
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinuclear iridium(III) complexes for efficient near-infrared light-emitting electrochemical cells with electroluminescence up to 800 nm†
Lavinia
Ballerini
 a,
Wei-Min
Zhang
b,
Thomaz
Groizard
c,
Christophe
Gourlaouen
c,
Federico
Polo
de,
Abdelaziz
Jouaiti
*f,
Hai-Ching
Su
*b and
Matteo
Mauro
*a
a,
Wei-Min
Zhang
b,
Thomaz
Groizard
c,
Christophe
Gourlaouen
c,
Federico
Polo
de,
Abdelaziz
Jouaiti
*f,
Hai-Ching
Su
*b and
Matteo
Mauro
*a
aInstitut de Physique et Chimie des Matériaux de Strasbourg (IPCMS) UMR7504, Université de Strasbourg & CNRS, 23 rue du Loess, 67083 Strasbourg, France. E-mail: mauro@unistra.fr
bInstitute of Lighting and Energy Photonics, National Yang Ming Chiao Tung University, Tainan 71150, Taiwan. E-mail: haichingsu@nycu.edu.tw
cLaboratoire de Chimie Quantique, Institut de Chimie de Strasbourg UMR7177, Université de Strasbourg & CNRS, Rue Blaise Pascal, 67008 Strasbourg, France
dDepartment of Molecular Sciences and Nanosystems, Ca’ Foscari University of Venice Via Torino 155, 30172 Venezia, Italy
eEuropean Centre for Living Technology (ECLT), Ca' Bottacin, 30124, Venice, Italy
fLaboratoire de Synthèse et Fonctions des Architectures Moléculaires, UMR7140 Chimie de la Matiere Complexe, Université de Strasbourg & CNRS 4 rue Blaise, Pascal 67000 Strasbourg, France. E-mail: jouaiti@unistra.fr
First published on 9th July 2024
Abstract
Near infrared (NIR) emitting optoelectronic devices have great potential for applications in communication, encryption technologies, night-vision display and photodynamic biomedical devices. Nevertheless, their development is currently hampered by the lack of efficient NIR-emissive materials. Herein, a novel class of cationic binuclear Ir(III) emitters (Ir-D1 and Ir-D2) based on a ditopic coordinating scaffold featuring the π-deficient thiazolo[5,4-d]thiazole and π-accepting moiety (either pyridine or pyrazine), is described and fully characterized using photophysical and computational techniques. Comparison with the parental mononuclear derivatives Ir-M1 and Ir-M2 is provided as well. Remarkably, the binuclear complexes display NIR photoluminescence in solution with a maximum up to λem = ca. 840 nm, which represent some rare examples of metal complexes emitting in this spectral region. Interestingly, NIR photoluminescence is retained in polymer-matrix thin-film for the binuclear counterparts. These findings prompt the successful use of these NIR emitters as electroactive materials in light emitting electrochemical cells (LECs). Binuclear complexes Ir-D1 and Ir-D2 yield electroluminescence peaking at λEL = 750 and 800 nm, respectively, and device performances that are the highest reported for LECs in this spectral region to date for molecular (i.e. non-excimer) emitters. This work demonstrates the superior performances of the binuclear design strategy for achieving efficient NIR electroluminescence.
Introduction
Light-emitting electrochemical cells (LECs) are an alternative and appealing class of optoelectronic devices with potential application in low-cost, large-area display technology and disposable devices.1–4 Efficient LECs make use of mobile ions that are able to migrate toward the oppositely charged electrodes under an applied electrical bias and form the p–i–n junction where light-emission takes place. Toward this aim, ionic transition metal complexes (iTMCs) are typically used as electroluminescent materials, particularly cyclometalated IrIII-based complexes,5 due to their outstanding redox and photophysical properties. Furthermore, the use of charged emitters may render the addition of additive salts unnecessary, minimizing detrimental segregation processes and device complexity. While the successful fabrication of blue- to red-emitting LEC devices with good efficiency has been demonstrated to date,6–11 targeting deep-red to near infrared (NIR) electroluminescence (λEL >650 nm) in LECs is highly challenging.12a Nevertheless, these NIR emitting devices find promising applications in night-vision technology, telecommunication, data encryption, counterfeiting technology, and as bio-medical phototherapy devices.12b–e The main limiting factor is the availability of efficient and bright deep-red to NIR emitters as electroactive materials due to the intrinsic difficulty to achieve a suitable molecular design.12,13 Indeed, long-wavelength emitters typically suffer from low to near-zero photoluminescence quantum yield (PLQY) due to a combination of two factors: (i) the efficient non-radiative vibronic coupling between the potentially emitting lowest-lying triplet (T1) and ground (S0) state that shows a natural logarithm dependence of the non-radiative rate constant (knr) with the T1–S0 energy gap, as ruled by the energy gap law;14 (ii) the third-power dependency of the radiative rate constant (kr) on the energy of the emitted photon. Designing emitters that possess a combination of high kr and low knr is of utmost importance to achieve radiative recombination that is competitive with the radiationless channels, and it is therefore the key for achieving efficient photo- and electro-luminescence into the NIR spectral region. This goal is even more challenging when cationic iTMC are targeted since they often display even lower PLQY compared to the neutral counterparts. A typical strategy to shift the emission toward a longer wavelength region relies on the extension of the π-system of the chromophoric ligand. Regrettably, this is accompanied by an increase in the triplet ligand centred (3LC) character of the excited state, which in turn slows down SOC processes and lowers the kr. The vast majority of the examples deal with mononuclear iridium(III) derivatives15–27 and the formation of excimers was demonstrated as being an appealing approach as well.20 However, excimer-based emission often has the drawback of displaying emission and device performances strongly dependent on the concentration and fabrication process. Alternatively, a dinuclearization approach has recently emerged to be a superior chemical design strategy for addressing longer wavelength emission as demonstrated by other groups28–33 as well as by us.34–37 This is due to the fact that the presence of two (or more) heavy metal atoms helps to improve SOC processes, also by increasing the metal character of the emitting states, and mixing via configurational interaction.38 The higher rigidity of the binuclear structure as well as chromophoric scaffolds also reduce the radiationless quenching and slow the knr, thus enabling competitive radiative recombination into the deep-red to NIR region.Remarkable examples of neutral deep-red to NIR binuclear emitters were recently reported by the group of Kozhevnikov that feature either a cyclometalated thieno[3,2-b]thiophene (IrIr)28 or a thiazolo[5,4-d]thiazole (Ir1)29 chromophoric rigid ligand and two “Ir(N^C^N)” fragments bearing a bis-cyclometalating terdentate N^C^N ligand (see Fig. 1 for the chemical structure). In particular, the IrIr complex displays NIR emission with λem = 714 nm and PLQY = 3.5% in CH2Cl2, which despite the apparently modest value represents one of the highest in the field of TMC NIR emitters. On the other hand, the Ir1 complex displays a less extended LUMO level, but it features a more π-accepting thiazolo[5,4-d]thiazole moiety.
 | ||
| Fig. 1 Chemical structure of the NIR-emitting binuclear Ir-D1 and Ir1 complexes described here along with the parental IrIr and Ir1 complexes previously described. Photo- and electroluminescence characteristics are provided for comparison purposes. | ||
A few reports describe TMC-based NIR OLEDs with λem, EL >700 nm,15–19,39–42 but only a limited number of examples display pure NIR emission. As far as solution-processed devices are concerned, the first example of pure NIR OLED was only recently reported by Qiao and co-workers, and showed λem, EL = 850 nm and EQE max = 0.17%.18 Zhu et al. reported NIR OLED with λem, EL = 760–764 nm and EQE max = 0.6–1.45%.17 To date, examples of LEC devices that display EL spectra squarely falling into the NIR region are even rarer.12a While several multinuclear complexes have indeed been reported for use in LECs, only few examples exhibiting EL peak wavelength >750 nm were reported to date and with very low EQEs values of 10−3–10−6% (see the ESI† for a comparison list).
Hence, we wondered whether the combination of the good electronic properties of the π-accepting thiazolo[5,4-d]thiazole scaffold decorated with two lateral N-containing π-accepting rings (e.g. pyridine and pyrazine) would provide a suitable ditopic chromophoric ligand for efficiently shifting the emission of binuclear iridium(III) iTMCs into the NIR region and thus enable their use in NIR LECs. Finally, we compared the herein proposed binuclear complexes with the parental mononuclear derivatives to confirm the superior performances of the former compared to the latter, yielding LEC devices with electroluminescence fully into the NIR region with the highest performances reported to date arising from molecular emission in the region 750–800 nm, to the best of our knowledge.
Results and discussion
Synthesis and characterization
A schematic of the synthetic pathway used for the synthesis of the mononuclear Ir-M1 and Ir-M2 complexes as well as their binuclear counterparts Ir-D1 and Ir-D2 is depicted in Scheme 1. Full details of the synthetic procedures used for the preparation of ligands L1–L4 are reported in the ESI† (for L2 and L4) and elsewhere by us (for L1 and L3).37 Monochelating ligand 2-(5-(3,5-di-tert-butylphenyl)pyrazin-2-yl)benzo[d]thiazole (L2) was straightforwardly prepared via Pd-catalysed Suzuki coupling between 2-(5-bromopyrazin-2-yl)benzo[d]thiazole and (3,5-di-tert-butylphenyl)boronic acid. On the other hand, bis-chelating ligand 2,5-bis(5-(3,5-di-tert-butylphenyl)pyrazin-2-yl)thiazolo[5,4-d]thiazole (L4) was obtained as a yellow solid by reacting the 5-(3,5-di-tert-butylphenyl)pyrazine-2-carbaldehyde with dithiooxamide in DMF under reflux.43,44 Parental mononuclear derivatives bearing the 2-(pyridin-2-yl)benzo[d]thiazole ligand were reported previously by Bolink and co-workers.22 Nevertheless, it is foreseen that the introduction of bulkier (3,5-di-tert-butylphenyl) moieties of both mono- and binuclear metal complexes would be beneficial for improving solubility and reducing aggregation-caused quenching phenomena in the solid-state. The synthesis of the starting chloro-bridged dimer [Ir(ppy)2(μ-Cl)]2, where ppy is a cyclometalating 2-phenylpyridine ligand, was carried out following procedures reported elsewhere by Nonoyama.45 The dimer was subsequently reacted with Ag(I) for halogen abstraction to obtain the bis-solvato complex, followed by the addition of 2 equiv. of the mono-chelating N^N ligand (L1–L2) in a CH2Cl2/MeOH mixture under reflux to give mononuclear complexes Ir-M1 and Ir-M2, respectively. Similarly, a reaction of the bis-solvato complex with bis-chelating ligand either L3 or L4 in an equimolar ratio yielded the corresponding binuclear complexes Ir-D1 and Ir-D2, respectively, as a mixture of stereoisomers comprising the two enantiomers (Λ,Λ) and (Δ,Δ) as well as the meso-form in an overall 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1.2:1 ratio in the reaction crude. The target complexes were obtained in good to excellent yields as orange to deep-red powders after purification on SiO2 column chromatography and precipitation with KPF6. Experimental synthetic details are available in the ESI† along with their chemical characterization carried out by means of 1H, 13C{1H} NMR spectroscopy and high-resolution electrospray ionization mass (HR-ESI-MS) spectrometry. The stereoselective synthesis was achieved as well and the (chiro-)optical properties of the enantiopure version of these derivatives is out of the scope of the present work and they will be reported in due course.
:1 and 1.2:1 ratio in the reaction crude. The target complexes were obtained in good to excellent yields as orange to deep-red powders after purification on SiO2 column chromatography and precipitation with KPF6. Experimental synthetic details are available in the ESI† along with their chemical characterization carried out by means of 1H, 13C{1H} NMR spectroscopy and high-resolution electrospray ionization mass (HR-ESI-MS) spectrometry. The stereoselective synthesis was achieved as well and the (chiro-)optical properties of the enantiopure version of these derivatives is out of the scope of the present work and they will be reported in due course.
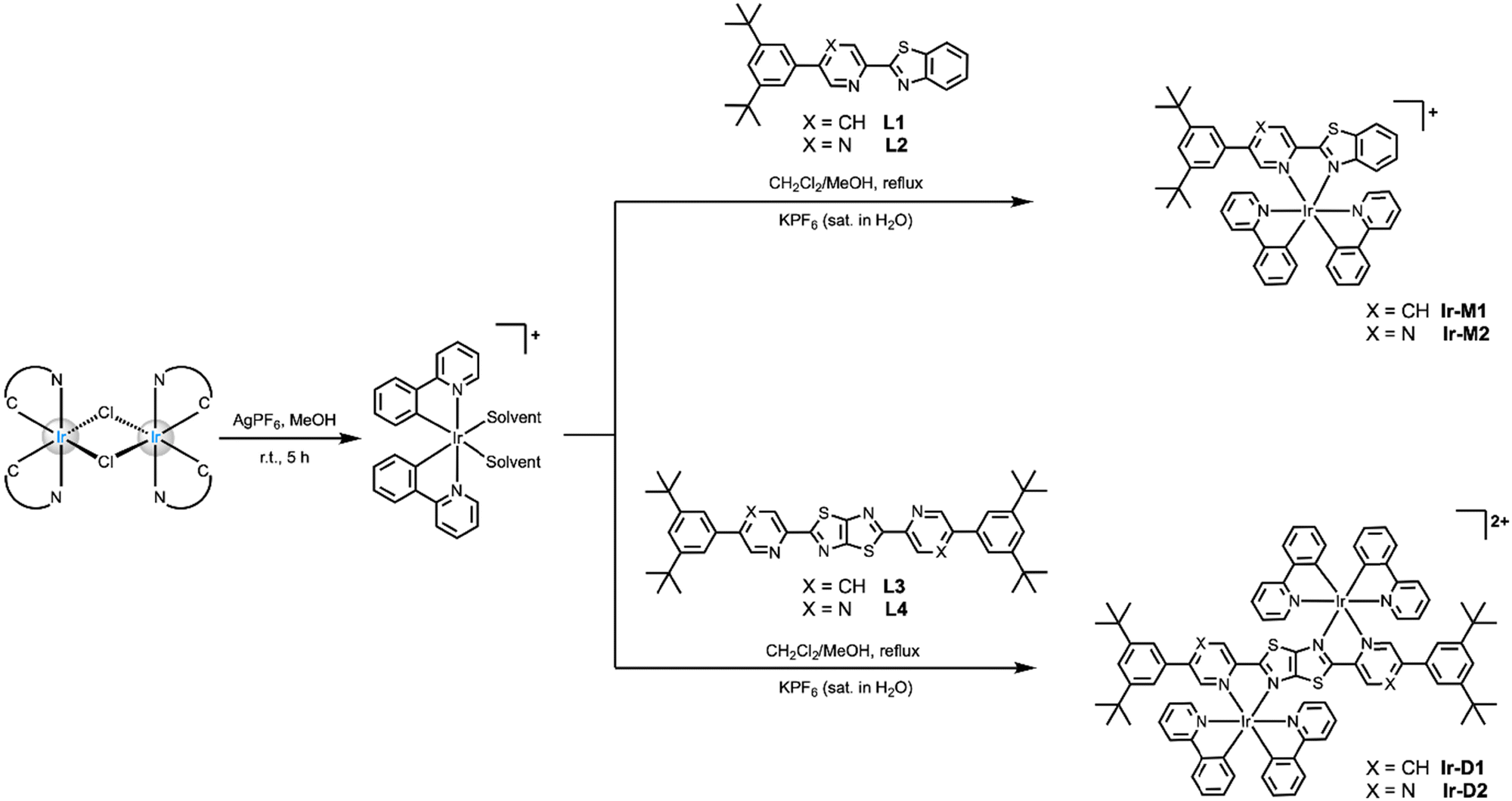 | ||
| Scheme 1 Synthetic procedure used for the preparation of monometallic complexes Ir-M1 and Ir-M2, and their binuclear counterparts Ir-D1 and Ir-D2. Cationic complexes were prepared as PF6− salts. | ||
All the complexes were crystallized and obtained as single crystals suitable for X-ray diffractometric analysis and the corresponding ORTEP diagrams are displayed in Fig. 2 for Ir-D1 and Ir-D2 and Fig. S1, S2 of the ESI,† for derivative Ir-M1 and Ir-M2, respectively. Crystallographic refinement parameters are provided in Tables S1–S4 of the ESI.† In all the crystalline structures, the iridium(III) center adopts a distorted octahedral geometry, expectedly, with the two nitrogen atoms of the 2-ppy ligand arranged in a N,N-trans fashion. For monometallic derivatives the coordination sphere is completed by the monochelating ligand, either L1 or L2, which coordinates via the N^N motif. On the other hand, the structure of the binuclear counterparts confirmed the coordination of “Ir(ppy)2” motifs onto the bis-chelating motif of either ligand L3 or L4. The latter bridges each metal center via one of the two Npyridine^NTzTz (TzTz = thiazolo[5,4-d]thiazole core) coordination motifs. Amongst the three possible stereoisomers, only the achiral meso-form was isolated by crystallization containing an inversion center i located in between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the two fused pentatomic rings of the thiazolo[5,4-d]thiazole scaffold.
C bond of the two fused pentatomic rings of the thiazolo[5,4-d]thiazole scaffold.
 | ||
| Fig. 2 ORTEP diagram of the meso-Ir-D1 (top) and meso-Ir-D2 (bottom) compounds with thermal ellipsoids shown at the 50% probability level obtained by single-crystal X-ray diffractometric analysis. Hydrogen atoms, PF6− counter-anions and solvent molecules are omitted for clarity. Atom color code: carbon (gray), nitrogen (light blue), iridium (dark blue), sulfur (yellow). | ||
The thermal properties of all the compounds were assessed by thermogravimetric analysis (TGA) and the corresponding data are plotted in Fig. S3–S6 of the ESI.† The decomposition temperature, defined as the temperature at which 5% weight loss occurs (T5%), are ca. 320–325 °C and 340 °C for the mononuclear and binuclear derivatives, respectively, implying a high thermal stability for these complexes. The electrochemical properties of all the investigated complexes were assessed by means of cyclic voltammetry in CH2Cl2 solution. The data are listed in Table 1 and the corresponding voltammograms are depicted in Fig. 3, where indication of the electrochemical HOMO–LUMO band gap is also provided (dashed lines). More details concerning the electrochemical characterization are provided in the ESI.†
| Complex |
|
ΔEp,O1,ib [mV] |
|
ΔEp,R1,ib [mV] |
e-ΔEH-Lc [eV] |
|---|---|---|---|---|---|
| a Reversible process. The formal potential, E0, was calculated as the average of the cathodic and anodic peak of the process. b ΔEp refers to the scan rate 0.1 V s−1. c Electrochemical band gap calculated as follows: E0O1,i − E0R1,i. | |||||
| Ir-M1 | 1.03 | 120 | −1.43 | 110 | 2.46 |
| Ir-M2 | 1.06 | 100 | −1.25 | 100 | 2.31 |
| Ir-D1 | 0.95 | 110 | −1.03 | 110 | 1.98 |
| −1.54 | 90 | ||||
| Ir-D2 | 1.00 | 120 | −0.83 | 110 | 1.82 |
| −1.28 | 100 | ||||
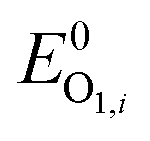

 | ||
| Fig. 3 Blank-subtracted cyclic voltammograms of 1 mM complexes Ir-M1 (orange trace), Ir-M2 (red traces), Ir-D1 (bordeaux trace) and Ir-D2 (black trace). Data recorded in CH2Cl2 using a three neck electrochemical cell, glassy carbon as the working electrode, Pt wire as the counter electrode, Ag wire as the quasi-reference electrode and 0.1 M TBAPF6 as the supporting electrolyte. Scan rate of 0.1 V s−1. | ||
In the positive-going scan, all compounds showed a reversible oxidation process, O1,i (with i denoting the investigated compound), whose standard potentials 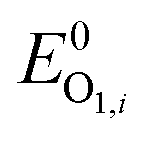 fall within the range +0.95 and +1.06 V vs. the ferrocene/ferricenium (Fc0|Fc+) redox couple, used as the internal standard.
fall within the range +0.95 and +1.06 V vs. the ferrocene/ferricenium (Fc0|Fc+) redox couple, used as the internal standard.  slightly depends on the nature of either the chromophoric N^N (L1–L2) or N^N–N^N (L3–L4) ligand. This redox process can confidently be ascribed to the oxidation that is mainly centered onto the metal and the cyclometalating ring of the “Ir(ppy)2” fragment and that can be described as a Ir(III)/Ir(IV) oxidation. The oxidation process for Ir-M2, however, possesses a less pronounced reversibility compared to the other metal complexes. It is also worth mentioning that the O1 process for Ir-D1 and Ir-D2 is expected to be bielectronic, as two Ir metal centers are involved, with a distinctive peak-to-peak separation of 29 mV and a higher current intensity. However, when two identical moieties connected by a bridge are (although poorly) interacting, a difference in their E0 lead to a larger peak-to-peak difference. This is a well-known effect46 and was previously observed for other compounds.47 Instead, in the negative-going scan, one or two reversible reduction processes Rn,i (where n denotes the process number and i the investigated compound) occurred, whose standard potential values
slightly depends on the nature of either the chromophoric N^N (L1–L2) or N^N–N^N (L3–L4) ligand. This redox process can confidently be ascribed to the oxidation that is mainly centered onto the metal and the cyclometalating ring of the “Ir(ppy)2” fragment and that can be described as a Ir(III)/Ir(IV) oxidation. The oxidation process for Ir-M2, however, possesses a less pronounced reversibility compared to the other metal complexes. It is also worth mentioning that the O1 process for Ir-D1 and Ir-D2 is expected to be bielectronic, as two Ir metal centers are involved, with a distinctive peak-to-peak separation of 29 mV and a higher current intensity. However, when two identical moieties connected by a bridge are (although poorly) interacting, a difference in their E0 lead to a larger peak-to-peak difference. This is a well-known effect46 and was previously observed for other compounds.47 Instead, in the negative-going scan, one or two reversible reduction processes Rn,i (where n denotes the process number and i the investigated compound) occurred, whose standard potential values  fall within the range from –0.83 to –1.43 V vs. Fc0|Fc+ (see Fig. 3). Clear differences are observed in the negative going scan between the different complexes. Mononuclear derivatives Ir-M1 and Ir-M2 show electrochemical waves whose
fall within the range from –0.83 to –1.43 V vs. Fc0|Fc+ (see Fig. 3). Clear differences are observed in the negative going scan between the different complexes. Mononuclear derivatives Ir-M1 and Ir-M2 show electrochemical waves whose 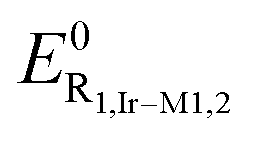 are –1.43 and –1.25 V, respectively, and that can be ascribed to a reversible, one-electron, reduction process located onto the coordinated L1 and L2 ligand, respectively. This is further corroborated by the fact that this difference agrees with the more electron accepting character of the pyrazine unit in derivative Ir-M2 compared to Ir-M1 that lowers the lowest unoccupied molecular orbital (LUMO) level, thus making reduction processes occur at a less negative potential. Moving from the mononuclear to the binuclear derivatives Ir-D1 and Ir-D2 yields to cyclic voltammograms that are characterized by two distinct, mono-electronic, reduction waves. The first process at
are –1.43 and –1.25 V, respectively, and that can be ascribed to a reversible, one-electron, reduction process located onto the coordinated L1 and L2 ligand, respectively. This is further corroborated by the fact that this difference agrees with the more electron accepting character of the pyrazine unit in derivative Ir-M2 compared to Ir-M1 that lowers the lowest unoccupied molecular orbital (LUMO) level, thus making reduction processes occur at a less negative potential. Moving from the mononuclear to the binuclear derivatives Ir-D1 and Ir-D2 yields to cyclic voltammograms that are characterized by two distinct, mono-electronic, reduction waves. The first process at  for Ir-D1 and Ir-D2, respectively, is anodically shifted compared to the mononuclear counterparts and are associated with reduction of the N^N–N^N ligands L3 and L4, respectively. Indeed, this lower potential is due to the even stronger π-accepting ability of the ditopic thiazolo[5,4-d]thiazole scaffold compared to the benzo[d]thiazole-based ligand L1 and L2, which further lowers the LUMO energy level. A subsequent process is observed at
for Ir-D1 and Ir-D2, respectively, is anodically shifted compared to the mononuclear counterparts and are associated with reduction of the N^N–N^N ligands L3 and L4, respectively. Indeed, this lower potential is due to the even stronger π-accepting ability of the ditopic thiazolo[5,4-d]thiazole scaffold compared to the benzo[d]thiazole-based ligand L1 and L2, which further lowers the LUMO energy level. A subsequent process is observed at 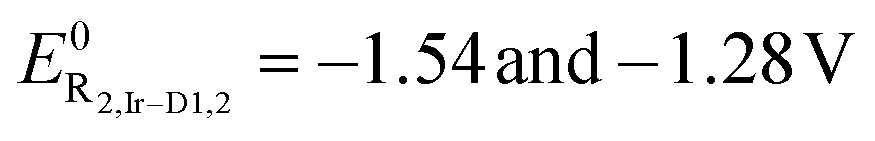 , for Ir-D1 and Ir-D2 respectively, and it is attributed to the second reduction process of the ditopic thiazolo[5,4-d]thiazole-based ligand. Overall, a steady narrowing of the HOMO–LUMO gap is observed within the series along Ir-M1 → Ir-M2 → Ir-D1 → Ir-D2, as determined by the electrochemical energy band gap (e-ΔEH-L). Furthermore, as previously observed for other bimetallic complexes,37 by taking into account the diffusion coefficient of Ir-M1, Ir-M2, Ir-D1 and Ir-D2 and by comparing the current intensities of their R1-2,i and O1,i processes with the oxidation of Fc, one can confidently state that the redox processes are all monoelectronic, except for O1,Ir-D1/2 that are bielectronic (see the ESI† for more details). Overall, these findings agree well with the optical properties (see below) and support the idea that these complexes are potentially suitable candidates in the field of solid-state electroluminescence devices.
, for Ir-D1 and Ir-D2 respectively, and it is attributed to the second reduction process of the ditopic thiazolo[5,4-d]thiazole-based ligand. Overall, a steady narrowing of the HOMO–LUMO gap is observed within the series along Ir-M1 → Ir-M2 → Ir-D1 → Ir-D2, as determined by the electrochemical energy band gap (e-ΔEH-L). Furthermore, as previously observed for other bimetallic complexes,37 by taking into account the diffusion coefficient of Ir-M1, Ir-M2, Ir-D1 and Ir-D2 and by comparing the current intensities of their R1-2,i and O1,i processes with the oxidation of Fc, one can confidently state that the redox processes are all monoelectronic, except for O1,Ir-D1/2 that are bielectronic (see the ESI† for more details). Overall, these findings agree well with the optical properties (see below) and support the idea that these complexes are potentially suitable candidates in the field of solid-state electroluminescence devices.
Photophysical investigation
The optical properties of the four complexes were firstly investigated in dilute CH2Cl2 solution under both air-equilibrated and degassed conditions at a concentration of 2 × 10−5 and of 3 × 10−5 M for mononuclear and binuclear derivatives, respectively. The spectra are displayed in Fig. 4 and the corresponding photophysical data are listed in Table 2. | ||
| Fig. 4 Left: UV-vis absorption (solid traces) and photoluminescence (symbols: square for Ir-M1, circle for Ir-M2, triangle for Ir-D1, star for Ir-D2) spectra of complexes Ir-M1 (orange traces), Ir-M2 (red traces), Ir-D1 (bordeaux traces) and Ir-D2 (black traces) in CH2Cl2 at room temperature. Emission spectra were recorded upon excitation at λexc = 500 (Ir-M1), 560 (Ir-M2) and 580 nm (Ir-D1 and Ir-D2). Right: Zoom-in of the lower-energy side of the UV-vis spectra. | ||
| Complex | λ max (ε) [nm, (103 M−1 cm−1)] | λ em [nm] | PLQY (%) | τ [ns] | PLQY (%) | τ [ns] | k r [105 s−1] | k nr [107 s−1] | λ em [nm] | τ [ns] |
![[small tau, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d4.gif) [μs] [μs] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Air-equilibrated | Degassed | 77 K | |||||||||
| sh denotes a shoulder. | |||||||||||
| Ir-M1 | 262sh, (41.6) | 650 | 7.3 | 258 | 10 | 405 | 2.47 | 0.22 | 596, 641, 707sh | 2479 (87%) | 2.8 |
| 297, (27.1) | 4115 (13%) | ||||||||||
| 358sh, (33.5) | |||||||||||
| 373, (36.5) | |||||||||||
| 516, (0.7) | |||||||||||
| Ir-M2 | 263, (40.4) | 718 | 2.1 | 72 | 2.3 | 76 | 3.03 | 1.29 | 659, 700sh | 1191 (82%) | 1.5 |
| 305, (25.7) | 2154 (18%) | ||||||||||
| 381sh, (35.1) | |||||||||||
| 395, (40.0) | |||||||||||
| 560, (0.7) | |||||||||||
| Ir-D1 | 261sh, (76.9) | 760 | 1 | 52 | 1.1 | 54 | 2.04 | 1.83 | 674, 743, 828 | 750 | — |
| 412, (60.5) | |||||||||||
| 442, (47.7) | |||||||||||
| 570, (1.3) | |||||||||||
| Ir-D2 | 260sh, (78.5) | 839 | 0.2 | 10 | 0.2 | 10 | 2.00 | 9.98 | 745, 777, 823sh | 258 (88%) | 0.3 |
| 290sh, (49.6) | 409 (12%) | ||||||||||
| 437, (64.0) | |||||||||||
| 467, (62.7) | |||||||||||
| 500sh, (27.8) | |||||||||||
| 610, (1.3) | |||||||||||
As far as the mononuclear complexes Ir-M1 and Ir-M2 are concerned, their UV-vis spectra are characterized by three main absorption bands. On the higher energy side, the intense and slightly structured bands at about λabs = 300 nm (ε = 2.5–2.7 × 104 M−1 cm−1) and 358–395 nm can be ascribed to electronic transitions with a strongly allowed character and of mainly single-manifold ligand centered (1LC) nature located on the phenylpyridine with a partial charge transfer (CT) character involving the metal and the coordinated ligands (see the computational section for a more detailed description). The spectra extend nicely into the visible region with a shoulder at ca. 425 and 450 nm that can be attributable to transitions with partial intraligand charge transfer character (1ILCT); whereas, the much weaker (ε = ca. 7 × 102 M−1 cm−1) broader and featureless band centered at λabs = 518 and ca. 560 nm for Ir-M1 and Ir-M2, respectively, can be assigned with confidence to the convolution of electronic processes of singlet and triplet character via spin–orbital coupling (SOC) with a metal-to-ligand and intraligand charge transfer (1,3MLCT/1,3ILCT) nature (see Fig. 4).
Expectedly, the absorption spectra of binuclear counterparts Ir-D1 and Ir-D2 display a two-fold increase of the intensity in the higher energy side at λabs = ca. 300 as a consequence of the presence of two Ir(ppy)2 moieties. The structured and intense band at λabs = 412 and 442 nm (for Ir-D1), and at 437 and 467 nm (for Ir-D2) can be ascribed to the π–π transitions involving the thiazolo[5,4-d]thiazole ligand scaffold mainly. This attribution is supported also by a bathochromic shift as large as 2540 and 2430 cm−1 when going from the mononuclear Ir-M1 and Ir-M2 to the corresponding binuclear derivatives Ir-D1 and Ir-D2, respectively, along with the concomitant and sizeable increase in the absorption intensity. It is worth noticing that the adopted dinuclearization strategy yields an extension of the absorption spectral onset attributable to transitions with important 1,3MLCT character far into the visible with a tail extending into the NIR region for derivative Ir-D2 (see Fig. 4). Absorption maxima are centered at λabs = 570 and 610 nm for Ir-D1 and Ir-D2, respectively. This large bathochromic shift is due to the energetic stabilization of the lowest unoccupied molecular orbitals (LUMOs) involved in the electronic absorption process and to an increase in the state mixing induced by a stronger SOC (see the computational section).
Upon photoexcitation into the MLCT band, dilute CH2Cl2 samples of all the complexes display luminescence with a broad and featureless profile. Remarkably, emissions span from the deep red to the NIR region, with the maxima centered at λem = 650, 718, 760 and 839 nm for derivatives Ir-M1, Ir-M2, Ir-D1 and Ir-D2, respectively. The emission profiles were insensitive to the presence of triplet dioxygen and appeared to be wavelength independent. The photoluminescence quantum yield (PLQY) and excited-state lifetime, τ, increased sizably upon degassing the samples of mononuclear derivative Ir-M1, that display an increase in PLQY from 7% to 10% and a concomitant prolongation of the lifetime from 258 to 405 ns. The other derivatives of the series were affected to a much smaller extent by the presence of oxygen as expected for a diffusion-controlled quenching process competing with a relatively fast radiative process observed for these derivatives, being τ = 76, 54 and 10 ns for degassed CH2Cl2 samples of Ir-M2, Ir-D1 and Ir-D2, respectively. These data allow us to estimate the radiative (kr) and non-radiative (knr) rate constants characterizing the emissive excited state by using the following equations (eqn (1) and (2)):
| kr = PLQY/τ | (1) |
| knr = (1 − PLQY)/τ | (2) |
Even more interestingly, kr values of the binuclear derivatives Ir-D1 and Ir-D2 are one order of magnitude higher than the related counterparts IrIr and Ir1 reported previously (see Fig. 1),28,29 indicative of a more efficient SOC process populating the emitting triplet state T1 and the larger 3MLCT character of the former compared to the latter. This is further corroborated by the broader and less structured emission band associated with the T1 → S0 transition observed for Ir-D1 and Ir-D2 compared to IrIr and Ir1 at both room and low temperature (see below).
The PLQY values decreased steadily when moving to derivatives emitting into longer wavelength regions within the series, namely from complex Ir-M1 to Ir-D2, and this effect is paralleled by a large increase of the knr from 0.22 × 107 to 9.98 × 107 s−1, respectively, due to the more efficient radiation-less processes taking place upon narrowing the T1–S0 energy gap, as ruled by the energy gap law (see Table 2 for the corresponding data).14
The emission spectra of the samples measured at low temperature in a frozen CH2Cl2 matrix at 77 K display a hypsochromic shift by about 1250–1680 cm−1 compared to the fluid solution, with the appearance of a moderate vibronic structure. The spectra are displayed in Fig. 5 and the data are listed in Table 2. Time-resolved emission decays could be fitted by using a bi-exponential model and provided average lifetimes of = 2.8 μs, 1.5 μs, and 0.3 μs, for Ir-M1, Ir-M2, and Ir-D2, respectively. On the other hand, a mono-exponential decay was observed for Ir-D1 with τ = 750 ns. Overall, these findings are indicative of a mainly 3MLCT character of the emissive excited state.
 | ||
| Fig. 5 Emission spectra recorded for samples of complexes Ir-M1 (orange trace), Ir-M2 (red trace), Ir-D1 (bordeaux trace) and Ir-D2 (black trace) in frozen CH2Cl2 at 77 K. The samples were excited at λexc = 470 nm (Ir-M1), 530 (Ir-M2), 580 (Ir-D1) and 600 nm (Ir-D2). | ||
Photoluminescence spectra of thin-film samples fabricated by spin coating from a CH2Cl2 solution of poly(methylmetacrylate) (PMMA) at 10 wt% doping level of complex are displayed in Fig. 6 and the data are listed in Table 3. The emission profiles display a hypsochromic shift in the order of 530–2500 cm−1 compared to the CH2Cl2 solution samples as typically observed for cationic complexes, due to the low polarity environment imparted by the PMMA polymeric network and as a consequence of the rigidochromic effect. Larger shifts are observed for the binuclear species most likely due to their doubly charged nature. Remarkably, PMMA thin-film samples display much larger PLQYs than solution samples, with values of 24%, 10%, 4.5% and 1.6% and a longer lifetime of = 1.3 μs, 0.6 μs, 0.8 μs and 1.2 μs, for derivatives Ir-M1, Ir-M2, Ir-D1 and Ir-D2, respectively. To ascertain the suitability of these novel emitters as electroluminescent materials in LEC devices the photophysical properties were further investigated in thin films of BMIMPF6:complex at the 80:20 wt% doping level, where BMIMPF6 is 1-butyl-3-methylimidazolium hexafluorophosphate ionic liquid (IL). The data are listed in Table 4 and the spectral comparison is displayed in Fig. 6. Due to the higher polarity of the environment imparted by the IL, for all the samples the emission profiles are bathochromically shifted by 350–1050 cm−1 compared to the PMMA matrix. Whereas the lifetime is shortened and the PLQY is lower by about a factor of two, in agreement with the energy gap law. At this stage, it is important to notice that the emission of binuclear complexes Ir-D1 and Ir-D2 retains the spectral profile into the longer wavelength region being fully into the NIR for the latter. Hence, they represent suitable and highly appealing candidates for the fabrication of NIR emissive LEC devices (see below).
 | ||
| Fig. 6 Emission spectra recorded for samples of complexes Ir-M1 (orange traces), Ir-M2 (red traces), Ir-D1 (bordeaux traces) and Ir-D2 (black traces) in PMMA thin-film at 10 wt% doping (dotted lines) and thin-film with 80:20 wt% BMIMPF6:complex (solid lines with coloured areas) upon excitation at 420 (for Ir-M1), 460 (for Ir-M2), and 480 nm for (for Ir-D1 and for Ir-D2). | ||
| Compound | λ em [nm] | PLQY (%) | τ [ns] |
[μs] |
k r [105 s−1] | k nr [105 s−1] |
|---|---|---|---|---|---|---|
| Ir-M1 | 623 | 24 | 952 (62%) | 1.3 | 1.85 | 5.85 |
| 1637 (38%) | ||||||
| Ir-M2 | 668 | 10 | 765 (32%) | 0.6 | 1.67 | 1.50 |
| 377 (68%) | ||||||
| Ir-D1 | 683, 735 | 4.5 | 370 (58%) | 0.8 | 0.56 | 11.9 |
| 1036 (37%) | ||||||
| Ir-D2 | 726, 770 | 1.6 | 157 (60%) | 1.2 | 0.13 | 8.2 |
| 2124 (11%) | ||||||
| 762 (29%) |
:20 wt% BMIMPF6:complex doping level
| Compound | λ em [nm] | PLQY (%) | τ [ns] |
[ns] |
|---|---|---|---|---|
| Ir-M1 | 637 | 12.3 | 342 (81%) | 422 |
| 614 (19%) | ||||
| Ir-M2 | 698 | 3.7 | 133 (43%) | 106 |
| 62 (57%) | ||||
| Ir-D1 | 732 | 2.9 | 248 (23%) | 171 |
| 123 (75%) | ||||
| Ir-D2 | 786 | 0.8 | 32 (38%) | 25 |
| 15 (62%) |
Computational study
The structures of the different complexes have been optimized using density functional theory (DFT) methods. Complexes Ir-D1 and Ir-D2 were optimized by either chiral (Δ, Δ or Λ, Λ) or achiral (meso) form within both C2 and Ci symmetry constraints, respectively. For both bimetallic species, the optimized structures of the meso form fit well with the corresponding experimental X-ray structure with a notable difference in the dihedral angle between the central π-conjugated ligand (either L3 or L4) and the two 3,5-di-tert-butylphenyl groups (Tables S5 and S6, ESI†). The rotation of aryls of Ir-D1 (Ir-D2) is governed by a low barrier estimated at about 4.1 kcal mol−1 (1.3 kcal mol−1) for a dihedral angle ∠(X–C6–C8–C9) of 0° between the aryl and the pyridine (pyrazine) ring, and a larger barrier of ca. 9.6 kcal mol−1 (13.6 kcal mol−1) for a dihedral angle of 90° (see Fig. S7 of the ESI† for atom labelling). These results imply a relatively free rotation of the aryls at room temperature and suggest that the difference between the crystal structure and the free molecule may come solely from the packing effect. Similar observation can be made for the monomers (Tables S7 and S8, ESI†). Given the negligible differences in terms of structural and optical properties computed for the chiral and achiral form of the binuclear derivatives, only the results of the meso stereoisomer of Ir-D1 and Ir-D2 will be further detailed in the following section.To gain better insights into the optical properties of these novel complexes, electronic absorption spectra were computed by means of time-dependent DFT (TD-DFT) without and with spin–orbit coupling (SOC) perturbation. The results are displayed in Fig. 7 and the data are listed in Table 5 and Table S9 of the ESI.†
 | ||
| Fig. 7 Computed absorption spectra of Ir-M1 (upper left), Ir-M2 (upper right), Ir-D1 (bottom left) and Ir-D2 (bottom right) with (solid traces) and without (dashed traces) spin–orbit coupling perturbation. See Tables S12 and S13 of the ESI† for details. | ||
| Complex | SOC state | E [eV] | λ calc (f) [nm] | % Contributing state |
|---|---|---|---|---|
| Ir-M1 | E3 | 2.193 | 565 (0.0063) | 88% T1, 3% T6, 2% S3 |
| E28 | 3.243 | 382 (0.7535) | 74% S6, 11% S7, 4% S5 | |
| E42 | 3.363 | 369 (0.0099) | 38% S9, 11% T12, 9% T9, 9% T13 | |
| E103 | 4.101 | 302 (0.1647) | 52% S22, 15% T25, 14% T24 | |
| E158 | 4.528 | 274 (0.1750) | 67% S37, 20% S39, 4% S38 | |
| Ir-M2 | E2 | 1.99 | 623 (0.0066) | 83% T1, 4% S1, 4% T3 |
| E26 | 3.004 | 413 (0.4185) | 38% S5, 28% S4, 20% S6 | |
| E33 | 3.08 | 403 (0.3208) | 39% S6, 35% S5, 6% T10 | |
| E103 | 3.959 | 313 (0.0791) | 38% S21, 14% S20, 9% T27 | |
| E127 | 4.14 | 300 (0.0588) | 45% S28, 31% T34, 17% S29 | |
| Ir-D1 | E5 | 1.825 | 679 (0.0247) | 84% T2, 5% S2, 4% T4 |
| E12 | 2.466 | 503 (0.3907) | 48% T4, 38% S3, 8% T2 | |
| E20 | 2.62 | 473 (0.5389) | 50% S3, 33% T4, 3% T7 | |
| E38 | 2.789 | 445 (0.1614) | 31% T11, 27% S7, 16% S8 | |
| E70 | 2.953 | 420 (0.0967) | 44% T17, 27% S13, 16% S10 | |
| Ir-D2 | E5 | 1.681 | 737 (0.0238) | 88% T2, 3% S2 |
| E15 | 2.344 | 529 (0.5931) | 42% T5, 40% S3, 6% T2 | |
| E23 | 2.462 | 503 (0.4547) | 34% T5, 30% S3, 27% T7 | |
| E48 | 2.686 | 462 (0.4406) | 71% S9, 5% T5, 4% S15 | |
| E58 | 2.81 | 441 (0.1453) | 31% S15, 29% T15, 22% S10 |
The absorption spectrum of compound Ir-M1 shows a main peak at λ = 384 nm (S6,M1) which corresponds to a LC transition (see Fig. 7 and Table S9, ESI†). An intense and relatively large band is observed between 340 and 270 nm arising from two major contributions computed at λ = 275 nm (S37,M1) and 304 nm (S22,M1) with a mixed MLCT/LC and LC nature, respectively, both involving the “Ir(ppy)2” moiety. The spectrum of complex Ir-M2 displays a main peak at λ = 404 nm (S5,M2) of mixed MLCT/LLCT/LC nature and a large band between 350 nm and 270 nm with also two major contributions computed at λ = 292 (S32,M2) and 317 nm (S21,M2). S32,M2 corresponds to a 1LC centered on the Ir(ppy)2 moiety, whereas S21,M2 possesses a mixed nature. A shoulder is also visible at λ = ca. 454 nm due mainly to a transition (S2,M2) of MLCT character. The theoretical spectra of both mononuclear complexes are in overall good agreement with the experimental ones and display a bathochromic shift of about 15–20 nm as shown in Fig. 7. The introduction of SOC perturbation has a small effect on the absorption spectra. The only significant point to mention is the appearance of a weak band in the spectra of the mononuclear compounds at around λ = 565 nm for Ir-M1 (623 nm for Ir-M2) which is the result of the convolution of the lower-lying singlet and triplet states.
The absorption spectrum of binuclear species Ir-D1 mainly consists of three peaks centered at λ = 486 nm (S3,D1), λ = 444 nm (S8,D1) and λ = 393 nm (S20,D1), respectively. The first peak at λ = 486 nm is associated with a transition of mixed LC/MLCT nature centered on the π-extended L3 ligand while the second is a mixed MLCT/LLCT (Fig. 8 and 9, and Fig. S9 of the ESI†). As for the mononuclear counterparts, the transitions at lower energy are of CT character mainly towards ligand L3, while transitions at higher energy involve the ppy moieties to a large extent. Due to too large computational cost, higher energy states could not be explored for the binuclear species. Thus, the theoretical electronic transitions corresponding to the experimental intense band at around λ = 250–350 nm (Fig. 3) were not computed. Moreover, it is worth mentioning that a quasi-degeneracy is observed for the lowest-lying excited singlets and of the lowest triplet state in all four complexes (see Tables S12 and S13 of the ESI†). Similar results were obtained by computing the spectrum of Ir-D2, with excited states exhibiting the similar nature as computed for Ir-D1 (Fig. 8).
 | ||
| Fig. 8 TheoDORE analysis showing the character of the first three singlet and triplet excited states of complexes Ir-M1, Ir-M2, Ir-D1 and Ir-D2. Colour code for the character: MLCT (yellow), LLCT (blue) and LC (red). The gray colour is used for the residual character components. For the bimetallic derivatives, meso-strutures with Ci symmetry and optimized without symmetry constraint were used for operational reasons of the TheoDORE code. | ||
 | ||
| Fig. 9 Electronic density difference maps (EDDMs) between S0 and T1, T2 or T3, at Franck–Condon geometry for meso-Ir-D1. Electronically enriched and depleted areas are colored in green and red, respectively. Color code for atoms: Ir (orange), S (yellow), N (blue), C (grey) and H (white). | ||
Overall, experimental and theoretical spectra computed for the binuclear derivatives without SOC are in fairly good agreement in terms of the energy of the bands (cf.Fig. 3 and 7), although the computed spectra do not show the lower energy shoulder present in the experimental ones. It is worth noting that the introduction of SOC has a much stronger effect on the spectra of the binuclear complexes compared to that of the mononuclear parental derivatives. This leads to a significantly improved agreement between computed and experimental absorption spectra. The presence of two heavy metal atoms makes the scalar relativistic treatment of the spectra insufficient for these kinds of complexes.
For complexes Ir-D1 (Ir-D2) spin–orbit perturbations lead to the splitting of the main singlet manifold transition peaking around λ = 490 nm (Ir-D2: λ = 515 nm) into two peaks at λSOC,1 = 473 and λSOC,2 = 503 nm (Ir-D2: λSOC,1 = 503 nm and λSOC,2 = 529 nm). This results from the mixing of the S3,D1 singlet of LC character with the T4,D1 of mixed MLCT/LLCT character (LC S3,D2 with mixed MLCT/LLCT T5,D2 and T7,D2). This corresponds to the main band of the experimental spectrum centered at λmax = 442 nm (Ir-D2: λmax = 467 nm) and its low energy shoulder at λ = ca. 480 nm (λ = 500 nm), respectively. Furthermore, a small peak appears around 679 nm for meso-Ir-D1 and 737 nm for meso-Ir-D2 due to the mixing of S2 and T2 for both complexes (Table 5). For both Ir-D1 and Ir-D2, a bathochromic shift of 20∼30 nm is observed between computed spectra with SOC and the experimental data (see Tables 2 and 5).
Upon introduction of spin–orbit perturbations into the calculations, the lowest computed SOC states retain an almost pure 3MLCT character similar to those observed for the non-perturbed counterparts. In the case of meso-Ir-D1, T1,D1 and T2,D1 are two quasi-degenerate triplet states, which correspond to the HOMO→LUMO (1.881 eV) and HOMO–1→LUMO (1.891 eV) transitions, respectively (see Fig. S8, ESI†). Both T1,D1 and T2, D1 share a mixed MLCT/LLCT character as shown by TheoDORE excited state composition analysis (see Fig. S9, ESI†) and in agreement with the experimental findings. At Franck–Condon (FC) geometry and as far as the T1 and T2 excited state are concerned, they are delocalized on both iridium centers of the binuclear complex (Fig. 8). T3,D1 corresponds to a HOMO–2→LUMO transition (2.127 eV) and shows a strong LCL3 character. Similar results can be observed for meso-Ir-D2 (see Fig. 8).
On the other hand, T1,M1 and T1,M2 are triplets of mixed MLCT/LLCT nature for the mononuclear complexes. They consist of 98% HOMO→LUMO transition, corresponding to a transition energy of 2.27 and 2.07 eV for T1,M1 and T1,M2, respectively. T2,M1 and T2,M2 are triplets with LC character and correspond to a 55% HOMO–1/33% HOMO–2 → LUMO transition (2.541 eV for Ir-M1). As expected, the absorption spectra of the mononuclear complexes are hypsochromically shifted compared to those of the binuclear counterparts due to the lower energy of the LUMO levels for the latter (cf. Fig. S8 and S10, ESI†). This effect arises from the smaller size of the accepting π-system of the mononuclear derivatives. Furthermore, we observe a greater stabilization of the 3LC state compared to the 3MLCT in the binuclear derivatives compared to the mononuclear counterparts, with the ΔE(3LC − 3MLCT) energy gap being 0.236 eV and 0.272 eV, respectively.
To investigate the emission properties of meso-Ir-D1 and meso-Ir-D2, triplet states were optimized without symmetry constraints. No significant evolution of the geometry was observed. Comparison of geometrical parameters is provided in Tables S5 and S6 (ESI†). For the bimetallic compounds, an overall breaking of the symmetry is observed as a consequence of the lifting of the degeneracy between T1 and T2 states and the localization of the excited state onto one of the two formally identical iridium motifs. This loss of symmetry of the excited state is clearly displayed by the electron density difference maps (EDDMs) for T1 and T2 as shown in Fig. 10. The results obtained for the excited singlet state are very similar.
 | ||
| Fig. 10 Electronic density difference maps (EDDMs) between S0 and either T1 or T2 at T1 optimized geometry for meso-Ir-D1. Electronically enriched and depleted areas are colored in green and red, respectively. Color code for atoms: Ir (orange), S (yellow), N (blue), C (grey) and H (white). | ||
The calculated emission wavelengths of the different complexes appeared to be bathochromically shifted by 30 to 62 nm when compared to the experimental maxima (see Tables 2, 4 and 6). This difference is slightly larger for the bimetallic complexes compared to the mononuclear counterparts, but the computed data agree well with the experimental findings. As abovementioned, the investigated binuclear complexes display a higher degree of structural flexibility, which can explain the slightly larger spectral shift between experimental and computed emission. Indeed, upon modification of the dihedral angles between the TzTz motif and the peripheral aryls on the L4 ligand, the emission wavelengths vary between 888 and 931 nm for meso-Ir-D2. Another aspect to be taken into account is that the overall planarity of the L3–L4 ligand may also play a role as we see that meso-Ir-D2 both display the largest difference and possesses the T1 with the most distorted geometry for the chromophoric TzTz ligand (Table S6, ESI†). Inclusion of spin orbit coupling does not significantly modify the above analysis (Table S11, ESI†).
| Complex | State | λ em [nm] | Oscillator strength | 105kr |
|---|---|---|---|---|
| Ir-M1 | S1 | 666 | 8.09 × 10−4 | 1.2 |
| T1 | 679 | |||
| Ir-M2 | S1 | 733 | 8.09 × 10−5 | 0.1 |
| T1 | 749 | |||
| Ir-D1 | S1 | 792 | 2.73 × 10−3 | 2.9 |
| T1 | 804 | |||
| Ir-D2 | S1 | 877 | 7.61 × 10−4 | 0.66 |
| T1 | 894 |
Electroluminescent characteristics of LECs
The EL spectra of LECs based on complexes Ir-M1, Ir-M2, Ir-D1, and Ir-D2 are depicted in Fig. 11. The PL spectra of the emissive layers of these LECs are also shown for comparison. In spite of the slightly altered microcavity effect influenced by varied emissive-layer thickness,49,50 for each complex both EL and PL spectra were similar, indicative of the fact that the same emitting excited state is involved in both photo- and electro-luminescence processes. Noteworthily, the Ir-D2 complex revealed the longest EL λmax = ca. 800 nm, which has still been rare in the published reports on the NIR LECs using iridium complexes.12a,20,25,51 It is noted that this long-wavelength emission does not result from optical interference51 and excimer formation20 since the solution PL spectrum of complex Ir-D2 also shows similar λmax = ca. 840 nm. | ||
| Fig. 11 Thickness-dependent EL spectra of the LECs based on the (a) Ir-M1, (b) Ir-M2, (c) Ir-D1, and (d) Ir-D2 complexes. The driving voltage of each device is shown in the inset. The PL spectra of the emissive layers are also shown for comparison. | ||
The time-dependent current, light output, and EQE of the LECs based on the Ir-M2 complex with various emissive-layer thicknesses are shown in Fig. 12. As a constant bias was applied on the device, the mobile ions in the emissive layer of the LEC drifted toward electrodes and gradually formed electrochemically doped layers. The latter promote carrier injection and cause increasing device current with time (Fig. 12a). A higher bias voltage or thinner device thickness resulted in a higher device turn-on speed. The light output generally followed the temporal evolution of the device current (Fig. 12b). Nevertheless, after reaching the maximum value, the light output decreased faster than the device current (cf.Fig. 12a and b). The faster decay rate of the light output may be associated with material degradation and exciton quenching near the ionic species in the extending doped layers.52 With the balanced carrier injection facilitated by the doped layers, the EQE increased rapidly shortly after a bias was applied (Fig. 12c). However, after approaching the peak value, the EQE decreased gradually due to similar reasons responsible for the decreasing light output. The optimized device thickness for the LECs based on the Ir-M2 complex to achieve the best EQE (0.65%) was 132 nm. Thinner or thicker devices showed lower EQEs because the overall contribution of the light outcoupling efficiency and carrier balance was inferior to that of the optimized device.10 This strategy was used to optimize the LECs based on the other complexes (Ir-M1, Ir-D1, and Ir-D2). The time-dependent EL characteristics of LECs based on complexes Ir-D2, Ir-M1 and Ir-D1, are shown in Fig. 13 and Fig. S11, S12 of the ESI,† respectively. They also showed similar temporal evolution trends in the EL characteristics to those of the LEC based on complex Ir-M2. For both mononuclear (Ir-M2 and Ir-M1) and binuclear complex pairs (Ir-D1 and Ir-D2), the complex possessing shorter EL emission wavelength showed higher EL efficiency (Table 7). This is in line with the trend in the PLQYs of these complexes (Tables 2–4). These data revealed that the proposed iridium complexes exhibited efficient deep-red to NIR EL emission. It is worth mentioning that the LEC device comprising Ir-D2 displays the combination of an EL spectrum that squarely falls into the NIR region λmax (ca. 800 nm), with a maximum EQE of 0.14% and peak power efficiency as high as 0.64–0.86 mW W−1. To the best of our knowledge, these data represent the highest performances for LECs from monomeric (i.e. non-excimeric) complex emitters in this spectral region (λmax >750 nm) to date.
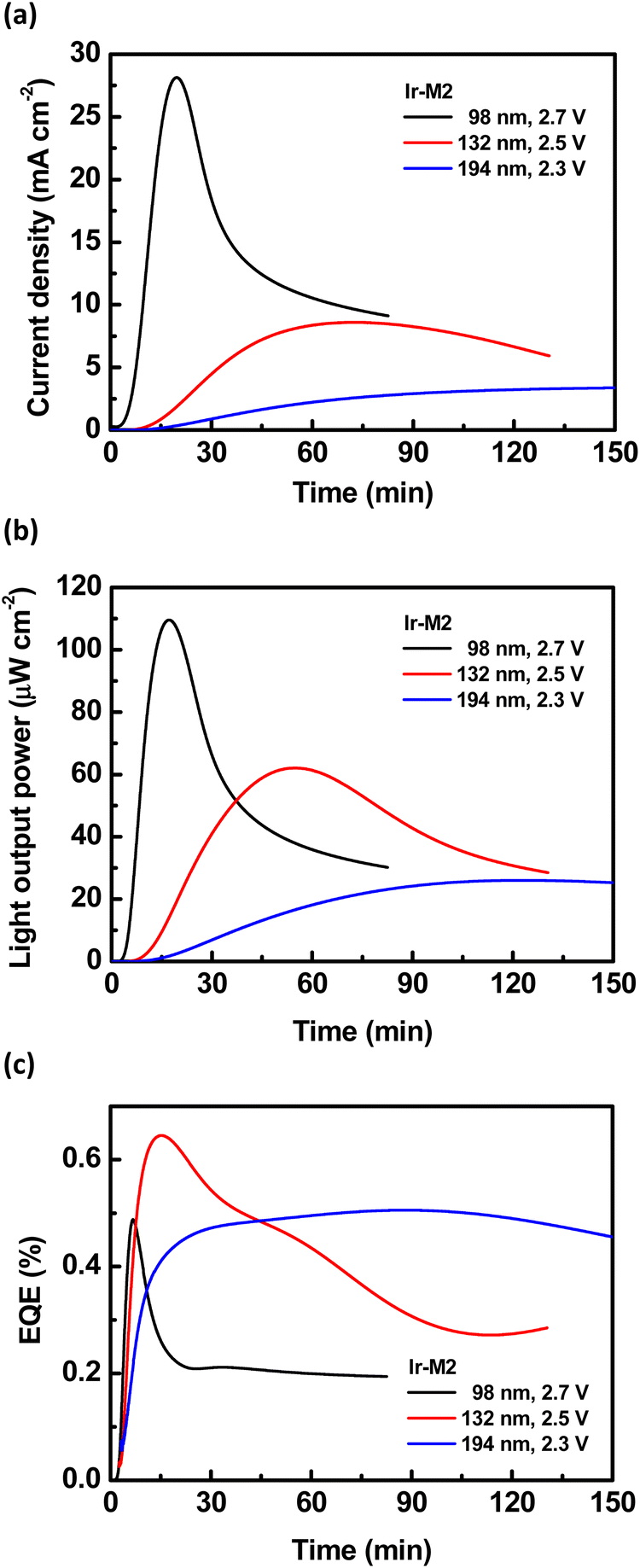 | ||
| Fig. 12 Time-dependent (a) current density, (b) light output, and (c) EQE of the LECs based on the Ir-M2 complex. | ||
 | ||
| Fig. 13 Time-dependent (a) current density, (b) light output, and (c) EQE of the LECs based on the Ir-D2 complex. | ||
| Complex | Concentrationa [mg mL−1] | Thickness [nm] | Bias [V] | ELmaxb [nm] | L max [μW cm−2] | η ext,max (%) | η P,max [mW W−1] |
|---|---|---|---|---|---|---|---|
| a Solution concentration for spin coating. b Stabilized EL emission peak wavelength. c Maximal light output power. d Maximal external quantum efficiency. e Power efficiency. | |||||||
| Ir-M1 | 40 | 107 | 2.3 | 642 | 39.9 | 2.30 | 18.31 |
| 60 | 149 | 2.5 | 656 | 129.6 | 2.67 | 19.46 | |
| 80 | 212 | 3.5 | 657 | 520.4 | 2.15 | 11.11 | |
| Ir-M2 | 40 | 98 | 2.7 | 692 | 109.6 | 0.49 | 3.08 |
| 60 | 132 | 2.5 | 699 | 62.1 | 0.65 | 4.35 | |
| 80 | 194 | 2.3 | 726 | 26.0 | 0.51 | 3.62 | |
| Ir-D1 | 60 | 124 | 1.9 | 738 | 11.6 | 0.57 | 4.80 |
| 80 | 151 | 1.8 | 744 | 2.2 | 0.74 | 6.58 | |
| 100 | 204 | 1.9 | 751 | 8.0 | 0.74 | 6.17 | |
| Ir-D2 | 40 | 107 | 2.5 | 793 | 39.8 | 0.11 | 0.64 |
| 60 | 156 | 2.5 | 788 | 43.3 | 0.14 | 0.86 | |
| 80 | 205 | 2.5 | 804 | 29.9 | 0.13 | 0.75 | |
To further study the effect of the nitrogen substitution in the complex on the device performance, the equation (eqn 3) governing EQE shown below is considered.
| ηEQE = ηout × γ × ηS,T × ηQY | (3) |
In this equation, ηEQE is the measured device EQE, ηout is the optical outcoupling efficiency, γ is the factor of carrier balance in the device, ηS,T is the emissive exciton generation efficiency of the complex and ηQY is the thin-film PLQY of the complex. For phosphorescent materials, both singlet and triplet excitons can be harvested (ηS,T = 100%). The optical outcoupling efficiency (ca. 20–30%) of a LEC device7,10 and the PLQY of the emissive layer (Table 4) are used for estimation. Even when ηout reached 30%, the LECs based on complexes Ir-M1 and Ir-D1 showed good factors of carrier balance (γ = 0.72 and 0.85 for complexes Ir-M1 and Ir-D1, respectively). On the other hand, the LECs based on complexes Ir-M2 and Ir-D2 exhibited inferior factors of carrier balance (γ = 0.59 and 0.58 for complexes Ir-M2 and Ir-D2, respectively) when the same ηout of 30% was employed. The inferior factors of carrier balance of the LECs using nitrogen substituted complexes Ir-M2 and Ir-D2 reveal that the nitrogen substitution in the complex red-shifts the EL spectrum (Table 7) at the expense of deteriorated device carrier balance.
To highlight the superior performance of the proposed NIR LECs, the EL characteristics of the reported NIR LECs showing an EL peak wavelength >750 nm are summarized in Table S10 (ESI†). The NIR LECs using the Ir-D2 complex showed the longest EL peak wavelength at ca. 800 nm and moderate light output of 30–40 μW cm−2. This light output is comparable with those obtained from the LECs exhibiting EL peak wavelength >750 nm. Furthermore, the peak EQE of the LECs based on complex Ir-D2 reached 0.14%, which is high among the reported NIR LECs delivering similar EL spectra (Table S10, ESI†). The LECs using the Ir-D2 complex showed device lifetime, which is defined by the time required for the light output of the device to decay from the maximum value to half of the maximum value, of <0.5 h. This device lifetime is long among the reported NIR LECs based on mononuclear and multinuclear complexes.20,25,53–55 However, the EL stability of complex Ir-D2 is inferior to that of the reported small molecules and conjugated polymers.56–62 Further improvement in the EL stability of cationic iridium complexes used for NIR LECs is still highly required.
Conclusions
A novel class of binuclear iridium(III) complexes featuring π-accepting chromophoric ligands based on the π–electron poor thiazolo[5,4-d]thiazole scaffold has been presented. In diluted CH2Cl2 solution the complexes display photoluminescence into the NIR region with λem maximum up to 840 nm arising from an excited state with strong 3MLCT character. Interestingly, the proposed binuclear design strategy yielded a bathochromic shift of the emission spectra compared to the mononuclear parental complexes. This also allowed us to keep the photoluminescence profile squarely in the NIR region also in the doped polymeric matrix. The optical properties were further elucidated by means of TD-DFT calculation also including SOC perturbation. Optimization of the triplet excited state indicates that the NIR radiative process arises from an excited state with a symmetry-broken 3MLCT character, whereas both cyclometalated metal centers are involved in the lowest-lying MLCT state at the Franck–Condon point. The appealing optical and redox properties prompted their investigation as electroactive materials in solution processed LEC devices. While the mononuclear derivatives displayed EL spectra only partially in the NIR region, the binuclear counterparts showed EL spectra whose profile largely shifted into the NIR, being λEL,max up to 751 and 804 nm, for Ir-D1 and Ir-D2, respectively. These findings represent some rare examples of NIR LECs to date with overall device performances recorded for Ir-D1 and Ir-D2 which are the best reported to date for LEC with EL maxima in the region 750–800 nm. These findings confirm that the dinuclearization strategy is a promising molecular design approach for achieving efficient emission in this challenging, longer wavelength region and will help to pave the way for achieving efficient NIR emitters for optoelectronic devices.Data availability statement
The computational data supporting this article have been included as part of the ESI.† CCDC 2338499–2338502† contains the supplementary crystallographic data for this paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
M. M. gratefully acknowledge the Université de Strasbourg and CNRS for financial support, and the French Agence Nationale de Recherche (ANR) for funding the grants ANR-20-CE29-0021 “PhotoMecha”, ANR-21-CE29-0015 “ChirON” ANR-22-CE07-0049-02 “BoostOLED”.C. G. and T. G. thank the HPC center of Strasbourg for the computational time. Molecular graphics and analyses were performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. This work was supported by the Higher Education Sprout Project of the National Yang Ming Chiao Tung University and Ministry of Education (MOE), Taiwan. N. Kyritsakas of the Service de Radiocristallographie, Fédération de chimie Le Bel – FR2010, Université de Strasbourg & CNRS is kindly acknowledged for the help in solving the X-ray structures. Dr Benoît Heinrich is kindly acknowledged for the technical help with TGA analyses and melting point determinations.
References
- S. B. Meier, D. Tordera, A. Pertegás, C. Roldán-Carmona, E. Ortí and H. J. Bolink, Mater. Today, 2014, 17, 217 CrossRef CAS.
- H. Su, Y. Chen and K. Wong, Adv. Funct. Mater., 2020, 30, 1906898 CrossRef CAS.
- R. Bai, X. Meng, X. Wang and L. He, Adv. Funct. Mater., 2020, 30, 1907169 CrossRef CAS.
- Light-Emitting Electrochemical Cells, ed., R. D. Costa, Springer International Publishing, 2017 Search PubMed.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef CAS PubMed.
- M.-C. Hou, D. Luo, Y.-T. Huang, S.-W. Liu, C.-W. Lu, C.-H. Chang and H.-C. Su, Org. Electron., 2024, 124, 106957 CrossRef CAS.
- Y.-Z. Chen, D. Luo, C.-H. Hsiang, R.-H. Yi, C.-H. Lin, C.-W. Lu, S.-W. Liu, C.-H. Chang and H.-C. Su, Org. Electron., 2020, 77, 105515 CrossRef CAS.
- R.-H. Yi, Y.-H. Lee, Y.-T. Huang, X.-J. Chen, Y.-X. Wang, D. Luo, C.-W. Lu and H.-C. Su, Inorg. Chem., 2024, 63, 4828 CrossRef CAS PubMed.
- H. J. Bolink, E. Coronado, R. D. Costa, N. Lardiés and E. Ortí, Inorg. Chem., 2008, 47, 9149 CrossRef CAS PubMed.
- R.-H. Yi, C.-L. Lo, D. Luo, C.-H. Lin, S.-W. Weng, C.-W. Lu, S.-W. Liu, C.-H. Chang and H.-C. Su, ACS Appl. Mater. Interfaces, 2020, 12, 14254 CrossRef CAS PubMed.
- K. S. Bejoymohandas, H. U. Kim, S. Sohn, W. Choi, S. Jung, F. Monti and T. Park, Inorg. Chem., 2023, 62, 43 CrossRef CAS PubMed.
- (a) Y. Lin, C. Lu and H. Su, Chem. – Eur. J., 2023, 29, e202202985 CrossRef CAS PubMed; (b) S. Yoon and T. S. Teets, Chem. Commun., 2021, 57, 1975 RSC; (c) P. L. dos Santos, P. Stachelek, Y. Takeda and P. Pander, Mater. Chem. Front., 2024, 8, 1731 RSC; (d) M. Vasilopoulou, A. Fakharuddin, F. Pelayo García de Arquer, D. G. Georgiadou, H. Kim, Abd Rashid bin Mohd Yusoff, F. Gao, M. K. Nazeeruddin, H. J. Bolink and E. H. Sargent, Nat. Photonics, 2021, 15, 656 CrossRef CAS; (e) A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 29, 1807623 CrossRef.
- Y. Zhang and J. Qiao, iScience, 2021, 24, 102858 CrossRef CAS PubMed.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952 CrossRef CAS.
- J. Xue, L. Xin, J. Hou, L. Duan, R. Wang, Y. Wei and J. Qiao, Chem. Mater., 2017, 29, 4775 CrossRef CAS.
- R. Tao, J. Qiao, G. Zhang, L. Duan, C. Chen, L. Wang and Y. Qiu, J. Mater. Chem. C, 2013, 1, 6446 RSC.
- C. You, D. Liu, M. Zhu, J. Yu, B. Zhang, Y. Liu, Y. Wang and W. Zhu, J. Mater. Chem. C, 2020, 8, 7079 RSC.
- Y. Zhang, Q. Li, M. Cai, J. Xue and J. Qiao, J. Mater. Chem. C, 2020, 8, 8484 RSC.
- Z. Chen, H. Zhang, D. Wen, W. Wu, Q. Zeng, S. Chen and W.-Y. Wong, Chem. Sci., 2020, 11, 2342 RSC.
- Y.-X. Liu, R.-H. Yi, C.-H. Lin, Z.-P. Yang, C.-W. Lu and H.-C. Su, J. Mater. Chem. C, 2020, 8, 14378 RSC.
- P.-N. Lai, C. H. Brysacz, M. K. Alam, N. A. Ayoub, T. G. Gray, J. Bao and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 10198 CrossRef CAS PubMed.
- C. D. Ertl, C. Momblona, A. Pertegás, J. M. Junquera-Hernández, M.-G. La-Placa, A. Prescimone, E. Ortí, C. E. Housecroft, E. C. Constable and H. J. Bolink, J. Am. Chem. Soc., 2017, 139, 3237 CrossRef CAS PubMed.
- C. Momblona, C. D. Ertl, A. Pertegás, J. M. Junquera-Hernández, H. J. Bolink, E. C. Constable, M. Sessolo, E. Ortí and C. E. Housecroft, J. Mater. Chem. C, 2018, 6, 12679 RSC.
- H.-F. Chen, K.-T. Wong, Y.-H. Liu, Y. Wang, Y.-M. Cheng, M.-W. Chung, P.-T. Chou and H.-C. Su, J. Mater. Chem., 2011, 21, 768 RSC.
- G. Chen, B. Chang, T. Shih, C. Lin, C. Lo, Y. Chen, Y. Liu, Y. Li, J. Guo, C. Lu, Z. Yang and H. Su, Chem. – Eur. J., 2019, 25, 5489 CrossRef CAS.
- G. Yu, C. Lin, Y. Liu, R. Yi, G. Chen, C. Lu and H. Su, Chem. – Eur. J., 2019, 25, 13748 CrossRef CAS PubMed.
- Q. Zeng, F. Li, Z. Chen, K. Yang, Y. Liu, T. Guo, G.-G. Shan and Z. Su, ACS Appl. Mater. Interfaces, 2020, 12, 4649 CrossRef CAS PubMed.
- M. Z. Shafikov, A. V. Zaytsev, A. F. Suleymanova, F. Brandl, A. Kowalczyk, M. Gapińska, K. Kowalski, V. N. Kozhevnikov and R. Czerwieniec, J. Phys. Chem. Lett., 2020, 11, 5849 CrossRef CAS PubMed.
- M. Z. Shafikov, A. V. Zaytsev and V. N. Kozhevnikov, J. Phys. Chem. C, 2021, 125, 20531 CrossRef CAS.
- R. E. Daniels, S. Culham, M. Hunter, M. C. Durrant, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Dalton Trans., 2016, 45, 6949 RSC.
- M. Z. Shafikov, R. Martinscroft, C. Hodgson, A. Hayer, A. Auch and V. N. Kozhevnikov, Inorg. Chem., 2021, 60, 1780 CrossRef CAS PubMed.
- M. Z. Shafikov, P. Pander, A. V. Zaytsev, R. Daniels, R. Martinscroft, F. B. Dias, J. A. G. Williams and V. N. Kozhevnikov, J. Mater. Chem. C, 2021, 9, 127 RSC.
- M. Z. Shafikov, R. Daniels and V. N. Kozhevnikov, J. Phys. Chem. Lett., 2019, 10, 7015 CrossRef CAS PubMed.
- M. Mauro, C. Yang, C. Shin, M. Panigati, C. Chang, G. D’Alfonso and L. De Cola, Adv. Mater., 2012, 24, 2054 CrossRef CAS PubMed.
- A. Bonfiglio, L. Pallova, V. César, C. Gourlaouen, S. Bellemin-Laponnaz, C. Daniel, F. Polo and M. Mauro, Chem. – Eur. J., 2020, 26, 11751 CrossRef CAS PubMed.
- A. Bonfiglio, P.-W. Hsiao, Y. Chen, C. Gourlaouen, Q. Marchand, V. César, S. Bellemin-Laponnaz, Y.-X. Wang, C.-W. Lu, C. Daniel, F. Polo, H.-C. Su and M. Mauro, Chem. Mater., 2022, 34, 1756 CrossRef CAS.
- A. Jouaiti, L. Ballerini, H. Shen, R. Viel, F. Polo, N. Kyritsakas, S. Haacke, Y. Huang, C. Lu, C. Gourlaouen, H. Su and M. Mauro, Angew. Chem., Int. Ed., 2023, 62, e202305569 CrossRef CAS PubMed.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622 CrossRef CAS.
- S. Kesarkar, W. Mróz, M. Penconi, M. Pasini, S. Destri, M. Cazzaniga, D. Ceresoli, P. R. Mussini, C. Baldoli, U. Giovanella and A. Bossi, Angew. Chem., Int. Ed., 2016, 55, 2714 CrossRef CAS PubMed.
- H. U. Kim, S. Sohn, W. Choi, M. Kim, S. U. Ryu, T. Park, S. Jung and K. S. Bejoymohandas, J. Mater. Chem. C, 2018, 6, 10640 RSC.
- S. F. Wang, Y. Yuan, Y. Wei, W. Chan, L. Fu, B. Su, I. Chen, K. Chou, P. Chen, H. Hsu, C. Ko, W. Hung, C. Lee, P. Chou and Y. Chi, Adv. Funct. Mater., 2020, 30, 2002173 CrossRef CAS.
- K. Tuong Ly, R.-W. Chen-Cheng, H.-W. Lin, Y.-J. Shiau, S.-H. Liu, P.-T. Chou, C.-S. Tsao, Y.-C. Huang and Y. Chi, Nat. Photonics, 2017, 11, 63 CrossRef CAS.
- M. R. Pinto, Y. Takahata and T. D. Z. Atvars, J. Photochem. Photobiol., A, 2001, 143, 119 CrossRef CAS.
- N. A. Sayresmith, A. Saminathan, J. K. Sailer, S. M. Patberg, K. Sandor, Y. Krishnan and M. G. Walter, J. Am. Chem. Soc., 2019, 141, 18780 CrossRef CAS PubMed.
- M. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 767 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Fundamentals and Applications, 2nd edn, John Wiley & Sons, New York, 2001 Search PubMed.
- F. Rizzo, F. Polo, G. Bottaro, S. Fantacci, S. Antonello, L. Armelao, S. Quici and F. Maran, J. Am. Chem. Soc., 2017, 139, 2060 CrossRef CAS PubMed.
- A. K. Pal, D. B. Cordes, A. M. Z. Slawin, C. Momblona, A. Pertegás, E. Ortí, H. J. Bolink and E. Zysman-Colman, RSC Adv., 2017, 7, 31833 RSC.
- J.-H. Hsu and H.-C. Su, Phys. Chem. Chem. Phys., 2016, 18, 5034 RSC.
- H.-C. Su, ChemPlusChem, 2018, 83, 197 CrossRef CAS PubMed.
- Y.-D. Lin, P.-W. Hsiao, W.-Y. Chen, S.-Y. Wu, W.-M. Zhang, C.-W. Lu and H.-C. Su, Chem. Eng. J., 2023, 469, 144055 CrossRef CAS.
- S. van Reenen, R. A. J. Janssen and M. Kemerink, Adv. Funct. Mater., 2015, 25, 3066 CrossRef CAS.
- S. Xun, J. Zhang, X. Li, D. Ma and Z. Y. Wang, Synth. Met., 2008, 158, 484 CrossRef CAS.
- S. Wang, X. Li, S. Xun, X. Wan and Z. Y. Wang, Macromolecules, 2006, 39, 7502 CrossRef CAS.
- D. Asil, J. A. Foster, A. Patra, X. de Hatten, J. del Barrio, O. A. Scherman, J. R. Nitschke and R. H. Friend, Angew. Chem., Int. Ed., 2014, 53, 8388 CrossRef CAS PubMed.
- C.-C. Ho, H.-F. Chen, Y.-C. Ho, C.-T. Liao, H.-C. Su and K.-T. Wong, Phys. Chem. Chem. Phys., 2011, 13, 17729 RSC.
- M. Mone, S. Tang, Z. Genene, P. Murto, M. Jevric, X. Zou, J. Ràfols-Ribé, B. A. Abdulahi, J. Wang, W. Mammo, M. R. Andersson, L. Edman and E. Wang, Adv. Opt. Mater., 2021, 9, 2001701 CrossRef CAS.
- M. Rémond, J. Hwang, J. Kim, S. Kim, D. Kim, C. Bucher, Y. Bretonnière, C. Andraud and E. Kim, Adv. Funct. Mater., 2020, 30, 2004831 CrossRef.
- K. T. Weber, K. Karikis, M. D. Weber, P. B. Coto, A. Charisidis, D. Charitaki, G. Charalamidis, P. Angaridis, A. G. Coutsolelos and R. D. Costa, Dalton Trans., 2016, 45, 13284 RSC.
- M. Mone, S. Tang, P. Murto, B. A. Abdulahi, C. Larsen, J. Wang, W. Mammo, L. Edman and E. Wang, Chem. Mater., 2019, 31, 9721 CrossRef CAS.
- E. Fresta, A. Charisiadis, L. M. Cavinato, N. Palandjian, K. Karikis, V. Nikolaou, G. Charalambidis, A. G. Coutsolelos and R. D. Costa, Adv. Photonics Res., 2021, 2, 2000188 CrossRef CAS.
- S. Tang, P. Murto, J. Wang, C. Larsen, M. R. Andersson, E. Wang and L. Edman, Adv. Opt. Mater., 2019, 7, 1900451 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2338499–2338502. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc02040e |
| This journal is © The Royal Society of Chemistry 2024 |