
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroduction of a mesityl substituent on pyridyl rings as a facile strategy for improving the performance of luminescent 1,3-bis-(2-pyridyl)benzene platinum(II) complexes: a springboard for blue OLEDs†
Alessia
Colombo
 a,
Giulia
De Soricellis
ab,
Claudia
Dragonetti
a,
Francesco
Fagnani
*a,
Dominique
Roberto
a,
Bertrand
Carboni
c,
Véronique
Guerchais
c,
Thierry
Roisnel
c,
Massimo
Cocchi
d,
Simona
Fantacci
e,
Eros
Radicchi
ef and
Daniele
Marinotto
g
a,
Giulia
De Soricellis
ab,
Claudia
Dragonetti
a,
Francesco
Fagnani
*a,
Dominique
Roberto
a,
Bertrand
Carboni
c,
Véronique
Guerchais
c,
Thierry
Roisnel
c,
Massimo
Cocchi
d,
Simona
Fantacci
e,
Eros
Radicchi
ef and
Daniele
Marinotto
g
aDipartimento di Chimica dell’Università degli Studi di Milano and UdR-INSTM di Milano, Via C. Golgi 19, I-20133 Milan, Italy. E-mail: francesco.fagnani@unimi.it
bDipartimento di Chimica, Università di Pavia, Via Taramelli 12, 27100 Pavia, Italy
cUniversité de Rennes 1, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France
dIstituto per la Sintesi Organica e la Fotoreattività (ISOF), Consiglio Nazionale delle Ricerche (CNR), via P. Gobetti 101, 40129 Bologna, Italy
eIstituto di Scienze e Tecnologie Chimiche “Giulio Natta” SCITEC, Consiglio Nazionale delle Ricerche (CNR), Computational Laboratory for Hybrid/Organic Photovoltaics (CLHYO), via Elce di Sotto 8, 06213, Perugia, Italy
fNRG – Dipartimento di Biotecnologie, Università degli Studi di Verona e INSTM, RU di Verona, Strada le Grazie 15, 37134 Verona, Italy
gIstituto di Scienze e Tecnologie Chimiche “Giulio Natta” (SCITEC), Consiglio Nazionale delle Ricerche (CNR), via C. Golgi 19, 20133 Milan, Italy
First published on 6th June 2024
Abstract
While the development of red and green phosphorescent organic light-emitting diodes (OLEDs) has seen rapid progress, that of efficient blue phosphorescent OLEDs remains a challenge. In the present report, the introduction of a bulky substituent on the pyridyl rings of a cycloplatinated 1,3-bis(pyridine-2-yl)-4,6-difluoro-benzene appears as a facile strategy to hinder strong Pt⋯Pt interactions, allowing the fabrication of efficient blue OLEDs. Thus, the preparation and characterization of a chlorido platinum(II) complex bearing a well-designed new N^C^N-cyclometalating ligand, namely 1,3-bis(4-mesityl-pyridin-2-yl)-4,6-difluoro-benzene, are reported. Its structure, along with that of the related pro-ligand, is determined by X-ray diffraction studies on a single crystal. The shortest Pt⋯Pt distance is much longer (8.59 Å) than that observed for other N^C^N-platinum(II) chlorido complexes including one with the bulky mesityl group on the cyclometalated benzene ring (4.4 Å). This new complex exhibits intense blue phosphorescence (470–471 nm) in dichloromethane solution (Φlum = 0.97) and in the PMMA film (1 wt% complex, Φlum = 0.95) whereas red phosphorescence (672 nm) is observed in a neat film (Φlum = 0.72). Even in the solid state, the novel complex is highly luminescent suggesting that the introduction of mesityl groups on the pyridine rings is a way to inhibit self-quenching both in the PMMA matrix and in neat films. It represents a useful tool for the fabrication of efficient blue OLEDs (8% wt complex) with CIE coordinates (0.13, 0.29) approaching true blue. The molecular geometry, ground state, electronic structure, and excited electronic states of the complex, both as a monomer and dimer aggregate in solution, are calculated using density functional theory (DFT) and time-dependent (TD) DFT approaches, giving insight into the electronic origin of the absorption spectra.
1. Introduction
Platinum complexes have attracted much attention for many applications such as nonlinear optics,1–6 bioimaging,7–14 sensing,15–17 and electroluminescent devices.18–38 Indeed, among a large number of organometallic materials, phosphorescent platinum(II) complexes have gained interest due to the important spin–orbit coupling associated with the metal which promotes intersystem crossing, and therefore the emission of light from triplet excited states, a behavior that is further improved by the presence of platinum–carbon bonds.39Thus, state-of-the-art organic light-emitting diodes (OLEDs), which are transforming the lighting and display industries, rely on phosphorescent metal complexes such as iridium(III) and platinum(II) for red and green emitters whereas fluorescent compounds are usually preferred for blue emitters.27,40 As a matter of fact, whereas the development of red and green phosphorescent OLEDs has seen rapid progress, that of efficient blue phosphorescent OLEDs remains a challenge.41–47
In the last 15 years, platinum(II) chlorido complexes with a cyclometalated 1,3-bis(pyridin-2-yl)-benzene (bispyb) ligand have been used as emitters in OLEDs, representing a precious tool for tailoring the emission colour and allowing high external quantum efficiencies.48 They show two phosphorescence bands, situated in the bluish green (monomeric emission) and red (excimer/aggregate emission) regions of the visible spectrum, in which contributions to the global emission can be controlled by the amount of the platinum complex in the host matrix used for the preparation of the OLED's blend emissive layer.48 The ability of these complexes to form excimers or aggregates through attractive intermolecular interactions like Pt–Pt or ligand–ligand or a combination of these,48–51 thanks to their square planar structure, offers attraction even for the preparation of near-infrared OLEDs.52
These platinum(II) complexes bearing a cyclometalated bispyb ligand, which allows rigid N^C^N coordination surroundings, are among the brightest emitters in solution at room temperature.53–58 Their appeal is further enriched by the possibility of controlling the emission color by the introduction of substituents into the pyridyl or aryl rings, maintaining excellent quantum yields.25 The choice of suitable groups to obtain blue emission has been investigated because one significant fundamental challenge in the study of phosphorescent metal complexes, which also impacts the development of OLED color displays, is the lack of blue-phosphorescent complexes with good photoluminescence quantum yields and high photostability.43 Remarkably, in [Pt(bispyb)Cl] complexes, the introduction of electron-withdrawing fluorine atoms at positions 4 and 6 of the central phenyl ring leads to a blue shift of the emission.26,49,56,59–61 An important aspect is that with the tridentate N^C^N-coordinating bispyb ligand, the luminescence of a cyclometalated Pt(II) complex can be shifted into the blue region, without the problematic drop-off in the quantum yield observed for bidentate analogues.59
On the other hand, it is worth pointing out that the introduction of a bulky aryl group at position 5 of the cycloplatinated phenyl ring of the 1,3-bis(pyridin-2-yl)-benzene ligand is a way to reduce the tendency to form excimers compared to the parent complex, owing to steric inhibition by the pendent aryl group preventing the necessary face-to-face approach of a complex in its excited state to a ground-state molecule.54
Surprisingly, the effect of the introduction of highly hindered aromatic substituents on position 4 of the pyridyl rings has not been reported. Yet, in the case of the complex with a fluorinated cycloplatinated 1,3-bis(pyridin-2-yl)-4,6-difluoro-benzene ligand ([Pt(bispy-4,6-dFb)Cl], for which the presence of the fluorine atoms leads to a blue shift of the emission,26,49,56 the knowledge of such an effect would be of particular interest because excimer or aggregate formation, and therefore the phosphorescence properties could be controlled achieving the appropriate spatial arrangement of bulky functional groups. Thus, it could be a simple way to decrease Pt–Pt interactions and/or π–π stacking with the aim of obtaining efficient blue OLEDs. Besides, the presence of bulky aromatic groups could not only give a brighter emission both in solution and in the device but it would also improve solubility to allow the formation of high-quality spin-coated thin films from solution.
These observations prompted us to prepare a simple complex with a mesityl group on each pyridine of the cycloplatinated 1,3-bis(pyridin-2-yl)-4,6-difluoro-benzene ligand ([Pt(bis(4-Mes-py)-4,6-dFb)Cl], Chart 1). The introduction of these bulky π-delocalized polarizable groups turned out to be a useful tool to achieve a huge luminescence quantum yield, both in solution and thin films, and to prepare efficient blue OLEDs.
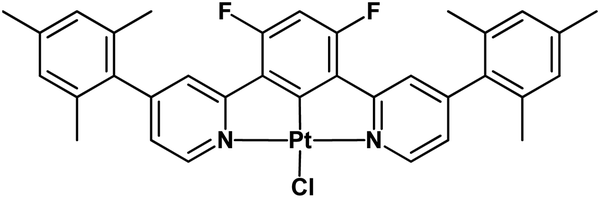 | ||
| Chart 1 Investigated novel complex ([Pt(bis(4-Mes-py)-4,6-dFb)Cl]). | ||
2. Results and discussion
2.1 Synthesis
The novel pro-ligand 2,2′-(4,6-difluoro-1,3-phenylene)bis(4-mesitylpyridine) was prepared by cross-coupling of 2-chloro-4-mesitylpyridine62 with 2,2′-(4,6-difluoro-1,3-phenylene)bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane)59 (Scheme 1). A further reaction of this pro-ligand with K2PtCl4 in MeCN/H2O led to [Pt(bis(4-Mes-py)-4,6-dFb)Cl]. The ligand and the product were fully characterized by NMR spectroscopy and elemental analysis (details are given in the ESI†).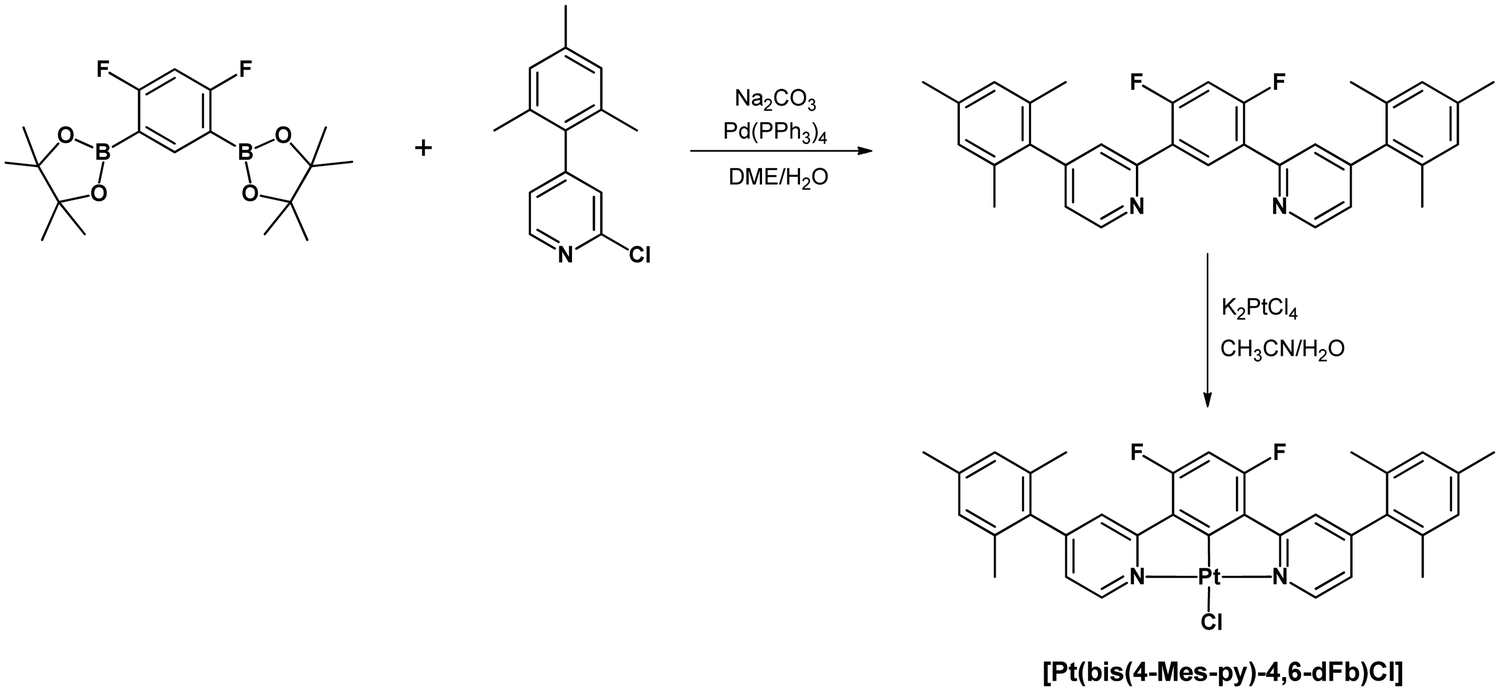 | ||
| Scheme 1 Synthetic pathway for the preparation of [Pt(bis(4-Mes-py)-4,6-dFb)Cl]. | ||
2.2 X-ray structure determination
In order to achieve better characterization of the novel pro-ligand and the related platinum complex, their X-ray structure was investigated. Crystals were grown by gradually evaporating a CH2Cl2 solution, resulting in the formation of a dichloromethane solvate of the platinum complex. The structure of the pro-ligand is presented in Fig. 1.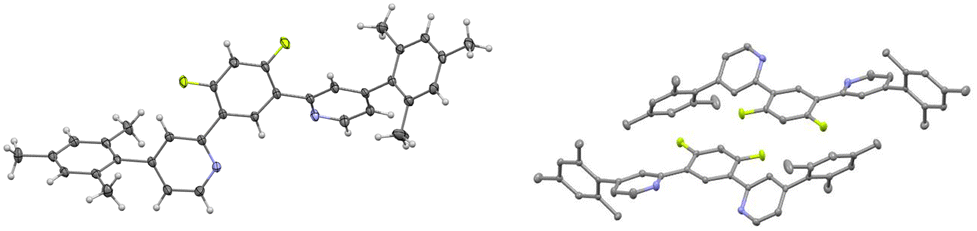 | ||
| Fig. 1 Views of the pro-ligand 2,2′-(4,6-difluoro-1,3-phenylene)bis(4-mesitylpyridine). | ||
The three aromatic rings of the dipyridyl benzene moiety are not coplanar, with angles of 17.07/19.23° and 33.01/36.43° between the planes of the lateral pyridyl rings and that of the central benzene. Contrarily to the other uncoordinated 1,3-bis(pyridin-2-yl)-benzenes reported in the Cambridge Crystallographic Data Centre, which have no substituents on the pyridine rings,54 there is no head-to-tail disposition of the two pyridyl rings which would minimize unfavorable electrostatic interactions between the lone pairs like in the transoid arrangement of the pyridine units about the interannular bonds in 2,2′:6′,2′′-terpyridines.63 This difference can be explained by the presence of bulky substituents on the pyridines in the pro-ligand L. The mesityl groups are close to perpendicular to the pyridine ring with angles of 103.47/102.61° and 104.47/90.79°, as would be expected because of the steric bulk of the substituents.
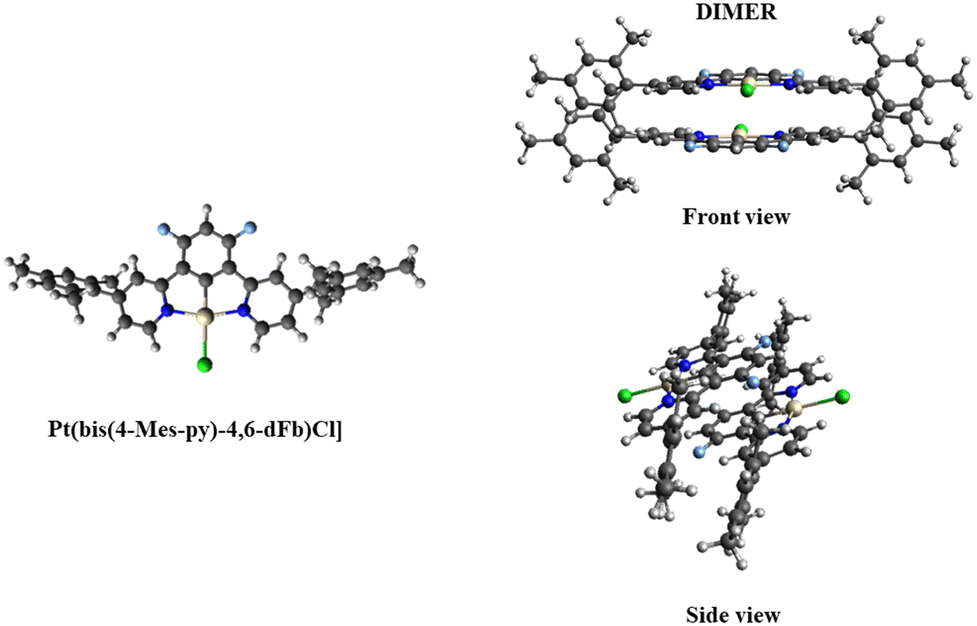
In complex [Pt(bis(4-Mes-py)-4,6-dFb)Cl], the coordination environment around the platinum center is distorted square planar, comprising two nitrogen atoms and one carbon atom from the terdentate ligand, along with one chlorine atom (Fig. 2). As revealed by the crystal structure, there is one distinct complex per two dichloromethane molecules within the asymmetric unit. The Pt1–C1 bond distance is 1.918(3) Å, Pt1–N11 = 2.024(3) Å, Pt1–N31 = 2.027(3), Pt1–Cl1 = 2.3906(9) Å (Fig. 2(A)). These distances are in the range of those reported for other platinum(II) chlorido complexes with a cyclometalated 1,3-bis(pyridin-2-yl)-benzene.53,54,58,64,65 The C1–Pt1–Cl1 axis is almost linear [179.6(1)°], whereas N11–Pt1–N31 is more distorted at 161.9(1)°, as a consequence of the chelate ring strain. The torsion angle between the mesityl groups and the pyridines is 62.7/60.7(5) and 61.6(5)/61.7(5). Interestingly, the shortest Pt⋯Pt distance is 8.59 Å (Fig. 2(B)) much longer than that previously reported for the related platinum(II) chlorido complex with a cyclometalated 5-mesityl-1,3-bis(pyridin-2-yl)-benzene ligand (4.4 Å).64 Clearly, the presence of mesityl groups on position 4 on the pyridines hinders the neighbouring of the metal atoms and therefore the Pt⋯Pt interactions. The strongest π–π interaction could occur between two difluorobenzene rings of pairs of molecules, at a relatively short distance of 3.61 Å (Fig. 2(C)).
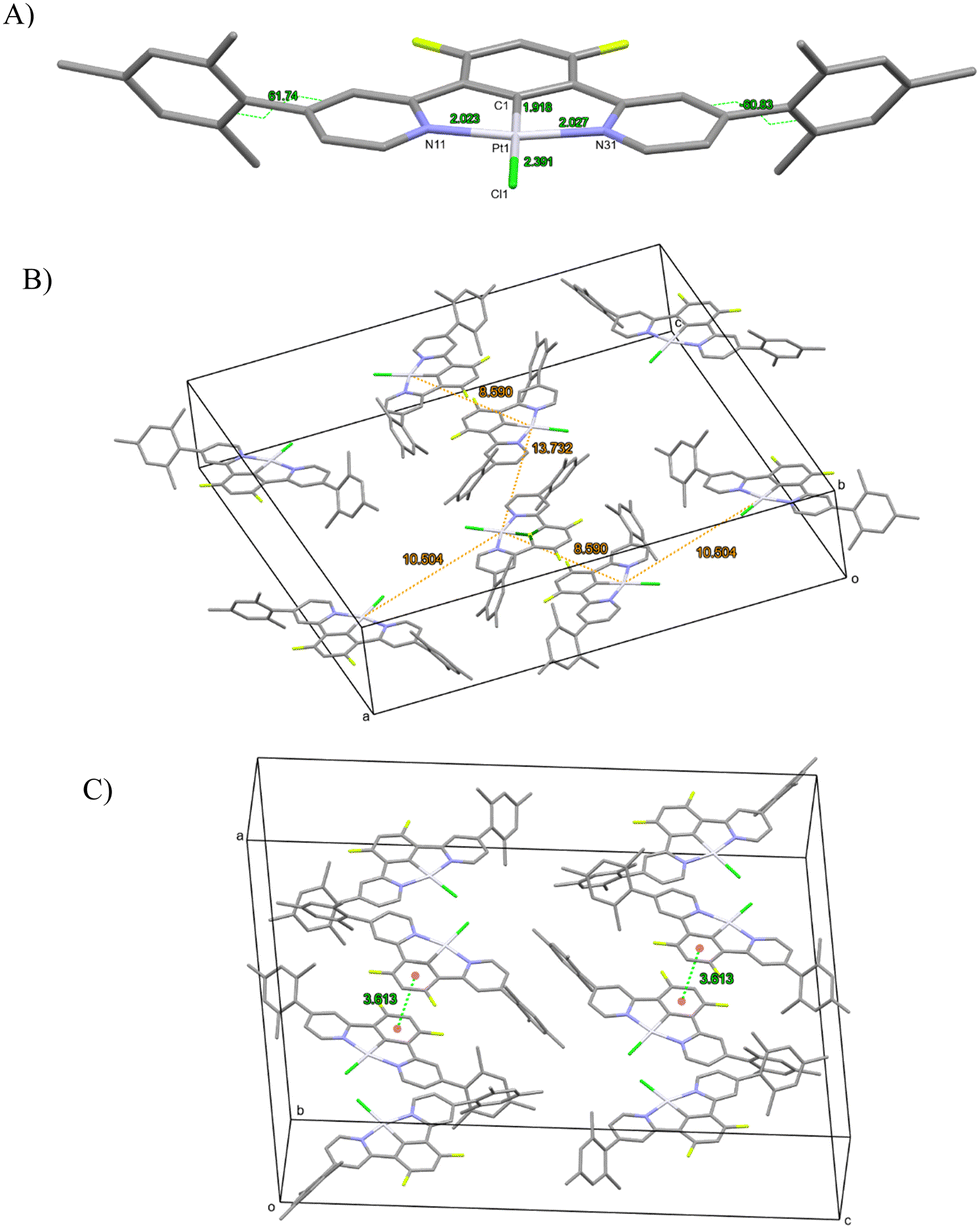 | ||
| Fig. 2 [Pt(bis(4-Mes-py)-4,6-dFb)Cl] complex: (A) molecular view with atomic distances around the Pt center and dihedral angle between the mesityl and the pyridyl rings; (B) unit cell with various Pt⋯Pt distances; and (C) unit cell with distances between difluorobenzene rings of the terdentate ligand. | ||
2.3 Photophysical properties in solution
The absorption spectrum of [Pt(bis(4-Mes-py)-4,6-dFb)Cl] in dichloromethane solution is shown in Fig. 3(A) (see the ESI,† for the calculation of molar extinction coefficient ε). The absorption spectra of the novel complex display an intense absorption band at 250–330 nm (ε263 = 2.3 × 104 L mol−1 cm−1) attributed to singlet intraligand transitions (1IL) of the cyclometalated N^C^N ligand and a less intense band at 350–420 nm (ε376 = 7.8 × 103 L mol−1 cm−1) assigned to singlet charge-transfer transitions involving the metal and the cyclometalated ligand (1MLCT).50,56,58 At wavelengths above 420 nm, weak absorption bands with peaks at 438 and 467 nm (ε438 = 1.4 × 102 M−1 cm−1, ε467 = 1.2 × 102 M−1 cm−1) can be observed (inset Fig. 3(A)). These absorption bands are attributed to triplet metal-to-ligand charge transfer transitions (3MLCTs) of the monomeric complex, as noted for other parent chloride derivatives.48,53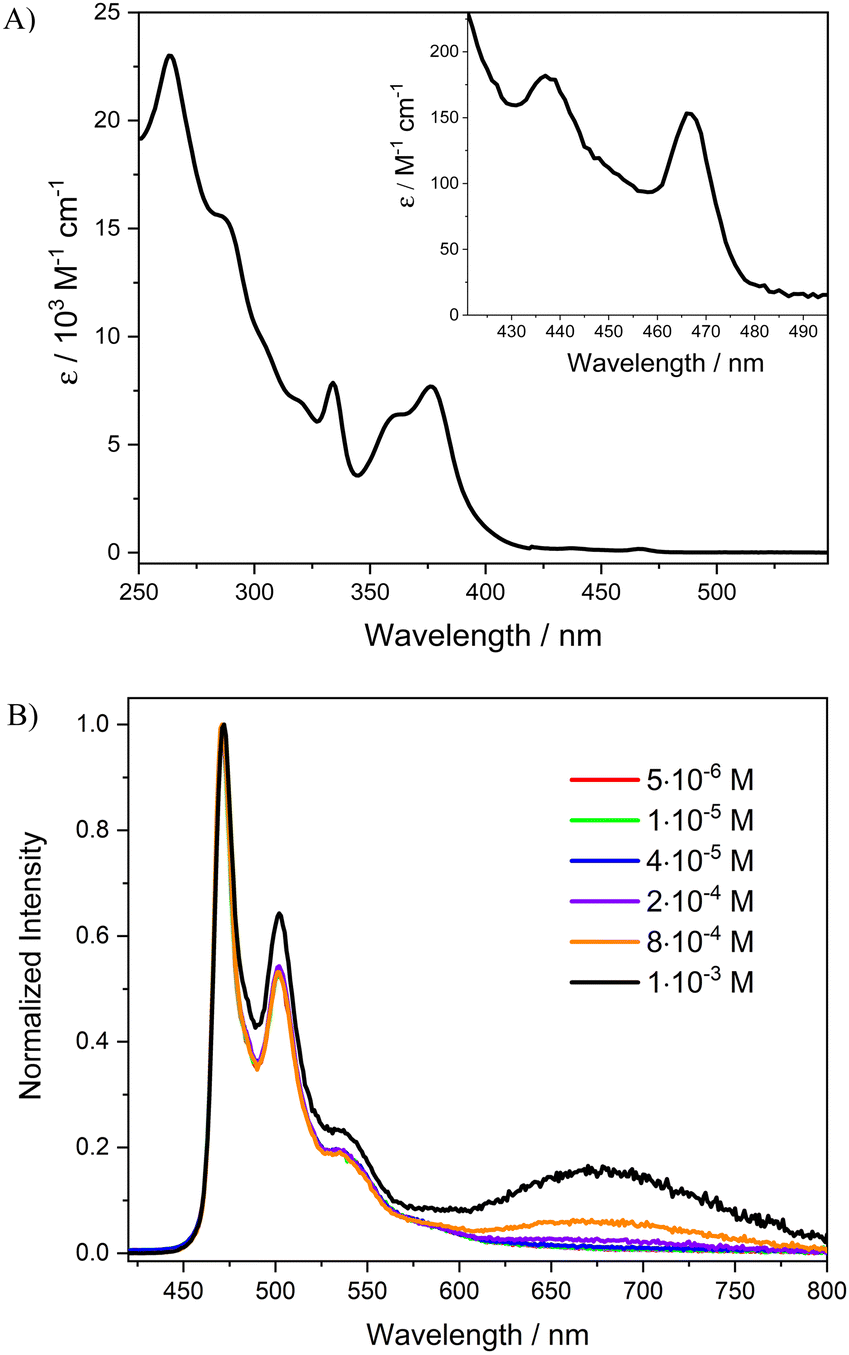 | ||
| Fig. 3 (A) Absorption spectrum of [Pt(bis(4-Mes-py)-4,6-dFb)Cl] in CH2Cl2. The weak bands at longer wavelengths are shown on an expanded scale for clarity. (B) Photoluminescence spectra at room temperature of [Pt(bis(4-Mes-py)-4,6-dFb)Cl] in deaerated CH2Cl2 at different concentrations (an excitation wavelength of 334 nm). | ||
Photoluminescence spectra of [Pt(bis(4-Mes-py)-4,6-dFb)Cl] in dichloromethane solution at various concentrations excited at 334 nm are shown in Fig. 3(B), whereas relevant values are presented in Table 1. In dilute dichloromethane solution (5 × 10−6 M) the complex is intensely phosphorescent in the blue region with a vibrationally structured emission spectrum having a high-energy emission maximum (λmax) at 471 nm. This emission can be attributed to the electronic transitions from the lowest triplet states to the ground state of the monomeric complex. Moreover, the λmax of the complex is similar to that of the parent complex without the mesityl substituents [Pt(bis(4-H-py)-4,6-dFb)Cl] (λmax = 472 nm),56 longer than that of the complex with NMe2 groups in position 4 on the pyridines ([Pt(bis(4-NMe2-py)-4,6-dFb)Cl], λmax = 453 nm)59 as expected since the introduction of electron-donor groups on the pyridines leads to a blue shift of the emission,59,60 but much shorter than that of the complex with triphenylamino groups ([Pt(bis(4-TPA-py)-4,6-dFb)Cl], λmax = 562 nm).50 Therefore, the introduction of the very bulky mesityl groups on the pyridine rings of the cyclometalated ligand does not affect the emission color of the platinum complex, in sharp contrast with the introduction of triphenylamine substituents. When the concentration of the monomer is increased up to 1 × 10−3 M, a new broad structureless band centered at 680 nm is detected (Fig. 3(B) and Table 1). This band at a lower energy can be mainly ascribed to the emission from aggregates, such as dimers of the complex, as evidenced by the continuous variation of the excitation spectrum with an increase in concentration (see Fig. S6 and S7, ESI†), by the deviation of the Lambert–Beer law (see Fig. S5, ESI†), and DFT calculations. However, an underlying excimer emission cannot be ruled out, as previously reported for related platinum(II) complexes.50 In any case, the presence of the band at 680 nm puts evidence that the introduction of the bulky mesityl groups on the pyridine rings does not block completely the formation of excimers/aggregates.
| λ max,em/nm monomer [excimer/aggregate] | Φ lum | τ/μs | k r /s−1 | k nr /s−1 | |
|---|---|---|---|---|---|
| a Excitation at 330 nm. b Radiative and nonradiative rate constants are calculated from the quantum yields and emission decay times according to Φlum = kr·τem = kr/(kr + knr). c In aerated solution. d Excimers/aggregates. e Film thickness = 2.49 μm. f Film thickness = 50 nm. | |||||
| 5 × 10−6 M, degassed CH2Cl2 | 471 | 0.97 | 4.77 | 2.0 × 105 | 6.3 × 103 |
| 0.18 c | |||||
| 2 × 10−4 M, degassed CH2Cl2 | 471 [680]d | 0.62 | 2.41 | 2.6 × 105 | 1.6 × 105 |
| 0.12 c | |||||
| In PMMA film 1 wt%e | 472 [680]d | 0.95 | 3.85 | 2.5 × 105 | 1.3 × 104 |
| Neat filmf | [672]d | 0.72 | 1.47 | 4.9 × 105 | 1.9 × 105 |
Complex [Pt(bis(4-Mes-py)-4,6-dFb)Cl] is highly luminescent in dilute deaerated dichloromethane solution (5 × 10−6 M), with an absolute luminescence quantum yield (Φlum) of 0.97, superior to that recently reported by our group for the parent [Pt(bis(4-TPA-py)-4,6-dFb)Cl] complex (Φlum = 0.90)50 or for [Pt(bis(4-H-py)-4,6-dFb)Cl] (Φlum = 0.85).49 As expected from the behaviour of similar platinum compounds,50 the luminescence of the novel complex is efficiently quenched by oxygen, being its quantum yield of 0.18 in air-equilibrated dichloromethane solution. In addition, there is quenching of the luminescence quantum yield upon increasing the complex concentration at 2 × 10−4 M in deaerated dichloromethane solution (Φlum = 0.62). Interestingly, this decrease of the quantum yield upon increasing the complex concentration is much lower than that observed for other N^C^N-platinum(II) complexes.51,58 Such superior performance could be reasonably attributed to the steric hindrance, caused by the mesityl substituents on the para position of the pyridines, which makes more difficult the formation of excimers and aggregates.
Excited state decay measurements of the [Pt(bis(4-Mes-py)-4,6-dFb)Cl] solutions at different concentrations were performed at an emission wavelength of 471 nm, excited at 374 nm. A mono-exponential function is used to fit all time decay luminescence curves (Table S2 and Fig. S8–S13 in the ESI†). In a dilute dichloromethane solution (5 × 10−6 M), a lifetime (τ) of 4.77 μs is obtained for the monomer complex, a value which decreases by 2.41 μs as the concentration is increased up to 2 × 10−4 M and by 0.85 μs with a further increase of the concentration to 1 × 10−3 M. Such results are compatible with the formation of bimolecular emissive excited states (excimers and aggregates) of the complex which causes diminution of the lifetime. This is also supported by the increase of the non-radiative rate constant (knr), which passes from 6.3 × 103 to 1.6 × 105 s−1 as the concentration increases from 5 × 10−6 M to 2 × 10−4 M, respectively (Table 1). Moreover, it is worth noting that the radiative rate constant (kr) of the complex at 5 × 10−6 M is 2.0 × 105 s−1, which is almost twice the one reported for the complex without mesityl substituents (1.2 × 105 s−1).56 Such a result is in accordance with the higher molar extinction coefficient of the electronic S0 → T1 transition found for the novel compound (1.8 × 102 L mol−1 cm−1) with respect to the parent complex (1.4 × 102 L mol−1 cm−1),56 reasonably due to slightly more effective spin–orbit coupling from the higher lying 1MLCT states to 3MLCT states in the [Pt(bis(4-Mes-py)-4,6-dFb)Cl] complex.
2.4 Photophysical properties in the solid state
For many applications, such as lighting devices or sensing, safeguarding luminescence in the solid state is a sought-after property.66,67 However, highly luminescent compounds in dilute solutions often have the problem of “aggregation-caused quenching” in concentrated solutions or in the solid state. For example, [Ir(phenylpyridine)3] is characterized by excellent luminescence in dilute solution (0.97) but the value drops below 0.03 in the neat film due to self-quenching by intermolecular π–π stacking interactions.67 Similarly, we recently reported that the luminescence quantum yield of the parent [Pt(bis(4-TPA-py)-4,6-dFb)Cl] complex decreases on going from CH2Cl2 solution (Φlum = 0.90) to the polymethylmetacrylate (PMMA) film (Φlum = 0.56) and drops drastically as the neat film (Φlum = 0.05); although a significant quantum yield was obtained in the PMMA film, the higher presence of excimers and aggregates in the neat film caused strong quenching of the quantum yield.50 We were curious to see the effect of the substitution of the triphenylamine substituents by the mesityl groups on the luminescence properties in the solid state. Therefore, we prepared a thin film in PMMA (1 wt%) and a neat film of the [Pt(bis(4-Mes-py)-4,6-dFb)Cl] complex, characterized by high solubility in dichloromethane (see the preparation and characterization of the films in the ESI†).The normalized absorption spectra of the PMMA and neat thin film of the [Pt(bis(4-Mes-py)-4,6-dFb)Cl] complex are shown in Fig. S14 (ESI†). The absorption spectrum of the PMMA film is quite similar to that measured in solution, probably due to the weak intermolecular interactions felt by the platinum complex in the polymeric matrix. Instead, broader absorption bands are observed in the neat film where there are stronger interactions between the molecules of the complex. Upon excitation at 334 nm, the blend of the platinum complex in the PMMA matrix shows a vibrationally structured emission spectrum with a maximum at 472 nm (Fig. 4(A)) and a τ of 3.85 μs (Fig. S16, ESI†), which resembles the one measured in the dilute dichloromethane solution (Fig. 3(B)) but with a shorter lifetime. Besides, a very weak band is detected at lower energy ca. 680 nm which can be ascribed to some bimolecular emissive excited states (excimers and aggregates) of the complex.
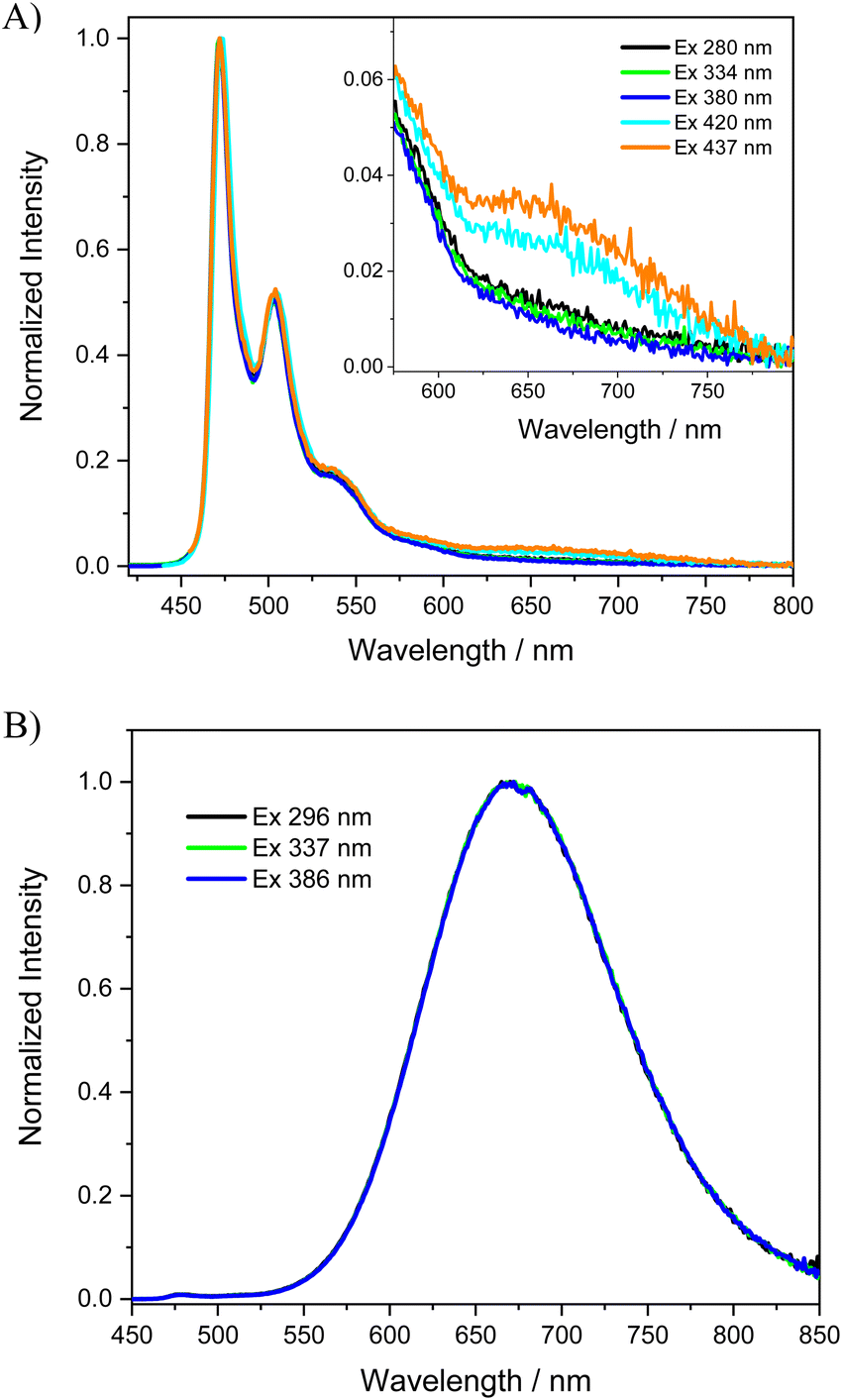 | ||
| Fig. 4 Photoluminescence spectra of the [Pt(bis(4-Mes-py)-4,6-dFb)Cl] complex (A) as a 1 wt% PMMA film, with a weak band at 680 nm shown in the expanded scale for clarity; (B) as a neat film. | ||
Completely different emission spectra are recorded for the neat film of the [Pt(bis(4-Mes-py)-4,6-dFb)Cl] complex, see Fig. 4(B). Irrespective of the excitation wavelength, the neat film is characterized by only one broad phosphorescent band with a maximum emission wavelength at 672 nm (τ = 1.47 μs, Fig. S18, ESI†). The spectral position of this band is similar to that recorded for the most concentrated solutions of the complex (Fig. 3(B)); therefore, it is reasonable to assume that it is exclusively produced by bimolecular emissive excited states, which are easily formed in the neat film by the shorter intermolecular distances and/or favourable intermolecular configurations of the molecules.28
Remarkably, as the PMMA thin film, [Pt(bis(4-Mes-py)-4,6-dFb)Cl] is characterized by an impressive phosphorescence quantum yield of 0.95, much higher than that reported for the parent [Pt(bis(4-TPA-py)-4,6-dFb)Cl] and standing among the best quantum yields reported for Pt complexes in polymeric films.50 Even as a neat film, the novel complex is highly luminescent (Φlum = 0.72). Therefore, the introduction of mesityl groups is a way to inhibit self-quenching not only in the PMMA matrix but also as a neat film. This self-quenching reduction is an attractive feature for practical applications such as OLED technology, where local concentrations may be quite high, and self-quenching is a wasteful energy sink that diminishes device efficiencies.54
2.5 Computational modelling
To comprehend the intriguing photochemical behavior in a solution of [Pt(bis(4-Mes-py)-4,6-dFb)Cl], calculations based on DFT were performed both on the isolated complex and on the dimer, the simplest representative species of the aggregation phenomenon in solution, which is able to explain bimolecular optical properties revealed by spectroscopic measurements. We started by optimizing the monomer and the dimer geometries in CH2Cl2 (the solvent employed in the spectroscopic measurements), and then the computed electronic structure in terms of energy and nature of the frontier orbitals were analyzed and eventually the lowest 80/5 (150/10) excited singlet–singlet/singlet–triplet states for the monomer (dimer) were computed, characterized, and convoluted to simulate the absorption spectra. The calculations were performed with the Gaussian09 program package (G09)68 by using the B3LYP exchange–correlation functional69 integrated with the D3-BJ model70 to include the dispersion interactions in the geometry optimizations. The 6-31G** basis set71–73 was utilized for all the atoms except for Pt, which has been described with the LANL2DZ basis set along with the corresponding pseudopotentials.74 It is worth noting that TDDFT calculation of the singlet–triplet excitations in G09 does not include Spin–Orbit Coupling (SOC) effects, thus providing only the energy of the transitions with null oscillator strength. Therefore, relativistic calculations with the B3LYP exchange–correlation functional were performed in the G09 optimized geometries of the monomer and dimer by means of the Zeroth Order Regular Approximation (ZORA)75–77 to the Dirac equation as implemented in Amsterdam Density Functional (ADF) software,78,79 allowing to include the singlet–triplet excitations of the absorption spectra through TDDFT80,81 and to comprehend the effect of the SOC on [Pt(bis(4-Mes-py)-4,6-dFb)Cl], eventually considering the Tamm–Dancoff approximation to speed up the calculations. The Slater-type TZP basis set82 was employed for all the atoms (1s core was kept frozen for C, N and F, while for Pt and Cl the cores 1s–4f and 1s–2p were kept frozen, respectively). The CH2Cl2 solvation effects were included in both cases in an implicit way (conductor-like continuum polarizable model, C-PCM,83–86 in G09 and the conductor-like screening model, COSMO,87 in ADF).In Fig. 5 the optimized molecular structure of both the monomer and the dimer is reported. It is interesting that in the optimized [Pt(bis(4-Mes-py)-4,6-dFb)Cl] structure in solution, the mesityl groups do not orient completely perpendicular to the pyridine, being the average dihedral angles between the mesityl groups and the pyridine rings 77°, a value similar to that observed in the X-ray structure (Fig. 2). In the dimer, the two monomers are arranged in a head-to-tail configuration with respect to the Pt centers being staggered from the other. The computed Pt–Pt distance is 6.31 Å, 2.24 Å smaller than that measured by X-rays. As expected, the optimized dimer in CH2Cl2 is the smallest model for aggregation in solution and it is not directly comparable to the dimer geometry taken from the X-ray structure: in the crystal, pillars of packed head-to-tail monomers are observed (see Fig. 2) and the monomeric unit experiences a different environment with respect to the CH2Cl2 solution. The aggregation energy of two monomeric units in solution has been computed to be −47.4 kcal mol−1, measuring the strength of the π–π stacking interaction which drives the aggregation.
 | ||
| Fig. 5 Optimized molecular geometry in CH2Cl2 of [Pt(bis(4-Mes-py)-4,6-dFb)Cl] (left) and its dimer (right) in the front and side views. | ||
The electronic structure of both the monomer and dimer in CH2Cl2 solution was analyzed and their absorption spectra were simulated and characterized to assign the main optical features. The energy levels of both the monomer (left) and dimer (right), evaluated with (w) and without (w/o) the inclusion of SOC effects, are shown in Fig. 6, along with the electron density plots of the HOMO and LUMO. The plots of the molecular orbitals involved in the main excited state transitions for both the monomer and dimer are reported in the ESI† (Fig. S19 and S20).
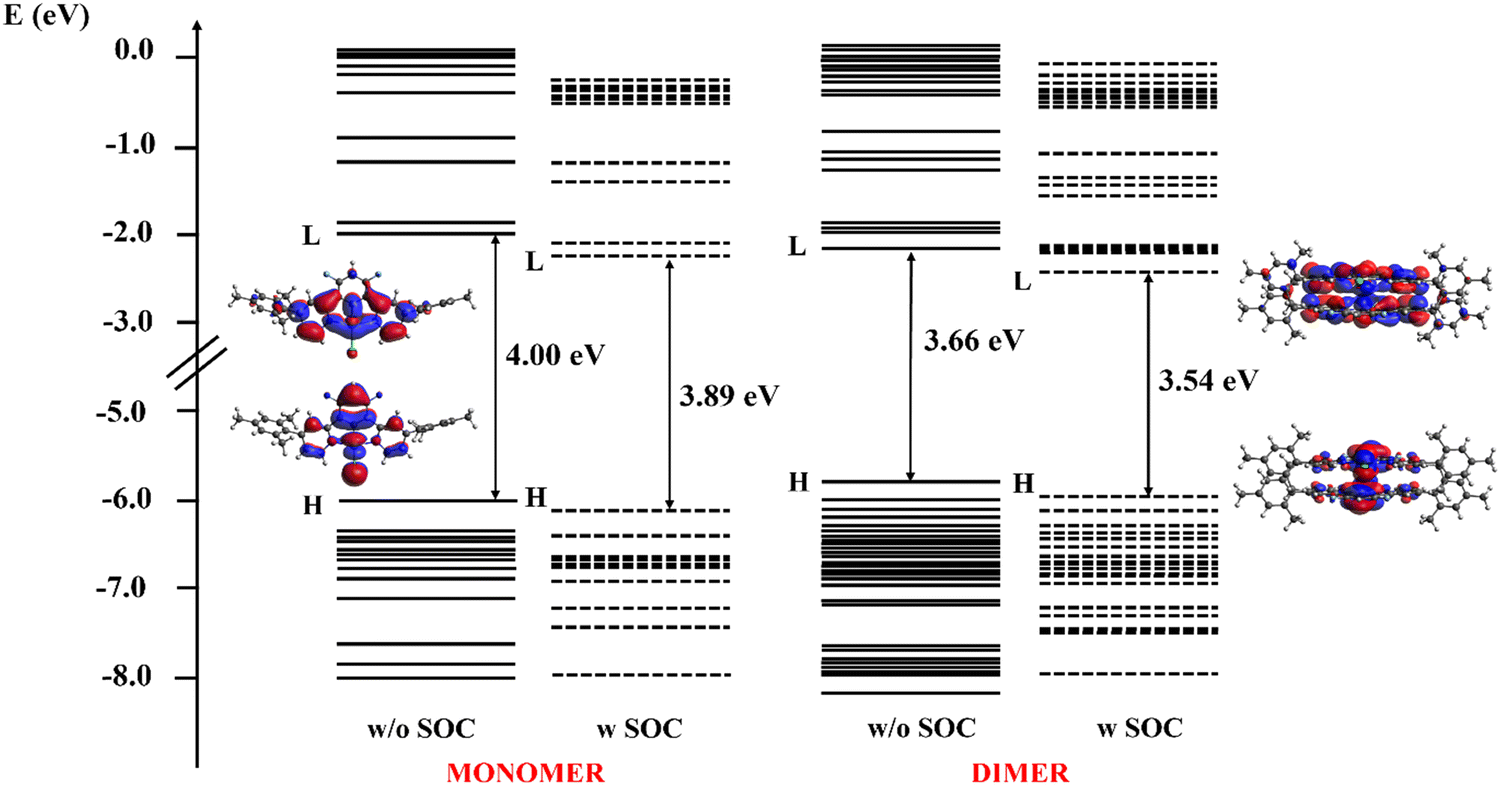 | ||
| Fig. 6 Energy levels of the occupied and unoccupied molecular orbitals of [Pt(bis(4-Mes-py)-4,6-dFb)Cl] (left) and its dimer (right) in the −8.0 to 0.2 eV range. Both for the monomer and dimer, energy levels orbitals without (w/o) and with (w) SOC are plotted using solid and dashed lines, respectively. The isodensity plot (isodensity contour = 0.02) of the HOMO (H) and LUMO (L) of both systems are reported, for which no differences were seen including or not SOC effects. | ||
The orbitals density of the dimer in the −8.0 to 0.2 eV energy range is higher than that of the monomer, as expected, due to the larger number of the resulting eigenfunctions from the combinations of the atomic orbitals of the doubled system. Moreover, the HOMO–LUMO gap decreases going from the monomer to the dimer: in the latter, the orbitals of the monomer are replaced by a pair of orbitals, one at a higher energy and one at a lower energy with respect to the monomer reference, giving rise to the destabilization of the HOMO and the stabilization of the LUMO that closes the energy gap. The HOMO, both for the monomer and the dimer, is delocalized along the Pt–Cl axis and is the result of a combination of the Pt dyz orbital, the Cl pz orbital, and the bonding combinations of p orbitals of the fluorinated ring with smaller anti-bonding contributions coming from the pyridyl rings. The LUMO is the combination of the Pt dyz and the pz orbitals of both the pyridyl nitrogen, with smaller contributions from the C pz orbital of the phenyl and pyridyl rings that lie in the Pt-pyridyl plane. When SOC is included, the HOMO–LUMO gap maintains the same trend but slightly decreases to 3.89 and 3.54 eV for the monomer and the dimer, respectively. Notably, the isodensity plots of both the HOMO and the LUMO orbitals were practically the same also when SOC effects are included and are thus not reported in Fig. 6.
We have computed 80/150 TDDFT singlet excited states for the monomer/dimer to simulate its UV-vis absorption spectra and to assign the main optical features (details of the computed transitions are reported in Tables S3 and S4, ESI†). For the main excitations of the simulated spectra, the eigenvectors have been analyzed and the related excited state densities have been plotted (see Fig. S21 and S22, ESI†). This visualization allows us to follow where the electrons move (blue color) and where they arrive (red color) in optical transitions. It is worth noting that the excitations of the dimer with a significative oscillator strength are more numerous than those of the monomer and the lowest one, S0 → S1 (f = 0.03) computed at 408 nm, is red shifted with respect to the corresponding one of the monomer at 375 nm and characterized by an oscillator strength of 0.01. Overall, both the simulated spectra show the main experimental features; however, the calculated spectrum of the monomer is correlated with the UV-vis spectrum recorded at low concentrations, while that of the dimer, showing low intensity features at lower energies, is related to the spectrum recorded at higher concentrations, see Fig. 7, panels (a) and (b), respectively. Interestingly, the most intense band of the simulated absorption spectrum of the monomer (270–250 nm), the light blue line in panel (a) in Fig. 7, can be assigned as a 1IL band, since the most intense transitions in that energy region have as starting and ending states the orbitals delocalized on the [(4-Mes-py)-4,6-dFb] ligand, in agreement with the experimental assignment, see Table S3 and Fig. S19 in the ESI.†
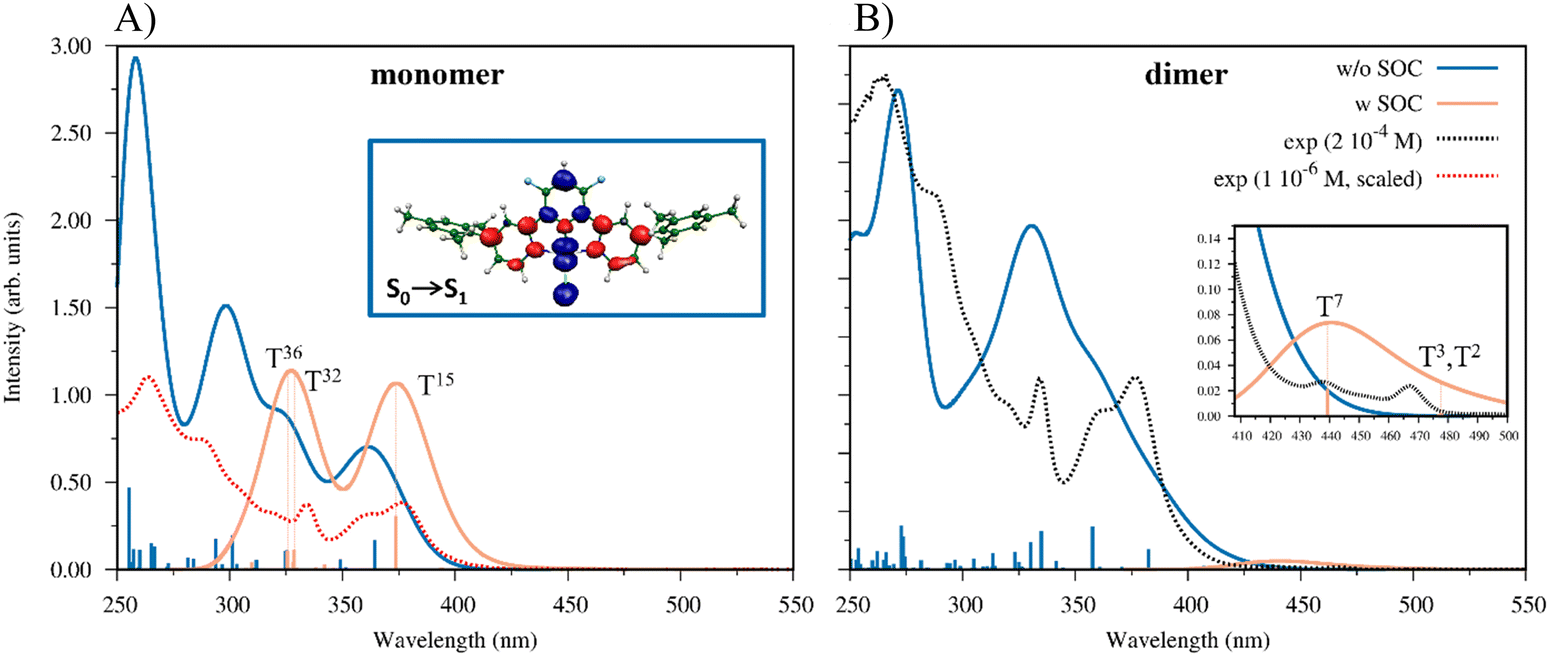 | ||
| Fig. 7 (A) Comparison between the simulated absorption spectra of the monomer without and with SOC (light blue and light orange line, respectively) and the UV-Vis spectrum recorded at 1 × 10−6 M (dotted red line). In the inset, the difference between the density plots of S1 and S0 is reported. (B) Comparison between the simulated absorption spectra of the dimer w/o and w SOC effects (light blue and light orange line, respectively) and the UV-Vis spectrum measured at 2 × 10−4 M (dotted black line). In the inset, the weak bands at longer wavelengths are reported in the expanded scale for clarity. The Tn labels indicate the most important transitions as obtained from the output of the relativistic calculations. | ||
The experimental spectral features are registered at lower energies than the excitations we have computed, probably because the present methodology neglects the SOC, while it plays a relevant role in describing the absorption spectra due to the presence of the heavy Pt center.
A first approach, to indirectly consider this, has been then to compute the lowest 5(10) singlet–triplet transitions for the monomer (dimer) by G09 non-relativistic calculations, even though they have a null oscillator strength as forbidden transitions. Interestingly, for the monomer the first 5 singlet–triplet states are computed in the range 370–453 nm and for the dimer the 10 states are in the range 424–465 nm, red-shifted with respect to the singlet–singlet ones. The singlet–triplet transitions are reported in Tables S5 and S6 in the ESI† material. In light of these results, and to comprehend the low energy region spectral features, TDDFT calculations were performed with ZORA by using ADF software, thus taking into account the SOC effect on both the monomer and dimer optimized structures at the non-relativistic level, see the simulated spectra of the monomer and dimer in light orange, reported in panels (a) and (b) of Fig. 7, respectively. Noteworthily, to probe the effect of SOC on the geometry of the complex, we optimized it at the same relativistic level employed for the spectral calculation, noting non-significant differences (like Pt–N and Pt–C bond distance differences in the second significant figures, in Å) with the optimized geometry computed at a non-relativistic level. Therefore, we considered it reasonable to use both the monomer and dimer non-relativistic optimized geometries for the spectral calculations. In both the monomer and dimer cases, the absorption bands are slightly red-shifted with respect to the non-relativistic calculations, with absorption maxima centered at 327 and 374 nm (monomer) and 440 nm (dimer), as shown in Fig. 7, light orange profiles in panels (a) and (b), respectively. The simulated absorption spectra of the monomer w/o and w SOC (light blue and light orange line, respectively) are compared with the experimental absorption spectrum at the lowest solution concentration (1 × 10−6 M), red dotted line, in panel (a) of Fig. 7, along with the plot of the difference of density associated with the S0 → S1 transition. On the other hand, the computed absorption spectrum of the dimer without and with SOC (light blue and light orange line, respectively) is compared with the experimental UV-vis spectrum at a higher concentration (2 × 10−4 M), black dotted line, in panel (b) of Fig. 7. It is worth underlining that how the features simulated at the relativistic level for the monomer match the experimental spectra. The band centered at 327 nm comes from two different excitations, which are characterized by inner orbitals (HOMO−7, HOMO−3) to LUMO transitions, while the peak at 373 nm shows a predominant HOMO to LUMO character, as reported in Table 2. In the case of the dimer, besides the absorption at 440 nm – which is already indicative of the red-shift from the monomer spectrum – we observe a small absorption band at around 477 nm, in agreement with the low energy experimental features that arise at high concentrations (see Fig. 3(A), inset), confirming the reliability of our hypothesis. In this case, all the involved excited states are strongly characterized by HOMO to LUMO transition. The plots of the molecular orbitals involved in the main excited state transitions for both the monomer and dimer are reported in the ESI† (Fig. S23 and S24). The simulation of a dimer spectrum able to span the same energy region as that of the monomer should include a larger number of excitations that are highly time demanding. However, the limited number of the excitations computed including SOC effects (60/8 for the monomer/dimer, respectively) allows us to reproduce the low-energy feature of the UV-vis spectra at different concentrations, showing the relevance to include spin–orbit coupling to adequately reproduce all the spectral features for Pt-based complexes.
| SOC excited statesa | E (eV)/λ (nm) | f (oscillator strength) | Characterb |
|---|---|---|---|
| a The reported excited states are characterized by f ≥ 0.001. b Only the components ≥5% are reported in the excited state character. | |||
| T15 (monomer) | 3.32 eV/373 nm | 0.307 | 81% (H → L); 7% (H−1 → L+1); 6% (H−1 → L) |
| T32 (monomer) | 3.77 eV/329 nm | 0.114 | 38% (H−7 → L); 13% (H−3 → L); 6% (H → L+2); 6% (H−2 → L) |
| T36 (monomer) | 3.81 eV/325 nm | 0.108 | 45% (H−3 → L); 13% (H−7 → L); 13% (H−2 → L+1); 9% (H → L+2) |
| T2 (dimer) | 2.60 eV/477 nm | 0.003 | 72% (H → L); 7% (H−1 → L+1) |
| T3 (dimer) | 2.60 eV/477 nm | 0.001 | 74% (H → L) |
| T7 (dimer) | 2.82 eV/440 nm | 0.021 | 31% (H → L); 16% (H → L+2); 14% (H−3 → L) |
2.6 OLEDs
Organic light emitting diodes (OLEDs) are becoming the main technology for high-quality flat panel displays as well as for solid-state lighting due to their unique features such as energy-efficient, fast response, high contrast, high color purity, and wide view-angle. Thus, the market of OLED displays has grown fast due to its potential applications in televisions, smartphones, tablets, computer monitors, automobiles, head-up-display, smartwatches, and so on:88 it was valued at 42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 490 million US$ in 2020 and is expected to reach 185830 million US$ by the end of 2026.89 Multicolor luminescent materials are essential to guarantee the quality of the devices, because a flat panel display should be able to produce all the colors from the real world. Three kinds of luminescent materials emitting primary colors red, green, and blue are usually employed according to the primary-color theory, although different emitting colors can affect the quality of displays. Unfortunately, the blue emitters generally show a markedly inferior electroluminescence performance with regard to lifespan, efficiencies, color quality, and charge-carrier injection/transport.90 Therefore, there is an imperative need for the discovery of blue emitters, in particular phosphorescent organometallic complexes with long-lived excited state lifetimes that could efficiently harvest both singlet and triplet electro-generated excitons, thus opening the possibility of reaching theoretically 100% internal quantum efficiency in such devices.31
490 million US$ in 2020 and is expected to reach 185830 million US$ by the end of 2026.89 Multicolor luminescent materials are essential to guarantee the quality of the devices, because a flat panel display should be able to produce all the colors from the real world. Three kinds of luminescent materials emitting primary colors red, green, and blue are usually employed according to the primary-color theory, although different emitting colors can affect the quality of displays. Unfortunately, the blue emitters generally show a markedly inferior electroluminescence performance with regard to lifespan, efficiencies, color quality, and charge-carrier injection/transport.90 Therefore, there is an imperative need for the discovery of blue emitters, in particular phosphorescent organometallic complexes with long-lived excited state lifetimes that could efficiently harvest both singlet and triplet electro-generated excitons, thus opening the possibility of reaching theoretically 100% internal quantum efficiency in such devices.31
Since the first example of a platinum(II) phosphorescent emitter for the fabrication of an efficient OLED device,91 a remarkable amount of work has been devoted to looking for phosphorescent platinum(II) complexes that display improved device performances.31
In this panorama, platinum(II) chlorido complexes with a cyclometalated terdentate 1,3-bis(pyridin-2-yl)benzene ligand certainly play an important role as emitters in light-emitting devices and have potential even as blue emitters. Thus, it was reported that [Pt(1,3-bis(pyridin-2-yl)-4,6-difluoro-benzene)Cl] (5 wt% of the complex in the emitting layer) allows the preparation of bluish green OLEDs with CIE coordinates (0.19, 0.42), a quantum efficiency (QE) of 7.8% ph e−1, a power efficiency (PE) of 6.9 lm W−1 and a luminance efficiency (LE) of 15.9 cd A−1.49 The introduction of CH3 groups in the para position of the pyridine rings leads to better QE (13.0% ph e−1), PE (9.3 lm W−1) and LE (26.0 cd A−1) with CIE co-ordinates of (0.18, 0.35)49 closer to that of the true blue CIE coordinates which are (0.16, 0.13).46 Substitution of the methyl groups with electron-donating methoxy60 or dimethylamino59 substituents allows to go closer to the true blue, with CIE coordinates of (0.18, 0.27) and (0.20, 0.30), respectively, but with a drop of QE, PE and LE. The use of a pure film of these complexes as an emitter led to the preparation of red OLEDs.49,59,60 We were curious to see the effect of the introduction of the bulky mesityl group in position 4 on the pyridines on the characteristics and efficiency of the OLEDs.
Therefore, in the present work, OLEDs were fabricated using an emitting layer either a bis-4-(N-carbazolyl)phenyl)phenylphosphine oxide (BCPO) matrix hosting the [Pt(bis(4-Mes-py)-4,6-dFb)Cl] complex (8% wt) or a film of the pure complex. Holes were injected from the indium tin oxide anode and passed through a 50 nm thick transporting layer made of 4,4′,4′′-tris(N-carbazolyl)triphenylamine TCTA. Electrons were injected from an Al/LiF cathode and transported to the emitting layer (EML) by means of a layer of 2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole) (TPBi, 30 nm thick). Charges recombined in the 30 nm thick EML made of pure [Pt(bis(4-Mes-py)-4,6-dFb)Cl] or of a BCPO matrix hosting the platinum complex (8% wt). EL spectra of the OLEDs are shown in Fig. 8(A): OLED emissions are in the blue (CIE = 0.13, 0.29) and red (CIE = 0.56, 0.36) regions, for [Pt(bis(4-Mes-py)-4,6-dFb)Cl] 8 wt% and pure [Pt(bis(4-Mes-py)-4,6-dFb)Cl], respectively.
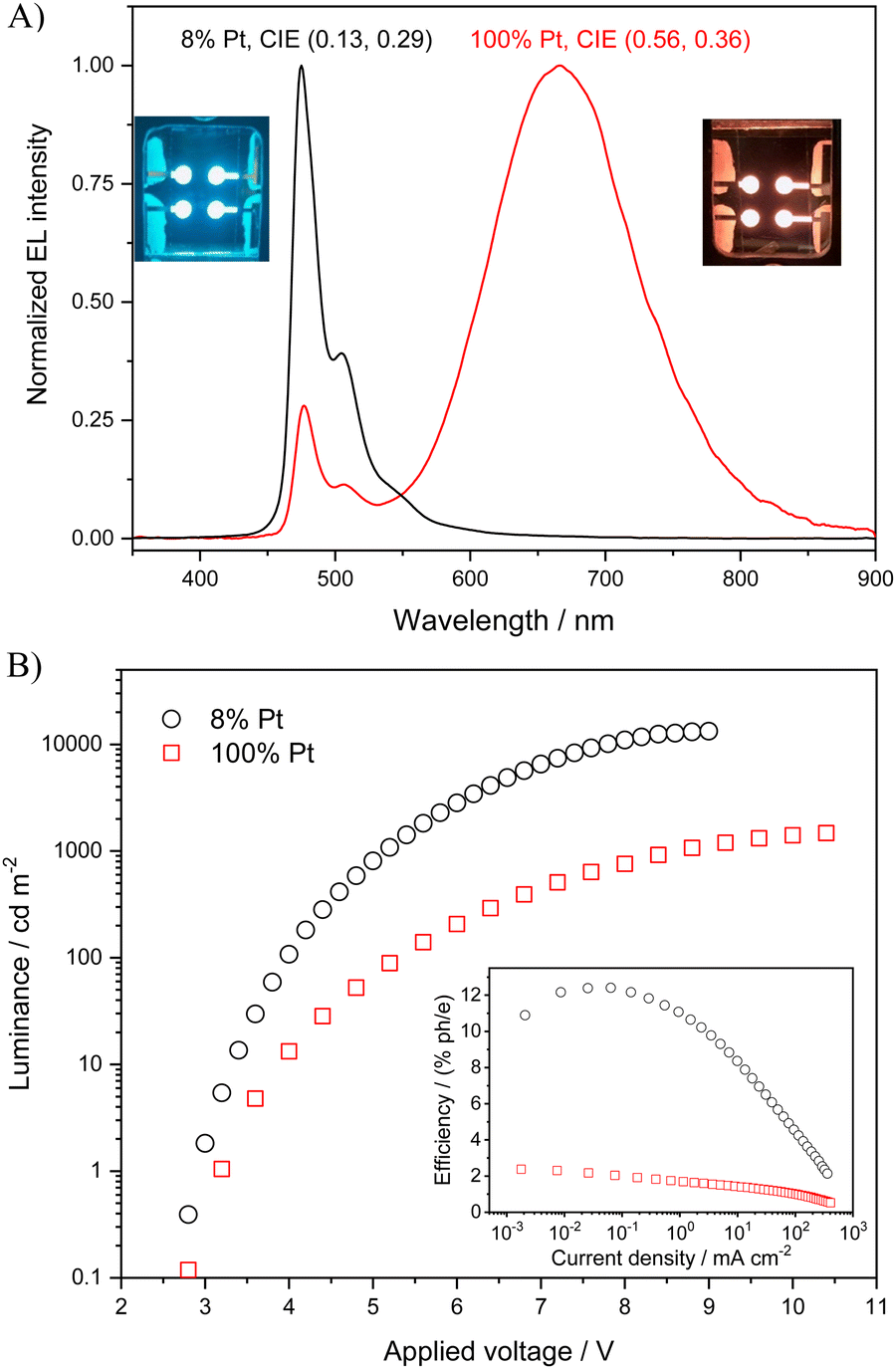 | ||
| Fig. 8 (A) EL spectra, images and CIE coordinates of the OLED devices having 8% and 100% Pt complex. (B) Luminance vs. applied voltage and external EL efficiency vs. current density for the two OLEDs. | ||
There is no substantial contribution to the EL emission bands from the electron-transporting (hole-blocking) or TCTA binder layers, in accordance with a good charge carrier confinement within the EML and complete energy transfer from the excited states of BCPO (formed by charge carrier recombination) to the Pt complex.
The luminance and external EL efficiency as a function of current density and applied voltage of the two OLEDs are shown in Fig. 8(B). The EL efficiency shows the typical roll-off at high currents due to exciton–exciton and/or exciton/charge interaction and to high field induced exciton dissociation.92
In Table 3, the performance of blue and red OLEDs based on previously reported N^C^N platinum complexes are reported for comparison with that of the novel emitter prepared in the present work.
| Pt(bis(4-R-py)-4,6-dFb)Cl | J (mA cm−2) | QE (% ph e−1) | PE (lm W−1) | LE (cd A−1) | CIE (x,y) | |
|---|---|---|---|---|---|---|
| R = H49 | 5% | 3.1 | 7.8 | 6.9 | 15.9\ | 0.19, 0.42 |
| 100% | 31.3 | 5.3 | 0.47 | 1.6 | 0.63, 0.35 | |
| R = Me49 | 5% | 1.5 | 13.0 | 9.3 | 26.0 | 0.18, 0.35 |
| 100% | 3.7 | 14.7 | 5.0 | 13.9 | 0.61, 0.38 | |
| R = OMe60 | 5% | 4.7 | 6.1 | 2.8 | 10.7 | 018, 0.27 |
| 100% | 1.8 | 16 | 8.1 | 27.7 | 0.58, 041 | |
| R = NMe259 | 3% | 6.0 | 3.9 | 2.4 | 6.9 | 0.20, 0.30 |
| 100% | 3.9 | 6.4 | 5.7 | 14 | 0.52, 0.47 | |
| R = Mes [this work] | 8% | 2.4 | 10.2 | 12.0 | 18.3 | 0.13, 0.29 |
| 100% | 67.2 | 1.1 | 0.3 | 0.8 | 0.56, 0.36 | |
The red OLED with [Pt(bis(4-Mes-py)-4,6-dFb)Cl] shows relatively poor performance compared to its parent complexes, reasonably due to the lower luminescence of the Pt–Pt aggregates. On the other hand, remarkably, the OLED with the [Pt(bis(4-Mes-py)-4,6-dFb)Cl] complex (8% wt) exhibits a blue emission with CIE coordinates (0.13, 0.29) closer to that of the true blue and better performance (in terms of QE, PE, and LE) than its predecessors with a methoxy or dimethylamino substituent instead of the mesityl group. It shows an ELQE max of 12.5% and reaches a luminance of about 13500 cd m−2 at 9 V. This performance is excellent when compared to that recently reached with blue OLEDs based on other efficient platinum complexes such as those featuring cyclometalated N-pyridyl carbazole ligands with monocarborane clusters (EQEmax = 6.2% at 1010 cd m−2)45 or with the trans-N-heterocyclic carbene platinum(II) acetylide complex bearing phenyl acetylene ligands (EQEmax = 8% at low luminance, with significant efficiency roll-off at higher luminance, approaching 2% at 500 cd m−2).41 The results obtained with [Pt(bis(4-Mes-py)-4,6-dFb)Cl] are really encouraging, showing the great potential of N^C^N platinum complexes for the fabrication of blue OLEDs.
3. Conclusion
In conclusion, a novel complex, namely [Pt(bis(4-mesityl-pyridin-2-yl)-4,6-difluorobenzene)Cl], was easily prepared and well characterized. Remarkably, it exhibited intense blue phosphorescence in dichloromethane solution and in the PMMA film (1 wt% complex), with quantum yields approaching unity, whereas red phosphorescence is observed in a neat film, maintaining an excellent quantum yield. The origin of these intriguing optical properties was further analysed by means of DFT and TDDFT calculations, eventually involving SOC effects, which accurately describe the absorption spectrum of the complex and the effect of its concentration increase in CH2Cl2 solution, which was explained in terms of the formation of aggregated species, such as dimers. Indeed, in the UV-vis absorption spectra, the formation of aggregated species cannot be ruled out, as shown by the low-energy spectral features measured at high concentrations.The introduction of a bulky mesityl substituent on the pyridyl rings of the cycloplatinated ligand appears as a facile strategy to increase the Pt⋯Pt distance, reaching a value (8.59 Å) much longer than that observed for previously reported chlorido N^C^N-platinum(II) complexes.
This tactic represents a springboard for the fabrication of efficient blue OLEDs (8% wt complex) with CIE coordinates of (0.13, 0.29) approaching true blue. In the near future, it is expected that the presence of a bulky substituent combined with the introduction of electron-donor groups on the pyridines will allow us to reach not only true blue but also deep blue OLEDs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Fondazione Cariplo and Regione Lombardia are acknowledged for the instrumentation bought during the SmartMatLab Centre project (2014). The work was supported by the National Interuniversity Consortium of Materials Science and Technology (project INSTM21MIROBERTO), Università degli Studi di Milano (project PSR2022_DIP_005_PI_LCARL) and CNR. This work has been funded by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY. CUP: B43C22000470005.Notes and references
- A. Colombo, C. Dragonetti, D. Marinotto, S. Righetto, D. Roberto, S. Tavazzi, M. Escadeillas, V. Guerchais, H. Le Bozec, A. Boucekkine and C. Latouche, Cyclometalated 4-Styryl-2-phenylpyridine Platinum(II) Acetylacetonate Complexes as Second-Order NLO Building Blocks for SHG Active Polymeric Films, Organometallics, 2013, 32, 3890–3894 CrossRef CAS.
- H. Zhao, E. Garoni, T. Roisnel, A. Colombo, C. Dragonetti, D. Marinotto, S. Righetto, D. Roberto, D. Jacquemin, J. Boixel and V. Guerchais, Photochromic DTE-Substituted-1,3-di(2-pyridyl)benzene Platinum(II) Complexes: Photomodulation of Luminescence and Second-Order Nonlinear Optical Properties, Inorg. Chem., 2018, 57(12), 7051–7063 CrossRef CAS PubMed.
- S. Attar, F. Artizzu, L. Marchik, D. Espa, L. Pilia, M. F. Casula, A. Serpe, M. Pizzotti, A. Orbelli-Biroli and P. Deplano, Uncommon Optical Properties and Silver-Responsive Turn-Off/On Luminescence in a PtII Heteroleptic Dithiolene Complex, Chem. – Eur. J., 2018, 24, 10503–10512 CrossRef CAS PubMed.
- A. Colombo, C. Dragonetti, V. Guerchais and D. Roberto, An excursion in the second-order nonlinear optical properties of platinum complexes, Coord. Chem. Rev., 2021, 446, 214113 CrossRef CAS.
- S. S. Attar, L. Pilia, D. Espa, F. Artizzu, A. Serpe, M. Pizzotti, D. Marinotto, L. Marchiò and P. Deplano, Insight into the Properties of Heteroleptic Metal Dithiolenes: Multistimuli Responsive Luminescence, Chromism, and Nonlinear Optics, Inorg. Chem., 2021, 60, 9332–9344 CrossRef CAS PubMed.
- J. Boixel, A. Colombo, F. Fagnani, P. Matozzo and C. Dragonetti, Intriguing Second-Order NLO Switches Based on New DTE Compounds, Eur. J. Inorg. Chem., 2022, e202200034 CrossRef CAS.
- Q. Zhao, C. Huang and F. Li, Phosphorescent heavy-metal complexes for bioimaging, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Lighting the way to see inside the live cell with luminescent transition metal complexes, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS.
- E. Baggaley, S. W. Botchway, J. W. Haycock, H. Morris, I. V. Sazanovich, J. A. G. Williams and J. A. Weinstein, Long-lived metal complexes open up microsecond lifetime imaging microscopy under multiphoton excitation: from FLIM to PLIM and beyond, Chem. Sci., 2014, 5, 879–886 RSC.
- A. Colombo, F. Fiorini, D. Septiadi, C. Dragonetti, F. Nisic, A. Valore, D. Roberto, M. Mauro and L. De Cola, Neutral N^C^N terdentate luminescent Pt(II) complexes: their synthesis, photophysical properties, and bio-imaging applications, Dalton Trans., 2015, 44, 8478–8487 RSC.
- F. Fagnani, G. De Soricellis, A. Colombo, C. Dragonetti, D. Roberto, A. di Biase, S. Fantacci and D. Marinotto, Photophysical investigation of highly phosphorescent N^C^N platinum(II) azido complexes and their triazole derivatives, Dyes Pigm., 2024, 225, 112064 CrossRef CAS and references therein.
- A. S.-Y. Law, L. C.-C. Lee, K. K.-W. Lo and V. W.-W. Yam, Aggregation and Supramolecular Self-Assembly of Low-Energy Red Luminescent Alkynylplatinum(II) Complexes for RNA Detection, Nucleolus Imaging, and RNA Synthesis Inhibitor Screening, J. Am. Chem. Soc., 2021, 143, 5396–5405 CrossRef CAS PubMed.
- G. De Soricellis, F. Fagnani, A. Colombo, C. Dragonetti and D. Roberto, Exploring the potential of N^C^N cyclometalated Pt(II) complexes bearing 1,3-di(2-pyridyl)benzene derivatives for imaging and photodynamic therapy, Inorg. Chim. Acta, 2022, 541, 121082 CrossRef CAS.
- J. Berrones Reyes, P. S. Sherin, A. Sarkar, M. K. Kuimova and R. Vilar, Platinum(II)-Based Optical Probes for Imaging Quadruplex DNA Structures via Phosphorescence Lifetime Imaging Microscopy, Angew. Chem., Int. Ed., 2023, e202310402 CAS.
- A. Haque, H. El Moll, K. M. Alenezi, M. S. Khan and W. Y. Wong, Functional materials based on cyclometalated platinum (II) β-diketonate complexes: A Review of structure–property relationships and applications, Materials, 2021, 14, 4236–4261 CrossRef CAS PubMed.
- A. S.-Y. Law, M. C.-L. Yeung and V. W.-W. Yam, Arginine-Rich Peptide-Induced Supramolecular Self-Assembly of Water-Soluble Anionic Alkynylplatinum(II) Complexes: A Continuous and Label-Free Luminescence Assay for Trypsin and Inhibitor Screening, ACS Appl. Mater. Interfaces, 2017, 9, 41143–41150 CrossRef CAS PubMed.
- Y. Y. Ning, G. Q. Jin, M. X. Wang, S. Gao and J. L. Zhang, Recent progress in metal-based molecular probes for optical bioimaging and biosensing, Curr. Opin. Chem. Biol., 2022, 66, 102097 CrossRef CAS PubMed.
- V. Adamovich, J. Brooks, A. Tamayo, A. M. Alexander, P. I. Djurovich, B. W. D’Andrade, C. Adachi, S. R. Forrest and M. E. Thompson, High efficiency single dopant white electrophosphorescent light emitting diodes, New J. Chem., 2002, 26, 1171–1178 RSC.
- W. Sotoyama, T. Satoh, N. Sawatari and H. Inoue, Efficient organic light-emitting diodes with phosphorescent platinum complexes containing N^C^N-coordinating tridentate ligand, Appl. Phys. Lett., 2005, 86, 153505 CrossRef.
- M. Cocchi, D. Virgili, V. Fattori, D. L. Rochester and J. A. G. Williams, N^C^N-Coordinated Platinum(II) Complexes as Phosphorescent Emitters in High-Performance Organic Light-Emitting Devices, Adv. Funct. Mater., 2007, 17, 285–289 CrossRef CAS.
- X. Yang, Z. Wang, S. Madakuni, J. Li and G. E. Jabbour, Efficient Blue- and White-Emitting Electrophosphorescent Devices Based on Platinum(II) [1,3-Difluoro-4,6-di(2-pyridinyl)benzene] Chloride, Adv. Mater., 2008, 20, 2405–2409 CrossRef CAS.
- W. Y. Wong and C. L. Ho, Heavy metal organometallic electrophosphors derived from multi-component chromophores, Chem. Soc. Rev., 2009, 253, 1709–1758 CAS.
- C. M. Che, C. C. Kwok, S. W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Y. Zhu and H. Yersin, Photophysical properties and OLED applications of phosphorescent platinum(II) Schiff base complexes, Chem. – Eur. J., 2010, 16, 233–247 CrossRef CAS PubMed.
- G. Zhou, Q. Wang, X. Wang, C.-L. Ho, W.-Y. Wong, D. Ma, L. Wang and Z. Line, Metallophosphors of platinum with distinct main-group elements: a versatile approach towards color tuning and white-light emission with superior efficiency/color quality/brightness trade-offs, J. Mater. Chem., 2010, 20, 7472–7484 RSC.
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Light-emitting devices based on organometallic platinum complexes as emitters, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS.
- E. Rossi, L. Murphy, P. L. Brothwood, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, M. Cocchi and J. A. G. Williams, Cyclometallated platinum(II) complexes of 1,3-di(2-pyridyl)benzenes: tuning excimer emission from red to near-infrared for NIR-OLEDs, J. Mater. Chem., 2011, 21, 15501–15510 RSC.
- L. F. Gildea and J. A. G. Williams, Iridium and platinum complexes for OLEDs, in Organic Light-Emitting Diodes: Materials, Devices and Applications, ed. A. Buckley, Woodhead, Cambridge, 2013 Search PubMed.
- P. Brulatti, V. Fattori, S. Muzzioli, S. Stagni, P. P. Mazzeo, D. Braga, L. Maini, S. Milita and M. Cocchi, Tuning the colour and efficiency in OLEDs by using amorphous or polycrystalline emitting layers, J. Mater. Chem. C, 2013, 1, 1823–1831 RSC.
- X. Yang, C. Yao and G. Zhou, Highly Efficient Phosphorescent Materials Based on Platinum Complexes and Their Application in Organic Light-Emitting Devices (OLEDs), Platinum Met. Rev., 2013, 57, 2–16 CrossRef CAS.
- Q. Wang, I. W. H. Oswald, X. Yang, G. Zhou, H. Jia, Q. Qiao, Y. Chen, J. Hoshikawa-Halbert and B. E. Gnade, A Non-Doped Phosphorescent Organic Light-Emitting Device with Above 31% External Quantum Efficiency, Adv. Mater., 2014, 26, 8107–8113 CrossRef CAS PubMed.
- C. Cebrian and M. Mauro, Recent advances in phosphorescent platinum complexes for organic light-emitting diodes, Beilstein J. Org. Chem., 2018, 14, 1459–1481 CrossRef CAS PubMed.
- X. Yang, H. Guo, X. Xu, Y. Sun, G. Zhou, W. Ma and Z. Wu, Enhancing Molecular Aggregations by Intermolecular Hydrogen Bonds to Develop Phosphorescent Emitters for High-Performance Near-Infrared OLEDs, Adv. Sci., 2019, 6, 1801930 CrossRef PubMed.
- W.-C. Chen, C. Sukpattanacharoen, W.-H. Chan, C.-C. Huang, H.-F. Hsu, D. Shen, W.-Y. Hung, N. Kungwan, D. Escudero, C.-S. Lee and Y. Chi, Modulation of Solid-State Aggregation of Square-Planar Pt(II) Based Emitters: Enabling Highly Efficient Deep-Red/Near Infrared Electroluminescence, Adv. Funct. Mater., 2020, 30, 2002494 CrossRef CAS.
- A. Haque, L. Xu, R. A. Al-Balushi, M. K. Al-Suti, R. Ilmi, Z. Guo, M. S. Khan, W.-Y. Wong and P. R. Raithby, Cyclometalated tridentate platinum(II) arylacetylide complexes: Old wine in new bottles, Chem. Soc. Rev., 2019, 48, 5547–5563 RSC.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, The triplet state of organo-transition metal compounds. Triplet harvesting and singlet harvesting for efficient OLEDs, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- P. T. Chou, Y. Chi, M. W. Chung and C. C. Lin, Harvesting luminescence via harnessing the photophysical properties of transition metal complexes, Coord. Chem. Rev., 2011, 255, 2653–2665 CrossRef CAS.
- D. Roberto, A. Colombo, C. Dragonetti, F. Fagnani, M. Cocchi and D. Marinotto, A Novel Class of Cyclometalated Platinum(II) Complexes for Solution-Processable OLEDs, Molecules, 2022, 27, 5171 CrossRef CAS PubMed.
- Y. C. Wei, S. F. Wang, Y. Hu, L. S. Liao, D. G. Chen, K. H. Chang, C. W. Wang, S. Liu, W. H. Chan, J. L. Liao, W. Y. Hung, T. H. Wang, P. T. Chen, H. Hsu, F. Y. Chi and P. T. Chou, Overcoming the energy gap law in near-infrared OLEDs by exciton–vibration decoupling, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
- E. A. Wood, L. F. Gildea, D. S. Yufit and J. A. G. Williams, Synthesis, structures and luminescence properties of N^C^N-coordinated platinum(II) complexes based on an anthracene core: A red shift and a twist, Polyhedron, 2021, 207, 115401 CrossRef CAS.
- A. K. Pal, S. Krotkus, M. Fontani, C. F. R. Mackenzie, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, High-Efficiency Deep-Blue-Emitting Organic Light-Emitting Diodes Based on Iridium(III) Carbene Complexes, Adv. Mater., 2018, 30, 1804231 CrossRef PubMed.
- J. D. Bullock, A. Salehi, C. J. Zeman IV, K. A. Abboud, F. So and K. S. Schanze, In Search of Deeper Blues: Trans-N-Heterocyclic Carbene Platinum Phenylacetylide as a Dopant for Phosphorescent OLEDs, ACS Appl. Mater. Interfaces, 2017, 9, 41111–41114 CrossRef CAS PubMed.
- X. Li, J. Zhang, Z. Zhao, L. Wang, H. Yang, Q. Chang, N. Jiang, Z. Liu, Z. Bian, W. Liu, Z. Lu and C. Huang, Deep Blue Phosphorescent Organic Light-Emitting Diodes with CIEy Value of 0.11 and External Quantum Efficiency up to 22.5%, Adv. Mater., 2018, 30, 1705005 CrossRef PubMed.
- Y. Wu, Z. Wen, J. I.-C. Wu and T. S. Teets, Efficient Deep Blue Platinum Acetylide Phosphors with Acyclic Diaminocarbene Ligands, Chem. – Eur. J., 2020, 26, 16028–16035 CrossRef CAS PubMed.
- R. He, Z. Xu, S. Valandro, H. D. Arman, J. Xue and K. S. Schanze, High-Purity and Saturated Deep-Blue Luminescence from trans-NHC Platinum(II) Butadiyne Complexes: Properties and Organic Light Emitting Diode Application, ACS Appl. Mater. Interfaces, 2021, 13, 5327–5337 CrossRef CAS PubMed.
- Y. Shen, X. Kong, F. Yang, H.-D. Bian, G. Cheng, T. R. Cook and Y. Zhang, Deep Blue Phosphorescence from Platinum Complexes Featuring Cyclometalated N-Pyridyl Carbazole Ligands with Monocarborane Clusters (CB11H12−), Inorg. Chem., 2022, 61, 16707–16717 CrossRef CAS PubMed.
- T. Maganti and K. Venkatesan, The Search for Efficient True Blue and Deep Blue Emitters: An Overview of Platinum Carbene Acetylide Complexes, ChemPlusChem, 2022, 87, e202200014 CrossRef CAS PubMed.
- Y. H. Nguyen, V. Q. Dang, J. V. Soares, J. I. Wu and T. S. Teets, Efficient blue-phosphorescent trans-bis(acyclic diaminocarbene) platinum(II) acetylide complexes, Chem. Sci., 2023, 14, 4857–4862 RSC.
- F. Fagnani, A. Colombo, C. Dragonetti, D. Roberto and D. Marinotto, The intriguing effect of thiolates as co-ligands in platinum(II) complexes bearing a cyclometalated 1,3-di(2-pyridyl)benzene, Inorg. Chim. Acta, 2022, 532, 120744 CrossRef CAS.
- M. Cocchi, J. Kalinowski, V. Fattori, J. A. G. Williams and L. Murphy, Color-variable highly efficient organic electrophosphorescent diodes manipulating molecular exciton and excimer emissions, Appl. Phys. Lett., 2009, 94, 073309 CrossRef.
- A. Colombo, G. De Soricellis, F. Fagnani, C. Dragonetti, M. Cocchi, B. Carboni, V. Guerchais and D. Marinotto, Introduction of a triphenylamine substituent on pyridyl rings as a springboard for a new appealing brightly luminescent 1,3-di-(2-pyridyl)benzene platinum(II) complex family, Dalton Trans., 2022, 51, 12161–12169 RSC.
- F. Fagnani, A. Colombo, C. Dragonetti, D. Roberto and D. Marinotto, New members of a class of cyclometalated 1,3-di-(2-pyridyl)benzene platinum(II) complexes bearing a tetrazole-thiolate ancillary ligand, Inorg. Chim. Acta, 2023, 550, 121446 CrossRef CAS.
- R. J. Salthouse, P. Pander, D. S. Yufit, F. B. Dias and J. A. G. Williams, Near-infrared electroluminescence beyond 940 nm in Pt(N^C^N)X complexes: influencing aggregation with the ancillary ligand X, Chem. Sci., 2022, 13, 13600 RSC.
- J. A. G. Williams, A. Beeby, S. Davies, J. A. Weinstein and C. Wilson, An Alternative Route to Highly Luminescent Platinum(II) Complexes: Cyclometalation with N^C^N-Coordinating Dipyridylbenzene Ligands, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS PubMed.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Controlling Emission Energy, Self-Quenching, and Excimer Formation in Highly Luminescent N^C^N-Coordinated Platinum(II) Complexes, Inorg. Chem., 2005, 44, 9690–9703 CrossRef CAS PubMed.
- J. A. G. Williams, The coordination chemistry of dipyridylbenzene: N-deficient terpyridine or panacea for brightly luminescent metal complexes?, Chem. Soc. Rev., 2009, 38, 1783–1801 RSC.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Improving the Performance of Pt(II) Complexes for Blue Light Emission by Enhancing the Molecular Rigidity, Inorg. Chem., 2012, 51, 312–319 CrossRef CAS PubMed.
- A. Rodrigue-Witchel, D. L. Rochester, S. B. Zhao, K. B. Lavelle, J. A. G. Williams, S. Wang, W. B. Connick and C. Reber, Pressure-induced variations of MLCT and ligand-centered luminescence spectra in square-planar platinum(II) complexes, Polyhedron, 2016, 108, 151–155 CrossRef CAS.
- C. Dragonetti, F. Fagnani, D. Marinotto, A. di Biase, D. Roberto, M. Cocchi, S. Fantacci and A. Colombo, First member of an appealing class of cyclometalated 1,3-di-(2-pyridyl)benzene platinum(II) complexes for solution-processable OLEDs, J. Mater. Chem. C, 2020, 8, 7873–7881 RSC.
- L. Murphy, P. Brulatti, V. Fattori, M. Cocchi and J. A. G. Williams, Blue-shifting the monomer and excimer phosphorescence of tridentate cyclometallated platinum(II) complexes for optimal white-light OLEDs, Chem. Commun., 2012, 48, 5817–5819 RSC.
- M. Cocchi, J. Kalinowski, L. Murphy, J. A. G. Williams and V. Fattori, Mixing of molecular exciton and excimer phosphorescence to tune color and efficiency of organic LEDs, Org. Electron., 2010, 11, 388–396 CrossRef CAS.
- W. Sotoyama, T. Satoh, H. Sato, A. Matsuura and N. Sawatari, Excited States of Phosphorescent Platinum(II) Complexes Containing N^C^N-Coordinating Tridentate Ligands: Spectroscopic Investigations and Time-Dependent Density Functional Theory Calculations, J. Phys. Chem. A, 2005, 109, 9760–9766 CrossRef CAS PubMed.
- F. Henwood, A. K. Bansal, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Solubilised bright blue-emitting iridium complexes for solution processed OLEDs, J. Mater. Chem. C, 2016, 4, 3726 RSC.
- W. Leslie, A. S. Batsanov, J. A. K. Howard and J. A. G. Williams, Cross-couplings in the elaboration of luminescent bis-terpyridyl iridium complexes: the effect of extended or inhibited conjugation on emission, Dalton Trans., 2004, 623–631 RSC.
- E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, F. Demartin, M. Cocchi, P. Brulatti, V. Fattori and J. A. G. Williams, From red to near infra-red OLEDs: the remarkable effect of changing from X = –Cl to –NCS in a cyclometallated [Pt(N^C^N)X] complex {N^C^N = 5-mesityl-1,3-di-(2-pyridyl)benzene}, Chem. Commun., 2012, 48, 3182–3184 RSC.
- D. J. Cardenas, A. M. Echavarren and M. C. Ramìrez de Arellano, Divergent Behavior of Palladium(II) and Platinum(II) in the Metalation of 1,3-Di(2-pyridyl)benzene, Organometallics, 1999, 18, 3337–3341 CrossRef CAS.
- H. Imoto, S. Tanaka, T. Kato, S. Watase, K. Matsukawa, T. Yumura and K. Naka, Highly Efficient Solid-State Phosphorescence of Platinum Dihalide Complexes with 9-Phenyl-9-arsafluorene Ligands, Organometallics, 2016, 35, 364–369 CrossRef CAS.
- P. H. Lanoë, A. Moreno-Betancourt, L. Wilson, C. Philouze, C. Monnereau, H. Jamet, D. Jouvenot and F. Loiseau, Neutral heteroleptic cyclometallated Platinum(II) complexes featuring 2-phenylbenzimidazole ligand as bright emitters in solid state and in solution, Dyes Pigm., 2019, 162, 967–977 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. D. McLean and G. S. Chandler, Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- A. J. H. Wachters, Gaussian basis set for molecular wavefunctions containing third-row atoms, J. Chem. Phys., 1970, 52, 1033 CrossRef CAS.
- A. Petersson and M. A. Al-Laham, A Complete Basis Set Model Chemistry. II. Open-Shell Systems and the Total Energies of the First-Row Atoms, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- E. V. Lenthe, E. J. Baerends and J. G. Snijders, Relativistic regular two-component Hamiltonians, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef.
- E. V. Lenthe, E. J. Baerends and J. G. Snijders, Relativistic total energy using regular approximations, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef.
- E. V. Lenthe, A. E. Ehlers and E. J. Baerends, Geometry optimizations in the zero order regular approximation for relativistic effects, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef.
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- ADF 2014.04 and ADF 2017.101, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com Search PubMed.
- S. J. A. van Gisbergen, J. G. Snijders and E. J. Baerends, Implementation of time-dependent density functional response equations, Comput. Phys. Commun., 1999, 118, 119–138 CrossRef CAS.
- F. Wang, T. Ziegler, E. van Lenthe, S. van Gisbergen and E. J. Baerends, The calculation of excitation energies based on the relativistic two-component zeroth-order regular approximation and time-dependent density-functional with full use of symmetry, J. Chem. Phys., 2005, 122, 204103 CrossRef PubMed.
- E. van Lenthe and E. J. Baerends, Optimized Slater-type basis sets for the elements 1–118, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- S. Miertus, E. Scrocco and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Ab initio study of solvated molecules: a new implementation of the polarizable continuum model, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- V. Barone and M. Cossi, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- C. C. Pye and T. Ziegler, An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package, Theor. Chem. Acc., 1999, 101, 396–408 Search PubMed.
- The Display Market, https://oled.com/oled-markets/oleddisplays/.
- S. S. Swayamprabha, D. K. Dubey, Shahnawaz, R. A. K. Yadav, M. R. Nagar, A. Sharma, F.-C. Tung and J.-H. Jou, Approaches for Long Lifetime Organic Light Emitting Diodes, Adv. Sci., 2021, 8, 2002254 Search PubMed.
- X. Yang, X. Xu and G. Zhou, Recent advances of the emitters for high performance deep-blue organic light-emitting diodes, J. Mater. Chem. C, 2015, 3, 913 RSC.
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Highly efficient phosphorescent emission from organic electroluminescent devices, Nature, 1998, 395, 151–154 CrossRef CAS.
- J. Kalinowski, W. Stampor, J. Szmytkowski, D. Virgili, M. Cocchi, V. Fattori and C. Sabatini, Coexistence of dissociation and annihilation of excitons on charge carriers in organic phosphorescent emitters, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 85316 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, preparation and characterization of thin films, photoluminescence properties in solution and in the solid state, and fabrication of OLEDs. CCDC 2087751 and 2087747. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc01458h |
| This journal is © The Royal Society of Chemistry 2024 |