
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ monitoring drying process to disclose the correlation between the molecular weights of a polymer acceptor with a flexible spacer and the performance of all-polymer solar cells†
Jiale
Xiang
ab,
Sven
Englund
a,
Zewdneh
Genene
 c,
Guanzhao
Wen
d,
Yanfeng
Liu
*ae,
Nannan
Yao
af,
Rui
Zhang
a,
Leiqiang
Qin
a,
Lei
Wang
a,
Ergang
Wang
*c,
Wei
Zhang
*d and
Fengling
Zhang
*ab
c,
Guanzhao
Wen
d,
Yanfeng
Liu
*ae,
Nannan
Yao
af,
Rui
Zhang
a,
Leiqiang
Qin
a,
Lei
Wang
a,
Ergang
Wang
*c,
Wei
Zhang
*d and
Fengling
Zhang
*ab
aDepartment of Physics, Chemistry and Biology, Linköping University, 58183 Linköping, Sweden. E-mail: fengling.zhang@liu.se
bWallenberg Initiative Materials Science for Sustainability, Department of Physics, Chemistry and Biology, Linköping University, 58183 Linköping, Sweden
cDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-41296 Göteborg, Sweden. E-mail: ergang@chalmers.se
dSchool of Physics and Materials Science, Guangzhou University, Guangzhou 510006, P. R. China. E-mail: wzhang@gzhu.edu.cn
eCollege of Materials and Textile Engineering, Nanotechnology Research Institute, Jiaxing University, Jiaxing 314001, P. R. China. E-mail: yanfeng.liu@zjxu.edu.cn
fState Key Laboratory of Silicon and Advanced Semiconductor Materials, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, P. R. China
First published on 17th July 2024
Abstract
Molecular weight (Mn) and conjugation of polymers can profoundly influence the performance of all-polymer solar cells (all-PSCs) via nanostructures of bulk heterojunctions (BHJs). To study the correlation between Mn and the performance of all-PSCs based on an acceptor with a flexible conjugation-break spacer (FCBS), three batches of acceptors, named PYTS, were synthesized with different number-average Mn from 9, 13 to 19 kDa. Blends with a polymer donor PBDB-T, the all-PSCs based on PYTS with Mn of 9 kDa and 19 kDa, exhibit power conversion efficiencies (PCEs) of 5.99% and 9.43%, respectively, primarily due to the increased short-circuit current density (Jsc) from 13.02 to 18.73 mA cm−2. To disclose the impact of Mn on device performance, dynamics of mixed PBDB-T:PYTS solutions to solid BHJs is studied by monitoring the drying process with home-made in situ multifunctional spectroscopy, which demonstrates that PYTS with Mn of 19 kDa has a longer drying time than the PYTS with Mn of 9 kDa. Prolonged drying of the BHJs with higher Mn PYTS facilitates more tightly packed structures with higher crystallinity. A systematic investigation on the nanostructures of BHJs, charge generation, transport and recombination is carried out with grazing-incidence wide-angle X-ray scattering (GIWAXS), transient absorption spectroscopy (TAS) and characterization of all-PSCs. The results indicate that increased crystallinity in the BHJs benefits exciton dissociation, electron transport, prolonged carrier lifetimes, and decreased non-geminate recombination rate constants in the corresponding devices. Combining the in situ study of drying and the investigation on films and devices provides us a comprehensive understanding of the interplay between Mn, the drying process, the nanostructures of BHJs and device performance. This work not only emphasizes the essential role of Mn in governing the device performance, but also exhibits recorded film formation through the in situ spectroscopy, enabling us to manipulate the nanostructure of BHJs by optimizing Mn of polymers and processing parameters.
Introduction
All-polymer solar cells (all-PSCs), featuring the incorporation of polymeric donors (PDs) and acceptors (PAs) was first demonstrated in 1995 by A. J. Heeger's and A. B. Holmes’ group, respectively.1,2 An n-type polymer, with poly(pyridopyrazine vinylene) (EHH-PPyPzV) as the electron acceptor was synthesized by our collaborators M. R. Andersson and coworkers.3 We fabricated all-PSCs using poly(2-methoxy-5-(20-ethylhexyloxy)-1,4-phenylenevinylene) (MEH-PPV) as the electron donor and EHH-PPyPzV as the electron acceptor, which presented an external quantum efficiency of 7% and an open circuit voltage of 900 mV under low-intensity monochromatic light.4 Though some effort has been made, the development of all-PSCs has been hindered because the optical and electrical properties of available acceptor polymers could not compete with dominating fullerene derivatives, such as P71CBMs and other non-fullerene acceptors (NFAs).5–7With the emergence of NFAs, all-PSCs have regained considerable attention due to their intrinsic benefits, including their enhanced morphological stability, and improved mechanical properties over small molecular acceptor-based organic solar cells (OSCs).8–13 Utilizing polymerized small-molecule acceptors (PSMAs), the power conversion efficiencies (PCEs) of all-PSCs have reached over 18%.14–17 To increase the performance of PSCs, the development of an interlayer could also be important.18–20 However, the unsatisfactory charge separation and transportation, which mainly originate from the undesirable bulk heterojunction (BHJ) morphology of active layers, lead to the low short-circuit current density (Jsc) and small fill factor (FF), hindering further advances in the photovoltaic performance of all-PSCs.10,21–23 The morphology of BHJs is determined not only by the molecular aggregation of donors and acceptors, but also by their solubility in the processing solvent, aggregation rate and crystallization behaviors in solutions.24,25 Given the solubility of conjugated polymers, the thermodynamics and crystallization kinetics26–28 of all-polymer blends are profoundly affected by their number-average molecular weight (Mn) in contrast to small molecules,29 elucidating the interplay between Mn, blend morphology and device performance is of crucial importance.
Most of the efforts made to reveal the effects of the Mn of PDs and PAs on the photovoltaic properties of all-PSCs mainly studied the morphologies of dried solid films and provided some interesting results.30–35 It was found that increased Mn promoted the strong face-on packing of the polymers and reduced the domain sizes of the blends, resulting in increased Jsc and FF, thus enhancing the PCE.36 In contrast, others pointed out that polymers with high Mn lead to coarser domains in BHJ films due to large liquid–liquid phase separations, which could cause increased charge recombination and suppressed charge extraction.34 It seems that some contradictions appear. Moreover, the emerging class of acceptor polymers with conjugation breakers have not been well studied in terms of the impact of Mn on their photovoltaic performance.37 To fully understand the issue, the transition from solution to solid-state should also be taken into consideration. It is a commonly accepted view that the drying process determines the final morphology on the micro/nano-scale level, such as the assembly of donor and acceptor molecules, the distribution of “pure” donor, acceptor, intermixed domains, and their domain sizes.24,25 The pure domains govern the charge transportation behaviours,38,39 and the intermixed amorphous part decides the charge separation efficiency.40–42 The drying process can also affect the packing and alignment of polymer chains.43,44
Previously, to understand how the drying process impacts the blend morphology in organic solar cells, we studied the film formation processes of three representative BHJs composed of donor PBDB-T with acceptors PC71BM, IT-M, and N2200 by monitoring the drying process from liquids to films with in situ ultraviolet-visible (UV-vis) absorption spectra and photoluminescent (PL) spectroscopy.45 The results indicate that higher molecular weight components dominate the drying process and final morphology of the BHJs. Furthermore, we investigated solvent impacts on the morphology of blend films and performance of the OSCs by monitoring the drying process of PBDBT:PF5–Y5 blends in chlorobenzene (CB) and ortho-xylene (o-XY) with in situ multifunctional spectroscopy.46 Finer-mixed donor/acceptor nanostructures are obtained in CB-cast film corresponding to more efficient charge generation in the corresponding solar cells. In addition, we also revealed the function of a commonly used solvent additive 1-chloronaphthalene (CN) in enhancing the performance of all-PSCs based on PBDB-T:PF5-Y5 by studying the drying process, which suggests that the improved performance of PBDB-T:PF5-Y5 solar cells originated from enhanced crystallinity and hole mobility since CN promotes self-aggregation of PBDB-T during the drying process.47
Recently, a kind of PAs with a flexible conjugation-break spacer (FCBS) has been developed. With the introduction of FCBS, the molecular rigidity and aggregation behavior of PAs could be regulated to achieve all-PSCs with improved mechanical properties and PV performance synergetically.37,48 The reduced backbone rigidity helps improve the miscibility with PDs and extending the long-term stability of devices. Nevertheless, the effect of the molecular weight of these polymers on the morphology and performance in all-PSCs has not been explored, particularly its effects on crystallinity behavior and drying properties.
In this work, we systematically investigated the impact of the Mn of PYTS, a polymer acceptor with FCBS demonstrating a high PCE of 14.68% and excellent mechanical stretchability with a crack onset strain of 21.64% and toughness of 3.86 MJ m−3,49 using PBDB-T (Mn = 35 kDa) as the polymer donor. Three PYTS polymers with different Mn were examined, including low Mn (∼9 kDa), medium Mn (∼13 kDa), and high Mn (∼19 kDa), denoted as PYTSL, PYTSM, and PYTSH, respectively. The devices based on PYTSH achieved the highest values of 9.43%, while those with PYTSL exhibited a significantly lower PCE of 5.99%. The morphological development in blends was monitored by in situ multi-spectroscopy, revealing that a blend consisting of PYTSH exhibits an extended drying time with the same material concentrations, resulting in more tightly packed structures with higher crystallinity, which are beneficial for more efficient charge carrier transport. Furthermore, with the help of transient absorption (TA), we found that the PBDB-T:PYTSH blend film shows a larger phase separation size and a longer lifetime of charge recombination, with which a higher possibility of charge extraction by electrodes can be reached. In summary, this study gives a cohesive comprehension regarding the influence of the polymer component molecular weight in all-PSCs on blend drying kinetics, charge transport characteristics, blend morphology, and photovoltaic characteristics, thereby establishing new correlations between molecular weight and device performance for more advanced all-PSC systems.
Experiment
Materials
All materials except the PYTS were purchased from commercial suppliers. PEDOT:PSS was purchased from Heraeus, and ZnO nanoparticles (NPs) were purchased from Avantama AG. The donor polymer PBDB-T was purchased from Ossila B.V. with a molar mass of Mn = 35 kDa and Đ = 2.2. PYTS was synthesized by Stille coupling reaction according to previously reported work.49 Unless otherwise specified, such chemicals were used without any further purification.Cyclic voltammetry
Cyclic voltammetry (CV) was measured to study the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) energy levels of the polymers. The samples drop-coated on the glassy carbon electrode were used as the working electrode. The counter electrode was platinum wire, and the reference electrode was an Ag/Ag+ (0.01 M AgNO3 + 0.1 M TBAPF6) electrode. The electrolyte used in this work was acetonitrile containing 0.1 M TBAPF6, at the scan rate of 50 mV s−1.In situ PL and absorption
The in situ PL setup is similar to our previous work.45 In short, the polymer inks were coated on a pre-cleaned glass substrate using a blade coater (Erichsen 510) with a moving blade (ZFR 2040). A 532 nm continuous wave laser diode was used to excite the wet polymer films and an optical fiber was mounted on top of the resulting films, to continuously collect emission signals to the spectrometer (QE-Pro, Ocean Optics). The integration time for every PL spectrum was set to be 100 ms.For the in situ absorption setup, a home-build stage was used to lift the glass substrate and the blade on top of the blade coater surface. An optical fiber was connected to a tungsten lamp (DH-2000, Ocean Optics) and mounted above the substrate, to shine white light onto the polymer films. The transmitted signals from the polymer films were guided by another optical fiber, which was mounted below the substrate, to the spectrometer. The integration time for every absorption spectrum was set to be 100 ms.
Transient absorption measurements
Femtosecond transient absorption measurements were carried out using a home-built transient absorption (TA) system. The light source of the TA system is a femtosecond laser coming from a regenerative amplifier (Legend Elite F 1K HE + II, Coherent), operating at 800 nm with a repetition rate of 1 kHz. The fundamental light is divided into two parts. One part of the light is used directly as the pump light. The other part is used to excite a sapphire plate for the generation of supercontinuum white light, which is further used as the probing light for differential absorption measurement. The probing light was guided into a monochromator (Omni-λ200i, Zolix) and detected using a CCD detector (Pascher Instrument). The pump “on” and “off” for a pair of sequential actinic pulses was regulated by a mechanical chopper (500 Hz, MC2000B-EC, Thorlabs) in the pump beam. With the help of a mechanical delay line, the probe is time-delayed relative to the pump. The TA measurements were performed at room temperature in ambient air.Atomic force microscopy
Atomic force microscopy (AFM) measurements were obtained using a Digital Instruments Dimension 3100 atomic force microscope operated in tapping mode, using a cantilever with a nominal spring constant of 40 N m−1. The scan size was 1 × 1 μm. Samples were blade-coated with the CB solutions of PBDB-T:PYTSL, PBDB-T:PYTSM, and PBDB-T:PYTSH with 16 mg mL−1 (PBDB-T![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PYTS = 1:1 weight ratio) on cleaned glass substrates at a speed of 11, 12, and 14 mm s−1, respectively, under ambient conditions.
:PYTS = 1:1 weight ratio) on cleaned glass substrates at a speed of 11, 12, and 14 mm s−1, respectively, under ambient conditions.
Grazing incident wide-angle X-ray scattering (GIWAXS)
The grazing incident wide-angle X-ray scattering (GIWAXS) measurements were performed at beamline 7.3.3 at the advanced light source. Samples were prepared on Si substrates using identical blend solutions as used in the devices. The 10 keV X-ray beam was incident at a grazing angle of 0.12° and 0.14°. The scattered X-ray was detected using a Dectris Pilatus 2 M photon counting detector.Ellipsometry
Spectral Mueller matrices were recorded using a dual rotating compensator ellipsometer (RC2, J. A. Woollam Co., Inc.) in the spectral range 210–1690 nm at angles of incidences 45° to 75° in steps of 5°. Data analysis was performed using the CompleteEASE software (J. A. Woollam Co., Inc.). A measurement was done on a film-free glass sample and its refractive index was modeled with a Cauchy dispersionwhere A, B and C are fit parameters. The backside of the glass substrate was roughened mechanically to reduce effects of backside reflections. The best fit n(λ) spectrum was then used as the substrate refractive index for all samples with polymer films. For the polymer samples a three-phase model (substrate/film/ambient) was used. The film refractive index was fitted with non-linear regression using a Kramer–Kronig consistent B-spline dispersion model50 simultaneously as the film thickness d was fitted. The thickness values reported have 90% confidence values of ±0.5 nm.
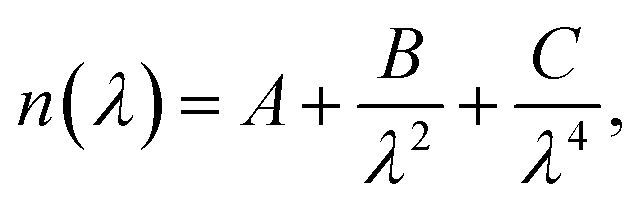
Device fabrication
Devices were fabricated with a structure of glass/indium tin oxide (ITO)/PEDOT:PSS/active layer/PFN-Br/Al. The glass/ITO substrate was first sonicated in DI water with detergent for 30 min, followed by cleaning with acetone and isopropyl alcohol (IPA) in an ultrasonic bath. This was followed by UV-ozone plasma for 20 min. PEDOT:PSS with a thickness of approximately 30 nm was deposited by spin-coating at 4000 rpm for 40 s. Later it was annealed in air for 20 min at 120 °C.For spin-coating devices, after transferring substrates to a nitrogen glovebox, a first set of active layers were then spin-coated atop ITO/PEDOT:PSS from a solution of PBDB-T:PYTSL, PBDB-T:PYTSM, and PBDB-T:PYTSH with 16 mg mL−1 (PBDB-T:PYTS = 1:1 weight ratio) in CB, at 3500 rpm for 30 s. A second set of spin-coating devices from a solution of PBDB-T:PYTSL, PBDB-T:PYTSM, and PBDB-T:PYTSH with concentrations of 13 mg mL−1, 16 mg mL−1, and 19 mg mL−1 were made, with everything else alike. The samples were annealed at 120 °C for 10 min.
For blade-coating devices, the solution of PBDB-T:PYTSL, PBDB-T:PYTSM, and PBDB-T:PYTSH at a concentration of 16 mg mL−1 (PBDB-T:PYTS = 1:1 weight ratio) in CB was blade-coated atop the PEDOT:PSS layer at a speed of 11, 12, and 14 mm s−1 respectively under ambient conditions. Then, the samples were transferred into a glove box and annealed at 120 °C for 10 min under nitrogen. The thicknesses of the active layer were optimized between 85 and 105 nm.
Later, deposition of PFN-Br (0.5 mg mL−1 in methanol) was completed at 4000 rpm for 30 s atop the active layer. Finally, the top Al electrode of 100 nm thickness was thermally evaporated through a mask with a background pressure of ∼2 × 10−7 mbar. The areas of the all-PSCs are about 0.04 cm2. The devices were encapsulated using glass and epoxy adhesive before removal from the glove box for measurements.
Characterization
The current density–voltage (J–V) characteristics of the solar cells were measured using a Keithley 2400 source meter unit under AM1.5G (100 mW cm−2) irradiation from a solar simulator (Newport MiniSol™ model LSH-7320). The J–V curves were measured from −2 to 2 V. The scan speed and dwell times were fixed at 0.05 V per step and 2 ms, respectively. The EQE spectra were measured using a QE-R solar cell spectral response measurement system (Enli Technology Co., Ltd, Taiwan).Space-charge limited current (SCLC) measurement
The electron and hole mobilities of BHJ films were measured by using the method of SCLC. The electron-only device with an architecture of ITO/ZnO nanoparticles (NPs)/PFN-Br/PBDB-T:PYTS/LiF/Al and the hole-only device with an architecture of ITO/PEDOT:PSS/PBDB-T:PYTS/MoOx/Al were fabricated. The mobility was determined by fitting the dark current to the model of a single carrier SCLC, described by the equation:where J is the current density, ε0 is the permittivity of free space, εr is the relative dielectric constant of the transport medium, μ is the charge mobility and L is the thickness of the active layer. V = Vapp − Vbi, where Vapp is the applied voltage, and Vbi is the offset voltage. The mobility can be calculated from the slope of the J0.5 ∼ V curves.
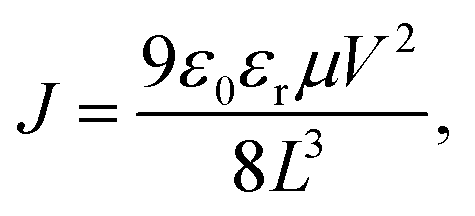
Results and discussion
Electrochemical and optical properties
The chemical structures of PBDB-T and PYTS are shown in Fig. 1a. The molecular weight and polydispersity index of the PYTS samples were characterized by gel permeation chromatography (GPC). The data are summarized in Fig. S1 (ESI†) and Table 1. The energy levels of PBDB-T and PYTS series were calculated using CV (Fig. S2, ESI†), as shown in Fig. 1b. The LUMO and HOMO energy levels of PYTS with different Mn only have a tiny gap (Table 1), suggesting the Mn has little impact on energy levels, as expected due to their similarity in chemical composition. | ||
| Fig. 1 (a) Molecular structures of donor polymer (PBDB-T) and acceptor polymer (PYTS). (b) HOMO/LUMO energy levels of the acceptor polymer of varying Mn. (c) Thin-film optical absorption spectra of the PBDB-T:PYTS blend films, with different Mn of PYTS polymers. | ||
| PAs | M n (kDa) | M w (kDa) | PDI | λ max (nm) | E g (eV) | E HOMO (eV) | E LUMO (eV) |
|---|---|---|---|---|---|---|---|
| PYTSL | 9 | 25 | 2.7 | 787 | 1.4 | −5.66 | −3.73 |
| PYTSM | 13 | 37 | 2.9 | 791 | 1.4 | −5.66 | −3.75 |
| PYTSH | 19 | 99 | 5.1 | 791 | 1.4 | −5.66 | −3.76 |
Three polymer acceptors PYTSL, PYTSM, and PYTSH were characterized for their optical properties as blend films with PBDB-T. The UV-vis absorption spectra measured in films are shown in Fig. 1c, and the corresponding data are summarized in Table 1. In the films, as Mn increased, the absorption maximum (λmax) showed a red shift from 787 nm of PYTSL to 791 nm of PYTSH, while the λmax of PYTSM remained consistent with PYTSH, suggesting that PYTSH and PYTSM demonstrated stronger π–π interactions compared to PYTSL.
The ellipsometer was used to measure the refractive index (n) and extinction coefficient (k) values of three PYTS acceptors. As shown in Fig. S3 (ESI†), all the blend films show similar n and k values and show an absorption onset at 1.4 eV, with contributions from both the donor and the acceptor. It indicated that the Mn has little impact on the optical bandgap of this semiconductor, consistent with the absorption results.
Photovoltaic performance
To explore the influence of the PYTS series with different Mn on the photovoltaic performance, all-PSCs with a regular structure of ITO/PEDOT:PSS/PBDB-T:PYTS/PFN-Br/Al were fabricated via blade-coating. The detailed procedures for the device fabrication are described in the experimental section. The devices with the same concentration of 16 mg mL−1 were fabricated via spin-coating. The distributions of open circuit voltage (Voc), Jsc, FF and PCE were depicted in Fig. S4 (ESI†) and the photovoltaic parameters are listed in Table S1 (ESI†). In comparison to devices based on PYTSL, those based on PYTSM and PYTSH have attained a much higher efficiency, where the optimal device reaches maximum PCEs of 8.16% and 8.14%, respectively, accompanied by synergistic enhancements in Voc, Jsc, and FF. However, it was observed that the same concentration for blends with different Mn gives different viscosity of the active layer solution. Specifically, a higher molecular weight results in greater solution viscosity, which not only directly influences the film formation dynamics and morphology during spin coating, but also leads to a thicker active layer. Different viscosity leads to different layer thicknesses, which may not be the optimized conditions for each device. The thickness-dependent performance of all-PSCs is shown in Fig. S5 (ESI†). In addition, viscosity influences the competitive relationship between solid–liquid (S–L) demixing and liquid–liquid (L–L) demixing. Lower solution viscosity would extend the polymer crystal growth time, contributing to increased coarseness. Conversely, increased viscosity prevents large-scale L–L phase separation.24,51 To optimize the device performance, we used different material concentrations for controlled solution viscosity for device fabrication and the detailed photovoltaic performance of the investigated PBDB-T:PYTS all-PSCs are shown in Fig. S6 (ESI†). Additionally, Fig. 2a plots the J–V curves of the best-performing solar cells based on the PYTS series as acceptors, with corresponding photovoltaic parameters summarized in Table 2. All-PSCs based on PBDB-T:PYTSH exhibit the highest PCE of 9.43% with a Voc of 0.90 V, a Jsc of 18.55 mA cm−2, and an FF of 56.32%. Slightly lower performance is found for the PBDB-T:PYTSM devices with a PCE of 8.16%, accompanied by a Voc of 0.90 V, a Jsc of 17.10 mA cm−2, and an FF of 53.30%. However, a much lower PCE of 5.99% with a Voc of 0.89 V, a Jsc of 13.02 mA cm−2, and an FF of 51.57% was obtained for the PBDB-T:PYTSL devices, mainly due to the drastically reduced Jsc and FF values. The external quantum efficiency (EQE) results demonstrated an increased photon response in the entire absorption range (Fig. 2b), which is consistent with the improved Jsc values for the corresponding PYTSH-based devices. Furthermore, the all-PSCs via blade-coating were fabricated as well, using different blade moving speeds to achieve optimized thicknesses. It yields similar trends in variation, with relevant photovoltaic parameters provided in the ESI† (Fig. S7 and Table S2). Impressively, the dark-current values systematically decreased with increasing Mn of PAs, which indicated PBDB-T:PYTSH all-PSCs possessed increased shunt resistance and suppressed leakage current, compared to those of PYTSL- and PYTSM-based devices (Fig. 2c). | ||
| Fig. 2 The PYTS Mn dependence of photovoltaic properties of optimized PBDB-T:PYTS devices. (a) J–V curves, (b) EQE spectra, (c) dark J–V curves and (d) charge mobilities. | ||
| V oc (V) | J sc (mA cm−2) | FF (%) | PCEa (%) | |
|---|---|---|---|---|
| a The values in parenthesis are the standard deviation obtained from 24 devices. | ||||
| PBDB-T:PYTSL | 0.89 (0.88 ± 0.008) | 13.02 (11.59 ± 0.67) | 51.57 (49.84 ± 5.48) | 5.99 (5.10 ± 0.75) |
| PBDB-T:PYTSM | 0.90 (0.89 ± 0.004) | 17.10 (15.60 ± 1.17) | 53.30 (53.79 ± 2.58) | 8.16 (7.64 ± 0.39) |
| PBDB-T:PYTSH | 0.90 (0.89 ± 0.010) | 18.55 (16.73 ± 1.25) | 56.32 (57.57 ± 1.75) | 9.43 (8.60 ± 0.63) |
To gain a deeper insight into the FF and Jsc changes between devices, the charge mobilities for blends were evaluated by the space charge limited current (SCLC) method (Fig. 2d and Fig. S8, ESI†). The hole mobilities (μhs), which were estimated from hole-only devices with a structure of ITO/PEDOT:PSS/PBDB-T:PYTS/MoOx/Al, are 5.78 × 10−4 cm2 V−1 s−1 (PBDB-T:PYTSL), 5.92 × 10−4 cm2 V−1 s−1 (PBDB-T:PYTSM) and 5.89 × 10−4 cm2 V−1 s−1 (PBDB-T:PYTSH), respectively. The μh remains independently unchanged in different blend films due to the same donor. The μes from electron-only devices of ITO/ZnO NPs/PFN-Br/PBDB-T:PYTS/LiF/Al were 7.91 × 10−5 cm2 V−1 s−1 (PBDB-T:PYTSL), 9.97 × 10−5 cm2 V−1 s−1 (PBDB-T:PYTSM) and 1.67 × 10−4 cm2 V−1 s−1 (PBDB-T:PYTSH), respectively. The improved electron mobilities of PBDB-T:PYTSH, which could be ascribed to the enhanced crystallinity of the acceptor, balance the charge transport of blends, presenting a minimum μh/μe value of 3.52, while that of PBDB-T: PYTSL and PBDB-T:PYTSM are 5.94 and 7.30, respectively. The enhancement of charge carrier transport property allowed the PYTSH-based devices to achieve an improved Jsc and FF. It is well recognized that FF mainly depends on free charge recombination and charge carrier transport. We noticed that the devices processed active layers from solutions at RT exhibit relatively smaller FFs than those of all-PSCs based on PBDB-T:PYTSM spin-coated from hot solutions at 80 °C.49 By comparing the above electron mobilities (from 7.91 × 10−5 cm2 V−1 s−1 to 1.67 × 10−4 cm2 V−1 s−1) with those (4.8 × 10−4 cm2 V−1 s−1) in ref. 49, we believe that the smaller FFs in our devices could be dominantly attributed to the relatively smaller μes of PYTS acceptors and their imbalance with the μh (∼5.8 × 10−4 cm2 V−1 s−1) of the donor PBDB-T. Slowly electron transport from the BHJs to the collecting electrode would increase the possibility of charge recombination and reduce FFs.
We further evaluated the correlation between Mn and the storage stability of all-PSCs. As shown in Fig. S9 (ESI†), all devices exhibit two stage degradations, a fast decay in 72 hours, then followed by a slow decay. The stabilities present an opposite tendence with device performance and Mn, that is, the PYTSL-based devices showed the best stability, remaining 72.7% of the initial PCE after 288 hours, while PYTSM and PYTSH-based devices decayed to 64.4% and 60.4% of the initial PCE, respectively. The stability result suggests that a higher Mn of PYTS acceptors could not enhance the stability of all-PCSs.
In situ monitoring of film-formation and morphology development
Fully comprehending and regulating morphology can be obtained by monitoring the film formation process of BHJ films. In this study, we used in situ UV-vis and PL spectroscopic recording film drying through blade-coating with a custom-built setup45 to discern the effects of the molecular weight of PAs on morphological evolution.46,47 Fig. S10 (ESI†) depicts the temporal evolutions of UV-vis absorption contour maps of blends with PAs of different Mn, where the colors represent the intensity of absorption signals at a specific time interval and wavelength. According to our previous work, the drying process can be divided into three temporal stages, i.e., the liquid film stage (t0 < t < t1), the liquid–solid transition stage (t1 < t < t2), and eventually the dried film stage (t2 < t).47 As seen in Fig. S11 (ESI†), the absorption peaks of PYTS in all blend films present different degrees of red shifts (from 773 nm to 788 nm for PYTSL, from 776 nm to 787 nm for PYTSM, and from 776 nm to 782 nm for PYTSH), implying the presence of ordered packing in these molecules and the formation of π–π interactions in the solid films. After annealing, the peak positions of PYTSM and PYTSH exhibit a further red-shift to 791 nm (Fig. S12, ESI†), which is attributed to the further aggregation of the longer polymer chains under thermal stress. In the meantime, the peak position of PBDB-T remained almost unchanged along with time in all three systems (∼620 nm). In our previous work, a similar phenomenon was observed. This indicates that self-aggregation of PBDB-T was restricted by blending with polymer acceptors.29,31 By increasing Mn, the crystallization of the blend film tended to slow down, as revealed by the extended drying time from 33 s (PYTSL), to 43 s (PYTSM) and to 51 s (PYTSH) (Fig. S11, ESI†). With longer crystallization time, the crystal size could be larger, forming more ordered crystallinity structures. These behaviors are in line with the observation that the film morphology of the blends containing PYTSH exhibits strongest crystalline order shown in GIWAXS measurements, compared to that of PYTSL, which will be discussed later.We proceeded to assess the quenching kinetics through in situ PL characterization. The contour maps demonstrating PL evolution are presented in Fig. S13 (ESI†). Fig. 3 displays the time-dependent PL peak location and intensity of blends consisting of PAs with different Mn. The extracted temporal evolutions of the PL peak position and integrated intensity are associated with the spatial distribution of fluorophores and quenchers in the blends.52 The PL peak position exhibited a similar evolution trend to that observed in the in situ absorption spectra, characterizing three stages as well. Within the liquid film stage, the emission from PYTS undergoes a gradual quenching with a weak red shift, indicating the slight enhancement of interactions between polymer chains. Besides, compared to PYTSL, PYTSM and PYTSH show a greater redshift of the original PL peak, implying stronger preaggregation of acceptors in solutions. The PBDB-T:PYTSL blend film shows a liquid film stage time of 12 s, which is much shorter than that of PBDB-T:PYTSM (21 s) and PBDB-T:PYTSH (24 s). It indicates a more rapid formation of interactions between PBDB-T and PYTSL, accelerating the growth rate of the film. In the second stage, the PYTS shows a more rapid quenching, accompanied by a rapid red shift. The spectral red-shift contributes to the polymer aggregation and the rapid drops in the PL signals arise from the fluorophore-quencher interaction.53 This suggested the proximity between the donor and acceptor molecules decreases as the solvent evaporates. Compared with the PBDB-T:PYTSL blend film (t2 ∼ 17 s), the prolonged PL quenching of PBDB-T:PYTSH (t2 ∼ 30 s) can be assigned to a slower interdiffusion process between PBDB-T and PYTSH, implying a lower miscibility between these two polymers. Therefore, a larger phase separation was formed in the PBDB-T:PYTSH film. With the larger pure phase of donor and acceptor, the charge carrier transportation can be enhanced, in line with the improved mobility values. After that, the PL intensities and peak positions are constant, suggesting the complete formation of solid films.
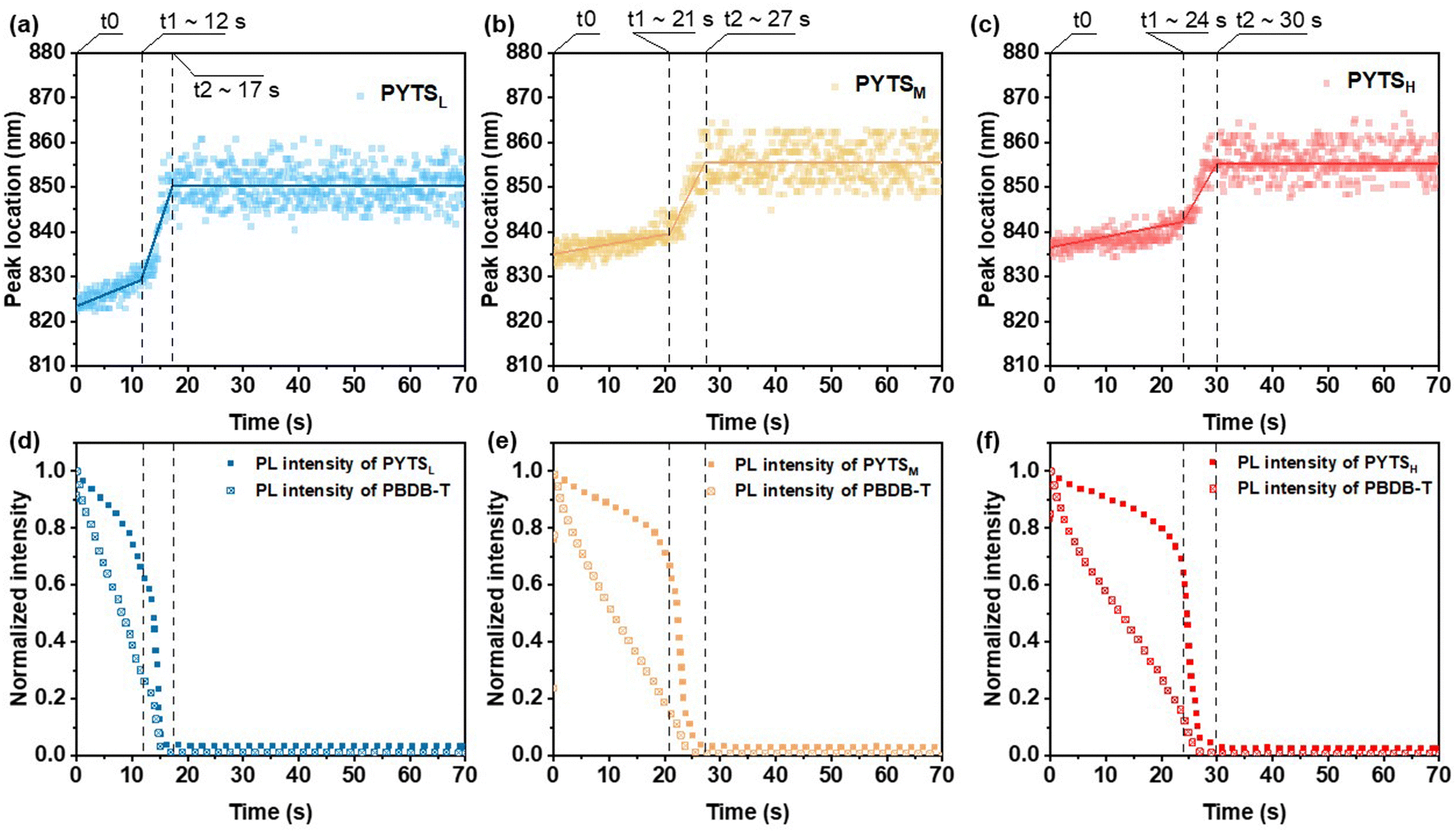 | ||
| Fig. 3 In situ PL results of the PBDB-T:PYTS blend film via blade-coating, as a function of drying time. Locations of emission peaks of (a) PYTSL, (b) PYTSM, and (c) PYTSH, respectively. Peak intensity for PBDB-T and PYTS emission peaks (at 670 and 850 nm, respectively) of (d) PYTSL, (e) PYTSM, and (f) PYTSH, respectively. | ||
TA spectroscopy
To investigate the role of molecular weight on charge photogeneration and recombination processes in an ultrafast timescale, we performed the transient absorption (TA) measurements of blend films. Fig. 4 shows the TA spectra of the blend films at the indicated delay time by selecting to excite the acceptor materials with the excitation wavelength of 800 nm.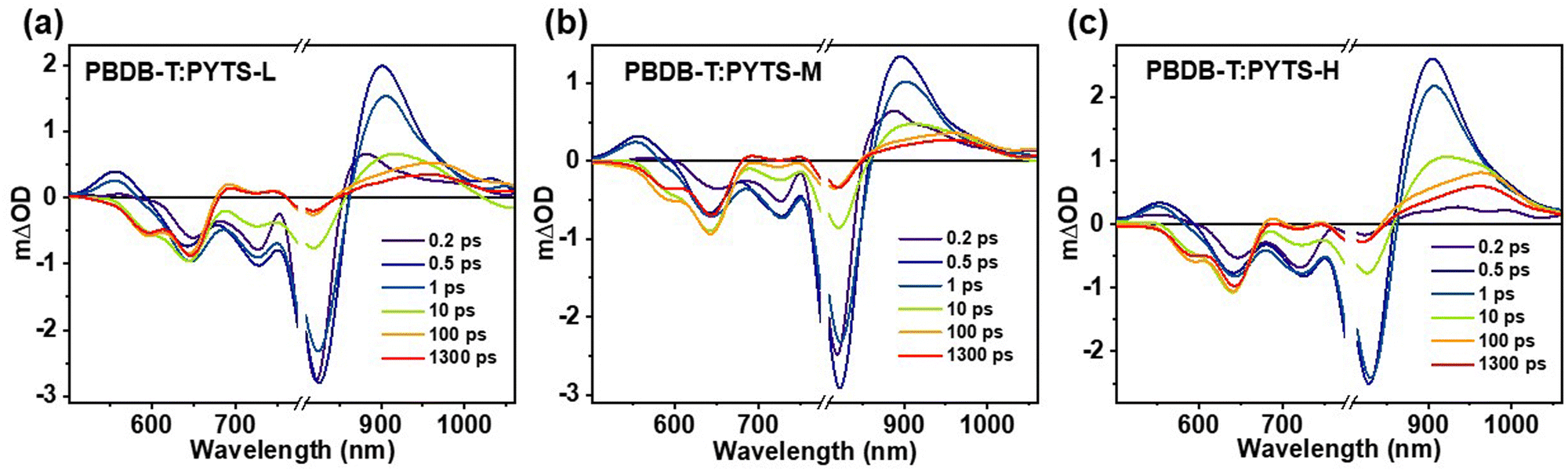 | ||
| Fig. 4 The representative TA spectra of (a) PBDB-T:PYTSL, (b) PBDB-T:PYTSM and (c) PBDB-T:PYTSH blend films after excitation at 800 nm with an excitation fluence of 3.6 × 1013 photons cm−2 pulse−1. | ||
The TA kinetics of ∼645 nm were extracted to estimate the charge photogeneration and recombination processes in the blend films, which corresponds to the ground state bleaching process of the donor material, as shown in Fig. S14 (ESI†). The kinetics of 645 nm in the PBDB-T:PYTSL blend film display a fast generation and a slow relaxation process. However, an instantaneous generation, a slow rise and a slow relaxation process is exhibited in the kinetics of ∼645 nm in PBDB-T:PYTSM and PBDB-T:PYTSH blend films. The dynamics of hole-transfer involve both an ultrafast charge-transfer process and an exciton diffusion-mediated charge transfer process, that can be fitted by a biexponential function:46,54
 ps than PBDB-T:PYTSM (≈5.7ps) and PBDB-T:PYTSL (within instrument response function), indicating a larger acceptor domain size in PBDB-T:PYTSH films.55–57 The phase domain size increases with the increasing molecular weight of PA, which corresponds with the results of AFM measurements (Fig. S16, ESI†).
ps than PBDB-T:PYTSM (≈5.7ps) and PBDB-T:PYTSL (within instrument response function), indicating a larger acceptor domain size in PBDB-T:PYTSH films.55–57 The phase domain size increases with the increasing molecular weight of PA, which corresponds with the results of AFM measurements (Fig. S16, ESI†).
Besides, we found the TA kinetics of 645 nm recovered with a delay time longer than 30 ps, which can be attributed to the charge recombination process. Accordingly, the charge recombination rate is independent and dependent on the excitation fluency in geminate recombination and bimolecular recombination, respectively. In this work, we performed the TA measurements of blend films with different fluencies, as shown in Fig. S15 (ESI†). It can be seen that the charge recombination mechanism for PBDB-T:PYTSL, PBDB-T:PYTSM and PBDB-T:PYTSH blend films can be attributed to geminate, geminate, and bimolecular recombination, respectively. Besides, by using the single-exponential function to fit the charge recombination kinetics of 645 nm, we can estimate the lifetime of charge recombination to ∼3.7 ns, ∼3.9 ns, and ∼5.4 ns for PBDB-T:PYTSL, PBDB-T:PYTSM and PBDB-T:PYTSH blend films, respectively, and the detailed fitting parameters can be found in Table S4 (ESI†). This indicates the increasing molecular weight of the acceptor material can suppress the charge recombination process in the blend film. Combining the TA measurement and the performance of devices, it is concluded that the PBDB-T:PYTSH blend film shows a larger acceptor phase separation, a longer lifetime of charge recombination and thus a higher PCE of ∼9.43% over those of PBDB-T:PYTSL blend film, indicating the Mn plays a critical role in charge transportation and recombination processes and the performance of devices.
Morphological properties
To elucidate the origins of the different photovoltaic performance of the PBDB-T:PYTS blends, we examined the morphological properties of the blend films using the combined measurements of grazing incident wide-angle X-ray diffraction (GIWAXS) and atomic force microscopy (AFM). In the AFM images, as Mn increased from 9 kDa to 19 kDa, the root-mean-square averaged surface roughness (Rq) of the blends increased from 1.24 nm to 1.75 nm (Fig. S16, ESI†), and the sizes of the domains became larger with increasing Mn. This indicates that the longer polymer chains increased the phase segregation degree between PD and PA. The PBDB-T:PYTSL films lack phase separated domains, which suppressed the exciton dissociation and promoted charge recombination, thus resulting in reduced Jsc and FF. Fig. 5a–c presents the 2D GIWAXS images of PBDB-T:PYTS blends, with Fig. 5d showing the 1D scattering profiles of the blends taken along the in-plane and out of plane directions. Table S5 (ESI†) presents the peak fitting results for the out-of-plane π stacking peaks (010) and in-plane (100) peaks in the blends. The PBDB-T:PYTSL blend film shows a broad (010) peak in the out-of-plane (OOP) direction at 1.66 Å−1 (d = 3.78 Å) with a crystal coherence length (CCL) of 1.61 nm and a (100) peak in the in-plane (IP) direction at 0.28 Å−1 (d = 22.44 Å) with a CCL of 7.84 nm. The PBDB-T:PYTSM and PBDB-T:PYTSH blend film exhibits enhanced molecular co-facial packing, as indicated by the sharper (010) peak in the OOP direction at 1.68 Å−1 (d = 3.74 Å) with an increased CCL of 1.77 and 2.19 nm, respectively, indicating the π–π stacking and crystal size of polymers were further improved. Generally, the π–π stacking distance and CCL results affect the charge transport characteristics of the polymer materials.39,58 By increasing Mn, the blends utilize a better transport feature of PYTSH than PYTSL due to larger pure polymer domains, which is consistent with the mobility values. | ||
| Fig. 5 2D GIWAXS patterns of PBDB-T:PYTS blend films with different Mn. (a) PBDB-T:PYTSL, (b) PBDB-T:PYTSM, and (c) PBDB-T:PYTSH. (d) Linecuts of GIWAXS patterns for the three blend films. | ||
Conclusions
In this work, from liquids to films and devices, the Mn effect of PYTS on PCEs of all-PSCs based on PBDB-T:PYTS blends with three different Mn has been comprehensively investigated using in situ spectroscopy and several ex situ techniques. The Jsc of devices increases with the Mn (9, 13 and 19 kDa) of PYTS from 13.02, 17.16 to 18.73 mA cm−2, which results in PCE enhancement from 5.99%, 8.16% to 9.43%. The morphology evolutions from liquids to solid BHJs of the three PBDB-T:PYTS systems were monitored using in situ multifunctional spectroscopy. Furthermore, the nanostructures of PBDB-T:PYTS films, charge generation, transport and recombination of corresponding all-PSCs were characterized with GIWAXS, AFM and TAS. The results indicate that the increased PCE of all-PSCs with Mn of PYTS was mainly attributed to the improved crystallinity in PBDB-T:PYTSH during drying, which facilitates electron transport, prolongs carrier lifetimes and decreases the non-geminate recombination rate constants. This study deepens our understanding of the interplay between Mn, morphology evolutions, nanostructure of BHJs, and the photovoltaic performance in the corresponding devices. It also highlights the irreplaceable function of in situ spectroscopy to record the nanostructure evolution from liquid to BHJ for further improving the PCEs of full PSCs.Author contributions
Y. L. conceived the topic and guided S. E. on the in situ setup. J. X. and S. E. conducted the experiments of in situ analysis, fabrication and characterization of all-PSCs. Z. G. and E. W. provided polymer acceptors. G. W. and W. Z. conducted and analysed TA measurement, R. Z. carried out GIWAXS and interpretation. L. Q. took voltammetry measurements. L. W. imaged the surfaces of blend films with AFM, Y. L. and N. Y. assistant S. E. work in the lab, J. X. and F. Z. composed the draft of the manuscript, integrated contributions from all co-authors and finalized the manuscript. F. Z. supervised the work and coordinated the collaborations.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Wallenberg Initiative Materials Science for Sustainability (WISE) and the project “Mastering Morphology for Solution-borne Electronics” (2016.0059) funded by the Knut and Alice Wallenberg Foundation. S. E. and F. Z. acknowledge funding from Swedish Government Research Area in Materials Science on Functional Materials at Linköping University Faculty Grant SFO-Mat-LiU No. 200900971, the Swedish Research Council (2017-04123) and the Swedish Energy Agency (52485-1) for financial support. J. X., F. Z., S. E., G. W. and W. Z. acknowledge funding from Guangzhou University of China. W. Z. acknowledges funding from the National Natural Science Foundation of China (No. 21903017), the Guangdong Basic and Applied Basic Research Foundation (No. 2023A1515011500), and On Campus Research Projects at Guangzhou University (No. ZH2023005). E. W. acknowledges funding from the Knut and Alice Wallenberg foundation under contract 2022.0192, the Swedish Research Council (2019-04683), and the Swedish Research Council Formas (2020-01201). Y. L. acknowledges funding from Jiaxing University (No. CD70523003), and the Open Project Program of Key Laboratory of Yarn Materials Forming and Composite Processing Technology of Zhejiang Province (No. MTC2023-02).Notes and references
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS
.
- G. Yu and A. J. Heeger, J. Appl. Phys., 1995, 78, 4510–4515 CrossRef CAS
- M. Jonforsen, T. Johansson, O. Inganäs and M. R. Andersson, Macromolecules, 2002, 35, 1638–1643 CrossRef CAS
- F. Zhang, M. Jonforsen, D. M. Johansson, M. R. Andersson and O. Inganäs, Synth. Met., 2003, 138, 555–560 CrossRef CAS
- M. Granström, K. Petritsch, A. C. Arias, A. Lux, M. R. Andersson and R. H. Friend, Nature, 1998, 395, 257–260 CrossRef
- A. C. Arias, J. D. MacKenzie, R. Stevenson, J. J. M. Halls, M. Inbasekaran, E. P. Woo, D. Richards and R. H. Friend, Macromolecules, 2001, 34, 6005–6013 CrossRef CAS
- C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 15–26 CrossRef CAS
- J. Yi, G. Zhang, H. Yu and H. Yan, Nat. Rev. Mater., 2024, 9, 46–62 CrossRef CAS
- H. Yu, Y. Wang, X. Zou, J. Yin, X. Shi, Y. Li, H. Zhao, L. Wang, H. M. Ng, B. Zou, X. Lu, K. S. Wong, W. Ma, Z. Zhu, H. Yan and S. Chen, Nat. Commun., 2023, 14, 2323 CrossRef CAS PubMed
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed
- G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Angew. Chem., Int. Ed., 2019, 58, 4129–4142 CrossRef CAS PubMed
- Z. Genene, W. Mammo, E. Wang and M. R. Andersson, Adv. Mater., 2019, 31, 1807275 CrossRef PubMed
- Q. Fan, W. Su, S. Chen, W. Kim, X. Chen, B. Lee, T. Liu, U. A. Méndez-Romero, R. Ma, T. Yang, W. Zhuang, Y. Li, Y. Li, T.-S. Kim, L. Hou, C. Yang, H. Yan, D. Yu and E. Wang, Joule, 2020, 4, 658–672 CrossRef CAS
- R. Zeng, L. Zhu, M. Zhang, W. Zhong, G. Zhou, J. Zhuang, T. Hao, Z. Zhou, L. Zhou, N. Hartmann, X. Xue, H. Jing, F. Han, Y. Bai, H. Wu, Z. Tang, Y. Zou, H. Zhu, C.-C. Chen, Y. Zhang and F. Liu, Nat. Commun., 2023, 14, 4148 CrossRef CAS PubMed
- P. Wu, Y. Duan, Y. Li, X. Xu, R. Li, L. Yu and Q. Peng, Adv. Mater., 2024, 36, 2306990 CrossRef CAS PubMed
- T. Zhang, Y. Xu, H. Yao, J. Zhang, P. Bi, Z. Chen, J. Wang, Y. Cui, L. Ma, K. Xian, Z. Li, X. Hao, Z. Wei and J. Hou, Energy Environ. Sci., 2023, 16, 1581–1589 RSC
- Z. Ge, J. Qiao, Y. Li, J. Song, C. Zhang, Z. Fu, M. H. Jee, X. Hao, H. Y. Woo and Y. Sun, Adv. Mater., 2023, 35, 2301906 CrossRef CAS PubMed
- J. Wen, R. Lin, Y. Wu, H. Zhang, H. Zhou, Z. Liu, Y. Xie, D. Ye and X. Ouyang, EcoMat, 2023, 5, e12318 CrossRef CAS
- J. Xiang, Z.-X. Liu, H. Chen and C.-Z. Li, Adv. Mater., 2023, 35, 2303729 CrossRef CAS PubMed
- J. Wen, R. Lin, Y. Wu, H.-C. Hu, Z. Liu, H. Zhou and X. Ouyang, Chem. Eng. J., 2022, 450, 138169 CrossRef CAS
- Z.-G. Zhang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 4422–4433 CrossRef CAS PubMed
- D. Mori, H. Benten, H. Ohkita and S. Ito, Adv. Energy Mater., 2015, 5, 1500304 CrossRef
- R. S. Gurney, D. G. Lidzey and T. Wang, Rep. Prog. Phys., 2019, 82, 036601 CrossRef CAS PubMed
- C. McDowell, M. Abdelsamie, M. F. Toney and G. C. Bazan, Adv. Mater., 2018, 30, 1707114 CrossRef PubMed
- N. D. Treat and M. L. Chabinyc, Annu. Rev. Phys. Chem., 2014, 65, 59–81 CrossRef CAS PubMed
- J. Wang, Y. Wang, K. Xian, J. Qiao, Z. Chen, P. Bi, T. Zhang, Z. Zheng, X. Hao, L. Ye, S. Zhang and J. Hou, Adv. Mater., 2024, 36, 2305424 CrossRef CAS PubMed
- J. J. van Franeker, G. H. L. Heintges, C. Schaefer, G. Portale, W. Li, M. M. Wienk, P. van der Schoot and R. A. J. Janssen, J. Am. Chem. Soc., 2015, 137, 11783–11794 CrossRef CAS PubMed
- A. Khasbaatar, Z. Xu, J.-H. Lee, G. Campillo-Alvarado, C. Hwang, B. N. Onusaitis and Y. Diao, Chem. Rev., 2023, 123, 8395–8487 CrossRef CAS PubMed
- Q. Fan, Q. An, Y. Lin, Y. Xia, Q. Li, M. Zhang, W. Su, W. Peng, C. Zhang, F. Liu, L. Hou, W. Zhu, D. Yu, M. Xiao, E. Moons, F. Zhang, T. D. Anthopoulos, O. Inganäs and E. Wang, Energy Environ. Sci., 2020, 13, 5017–5027 RSC
- K. Zhou, K. Xian and L. Ye, InfoMat, 2022, 4, e12270 CrossRef CAS
- Z. Wang, Y. Guo, X. Liu, W. Shu, G. Han, K. Ding, S. Mukherjee, N. Zhang, H.-L. Yip, Y. Yi, H. Ade and P. C. Y. Chow, Nat. Commun., 2024, 15, 1212 CrossRef CAS PubMed
- D. K. Tran, A. Robitaille, I. Jo Hai, X. Ding, D. Kuzuhara, T. Koganezawa, Y.-C. Chiu, M. Leclerc and S. A. Jenekhe, J. Mater. Chem. A, 2020, 8, 21070–21083 RSC
- N. Zhou, A. S. Dudnik, T. I. N. G. Li, E. F. Manley, T. J. Aldrich, P. Guo, H.-C. Liao, Z. Chen, L. X. Chen, R. P. H. Chang, A. Facchetti, M. Olvera de la Cruz and T. J. Marks, J. Am. Chem. Soc., 2016, 138, 1240–1251 CrossRef CAS PubMed
- K. D. Deshmukh, R. Matsidik, S. K. K. Prasad, L. A. Connal, A. C. Y. Liu, E. Gann, L. Thomsen, J. M. Hodgkiss, M. Sommer and C. R. McNeill, Adv. Funct. Mater., 2018, 28, 1707185 CrossRef
- Z. Li, W. Zhong, L. Ying, F. Liu, N. Li, F. Huang and Y. Cao, Nano Energy, 2019, 64, 103931 CrossRef CAS
- H. Kang, M. A. Uddin, C. Lee, K.-H. Kim, T. L. Nguyen, W. Lee, Y. Li, C. Wang, H. Y. Woo and B. J. Kim, J. Am. Chem. Soc., 2015, 137, 2359–2365 CrossRef CAS PubMed
- Q. Fan, W. Su, S. Chen, T. Liu, W. Zhuang, R. Ma, X. Wen, Z. Yin, Z. Luo, X. Guo, L. Hou, K. Moth-Poulsen, Y. Li, Z. Zhang, C. Yang, D. Yu, H. Yan, M. Zhang and E. Wang, Angew. Chem., Int. Ed., 2020, 132, 20007–20012 CrossRef
- S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Nat. Mater., 2020, 19, 491–502 CrossRef CAS PubMed
- K. Gu and Y.-L. Loo, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1559–1571 CrossRef CAS
- A. J. Gillett, A. Privitera, R. Dilmurat, A. Karki, D. Qian, A. Pershin, G. Londi, W. K. Myers, J. Lee, J. Yuan, S.-J. Ko, M. K. Riede, F. Gao, G. C. Bazan, A. Rao, T.-Q. Nguyen, D. Beljonne and R. H. Friend, Nature, 2021, 597, 666–671 CrossRef CAS PubMed
- K. Gu, C. R. Snyder, J. Onorato, C. K. Luscombe, A. W. Bosse and Y.-L. Loo, ACS Macro Lett., 2018, 7, 1333–1338 CrossRef CAS PubMed
- D. T. Duong, V. Ho, Z. Shang, S. Mollinger, S. C. B. Mannsfeld, J. Dacuña, M. F. Toney, R. Segalman and A. Salleo, Adv. Funct. Mater., 2014, 24, 4515–4521 CrossRef CAS
- J. Jia, Q. Huang, T. Jia, K. Zhang, J. Zhang, J. Miao, F. Huang and C. Yang, Adv. Energy Mater., 2022, 12, 2103193 CrossRef CAS
- I. Jalan, C. F. N. Marchiori, Z. Genene, A. Johansson, C. Moyses Araujo, E. Wang, J. van Stam and E. Moons, J. Mater. Chem. C, 2023, 11, 9316–9326 RSC
- Y. Liu, A. Yangui, R. Zhang, A. Kiligaridis, E. Moons, F. Gao, O. Inganäs, I. G. Scheblykin and F. Zhang, Small Methods, 2021, 5, 2100585 CrossRef CAS PubMed
- N. Yao, Q. Fan, Z. Genene, H. Liu, Y. Xia, G. Wen, Y. Yuan, E. Moons, J. van Stam, W. Zhang, X. Lu, E. Wang and F. Zhang, Sol. RRL, 2023, 7, 2201134 CrossRef CAS
- Y. Liu, Q. Fan, H. Liu, I. Jalan, Y. Jin, J. van Stam, E. Moons, E. Wang, X. Lu, O. Inganäs and F. Zhang, J. Phys. Chem. Lett., 2022, 13, 11696–11702 CrossRef CAS PubMed
- Q. Fan, R. Ma, T. Liu, J. Yu, Y. Xiao, W. Su, G. Cai, Y. Li, W. Peng, T. Guo, Z. Luo, H. Sun, L. Hou, W. Zhu, X. Lu, F. Gao, E. Moons, D. Yu, H. Yan and E. Wang, Sci. China: Chem., 2021, 64, 1380–1388 CrossRef CAS
- Z. Genene, J.-W. Lee, S.-W. Lee, Q. Chen, Z. Tan, B. A. Abdulahi, D. Yu, T.-S. Kim, B. J. Kim and E. Wang, Adv. Mater., 2022, 34, 2107361 CrossRef CAS PubMed
- B. Johs and J. S. Hale, Phys. Status Solidi A, 2008, 205, 715–719 CrossRef CAS
- Z. Hamid, A. Wadsworth, E. Rezasoltani, S. Holliday, M. Azzouzi, M. Neophytou, A. A. Y. Guilbert, Y. Dong, M. S. Little, S. Mukherjee, A. A. Herzing, H. Bristow, R. J. Kline, D. M. DeLongchamp, A. A. Bakulin, J. R. Durrant, J. Nelson and I. McCulloch, Adv. Energy Mater., 2020, 10, 1903248 CrossRef CAS
- W. Zhang, Y. Yue, R. Yang, Y. Zhang, W. Du, G. Lu, J. Zhang, H. Zhou, X. Zhang and Y. Zhang, Energy Environ. Sci., 2024, 17, 2182–2192 RSC
- Y. Wang, X. Wang, B. Lin, Z. Bi, X. Zhou, H. B. Naveed, K. Zhou, H. Yan, Z. Tang and W. Ma, Adv. Energy Mater., 2020, 10, 2000826 CrossRef CAS
- C. Chen, G. Wen, Z. Xiao, J. Peng, R. Hu, Z. Chen, C. Zhang and W. Zhang, Photonics, 2022, 9, 892 CrossRef CAS
- G. Wen, R. Hu, J. Feng, J. Peng, Z. Chen, C. Zhang, N. Zhao, X. Zou, Z. Chen, C. Liu and W. Zhang, Chem. Eng. J., 2023, 475, 145939 CrossRef CAS
- Z. Chen and H. Zhu, J. Phys. Chem. Lett., 2022, 13, 1123–1130 CrossRef CAS PubMed
- J. Feng, G. Wen, R. Hu, J. Peng, H. Lu, N. Zhao, X. Zou and W. Zhang, Sol. RRL, 2023, 7, 2300372 CrossRef CAS
- Z. Xu, K. S. Park, J. J. Kwok, O. Lin, B. B. Patel, P. Kafle, D. W. Davies, Q. Chen and Y. Diao, Adv. Mater., 2022, 34, 2203055 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc02217c |
| This journal is © The Royal Society of Chemistry 2024 |