
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of counterion tether length on stability, work function and application of a self-compensated, hole-doped triarylamine-alt-fluorene model polymer†
Qi-Mian
Koh
a,
Kevin Christopher
Boellaard
a,
Yu
Wang
a,
Cindy G.
Tang
b,
Qiu-Jing
Seah
a,
Peter. K. H.
Ho
b,
Rui-Qi
Png
 *b and
Lay-Lay
Chua
*ab
*b and
Lay-Lay
Chua
*ab
aDepartment of Chemistry, National University of Singapore, Lower Kent Ridge Road, Singapore, S117552, Singapore. E-mail: chmcll@nus.edu.sg
bDepartment of Physics, National University of Singapore, Lower Kent Ridge Road, Singapore, S117550, Singapore. E-mail: phypngrq@nus.edu.sg
First published on 30th October 2024
Abstract
Self-compensated, hole-doped polymers with ultrahigh workfunction can provide ohmic hole contacts even for deep-ionization-energy semiconductors. The self-compensation is usually imposed by an anion tethered to a flexible –(CH2)n– chain, but the effect of tether length n is unknown. Here, using the mTFF triarylamine–fluorene as a model semiconductor, we have designed, synthesized and characterized a family of mTFF-CnSISC2F5 polymers, where C2F5SIS is the pentafluoroethanesulfonylimidosulfonyl anion, tethered to a Cn alkylene chain that is systematically varied between C2 and C6 in length. Solution doping yields self-compensated films with a work function of ca. 5.75 eV, which is ca. 0.15 eV higher than that obtained by film doping and higher than that of the corresponding hole-doped mTFF films counterbalanced by SbF6− anions. We attribute the higher work function to a frustrated packing of the counteranions about the holes. The ultrahigh work function is consistent with electroabsorption measurements and the ability of the films to inject holes into PFOP, a model deep-ionization-energy semiconductor, without bias pre-conditioning. While the tether length only weakly influences the work function, it strongly influences hygroscopicity, processability, and thermal stability of the hole-doped polymers. OPLS4 molecular dynamics simulations suggest that short tether lengths (C2 and C3) result in interchain charge compensation, but medium and long tether lengths (C4 and C6) result in mixed interchain/intrachain charge compensation. Overall, the C3 tether provides the best thermal and ambient stability. These results reveal new aspects regarding the role of tether length in self-compensated, charge-doped polymers.
Introduction
Optimization of the performance of organic semiconductor (OSC) devices, such as organic light-emitting diodes and solar cells, requires tailoring the work function of charge injection or collection layers. This is significantly aided by the recent development of self-compensated (SC) charge-doped polymers of both charge signs, where the free carriers on the polymer backbone are counterbalanced by tethered ions.1,2 This self-compensation stabilizes the doping level, preventing dopant migration and thus enabling precise control of the doping profile in the device.1,2 Furthermore, these materials offer tunable work functions spanning an ultrawide range (2.5 to 6.0 eV) through the span of ionization energies (IE) and electron affinities (EA) available by chemical design, further fine-tuned through the local Madelung potential effect by counterion and spectator-ion selection. This immense flexibility contrasts with traditional “self-doped” polymers that are limited to materials with low IE for self-hole-doping, or high EA for self-electron-doping.3–6 The versatility of SC polymers is associated with separation of the charge-doping and charge-compensation processes. The polymer is doped by electron transfer with strong oxidants or reductants, and then compensated by association with tethered counterions by elimination of the dopant by-product. This approach has enabled the fabrication of high-performance solution-processed devices across various semiconductors,7,8 including blue thermally activated delayed fluorescence light-emitting diodes,9–11 perovskites,12 and quantum dots.13Using hole-doped triarylamine-alt-fluorene (TAF) copolymers as model systems,14 the influence of various factors on the properties of SC polymers has been investigated.1,15,16 These factors include the counteranion (e.g., trifluoromethanesulfonylimidosulfonyl (CF3SIS) vs. pentafluoroethanesulfonylimidosulfonyl (C2F5SIS)) and spectator cation (e.g., Li+vs. Cs+). Larger counteranions lead to weaker coulombic attraction of the hole carriers but stronger ion surface layering, which raises work function, while smaller spectator cations lead to stronger coulombic repulsion of the hole carriers, which also raises work function.1,15,16 However, one critical aspect remains unexplored: the impact of length of the –(CH2)n– flexible chain that tethers the counterion to the polymer backbone. We expect that short tethers would lead to interchain charge compensation, where the carrier on the polymer backbone is charge-balanced primarily by a counterion tethered to the adjacent polymer backbone, while long tethers would lead to mixed interchain/intrachain charge compensation, where the carrier is charge-balanced by a counterion tethered to the same or adjacent polymer backbone. This may affect ion cluster morphology and associated properties in non-trivial ways. Moreover, increasing the value of n increases the hydrophobic volume of the polymer and dilutes the semiconductor core fraction, which may also have unexpected effects.
In this work, we elucidate some of these effects, demonstrating that the tether length influences not only hygroscopicity and processability, but also stability of the SC hole-doped state. We studied poly[9,9-bis(ω-pentafluoroethanesulfonylimidosulfonylalkyl)fluorene-2,7-diyl-alt-1,4-phenylene-N-(m-trifluoromethylphenyl)imino-1,4-phenylene] with different tether lengths, denoted mTFF-CnSISC2F5, where n = 2, 3, 4 and 6, with sodium as the spectator cation. The spectator cation, together with doped hole if any, provides charge balance to the tethered anion. As a member of TAF polymers, mTFF exhibits non-dispersive and well-resolved frontier π-electron bands,14 with relatively well-characterised ion clusters and surface layering behaviors,15,16 and relatively well-understood electronic transition and transport properties.17 Therefore, mTFF provides an excellent model to investigate the effects of tether length here. Our selection of C2F5SIS as the anion is driven by its stability18 and low hygroscopicity.19 Our selection of Na+ as the spectator cation is driven by its favourable effect on work function15 and processability.16
Results and discussion
Synthesis
The mTFF-CnSISC2F5 polymers were synthesized by microwave Suzuki coupling of the 9,9-bis(ω-pentafluoroethanesulfonylimidosulfonylalkyl)-2,7-dibromofluorene (diBr-FCnSISC2F5) monomer and the 4,4′-N-(m-trifluoromethylphenyl)iminobis(phenylboronic acid pinacol ester) (mTF-diEs) monomer to give the respective polymers following the procedure reported in Tang et al.1 and Ang et al.16Fig. 1 shows the generic chemical structure of the polymers. | ||
| Fig. 1 Chemical structure of the self-compensated, hole-doped mTFF-CnSISC2F5 polymer. n denotes the number of CH2 methylene units in the side-chain spacer (2, 3, 4 or 6). y denotes the doping level, in fraction of hole per repeat unit, where 0 represents the undoped polymer and 1 represents the fully hole-doped polymer. | ||
Attaching the C2F5SIS anion to the fluorene moiety requires bespoke approaches for each tether length. For propyl and butyl tethers, syntheses of diBr-FC3SO3Na and diBr-FC4SO3Na were achieved by base-promoted 9-deprotonation of 2,7-dibromofluorene, followed by nucleophilic attack by the fluorene anion on the electrophilic carbon adjacent to the oxygen atom of commercially available sultones, i.e. 1,3-propanesultone and 1,4-butanesultone, to give diBr-FC3SO3Na and diBr-FC4SO3Na, respectively (Scheme 1a). However, β-sultone is unstable,20 while 1,6-sultone is not commercially available. Hence, the syntheses of C2 and C6 tethered anions were achieved by different pathways. For diBr-FC6SO3Na, the fluorene anion was reacted with 1,6-dibromohexane to give 9,9′-bis(bromohexyl)-2,7-dibromofluorene, and the tethered bromine then converted to iodine via Finkelstein reaction to provide a better leaving group for nucleophilic substitution reaction with sodium metabisulfite (Scheme 1b). This reaction could not be used for diBr-FC2SO3Na due to facile competitive deprotonation of 1,2-dibromoethane to bromoethene. Instead, the monomer was synthesized by nucleophilic substitution on methyl bromoacetate by the fluorene anion, followed by reduction to the hydroxyethyl moiety,21 substitution with iodide, then nucleophilic displacement with thiourea to yield isothiouronium, followed by acidification to cleave to hydrosulfide, and oxidation of the resultant diBr-FC2SH by performic acid to diBr-FC2SO3H (Scheme 1c). This reaction is adapted from the synthesis of amino alkyl sulfonic acids.22
 | ||
| Scheme 1 (a) Synthesis path for diBr-FCnSO3H. 1,3-Propanesultone is used for n = 3; 1,4-butanesultone is used for n = 4. (b) Synthesis path for diBr-FC6SO3H. (c) Synthesis path for the diBr-FC2SO3H monomer. | ||
In general, the ω-sulfonic acid monomers were subsequently converted to the corresponding C2F5SIS-tethered monomers, and polymerized with the diEs-mTFF monomer to give the mTFF-CnSISC2F5 polymers (see Experimental). Elemental analyses of the polymers gave satisfactory results (elemental ratio, ±5 atom%). The number-average molar masses by gel permeation chromatography were determined to be 26, 23, 14 and 19 kDa for n = 2, 3, 4 and 6, respectively. Thus, sufficiently long polymers were obtained with more or less similar molar weight.
Solution processability
The undoped mTFF-CnSISC2F5 polymers are generally soluble in polar organic solvents, such as acetonitrile (ACN), tetrahydrofuran (THF), nitromethane (NM), propylene carbonate (PC), dimethylformamide (DMF) and dimethylsulfoxide (DMSO). Thus, the variation in tether length over the selected range does not significantly alter the solubility characteristics of the polymers.Hygroscopicity
The undoped polymers were evaluated using a “segmented thermogravimetry” protocol with repeated measurements, as described in ref. 19. The thermograms are shown in Fig. 2. They exhibit three distinct weight loss steps, confirming the presence of three types of sorbed water as expected:19 type-I water desorbing below 35 °C, type-II water between 35 °C and 130 °C, and type-III water (present only in the first scan) between 130 °C and 230 °C. These water species correspond to hydrogen-bonded water in the secondary hydration shell, anion-bound water in the primary hydration shell – located at the surface of the ion clusters – and caged water, respectively.19 The hydration number for type-I water was found to be nw ≈ 1.0 H2O per ion pair, independent of tether length (data taken from the second scan), but this is a significant underestimate.19 In contrast, the hydration number for type-II water decreases systematically with tether length, that is, nw decreases from 0.54 to 0.15 H2O per ion pair as the tether length increases from n = 2 to 6. The hygroscopicity of a polyelectrolyte depends not only on the nature of the ion pair, but also on the morphology of the ion cluster formed by the ion pair.19 The observed trend here suggests that the effective surface area per ion pair in the ion cluster decreases as its tether becomes longer. This indicates that the ions can assemble into larger aggregates with increasing tether length.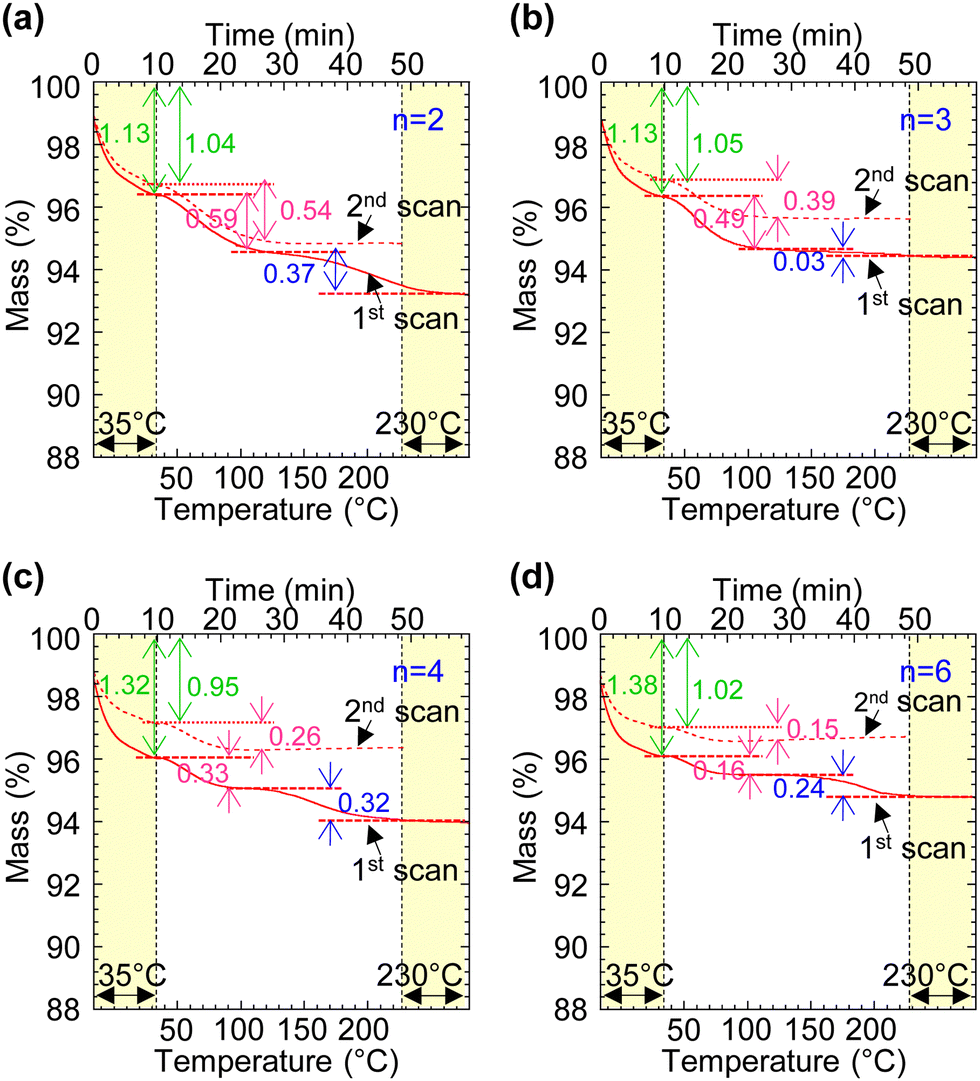 | ||
| Fig. 2 Thermogravimetry analysis of mTFF-CnSISC2F5 powders. (a) n = 2, (b) 3, (c) 4, and (d) 6. After the first scan (solid line), the sample was cooled to room temperature, equilibrated under ambient conditions for 2 h, and reloaded into the thermoanalyzer for a second scan (dashed line). Conditions: ramp rate, 5 K min−1; atmosphere, N2. The weight loss steps correspond to water desorption: type-I water (below 35 °C, green annotation), type-II water (35–130 °C, magenta), and type-III water (130–230 °C, blue). The corresponding hydration number is annotated in the plot (nw in H2O per ion pair). Note that nw for type-I H2O content is significantly underestimated due to rapid desorption in N2 during the thermoanalyzer dead time. | ||
Hole doping
The hole-doped polymers were generated by quantitative oxidation with nitrosonium hexafluoroantimonate (NOSbF6) or tris(p-bromophenyl)aminium hexachloroantimonate (TBPASbCl6; also known as “Magic Blue”) in a nitrogen glovebox. We call this the “solution doping” method. Both NO+ and TBPA˙+ are strong one-electron oxidants (ε° (vs. SCE) in ACN: NO+/NO˙, 1.26 V; TBPA˙+/TBPA, 1.06 V). All the hole-doped polymers, except mTFF-C6SISC2F5, are soluble in polar solvents, including ACN, NM and PC, which have sufficiently high oxidation potentials to be compatible with the doped polymers. However, hole-doped mTFF-C6SISC2F5 undergoes gelation when oxidized by 0.7 equivalent or more of NOSbF6, which cannot be overcome by dilution or heating. We speculate that this gelation arises from intrachain charge compensation, which binds the C2F5SIS anion to the doped hole, raising the overall hydrophobicity of the polymer. Thus, near-saturation hole-doped mTFF-C6SISC2F5, i.e. with a doping level (DL) near to the maximum value of 1 hole per repeat unit (h+ per r.u.), cannot be obtained by solution doping. In contrast, near-saturation hole-doped mTFF-CnSISC2F5 can be readily obtained for n between 2 and 4 inclusive. Their SC forms were then generated through purification by repeated precipitation–redissolution cycles, during which the DL of the polymer drops marginally.The near-saturation hole-doped state can also be generated by a different approach called the “film doping” method. This method involves contacting a cast film of the undoped polyelectrolyte with a dilute solution of the dopant, e.g., 0.4 mM TBPASbCl6 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ACN:dimethyl carbonate (DMC), in a nitrogen glovebox. DMC is a nonsolvent for both undoped and hole-doped states of the polymer. Nevertheless, the presence of ACN in the solvent mixture is sufficient to swell the film, facilitating chemical injection of holes and in-diffusion of counteranions, i.e., SbCl6−. The SC forms were then generated through purification by a spin-rinse cycle to remove the excess counteranions and spectator cations as NaSbCl6, during which the DL of the polymer also drops marginally.
:10 ACN:dimethyl carbonate (DMC), in a nitrogen glovebox. DMC is a nonsolvent for both undoped and hole-doped states of the polymer. Nevertheless, the presence of ACN in the solvent mixture is sufficient to swell the film, facilitating chemical injection of holes and in-diffusion of counteranions, i.e., SbCl6−. The SC forms were then generated through purification by a spin-rinse cycle to remove the excess counteranions and spectator cations as NaSbCl6, during which the DL of the polymer also drops marginally.
Electronic spectroscopy
We compared the UV-Vis-NIR spectra of the polymer films to evaluate their DL and whether the tether length influences disorder in the film (Fig. 3). Doping of TAF polymers leads to bleaching of the N1 and N2 absorption bands of the neutral polymer segments, and formation of the P1, P2 and P3 polaron absorption bands. The one-electron excitations that contribute to these transitions have been reported in ref. 17. The results here show that increasing tether length induces a small blueshift of the N1 absorption band (at ca. 3.3 eV), which is a HOMO → LUMO excitation of the neutral polymer (i.e., undoped) segments, from the triphenylamine unit to the flanking fluorene units. The blueshift is ca. 100 meV from C2 to C6. In contrast, the blueshift is only 10–20 meV when dioctyl side chains on the fluorene unit are substituted by C3SISCF3M side chains.17 Thus, the larger blueshift here cannot be attributed solely to further changes in polarizability. Instead, it is attributed to assembly of the C2F5SIS− M+ ion pairs into larger clusters with tether lengthening, which causes stronger twisting of the polymer backbone, i.e., increasing its dihedral angles.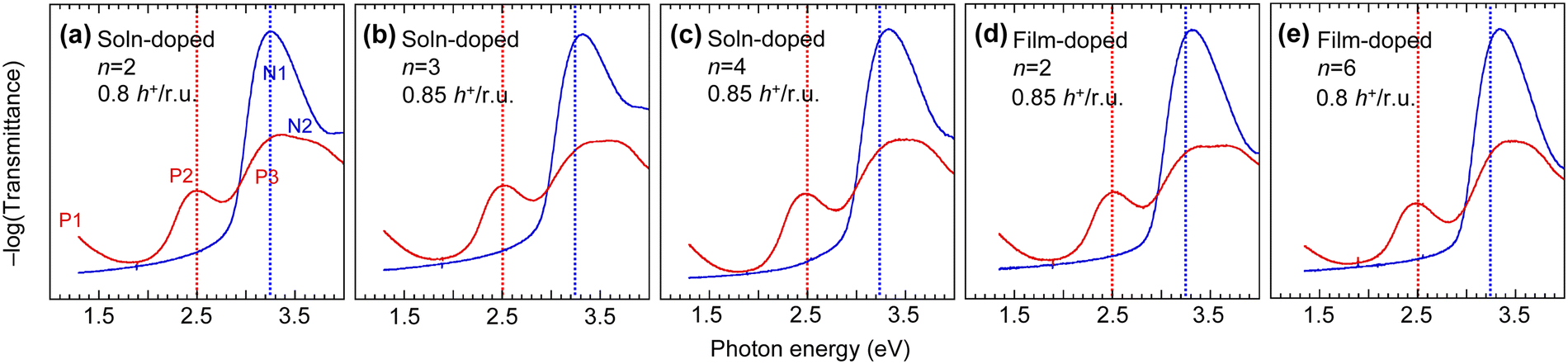 | ||
| Fig. 3 Electronic spectroscopy of mTFF-CnSISC2F5 films in the neutral and nearly saturation hole-doped state. Solution-doped films: (a) n = 2, (b) 3, and (c) 4; and film-doped films: (d) n = 2 and (e) 6. Spectra: (blue) neutral, (red) hole-doped, with doping level in hole per repeat unit annotated. Vertical dotted lines as guide-to-the-eye: (blue) 3.25 eV and (red) 2.50 eV, aligned to peak centres in (a). 25–35-nm-thick films were prepared by spin-casting on O2-plasma-cleaned quartz substrates in a nitrogen glovebox. | ||
The P2 polaron absorption band (at ca. 2.5 eV) corresponds to a HOMO → LUMO excitation of the hole-doped segments, admixed with HOMO−m → SOMO* excitations.17 This band redshifts ca. 30 meV from C4 to C6, which suggests that the conformational order of the hole-doped polymer backbone improves with tether length. The DL can be accurately evaluated from the intensities of the N1 and P2 bands relative to those of the undoped film and an authentic fully doped film (without the purification step) as ref. 17. Thus, the SC hole-doped films were found to exhibit a DL of 0.80–0.85 h+ per r.u. The polaron spectra are practically identical for solution-doping and film-doping methods for the same polymer. Thus, the electronic state does not strongly depend on the method of doping.
Atomic force microscopy analysis reveals that both solution-doped and film-doped samples exhibit similar surface morphologies (Fig. S1, ESI†). The surfaces are smooth, with a root-mean-square roughness of ca. 10 Å. Thus, both solution-doping and film-doping methods can produce high-quality doped films.
Surface composition
We characterized the surface compositions of the films by X-ray photoemission spectroscopy (Fig. 4). This allows evaluation of the chemical species and DL present at the film surface. The atomic stoichiometries of the undoped films are in good agreement with theory (±10 atom%), except for carbon, which is high by ca. 20% (Table 1). The shapes of the C1s, S2p and F1s spectra do not change significantly with tether length. The enhanced detection of carbon indicates that the probed surface layer (ca. 25 Å deep) is selectively enriched with the polymer backbone relative to the C2F5SIS anion.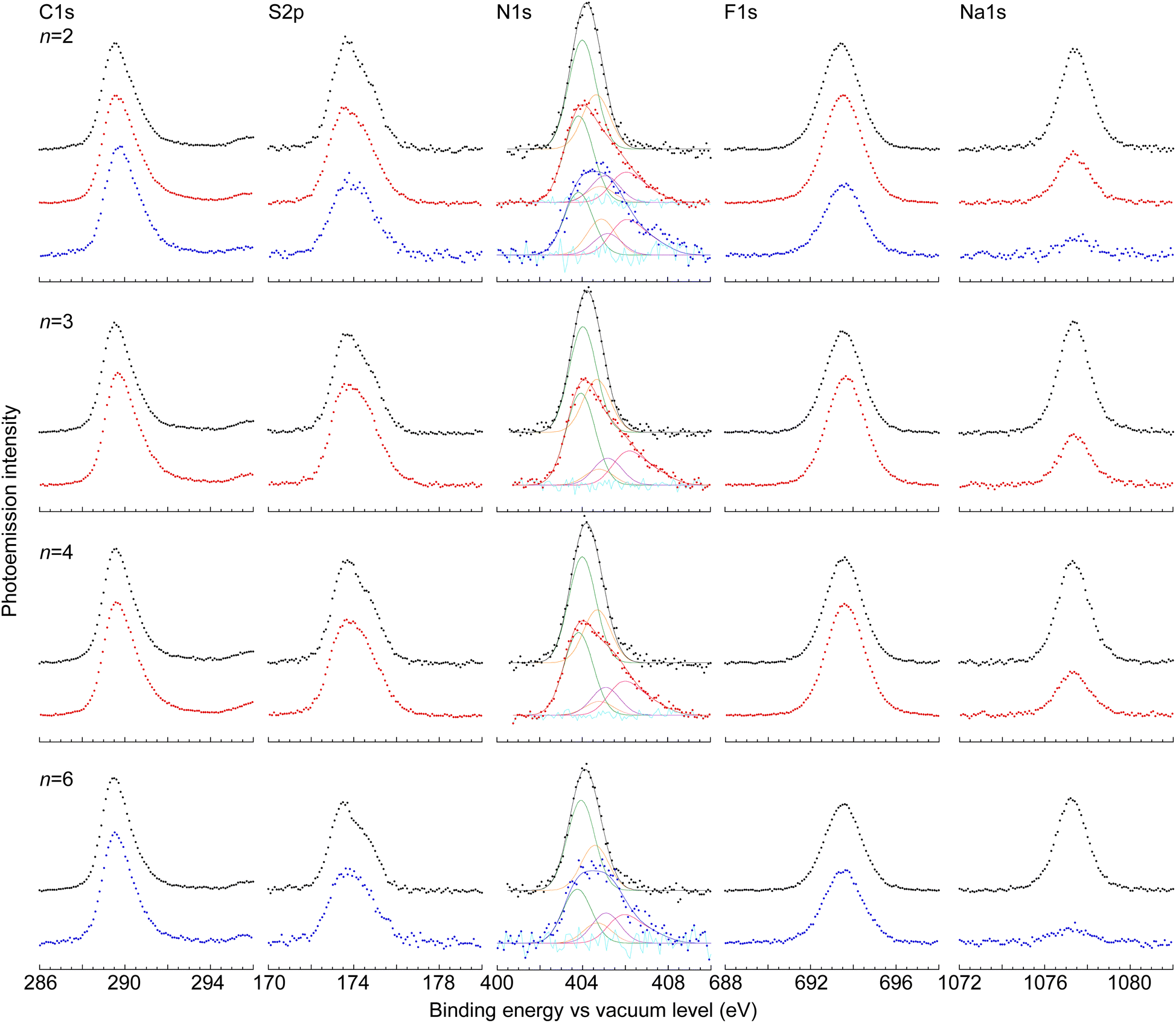 | ||
| Fig. 4 Core-level X-ray photoemission spectra of undoped and self-compensated, saturation-hole-doped mTFF-CnSISC2F5 films. C1s, S2p, N1s, F1s and Na1s spectra plotted against binding energy measured from the vacuum level, for n = 2 (top panel), 3, 4, and 6 (bottom panel). Legend: circles, data – (black) undoped polymer, (red) solution-doped polymer, (blue) film-doped polymer. The N1s spectra were self-consistently fitted with components representing different species in the surface layer of the film: (green) C2F5SIS anion, (magenta) protonated C2F5SIS, (violet) amine, and (magenta) aminium moieties. | ||
:1, as theoretically expected. N(+1) is triarylamininium, fitted with an asymmetric bandshape function with tailing towards higher binding energy, defined by a fundamental component at 405.85 eV and two higher components at 0.9 eV (45% of fundamental) and 1.8 eV (25% of fundamental). N(0) is triarylamine, at 404.8 eV. N(−1) is C2F5SIS anion, at 403.8 eV. NH is protonated C2F5SIS, at 405.1 eV. The doping level is 75–80%, slightly lower than that in the bulk. Film-doped films bind TBPA˙+ to the surface, which contributes ‘excess’ intensity to N(+1) and N(0) components, the latter due to inadvertent reduction. Atomic stoichiometry ratios are normalized to S2p = 4.0. Theoretical ratios for the undoped polymers are C:S:F:Na:N = m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :4:13:2:3, where m = 40, 42, 44, and 48, for n = 2, 3, 4, and 6, respectively
:4:13:2:3, where m = 40, 42, 44, and 48, for n = 2, 3, 4, and 6, respectively
| n | C1s | S2p | F1s | Na1s | N1s | N1s: C2F5SIS− | N1s: amine | ||
|---|---|---|---|---|---|---|---|---|---|
| N(−1)a | NHb | N(0)c | N(+1)d | ||||||
| Footnotes: relative binding energies.a N(−1) BE, ca. 403.8 eV.b NH BE, ca. 405.1 eV.c N(0) BE, ca. 404.8 eV.d N(+1) BE, ca. 405.85 eV. | |||||||||
| Undoped | |||||||||
| 2 | 49 | 4.0 | 12 | 2.0 | 2.8 | 1.8 | — | 0.9 | — |
| 3 | 51 | 4.0 | 12 | 2.3 | 2.9 | 1.9 | — | 0.95 | — |
| 4 | 52 | 4.0 | 12 | 2.0 | 2.8 | 1.8 | — | 0.9 | — |
| 6 | 58 | 4.0 | 12 | 2.2 | 3.0 | 1.9 | — | 0.95 | — |
| Solution-doped | |||||||||
| 2 | 49 | 4.0 | 12 | 1.0 | 3.0 | 1.55 | 0.45 | 0.25 | 0.75 |
| 3 | 51 | 4.0 | 11 | 0.8 | 2.9 | 1.45 | 0.4 | 0.25 | 0.75 |
| 4 | 52 | 4.0 | 12 | 0.8 | 2.9 | 1.45 | 0.45 | 0.25 | 0.8 |
| Film-doped | |||||||||
| 2 | 52 | 4.0 | 10 | 0.25 | 3.9 | 1.45 | 0.5 | 0.8 | 1.15 |
| 6 | 57 | 4.0 | 11 | 0.4 | 3.4 | 1.3 | 0.7 | 0.45 | 0.95 |
The N1s peak of the C2F5SIS anion emerges at ca. 404.15 eV in the undoped film, and that of the triphenylamine moiety at 404.6 eV. Doping broadens the N1s envelope: the C2F5SIS anion peak downshifts to 403.8 eV, the amine peak upshifts to 404.8 eV, and a new peak emerges at a higher binding energy of 405.85 eV, with strong tailing towards even higher binding energy. This last species is characteristic of the triphenylaminium moiety. Curve-fitting reveals an additional fourth component at 405.1 eV, which we assign to the protonated C2F5SIS species. Thus, we deconvolute the N1s spectrum into four components, corresponding to the C2F5SIS anion (denoted “N(−1)”), protonated C2F5SIS (“NH”), amine (“N(0)”) and aminium (“N(+1)”). The area ratio of the curve-fitted aminium to the total amine and aminium, i.e., [N(+1)]/([N(+1)] + [N(0)]), gives the DL at the film surface. Thus, the SC hole-doped films were found to exhibit a DL of 0.75–0.80 h+ per r.u. at their surfaces, which is marginally lower than the DL in the bulk of the film.
XPS reveals a significant difference between the solution-doped and film-doped samples. While solution doping and self-compensation reduce the amount of Na+ to half of its original value, as expected, film doping and self-compensation reduce the amount much more to 10–20% of its original value. Furthermore, excess amine and aminium species emerge (Table 1). These results indicate the displacement of Na+ by TBPA˙+ at the film surface. The surface binding of TBPA˙+ to polyelectrolyte films has previously been established and utilized to introduce dopants for monolayer doping of organic semiconductor interfaces.23 This TBPA˙+ is expected to oxidize moisture over time, releasing H+ to protonate the C2F5SIS anion. The products of this reaction are detected, TBPA and SO2NHSO2C2F5 as N(0) and NH, respectively. Thus, while both solution doping and film doping can yield high quality hole-doped films, the latter tends to bind excess oxidant to the film surface.
Vacuum work function
We characterized the work function φ and valence band structure of the films using ultraviolet photoemission spectroscopy. The spectra of the undoped films show the HOMO band, which arises from the triphenylamine units, and the HOMO−1 band, which arises from the fluorene units (Fig. 5a).14,17 The work function is given by φ = Ek,LECO + hv − Ek,FL, where hv is the photon energy (He I, 21.22 eV), and Ek,FL and Ek,LECO are the kinetic energies of the photoelectrons emitted from the Fermi level (FL) established by a sputtered-clean Au reference and at the low-energy cutoff (LECO) of the film, respectively. | ||
| Fig. 5 Ultraviolet photoemission spectra of undoped and self-compensation, near-saturation hole-doped mTFF-CnSISC2F5 films. (a) Undoped, (b) solution-doped and (c) film-doped. The inset in (b) compares the solution-doped with a film-doped C2 sample fabricated in the same run. The film-doped sample shows the VB downshifts (i.e., away from the vacuum level) by 0.05 eV but the Fermi level (EF) upshifts by 0.2 eV. (d) Plot of work function against tether length. The doping level is 0.75–0.8 h+ per r.u. for all films. Work functions of near-saturation hole-doped mTFF counterbalanced by BArF and SbF6 are also shown for reference. | ||
Hole doping bleaches out the HOMO band and deepens the Fermi level (Fig. 5b). Thus, the solution-doped SC films exhibit φ values of ca. 5.75 eV, which show a weak increasing trend from C2 to C4. These φ values are midway between those of mTFF:SbF6, counterbalanced by the small SbF6− (3.35-Å radius), and mTFF:BArF, counterbalanced by the large tetrakis(3,5-bis(trifluoromethyl)phenyl)borate anion (8-Å radius) (Fig. 5d).14 In general, φ increases weakly with DL in the heavily doped regime, but strongly with counteranion size due to loss of Madelung stabilization.14 The tethered C2F5SIS anion here contacts the hole on the triphenylaminium unit over a distribution of distances between 4.5 and 6.5 Å (vide infra) due to its shape, orientation and tether, which gives apparent radii between 3.2 and 5.2 Å. This explains the enhanced φ values.
On the other hand, the film-doped SC films exhibit significantly lower φ values of ca. 5.55 eV. The shape and position of their valence band are practically identical to those of the solution-doped films, but their Fermi level is ca. 0.2 eV shallower (inset, Fig. 5b). This rules out any loss of doping or formation of unfavourable dipole at the surface, but indicates closer packing of counteranions to give stronger stabilisation of the holes (Fig. 5c). Thus, the counterions seem to relax more completely about the holes for film doping than solution doping. Yet, the lack of φ dependence on tether length suggests that the packing of these counterions does not change markedly with tether length within each doping method (Fig. 5d).
Molecular modeling: isolated repeat units
To investigate the charge-compensation structure of the counterions theoretically, we computed the PM3 molecular model of the hole-doped polymer repeat unit in the gas phase (Fig. 6; bond lengths and angles in Table 2). These calculations reveal that the tethered CnSISC2F5 anions position nearer to one edge of the polymer backbone due to their 9,9-fluorene attachment. This would enhance the visibility of the polymer backbone to XPS, as found experimentally. The calculations also reveal that the C2 and C3 tethers are not long enough for the C2F5SIS imide nitrogen to contact the triphenylaminium unit, but the C6 tether is long enough, i.e., the N(+1)⋯N(−1) distance decreases to ca. 5 Å, depending on the conformation of the tether. Thus, intrachain charge compensation, where the counterion is tethered to the same polymer, may occur in the solid film for the C4 and C6 tethers, but not the shorter ones. Interchain compensation, where the counterion is tethered to an adjacent polymer, must thus occur for the shorter tethers. | ||
| Fig. 6 Representative PM3 molecular models for the mTFF-CnSISC2F5 intrachain self-compensated hole-doped state. (a) n = 2, (b) 3, (c) 4, and (d) 6. Atom legend: (grey) carbon, sodium; (white) hydrogen; (blue) nitrogen; (red) oxygen; (yellow) fluorine, sulfur. Ball-and-stick model and space-filling model. | ||
| R-SIS-RF | PM3 length (Å) | OPLS4 length (Å) |
|---|---|---|
| R: C–C | 1.52 | 1.55 |
| R: C–SO2 | 1.82 | 1.88 |
| RSO2–N (imide) | 1.68 | 1.54 |
| (imide) N–SO2RF | 1.64 | 1.56 |
| RF: SO2–C | 2.03 | 1.92 |
| RF: C–C | 1.61 | 1.54 |
| R-SIS-RF | PM3 angle (°) | OPLS4 angle (°) |
|---|---|---|
R: C–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –C –C |
112 | 120 |
R: C–![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) O2–N O2–N |
101 | 103 |
SO2–![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) (imide)–SO2 (imide)–SO2 |
143 | 120 |
| RF: N–O2–C |
102 | 107 |
Molecular modeling: solid state
To investigate the crossover from interchain to intrachain compensation, we computed the polymer morphology in the amorphous solid state using the OPLS4 force field,24 and extracted the pair correlation functions g(r), as shown in Fig. 7a. The results reveal that the imide nitrogen and the Na+ spectator cation are distributed over a range of distances from the hole (aminium nitrogen). The holes are indeed counterbalanced exclusively by interchain anions for the C2 and C3 tethers, but a mixture of intrachain and interchain anions for C4 and C6 tethers (Fig. 7b). As the tether length increases, it appears that interchain compensation dominates again, presumably due to entropy effects. The closest approach counteranion distance is ca. 4.5 Å for the C2 and C3 tethers, but ca. 4.9 Å for the C4 and C6 tethers due to stronger ion clustering with associated Na+ distribution. | ||
| Fig. 7 OPLS4 molecular dynamics simulation of saturated-doped SC mTFF-CnSISC2F5. (a) Pair correlation functions g(r) for triphenylaminium nitrogen: red, to C2F5SIS imide nitrogen; blue, to Na+; and green, to next triphenylaminium nitrogen. (b) g(r) for triphenylaminium nitrogen: open circles, to C2F5SIS imide nitrogen tethered to the adjacent chain (interchain compensation); open squares, tethered to the same chain (intrachain compensation). | ||
As a consequence, the electrochemical potential of the holes due to the ion distribution14,25 turns out to vary little with tether length. To estimate the local Madelung potential at the hole site, we computed.

Effective work function and PFOP diode characteristics
To check whether the high φ values of mTFF-CnSISC2F5 as the hole injection layer (HIL) persist in devices, we measured the built-in potential (Vbi) and current density–voltage (JV) characteristics of diodes fabricated with poly(9,9-bis(4-octylphenyl)fluorene-2,7-diyl) as the semiconductor (PFOP, IE 5.8 eV; chemical structure in the inset of Fig. 9) and Al as the opposite electrode in the following structure: glass/ITO/30-nm HIL/90-nm PFOP/Al. This experiment provides a demanding test of the ability of the HIL to form ohmic hole contacts. We measured the dc-bias dependent Stark spectrum of the polymer,26,27 as well as the subgap absorption spectrum from electroabsorption.28 The modulated reflectance spectra ΔR/R varies linearly with the applied dc bias (Vdc), where ΔR is the modulated root-mean-square reflectance and R is the dc reflectance (Fig. 8). This shows that Vbi is well-behaved, which enables its accurate determination from the interpolated Vdc required to null the Stark absorption feature. A well-behaved Vbi in turn indicates well-behaved electrode φ values inside the devices.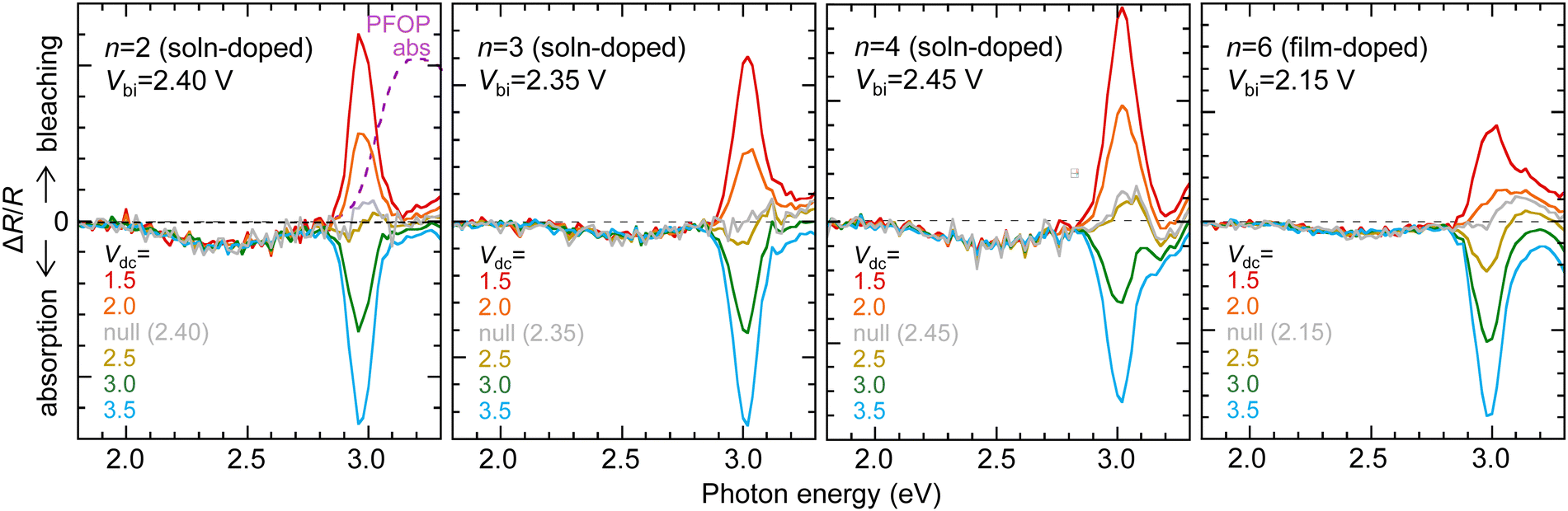 | ||
| Fig. 8 Stark and subgap absorption by electroabsorption spectroscopy. Normalized reflectance spectra ΔR/R for ITO/hole-doped mTFF-CnSISC2F5/PFOP/Al. Upward features correspond to induced bleaching in-phase with the forward-bias half-cycle. The null spectrum is indicated by the grey line. Measurements were performed at 30 K, with a root-mean-square a.c. modulation of 0.5 V and a frequency of 535 Hz. The Stark peak occurs at 3.0 eV. The Fermi level of the Al/PFOP contact lies 3.3 eV below the effective vacuum level of PFOP. | ||
Thus, the diodes with solution-doped mTFF-CnSISC2F5 films having a φ of ca. 5.75 eV were found to exhibit Vbi values of 2.40 ± 0.05 V. Vbi relates to the difference in effective φ between the anode and cathode contacts: eVbi = φan − φcat.29 Thus, the effective φ of the contact under study can be evaluated if the effective φ of the other contact is known.29 Since the effective φ of the Al contact with PFOP (φcat) is 3.30 ± 0.05 eV,29 the effective φ of the HIL inside the diode is evaluated to be 5.70 ± 0.07 eV, given by φan = eVbi + 3.30 eV, which agrees with the φ value measured by UPS. This finding affirms two conclusions: (1) the work function of the film does not change in the device and (2) the Fermi level of the HIL can closely approach the HOMO band edge of the PFOP test semiconductor. The latter conclusion corresponds to shallow Fermi level pinning, similar to the situation at the electron contact.2 Shallow pinning provides better energy alignment for hole injection. Indeed, the strong modulated subgap absorption over the spectral range of 2.1–2.8 eV reveals the accumulation of a high density of holes in a δ-doped layer at the PFOP interface.28 The subgap absorption is one order of magnitude stronger than for poly(3,4-ethylenedioxythiophene):poly(styrenesulfonic acid) (PEDT:PSSH) HIL films.30 This demonstrates that a true ohmic hole contact is indeed formed. The diodes with film-doped mTFF-CnSISC2F5 films having a φ of ca. 5.55 eV were found to exhibit Vbi values of 2.15 V, which corresponds to an effective φ value for the HIL of 5.45 eV.
We demonstrate that the mTFF-CnSISC2F5 films with the high but not the low φ can efficiently inject holes into PFOP (Fig. 9). Diodes fabricated with solution-doped mTFF-CnSISC2F5 HILs give the highest current density. Diodes with film-doped mTFF-CnSISC2F5 HILs give 80% lower current density. Earlier work with different TAF copolymers has shown that efficient hole injection is attained when φ exceeds about IE − 0.2 eV, i.e., the Fermi level of the doped organic electrode closely approaches or becomes deeper than the conventionally defined HOMO band edge of the semiconductor.14,16
 | ||
| Fig. 9 JV curve of the hole-dominated PFOP diode with self-compensated hole-doped mTFF-CnSISC2F5 as the hole injection layer. Device structure: ITO/30-nm HIL/90-nm PFOP/Al. Data from the forward scan of a representative diode are shown. For solution-doped, n = 2 (red, solid); n = 3 (orange, solid); n = 4 (green, solid); for film-doped, n = 6 (blue, dash); n = 2 (red, dash). Inset: Chemical structure of PFOP. | ||
Since the mTFF-CnSISC2F5 HILs are bulk-doped, they form true ohmic contact that does not depend on confinement of opposite carriers.31–33 In contrast, PEDT:PSSH modified with perfluorinated ionomers (PFI) can also give ultrahigh work functions due to surface segregation of an insulating PFI monolayer, but this imposes a tunnel barrier that impedes formation of a true ohmic contact under demanding conditions.34
Thermal stability
To evaluate the thermal stability of the SC hole-doped mTFF-CnSISC2F5 films, we measured the electronic spectra of the films as they are baked at increasing temperatures in a nitrogen glovebox. The results reveal that the C3-tether film can withstand short heating up to 120 °C but the others only up to 100 °C before thermal dedoping occurs (Fig. 10a). Nevertheless, hole-doped TAF polymers exhibit advantageously wide work function and conductivity plateaus. Their φ values lie within 80 meV of that of saturation doping for DL ≳ 0.25 h+ per r.u.14 Their electrical conductivity values lie above 40% of the peak value for 0.25 ≲ DL ≲ 0.75 h+ per r.u.17 Thus, the minimum DL may be specified at 0.25 h+ per r.u. for hole injection applications. For this threshold, mTFF-CnSISC2F5 films are able to tolerate short baking of up to 180 °C for the C6-tether film, and up to 210 °C for the C3-tether film, and temperatures between these two for the other films. Film doping gives similar stability results as solution doping, which suggests that thermal stability is determined by tether length rather than processing. | ||
| Fig. 10 Stability of self-compensated hole-doped mTFF-CnSISC2F5. (a) Thermal stability for 5-min bake at each temperature in the glovebox. (b) Ambient stability (22 °C, relative humidity 65%). Film thickness, 25–35 nm. Stability in the nitrogen glovebox is 100 times as long. | ||
Ambient stability
The C2–C4-tether films also exhibit excellent ambient stability of their ultrahigh workfunction state despite the potential to oxidize moisture.18 Their DL values are stable for up to 1 h under ambient conditions (22 °C, 65% relative humidity), which provides sufficient time for processing without protection by inert gas. But the DL values decline significantly after a few hours. The C3-tether film stabilizes at a DL of about 0.45 h+ per r.u. after 1 month, which is remarkable for an ultrahigh-workfunction film.18 Thus, the C3 tether confers best overall stability to the doped film. This conclusion also appears consistent with its marginally higher DL after self-compensation. The enhanced stability may be related to its ion cluster morphology that may better exclude moisture while providing sufficient Coulomb stabilization for the holes.Conclusion
In summary, we have designed, synthesized and characterized mTFF-CnSISC2F5 polymers with n = 2, 3, 4 and 6. We found that the material with the C6 tether becomes intractable in the fully hole-doped state, likely due to an enhanced hydrophobicity. The work function of the self-compensated hole-doped state can reach 5.75 eV, largely independent of tether length, which reflects a switchover from interchain to mixed interchain/intrachain self-compensation as the tether length increases, to maintain a similar local ion cluster environment in the vicinity of the holes. This enables unprecedentedly efficient hole injection from the hole-doped mTFF semiconductor core into PFOP. Yet, the C3 tether confers higher thermal and ambient stability than the other tethers, which we attribute to a more optimal ion cluster morphology.Experimental
Materials
Poly(9,9′-bis(4-octylphenyl)fluorene-2,7-diyl) (PFOP) was obtained from Cambridge Display Technology Ltd/Sumitomo Chemical Co., Ltd.Monomer synthesis
Into a 10-mL 1-neck rbf, N,N-diphenyl-(m-trifluoromethyl)aniline (2.04 g, 6.52 mmol) and anhydrous dimethylformamide (DMF, 2.8 mL) were added. The reaction mixture was stirred under inert conditions for 15 min. N-Bromosuccinimide (2.44 g, 13.7 mmol) in anhydrous DMF (2.8 mL) solution was added dropwise into the rbf maintained at 5–7 °C. The reaction mixture was warmed to room temperature and stirred for 1 h, followed by precipitated in water. The crude product was extracted with hexane and dried over sodium sulphate. The extract was concentrated via rotary evaporation and purified by column chromatography with hexane as the eluent to yield pure N,N-di-p-bromophenyl-(m-trifluoromethyl)aniline (2.47 g, 81%). 1H NMR (300 MHz, CDCl3), δ (ppm): 7.42–7.36 (m, 4H), 7.35–7.32 (d, 1H), 7.26–7.24 (m, 2H), 7.20–7.17 (d, 1H), 6.97–6.92 (m, 4H).
To a 250-mL 1-neck rbf, N,N-di-p-bromophenyl-(m-trifluoromethyl)aniline (5.0 g, 10.6 mmol) was added. The rbf was pumped down to vacuum and backfilled with argon thrice. Distilled tetrahydrofuran (THF, 70 mL) was added to dissolve the monomer. The reaction mixture was cooled to −78 °C before dropwise addition of 2.0 M n-butyl lithium in cyclohexane (12 mL, 24 mmol). The resulting reaction mixture was stirred at −78 °C for 2 h before the addition of 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17 mL, 83.3 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 24 h. The reaction was then quenched with water and concentrated via rotary evaporation. The crude product was extracted with dichloromethane (DCM) thrice and dried over sodium sulphate. Lastly, the extract was concentrated via rotary evaporation and purified by column chromatography with (19:1 ratio) hexane:ethyl acetate solvent mixture as the eluent to yield pure diEs-mTFF as a white solid (2.2 g, 37%). 1H NMR (400 MHz, CDCl3), δ (ppm): 7.72–7.69 (d, 4H), 7.36–7.31 (m, 2H), 7.26–7.22 (m, 2 H), 7.07–7.04 (d, 4H), 1.34 (s, 24H).
:1 hexane:ethyl acetate solvent mixture as the eluent to yield pure diBr-FC2OH as a white solid (3.86 g, 93.7%). 1H NMR (400 MHz, DMSO-d6), δ (ppm): 7.78 (m, 4H), 7.53 (dd, 2H), 4.18 (t, 2H), 2.63 (m, 4H), 2.24 (m, 4H). 13C NMR (400 MHz, CDCl3), δ (ppm): 150.8, 138.5, 131.2, 126.7, 122.1, 121.7, 58.8, 52.2, 42.7.
:1 hexane:ethyl acetate solvent mixture as the eluent to yield pure diBr-C2I as a white solid (1.39 g, 84.3%). 1H NMR (400 MHz, DMSO-d6), δ (ppm): 7.95 (d, 2H), 7.84 (d, 2H), 7.61 (dd, 2H), 2.72 (m, 4H), 2.28 (m, 4H). 13C NMR (400 MHz, CDCl3), δ (ppm): 148.1, 139.3, 131.8, 126.3, 122.5, 121.9, 59.4, 44.4, −2.66. HRMS (APCI) m/z 629.7548 (calcd for M+ 629.7552).
:1 hexane:ethyl acetate solvent mixture as the eluent to yield pure diBr-FC2SH as a white solid (82.8 mg, 80.1%). 1H NMR (400 MHz, DMSO-d6), δ (ppm): 7.83 (m, 4H), 7.58 (dd, 2H), 2.33 (m, 4H), 2.27 (t, 2H), 1.59 (m, 4H). 13C NMR (400 MHz, CDCl3), δ (ppm): 150.2, 138.6, 130.7, 126.4, 122.3, 121.3, 55.9, 42.6, 18.5. HRMS (APCI) m/z 440.8926 (calcd for M− 440.8987).
In a 100-mL Nalgene bottle, 3.5 g (5.5 mmol) of diBr-FC4SO3Na was dissolved in MeOH. The Amberlyst 15 hydrogen form H+ resin at 20 molar equivalents (23.3 g, 109.3 mmol) was washed to pH 7 with deionized water and pre-conditioned with MeOH overnight before adding to the solution. The solution was left on a mechanical roller for 6 h at room temperature. The solution was then filtered through a 0.45-μm nylon syringe filter into a rbf. The filtered solution was dried via rotary evaporation, yielding the 9,9′-bis(3′-sulfonatobutyl)-2,7-dibromofluorene acid form (diBr-FC4SO3H) as an off-white solid. Upon drying, diBr-FC4SO3H (2.50 g, 4.2 mmol) was added into a 150-mL 2-neck rbf fitted with a stopcock valve adaptor. The rbf was pumped down to vacuum and backfilled with argon thrice. Distilled THF (80 mL) was added to dissolve the monomer. Catalytic anhydrous DMF (0.25 mL) was added, followed by dropwise addition of oxalyl chloride (7.1 mL, 84.0 mmol). The reaction was stirred at room temperature for 6 h. Upon completion of reaction, the 9,9′-bis(3′-chlorosulfonylbutyl)-2,7-dibromofluorene (diBr-FC4SO2Cl) intermediate was dried in situ under vacuum to remove all solvents and oxalyl chloride, and re-dissolved in anhydrous chloroform (40 mL). In a separate rbf, pentafluoroethanesulfonamide (PFES, 16.7 g, 84.0 mmol) was mixed with anhydrous triethylamine (TEA, 11.7 mL, 84.0 mmol) in anhydrous chloroform (30 mL). This solution was added to diBr-FC4SO2Cl solution and left to stir at room temperature for 1 h. Upon completion of reaction, the mixture was concentrated by rotary evaporation and precipitated in diethyl ether. The solid was re-dissolved in chloroform and precipitated in diethyl ether twice. The crude product was re-dissolved in chloroform extracted against deionized water. The organic phase was concentrated and then precipitated in diethyl ether, yielding pure 9,9′-bis(3′-pentafluoroethylsulfonylimidobutyl)-2,7-dibromofluorene triethylammonium salt (diBr-FC4SISC2F5-TEAH) as a brown oil (yield = 82%). 1H NMR (400 MHz, DMSO-d6), δ (ppm): 7.79–7.77 (d, 2H), 7.74–7.72 (d, 2H), 7.52–7.50 (d, 2H), 3.00–2.90 (q, 12H), 2.71–2.67 (t, 4H), 2.05–2.01 (t, 4H), 1.46–1.40 (m, 4H), 1.20–1.10 (t, 18H), 0.54–0.49 (m, 4H). 19F NMR (400 MHz, DMSO-d6), δ (ppm): −78.6 (s, 3F), −116.8 (s, 2F).
:1 MeOH:H2O (10 mL). The Amberlyst 15 hydrogen form H+ resin at 20 molar equivalents (2.22 g, 10.4 mmol) was washed to pH 7 with deionized water and pre-conditioned with MeOH overnight before adding to the solution. The solution was left on a mechanical roller for 6 h at room temperature. The solution was then filtered through a 0.45-μm nylon syringe filter into a rbf and dried via rotary evaporation to yield the crude product as an orange solid. The crude solid was washed with hexane to yield the 9,9′-bis(3′-sulfonatohexyl)-2,7-dibromofluorene acid form (diBr-FC6SO3H) as an off-white solid. Into a 50-mL 2-neck rbf fitted with a stopcock valve adaptor, diBr-FC6SO3H) (200 mg, 0.3065 mmol) was loaded. The rbf was pumped down to vacuum and backfilled with argon thrice. Distilled tetrahydrofuran (3.4 mL) and anhydrous chloroform (3.4 mL) were added to dissolve the monomer. Catalytic anhydrous DMF (0.01 mL) was further added, followed by dropwise addition of oxalyl chloride (0.52 mL, 6.13 mmol). The reaction was stirred at room temperature for 6 h. Upon completion of reaction, the 9,9′-bis(3′-chlorosulfonylhexyl)-2,7-dibromofluorene (diBr-FC6SO2Cl) intermediate was dried in situ under vacuum to remove all solvents and oxalyl chloride, and re-dissolved in anhydrous chloroform (3 mL). In a separate rbf, pentafluoroethanesulfonamide (PFES, 1.220 g, 6.13 mmol) was mixed with anhydrous triethylamine (TEA, 0.82 mL, 6.13 mmol) in anhydrous chloroform (3 mL). This solution was added to the diBr-FC6SO2Cl solution and left to stir at room temperature for 3 h. Upon completion of reaction, the mixture was concentrated by rotary evaporation and precipitated in diethyl ether. The precipitate was re-dissolved in chloroform and precipitated twice in diethyl ether. The crude product was re-dissolved in chloroform and extracted against water and brine thrice. The extract was dried over sodium sulfate and precipitated in diethyl ether, yielding impure diBr-FC6SISC2F5-TEAH as a brown oil.
:1 THF:DMF mixture (6.2 mL) was added to dissolve the monomers, followed by addition of 0.6 M Na2CO3 solution (3.75 mL, 2.25 mmol). The solution was purged with Ar for a further 15 min before heating in a microwave at 130 °C for 15 min and then cooled. The solution was then precipitated in deionized water. The resulting precipitate was filtered over a 0.45-μm nylon membrane filter. The precipitate was then dissolved in 1:1 ACN:MeOH (typically about 5 mL) and added to a solution of sodium diethyldithiocarbamate in 5 mL of 1:1 ACN:MeOH (2.0 g, 9.2 mmol). The solution was purified by dialysis through a regenerated cellulose membrane (MWCO = 12–14 kDa), using 1:1 ACN:MeOH as a dialysate for 30 min. The volume ratio of the dialyzed polymer solution to the solvent bath was about 1:50. This step was repeated with fresh dialysate 2 more times for 30 min, and another 2 times for 1 h. The dialyzed solution was precipitated in diethyl ether and washed with water to yield poly(9,9′-bis(3′-pentaethylsulfonylimidoethyl)fluorene-2,7-diyl-1,4-phenylene-N-(m-trifluoro-methylphenyl)-amino-1,4-phenylene) sodium salt (mTFF-C2SISC2F5) as an off-white solid. 1H NMR (400 MHz, DMSO-d6), δ (ppm): 7.94–7.20 (m, 18H), 2.88–2.66 (m, 4H), 2.57–2.30 (m, 4H). GPC in DMF + 0.1 M LiBr: Mn = 26 kDa, Mw = 59 kDa, PDI = 2.3. Elemental analysis: C:H:N:S = 43.11%:2.73%:3.91%:10.69% = 3.58:2.73:0.28:0.33 = 40:30.5:3.12:3.68 (theo 40:28:3:4).
:1 THF:DMF mixture (6.3 mL) was added to dissolve the monomers, followed by addition of 0.6 M Na2CO3 solution (3.75 mL, 2.25 mmol). The solution was purged with Ar for a further 15 min before heating in a microwave at 130 °C for 15 min and then cooled. The solution was precipitated in deionized water. The resulting precipitate was then filtered over a 0.45-μm nylon membrane filter. The precipitate was dissolved in 1:1 ACN:MeOH (typically about 5 mL) and added to a solution of sodium diethyldithiocarbamate in 5 mL of 1:1 ACN:MeOH (2.0 g, 9.2 mmol). The solution was purified by dialysis through a regenerated cellulose membrane (MWCO = 12–14 kDa), using 1:1 ACN:MeOH as a dialysate for 30 min. The volume ratio of the dialyzed polymer solution to the solvent bath was about 1:50. This step was repeated with fresh dialysate 2 more times for 30 min, and another 2 times for 1 h. The dialyzed solution was precipitated in diethyl ether and washed with deionized water to yield poly(9,9′-bis(3′-pentaethylsulfonylimidopropyl)fluorene-2,7-diyl-1,4-phenylene-N-(m-trifluoro-methylphenyl)-amino-1,4-phenylene) sodium salt (mTFF-C3SISC2F5) as an off-white solid. 1H NMR (400 MHz, DMSO-d6), δ (ppm): 7.98–7.11 (m, 18H), 2.85–2.69 (m, 4H), 2.30–2.10 (m, 4H), 1.06–0.88 (m, 4H). GPC in DMF + 0.1 M LiBr: Mn = 23 kDa, Mw = 47 kDa, PDI = 2.1. Elemental analysis: C:H:N:S = 43.32%:3.04%:3.77%:10.77% = 3.61:3.04:0.27:0.34 = 42:35.3:3.14:3.95 (theo 42:32:3:4).
:1 THF:DMF mixture (6.4 mL) was added to dissolve the monomers, followed by addition of 0.6 M Na2CO3 solution (3.6 mL, 2.15 mmol). The solution was purged with Ar for a further 15 min before heating in a microwave at 130 °C for 15 min and then cooled. The solution was precipitated in deionized water. The resulting precipitate was then filtered over a 0.45-μm nylon membrane filter. The precipitate was dissolved in 1:1 ACN:MeOH (typically about 5 mL) and added to a solution of sodium diethyldithiocarbamate in 5 mL of 1:1 ACN:MeOH (1.9 g, 8.6 mmol). The solution was purified by dialysis through a regenerated cellulose membrane (MWCO = 12–14 kDa), using 1:1 ACN:MeOH as a dialysate for 30 min. The volume ratio of the dialyzed polymer solution to the solvent bath was about 1:50. This step was repeated with fresh dialysate 2 more times for 30 min, and another 2 times for 1 h. The dialyzed solution was precipitated in diethyl ether and washed with deionized water to yield poly(9,9′-bis(3′-pentaethylsulfonylimidobutyl)fluorene-2,7-diyl-1,4-phenylene-N-(m-trifluoro-methylphenyl)-amino-1,4-phenylene) sodium salt (mTFF-C4SISC2F5) as an off-white solid. 1H NMR (400 MHz, DMSO-d6), δ (ppm): 8.00–7.05 (m, 18H), 2.90–2.65 (m, 4H), 2.40–2.00 (m, 4H), 1.60–1.40 (m, 4H), 0.80–0.50 (m, 4H). GPC in DMF + 0.1 M LiBr: Mn = 14 kDa, Mw = 20 kDa, PDI = 1.4. Elemental analysis: C:H:N:S = 43.39%:3.41%:3.69%:10.49% = 3.61:3.41:0.26:0.33 = 44:41.6:3.17:4.02 (theo 44:36:3:4).
:1 THF:DMF mixture (6.1 mL) was added to dissolve the monomers, followed by addition of 0.6 M Na2CO3 solution (3.9 mL, 2.35 mmol). The solution was purged with Ar for a further 15 min before heating in a microwave at 130 °C for 15 min and then cooled. The solution was precipitated in deionized water. The resulting precipitate was then filtered over a 0.45-μm nylon membrane filter. The precipitate was dissolved in 1:1 ACN:MeOH (typically about 5 mL) and added to a solution of sodium diethyldithiocarbamate in 5 mL of 1:1 ACN:MeOH (2.1 g, 9.4 mmol). The solution was purified by dialysis through a regenerated cellulose membrane (MWCO = 12–14 kDa), using 1:1 ACN:MeOH as a dialysate for 30 min. The volume ratio of the dialyzed polymer solution to the solvent bath was about 1:50. This step was repeated with fresh dialysate 2 more times for 30 min, and another 2 times for 1 h. The dialyzed solution was precipitated in diethyl ether and washed with deionized water to yield poly(9,9′-bis(3′-pentaethylsulfonylimidohexyl)fluorene-2,7-diyl-1,4-phenylene-N-(m-trifluoro-methylphenyl)-amino-1,4-phenylene) sodium salt (mTFF-C6SISC2F5) as an off-white solid. 1H NMR (400 MHz, DMSO-d6), δ (ppm): 8.21–7.00 (m, 18H), 2.89–2.73 (m, 4H), 2.30–1.95 (m, 4H), 1.56–1.38 (m, 4H), 1.21–0.95 (m, 8H), 0.74–0.40 (m, 4H). GPC in DMF + 0.1 M LiBr: Mn = 19 kDa, Mw = 30 kDa, PDI = 1.6. Elemental analysis: C:H:N:S = 46.03%:3.79%:3.56%:9.74% = 3.83:3.79:0.25:0.30 = 48:47.5:3.13:3.76 (theo 48:44:3:4).
Doping methods
:DMC (1:10) in the glovebox and excess dopant spun-off to dope the film. The excess ions and salt by-products were removed by two consecutive spin-rinse steps in the same solvent mixture to give the self-compensated, hole-doped film.
Author contributions
L. L. C. directed material development and characterization. Q. M. K. conducted the experiments. C. G. T. conducted the electroabsorption measurement. R. Q. P. performed the OPLS4 calculations. Q. M. K., K. C. B., Y. W. and Q. J. S. contributed to material development. P. H. K. H. provided advice. Q. M. K., R. Q. P. and L. L. C. wrote the manuscript. All authors discussed the experiments and results.Data availability
The data that support the findings of this study are all available in the article.Conflicts of interest
There is no conflict to declare.Acknowledgements
This research is partially supported by the National Research Foundation, Prime Minister's Office, Singapore, under its Competitive Research Programme (CRP Award No. NRF-CRP24-2020-0006: A-0008375-00-00, A-0008375-01-00).References
- C. G. Tang, M. C. Ang, K. K. Choo, V. Keerthi, J. K. Tan, M. N. Syafiqah, T. Kugler, J. H. Burroughes, R. Q. Png, L. L. Chua and P. K. H. Ho, Doped polymer semiconductors with ultrahigh and ultralow work functions for ohmic contacts, Nature, 2016, 539, 536–540 CrossRef CAS PubMed.
- C. G. Tang, M. N. Syafiqah, Q. M. Koh, C. Zhao, J. Zaini, Q. J. Seah, M. J. Cass, M. J. Humphries, I. Grizzi, J. H. Burroughes, R. Q. Png, L. L. Chua and P. K. H. Ho, Multivalent anions as universal latent electron donors, Nature, 2019, 573, 519–525 CrossRef CAS PubMed.
- S. A. Chen and M. Y. Hua, Structure and doping level of the self-acid-doped conjugated conducting polymers: poly[n-(3'-thienyl)alkanesulfonic acids], Macromolecules, 1993, 26, 7108–7110 CrossRef CAS.
- H. S. O. Chan, P. K. H. Ho, S. C. Ng, B. T. G. Tan and K. L. Tan, A new water-soluble, self-doping conducting polyaniline from poly(o-aminobenzylphosphonic acid) and its sodium salts: synthesis and characterization, J. Am. Chem. Soc., 1995, 117, 8517–8523 CrossRef CAS.
- T. H. Reilly III, A. W. Hains, H.-Y. Chen and B. A. Gregg, A self-doping, O2-stable, n-type interfacial layer for organic electronics, Adv. Energy Mater., 2012, 2, 455–460 CrossRef.
- C. K. Mai, H. Zhou, Y. Zhang, Z. B. Henson, T. Q. Nguyen, A. J. Heeger and G. C. Bazan, Facile doping of anionic narrow-band-gap conjugated polyelectrolytes during dialysis, Angew. Chem., Int. Ed., 2013, 52, 12874–12878 CrossRef CAS PubMed.
- S. Watanabe, R. Hakamatani, K. Yaegashi, Y. Yamashita, H. Nozawa, M. Sasaki, S. Kumagai, T. Okamoto, C. G. Tang, L. L. Chua, P. K. H. Ho and J. Takeya, Surface doping of organic single-crystal semiconductors to produce strain-sensitive conductive nanosheets, Adv. Sci., 2021, 8, 2002065 CrossRef CAS PubMed.
- C. Zhao, C. G. Tang, Z. L. Seah, Q. M. Koh, L. L. Chua, R. Q. Png and P. K. H. Ho, Improving organic photovoltaic cells by forcing electrode work function well beyond onset of Ohmic transition, Nat. Commun., 2021, 12, 2250 CrossRef CAS PubMed.
- K. Philipps, Y. Ie, B. van der Zee, R. Q. Png, P. K. H. Ho, L. L. Chua, E. del Pino Rosendo, C. Ramanan, G. J. A. H. Wetzelaer, P. W. M. Blom and J. J. Michels, Role of linker functionality in polymers exhibiting main-chain thermally activated delayed fluorescence, Adv. Sci., 2022, 9, 2200056 CrossRef CAS PubMed.
- X. Tan, D. Dou, L. L. Chua, R. Q. Png, D. G. Congrave, H. Bronstein, M. Baumgarten, Y. Li, P. W. M. Blom and G.-J. A. H. Wetzelaer, Inverted device architecture for high efficiency single-layer organic light-emitting diodes with imbalanced charge transport, Nat. Commun., 2024, 15, 4107 CrossRef CAS PubMed.
- B. vander Zee, S. Paulus, R. Q. Png, P. K. H. Ho, L. L. Chua, G. J. A. H. Wetzelaer and P. W. M. Blom, Role of singlet and triplet excitons on the electrical stability of polymer light-emitting diodes, Adv. Electron. Mater., 2020, 6, 2000367 CrossRef CAS.
- A. G. Ricciardulli, B. van der Zee, K. Philipps, G. A. H. Wetzelaer, R. Q. Png, P. K. H. Ho, L. L. Chua and P. W. M. Blom, Polymer–perovskite blend light-emitting diodes using a self-compensated heavily doped polymeric anode, APL Mater., 2020, 8, 021101 CrossRef CAS.
- L. X. Wang, C. G. Tang, Z. S. Tan, H. Y. Phua, J. Chen, W. Lei, R. Q. Png, L. L. Chua and P. K. H. Ho, Double-type-I charge-injection heterostructure for quantum-dot light-emitting diodes, Mater. Horiz., 2022, 9, 2147–2159 RSC.
- R. Q. Png, M. C. Ang, M. H. Teo, K. K. Choo, C. G. Tang, D. Belaineh, L. L. Chua and P. K. H. Ho, Madelung and Hubbard interactions in polaron band model of doped organic semiconductors, Nat. Commun., 2016, 7, 11948 CrossRef CAS PubMed.
- M. C. Y. Ang, Q. M. Koh, C. G. Tang, Q. J. Seah, Y. Wang, M. Callsen, Y. P. Feng, R. Q. Png and L. L. Chua, Spectator cation size effect on the work function and stability of self-compensated hole-doped polymers, J. Mater. Chem. C, 2020, 8, 124–131 RSC.
- M. C. Y. Ang, C. G. Tang, Q. M. Koh, C. Zhao, Q. J. Seah, Y. Wang, M. Callsen, Y. P. Feng, R. Q. Png and L. L. Chua, Bulk ion-clustering and surface ion-layering effects on work function of self-compensated charged-doped polymer semiconductors, Mater. Horiz., 2020, 7, 1073–1082 RSC.
- Q. M. Koh, N. S. Mazlan, Q. J. Seah, J. C. Yang, Y. J. Chen, R. Q. Png, P. K. H. Ho and L. L. Chua, Effects of planarization of the triphenylamine unit on the electronic and transport properties of triarylamine–fluorene copolymers in both doped and undoped forms, ACS Appl. Mater. Interfaces, 2024, 16, 39708–39716 CrossRef CAS PubMed.
- Q. M. Koh, C. G. Tang, M. C. Y. Ang, K. K. Choo, Q. J. Seah, R. Q. Png, L. L. Chua and P. K. H. Ho, Overcoming the water oxidative limit for ultra-high-workfunction hole-doped polymers, Nat. Commun., 2021, 12, 3345 CrossRef CAS PubMed.
- C. G. Tang, M. Nur Syafiqah, Q. M. Koh, M. C. Y. Ang, K. K. Choo, M. M. Sun, M. Callsen, Y. P. Feng, L. L. Chua, R. Q. Png and P. K. H. Ho, Water binding and hygroscopicity in π-conjugated polyelectrolytes, Nat. Commun., 2023, 14, 3978 CrossRef CAS PubMed.
- F. M. Koch and R. Peters, Catalytic enantio- and diastereoselective formation of β-Sultones: ring-strained precursors for enantioenriched β-hydroxysulfonyl derivatives, Angew. Chem., Int. Ed., 2007, 46, 2685–2689 CrossRef CAS PubMed.
- J. C. Yan, X. Cheng, Q. L. Zhou and J. Pei, Chiral polyfluorene derivatives: Synthesis, chiroptical properties, and investigation of the structure−property relationships, Macromolecules, 2007, 40, 832–839 CrossRef CAS.
- V. V. Sureshbabu, T. M. Vishwanatha and B. Vasantha, A simple synthesis of Nβ-Fmoc/Z-amino alkyl thiols and their use in the synthesis of Nβ-Fmoc/Z-amino alkyl sulfonic acids, Synlett, 2010, 1037–1042 CrossRef CAS.
- W. L. Seah, C. G. Tang, R. Q. Png, V. Keerthi, C. Zhao, H. Guo, J. G. Yang, M. Zhou, P. K. H. Ho and L. L. Chua, Interface doping for ohmic organic semiconductor contacts using self-aligned polyelectrolyte counterion monolayer, Adv. Funct. Mater., 2017, 27, 1606291 CrossRef.
- C. Lu, C. Wu, D. Ghoreishi, W. Chen, L. Wang, W. Damm, G. A. Ross, M. K. Dahlgren, E. Russell, C. D. Von Bargen, R. Abel, R. A. Friesner and E. D. Harder, OPLS4: Improving force field accuracy on challenging regimes of chemical space, J. Chem. Theory Comput., 2021, 17, 4291–4300 CrossRef CAS PubMed.
- P. J. Chia, S. Sivaramakrishnan, M. Zhou, R. Q. Png, L. L. Chua, R. H. Friend and P. K. H. Ho, Direct evidence for the role of the Madelung potential in determining the work function of doped organic semiconductors, Phys. Rev. Lett., 2009, 102, 096602 CrossRef PubMed.
- I. H. Campbell, M. D. Joswick and I. D. Parker, Direct measurement of the internal electric field distribution in a multilayer organic light-emitting diode, Appl. Phys. Lett., 1995, 67, 3171–3173 CrossRef CAS.
- V. Bodrozic, T. M. Brown, S. Mian, D. Caruana, M. Roberts, N. Phillips, J. J. Halls, I. Grizzi, J. H. Burroughes and F. Cacialli, The built-In potential in blue polyfluorene-based light-emitting diodes, Adv. Mater., 2008, 20, 2410–2415 CrossRef CAS.
- M. Zhou, L. L. Chua, R. Q. Png, C. K. Yong, S. Sivaramakrishnan, P. J. Chia, A. T. S. Wee, R. H. Friend and P. K. H. Ho, The role of delta-doped interfaces for Ohmic contacts to organic semiconductors, Phys. Rev. Lett., 2009, 103, 036601–036604 CrossRef PubMed.
- M. Zhou, R. Q. Png, S. H. Khong, S. Sivaramakrishnan, L. H. Zhao, L. L. Chua, R. H. Friend and P. K. H. Ho, Effective work functions for the evaporated metal/organic semiconductor contacts from in-situ diode flatband potential measurements, Appl. Phys. Lett., 2012, 101, 013501 CrossRef.
- Q. M. Koh, Q. J. Seah, J. C. Yang, N. S. Mazlan, Q. E. Hoh, Z. L. Seah, R. Q. Png, P. K. H. Ho and L. L. Chua, Evidence for morphology control by polyelectrolyte templating in PEDT:PSSH, Adv. Funct. Mater., 2024, 2407493 CrossRef.
- C. V. Hoven, A. Garcia, G. C. Bazan and T. Q. Nguyen, Recent applications of conjugated polyelectrolytes in optoelectronic devices, Adv. Mater., 2008, 20, 3793–3810 CrossRef CAS.
- F. Huang, H. Wu and Y. Cao, Water/alcohol soluble conjugated polymers as highly efficient electron transporting/injection layer in optoelectronic devices, Chem. Soc. Rev., 2010, 39, 2500–2521 RSC.
- J. H. Lee, B. H. Lee, S. Y. Jeong, S. B. Park, G. Kim, S. H. Lee and K. Lee, Radical cation–anion coupling-induced work function tunability in anionic conjugated polyelectrolytes, Adv. Energy Mater., 2015, 5, 1501292 CrossRef.
- D. Belaineh, J. K. Tan, R. Q. Png, P. F. Dee, Y. M. Lee, B. N. N. Thi, N. S. Ridzuan and P. K. H. Ho, Perfluorinated ionomer-modified hole-injection layers: ultrahigh workfunction but nonohmic contacts, Adv. Funct. Mater., 2015, 25, 5504–5511 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc02228a |
| This journal is © The Royal Society of Chemistry 2024 |