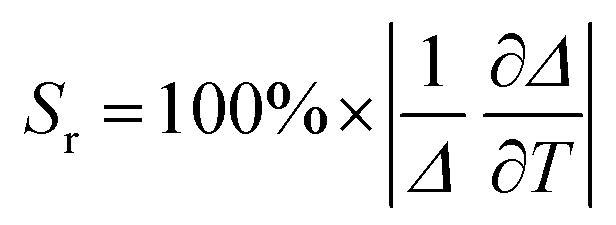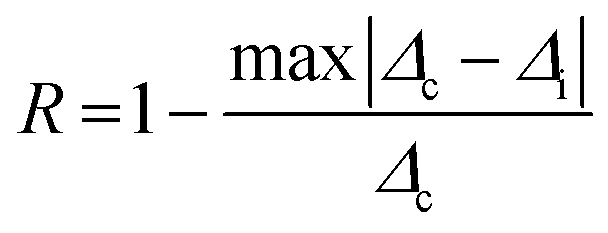DOI:
10.1039/D4TC01740D
(Paper)
J. Mater. Chem. C, 2024,
12, 11785-11802
Exploring the potential of lanthanide-doped oxyfluoride materials for bright green upconversion and their promising applications towards temperature sensing and drug delivery†
Received
28th April 2024
, Accepted 26th June 2024
First published on 29th June 2024
Abstract
The most efficient upconversion (UC) materials reported to date are based on fluoride hosts with low phonon energies, which reduce the amount of nonradiative transitions. In particular, NaYF4 doped with Yb3+ and Er3+ at appropriate ratios is known as one of the most efficient UC phosphors. However, its low thermal stability limits its use for certain applications. On the other hand, oxide hosts exhibit better thermal stability, yet they have higher phonon energies and are thus prone to lower UC efficiencies. As a result, developing host nanomaterials that combine the robustness of oxides with the high upconversion efficiencies of fluorides remains an intriguing prospect. Herein, we demonstrate the formation of ytrrium doped oxyfluoride (YOF:Yb3+,Er3+) particles, which are prepared by growing a NaYF4:Yb3+,Er3+ layer around SiO2 spherical particles and consecutively applying a high-temperature annealing step followed by the removal of SiO2 template. Our interest lies in employing these materials as Boltzmann type physiological range luminescence thermometers, but their weak green emission is a drawback. To overcome this issue, and engineer materials suitable for Boltzmann type thermometry, we have studied the effect of introducing different metal ion co-dopants (Gd3+, Li+ or Mn2+) into the YOF:Yb3+,Er3+ particles, focusing on the overall emission intensity, as well as the green to red ratio, upon 975 nm laser excitation. These materials are explored for their use as ratiometric thermometers, and further also as drug carriers, including their simultaneous use for these two applications. The investigation also includes examining their level of toxicity towards specific human cells – normal human dermal fibroblasts (NHDFs) – to evaluate their potential use for biological applications.
Introduction
A wide variety of host materials, including fluorides, oxides, chlorides, and bromides, have been subject to extensive investigation by researchers in pursuit of developing Ln3+-doped materials that exhibit exceptional upconversion (UC) efficiency.1–3 Many of these (nano)materials have received a lot of attention due to their potential employment in bioimaging, anti-counterfeiting, or as luminescent probes.4,5 Low-phonon energy fluoride hosts (300–500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−1), such as NaYF4, NaGdF4, and NaLuF4, have very often been used in these studies, because of their efficient near infrared (NIR)-to-visible UC, and well-established synthesis routes.6,7 On the other hand, oxide materials make excellent shell structured materials owing to their strong mechanical strength, photochemical stability, and environmentally friendly synthesis.8,9 However, despite possessing desirable properties, these matrices have higher phonon energies (>500cm−1) compared to the fluorides, and as a result, they are inherently susceptible to lower UC efficiencies.10,11 Therefore, oxide materials are not necessarily considered as the most suitable candidates for use as hosts for UC applications. In order to benefit from both the low phonon energy of fluorides and the stability of oxides, rare earth oxyfluorides, such as yttrium oxyfluoride (YOF), have been developed.11
cm−1), such as NaYF4, NaGdF4, and NaLuF4, have very often been used in these studies, because of their efficient near infrared (NIR)-to-visible UC, and well-established synthesis routes.6,7 On the other hand, oxide materials make excellent shell structured materials owing to their strong mechanical strength, photochemical stability, and environmentally friendly synthesis.8,9 However, despite possessing desirable properties, these matrices have higher phonon energies (>500cm−1) compared to the fluorides, and as a result, they are inherently susceptible to lower UC efficiencies.10,11 Therefore, oxide materials are not necessarily considered as the most suitable candidates for use as hosts for UC applications. In order to benefit from both the low phonon energy of fluorides and the stability of oxides, rare earth oxyfluorides, such as yttrium oxyfluoride (YOF), have been developed.11
Lanthanide-doped hollow nanomaterials have emerged as a promising class of nanomaterials for theranostic applications, owing to their exceptional fluorescence and paramagnetic properties facilitated by the presence of lanthanide ions.12 These attributes make them excellent candidates for multimodal bioimaging agents, with the added advantage of spacious interior cavities suitable for efficient storage and delivery of therapeutic agents. Previous contributions to this field include the facile template synthesis of Eu3+-doped Y2O3, YOF, La2O3, and LaOF hollow spheres, imparting a distinctive red luminescence.13 Another innovative approach involves the pyrolysis at 600 °C of liquid aerosols generated from aqueous solutions containing rare earth chlorides and trifluoroacetic acid, leading to the synthesis of micrometer-sized hollow YOF spheres with a cubic structure. This method allows for co-doping with Er3+ and Yb3+, yielding UC phosphors capable of emitting intense red light under near-infrared irradiation.14 Furthermore, a controlled ion exchange process has been employed for the facile synthesis of uniform and monodisperse GdOF:Yb3+,Er3+ hollow nanospheres, each approximately 100 nm in diameter. This enhances the versatility of applications in biolabeling and drug delivery domains.15 In general, rare-earth oxyfluorides (REOF) have already drawn attention for applications in the down-conversion and UC luminescence. For example, Tien et al.16 proposed a simple procedure to develop a heterogeneous YOF:Yb3+,Er3+ shell on a NaYF4:Yb3+,Er3+ core particles with good chemical stability through high-temperature annealing-induced reaction between O2 and NaYF4:Yb3+,Er3+.16 Consequently, the UC emission intensity of NaYF4:Yb3+,Er3+ was increased by over 25 times by such a “native” YOF layer. In addition, YOF:Yb3+,Er3+ nanocrystals have also been observed to exhibit exceptional red single-band luminescence performance with high color purity.17 Li et al.18 reported that YOF:Ln3+ (Ln = Eu, Tb, Dy, and Tm) submicrospheres exhibit bright multicolor UC photoluminescence, with red, green, yellow, and blue emission, respectively, upon 980 nm laser excitation. Moreover, it has been reported that when excited with NIR (980 nm) light, YOF:Ho3+,Yb3+ nanophosphors synthesized by the Pechini sol–gel method exhibit strong visible UC with a maximum relative sensitivity of 0.81% K−1.19 However, it is still a challenge to establish a successful approach to synthesize YOF with efficient green UC luminescence. For the preparation of hollow inorganic materials, silica, known for its cost-effectiveness and uniform structure, is a preferred hard template. The preparation of solid silica spheres utilizes the classical or modified Stöber method, allowing controlled growth through the hydrolysis of alkyl silicates like TEOS in the presence of catalysts such as ammonia. Furthermore, silica exhibits effective adsorption of rare earth cations (RE), forming Si–O–RE bonds at the interface or inducing Coulomb attraction between rare earth cations and the electronegative silica surface, particularly in higher pH conditions.20 Removal of silica templates is typically achieved with NaOH or HF solutions. For instance, Chang et al. showcased the controllability of silica core size by adjusting etching conditions in the synthesis of SiO2@TiO2:Eu3+,Sm3+. The variation in etching time led to a transition from the initial core–shelled structure to a completely hollow structure, illustrating the disappearance of the SiO2 core and the enlargement of the interior cavity.21
Increasing the effective concentration of Ln3+ dopants in a material can generally increase photoluminescence efficiency. High dopant concentrations could, however, result in potential cross-relaxation, which would result in quenching of the excitation energy.22 Therefore, the concentration of activator ions should be kept adequately low to prevent any quenching impact. The precise spacing between the lanthanide ion-occupied lattice sites determines the maximum concentration limit.23 Numerous approaches have been proven to be efficient for boosting the light emission intensity of lanthanide doped luminescence materials, which have been the focus of extensive research for many years.2 The effectiveness of luminescence is thought to be improved by metal ion co-doping, which is a simple and effective procedure. Doping optical activators into a host lattice, like lanthanide ions or transition-metal ions, presents a potential strategy for improved UC luminescence because energy transfer processes can be manipulated. The electron transition in the 4fn configuration, which consists of extremely intricate energy levels as a result of spin–orbit coupling and crystal field interaction, is the prime source of lanthanide luminescence. The main intra-4f electronic–dipole transitions of Ln3+ are prohibited, however by mixing opposite-parity configurations, the selection rule can be broken, leading to weakly allowed electric dipole transitions. The probability of electronic dipole transitions is significantly increased in response to an asymmetric crystal field.23,24 Therefore, manipulating the crystal structure to lower the symmetry around the lanthanide ions may enhance the intensity of luminescence emission. Also, metal ion co-doping can improve overall energy transfer efficiency and has the potential to overcome the phonon quenching impact by providing an effective pathway in the energy transfer process. For example, metal ions such as Gd3+, Li+, Ca2+, Mn2+, and Fe3+ have previously been employed as co-dopants for various UC host matrices such as MSc2O4:Yb3+,Er3+ (M = Mg, Ca, Sr, and Ba) or NaYF4:Yb3+,Er3+, Y2O3:Yb3+,Er3+ to enhance the green/red (G/R) or red/green (R/G) UC luminescence.23–27 It is known that the introduction of metal ion co-dopants into UC materials is an excellent approach to effectively utilize and modulate the excitation energy of Ln3+ ions, which can enhance energy-transfer efficiency. Additionally, it is also known that co-doping with metal ions causes a substitution of lanthanide ion sites within the host lattice, leading to distortion of the lattice and alteration of interplanar spacing, owing to the difference in ionic radii between the co-doped metal ions and lanthanide ions. Consequently, the asymmetric host lattice influences the surroundings of inner lanthanide ions, which often leads to an enhancement of their luminescence.23,28,29
Up to date, besides a few studies, the YOF material has not been investigated very often, and even less as a host matrix for UC luminescence. Our study introduces a novel approach involving the design of spherical YOF particles using SiO2 templates, resulting in the successful creation of hollow YOF structures. Additionally, no prior investigations have reported enhancements in green UC luminescence for YOF doped with Yb3+ and Er3+, nor have the biomedical applications of this material been thoroughly explored until now.
In our work we present the synthesis of core–shell SiO2@NaYF4:Yb3+,Er3+ spherical particles, following a high-temperature annealing step, where an in situ ion-exchange reaction between O2 and NaYF4 is induced, resulting in the formation of SiO2@YOF:Yb3+,Er3+ particles. Afterwards, in a subsequent step, the silica template can be removed to produce hollow YOF:Yb3+,Er3+ particles, which retain the spherical shape and size before annealing. In the study we have investigated the effect of Yb3+ ion concentration on the UC properties of YOF:1%Er3+, x%Yb3+ (x = 1%, 2%, 5%, 10% and 20%). We have also prepared and studied the effect of Gd3+, Mn2+ and Li+ co-doping in SiO2@YOF:Yb3+,Er3+, in an attempt to boost the green UC emission of the materials. The structures, morphologies and particle sizes of the as-obtained particles were evaluated by X-ray powder diffraction (PXRD), scanning electron microscope (SEM) and transmission electron microscope (TEM). Under 975 nm continuous wavelength (CW) laser excitation, the room temperature UC luminescence emission of the samples was determined. We observed that co-doping YOF:Yb3+,Er3+ with 5% Gd3+ ions resulted in the highest G/R UC luminescence intensity. Furthermore, the optical thermometric performance of the resultant doped YOF materials were studied and the relative sensitivity (Sr) was determined. Additionally, this host material in its hollow sphere form has been studied as a potential drug carrier by using doxorubicin hydrochloride (DOX·HCl) as a model anticancer drug, with the aim of combining both functions – thermometry and drug delivery – in one single partcile. Moreover, for a complete evaluation of these particles, we have investigated their cytotoxicity on healthy human dermal fibroblasts, as well as the stability of the YOF at different pH conditions.
Experimental
Materials and methods
Materials.
Yttrium(III) nitrate hexahydrate 99.9% trace metals basis (Y(NO3)3·6H2O), erbium(III) nitrate pentahydrate 99.9% trace metals basis (Er(NO3)3·5H2O), ytterbium(III) nitrate pentahydrate 99.9% trace metals basis (Yb(NO3)3·5H2O), gadolinium(III) nitrate hexahydrate 99.9% trace metals basis (Gd(NO3)3·6H2O), manganese(II) nitrate hydrate, 98% (Mn(NO3)2·xH2O), lithium nitrate 98+ (LiNO3), tetraethyl orthosilicate, 98% (TEOS) and doxorubicin hydrochloride (DOX·HCl) were purchased from Sigma Aldrich. Urea, 99.5% (CO(NH2)2), and sodium fluoride (NaF) were purchased from Thermo Scientific Chemicals. Sodium hydroxide, ≥99% was purchased from Carl Roth. Ethylene glycol a.r.,99.5 + % (EG) and ethanol a.r.,96% were purchased from Chem Lab. Deionized (DI) water was used as a solvent unless stated otherwise.
Synthesis of YOF:Yb3+,Er3+.
Uniform SiO2@NaYF4:Yb3+,Er3+ spherical particles were synthesized using a hard template method.30 First, the Stöber method was used to synthesize silica particles.31 A mixture of 74 mL ethanol, 10 mL distilled water, and 3.14 mL of 36–38% ammonia solution was stirred using a magnetic stirrer in a flask immersed in a water bath set at 30 °C. Subsequently, 6 mL of TEOS was added dropwise while stirring continuously, and the reaction was allowed to proceed for 1 h. Finally, the silica particles were centrifuged, washed thrice with ethanol, and dried overnight at 80 °C. Then, an amorphous Y(OH)CO3:Yb3+,Er3+ shell was constructed around the silica particles. In this synthesis procedure, 30 mg of silica particles were sonicated for 30 minutes in 100 mL of DI water. Then, still under sonication, 3 g urea and 0.5 mmol of rare earth precursors – Y(NO3)3·6H2O, Yb(NO3)3·5H2O and Er(NO3)3·5H2O (with a ratio of x%Y:y%Yb:1%Er, where x = 100 − (y%Yb + 1%Er) and y = 1%, 5%, 10%, 20%) were introduced. The solution was then transferred to a round bottom flask and heated for 2 h at 90 °C while being vigorously stirred in a sealed environment. The flask was gradually cooled and then the product was separated by centrifugation. The SiO2@Y(OH)CO3:Yb3+,Er3+ particles were purified three times with DI water and ethanol, then dried in an oven at 80 °C for 12 h.
The SiO2@NaYF4:Yb3+,Er3+ composite particles were prepared by incubating the obtained SiO2@Y(OH)CO3:Yb3+,Er3+ with NaF (0.2 mmol), 7.5 mL DI water, and 25 mL EG for 12 h at 180 °C in a 50 mL stainless steel autoclave with a Teflon lining. After cooling the autoclave down to room temperature, the product was separated by centrifugation (7000 rpm) and then washed with DI water three times. The product was next dried in an oven at 80 °C. Finally, the SiO2@NaYF4:Yb3+,Er3+ powder was heat treated in air at various temperatures: 400 °C, 600 °C, 650 °C, 700 °C, and 800 °C for 2 h in a muffle furnace, in an attempt to obtain SiO2@YOF:Yb3+,Er3+ material. In a last step, we also show that in order to obtain hollow YOF:Yb3+,Er3+ spheres, the silica template can be etched out using a 1 M NaOH solution (with heating at 70 °C for 3 h). Afterwards, the product was collected by centrifugation, and washed three times with DI water and ethanol and finally dried in drying oven at 80 °C for 12 h. Scheme 1 illustrates the synthesis procedure for forming hollow YOF:Yb3+,Er3+ particles.
 |
| | Scheme 1 Schematic overview of the different steps leading to the formation of YOF:Yb3+,Er3+ hollow particles. | |
The synthesis of SiO2@NaYF4:Yb3+,Er3+ materials additionally co-doped with either Gd3+, Mn2+ or Li+ was carried out in a similar way to that described above for the SiO2@NaYF4:Yb3+,Er3+, with the difference that certain amounts of Gd(NO3)3·6H2O, Mn(NO3)2·xH2O or LiNO3 were added to the precursor solution with the aim of obtaining different co-dopant concentrations (2%, 5%, 8% or 10%). Last, the material was annealed at 600 °C in air for 2 h in a furnace in order to obtain SiO2@YOF:Yb3+,Er3+.
Characterization
Transmission electron microscopy (TEM) images were recorded at the UGent TEM Core Facility using a JEOL JEM-2200FS TEM operating at 200 kV and equipped with a Cs corrector. 300-Mesh Holey Carbon copper grids were used to prepare the samples. A high-angle annular dark field (HAADF) detector was used to acquire scanning TEM (STEM) images. Energy dispersive X-ray (EDX) spectroscopy in HAADF-STEM mode was used to determine the composition of the material. Scanning Electron Microscopy (SEM) was carried out using an FEI Quanta 200 FSEM with an FEI Nova 600 Nanolab Dual-Beam focused ion beam in secondary electron mode. Powder X-ray diffraction (PXRD) patterns were collected using a Bruker D8 Advance X-ray diffractometer equipped with an autochanger and LynxEye XE-T Silicon strip Line detector, operated at 40 kV, 30 mA using Cu Kα radiation (λ = 1.5406 Å) in a Bragg–Brentano geometry and also a Rigaku Mini Flex benchtop PXRD equipped with a rotating sample holder and a D/tex Ultra Silicon strip detector which was operated at 40 kV, 15 mA using Cu-Kα radiation (λ = 1.5406 Å) in Bragg–Brentano (θ:2θ) geometry. Thermo-gravimetry Analysis (TGA) and Differential Thermal Analysis (DTA) were evaluated on a Netsch-STA 449 Jupiter F1 apparatus under air flow, temperature ranging from 20 °C to 1000 °C with a heating rate of 10 °C min−1. All XPS and sputter-profiling experiments were carried out in UHV on an ESCALAB (VG Scientific) spectrometer. XPS spectra were recorded from the as-received surfaces using an Al Kα (1486.6 eV) X-ray source. A low energy electron flood gun 3 eV was used as a neutralizer. Metal ions doping in the materials was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) using an iCap 7000 duo (ThermoFisher).
Thermometry and luminescence spectroscopy measurements were carried out using an Edinburgh FLS920 spectrofluorometer that was fitted with a Hamamatsu R928P photomultiplier tube to detect emission signals in the near UV to visible range. A 975 nm CW laser was used to obtain all emission spectra. A QPOD 2e (Quantum Northwest, USA) was placed in the sample chamber of the spectrometer to analyze the temperature-dependent photoluminescence behavior of the aqueous suspensions of the particles. To determine the power density, the laser power was measured using a power meter (PM160T-HP). The readings obtained were 1.35 W for the sample in DI and 925 mW for the solid sample. Subsequently, the BC106N-VIS/M CCD camera beam profiler was employed to ascertain the area of the elliptical beam profile. This area measurement was then used to calculate the power densities, resulting in 10.73 W cm−2 for the solid sample and 15.70 W cm−2 for the dispersed sample. The measurements were conducted while stirring at a 600 rpm speed. Some of the measurements were carried out employing an Edinburgh FLS1000 spectrofluorometer equipped with a Luma heating–cooling system (Quantum Northwest, USA). The temperature-dependent emission spectra of the powder sample, measured above room temperature (25–300 °C), were captured using a Linkam (Surrey, UK) THMS600 Microscope Stage (±0.1 °C temperature stability), which could be positioned within the spectrometer for analysis. All emission spectra in the manuscript have been corrected for detector response. The TeSen program (https://www.tesen.ugent.be)32 was used to process all temperature-dependent luminescence data. The luminescence lifetimes were recorded using the PM-2 pulse width control box coupled with the 975 nm CW laser (provided by Edinburgh Instruments).
Results and discussion
Phase, structure, morphology, and formation process
PXRD patterns were recorded to identify the composition and phase purity of the as-synthesized samples. The materials were analyzed before and after the different heat treatment steps and subsequent NaOH etching step. The diffraction pattern of SiO2@NaYF4:Yb3+,Er3+ composite recorded before annealing, in the 10° to 60° 2θ range, suggests that the material is mostly amorphous. A pronounced change in the diffraction pattern is observed after heat treatment of the SiO2@NaYF4:Yb3+,Er3+ particles. The crystallinity of the material is significantly enhanced after annealing at 400 °C (Fig. 2(A)). After heat treatment of the powder at this temperature the diffraction peaks can be assigned to the cubic phase of NaYF4 (60257-ICSD). When the heat treatment is carried out at 600 °C we can observe the formation of primarily the YOF cubic phase (28684-ICSD). However, some additional peaks are observed in the YOF PXRD patterns, which do not match the cubic phase. This can best be assigned to small mixtures of the tetragonal and trigonal YOF phase. We determined the presence of cubic, trigonal, and tetragonal phases through the utilization of the reference intensity ratio (RIR) method. The analysis revealed a predominance of the cubic phase at 90%, with the trigonal phase contributing 1%, and the tetragonal phase accounting for 9% of the composition. Also due to the very similar crystal structure and therefore similar peak positions of the cubic NaYF4 and cubic YOF, it cannot be completely excluded that some cubic NaYF4 is still present in the samples (both NaYF4 and YOF have a cubic Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, the unit cell of YOF is a bit smaller, hence PXRD peaks shift to higher 2 theta degrees). Schematic representations of the cubic structure of NaYF4 and YOF have been shown in Fig. 1. An annealing procedure in an air atmosphere at 600 °C activates the in situ ion exchange interaction between O2 molecules and the NaYF4 particles, where O2− ions replace F− ions in the NaYF4 lattice, enabling a transformation from NaYF4 to YOF (O2 + 2NaYF4 → 2YOF + 2F2 + 2NaF). When the annealing temperature is raised above 600 °C, the YOF transforms into a cubic Y2O3 (66242-ICSD) material. If the annealing temperature is further increased to 800 °C, the cubic Y2O3 transforms into monoclinic Y2O3 (192863-ICSD). Additionally, it should be noted that, the PXRD pattern for the SiO2@YOF:Yb3+,Er3+ material did not change after the silica template was removed (etched using a 1 M NaOH solution) in order to obtain the hollow YOF:Yb3+,Er3+ spheres (Fig. 2(B)). We have also studied all of the prepared SiO2@YOF:Yb3+,Er3+ materials co-doped with Gd3+, Mn2+ or Li+ and no changes to the PXRD diffractograms are observed even at a 10% co-dopant amount. Therefore, it can be stated that additional co-dopants do not impose any changes to the material phase and do not affect the annealing temperature significantly as the YOF material was still obtained at 600 °C (Fig. 2(C)).
m space group, the unit cell of YOF is a bit smaller, hence PXRD peaks shift to higher 2 theta degrees). Schematic representations of the cubic structure of NaYF4 and YOF have been shown in Fig. 1. An annealing procedure in an air atmosphere at 600 °C activates the in situ ion exchange interaction between O2 molecules and the NaYF4 particles, where O2− ions replace F− ions in the NaYF4 lattice, enabling a transformation from NaYF4 to YOF (O2 + 2NaYF4 → 2YOF + 2F2 + 2NaF). When the annealing temperature is raised above 600 °C, the YOF transforms into a cubic Y2O3 (66242-ICSD) material. If the annealing temperature is further increased to 800 °C, the cubic Y2O3 transforms into monoclinic Y2O3 (192863-ICSD). Additionally, it should be noted that, the PXRD pattern for the SiO2@YOF:Yb3+,Er3+ material did not change after the silica template was removed (etched using a 1 M NaOH solution) in order to obtain the hollow YOF:Yb3+,Er3+ spheres (Fig. 2(B)). We have also studied all of the prepared SiO2@YOF:Yb3+,Er3+ materials co-doped with Gd3+, Mn2+ or Li+ and no changes to the PXRD diffractograms are observed even at a 10% co-dopant amount. Therefore, it can be stated that additional co-dopants do not impose any changes to the material phase and do not affect the annealing temperature significantly as the YOF material was still obtained at 600 °C (Fig. 2(C)).
 |
| | Fig. 1 Schematic representation of the cubic structures of NaYF4 (left) and YOF co-doped with Gd (right). | |
 |
| | Fig. 2 PXRD patterns of (A) SiO2@NaYF4:20%Yb3+,Er3+ (annealing at different temperatures ranging from 400–800 °C) (B) SiO2@YOF:20%Yb3+,Er3+ (600 °C) and hollow YOF:20%Yb3+,Er3+, (C) SiO2@YOF:20%Yb3+,Er3+ co-doped with Gd3+, Mn2+ or Li+ at varying percentages (2%, 5%, 8%, 10%). | |
To further analyze the obtained materials, they were studied with TEM. In Fig. 3(A) it can be seen that the synthesized SiO2@YOF:Yb3+,Er3+ composite particles have spherical morphology (300–380 nm). The difference in contrast between the TEM images taken before and after the NaOH etching indicated that the silica core had been successfully removed (Fig. 3(B) and (C)) and a hollow structure was formed. Additionally, the measured shell thickness of the resulting hollow YOF is 29.5 ± 2.7 nm, significantly thinner than the average thickness of SiO2@YOF:Yb3+,Er3+, which is 50.7 ± 3.2 nm. This finding provides additional support for the effective removal of the silica core. The size of the hollow YOF particles (300–400 nm) indicates that the silica removal mostly does not affect the size and shape of the material. Fig. 3(D) depicts the morphology of the particles after heat treatment at 800 °C and shows that although the material transforms into yttrium oxide, the spherically morphology is not disrupted, although a small size shrinkage can be observed, as shown in the histograms below the TEM images. The size and morphology of the materials were additionally analyzed using SEM confirming that the pristine SiO2@NaYF4:Yb3+,Er3+ material is spherical, with good size distribution and no visible cracks in the spheres or broken up particles (Fig. 4(A)). Also, SEM images are shown for the SiO2@YOF:Yb3+,Er3+ (600 °C), hollow YOF:Yb3+,Er3+ after etching, and 5% Gd3+ co-doped SiO2@YOF:Yb3+,Er3+ (Fig. 4(B) and (D)). It is clear from these images that neither the heat treatment nor the etching process affects the spherical morphology and causes visible damage to the sphere morphology. Also, co-doping with Gd3+ ions does not show any effect on the particle morphology.
 |
| | Fig. 3 TEM images of (A) SiO2@NaYF4:Yb3+,Er3+, (B) SiO2@YOF:Yb3+,Er3+ (600 °C), (C) YOF:Yb3+,Er3+, and (D) Y2O3@ Yb3+,Er3+ (800 °C). Particle size distribution histograms are given underneath the TEM images. | |
 |
| | Fig. 4 SEM images of (A) SiO2@NaYF4:Yb3+,Er3+, (B) SiO2@YOF:Yb3+,Er3+ (C) YOF:Yb3+,Er3+, and (D) 5%Gd3+ co-doped SiO2@YOF:Yb3+,Er3+. | |
EDX maps of the SiO2@YOF:Yb3+,Er3+ and YOF:Yb3+,Er3+, that is the YOF materials before and after SiO2 template removal, are presented in Fig. 5(A) and (B) to further demonstrate the composition of the materials and the distribution of the elements (Y, F, O, Si, Yb, and Er) in the particles. The presence of O in element mapping additionally confirms the conversion of NaYF4 to YOF. The EDX maps for 5%Gd co-doped SiO2@YOF:Yb3+,Er3+ (Fig. 5(C)) indicate the presence of Y, F, O, Si, Gd, Yb and Er in the synthesized materials, therefore confirming that YOF was formed, as well as that the lanthanide co-dopants Gd, Yb and Er are evenly distributed throughout the YOF matrix.
 |
| | Fig. 5 HAADF-STEM images with EDX maps of (A) SiO2@YOF:Yb3+,Er3+ (B) YOF:Yb3+,Er3+ and (C) 5%Gd3+ co-doped SiO2@YOF:Yb3+,Er3+. | |
Thermal decomposition of the SiO2@NaYF4:Yb3+,Er3+ was measured to gain insights into its thermal properties and behavior as a function of temperature with a TGA-DTA analysis at a slow heating rate of 0.5 °C min−1 till 1000 °C. Graphs are shown in the ESI† (Fig. S3A–C). Three regions of mass loss are observed. Before 300 °C they are mainly assigned to volatile components, non-bound water-humidity, solvents and impurities. Between 300 °C and 600 °C a multistep decomposition with a mass loss of about 18% is observed. Between 700 °C and 1000 °C a second stepwise decomposition is observed, presumably correlated with defluorination.
Luminescence and thermometry properties
The UC luminescence spectra of the particles after annealing at different temperatures: 400 °C, 600 °C, 650 °C, 700 °C and 800 °C were recorded under 975 nm CW laser excitation and are shown in Fig. 6(A). It is worth noting that the ratio of the green to red emission significantly changed for the samples after annealing at different temperatures. At an annealing temperature of 400 °C, the cubic phase of NaYF4:Yb3+,Er3+ is recognized for its pronounced red emission.33–35 However, the green emission is relatively the strongest for the sample which were annealed at 600 °C (assigned to the YOF material based on PXRD). The YOF:Yb3+,Er3+ particles exhibit UC emission of Er3+, with peak maxima at 520 nm (2H11/2 → 4I15/2), 543 (4S3/2 → 4I15/2) and 654 nm (4F9/2 → 4I15/2) (Fig. 6). Generally, the populated 4F7/2 state predominately undergoes non-radiative relaxation to generate green UC luminescence via the 2H11/2 and 4S3/2 states. Concurrently, the presence of the populated 4F9/2 level elicits red UC emissions.36 It is known that the emission intensity and the relative ratio of green to red emission in UC Yb–Er materials can be influenced by a number of factors, including the crystallographic phase of the material, particle size and shape, rare earth ion concentration, impurities, and ligands. In this study we investigated three different co-dopants: Gd3+, Mn2+ or Li+ as a route to enhance the green emission, which is relevant for applications in luminescence thermometry.
 |
| | Fig. 6 (A) Emission spectra recorded in solid state after annealing the materials at different temperatures: 400 °C, 600 °C, 650 °C, 700 °C and 800 °C. (B) Emission spectra of SiO2@YOF:20%Yb3+,1%Er3+ before and after Gd3+ co-doping (excited using a 975 nm CW laser, different concentrations of Gd3+ were explored: 0%, 2%, 5%, 8%, 10%). | |
While Gd3+ ions themselves do not participate in energy transfer or exhibit luminescence in the visible spectral range, they can act as migration centers for energy transfer.37 The energy migration facilitated by the network of Gd3+ ions play a crucial role, particularly in applications that demand precise regulation of energy transfer within the core, shell, or surface of nanocrystals.38 Furthermore, the incorporation of Gd3+ ions through co-doping not only leads to the stabilization of crystal structures but also induces notable modifications in the morphology and size of (nano)crystals.39,40 Consequently, these alterations provide an additional degree of control for tuning the luminescent characteristics of the particles. The influence of Gd3+ concentration on the emission spectra of SiO2@YOF:Yb3+,Er3+,x%Gd3+ (x = 2%, 5%, 8% and 10%) is shown in Fig. 6(B). An initial increase in Gd3+ concentration caused an increase in overall emission intensity as well as an enhanced green emission in regard to the red emission. 5% Gd3+ co-doping yielded best results, however when the Gd3+ content increased further, the emission intensity and intensity of the green emission starts to decrease. Consequently, it can be inferred that the concentration of Gd3+ influences the emission intensity of 4S3/2 → 4I15/2 (Er3+) in SiO2@YOF:Yb3+,Er3+,x%Gd3+. In the Gd3+ co-doped SiO2@YOF:Yb3+,Er3+ system, the considerable energy gap (32000 cm−1) between the ground state and the first excited state of Gd3+ ions hinders their direct absorption of 975 nm photons. Consequently, Gd3+ ions can become involved in energy transfer processes from Er3+ to Gd3+ at high-energy excited states as reported in earlier literature.41 Therefore, the changes in emission intensity observed in host material co-doped with a higher concentration of Gd3+ can be attributed to a decrease in the average interatomic distance between Er3+ and Gd3+ ions, thus facilitating effective energy transfers between them. Consequently, through the introduction of varied concentrations of Gd3+ ions into a host matrix, one can effectively regulate the population processes associated with their high-energy excited states. This approach proves to be an effective method for studying the energy transfer processes among the Ln3+ ions in the co-doped system.42 To study if this energy transfer process is taking place in our YOF host material we have carried out additional measurements. To obtain the emission spectrum of Gd3+ in SiO2@YOF:Yb3+,Er3+,5%Gd3+, we excited the sample at 273 nm, resulting in emission peaks at 306 nm (corresponding to the 6P5/2 → 8S7/2 transition) and 311 nm (corresponding to the 6P7/2 → 8S7/2 transition) (Fig. S17A, ESI†). Additionally, under 975 nm excitation, we observed the upconversion luminescence spectra of SiO2@YOF:Yb3+,Er3+,5%Gd3+ in the 350–700 nm range, with emission peaks at 410 nm (2H9/2 → 4I15/2), 520 nm (2H11/2 → 4I15/2), 543 nm (4S3/2 → 4I15/2), and 654 nm (4F9/2 → 4I15/2) (Fig. S17B, ESI†). However, we do not detect any Gd3+ emission under these excitation conditions (975 nm), therefore there is no clear indication of the presence of energy transfer between the ions. There is a strong possibility that the enhancement in UC luminescence is caused by lattice distortion. Gd3+ co-doping causes crystal lattice defects, which lead to the replacement of the smaller Y3+ (1.159 Å) by the larger Gd3+ (1.193 Å) ions. A decrease in the average crystallite size or crystallinity is shown by the broadening of XRD peaks with increasing Gd3+ co-dopant levels. In addition, the XRD peaks slightly shift towards lower diffraction angles when compared to samples without Gd3+ co-doping, which could cause a considerable increase in lattice parameter owing to the complete substitution of smaller Y3+ ions by Gd3+ ions in the host lattice. As a result, the YOF host experiences crystal lattice defects by the addition of Gd3+ ions, altering the symmetry around the Yb3+ and Er3+ ions, which is advantageous for breaking the forbidden transitions of the lanthanide ions, leading to an increase in the luminescence intensity of Er3+. On the other hand, the lattice distortion might enhance the exchange interactions between Yb3+ and Gd3+, resulting in an increased energy transfer rate between Yb3+ and Er3+ and a stronger UC luminescence.39 It is clear the effect of Gd3+ co-doping is complex and can have multiple effects. On the other hand, the emission intensity of SiO2@YOF:Yb3+,Er3+ exhibits a decreasing trend as the Gd3+ dopant content increases. This outcome is primarily attributed to the reduction in particle size, which increases the number of surface quenching sites. This, in turn, facilitates nonradiative energy transfer processes among lanthanide ions and, consequently, suppressing upconversion luminescence.40,43 Moreover, a continued elevation in Gd3+ ions lead to concentration quenching.
Metal ions with different valence states, such as Mn2+ and Li+, were also investigated as co-dopants for SiO2@YOF:Yb3+,Er3+ particles in order to show the impact of the cationic dopant valence state on the emission of Er3+ ions through the manipulation of the local crystal field. For the YOF crystal lattice, Mn2+ (0.81 Å) ions are small enough to replace Y3+. Therefore, when Y3+ ions are substituted with an optimal concentration of Mn2+ ions, the resulting size mismatch should cause the lattice to shrink, lowering the crystal symmetry around the Yb3+ and Er3+ ions, which is also advantageous for breaking the forbidden transitions of the lanthanide ions and resulted in an increase in the luminescence intensity of Er3+.44 It is worth mentioning that the diffraction peaks of our materials experience a slight shift towards higher angles as the concentration of Mn2+ increases (Fig. 2(C)). This shift is primarily ascribed to the reduction in the unit cell volume of the YOF host, possibly attributed to the substitution of Y3+ ions with Mn2+ possessing a relatively smaller radius.45 The substitution introduces an additional F− ion on the grain surface, leading to the induction of transient electric dipoles with outward-pointing negative poles. These dipoles significantly impede the diffusion of necessary F− ions for crystal growth from the solution to the grain surface, primarily due to charge repulsion. As a consequence, this process hinders the growth of the host material.24 This is evident in the gradual narrowing of the diffraction peaks as the Mn2+ content increases. This alteration effectively disrupts the symmetry around the Ln3+ ions, leading to a substantial enhancement in luminescence intensities. The intense red UCL at 5 mol% Mn2+, as shown in Fig. 7(B) could potentially be attributed to non-radiative energy transfer from the Er3+ to Mn2+ (2H9/2/4S3/2 →4T1), followed by back-energy transfer to Er3+(4T1 →4F9/2).46,47 A quenching phenomenon induced when higher concentrations of Mn2+ ion were used in the synthesis was also demonstrated by the fact that the fluorescence intensity of SiO2@YOF:Yb3+,Er3+ decreased as Mn2+ concentration rose from 5 mol% to 10 mol%. In general, in our study, we observed a steady increase in the R/G ratio with rising Mn2+ co-dopant concentrations (0–5 mol%) employed in the synthesis. However, based on ICP-OES results (Table S2, ESI†) we know that actually only trace amounts of Mn2+ built into the lattice. Therefore, it is clear the enhancement is not attributed to the incorporation of Mn2+ ions into the SiO2@YOF:Yb3+,Er3+ matrix and energy transfer process between Er3+ and Mn2+ (or they are happening only at trace levels). The introduction of dopants or impurities can however affect the crystal growth and shape or phase even if they are not incorporated into the lattice of the host material. Therefore, the Mn2+ concentration used in the synthesis influences the G/R ratio, not because of significant Mn2+ incorporation, but due to its adsorption during the process. Furthermore, XPS analysis failed to detect Mn, resulting in an inability to determine its true oxidation state (Fig. S2 and Table S1, ESI†). Nonetheless, the co-doping procedure involving Mn2+ used in our experiment has been previously reported.46,48 Therefore, based on this information, we predicted the presence of Mn2+ ions, also evident in the enhancement of red emission with increasing concentration of Mn2+. The exact amount of the dopant and co-dopants embedded in the YOF particles was assessed by ICP-OES (see Table S2, ESI†) and as stated earlier the detection of only trace amounts of Mn2+ in the co-doped samples strongly suggests that Mn has been minimally incorporated into the host material. This also explains why Mn2+ could not be detected using XPS. Interestingly despite the limited concentration of Mn2+, noticeable changes in the R/G emission behavior are observed. The observed modifications in emission characteristics can be ascribed to the presence of structural defects introduced during the synthesis process of Mn2+ co-doped samples. It can be proposed that during the co-doping process, the incorporation of Mn2+ disrupts the regular crystal lattice of the material, leading to the formation of structural imperfections or defects. These defects can significantly influence the electronic and optical properties of the host material, thereby resulting in alterations in the emitted spectra.
 |
| | Fig. 7 Green/red (G/R) ratios for SiO2@YOF:20%Yb3+,1% Er3+ co-doped with different metal ion (A) Gd3+, (B) Mn2+ and (C) Li+. | |
In the periodic table of elements, the Li+ ion has the smallest cationic radius, which is advantageous for its mobility and site occupation in the host lattice. These benefits make them appealing for use in manipulating the host lattice local crystal field.3 When Li+ ions were co-doped at a concentration of 2 mol%, the major diffraction peak (around 2θ = 30°) shifted toward larger angles (Fig. 2(C)). When the Li+ ion concentration was 5 mol%, the main diffraction peak gradually shifted towards lower angles. Finally, for Li+ ion concentrations greater than 5 mol%, the main diffraction peak showed no shifts anymore. The peak shifting result could be linked to the Y3+-site substitution on the cell parameters is significant for Li+ ion concentrations of 0–2 mol% and on the other hand, the effect on the occupation of interstitial sites becomes substantial for Li+ ion concentrations of 2–5 mol%. The consistent peak position for Li+ ion concentrations above 5 mol% would suggest that no more Li+ ions can reach the YOF host lattice. However, once again here, as in the case of Mn2+, only trace amounts of Li+ could be detected in the host matrix employing ICP-OES (Table S2, ESI†). The observed emission characteristics, specifically the change in emission of R/G, can be therefore attributed to the alterations in the lattice structure that occur during the synthesis process in the presence of Li+ co-doping. These changes in the lattice structure led to the formation of structural defects, which significantly influence the emitted light and ultimately result in the observed spectral changes. Karan et al., reported that the process of doping is not solely influenced by the nature of host materials, instead, dopants have the ability to manipulate their surroundings to create a more energetically stable environment, which may involve modifying the lattice arrangement of the hosts.49 According to Guria et al., the introduction of dopants or impurities can affect crystal growth and shape or phase changes without necessarily incorporating into the lattice of the host materials. Consequently, the adsorption of dopants or impurities during the formation of particles plays a significant role in determining their characteristics.50 This supports our findings, suggesting that trace amounts of Li+ and Mn2+ can significantly influence the behaviour and properties of the host material. It should also be mentioned that the introduction of Mn2+ or Li+ into YOF matrix through the substitution of Y3+, even if at small amounts, disrupts the charge balance in YOF. This disturbance is a direct outcome of replacing Y3+ with Mn2+ or Li+. To restore the charge equilibrium, F− vacancies are generated, initiating subsequent lattice contraction. Additionally, the difference in ionic radius between Y3+ (1.159 Å) and Mn2+ (0.81 Å) or Li+ (0.68 Å) also contributes to lattice contraction. These two factors collectively induce a subtle shift in the diffraction peaks after the introduction of Mn2+ or Li+ co-doping. Furthermore, to ensure the reliability of our G/R findings in these co-doped materials, we synthesized the whole series of SiO2@YOF co-doped with Mn2+ and Li+ two additional times. Our repeated experiments also demonstrated that increasing the concentration of Mn2+ or Li+ from 0 to 5 mol% results in an increase in the overall G/R intensity. Concentrations above 5 mol%, lead to a decrease in overall G/R intensity, highlighting the significant role of Mn2+ and Li+ ion concentration in this process (Fig. S18A and B, ESI†). It is noticeable that the green emission is comparatively weaker for both Li+ and Mn2+ co-doping. In general, we have observed that samples with a low Gd3+, Mn2+ and Li+ co-doping concentration benefit from a significant luminescence enhancement during the annealing process. On the other hand, higher doping concentrations resulted most likely in defects and cross-relaxation processes that caused quenching of the UC luminescence. The (G/R) ratios corresponding to UC luminescence of SiO2@YOF:Yb3+,Er3+ with different metal ion (Gd3+, Mn2+ or Li+) co-doping concentration such as 2%, 5%, 8% and 10% are shown in Fig. 7(A)–(C). Only in the case of Gd3+ co-doping the desired green emission became dominant, which was the goal of our study.
Further, the decay curves of the green 4S3/2 → 4I15/2 (543 nm) and red 4F9/2 → 4I15/2 (654 nm) emission peaks of SiO2@YOF:Yb3+,Er3+,x% Gd3+ (x = 0%, 2%, 5%, 8%, 10%) were recorded upon pulsed laser excitation of 975 nm. All decay curves were fitted using a monoexponential function. The decay profiles have been presented in Fig. 8(A) and (B) and the calculated decay times have been overviewed in Table 1. It can be noticed that the decay time for the green emission increased with the introduction of Gd3+ concentration when co-doped with up to 5% of the ion, and then decrease for 8% to 10% Gd3+ co-doping, which is consistent with the observations made for emission intensities. When recording the decay times for the red 4F9/2 → 4I15/2 (654 nm) emission peak, it could be observed that the co-doping of Gd3+ enhances the decay time, however no clear trend was observed, as all values are quite similar.
 |
| | Fig. 8 Luminescence decay curves for (A) green 4S3/2 → 4I15/2 (543 nm) and (B) red 4F9/2 → 4I15/2 (654) emission peaks in SiO2@YOF:20%Yb3+,1%Er3+ before and after Gd3+ co-doping. | |
Table 1 Overview of decay curves and R2 of the green 4S3/2 → 4I15/2 (543 nm) and red 4F9/2 → 4I15/2 (654 nm) emission peaks for the Gd3+ co-doped SiO2@YOF:20%Yb3+,1%Er3+ samples
| SiO2@YOF:Yb3+,Er3+,x%Gd3+ |
τ
543 [μs] |
R
2
|
| 0% Gd3+ |
122 |
0.997 |
| 2% Gd3+ |
230 |
0.997 |
| 5% Gd3+ |
298 |
0.997 |
| 8% Gd3+ |
254 |
0.998 |
| 10% Gd3+ |
67 |
0.990 |
| SiO2@YOF:Yb3+,Er3+,x%Gd3+ |
τ
654 [μs] |
R
2
|
| 0% Gd3+ |
68 |
0.997 |
| 2% Gd3+ |
100 |
0.996 |
| 5% Gd3+ |
85 |
0.990 |
| 8% Gd3+ |
89 |
0.992 |
| 10% Gd3+ |
77 |
0.990 |
For investigating the thermometric performance of the SiO2@YOF:Yb3+,Er3+ we focused on the Gd3+ co-doped samples as they showed strongest green emission, which is used for Boltzmann thermometry.51 Thermometry based on the Yb3+–Er3+ UC system is most known in the visible emission region and relies on the ratio of the integrated emission intensities from the thermally coupled excited states 2H11/2 and 4S3/2. We have observed in the study that the G/R emission in the SiO2@YOF:Yb3+,Er3+ material could be tuned by changing the concentration of Gd3+ (2%, 5%, 8% and 10%) co-doping. However, the G/R ratio is relatively low at 10% Gd3+ co-doping, making this sample unsuitable for thermometry. The temperature-dependent and room-temperature luminescence properties of the Gd3+ co-doped materials were investigated in aqueous suspensions. The temperature-dependent emission maps (293.15–333.15 K; 20–60 °C) for SiO2@YOF:Yb3+,Er3+,x%Gd3+ (x = 0%, 2%, 5%, 8%) are presented in Fig. 9(A)–(D). We compared the temperature-dependent luminescence behavior of the material before and after co-doping of Gd3+ in these YOF-based UC materials. The population ratio of two thermally coupled excited states is determined by the Boltzmann distribution. The thermometric parameter Δ calculated based on the intensity ratio of the 520 nm (2H11/2 → 4I15/2) and 543 nm (4S3/2 → 4I15/2) radiative transitions is a very important parameter used to evaluate the performance of optical thermometers (see eqn (1)):
| |  | (1) |
where
α =
C g2/
g1. While
g2 and
g1 are the (2
J + 1)-fold degeneracies of the
2H
11/2 and the
4S
3/2 levels, respectively,
C provides information about the ratio of the radiative transition probabilities from the two thermally coupled levels to the addressed ground level and Δ
E is the effective energy gap between the two excited levels. The experimental data was fitted with the model given in
eqn (1) obtained from the integrated intensity ratios from the
2H
11/2 and
4S
3/2 levels, respectively, to yield Δ
E = (431.3 ± 1.9) cm
−1 (
R2 = 0.993) for of SiO
2@YOF:Yb
3+,Er
3+,0%Gd
3+. Whereas for, SiO
2@YOF:Yb
3+,Er
3+,2%Gd
3+ Δ
E = (652.1 ± 2.0) cm
−1 (
R2 = 0.990), SiO
2@YOF:Yb
3+,Er
3+,5%Gd
3+ Δ
E = (659.6 ± 0.1) cm
−1 (
R2 = 0.984), and SiO
2@YOF:Yb
3+,Er
3+,8%Gd
3+ Δ
E = (562.4 ± 0.5) cm
−1 (
R2 = 0.978). The increase in energy difference (Δ
E) following the co-doping of SiO
2@YOF:Yb
3+,Er
3+ with Gd
3+ is evident (
Fig. 10(A)–(D)). The Δ
E for the aforementioned particles agrees well with the spectroscopically expected value of the energy difference between the
2H
11/2 and
4S
3/2 level.
52,53
 |
| | Fig. 9 Emission maps for SiO2@YOF:Yb3+,Er3+ after Gd3+ co-doing recorded at 293.15–333.15 K (20–60 °C): (A) 0% Gd3+, (B) 2% Gd3+ (C) 5% Gd3+ and (D) 8% Gd3+. | |
 |
| | Fig. 10 Graphs overviewing the Δ parameters for SiO2@YOF:Yb3+,Er3+,x%Gd3+ co-doped samples. The points show the experimental Δ parameters, and the solid line shows the least squares fit to the experimental points. The following Gd3+ co-doping was used: (A) 0% Gd3+, (B) 2% Gd3+ (C) 5% Gd3+ and (D) 8% Gd3+. | |
Relative sensitivity Sr, which is independent of the operational principle of the thermometer, represents the relative change of the thermometric parameter per unit temperature change (in %K−1) (eqn (2)).52 This makes it possible to compare various thermometric materials directly and quantitatively.
| |  | (2) |
The maximum relative sensitivity was calculated using eqn (2) and is equal to 0.716% K−1 at 293.15 K for SiO2@YOF:Yb3+,Er3+. It is interesting to observe that while the Sr value improves as Gd3+ concentration rises from 0% to 5%, it begins to decline from 8% onward. This could possibly be due to the decrease in green intensity (also evident from the much lower R2 value in this case). The optimal Gd3+ co-doping concentration for strong green emission was 5%, which also exhibits the highest Sr value in comparison to other concentrations (Fig. 11(A)–(D)). These values have been overviewed in Table 2 together with the R2 fits. The incorporation of Gd3+ demonstrates an effective enhancement in optical thermometric response, which can be compared to the findings of various thermometric materials listed in Table 3. Temperature uncertainty (δT) is another very important thermometric parameter, as it considers both the relative sensitivity and the measurement error of the intensity ratio (δΔ) (eqn (3)).
| |  | (3) |
 |
| | Fig. 11 Plots of the relative sensitivity (Sr) at varying temperatures (293.15–333.15 K, step of 5 K) for SiO2@YOF:Yb3+,Er3+,x% Gd3+ where x = (A) 0% Gd3+, (B) 2% Gd3+ (C) 5% Gd3+ and (D) 8% Gd3+. | |
Table 2 Overview of R2 and Sr values for SiO2@YOF:20%Yb3+,1%Er3+,x%Gd3+ where different amounts of Gd3+ co-dopants were incorporated into the materials
| SiO2@YOF:Yb3+,Er3+,x% Gd3+ |
R
2
|
S
r (%K−1) (293.15 K) |
| 0% Gd3+ |
0.993 |
0.723 |
| 2% Gd3+ |
0.990 |
1.100 |
| 5% Gd3+ |
0.984 |
1.105 |
| 8% Gd3+ |
0.978 |
0.959 |
Table 3 Relative temperature sensitivity (Sr) of optical thermometers based on typical Yb3+–Er3+ doped of UC materials compared to our materials
| Compounds |
Temperature range (K) |
S
r (%K−1) |
Ref. |
| Na2GdMg2(VO4)3:Yb3+,Er3+ |
303–573 |
0.976 (303 K) |
54
|
| NaYF4:Yb3+,Er3+ |
223–403 |
0.36 (363 K) |
55
|
| YbF3:Er3+ |
303–500 |
0.46 (303 K) |
56
|
| Ba3Gd2F12:Yb3+,Er3+ |
296–600 |
1.12 (296 K) |
57
|
| La2O3:Yb3+,Er3+ |
303–600 |
0.91 (303 K) |
58
|
| NaGdF4:Er3+,Yb3+,Fe3+ |
303–503 |
0.781 (303 K) |
59
|
| Y2O3:Yb3+,Er3+ |
288–328 |
1.097 (288 K) |
53
|
| YF3:Yb3+,Er3+ |
293–473 |
1.06 (293 K) |
60
|
| SiO2@YOF:Yb3+,Er3+ |
293–333 |
0.723 (293 K) |
Our work |
| SiO2@YOF:Yb3+,Er3+,Gd3+ |
293–333 |
1.105 (293 K) |
Our work |
For SiO2@YOF:Yb3+,Er3+,0%Gd3+ throughout the whole temperature range δT< 0.32 K yielding a good result. While for SiO2@YOF:2%Gd (δT < 0.13 K), SiO2@YOF:5%Gd (δT < 0.082 K), and SiO2@YOF:8%Gd (δT < 0.12 K) (see Fig. 12(A)–(D)) showing even superior behavior of the materials.
 |
| | Fig. 12 Graph depicting the temperature uncertainty in the whole temperature range for SiO2@YOF:Yb3+,Er3+,x% Gd3+ where x = (A) 0% Gd3+, (B) 2% Gd3+ (C) 5% Gd3+ and (D) 8% Gd3+. | |
We have additionally investigated the high-temperature-dependent behavior of SiO2@YOF:Yb3+,Er3+,5%Gd3+ in solid form. This sample was selected due to its highest Sr value among the series of samples when investigated in the physiological range. These measurements were additionally carried out because it is known that YOF is more thermally stable than for example NaYF4, which is one of the most explored UC materials.18 We therefore sought to determine whether it could also be utilized as a high temperature UC thermometer for other future applications. We observed that the overall intensity decreases with an increase in temperature, as is expected due to thermal quenching. The thermometric parameter Δ of the 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions increased gradually with an increase in temperature (25–300 °C) and the calculated value of ΔE is 562.4 ± 0.5 cm−1 (R2 = 0.998) and, the maximum Sr is 0.912%K−1 (Fig. S10A–C, ESI†). Furthermore, the relative sensitivities calculated for the mentioned thermometer are comparable to those of NaYF4:Yb3+,Er3+ thermometers.61 To check if the material can be used as a reliable thermometer more than once, it is essential to conduct cycle tests and the repeatability is calculated using eqn (4).
| |  | (4) |
Where
Δc is the mean thermometric parameter, and
Δi is the value of each measurement of the thermometric parameter.
62
Finally, cycle tests of SiO2@YOF:Yb3+,Er3+,5%Gd3+ in DI water were carried out at two different heating–cooling cycles: 293.15 K to 333.15 K to determine whether the material could be utilized as a reliable thermometer more than once when used in the physiological temperature range (Fig. S10D, ESI†). The material showed repeatability between 96% to 98% using eqn (4). The thermometry properties of Mn2+ and Li+ co-doped SiO2@YOF:Yb3+,Er3+ materials were not explored any further due to the fact that their green emission was considerably weak and not suited for thermometric applications. Nonetheless such materials can be of value for applications where red UC emission is preferred. For example, the incorporation of Li+ and Mn2+ co-doping in UC nanoparticles such as Gd2O3, NaYF4, leading to enhanced single-band red emission, shows significant potential for optical bioimaging applications, especially in the domain of in vivo optical bioimaging, where improved tissue penetration into deeper layers is crucial.63–65 The Yb3+ and Er3+ doping concentration in the SiO2@YOF:20%Yb3+,1%Er3+ was selected based on the well-known best ratios for the NaYF4 host matrix. As in the first synthesis step NaYF4 host material was obtained this seemed like a logical choice. However, upon further investigation of this material, we have also considered whether the 20%Yb3+,1%Er3+ doping ratios are the most optimal for the YOF host matrix. As a result, to evaluate this, we have prepared a series of SiO2@YOF:Yb3+,Er3+ samples with various concentrations of Yb3+ ions (1%, 2%, 5%, 10% and 20%), while the concentration of Er3+ was always maintained at 1%. The PXRD phase, emission spectra, luminescence decay curves and the Sr of these SiO2@YOF:Yb3+,Er3+ samples with different Yb3+ ion concentrations are presented in Fig. S1A, S4, S5 and S6 (ESI†) as well as these results are also overviewed in Tables S3 and S4 (ESI†). The G/R ratio of intensity can be seen to increase as the concentration of Yb3+ increases up to 10%, then at 20%, a quenching effect is observed. These results highlight the significance of Yb3+ concentration on the G/R ratio of the YOF host matrix. Having observed a high G/R ratio in YOF with a 10%Yb3+ doping concentration, we sought to determine the effect of co-doping with Gd3+ at different concentrations (2%, 5%, 8% and 10%) on this ratio (Fig. S7, ESI†). The highest UC luminescence intensity was found to be achieved by co-doping YOF:10%Yb3+,Er3+ with 5%Gd3+ ions. This finding provides important insights into the optimal concentration of Gd3+ for achieving efficient UC luminescence in YOF:Yb3+,Er3+ co-doped with Gd3+. Detailed information is provided regarding the XRD pattern (Fig. S1B, ESI†), luminescence decay time (Fig. S8, ESI†) and the relative sensitivity (Sr) (Fig. S9, ESI†) of Gd3+ co-doping and is also overviewed in Tables S5 and S6 of the ESI.†
To better understand the stability of the material, especially as they are meant to be used for biological applications, we studied the degradation properties of YOF using SEM, PXRD and PL. We conducted an experiment involving the immersion of SiO2@YOF:20%Yb3+,1%Er3+,5%Gd3+ particles in phosphate buffer saline (PBS) with 2 mg mL−1 concentration at various pH levels, including pH 7.4, pH 5.5, and pH 4, for a duration of 48 hours. After soaking in PBS with a pH of 7.4, the YOF spheres exhibited a noticeable increase in surface roughness compared to the initial sample based on SEM analysis (Fig. S11B, ESI†). However, in the presence of an acidic buffer solution at pH 5.5, the YOF spheres experienced further corrosion and pronounced roughening (Fig. S11C, ESI†). Subsequent treatment in PBS at pH 4 resulted in even more substantial degradation of the YOF spherical morphology (Fig. S11D, ESI†). In addition to the aforementioned investigation, we performed XRD analysis on the samples subjected to the different pH treatments. However, no noticeable changes were observed in the XRD patterns. These findings highlight the pH-dependent degradation behavior of YOF, with increased surface roughness and deterioration at lower pH values. However, the XRD analysis did not indicate any structural changes, suggesting that the crystalline nature of the YOF material remained unaffected (Fig. S11E, ESI†). Additionally, we performed photoluminescence measurements with concentration 2 mg mL−1 and found no significant changes in the G/R ratio, indicating minimal impact of pH on the optical properties of the YOF (Fig. S11F, ESI†). This knowledge is important as further drug release experiments are carried out at varying pH, therefore, it is valid to know how pH affects the host matrix.
Cytotoxicity for SiO2@YOF:Yb3+,Er3+ particles
The cytotoxicity data are summarized in Fig. 13(A). The SiO2@YOF:Yb3+,Er3+ is non-toxic to NHDF cells in a very wide concentration range from 0–5 mg mL−1. Overall, the sample even slightly enhances the average cell viability as compared to the controls (those containing only seeded cells and PB-HS cell viability reagent and not treated with sample). Even at the highest tested concentration of 5 mg mL−1 which is a relatively high concentration for in vitro studies, the cell viability is not significantly affected or inhibited and ranges in the interval from 93–105%. This proves the excellent compatibility of the as-synthesized material with the healthy fibroblasts and makes it a favorable candidate for potential future in vivo studies in optimal concentration. The widefield microscopy images in Fig. 13(B) depict the cell visualizations after being treated with the varying concentrations and after being labelled with calcein–AM. Calcein–AcetoMethoxy is essentially a non-fluorescent, cell-permeable ester dye that is hydrolyzed intracellularly by esterases present in viable cells to the highly green fluorescent Calcein form.
 |
| | Fig. 13 (A) Bar chart (mean ± SD) summarizing the cytotoxicity test results for the SiO2@YOF:Yb3+,Er3+ investigated in the concentration range from 0–5 mg mL−1 on five technical replicates per sample concentration. A nonparametric statistical analysis, applying a Mann–Whitney U test yielded one statistically significant difference (* represents p < 0.05) between the median of the control group and the median of the 2.5 mg mL−1 group. The remaining statistically non-significant differences between the control group and the remaining concentration groups have been omitted for the sake of clarity. (B) Fluorescence microscopy images of the SiO2@YOF:Yb3+,Er3+ technical replicates containing NHDF cells stained with Calcein–AM cell labeling dye at a final well concentration of 1.5 μM. Scale bars are set to 500 μm. | |
Drug loading and release experiments
Doxorubicin hydrochloride (DOX·HCl), a well-known model chemotherapeutic drug was selected to evaluate the release behavior from the hollow YOF:Yb3+,Er3+ particles. To quantitatively analyze the DOX during the experiment, a calibration curve of DOX was prepared using a series of solutions with different concentrations of DOX, versus their absorbance at 480 nm. After the sample was loaded with DOX, we evaluated both the drug loading capacity (LC%), which was calculated to be 37.8% (eqn (S2) in the ESI†) and the drug encapsulation efficiency (EE%), which was estimated to be 60.9% (eqn (S3) in the ESI†) using both the calibration curves (Fig. S12–S14, ESI†) and the measured absorbance value. To explore the pH-responsive behavior of DOX-YOF systems as a function of release time, we have determined the absorbance at 480 nm as a function of time at pH 7.4, pH 5.5 and pH 4 in PBS buffer at 37 °C, over a time period of 72 h are shown in Fig. 12. The pH-sensitive DOX release may be advantageous at lower pH in intracellular lysosomes, endosomes, and some malignant tissues for targeted release and controlled therapy at the pathological areas.66 For DOX-YOF systems, about 8% of DOX was released in the neutral condition at pH 7.4, while ∼15% of DOX was released in the acidic PBS solution at pH 5.5, and around 20% DOX was released at pH 4, within 72 h showing a clear increase of drug release at lower pH (Fig. 14). The increased drug release patterns observed at acidic pH levels (5.5 and 4) in comparison to pH 7.4 can be ascribed to various factors. Firstly, the electrostatic interaction between YOF and DOX might weaken in acidic conditions. Another factor is the presence of protons in acidic buffer solutions can facilitate the penetration of pores, accelerating drug release by protonating the amino group of DOX.67 Additionally, deterioration of the drug carrier after a prolonged release period in an acidic buffer may contribute to accelerating drug release to some extent.68 However, the mechanism behind the in general rather slow release of DOX from YOF can possibly be attributed to the fact that the diffusion of the drug molecules occurs from the inner pores of the material rather than its surface.53,69 This would suggest that the DOX molecules are encapsulated within the particles of YOF rather than being adsorbed onto the material's exterior surface. As of now, no substantial work has been carried out on employing YOF host material for drug delivery. However, existing reports on inorganic fluoride host particles for DOX release serve as valuable references for comparing with our drug loading and release results. For instance, Zaho et al. reported that for NaYF4:Yb,Er,@NaGdF4@mSiO2–MnO2 the loading efficiency was calculated to be 56%.70 At pH 7.4, DOX release was around 10.0% after 80 hours. However, with the addition of 10 mM glutathione (GSH) at pH 7.4 and at pH 5.0, the release surpasses that at pH 7.4 without GSH, reaching about 19.5% after 80 hours and a remarkable 60.0% after 24 hours with 10 mM GSH at pH 5.0. This implies that without additional GSH functionalization, the drug release is slow, prompting consideration for further functionalization of our YOF host material to achieve faster drug release. Another example is NaYF4:Yb3+/Er3+@SiO2@P(NIPAM-co-MAA) hybrid microspheres, where the actual weight-based loading of DOX was calculated to be 4.9%. While only 22% of DOX was released after 72 hours at pH 7.4 and 37 °C, over 90% was released within 24 hours under mildly acidic conditions (pH 5.0, 37 °C).71 However, it is important to note that the DOX loading efficiency for these materials is considerably lower compared to our YOF host materials. Also, the relatively low drug loading capacity and rapid drug release significantly limit their suitability for biological applications, causing reduced therapeutic effectiveness and higher toxicity to normal cells.72 In the future, to improve the release kinetics of DOX from YOF particles, approaches that aim to enhance the diffusion of DOX can be explored. For instance, utilizing temperature-sensitive drug delivery systems, surface modifications of materials, synergistic combinations with other drugs, or ultrasound stimulation can be effective strategies. To investigate the impact of DOX on the green UC emission of the material, DOX was incorporated into hollow YOF:10%Yb3+,1%Er3+,5%Gd3+ particles at a 1:1 ratio, and the photoluminescence of hollow YOF:10%Yb3+,1%Er3+,5%Gd3+ and DOX@YOF:10%Yb3+,1%Er3+,5%Gd3+ at a concentration of 1 mg mL−1 was measured. Under 975 nm excitation, a decrease in the green UC emission of DOX@YOF:10%Yb3+,1%Er3+,5%Gd3+ (Fig. S15A, ESI†) was observed, while the red emission remained unaffected. This suggests a quenching effect resulting from energy transfer between the YOF:10%Yb3+,1%Er3+,5%Gd3+ donor and DOX acceptor, where the green emission is partially absorbed by the DOX.73 As demonstrated in Fig. S15B (ESI†), the UV-ViS spectroscopy of DOX absorbance showed an overlap between the absorption spectrum of DOX and the green emission of YOF:10%Yb3+,1%Er3+,5%Gd3+, indicating the close proximity of DOX to the Er3+ ions. Consequently, this fact could play a crucial role in monitoring the spectral changes associated with the loading and release of DOX from host materials employing luminescence emission.74 This suggests an additional advantage of potentially monitoring the progress of therapy through luminescence, enhancing its potential as a theranostic platform. Since the presence of DOX causes a decrease in the green UC emission of DOX@YOF:10%Yb3+,1%Er3+,5%Gd3+, we investigated the thermometric performance of DOX-loaded YOF:10%Yb3+,1%Er3+,5%Gd3+ by analysing their emission spectra under 975 nm excitation across a physiological temperature range (20–60 °C) to confirm that after drug loading it would still be useful as a temperature sensor. The thermometric parameter Δ of the 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions increased gradually with temperature, resulting in a calculated ΔE value of 621 cm−1 (R2 = 0.994) and a maximum Sr of 1.041%K−1 (Fig. S16A–C, ESI†). These findings suggest that the DOX loading did not significantly affect the thermometric performance of the material, as the Sr value remained comparable to that of YOF:10%Yb3+,1%Er3+,5%Gd3+ without DOX loading (Sr of 1.133% K−1, Fig. S9B, ESI†). This is because the thermometric behaviour of DOX@YOF:10%Yb3+,1%Er3+,5%Gd3+ depends on the ratio of emission intensities from thermally coupled levels of the rare-earth ions (Yb3+–Er3+), and not their absolute intensities. Given that these thermometric properties are intrinsic to the rare-earth ions and their interactions within the host lattice, as a result the presence of DOX on the surface does not alter this inherent behaviour. However, the decreased green emission resulting from DOX loading leads to higher temperature uncertainty in DOX@YOF:10%Yb3+,1%Er3+,5%Gd3+ (δT < 0.238 K, Fig. S16D, ESI†) compared to YOF:10%Yb3+,1%Er3+,5%Gd3+ (δT < 0.167 K), which is fully expected due to the lower overall green signal of the DOX loaded material. Nonetheless, the DOX loaded material still shows very good temperature uncertainty.
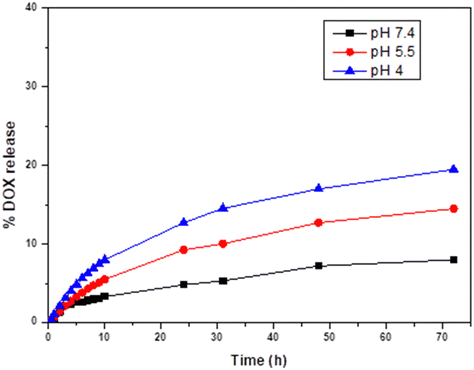 |
| | Fig. 14 Drug release profiles of DOX@YOF:Yb3+,Er3+ monitored at three different pH values (pH 4, 5.5 and 7.4). | |
Conclusions
In summary, this study presents novel findings in the field of YOF material development and their doping with Yb3+ and Er3+ ions. The investigation highlights the previously unexplored aspects of improving the green UC luminescence in YOF:Yb3+,Er3+ and its potential biomedical applications. The synthesis methodology involves the fabrication of SiO2@NaYF4:Yb3+,Er3+ core–shell particles, followed by a high-temperature annealing step to induce an in situ ion-exchange reaction between O2 and NaYF4, leading to the formation of SiO2@YOF:Yb3+,Er3+ particles. Subsequently, the removal of the silica template yields hollow YOF:Yb3+,Er3+ particles while retaining their spherical shape. The investigation aims to explore the effect of Yb3+ ion concentration on UC properties in the YOF host. It also explores co-doping with Gd3+, Mn2+, and Li+ ions to enhance green UC emission. The study reveals that co-doping with 5% Gd3+ ions result in the highest intensity of green-to-red UC luminescence. Moreover, the optical thermometric performance of the doped SiO2@YOF particles is investigated, and the relative sensitivity (Sr) is determined. Furthermore, the potential application of the synthesized material as a drug carrier is explored using doxorubicin as a model anticancer drug, aiming to combine thermometry and drug delivery functionalities within a single material. The findings contribute to the advancement of their potential biomedical applications, filling gaps in the current knowledge and paving the way for further research and development in this field.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.† Additional data that support the findings of this study can be requested directly from the corresponding author (Anna M. Kaczmarek) by contacting her through email.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is part of a project that has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 945945; acknowledged by A. M. K., S. M., M. L. and S. P.). A. M. K. also thanks the Special Research Fund (BOF) – UGent for funding (BOF/STG/202002/004). TEM measurements were performed at the UGent TEM Core Facility.
References
- H. Yao, H. Shen and Q. Tang, Mater. Res. Express, 2019, 6, 125017 CrossRef CAS.
- H. Dong, L.-D. Sun and C.-H. Yan, Nanoscale, 2013, 5, 5703–5714 RSC.
- S. Han, R. Deng, X. Xie and X. Liu, Angew. Chem., Int. Ed., 2014, 53, 11702–11715 CrossRef CAS PubMed.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- K. Du, J. Feng, X. Gao and H. Zhang, Light: Sci. Appl., 2022, 11, 222 CrossRef CAS PubMed.
- B. T. Huy, Z. Gerelkhuu, J. W. Chung, V.-D. Dao, G. Ajithkumar and Y.-I. Lee, Mater. Sci. Eng., B, 2017, 223, 91–97 CrossRef CAS.
- M. Lin, Y. Zhao, S. Wang, M. Liu, Z. Duan, Y. Chen, F. Li, F. Xu and T. Lu, Biotechnol. Adv., 2012, 30, 1551–1561 CrossRef CAS PubMed.
- R. Hoskins and B. Soffer, Appl. Phys. Lett., 1964, 4, 22–23 CrossRef CAS.
- D. Gao, J. Gao, D. Zhao, Q. Pang, G. Xiao, L. Wang and K. Ma, J. Mater. Chem. C, 2020, 8, 17318–17324 RSC.
- H. J. Ma, W. K. Jung, Y. Park and D. K. Kim, J. Mater. Chem. C, 2018, 6, 11096–11103 RSC.
- T. T. Ngo, E. Cabello-Olmo, E. Arroyo, A. I. Becerro, M. Ocaña, G. Lozano and H. Míguez, ACS Appl. Mater. Interfaces, 2021, 13, 30051–30060 CrossRef CAS PubMed.
- X. Kang, C. Li, Z. Cheng, P. A. Ma, Z. Hou and J. Lin, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 80–101 CrossRef CAS PubMed.
- H. Wang, R. Wang, X. Sun, R. Yan and Y. Li, Mater. Res. Bull., 2005, 40, 911–919 CrossRef CAS.
- E. Martínez-Castro, J. García-Sevillano, F. Cussó and M. Ocaña, J. Alloys Compd., 2015, 619, 44–51 CrossRef.
- Y. Wang, T. Liu, X. Wang and H. Song, J. Mater. Res., 2013, 28, 848–855 CrossRef CAS.
- L. Tian, Y. Shang, S. Hao, Q. Han, T. Chen, W. Lv and C. Yang, Adv. Funct. Mater., 2018, 28, 1803946 CrossRef.
- M. Kang, H. B. Kang, S. Park and H. S. Jang, Chem. Commun., 2019, 55, 13350–13353 RSC.
- R. Li, L. Li, W. Zi, J. Zhang, L. Liu, L. Zou and S. Gan, New J. Chem., 2015, 39, 115–121 RSC.
- S. Jakka, M. Soares, M. Graça, A. Neves, P. Nagajyothi and K. Pavani, Opt. Mater., 2022, 129, 112442 CrossRef CAS.
- L. Guo, Z. Mu, P. Da, Z. Weng, P. Xi and C.-H. Yan, EnergyChem, 2022, 100088 CrossRef CAS.
- M. Chang, Y. Song, J. Chen, L. Cui, Z. Shi, Y. Sheng and H. Zou, ACS Sustainable
Chem. Eng., 2018, 6, 223–236 CrossRef CAS.
- Y. Wang, Nanoscale, 2019, 11, 10852–10858 RSC.
- S. Mohanty and A. M. Kaczmarek, Chem. Soc. Rev., 2022, 51, 6893–6908 RSC.
- S. Zeng, Z. Yi, W. Lu, C. Qian, H. Wang, L. Rao, T. Zeng, H. Liu, H. Liu and B. Fei, Adv. Funct. Mater., 2014, 24, 4051–4059 CrossRef CAS.
- B. T. Huy, A. P. Kumar, T. T. Thuy, N. N. Nghia and Y.-I. Lee, Appl. Spectrosc. Rev., 2022, 57, 265–299 CrossRef CAS.
- J. Tang, L. Chen, J. Li, Z. Wang, J. Zhang, L. Zhang, Y. Luo and X. Wang, Nanoscale, 2015, 7, 14752–14759 RSC.
- Y. Shi, M. Yuan, J. Li, F. Li, W. Cui, X. Jiao, Y. Peng, Y. Huang and L. Chen, Inorg. Chem., 2022, 61, 5309–5317 CrossRef CAS PubMed.
- S. Ye, E. H. Song and Q. Y. Zhang, Adv. Sci., 2016, 3, 1600302 CrossRef PubMed.
- S. Pei, X. Ge and L. Sun, Front. Chem., 2020, 8, 610481 CrossRef CAS.
- S. M. ul Hassan and Y. Kitamoto, Electrochim. Acta, 2015, 183, 160–164 CrossRef.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- A. M. Kaczmarek, R. Van Deun and M. K. Kaczmarek, Sens. Actuators, B, 2018, 273, 696–702 CrossRef CAS.
- Y. Jia, T.-Y. Sun, J.-H. Wang, H. Huang, P. Li, X.-F. Yu and P. K. Chu, CrystEngComm, 2014, 16, 6141–6148 RSC.
- Y. Li, G. Wang, K. Pan, B. Jiang, C. Tian, W. Zhou and H. Fu, J. Mater. Chem., 2012, 22, 20381–20386 RSC.
- P. Du, A. M. Deng, L. Luo and J. S. Yu, New J. Chem., 2017, 41, 13855–13861 RSC.
- J. H. Kim, H. Choi, E. O. Kim, H. M. Noh, B. K. Moon and J. H. Jeong, Opt. Mater., 2014, 38, 113–118 CrossRef CAS.
- M. Misiak, O. Pavlosiuk, M. Szalkowski, A. M. Kotulska, K. A. Ledwa and A. Bednarkiewicz, Nanotechnology, 2023, 34, 345702 CrossRef PubMed.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS PubMed.
- C. Secu, C. Bartha, E. Matei, C. Negrila, A. Crisan and M. Secu, J. Phys. Chem. Solids, 2019, 130, 236–241 CrossRef CAS.
- F. Shi and Y. Zhao, J. Mater. Chem. C, 2014, 2, 2198–2203 RSC.
- A. A. Vidyakina, I. E. Kolesnikov, N. A. Bogachev, M. Y. Skripkin, I. I. Tumkin, E. Lähderanta and A. S. Mereshchenko, Materials, 2020, 13, 3397 CrossRef PubMed.
- K. Zheng, D. Zhao, D. Zhang, N. Liu, F. Shi and W. Qin, J. Alloys Compd., 2011, 509, 5848–5852 CrossRef CAS.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- Y. Peng, Z. Cheng, W. U. Khan, T. Liu, M. Shen, S. Yang and Y. Zhang, New J. Chem., 2021, 45, 3876–3885 RSC.
- D. Chen, Y. Yu, F. Huang, P. Huang, A. Yang and Y. Wang, J. Am. Chem. Soc., 2010, 132, 9976–9978 CrossRef CAS PubMed.
- H. Wang, W. Lu, Z. Yi, L. Rao, S. Zeng and Z. Li, J. Alloys Compd., 2015, 618, 776–780 CrossRef CAS.
- J. Wang, F. Wang, C. Wang, Z. Liu and X. Liu, Angew. Chem., Int. Ed., 2011, 50, 10369–10372 CrossRef CAS PubMed.
- Y. Hu, X. Liang, Y. Wang, E. Liu, X. Hu and J. Fan, Ceram. Int., 2015, 41, 14545–14553 CrossRef CAS.
- N. S. Karan, S. Sarkar, D. Sarma, P. Kundu, N. Ravishankar and N. Pradhan, J. Am. Chem. Soc., 2011, 133, 1666–1669 CrossRef CAS PubMed.
- A. K. Guria and N. Pradhan, Chem. Mater., 2016, 28, 5224–5237 CrossRef CAS.
- S. A. Wade, S. F. Collins and G. W. Baxter, J. Appl. Phys., 2003, 94, 4743–4756 CrossRef CAS.
- A. M. Kaczmarek, M. Suta, H. Rijckaert, A. Abalymov, I. Van Driessche, A. G. Skirtach, A. Meijerink and P. Van Der Voort, Adv. Funct. Mater., 2020, 30, 2003101 CrossRef CAS.
- S. Mohanty, S. Premcheska, J. Verduijn, H. Rijckaert, A. G. Skirtach, K. Van Hecke and A. M. Kaczmarek, RSC Adv., 2022, 12, 33239–33250 RSC.
- L. Li, Y. Tong, J. Chen, Y. Chen, G. Abbas Ashraf, L. Chen, T. Pang and H. Guo, J. Am. Ceram. Soc., 2022, 105, 384–391 CrossRef CAS.
- B. Dong, R. N. Hua, B. S. Cao, Z. P. Li, Y. Y. He, Z. Y. Zhang and O. S. Wolfbeis, Phys. Chem. Chem. Phys., 2014, 16, 20009–20012 RSC.
- Z. Sun, M. Jia, Z. Fu, M. Zhang, H. Wang and Y. Xu, Chem. Eng. J., 2021, 406, 126755 CrossRef CAS.
- L. Li, W. Wang, H. Chen, S. Li, Q. Zhang, Y. Pan and Y. Li, J. Non-Cryst. Solids, 2021, 573, 121142 CrossRef CAS.
- R. Dey and V. K. Rai, Dalton Trans., 2014, 43, 111–118 RSC.
- J. Tang, P. Du, W. Li and L. Luo, J. Lumin., 2020, 224, 117296 CrossRef CAS.
- A. Ćirić, J. Aleksić, T. Barudžija, Ž. Antić, V. Đorđević, M. Medić, J. Periša, I. Zeković, M. Mitrić and M. D. Dramićanin, Nanomaterials, 2020, 10, 627 CrossRef PubMed.
- M. Ding, C. Lu, L. Chen, W. Bai, Y. Yuan and Z. Ji, Ceram. Int., 2018, 44, 16379–16387 CrossRef CAS.
- A. M. Kaczmarek, Y. Y. Liu, M. K. Kaczmarek, H. Liu, F. Artizzu, L. D. Carlos and P. Van Der Voort, Angew. Chem., Int. Ed., 2020, 132, 1948–1956 CrossRef.
- G. Tian, Z. Gu, L. Zhou, W. Yin, X. Liu, L. Yan, S. Jin, W. Ren, G. Xing, S. Li and Y. Zhao, Adv. Mater., 2012, 24, 1226–1231 CrossRef CAS PubMed.
- S. Zeng, Z. Yi, W. Lu, C. Qian, H. Wang, L. Rao, T. Zeng, H. Liu, H. Liu, B. Fei and J. Hao, Adv. Funct. Mater., 2014, 24, 4051–4059 CrossRef CAS.
- W. Yin, L. Zhao, L. Zhou, Z. Gu, X. Liu, G. Tian, S. Jin, L. Yan, W. Ren and G. Xing, Chem. – Eur. J., 2012, 18, 9239–9245 CrossRef CAS PubMed.
- H. Li, R. Wei, G.-H. Yan, J. Sun, C. Li, H. Wang, L. Shi, J. A. Capobianco and L. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 4910–4920 CrossRef CAS PubMed.
- X. Hu, X. Hao, Y. Wu, J. Zhang, X. Zhang, P. C. Wang, G. Zou and X.-J. Liang, J. Mater. Chem. B, 2013, 1, 1109–1118 RSC.
- K. Ge, C. Zhang, W. Sun, H. Liu, Y. Jin, Z. Li, X.-J. Liang, G. Jia and J. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 25078–25086 CrossRef CAS PubMed.
- W. Feng, X. Zhou, C. He, K. Qiu, W. Nie, L. Chen, H. Wang, X. Mo and Y. Zhang, J. Mater. Chem. B, 2013, 1, 5886–5898 RSC.
- L. Zhao, X. Ge, H. Zhao, L. Shi, J. A. Capobianco, D. Jin and L. Sun, ACS Appl. Nano Mater., 2018, 1, 1648–1656 CrossRef CAS.
- Y. Dai, P. a Ma, Z. Cheng, X. Kang, X. Zhang, Z. Hou, C. Li, D. Yang, X. Zhai and J. Lin, ACS Nano, 2012, 6, 3327–3338 CrossRef CAS PubMed.
- B. Liu, Y. Chen, C. Li, F. He, Z. Hou, S. Huang, H. Zhu, X. Chen and J. Lin, Adv. Funct. Mater., 2015, 25, 4717–4729 CrossRef CAS.
- Y. Huang, E. Hemmer, F. Rosei and F. Vetrone, J. Phys. Chem. B, 2016, 120, 4992–5001 CrossRef CAS PubMed.
- K. Li, Q. Su, W. Yuan, B. Tian, B. Shen, Y. Li, W. Feng and F. Li, ACS Appl. Mater. Interfaces, 2015, 7, 12278–12286 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional PXRD data. Additional luminescence and thermometry data. XPS, TGA and ICP-OES results. SEM images. DOX calibration curves. See DOI: https://doi.org/10.1039/d4tc01740d |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Mirijam
Lederer
ab,
Mirijam
Lederer