4 (ESI†) and the interactions are presented in Fig. S7 (ESI†). These interactions can be easily visualized using the non-covalent interaction (NCI) index.30 NCI tool uses the reduced gradient of electron density (RDG) to highlight the low-density regime where weak and dispersive interactions can be observed. Small red or blue disks represent strong hydrogen bonds whereas broad green surfaces indicate weak non-covalent interactions. As shown in Fig. 3, the green multiform surfaces represent the π–π interactions found in VATyr, VATrp and VAHis crystal structures.
N bond of the oxime group participates in π–π interaction with L-Trp (3.40 Å). Similar interactions are observed in VAHis, where distances of Cg(C5AN5A)⋯Cg(3) and Cg(C5BN5B)⋯Cg(4) are 3.37 Å and 3.45 Å, respectively. Moreover, in VAHis, the O1W oxygen atom is located in close vicinity of L-His 5-membered ring with a Lp(O1W)⋯Cg(4) distance of 3.386 Å at an angle of 89.95°. Detailed geometrical description of all mentioned above interactions is given in Table S4 (ESI†) and the interactions are presented in Fig. S7 (ESI†). These interactions can be easily visualized using the non-covalent interaction (NCI) index.30 NCI tool uses the reduced gradient of electron density (RDG) to highlight the low-density regime where weak and dispersive interactions can be observed. Small red or blue disks represent strong hydrogen bonds whereas broad green surfaces indicate weak non-covalent interactions. As shown in Fig. 3, the green multiform surfaces represent the π–π interactions found in VATyr, VATrp and VAHis crystal structures.
 | ||
| Fig. 3 NCI isosurfaces for (a) VATyr, (b) VATrp and (c) VAHis. | ||
The observation of weak interactions in both dimers and larger assemblies substantiates their presence in the crystal structures. In VASer and VALys, no π–π interactions can be found.
UV-Vis and 1H NMR spectroscopy, pH measurements
Absorption spectra for solution samples of VASer, VATyr, VATrp, VALys and VAHis are shown in Fig. 4a, while those for the solid state are shown in Fig. 4b. VASer crystals appeared colourless with no absorption maximum in the visible range. The slightly yellow colour of the VATyr phase corresponds to the weak absorption maximum within the range of 440–460 nm (compare Fig. S8, ESI†). Crystal phases of VATrp and VATrp* are both orange with λmax at 428 nm. The absorption maxima for VALys and VAHis are at λmax = 552 nm and λmax = 586 nm (purple and violet colour of the crystals), respectively. In our previous study, the obtained salts of tyraminium violurates absorbed light within the range of 524–581 nm. At this stage, we can assume that the proton transfer between the oxime group of VA and AA results in the shift of the absorption maximum towards longer wavelengths. The UV-Vis spectra for solutions of VASer, VATyr, VATrp, VALys, and VAArg are presented in Fig. 3b. The weak absorption peak at λmax = 544 nm in VASer, VATyr, and VATrp solutions could be associated with the solvation effect of VA by water molecules. The strong absorption maximum at the same wavelength for VALys, VAHis and VAArg solutions can be associated with the salt formation between VA and basic AA, i.e.L-Arg, L-His and L-Lys. To confirm this, we have recorded the 1H NMR spectra of L-His, L-Lys and L-Arg mixtures with VA (at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) as well as the obtained products (VALys and VAHis), both in D2O. The salt formation was monitored via the chemical downfield shift for the proton signals: Ha (associated with the stereogenic centre at Cα) and diasterotopic protons Hb and Hc attached to the Cβ carbon atom. Formation of the NH3+ ammonium group at C5 of L-Lys in VALys and the L-Lys and VA mixture is manifested via downfield shifts and separation of signals for protons: Ha (3.24 ppm to 3.67 ppm), Hd (1.57 ppm to 1.82 ppm), He (1.57 ppm to 1.82 ppm), Hf (2.86 ppm to 2.94 ppm), and diasterotopic Hb and Hc (Fig. S9, ESI†). In VAHis, the protonation of the imine nitrogen atom in the L-His imidazole ring was accompanied by the shift of signals for protons of the heterocyclic system: Hd from 6.93 ppm to 7.24 ppm, and He from 7.63 ppm to 8.40 ppm (Fig. S10, ESI†). Protonation of L-Arg in D2O solution after addition of VA is apparent through downfield chemical shifts and signals for Ha (3.60 ppm), Ha′ (3.68 ppm), He, He′ (3.14 ppm), and diasterotopic protons (Fig. S11, ESI†). We have also registered 1H NMR spectra in D2O for dissolved co-crystals of VASer and VATrp and mixtures of VA with L-Ser, L-Tyr and L-Trp (Fig. S12–S14, ESI†). The co-crystal formation was not accompanied by changes in proton chemical shifts for L-Trp, L-Tyr, and L-Ser in 1H NMR data. Having established the proton transfer between VA and L-Lys, L-His, and L-Arg, along with the formation of salts in solutions, we aimed to evaluate the stability of these salts and quantify their binding constants (K). For this purpose, UV-Vis titration experiments were performed using the procedure described in the Experimental section. Fig. 5 shows the result of the spectrophotometric titrations and Fig. S15 (ESI†) presents their titration curves (absorbance as a function of components molar ratio). The Joe-Jones method38 was used to evaluate the system stoichiometry, which was obtained from the point where the titration curve changes its slope. In each case, the stoichiometry of VA:AA was found to be 1:1. The binding constant was estimated using ReactLabTM EQUILIBRIA software [https://www.jplusconsulting.com] with logK = 4.955 ± 0.016 (VAArg), logK = 4.581 ± 0.006 (VALys), and logK = 3.583 ± 0.004 (VAHis). So, the obtained binding constants are as follows: K = 9.0 × 104 M−1 for VAArg, K = 3.8 × 104 M−1 for VALys and K = 3.8 × 103 M−1 for VAHis. In comparison with other organic complexes,39,40 these values show a moderate to high affinity of VA towards basic AA in the following order L-His < L-Lys < L-Arg. Calculated limit of detection (LoD) values are 2.54 × 10−5 M, 6.97 × 10−6 M and 8.26 × 10−6 M for VAHis, VALys and VAArg, respectively. These values show that L-Lys and L-Arg are detectable by VA at the micro level in solution samples. Details regarding LoD calculations can be found in the ESI† (Fig. S16–S18).
:1 ratio) as well as the obtained products (VALys and VAHis), both in D2O. The salt formation was monitored via the chemical downfield shift for the proton signals: Ha (associated with the stereogenic centre at Cα) and diasterotopic protons Hb and Hc attached to the Cβ carbon atom. Formation of the NH3+ ammonium group at C5 of L-Lys in VALys and the L-Lys and VA mixture is manifested via downfield shifts and separation of signals for protons: Ha (3.24 ppm to 3.67 ppm), Hd (1.57 ppm to 1.82 ppm), He (1.57 ppm to 1.82 ppm), Hf (2.86 ppm to 2.94 ppm), and diasterotopic Hb and Hc (Fig. S9, ESI†). In VAHis, the protonation of the imine nitrogen atom in the L-His imidazole ring was accompanied by the shift of signals for protons of the heterocyclic system: Hd from 6.93 ppm to 7.24 ppm, and He from 7.63 ppm to 8.40 ppm (Fig. S10, ESI†). Protonation of L-Arg in D2O solution after addition of VA is apparent through downfield chemical shifts and signals for Ha (3.60 ppm), Ha′ (3.68 ppm), He, He′ (3.14 ppm), and diasterotopic protons (Fig. S11, ESI†). We have also registered 1H NMR spectra in D2O for dissolved co-crystals of VASer and VATrp and mixtures of VA with L-Ser, L-Tyr and L-Trp (Fig. S12–S14, ESI†). The co-crystal formation was not accompanied by changes in proton chemical shifts for L-Trp, L-Tyr, and L-Ser in 1H NMR data. Having established the proton transfer between VA and L-Lys, L-His, and L-Arg, along with the formation of salts in solutions, we aimed to evaluate the stability of these salts and quantify their binding constants (K). For this purpose, UV-Vis titration experiments were performed using the procedure described in the Experimental section. Fig. 5 shows the result of the spectrophotometric titrations and Fig. S15 (ESI†) presents their titration curves (absorbance as a function of components molar ratio). The Joe-Jones method38 was used to evaluate the system stoichiometry, which was obtained from the point where the titration curve changes its slope. In each case, the stoichiometry of VA:AA was found to be 1:1. The binding constant was estimated using ReactLabTM EQUILIBRIA software [https://www.jplusconsulting.com] with logK = 4.955 ± 0.016 (VAArg), logK = 4.581 ± 0.006 (VALys), and logK = 3.583 ± 0.004 (VAHis). So, the obtained binding constants are as follows: K = 9.0 × 104 M−1 for VAArg, K = 3.8 × 104 M−1 for VALys and K = 3.8 × 103 M−1 for VAHis. In comparison with other organic complexes,39,40 these values show a moderate to high affinity of VA towards basic AA in the following order L-His < L-Lys < L-Arg. Calculated limit of detection (LoD) values are 2.54 × 10−5 M, 6.97 × 10−6 M and 8.26 × 10−6 M for VAHis, VALys and VAArg, respectively. These values show that L-Lys and L-Arg are detectable by VA at the micro level in solution samples. Details regarding LoD calculations can be found in the ESI† (Fig. S16–S18).
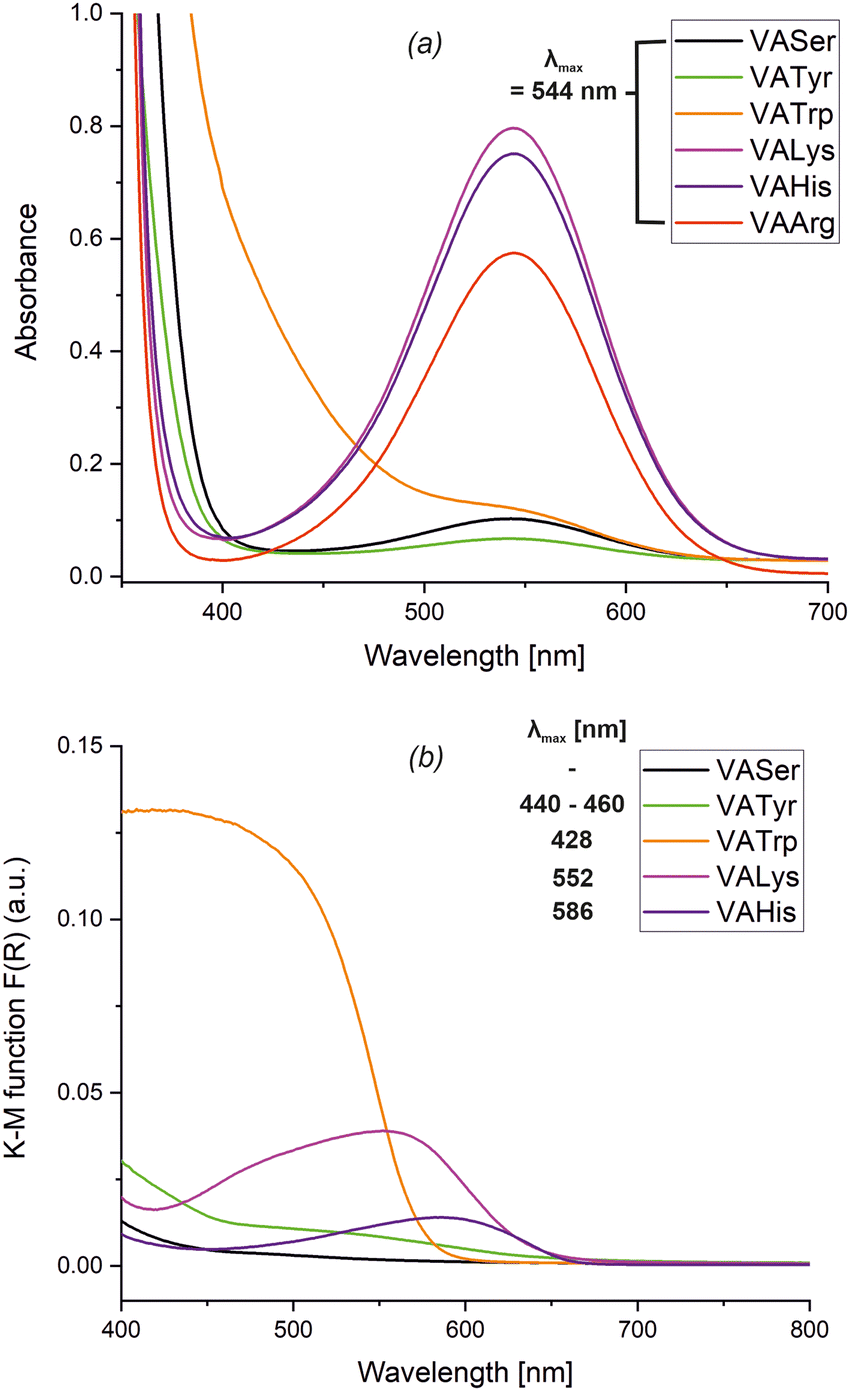 | ||
| Fig. 4 The UV-Vis spectra for (a) the aqueous solutions of VA (6.72 × 10−3 M in VATyr, 1.0 × 10−2 M in VAArg and 1.42 × 10−2 M in the remaining solutions) with L-Ser (1.42 × 10−2 M), L-Tyr (6.72 × 10−3 M), L-Trp (1.42 × 10−2 M), L-Lys (1.42 × 10−2 M), L-His (1.42 × 10−2 M) and L-Arg (1.42 × 10−2 M). (b) The powdered samples of VASer, VATyr, VATrp, VALys and VAHis. | ||
 | ||
| Fig. 5 Titration experiments for (a) L-Lys (b) L-Arg (c) L-His with VA. | ||
Hirshfeld surfaces and topological analysis of electron density
To determine the source of colour in crystalline materials, we conducted an extensive analysis of intermolecular interactions and characterized the structural features of the building blocks, as well as the crystal packing (Fig. S4, ESI†). For the comparison of intra- and intermolecular interactions of VA in co-crystals and VA anions in salts, Hirshfeld surface analysis41 was performed. This tool is particularly useful for studying polymorphs or multicomponent crystals where one of the building blocks remains consistent.The Hirshfeld surface of a molecule in a crystal is created by dividing the crystal space into regions where the electron density of the molecule (the promolecule), represented as a sum of spherical atoms, is greater than the electron density of the entire crystal (the procrystal). Once the Hirshfeld surface is defined, a fingerprint plot can be generated. It charts the distance from a point on the surface to the nearest external atom (de) against the distance to the nearest internal atom (di). The plot thus provides a detailed picture of how molecules in a crystal interact with their neighbors. Two-dimension fingerprint plots of VA molecule in VASer, VATyr, VATyr, VATrp and VATrp* as well as of VA anions in VALys and VAHis with percentage contribution of individual interactions are presented in Fig. 6.
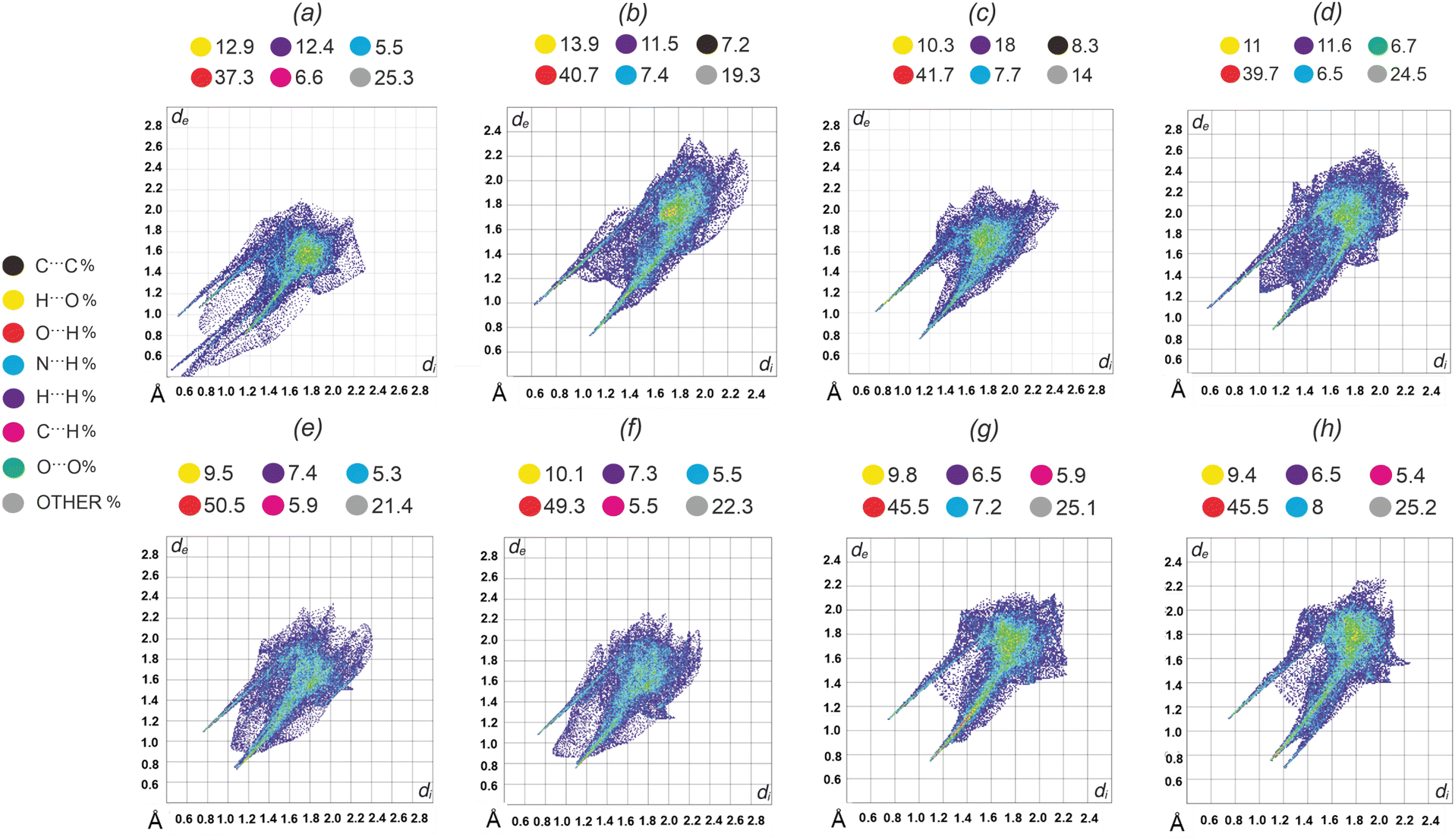 | ||
| Fig. 6 Two-dimensional Fingerprint plots of VA molecules in (a) VASer, (b) VATyr, (c) VATrp*, (d) VATrp, and VA anion in (e) VALys (A anion), (f) VALys (B anion), (g) VAHis (A anion) and (h) VAHis (B anion). Coloured dots above drawings denote the percentage contribution of a particular interaction. | ||
We can clearly see that each image is distinct; however, there can be found some similar features such as spikes in the left and right lower parts of the graphs representing O⋯H and H⋯O interactions or wing-like motifs from C⋯H and H⋯C contacts (upper part of the drawings). The main difference between co-crystals and salts can be seen comparing O⋯H and H⋯O interactions. The amount of O⋯H contacts in salts is ca. 8–13% larger than in co-crystals along with the slightly smaller contribution of H⋯O contacts. It can be also seen that in co-crystals the amount of H⋯H contacts (a set of dispersed points located between the spikes and in the center of the drawings) is almost as twice as that in salts. A quantitative analysis of the molecular structure and intermolecular interactions of the VA oxime group in the obtained phases was possible with QTAIM analysis.42 The electron density derived properties at the bond critical point (BCP) for the oxime bonds: C5–N5 and N5–O5 for the studied materials and the VA monohydrate crystal structure (for comparison purposes) are presented in Table S5 (ESI†). Molecular graphs, with marked BCPs for VA molecules and ions embedded in the crystal structures, are presented in Fig. 7 and Fig. S19 (ESI†). The major differences between the salt and co-crystal structures can be seen in the bond length analysis. The N5–O5 bond becomes shorter and C5–N5 longer going from the studied co-crystals to salts. The observed increasing values of electron density ρ(r) from 0.324 to 0.386 and more negative Laplacian ∇2ρ(r) from −0.262 to −0.440, for VASer and VAHis, respectively, can be correlated with the deprotonation of VA and an accumulation of negative charge at the BCP of the N5–O5 bond. However, there are no major differences in C–N and N–O bond characteristics between two salts VALys and VAHis and this is also true for the structures of the VASer, VATyr and VATrp co-crystals. To estimate the influence of the hydrogen bonds on the electronic changes within the oxime group we have calculated the interaction energies EO and EN for all studied phases (EN/EO denotes the energy of all possible hydrogen bonds in which the N5/O5 atom of VA oxime group participates, respectively; Table S6, ESI†).43EO has the largest contribution to the total energy (Etotal) in each crystalline phase and the highest value is observed for VA and its co-crystals, whereas the interaction energy decreases gradually when going from VA to VAHis (the summarized energy components are presented in Table 2). The obtained results are in agreement with some of our previous observations made for the polymorphs of tyraminium violurates, where the smallest interaction energies EO across polymorphs were found for violet crystals of form II (ca. −10.85 kcal mol−1). Only small variations in energy can be found across the co-crystals: largest values of interaction energies found for VATyr and the smallest one for VATrp. Note that values presented in Table 2 were calculated per VA molecule/ion and averaged where the disordered oxime group was present. This approach allowed us to compare the VA molecule/ion in all of the examined structures. Looking at the donors and acceptors involved in the strongest hydrogen bonds (Table S4, ESI†), we can clearly see that for the co-crystals, the O5 oxygen atom plays the role of a donor whereas in the strongest interactions in the salts, the O5 atom participates as an acceptor. Much weaker interactions (or none) are found for N5 nitrogen atoms. For the co-crystal solvates and salts, we can see a trend of decreasing interaction energies (Eint) with the bathochromic shift of the n–π* maximum (lower energy and longer wavelength). The case of VATrp is different as there are no solvent molecules stabilizing the structure through the hydrogen bond interactions. Instead, the hydrogen atom from the O5–H5 bond is accepted by the carboxylic oxygen atom of L-Trp. For comparison purposes, we have added a VATrp* structure which is a salt solvate (pseudopolymorph of anhydrous VATrp). In fact, we have already shown that there are no obvious changes in the colour of VATrp and VaTrp* as a result of the presence or absence of water molecules. This is the only case where we were able to obtain a crystal structure containing VA and amino acids without water molecules, although the crystals were of inferior quality.
 | ||
| Fig. 7 Selected molecular graphs with BCPs (green) showing different conformations of VA oxime group in co-crystals, salts and VA monohydrate. The a and b denote distinct molecules/ions in the asymmetric unit. | ||
| E O | E N | E total | |
|---|---|---|---|
| VA | −28.1 | −28.1 | |
| VASer | −21.2 | −21.2 | |
| VaTyr | −21.4 | −5.8 | −27.3 |
| VATrp | −19.2 | −19.2 | |
| VATrp* | −23.0 | — | −23.0 |
| VALys | −15.4 | −4.4 | −19.8 |
| VAHis | −13.3 | −2.7 | −16.0 |
Conclusions
We have designed, obtained and thoroughly analyzed six new crystal phases containing violuric acid (VA) and L-amino acids with diverse optical properties. Our research offers a novel perspective by demonstrating the crucial role of hydrogen bonding involving the oxime group on the absorption properties of a series of compounds. We confirmed that the intense coloration observed in some VA-based materials is linked to salt formation and the presence of VA in an anionic state. In particular, the L-amino acids with PI > 7 were found to form salts with VA whereas the ones with PI < 7 formed co-crystals (see Table 1). Additionally, our research resulted in the identification of various recurring synthons related to the presence of VA in both its neutral and deprotonated forms. In particular, we observed significant variations in the strength of interactions involving VA's oxime group across both salts and co-crystals. This observation supports the hypothesis that shifts in the absorption band within the visible spectrum are associated with weak interactions in these systems. Initially, one might assume that the coloration of these materials is due to π–π interactions. However, our findings reveal no direct correlation between the strength of these interactions and the position of the absorption band responsible for the colour. For instance, the intensely colored salts VALys and VAHis, with similar absorption bands (λmax = 552 nm and λmax = 586 nm, respectively), are different in this manner. In VALys, there are no π–π interactions, whereas in VAHis, we can find three with moderate strengths. VaSer crystals appear colourless with no absorption maximum in the visible range and no π⋯π interactions. Conversely, VATrp crystals, displaying a pale yellow hue (440–460 nm), exhibit the shortest Cg⋯Cg distances (3.24 Å) among the studied materials (see Table S4, ESI†). When examining hydrogen bonding, a different scenario emerges. We hypothesize that stronger interactions involving VA's oxime group shift the n–π* band towards the higher-energy end of the UV-Vis spectrum. This conclusion aligns with our previous research on tyraminium violurates. To confirm this hypothesis, we performed model TDDFT calculations using two clusters (Fig. S20, ESI†) constructed from the VAHis crystal structure. We selected the first cluster comprised of one VA anion, one L-His anion, and one water molecule. In this model, after optimizing the cluster, there is only one hydrogen bond, (H2O)H⋯O(oxime), with an H⋯A distance of 1.804 Å. Additionally, we constructed a second cluster by modifying the first one, removing the water molecule. This time, after optimization, there is only one hydrogen bond, (NH2)H⋯O(oxime), with an H⋯A distance of 2.109 Å. The subsequent TDDFT calculations (results summarized in Table S7, ESI†), indicate that changing the environment around the oxime group affects the shift of the absorption maximum towards shorter wavelengths, consistent with the strengthening of interactions involving the oxime oxygen atom. The two formed clusters demonstrate that the shift in absorption maximum correlates with the presence and nature of hydrogen bonds involving the oxime group. However, this hypothesis needs to be experimentally verified by exploring and studying the absorption properties of a single chromogen (violuric acid) with an expanded array of co-formers. This would help to assess the feasibility of designing multicomponent materials with customized chromic effects. Nonetheless, this study highlights the adaptability of VA as a versatile chromogen for designing and producing a variety of coloured materials, demonstrating its potential in material science.Experimental
Cocrystallisation
:1 molar ratio were dissolved in water. A mixture of VA and L-Lys was left for slow evaporation under ambient conditions. After a few days, purple needle-like crystals appeared in the vial.
Data collection and crystal structure refinement
Crystal structures of all obtained materials were determined using single crystal X-ray diffraction studies. SIR9244 was used to solve the structure of VASer and SHELXT45 for the remaining phases. The refinement procedure based on the F2 least-squares method was performed using the SHELXL2018 program.46 Aromatic and aliphatic hydrogen atoms were calculated at idealized positions and refined using a riding model. The remaining hydrogen atoms were found from different Fourier maps and refined freely with Uiso = 1.2 Uiso(parent atom). Crystal data, details of data collection, and structure refinement for all crystal structures are given in Table S2 (ESI†). In some of the crystal structures, the oxime group is disordered. In VASer, site occupancy factors for N5A–O5A and N5A1–O5A1 pairs are 0.58:0.42; in VATrp N5A–O5A and N5A1–O5A1, atom pairs are disordered over two positions in ratio 0.67:0.33; in VALys oxime O5A:O5A′ atoms are in ratio 0.87:0.13 and disordered CH2 groups in L-lysine are in ratio 0.12:0.88 (C2′/C3′:C2/C3). Additionally, in VAHis, O21W–H211 and O22W–H221 pairs of atoms in one of the water molecules are disordered in ratio 0.61:0.39, respectively.
Theoretical calculations
Non-covalent interaction (NCI) index calculations were performed using NCIPLOT software.30 The input wavefunctions were calculated with the M052X/6-311+G(2df,2p) level of theory using GAUSSIAN16 software (Revision C.01).47Test TDDFT calculations were carried out in Gaussian16 using B3LYP/6-31G(2d,2p).
The input wavefunctions for the theoretical electron density calculations were determined using the Crystal1731,32 software at the PBEsol0/POB-TZVP level of theory. The geometry of the molecules and ions was optimized with fixed lattice parameters. Topond33 was used for the topological analysis of electron density.
The M052X developed by Zhao and Truhlar48–51 has been shown to correctly describe noncovalent interactions.52 This functional was used for the NCI calculations that allowed a reliable description of weak, dispersive interactions.53
On the other hand, periodic calculations were performed with PBEsol0 functional which is a PBE0 revised specifically for solids.54,55 Both PBEsol and PBEsol0 were proven to correctly describe unit cell parameters as well as intermolecular interactions in solids.56,57
1H NMR spectroscopy
The 1H NMR spectra for 5 mg of L-Ser, L-Tyr, L-Trp, L-Lys, L-His and L-Arg, a mixture of VA and AA in 1:1 ratio (5 mg of VA, 3 mg of L-Ser, 5 mg of L-Tyr, 6 mg of L-Trp, 4 mg of L-His, 4 mg of L-Lys and 5 mg of L-Arg), and isolated 5 mg of VAHis and VALys salts and VATrp and VASer co-crystals in 1 mL D2O were recorded using a Bruker Avance III 600 at 300 K. The 1H NMR spectra were referenced internally to the residual proton resonance in D2O.
UV-Vis spectroscopy
All UV-Vis experiments were carried out using a PerkinElmer LAMBDA 365 Spectrophotometer at room temperature. Grinded solid-state samples of VASer, VATyr, VATrp, VALys and VAHis were mixed with barium sulphate, and spectra were registered using the 50 mm transmission/reflectance sphere whereas UV-Vis spectra for solutions were recorded in 1 cm quartz cells. The titration experiment for VAHis, VALys, and VAArg salts was performed as follows: the stock 2 mL 0.01 M solution of a particular amino acid (L-His, L-Lys, L-Arg) was titrated with 20 μL of VA solution (0.05 M) and the spectrum was recorded after each addition. The total number of added equivalents for each salt was 50.Author contributions
This publication was written with contributions from all authors.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for all the mentioned compounds has been deposited at the CCDC under CCDC deposition numbers 2008004–2008008.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out with equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract No. POIG.02.01.00–12-023/08) and the Ministry of Science and Higher Education, Warsaw, Poland (grant number 6903/IA/SP/2018). This research was supported by the National Science Centre, grant number 2018/30/E/ST5/00638. We gratefully acknowledge Polish high-performance computing infrastructure PLGrid (HPC Center: ACK Cyfronet AGH) for providing computer facilities and support within computational grant no. PLG/2023/016132.Notes and references
- A. Buryak and K. Severin, J. Am. Chem. Soc., 2005, 127, 3700–3701 CrossRef CAS PubMed.
- F. Sancenón, A. B. Descalzo, R. Martínez-Máñez, M. A. Miranda and J. Soto, Angew. Chem., Int. Ed., 2001, 40, 2640–2643 CrossRef.
- T. Minami, N. A. Esipenko, B. Zhang, L. Isaacs and P. Anzenbacher, Chem. Commun., 2014, 50, 61–63 RSC.
- H. Y. Fu, X. J. Liu and M. Xia, RSC Adv., 2017, 7, 50720–50728 RSC.
- Z. Zhao, H. Nie, C. Ge, Y. Cai, Y. Xiong, J. Qi, W. Wu, R. T. K. Kwok, X. Gao, A. Qin, J. W. Y. Lam and B. Z. Tang, Adv. Sci., 2017, 4, 1–8 Search PubMed.
- M. De Bastiani, M. I. Saidaminov, I. Dursun, L. Sinatra, W. Peng, U. Buttner, O. F. Mohammed and O. M. Bakr, Chem. Mater., 2017, 29, 3367–3370 CrossRef CAS.
- P. Bamfield and M. Hutchings, Chromic Phenomena, RSC, 3rd edn, 2018 Search PubMed.
- N. Bar and P. Chowdhury, ACS Appl. Electron. Mater., 2022, 4, 3749–3771 CrossRef CAS.
- M. Martínez-Junquera, E. Lalinde and M. T. Moreno, Inorg. Chem., 2023, 62, 11849–11868 CrossRef PubMed.
- F. Robert, P. L. Jacquemin, B. Tinant and Y. Garcia, CrystEngComm, 2012, 14, 4396–4406 RSC.
- D. A. Safin, M. Bolte and Y. Garcia, CrystEngComm, 2014, 16, 5524–5526 RSC.
- M. Morimoto, R. Kashihara, K. Mutoh, Y. Kobayashi, J. Abe, H. Sotome, S. Ito, H. Miyasaka and M. Irie, CrystEngComm, 2016, 18, 7241–7248 RSC.
- R. Hagihara, T. Umeno, S. Ueki, D. Yoshihara, Y. Fuchi, K. Usui, M. Sakuma, K. Ichi Yamada and S. Karasawa, Chem. – Eur. J., 2021, 27, 3039–3046 CrossRef CAS PubMed.
- R. Tan, Q. Lin, Y. Wen, S. Xiao, S. Wang, R. Zhang and T. Yi, CrystEngComm, 2015, 17, 6674–6680 RSC.
- K. Golka, S. Kopps and Z. W. Myslak, Toxicol. Lett., 2004, 151, 203–210 CrossRef CAS PubMed.
- B. Lellis, C. Z. Fávaro-Polonio, J. A. Pamphile and J. C. Polonio, Biotechnol. Res. Innov., 2019, 3, 275–290 CrossRef.
- K. Shiraki, N. Takata, R. Takano, Y. Hayashi and K. Terada, Pharm. Res., 2008, 25, 2581–2592 CrossRef CAS PubMed.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Int. J. Pharm., 2006, 320, 114–123 CrossRef CAS.
- J. R. Wang, X. Yu, C. Zhou, Y. Lin, C. Chen, G. Pan and X. Mei, Bioorganic Med. Chem. Lett., 2015, 25, 1036–1039 CrossRef CAS.
- S. Golob, M. Perry, M. Lusi, M. R. Chierotti, I. Grabnar, L. Lassiani, D. Voinovich and M. J. Zaworotko, J. Pharm. Sci., 2016, 105, 3626–3633 CrossRef CAS.
- X. L. Dai, J. Yao, C. Wu, J. H. Deng, Y. H. Mo, T. B. Lu and J. M. Chen, Cryst. Growth Des., 2020, 20, 5160–5168 CrossRef CAS.
- J. Wojnarska, M. Gryl, T. Seidler, A. Rydz, M. Oszajca, K. M. Stadnicka, M. Marzec, I. Matulková, I. Němec and P. Němec, Cryst. Growth Des., 2019, 19, 6831–6836 CrossRef CAS.
- M. Gryl, T. Seidler, J. Wojnarska, K. Stadnicka, I. Matulková, I. Němec and P. Němec, Chem. – Eur. J., 2018, 24, 8727–8731 CrossRef CAS PubMed.
- E. D. D’Silva, G. K. Podagatlapalli, S. V. Rao, D. N. Rao and S. M. Dharmaprakash, Cryst. Growth Des., 2011, 11, 5362–5369 CrossRef.
- B. Lu, Y. Zhang, X. Yang, K. Wang, B. Zou and D. Yan, J. Mater. Chem. C, 2018, 6, 9660–9666 RSC.
- L. Sun, W. Zhu, X. Zhang, L. Li, H. Dong and W. Hu, J. Am. Chem. Soc., 2021, 143, 19243–19256 CrossRef CAS PubMed.
- M. Gryl, A. Rydz, J. Wojnarska, A. Krawczuk, M. Kozieł, T. Seidler, K. Ostrowska, M. Marzec and K. M. Stadnicka, IUCrJ, 2019, 6, 226–237 CrossRef CAS PubMed.
- R. M. Awadallah, A. A. M. Belal, R. M. Issa and R. D. Peacock, Spectrochim. Acta, 1994, 47A, 1541–1546 Search PubMed.
- M. Gryl, M. Kozieł and K. M. Stadnicka, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 53–58 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D’Arco, M. Llunel, M. Causà, Y. Noël, L. Maschio, A. Erba, M. Rérat and S. Casassa, CRYSTAL17 User's Man., 2018, 211.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8, 1–36 CAS.
- C. Gatti and S. Casassa, TOPOND14 User's Manual, CNR- ISTM, Milano, Italy, 2013.
- A. J. Cruz-Cabeza, CrystEngComm, 2012, 14, 6362–6365 RSC.
- D. R. Lide, Handbook of Chemistry and Physics, CRC Press, 85th edn, 2004–2005 Search PubMed.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B, 1990, 46, 256–262 CrossRef PubMed.
- C. Janiak, J. Chem. Soc. Dalton Trans., 2000, 3885–3896 RSC.
- J. M. Bosque-Sendra, E. Almansa-Lopez, A. M. Garcia-Campana and L. Cuadros-Rodriguez, Anal. Sci., 2003, 19, 1431–1439 CrossRef CAS PubMed.
- A. A. Altaf, U. Hashmat, M. Yousaf, B. Lal, S. Ullah, A. A. Holder and A. Badshah, R. Soc. Open Sci., 2016, 3(11), 1–12, DOI:10.1098/rsos.160351.
- R. Ghosh and B. Singh, Indian J. Chem., Sect. A: Inorg., Phys., Theor. Anal., 1980, 19, 1102–1105 Search PubMed.
- J. J. McKinnon, D. Jayatilaka and M. Spackman, Chem. Commun., 2007, 3814 RSC.
- R. F. W. Bader, Preface, Oxford Clarendon Press, vol. 22, 2003 Search PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct, Chem., 2015, 71, 3–8 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao, N. E. Schultz and D. G. J. Truhlar, Chem. Phys., 2005, 123, 161103 Search PubMed.
- Y. Zhao, N. E. Schultz and D. G. J. Truhlar, Chem. Theory Comput., 2006, 2, 364–382 CrossRef PubMed.
- Y. Zhao and D. G. J. Truhlar, Chem. Phys., 2006, 125, 194101 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- E. G. Hohenstein, S. T. Chill and C. D. Sherrill, J. Chem. Theory Comput., 2008, 4(12), 1996–2000 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2007, 3(1), 289–300 CrossRef CAS PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100(13), 136406 CrossRef PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110(13), 6158–6170 CrossRef CAS.
- A. V. Terentjev, L. A. Constantin and J. M. Pitarke, Phys. Rev. B, 2018, 98, 214108 CrossRef CAS.
- J. Quertinmont, A. Carletta, N. A. Tumanov, T. Leyssens, J. Wouters and B. Champagne, J. Phys. Chem. C, 2017, 121, 6898–6908 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Summarized data of crystal structures included in the CSD search; crystal engineering procedures, crystal data and refinement details for studied crystalline materials; geometry of hydrogen bonds and π–π interactions observed in the studied crystals; 1H NMR data for the crystallisation solutions, their mixtures and crystallised dissolved products; details of UV-Vis measurements and titration studies; details of LoD calculations; and details of topological analysis of electron density. CCDC 2008004–2008008. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc03209h |
| This journal is © The Royal Society of Chemistry 2024 |