
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient triplet exciton phosphorescence quenching from a rhenium monolayer on silicon†
William H.
Banks‡
a,
Michael P.
Coogan
 a,
Tom
Markvart
bc and
Lefteris
Danos
*a
a,
Tom
Markvart
bc and
Lefteris
Danos
*a
aDepartment of Chemistry, Energy Lancaster, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: l.danos@lancaster.ac.uk
bSolar Energy Laboratory, Faculty of Engineering and the Environment, University of Southampton, Southampton, SO17 1BJ, UK
cCentre for Advanced Photovoltaics, Czech Technical University in Prague, Technická 2, 166 27 Prague 6, Czech Republic
First published on 15th August 2024
Abstract
We report efficient triplet exciton phosphorescence quenching from a Langmuir–Blodgett monolayer of a modified rhenium(I) fac-tricarbonyl bipyridine complex on the surface of crystalline silicon substrates indicating energy transfer. We monitor the luminescence quenching using phosphorescence lifetime imaging microscopy (PLIM) measurements as a function of distance of the monolayer to the silicon surface and have fitted the experimental phosphorescence lifetimes to a classical optical model. Our results show up to 95% phosphorescence quenching when the monolayer is close to the silicon surface (∼2 nm) indicative of efficient triplet resonance energy transfer from the rhenium monolayer to the silicon substrate. We believe this to be the first report of triplet sensitisation of silicon as a function of distance by a metal complex, and the most efficient triplet phosphorescence quenching from silicon reported to date.
The photosensitisation of crystalline silicon (c-Si) via excitons has been postulated as a method of exciting c-Si through excited-state resonance energy transfer from molecules close to the silicon surface.1–4 Dexter2 postulated that non-radiative energy (not electron) transfer from an organic monolayer at the surface of a semiconductor can generate electron–hole pairs in the latter. The mechanism can be approximated to a near-field dipole–dipole interaction similar to Förster resonance energy transfer (FRET). A distance proximity (0–5 nm) between the organic absorbing monolayer and the semiconductor surface is usually required for the direct generation of electron–hole pairs via FRET. A similar mechanism is utilized in light-harvesting in photosynthesis, and we can thus envisage an ultra-thin (∼1 μm) c-Si solar cell substituting the photosynthetic reaction centre and being sensitised by light-harvesting structures without loss in overall efficiencies, in effect splitting the photovoltaic process into two separate steps.5
Previous studies, involving evaporated dye layers,6 quantum dots,7,8 Langmuir Blodgett (LB) monolayers,9–11 and dye-loaded zeolites,12 have verified this sensitisation through fluorescence quenching of the excited state as a function of distance to the silicon surface. Significant fluorescence quenching is observed as the excited state emitters approach the silicon surface confirming efficient FRET. All these studies postulate that the presence of luminescence quenching is indicative of efficient energy transfer. The distance dependent emission intensities or lifetimes are fitted to a damping oscillating dipole model as a function of distance to the silicon surface developed for similar fluorescence quenching experiments carried out for molecular dye layers deposited on metal substrates.13
Most studies have focused on singlet excited states: to our knowledge, there has not been a study of distance dependence of a triplet metal complex emitter near the surface of silicon (previous studies of triplet states involved singlet fission rather than triplet emitting metal complexes).14–16 The longer exciton diffusion lengths usually observed from triplet excitons can photosensitise silicon and increase the efficiency of light-harvesting. The triplet exciton energy transfer observed can be augmented by recent advances in singlet fission14–18 and triplet–triplet annihilation19–21 and structures can be deposited on ultra-thin c-Si p/n junctions and potentially achieve efficiencies greater than the detailed balance single-junction Shockley–Queisser limit22 (33%) in addition to significant savings of the expensive semiconductor material.
We report phosphorescence lifetime imaging microscopy (PLIM) measurements for a rhenium fac-tricarbonyl bipyridine complex appended with a sixteen-carbon chain (ReC16) deposited as an LB monolayer on a c-Si substrate as a function of distance (2–250 nm). We fit our results to a modified classical model13 for molecular luminescence near the surface of silicon, showing significant exciton phosphorescence quenching (up to 95%) indicative of efficient triplet energy transfer from the ReC16 monolayer at close distance to the silicon surface (<5 nm).
The ReC16 tricarbonyl complex was synthesized according to a previous report (Fig. S1, ESI†) and has proven to be an excellent triplet emitter for biological imaging.23Fig. 1a shows a schematic of the deposited ReC16 monolayer assembly on fused silica or c-Si wafer substrates. The distance from the ReC16 layer to the silicon substrates is controlled by varying the number of monolayers of stearic acid (SA) deposited. For larger distances, thermal oxide grown silicon substrates were used and all multilayer thicknesses were measured with spectroscopic ellipsometry (Fig. S8, ESI†). The fused silica substrates are first covered with six layers of SA monolayers to avoid any unwanted quenching from the glass surface. After the deposition of ReC16 monolayer two or three layers of SA are added to ensure protection of the ReC16 monolayer from oxygen quenching and prevent chromophore aggregation.
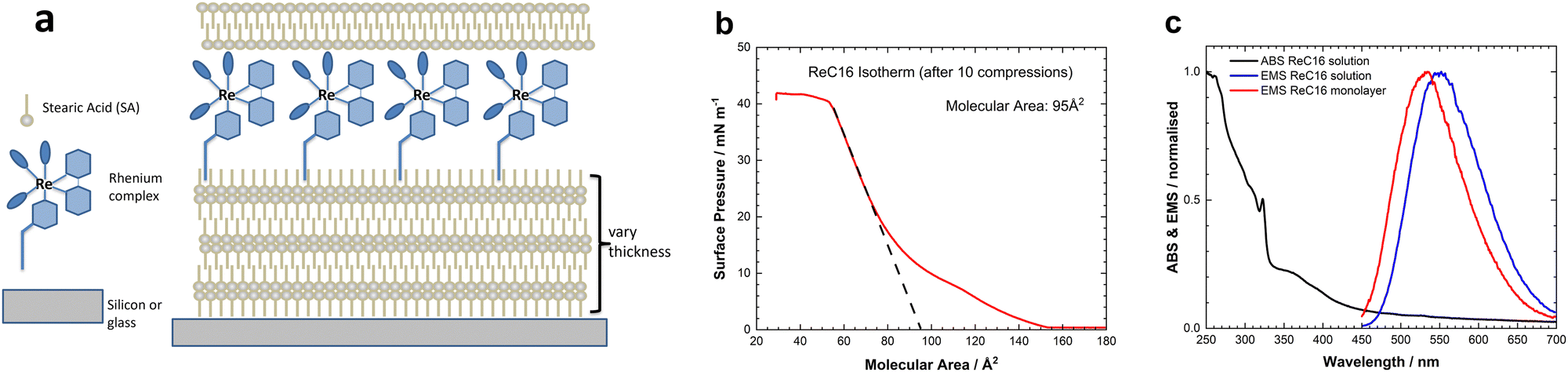 | ||
| Fig. 1 (a) Schematic of the fabrication of the ReC16 LB monolayer deposited on silicon or fused silica substrates. (b) Pressure–area isotherm of ReC16 on the surface of water. The extrapolated zero-pressure molecular area is shown in the figure inset. (c) Normalised absorption (black) and emission spectra from Re-C18 in solution (blue) (CHCl3) and deposited as monolayer (red) on a glass substrate. | ||
Fig. 1b shows the ReC16 pressure–area isotherm measured after an annealing 10 isotherm cycle (Fig. S2, ESI†). The curve shows the condensed region with surface pressure >20 mN m−1 and a collapse pressure of ∼40 mN m−1 resulting in a stable monolayer at the air–water interface. The shape of the isotherm agrees with previous pressure–area isotherms observed for similar rhenium complexes.24 The molecular area is estimated by extrapolating the condensed region to zero surface pressure (95 Å2 molecule−1) and is close to the calculated value (74 Å2) for a cross-sectional area (Fig. S3, ESI†). This confirms a near flat orientation of the Re moiety with the pyridine alkyl chain pointing away from the air–water interface in agreement with previous observations.25
Fig. 1c depicts the normalised absorption/emission spectra for the ReC16 complex in solution (CHCl3) and in LB films deposited on a fused silica substrate. The absorption spectrum shows the low-energy (dπ(Re) → π*) metal-to-ligand charge transfer (MLCT) transition (∼375 nm) together with the high-energy direct excitation of the bipyridine ligand (π → π*) transitions and do not show any shift from the LB film absorption (Fig. S4, ESI†). After excitation, there is a very fast intersystem crossing generating the triplet state which decays to the ground state via triplet emission. The emission spectrum from the solution and the LB monolayer shows similar behaviour with a slight blue shift observed in the LB monolayer spectra and an observed maximum at ∼535 nm (see also Fig. S5, ESI†). This blue shift is attributed to the different polarities of the lipophilic LB film and the more polarised chloroform solution. The excited state has charge transfer character and so is significantly more polar than the ground state. Thus it is stabilised by polar environments, and conversely its energy is raised in less polar media, giving the observed blue shift.26
Phosphorescence lifetime decay curves were obtained from both solution and the ReC16 monolayer deposited on fused silica substrates. The decay curves show triplet quenching from the presence of oxygen and subsequent freeze, pump and thaw cycles increase the triplet lifetime of the ReC16 in chloroform solution to approximately 450 ns. (Fig, S6, ESI†). The observed phosphorescence decay lifetime from the ReC16 monolayer is over 1000 ns with the presence of SA capping layers. We have used this lifetime as the unquenched lifetime of the emitter in calculations and fitting.
Fig. 2 shows the steady state emission spectra of the ReC16 monolayer deposited close to the silicon surface. There is significant phosphorescence quenching observed for the emission of ReC16 as the monolayer approaches the surface of silicon. The unquenched emission from the ReC16 deposited on fused silica substrate is shown as a reference together with the background emission from a bare silicon substrate (blank). Due to the low quantum yield of ReC16 (∼0.1%)23 we approach the limit of our instrument detection and the emission of the monolayer is completely quenched at a distance of approximately 2 nm from the surface of silicon. To improve the detection sensitivity, we used phosphorescence confocal microscopy and obtained PLIM images of the monolayers on the surface of silicon as a function of distance.
 | ||
| Fig. 2 Steady state emission spectra of ReC16 monolayer on a fused silica substrate (red) and as a function of distance with SA (1L – 2.2 nm) spacers on the silicon surface with native oxide present (∼2 nm). The excitation wavelength was 405 nm. | ||
Fig. 3 shows examples of PLIM images and the overall measured decay curves from a monolayer on glass and at various distances on silicon surfaces. We observe the formation of regular holes or dark states (∼5–10 μm) from the self-organization of the ReC16 molecules on the surface of silicon. The highly ordered structure of the ReC16 monolayer with apparent holes in a honeycomb-like network of emissive material is assigned to the need for the polar head groups, i.e., the complex and counterion (tetrafluoroborate) to pack in a low energy manner in the 2D plane orthogonal to the chains forming the monolayer. This is discussed in more detail in ESI† (Section 6).
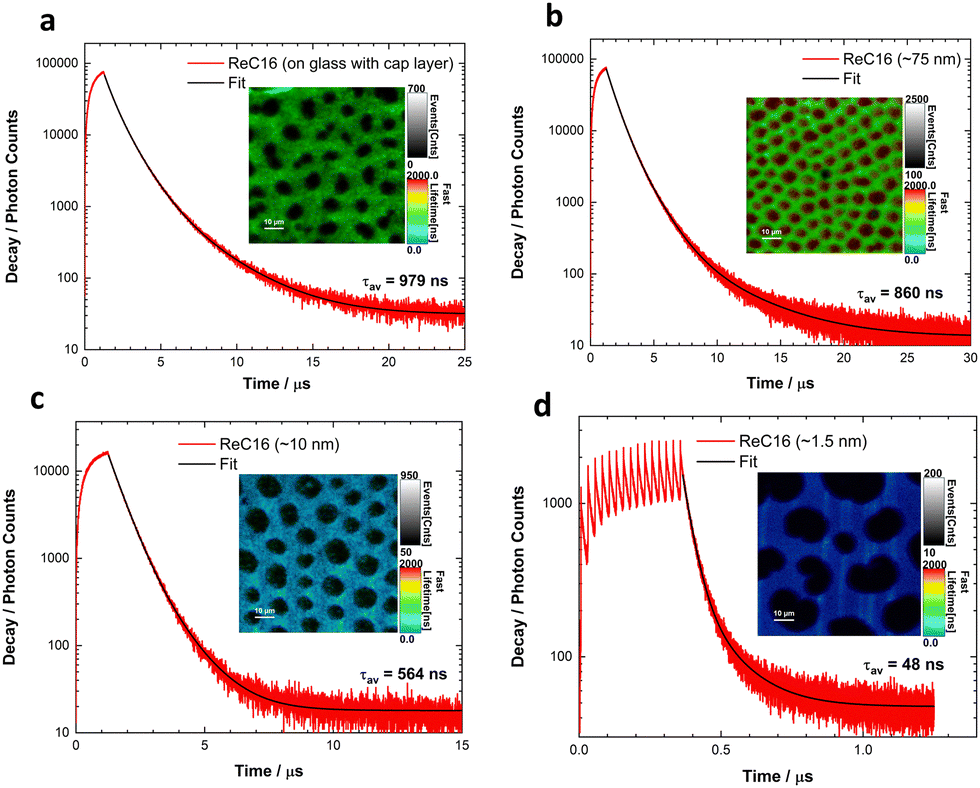 | ||
| Fig. 3 Examples of PLIM scans and corresponding decay curves from a ReC16 monolayer on (a) glass and (b)–(d) silicon substrates observed at different distances to the surface. Scale bars are all 10 μm for all figures. | ||
The overall decay curves are fitted to a two or three exponential model and the measured phosphorescence lifetime, τd normalised to the unquenched phosphorescence lifetime, τ0 obtained from a ReC16 monolayer deposited on fused silica substrates. We carried out these measurements for a distance range of 2–250 nm and fitted the lifetimes to a classical optical model developed by Chance, Prock and Silbey (CPS).13 In keeping with CPS, we use the value of τ0 for transitions originating from the triplet state.
Fig. 4 shows a plot of the normalised lifetime as a function of distance of the ReC16 monolayer to the silicon surface. We fit the measured lifetime ratios to a modified CPS model. The best fit is obtained for an emitter with a parallel (S) transition dipole moment orientation on the surface of silicon (Fig. S9, ESI†). At distances beyond 50 nm in the far field region, we observe the anticipated oscillations resulting from interference effects between the emitted light and the light reflected from the silicon surface. The CPS model fit aligns well with the experimental data in the far field region, indicating an adequate agreement between the two. However, our focus lies in the region near the silicon surface, specifically within distances less than 30 nm, where a notable decrease in the experimental phosphorescence lifetime is observed.
 | ||
| Fig. 4 Normalised phosphorescence lifetime versus distance for the ReC16 monolayer deposited on c-Si. Experimental points are fitted to a modified Chance–Prock–Silbey (CPS, full line) theory with a parallel dipole (S) orientation with QY = 75%. | ||
In our previous work,27,28 we investigated the quenching of molecular fluorescence through singlet emission and discovered the presence of ‘photon tunnelling’ within a specific range of 50 nm to 10 nm. This phenomenon was observed during the optical excitation of single crystalline silicon modes, induced by the fluorescence of dye monolayers. Earlier findings on singlet fluorescence quenching provided evidence for the existence of this optical coupling, attributed to the evanescent wave of the excited state. In our current study, we observe a reduction in the phosphorescence lifetime at distances below 30 nm. This optical mechanism injects energy via the evanescent field onto c-Si states that are otherwise forbidden from direct excitation by Snell's law.
As the ReC16 monolayer approaches closer to the silicon surface, we observe efficient phosphorescence quenching occurring for distances less than 10 nm. The experimental data fits well to a Förster-type energy transfer model, confirming that the mechanism of energy transfer is similar to a dipole–dipole non-radiative interaction. Although carrier generation could not be detected (see Section 9, ESI†), we observe an overall 95% phosphorescence quenching rate when the ReC16 monolayer is deposited 2 nm away from the surface. However, the presence of a native oxide layer (∼2 nm) on the silicon substrate prevents further proximity between the monolayer and the surface. Our work on c-Si surface chemistry has recently demonstrated the direct attachment of dyes on the surface of silicon at distances less than 1 nm exhibiting over 90% fluorescence quenching efficiencies.5,29 Further work is underway to remove the oxide and covalently attach rhenium complexes on the surface to c-Si in a similar fashion.
Overall, we observe a strong quantitative agreement between the measured lifetime ratios and the model without the need of additional fitting parameters but using only the quantum yield (QY) of the ReC16 monolayer. We obtain reasonable fits with a QY value of 75%, a reasonable value for transitions originating from the triplet state in accordance with the CPS model.13 A more detailed discussion of this point is available in ESI† (Section 9).
The demonstration of efficient exciton triplet phosphorescence quenching from a monolayer of ReC16 on the surface of silicon is important due to its implications for the observed efficiency of triplet energy transfer to c-Si. For efficient solar energy conversion, singlet fission, whereby one high energy singlet state generates two triplet states of approximately half the energy, is of great current interest as it potentially allows greater than the single junction limit solar cell efficiencies for silicon. However, the efficiency of such transfer from other systems has been much lower30,31 and indeed recent reports32,33 failed to realise this goal, highlighting the significance of this work in generating triplet excitons and studying their interaction with c-Si. Our proposal can be used as a model test bed for studying a simpler system to understand how triplets generated by singlet fission could potentially sensitise c-Si.
The phosphorescence of an LB monolayer from a rhenium complex was studied using PLIM on the surface of crystalline silicon substrates for distances between 2–250 nm to estimate the degree of quenching of phosphorescence emission. It was found that up to 95% quenching is observed as the monolayer approaches the surface of silicon indicative of efficient triplet energy transfer. This is the first time that triplet phosphorescence quenching is observed from organometallic complexes as a function of distance on the surface of silicon providing valuable additional information for the potential triplet excited states have to offer in silicon photosensitisation.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
WHB acknowledges financial support from the Department of Chemistry at Lancaster University for a PhD studentship. We would like to thank Dr George Adamopoulos for help with the ellipsometry measurements. TM wishes to acknowledge support from grant “Energy Conversion and Storage”, no. CZ.02.01.01/00/22_008/0004617, programme Johannes Amos Commenius, Excellent Research.Notes and references
- T. Markvart, Prog. Quantum Electron., 2000, 24, 107–186 CrossRef CAS.
- D. L. Dexter, J. Lumin., 1979, 18–19, 779–784 CrossRef CAS.
- V. M. Agranovich, Y. N. Gartstein and M. Litinskaya, Chem. Rev., 2011, 111, 5179–5214 CrossRef CAS PubMed.
- W. Peng, S. M. Rupich, N. Shafiq, Y. N. Gartstein, A. V. Malko and Y. J. Chabal, Chem. Rev., 2015, 115, 12764–12796 CrossRef CAS.
- L. Danos, N. R. Halcovitch, B. Wood, H. Banks, M. P. Coogan, N. Alderman, L. Fang, B. Dzurnak and T. Markvart, Faraday Discuss., 2020, 222, 405–423 RSC.
- T. Hayashi, T. G. Castner and R. W. Boyd, Chem. Phys. Lett., 1983, 94, 461–466 CrossRef CAS.
- H. M. Nguyen, O. Seitz, W. Peng, Y. N. Gartstein, Y. J. Chabal and A. V. Malko, ACS Nano, 2012, 6, 5574–5582 CrossRef CAS.
- M. T. Nimmo, L. M. Caillard, W. De Benedetti, H. M. Nguyen, O. Seitz, Y. N. Gartstein, Y. J. Chabal and A. V. Malko, ACS Nano, 2013, 7, 3236–3245 CrossRef CAS.
- M. I. Sluch, A. G. Vitukhnovsky and M. C. Petty, Phys. Lett. A, 1995, 200, 61–64 CrossRef CAS.
- L. Danos, R. Greef and T. Markvart, Thin Solid Films, 2008, 516, 7251–7255 CrossRef CAS.
- L. Danos and T. Markvart, Chem. Phys. Lett., 2010, 490, 194–199 CrossRef CAS.
- S. Huber and G. A. Calzaferri, Chem. Phys. Chem., 2004, 5, 239–242 CrossRef CAS.
- R. R. Chance, A. Prock and R. Silbey, Adv. Chem. Phys., 1978, 107, 1–64 Search PubMed.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS.
- J. Lee, P. Jadhav, P. D. Reusswig, S. R. Yost, N. J. Thompson, D. N. Congreve, E. Hontz, T. Van Voorhis and M. A. Baldo, Acc. Chem. Res., 2013, 46, 1300–1311 CrossRef CAS.
- A. J. Baldacchino, M. I. Collins, M. P. Nielsen, T. W. Schmidt, D. R. McCamey and M. J. Y. Tayebjee, Chem. Phys. Rev., 2022, 3, 021304 CrossRef CAS.
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS.
- R. W. MacQueen, M. Liebhaber, J. Niederhausen, M. Mews, C. Gersmann, S. Jäckle, K. Jäger, M. J. Y. Y. Tayebjee, T. W. Schmidt, B. Rech and K. Lips, Mater. Horizons, 2018, 5, 1065–1075 RSC.
- Y. C. Simon and C. Weder, J. Mater. Chem., 2012, 22, 20817–20830 RSC.
- J. de Wild, A. Meijerink, J. K. Rath, W. G. J. H. M. van Sark and R. E. I. Schropp, Energy Environ. Sci., 2011, 4, 4835–4848 RSC.
- Y. Y. Cheng, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66–71 RSC.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- V. Fernández-Moreira, F. L. Thorp-Greenwood, A. J. Amoroso, J. Cable, J. B. Court, V. Gray, A. J. Hayes, R. L. Jenkins, B. M. Kariuki, D. Lloyd, C. O. Millet, C. F. Williams and M. P. Coogan, Org. Biomol. Chem., 2010, 8, 3888 RSC.
- V. W. W. Yam, Y. Yang, H.-P. P. Yang and K.-K. K. Cheung, Organometallics, 1999, 18, 5252–5258 CrossRef CAS.
- J. Zhang, B. W. K. Chu, N. Zhu and V. W. W. Yam, Organometallics, 2007, 26, 5423–5429 CrossRef CAS.
- F. L. Thorp-Greenwood, J. A. Platts and M. P. Coogan, Polyhedron, 2014, 67, 505–512 CrossRef CAS.
- L. Fang, K. S. Kiang, N. P. Alderman, L. Danos and T. Markvart, Opt. Express, 2015, 23, A1528–A1532 CrossRef PubMed.
- L. Fang, L. Danos, T. Markvart and R. Chen, Opt. Lett., 2020, 45, 4618–4621 CrossRef PubMed.
- N. Alderman, L. Danos, L. Fang, M. C. Grossel and T. Markvart, Chem. Commun., 2017, 53, 12120–12123 RSC.
- M. Einzinger, T. Wu, J. F. Kompalla, H. L. Smith, C. F. Perkinson, L. Nienhaus, S. Wieghold, D. N. Congreve, A. Kahn, M. G. Bawendi and M. A. Baldo, Nature, 2019, 571, 90–94 CrossRef CAS PubMed.
- B. Daiber, S. Maiti, S. M. Ferro, J. Bodin, A. F. J. Van Den Boom, S. L. Luxembourg, S. Kinge, S. P. Pujari, H. Zuilhof, L. D. A. Siebbeles and B. Ehrler, J. Phys. Chem. Lett., 2020, 11, 8703–8709 CrossRef CAS PubMed.
- G. B. Piland, J. J. Burdett, T. Y. Hung, P. H. Chen, C. F. Lin, T. L. Chiu, J. H. Lee and C. J. Bardeen, Chem. Phys. Lett., 2014, 601, 33–38 CrossRef CAS.
- A. F. J. van den Boom, S. Ferro, M. Gelvez-Rueda, H. Zuilhof and B. Ehrler, J. Phys. Chem. Lett., 2023, 14, 4454–4461 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc02027h |
| ‡ Current address: Cancer Research UK Manchester Institute, The University of Manchester, Wilmslow Road, Manchester M20 4BX, UK. |
| This journal is © The Royal Society of Chemistry 2024 |