
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isomeric modulation of thermally activated delayed fluorescence in dibenzo[a,c]phenazine-based (deep) red emitters†
Sonny
Brebels
 abc,
Tom
Cardeynaels
abcd,
Louis
Jackers
abc,
Suman
Kuila
e,
Huguette
Penxten
abc,
Rebecca J.
Salthouse
f,
Andrew
Danos
*e,
Andrew P.
Monkman
e,
Benoît
Champagne
d and
Wouter
Maes
*abc
abc,
Tom
Cardeynaels
abcd,
Louis
Jackers
abc,
Suman
Kuila
e,
Huguette
Penxten
abc,
Rebecca J.
Salthouse
f,
Andrew
Danos
*e,
Andrew P.
Monkman
e,
Benoît
Champagne
d and
Wouter
Maes
*abc
aHasselt University, Institute for Materials Research (IMO-IMOMEC), Design & Synthesis of Organic Semiconductors (DSOS), Agoralaan 1, 3590 Diepenbeek, Belgium. E-mail: wouter.maes@uhasselt.be
bIMOMEC Division, IMEC, Wetenschapspark 1, 3590 Diepenbeek, Belgium
cEnergyville, Thorpark, 3600 Genk, Belgium
dUniversity of Namur, Laboratory of Theoretical Chemistry, Theoretical and Structural Physical Chemistry Unit, Namur Institute of Structured Matter, Rue de Bruxelles 61, 5000 Namur, Belgium
eDurham University, Department of Physics, OEM group, South Road, Durham DH1 3LE, UK. E-mail: andrew.danos@durham.ac.uk
fDurham University, Department of Chemistry, South Road, Durham DH1 3LE, UK
First published on 29th May 2024
Abstract
A series of four emissive regio-isomers are synthesized based on the dibenzo[a,c]phenazine-11,12-dicarbonitrile (DBPzCN) acceptor scaffold and a triphenylamine (TPA) donor. Density functional theory is utilized to compare the relative differences in molecular conformation, excited state distributions, and orbital interactions. Steady-state and time-resolved emission spectroscopy reveal strongly contrasting emissive properties and triplet harvesting of the four materials. In zeonex host emission maxima range widely, with differences of over 100 nm. Additionally, isomers 3-TPA-DBPzCN and 4-TPA-DBPzCN show photoluminescence quantum yields (PLQYs) of 46 and 62%, while 1-TPA-DBPzCN and 2-TPA-DBPzCN instead show values <1 and 24%, respectively. Relevant to thermally activated delayed fluorescence (TADF), very small singlet–triplet energy gaps are observed for isomers 2-TPA-DBPzCN and 4-TPA-DBPzCN, with corresponding reverse intersystem crossing (rISC) rates of 0.6 and 1.6 × 105 s−1, respectively. Unique in possessing both fast rISC and a relatively high PLQY, the unconventional 4-TPA-DBPzCN regio-isomer turns out to be an efficient TADF emitter, highlighting the important role of donor–acceptor substitution position in the design of efficient TADF materials targeting specific wavelength ranges.
1. Introduction
The importance of organic light-emitting diodes (OLEDs) in the display industry has risen from novelty to dominance in the last decade. Global interest in this technology is further encouraged by the appealing advantages it can provide in terms of reduced energy consumption, compatibility with flexible substrates, large-scale processability, and tuneability of material properties.1–3 Meanwhile, several challenges must still be overcome in the development of efficient OLED emitters, especially at the long wavelength edge of the visible spectrum. Indeed, emission in the deep red to near-infrared (NIR) region, commonly defined as the wavelength range between 650 and 1400 nm,4 is required to enable multiple interesting applications such as night vision, fingerprinting, security/cryptography, light-based communication, and extending further to biomedical imaging, biosensors, and optical therapies.5–11Unfortunately, the molecular design strategies for all-organic π-conjugated molecules that emit efficiently in this wavelength region are not straightforward. The absorption and emission bands of an organic emitter are typically red-shifted when the π-conjugated system is expanded, thereby lowering the optical gap.12 While effective, this approach often triggers two unfavorable phenomena that result in quenching of the fluorescence: the ‘energy gap’ law and the formation of ‘H-type’ aggregates. The energy gap law states that the narrowing of energy gaps between the excited singlet states and the vibrational manifolds of the ground state – an inescapable consequence of the increased density of states as the singlet energy decreases – increases the rate of non-radiative decay losses through molecular vibrations.13 This competes with the emissive decay mechanism, thereby reducing the fluorescence efficiency. While the rigidity of a π-conjugated system can help to combat vibrational losses, the resulting molecular planarity can often simultaneously induce the formation of the aforementioned H-aggregates via undesired cofacial stacking. Emission from the lower excited (red-shifted) state of such ‘excimer’ aggregates is forbidden due to the out-of-phase orientation of the molecular transition dipoles, again resulting in a significant loss of fluorescence efficiency at practical doping concentrations of such emitters.14
While it is clear that smart material design can help to suppress the effects of both phenomena, additional focus must also be placed on maximizing device emission efficiency. In parallel with the photoluminescence quantum yield (PLQY), it is vital for emissive materials to be able to generate emission from both singlet and triplet excitons in the context of OLED operation. For 3rd generation OLEDs, upconversion of excitons from the non-emissive triplet state(s) to the first excited emissive singlet state via reverse intersystem crossing (rISC) allows this. Both rISC and consequent thermally activated delayed fluorescence (TADF) are supported by the available thermal energy of the environment, and become active when the energy difference between singlet and triplet states (or singlet–triplet energy gap, ΔEST) is minimized.15,16 The design of charge-transfer (CT) TADF emitters that achieve such small ΔEST typically relies on the use of electron-donating and electron-accepting units, which maintain a twisted conformation along their connecting bond(s).17 This gives rise to a minimal overlap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) localized on the donor and acceptor part, respectively, and hence a low exchange energy and small ΔEST.18 However, some degree of overlap is still required as this supports the singlet emission oscillator strengths and higher quantum yields.19 As such, careful and balanced molecular design and structural finetuning are both crucial for the development of novel high-performance TADF materials.
One strategy to potentially improve TADF properties entails the investigation of positional or regio-isomers via ‘isomeric modulation’.20–31 In the case of red TADF emitters, this generally involves the relocation of donor units and/or auxiliary acceptor groups (e.g. nitriles) on a rigid acceptor core and assessing how this affects the emission wavelength, PLQY, and TADF efficiency.32–35 The dibenzo[a,c]phenazinedicarbonitrile (DBPzCN) acceptor scaffold is often employed in the design of red TADF emitters, and has multiple synthetically accessible sites for donor/acceptor attachment (Fig. 1).36–45
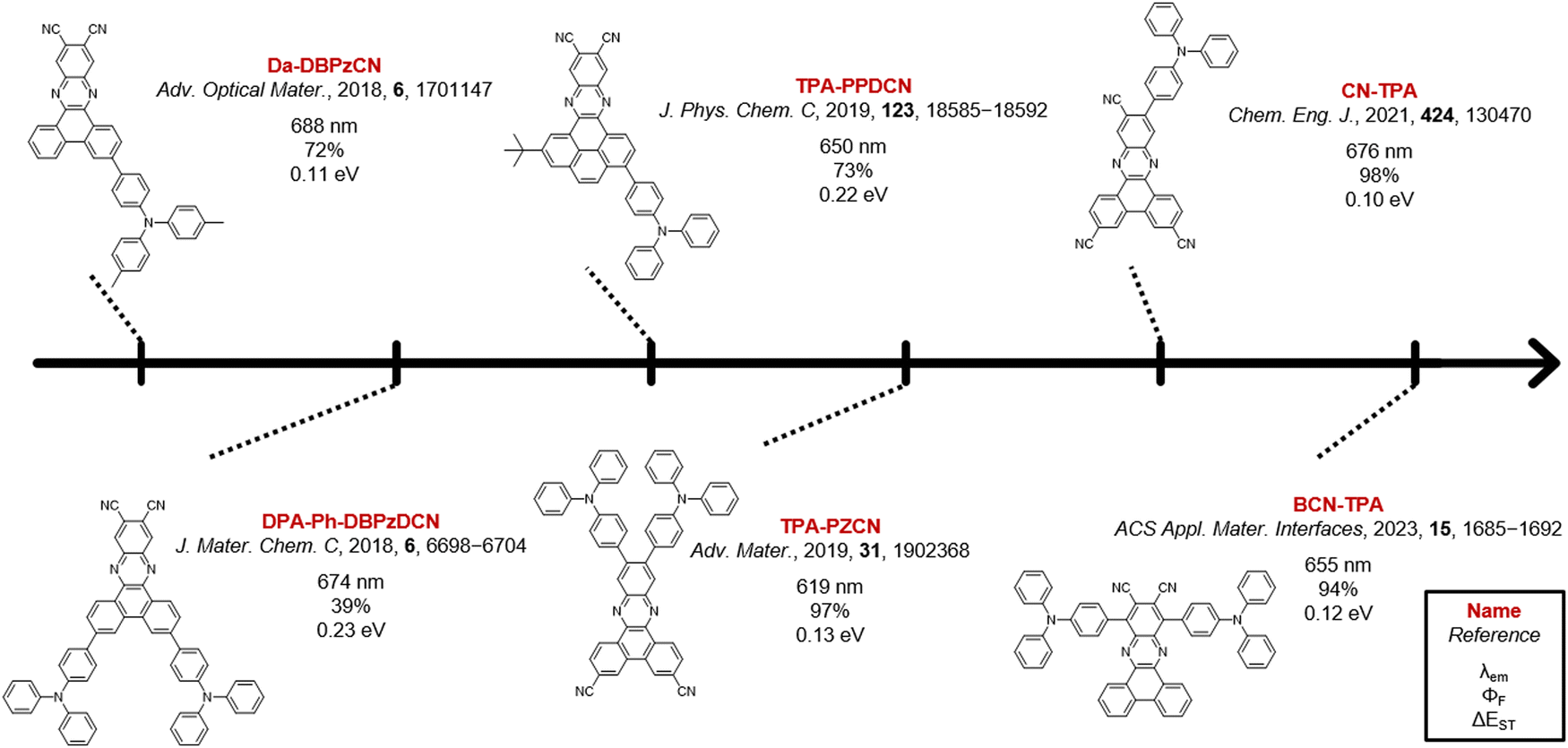 | ||
| Fig. 1 Examples of reported red TADF emitters based on the DBPzCN acceptor. The main photophysical characteristics (emission wavelength, fluorescence quantum yield, and singlet–triplet energy gap) are given for the materials in doped films. | ||
The extended π-conjugated system and significant rigidity of DBPzCN help to limit losses via non-radiative vibrational decay pathways.46 Additionally, the incorporation of the N-heterocyclic moiety and nitrile groups enhance the electron-withdrawing capability of the acceptor significantly, further redshifting the emission. By incorporating a pyrene moiety, Wang et al. and Shang et al. were recently able to access the previously unexplored 1(/8)-position of DBPzCN and thereby design novel TADF emitters.21,24 To expand upon this work and assess the full set of regio-isomers, we attach a donor unit on the remaining unexplored 4(/5)-position of DBPzCN and fully characterize and compare the emissive properties of the resulting novel materials (Fig. 2).
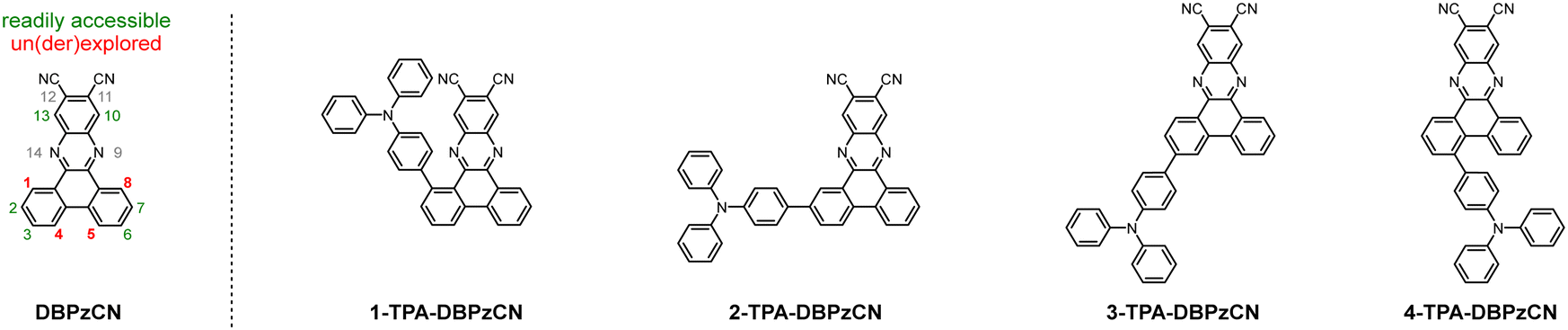 | ||
| Fig. 2 Structural overview of the DBPzCN template (with numbered core positions) and the explored emitters 1-TPA-DBPzCN, 2-TPA-DBPzCN, 3-TPA-DBPzCN, and 4-TPA-DBPzCN. | ||
In this study, four novel regio-isomers of (4-(diphenylamino)phenyl)dibenzo[a,c]phenazine-11,12-dicarbonitrile47 (TPA-DBPzCN; Fig. 2) are computationally analyzed, synthesized, and photophysically characterized. Triphenylamine (TPA) is employed as a strong electron-rich donor unit for which the precursor is readily available. Time-dependent density functional theory (TDDFT) calculations are used to simulate the molecular geometries of the molecules and derive their associated singlet and triplet energy levels. Following the preparation of the different bromo-substituted phenanthrene-9,10-dione precursors, two synthesis strategies are tested and utilized to obtain the final materials. Compounds 2-TPA-DBPzCN and 3-TPA-DBPzCN are readily synthesized from commercially available starting products, whereas 1-TPA-DBPzCN and 4-TPA-DBPzCN showcase the attachment of the donor unit on unconventional core positions. The four isomers are photophysically characterized in both solution and doped films, revealing significant differences between the isomers with regard to peak emission wavelength, PLQY, and TADF efficiency.
2. Results and discussion
2.1. Synthesis
2-Bromophenanthrene-9,10-dione and 3-bromophenanthrene-9,10-dione were purchased, while 1- and 4-bromophenanthrene were synthesized according to literature procedures and then oxidized to the respective phenanthrene-9,10-diones using chromium(VI) oxide (Scheme 1).48,49 Two different strategies – referred to as pathway ‘A’ and ‘B’ – were then investigated to synthesize the final materials. In pathway A, the brominated phenanthrene-9,10-diones were first coupled to the triphenylamine donor unit using a Suzuki–Miyaura cross-coupling reaction. Subsequently, the acquired TPA-substituted phenanthrene-9,10-diones were used in an acid-catalysed condensation reaction with 4,5-diaminophthalonitrile. This produced isomers 2-, 3-, and 4-TPA-DBPzCN in high yields, but was unsuccessful for the 1-TPA-DBPzCN isomer, even under harsher conditions (see ESI†, Section S2). This is thought to be a result of the large steric hindrance in combination with the strong electron-donating capacity of the TPA donor unit. Accordingly, pathway B was designed, in which the order of the previous reactions was reversed. Although this method is complicated by the limited solubility of the Br-DBPzCN intermediates, isomers 2- and 3-TPA-DBPzCN were nonetheless obtained in high yields. 1-TPA-DBPzCN was also successfully prepared, albeit in a lower yield of 20%, likely due to the still considerable steric hindrance and the poor solubility of the acceptor unit. The final materials underwent temperature-gradient vacuum sublimation to further purify them prior to their structural and photophysical characterization. Full details on the synthetic procedures and structural characterization data are provided in the Electronic ESI.†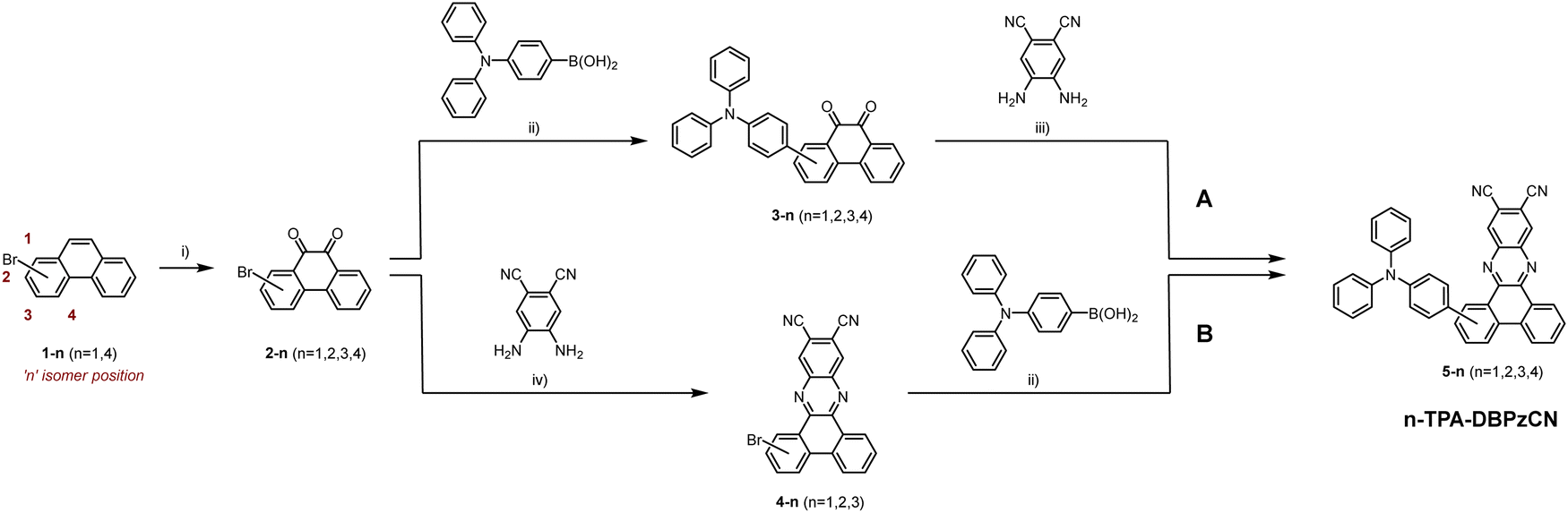 | ||
| Scheme 1 Synthesis protocol for 1-TPA-DBPzCN, 2-TPA-DBPzCN, 3-TPA-DBPzCN, and 4-TPA-DBPzCN: (i) CrO3, glacial acetic acid, reflux, 2 h; (ii) Pd(PPh3)4, Na2CO3, toluene, water, reflux, 24 h; (iii) HCl (37%), EtOH, THF, 40 °C, 24 h; (iv) glacial acetic acid, reflux, 24 h. | ||
2.2. Computational analysis and (TD)DFT calculations
DFT calculations using M06/6-311G(d) were performed to optimize the molecular geometries of isomer 1-, 2-, 3-, and 4-TPA-DBPzCN. Subsequently, singlet (S1–S2) and triplet (T1–T5) excited state energies were obtained using TDDFT calculations using a modified LC-BLYP (ω = 0.17 Bohr−1) exchange–correlation (XC) functional.50 These calculations were performed under the Tamm–Dancoff approximation51 (TDA) and the polarizable continuum model (PCM) in cyclohexane to simulate a non-polar environment.52 The Gaussian16 package was utilized to perform all calculations.53 The orbital spatial distributions were obtained from single-point calculations using the same LC-BLYP(17)/6-311G(d) method. The CT character of the involved states was investigated by looking at the differences between ground and excited state electron densities. These CT characters are described by the distance over which the electronic charge is transferred (dCT) and the related change in dipole moment (Δμ), calculated as described by Le Bahers and coworkers.54 Furthermore, the spin–orbit coupling (SOC) matrix elements were evaluated using the PySOC 23 program using the same XC functional, basis set, and PCM treatment as described above.55Optimization of the molecular geometries reveals clear similarities as well as differences in the relative orientation (and dihedral angle) of the TPA and DBPzCN subunits between the different isomers (Fig. 3). Molecules 2- and 3-TPA-DBPzCN give a smaller dihedral angle (θ ≈ 36°), not unexpected for a bond between TPA and a rigid but non-sterically constrained acceptor moiety.56 Accordingly, both isomers 2- and 3-TPA-DBPzCN retain a relatively planar conformation where the donor unit lies roughly in the same plane as the acceptor unit (Fig. S3, ESI†). Significant differences are observed for the geometry of 1- and 4-TPA-DBPzCN. The increased steric hindrance pushes the TPA away from the acceptor core in 1-TPA-DBPzCN, giving rise to a dihedral angle of around 71°. Meanwhile, 4-TPA-DBPzCN exhibits a slightly smaller steric repulsion with the acceptor core, resulting in a dihedral angle of 61°. Furthermore, both 1- and 4-TPA-DBPzCN are expected to assume a distorted conformation as the donor unit is pushed outside of the plane of the acceptor (Fig. S3, ESI†).
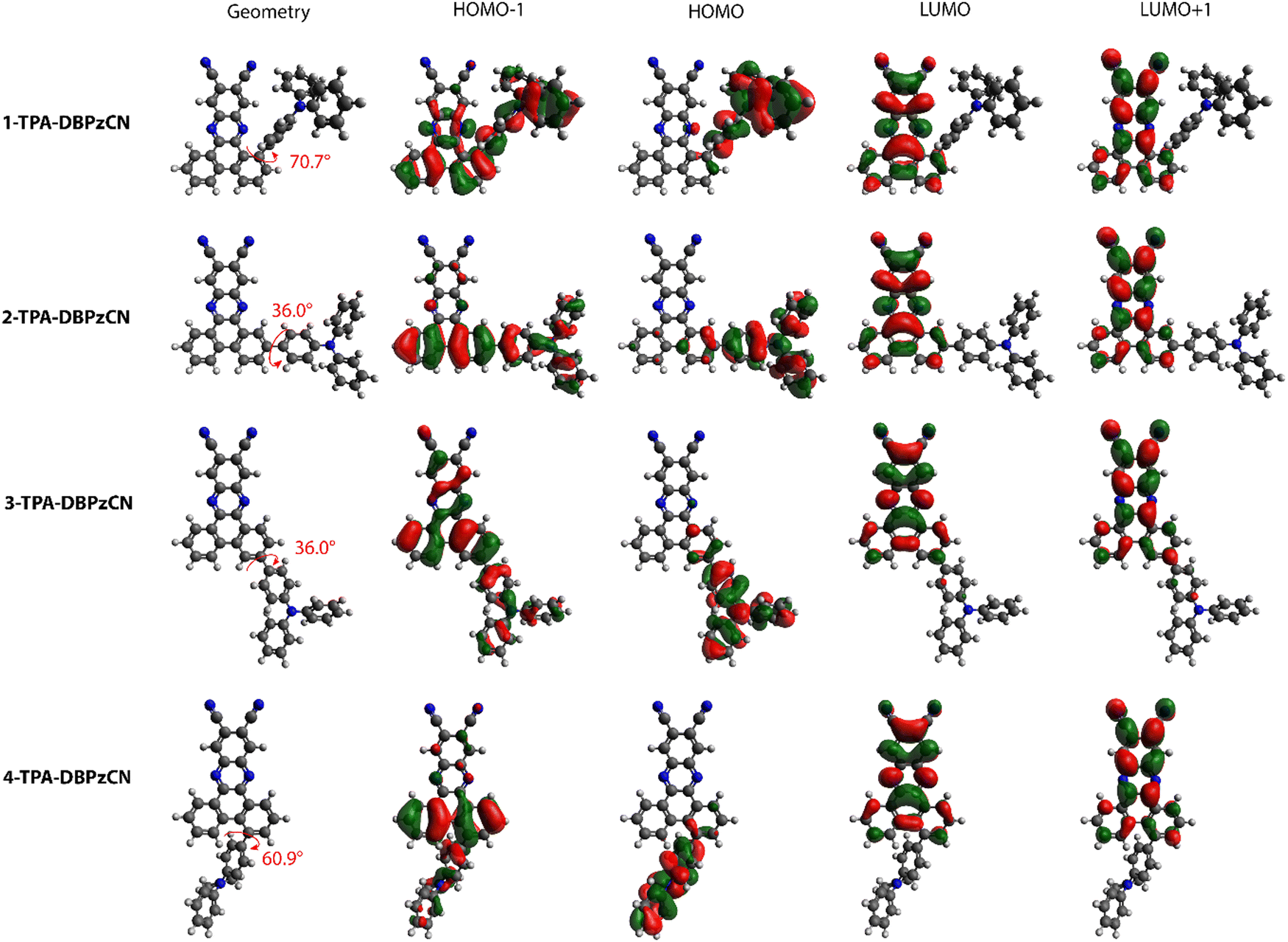 | ||
| Fig. 3 Optimized molecular geometries and orbital spatial distributions (LUMO+1, LUMO, HOMO, and HOMO−1) for 1-TPA-DBPzCN, 2-TPA-DBPzCN, 3-TPA-DBPzCN, and 4-TPA-DBPzCN. Values of dihedral angles are given in red. Isocontour values of 0.02 (a.u.) were used for all orbitals. | ||
Despite the clear differences in the calculated molecular geometries, the molecular orbital distributions are very similar for all four isomers. The HOMO is primarily localized on the TPA donor unit, whereas the LUMO and LUMO+1 can be found on the acceptor (Fig. 3). Superimposing the HOMO and LUMO distributions for each of the isomers shows a small overlap, thereby fulfilling the principal requirement of TADF molecular design. The main noteworthy difference in orbital distributions can be found for the HOMO−1. In 2- and 4-TPA-DBPzCN, this orbital is localized on the donor unit and the bottom part of the acceptor core, whereas 1- and 3-TPA-DBPzCN show a more delocalized orbital distribution onto the acceptor. As such, electron transitions that occur from the HOMO−1 might differ in their CT character.
From the TDDFT calculations, the nature, energy, and orbital distribution of the relevant excited states can be derived and compared. The first excited singlet states (S1) of all isomers are characterized mainly by a HOMO to LUMO transition, from which their CT character becomes apparent (Table S1, ESI†). Proper investigation of the CT character of other (higher-lying) excited states requires a more in-depth analysis of the ground and excited state electron density differences (Table S1, S2 and Fig. S1, ESI†). These show the change of electron density in a molecule upon a transition from the ground state to a specific excited state. Changing electron densities in both the donor and acceptor unit (decreasing and increasing, respectively) are thus indicative of the CT nature of the excited state. When the changing electron densities are confined to the same unit (similar molecular distribution), the energy state is characterized as a locally excited (LE) state instead. Additionally, the strength of the spin–orbit coupling between a singlet and triplet state can be determined from the calculated SOC matrix elements. Together with the relative positions and orderings of the energy levels and the singlet–triplet gap, these values help to express the likelihood of a transition to occur between two specific states. A comprehensive excited state energy diagram is presented in Fig. 4, and also tabulated in the ESI† (Table S3, ESI†).
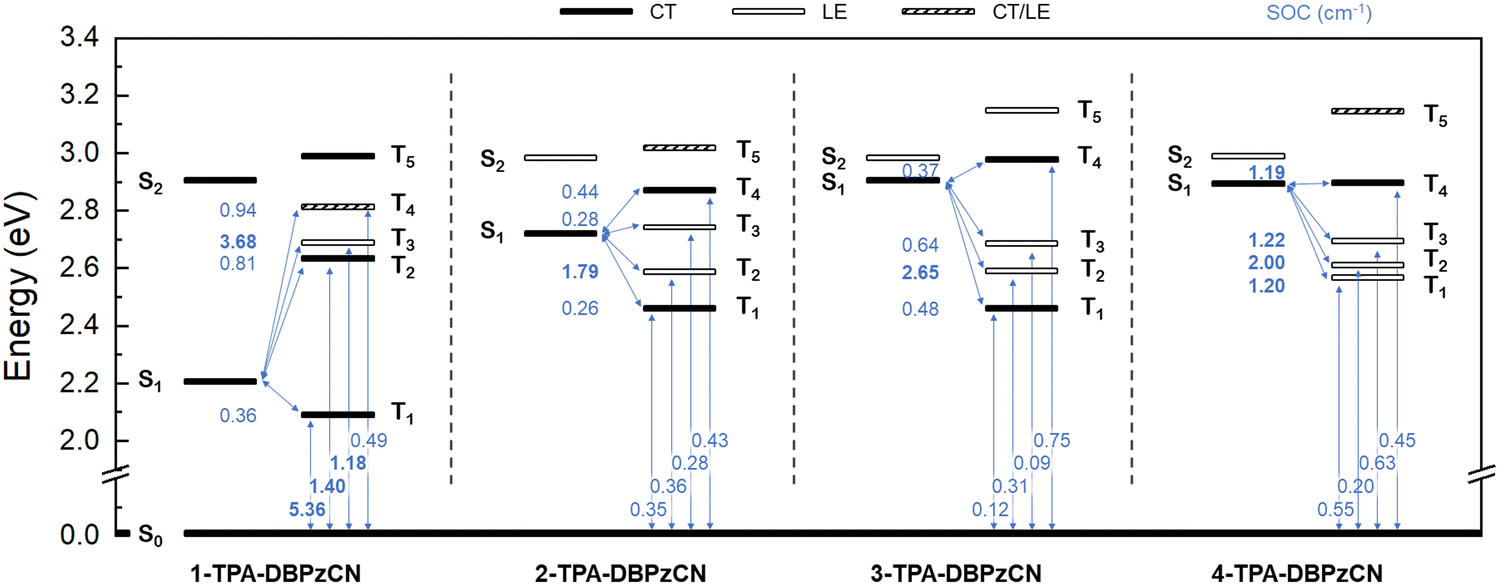 | ||
| Fig. 4 Schematic overview of the excited state energy levels (in eV) of all four isomers with their corresponding CT (black), LE (white), and mixed CT/LE (dashed) character as obtained through TDDFT calculations. Relevant S0–Tx and S1–Tx (x = 1–5) couplings and SOC values are shown in blue (SOC values > 1 cm−1 in bold). | ||
1-TPA-DBPzCN shows an interesting set of first excited (singlet and triplet) states which are relatively low in energy and noticeably separated from any higher states. With a small simulated ΔEST of 0.14 eV, good triplet upconversion could be envisaged. However, it is known that spin–orbit coupling between 1CT and 3CT states is forbidden under the requirement of total angular momentum conservation.57 Instead, intersystem crossing occurs more readily between 1CT and 3LE states (according to the El Sayed rule58), and is promoted by vibronic coupling (VC) in case of rISC for D-A TADF materials.59–61 As no neighbouring 3LE states are present in proximity to T1 (3CT) to enhance upconversion via vibronic coupling, T1 is expected to act as an exciton sink, despite the relatively small ΔEST.59 This is further corroborated by the large SOC value for the transition from T1 to the ground state (Fig. 4), allowing non-radiative deactivation and preventing efficient TADF.
A further comparison can be made between isomers 2-, 3-, and 4-TPA-DBPzCN. The theoretical ΔEST decreases from 0.45 eV in 3-TPA-DBPzCN to 0.32 eV in 4-TPA-DBPzCN, and down to 0.27 eV in 2-TPA-DBPzCN (Table S3, ESI†). In 2-TPA-DBPzCN, upconversion from T1 could be further enhanced by the presence of close-lying 3LE states through vibronic coupling, with T2 (LE) showing improved SOC and T3 (LE) being nearly isoenergetic to S1.62 Intermediate 3LE states between S1 and T1 are also present in 3-TPA-DBPzCN, but these exhibit a much larger gap to S1, which weakens the potential contribution of spin–vibronic coupling.63 Similarly to 2-TPA-DBPzCN, close-lying 3LE states and an S1-isoenergetic triplet state are also observed in 4-TPA-DBPzCN. Furthermore, it is also noted that 4-TPA-DBPzCN is the only isomer that has been simulated to have a T1 state with LE character. This could allow SOC and upconversion to S1 to compete with the decay from T1 to the ground state, thereby diminishing non-radiative losses.64 Furthermore, relatively large SOC values are observed for the transition between S1 and T1 (LE), T2 (LE), T3 (LE), and even T4 (CT), despite its apparent CT character. In this case, upconversion could occur directly from T1 (LE) – and potentially T2 (LE) and T3 (LE) – to S1 (CT) through a combination of SOC and VC.59,63 Alternatively, as the ΔEST of 4-TPA-DBPzCN is relatively large, second-order non-adiabatic coupling might occur between T1 (LE) and T4 (CT).65
2.3. Photophysical characterization
Steady-state absorption and emission spectra were measured in toluene and zeonex host (Fig. 5). Several high-energy absorption bands can be observed below 450 nm for all 4 isomers in toluene, which are attributed to the π–π* and n–π* transitions occurring in the TPA and DBPzCN moieties.47 The peak at around 360 nm is less defined for 1- and 4-TPA-DBPzCN, likely due to the distorted molecular geometry of the TPA unit. A relatively broad peak can be observed at longer wavelengths (>450 nm) which corresponds to the CT absorption band. The relative intensity of this CT band correlates with the calculated value of the oscillator strength for each isomer (3 ≫ 4 ≈ 2 > 1), which can be found in the ESI† (Table S3, ESI†). The emission spectra in toluene are comparable for each isomer, with exception of the shifted emission peak maximum for 2- and 4-TPA-DBPzCN compared to 1- and 3-TPA-DBPzCN (Fig. 5).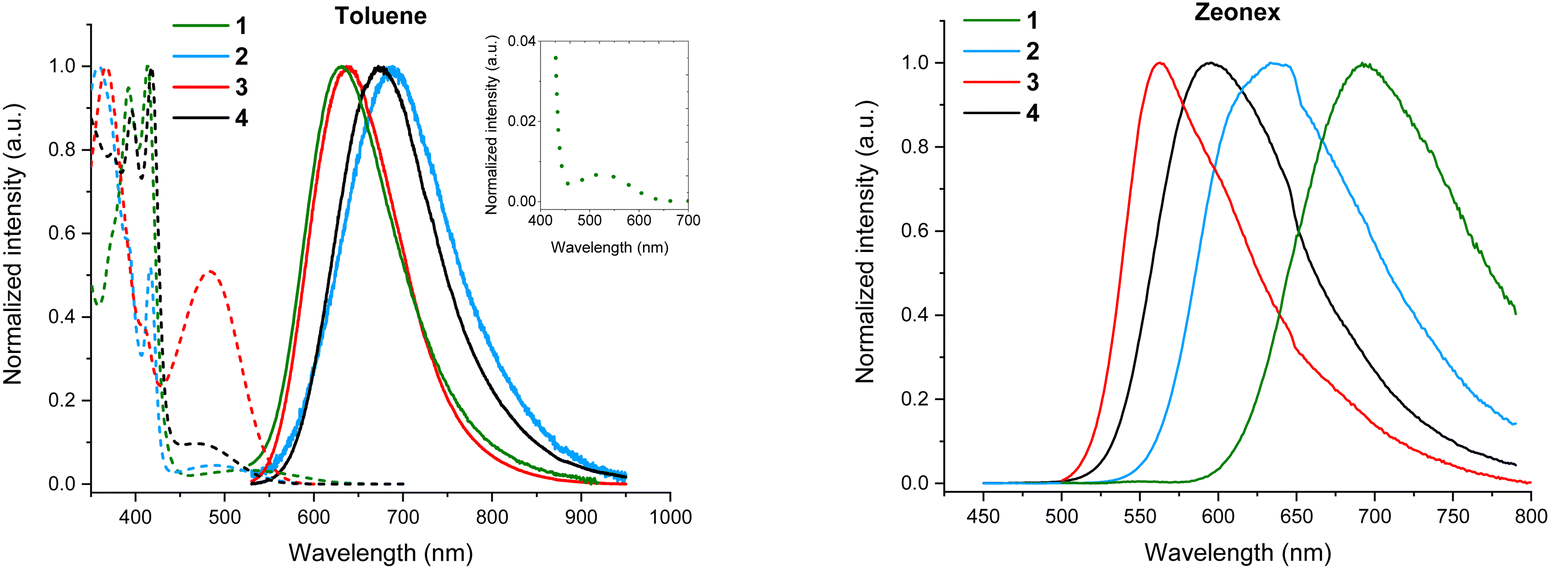 | ||
| Fig. 5 Normalized steady-state absorption (dashed lines) and emission spectra (solid lines) for all four isomers in 10−5 M toluene solution (left, λexc = 440 nm) and emission spectra for 0.1 w/w% zeonex solid films (right, λexc = 440 nm). Graph inset (left) shows the magnified absorption spectrum of 1-TPA-DBPzCN, revealing a weak CT absorption band. The inflexion around 650 nm in the emission spectra of all four isomers in zeonex film (right) is attributed to the onset of significant instrument-specific intensity correction at longer wavelengths. | ||
The fluorescence quantum yields (ΦF) of all four isomers were determined in toluene solution to ascertain and compare their luminescence efficiency (Table 1). These measurements were performed under inert (ΦF,inert) and ambient air conditions (ΦF,air) to evaluate the impact of an oxygen-quenched triplet upconversion mechanism (Fig. S4, ESI†). The quantum yields in toluene solution are relatively low, with the notable exception of 3-TPA-DBPzCN. The fluorescence improves from the “cis” molecular geometry in 1-TPA-DBPzCN (ΦF,air = 0.01) to the intermediate situation for 2-TPA-DBPzCN (ΦF,air = 0.05) to the “trans” molecular geometry in 3-TPA-DBPzCN (ΦF,air = 0.41). The unconventional geometry of 4-TPA-DBPzCN results in an intermediate quantum yield (ΦF,air = 0.11). Meanwhile, the quantum yields in oxygen-free toluene increase in tandem with the calculated oscillator strengths, improving the most for 3-TPA-DBPzCN (ΦF,inert = 0.52). Singlet oxygen quantum yields (ΦΔ) were also determined in toluene solution, to investigate the formation of triplet states under standard air atmosphere (Fig. S5, ESI†). Molecular oxygen (which exists in the triplet ground state) can quench the populated excited triplet states of emissive materials, quantitatively forming singlet oxygen. As such, the higher singlet oxygen quantum yield of 3-TPA-DBPzCN (ΦΔ = 0.58) is indicative of the formation of longer-lived triplet states which can undergo some rISC in solution.
| Compound | λ CT abs (nm) | λ em (nm) tol/zeonex | λ em (nm) CBP/DPEPO | Φ F, toluene air/inert | Φ F, zeonex air/inert | Φ Δ | HOMO/LUMOg (eV) |
|---|---|---|---|---|---|---|---|
| a CT band absorption maxima in toluene solution. b Fluorescence emission maxima in 10−5 M toluene solution and zeonex film (λexc = 440 nm). c Fluorescence emission maxima in CBP and DPEPO film (λexc = 440 nm). d Photoluminescence quantum yields in toluene solution under air and inert atmosphere relatively determined vs Coumarin 153 (ΦF = 0.38, λexc = 440 nm in ethanol). e Absolute photoluminescence quantum yields in zeonex determined using an integrating sphere under air and inert atmosphere at room temperature (λexc = 440 nm). f Singlet oxygen quantum yields in toluene solution determined vs coronene (ΦΔ = 0.90, λexc = 325 nm in toluene) by monitoring the absorbance of 1,3-diphenylisobenzofuran at 414 nm under emission from a secondary light source. g Determined from cyclic voltammetry (ESI, Section S7). | |||||||
| 1-TPA-DBPzCN | 536 | 632/692 | 707/727 | 0.01/0.02 | <0.01/<0.01 | 0.27 | −5.45/−3.61 |
| 2-TPA-DBPzCN | 495 | 694/634 | 641/700 | 0.05/0.07 | 0.17/0.24 | 0.30 | −5.51/−3.71 |
| 3-TPA-DBPzCN | 484 | 630/564 | 660/700 | 0.41/0.52 | 0.47/0.46 | 0.58 | −5.54/−3.64 |
| 4-TPA-DBPzCN | 480 | 672/592 | 641/667 | 0.11/0.14 | 0.49/0.62 | 0.39 | −5.57/−3.75 |
Additional steady-state absorption and emission spectra were measured in methylcyclohexane (MCH) and chloroform (CF) to investigate solvatochromic effects (Fig. S6, ESI†). The inherent broadness and observed red-shift of the emission bands in more polar solvents suggest a CT character for the emissive singlet state, as predicted by the TDDFT calculations. In chloroform, the strong stabilization of the CT state shifts the emission band beyond 700 nm, giving rise to NIR emission.
Next, the solid-state emission properties of doped films were investigated. Since the emitters show very low solubility (∼0.1 mg mL−1 in toluene) and hence an assumed tendency to aggregate, low doping concentrations were utilized throughout. Separately, considerable acceptor π-stacking (interplanar distances of 3.61 Å) was observed in the single crystal X-ray structure of 3-TPA-DBPzCN (the only material in this series that yielded usable crystals, Fig. S9, ESI†), lending credence to this assumed propensity towards aggregation. Firstly, 0.1 w/w% films in zeonex host were prepared. Zeonex has a non-conjugated polymer structure, allowing for the creation of smooth, homogenous films. Additionally, it exhibits a low polarizability (similar to MCH), which limits its influence on the stabilization of the CT states.66 It is therefore often used to investigate the intrinsic solid-state TADF properties of novel compounds. When going from the more polar toluene solution to a zeonex film, a blue-shift in the emission maximum is typically observed (Fig. 5). This is true for zeonex films of 2-, 3-, and 4-TPA-DBPzCN. Only 1-TPA-DBPzCN shows a significantly red-shifted emission in zeonex, which can be attributed to its large tendency to aggregate (as correlated with its exceptionally poor solubility). The luminescence efficiencies are noticeably different in the zeonex host. Compared to in toluene solution, the quantum yields remain roughly the same for 1-TPA-DBPzCN (ΦF,air = <0.01) and 3-TPA-DBPzCN (ΦF,air = 0.47), while a decent increase is observed for 2-TPA-DBPzCN (ΦF,air = 0.17) and especially 4-TPA-DBPzCN (ΦF,air = 0.49). With the exception of 3-TPA-DBPzCN, the quantum yield is even more enhanced in zeonex film when applying an inert atmosphere. This suggests an improvement of the triplet upconversion and the presence of a delayed fluorescence component, most pronounced for 2-TPA-DBPzCN (ΦF,inert = 0.24) and 4-TPA-DBPzCN (ΦF,inert = 0.62).
Two additional host materials were selected to investigate the effects of host nature and polarizability on the solid-state emission properties: 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP, ET = 2.6 eV)67 and bis[2-(diphenylphosphino)phenyl]ether oxide (DPEPO, ET = 3.0 eV).68 These small-molecule hosts are widely used in the development of OLEDs because of their excellent energy alignment and respective high hole or electron mobility. As the polar environment further stabilizes the CT emissive state relative to zeonex, a significant red-shift is observed in both 1 w/w% CBP and 1 w/w% DPEPO films, pushing the solid-state emission closer to the NIR (Fig. S10, ESI† and Table 1). Additional high-energy emission bands are revealed in both CBP and DPEPO films by changing the excitation wavelength from 400/450 to 350 nm. In CBP, these bands originate from (partial) excitation of the host, as confirmed by the measurement of a pure CBP film (Fig. S10, ESI†).69 In DPEPO, it is possible that these signals arise from a distorted conformation which might become more prevalent in the strongly polar host. The higher relative intensity of these undesired emission bands in 1-TPA-DBPzCN highlights its poor CT emission character, likely caused by the unfavourable twisted “cis”-geometry of the molecular structure.24
Time-resolved emission spectroscopy (TRES) experiments were then done to investigate the time-dependent emission mechanisms of the four emitters in zeonex films (as well as in CBP and DPEPO). Contour plots of the normalized time-resolved emission spectra of the zeonex films are shown in Fig. 6, while additional contour plots of the CBP and DPEPO films are provided in Fig. S11 and S12 (ESI†). For TADF emitters, three distinct emission phenomena can generally be identified at separate timescales, i.e. prompt fluorescence in the nanoseconds regime, delayed fluorescence in the micro- to milliseconds regime, and phosphorescence at the late milliseconds to seconds regime. Since classical phosphorescence is a forbidden transition, it is typically outperformed by non-radiative decay and thereby too weak to be observed at room temperature. Therefore, measurements are also done at 80 K to limit vibrational energy losses and analyse the phosphorescence emission to determine experimental T1 energies.
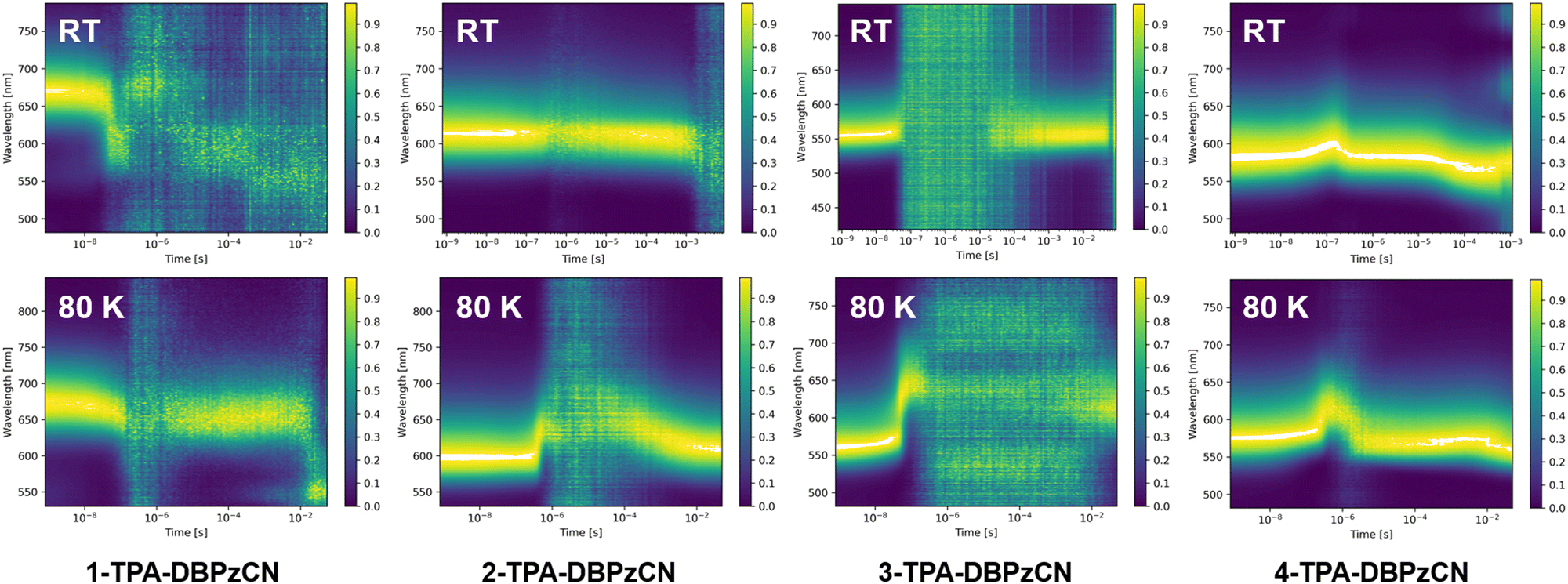 | ||
| Fig. 6 Normalized time-resolved emission spectra (contour plots) for all isomers in 0.1 w/w% zeonex films (λexc = 355 nm) at room temperature (top row) and 80 K (bottom row). | ||
At room temperature, initial fluorescence emission remains relatively constant for all four isomers up until a few hundred nanoseconds, when a small red-shift is observed for 3- and (most clear for) 4-TPA-DBPzCN. This shift is often attributed to the slower emission decay from shifted excited singlet states with slightly different molecular geometries, typically as a result of a changing dihedral angle between the donor and acceptor unit.70,71 At 80 K, this shift can be more pronounced due to the (limited) rotational freedom of the phenyl moieties of the TPA donor unit. For 1-TPA-DBPzCN, a rather unusual blue-shift appears to be observed at room temperature. However, investigation of the time-resolved emission spectra reveals the presence of a weak, second emission band which starts to dominate after the decay of the main prompt emission. Possibly linked to some rare molecular conformation of 1-TPA-DBPzCN, this band might only become visible due to the low PLQY of the material's primary emission channel.
In the micro- to milliseconds regime, delayed emission is observed at the same wavelength as the prompt emission for 2-, 3-, and 4-TPA-DBPzCN. This emission likely originates from the same singlet state (i.e. S1), suggesting TADF. Isomers 2- and especially 4-TPA-DBPzCN show relatively strong delayed fluorescence which persists for the entire time window of the measurement, while the delayed emission of 3-TPA-DBPzCN dips below the detection limit of the instrument for most of the early microseconds regime. Meanwhile, 1-TPA-DBPzCN shows only delayed fluorescence intensity comparable with the baseline of the instrument, again likely resulting from the extremely poor PLQY of this particular material.
At 80 K, the delayed emission of 2-, 3-, and 4-TPA-DBPzCN partially or completely disappears in the microseconds timeframe, as the triplet upconversion mechanism becomes thermally less accessible. Instead, a (small) red-shifted emission can be seen for 2- and 3-TPA-DBPzCN at a later timescale which represents the phosphorescence. Contrarily, a small blue-shift is observed at a later timescale for 4-TPA-DBPzCN, likely due to the LE nature of the T1 state (as predicted by TDDFT). This is experimentally confirmed by the phosphorescence spectrum at 80 K, which shows some vibronic fine structure (Fig. S13, ESI†). Since residual delayed fluorescence and phosphorescence are visible at roughly the same wavelength as the prompt fluorescence in the contour plot of 4-TPA-DBPzCN at 80 K, a minimal ΔEST is expected. This gap can be experimentally determined by subtracting the onset of the steady-state fluorescence spectrum at room temperature and the 80-millisecond delay-time phosphorescence spectrum at 80 K (Fig. S13, ESI† and Table 2). The smaller gap between the fluorescence and phosphorescence correlates with the increased intensity of the microsecond delayed fluorescence in the contour plots of 2-, 3- and 4-TPA-DBPzCN in zeonex film at room temperature. As such, this supports our previous hypothesis of TADF being the proposed triplet upconversion mechanism.
| Compound | ΔESTa (eV) | k F (107 s−1) | k ISC (106 s−1) | k rISC (105 s−1) |
|---|---|---|---|---|
| a Determined by taking the difference of the onset of the steady-state fluorescence band at room temperature and the 80-milliseconds delay-time phosphorescence band at 80 K. b Derived by applying a kinetic fitting model to the total emission decay curves.72 c Could not be determined due to absence of measurable phosphorescence. d Could not be determined due to the absence of measurable delayed fluorescence. | ||||
| 1-TPA-DBPzCN | /c | 2.46 | /d | /d |
| 2-TPA-DBPzCN | 0.027 | 0.91 | 9.33 | 0.19 |
| 3-TPA-DBPzCN | 0.124 | 3.23 | 3.69 | 0.12 |
| 4-TPA-DBPzCN | ∼0 | 1.35 | 18.3 | 1.59 |
Meanwhile, 2-, 3- and 4-TPA-DBPzCN show rather comparable contour plot profiles in both CBP and DPEPO (Fig. S11 and S12, ESI†). Here, the CT emission band is rather poorly defined due to the limited luminescence intensity which drops below the noise baseline of the instrument at longer emission lifetimes. Unfortunately, the contour plots of 1-TPA-DBPzCN in particular become quite unusable for characterising the CT emission bands in both CBP and DPEPO, as the high energy bands become more dominant.
Further comparison between the delayed emission properties can be made by plotting the total emission decay curves of the four isomers in zeonex. Fig. 7 shows the decrease of the total photon-count over time. The first descending curve represents the prompt fluorescence decay, whereas the second part shows the delayed fluorescence decay as a result of (delayed) triplet to singlet upconversion. The prompt fluorescence decay appears largely similar for all isomers, while the strongest and most rapid delayed emission component is observed for 4-TPA-DBPzCN. The relative contribution of the delayed emission to the total photon-count remains largely consistent between 2-, 3-, and 4-TPA-DBPzCN in CBP and DPEPO (Fig. S15, ESI†). However, the overall slope decreases when moving from zeonex to CBP and DPEPO films as a result of the larger polarizability and structural rigidity of the hosts.66 For 1-TPA-DBPzCN, only the prompt fluorescence decay could be plotted. This emission quickly disappears in zeonex and DPEPO, while a longer prompt emission is observed in CBP film.
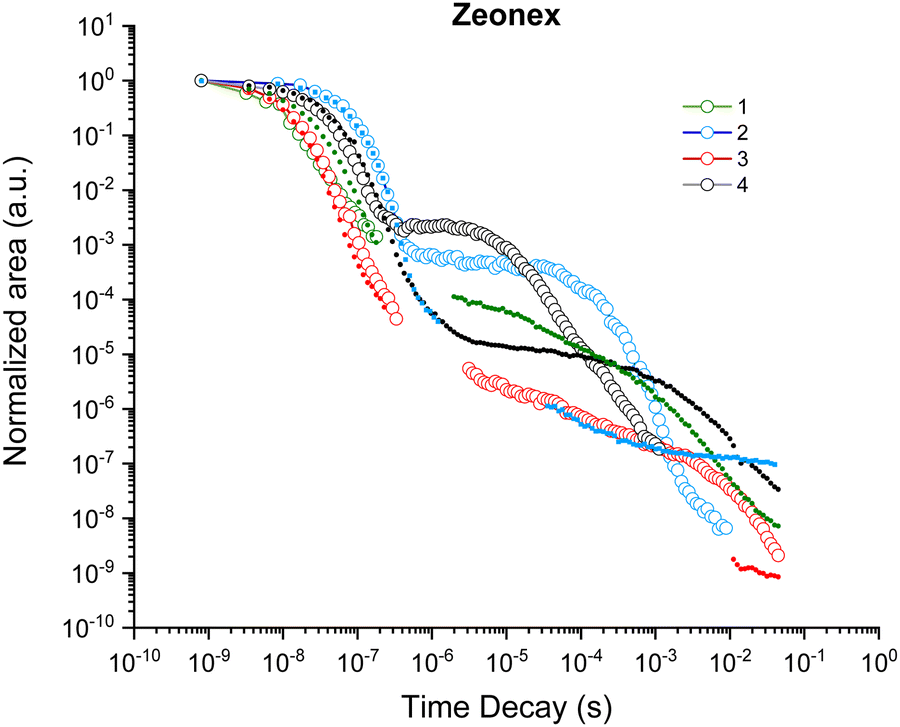 | ||
| Fig. 7 Total emission decay (calculated via the integrated area under the emission curve) for 1-TPA-DBPzCN (green), 2-TPA-DBPzCN (blue), 3-TPA-DBPzCN (red), and 4-TPA-DBPzCN (black) at room temperature (open circles) and 80 K (closed circles). Data points with signal below the noise baseline have been omitted from the figure. | ||
Applying a kinetic fitting model to the double decay curve allows for the determination of important kinetic properties such as the fluorescence rate contant (kF) and both the standard and reversed intersystem crossing rate constants (kISC and krISC, respectively).72 The fitted values for these parameters are given in Table 2 (and Table S7, ESI† for CBP and DPEPO). Highest values for kF are observed for 3-TPA-DBPzCN and correspond to faster prompt fluorescence. Meanwhile, the kISC of 3-TPA-DBPzCN appears to be smaller than those of 2- and 4-TPA-DBPzCN, indicating slower and less overall formation of populated triplet states following photo-excitation. This is particularly the case in zeonex films, where the largest differences between 3-TPA-DBPzCN and the other isomers are apparent. Its derived higher kF and lower kISC can be further corroborated by its high PLQY in film, which remains the same in both ambient air and inert atmosphere.
The most important difference in the kinetic properties of the isomer series can be found in the krISC values. krISC improves significantly going from 3-TPA-DBPzCN (∼1 × 104 s−1) to 2-TPA-DBPzCN (∼2 × 104 s−1) and finally to 4-TPA-DBPzCN (∼16 × 104 s−1) in zeonex. Since the rate (and efficiency) of the rISC process are intrinsically linked to the size of the singlet–triplet energy gap (in addition to the nature of the relevant states), a relative increase in krISC should be expected when the ΔEST is minimized further. This trend is clearly illustrated in Table 2, where the minimal ΔEST of 4-TPA-DBPzCN results in it possessing the best triplet upconversion in comparison to the larger ΔEST of 3-TPA-DBPzCN. The substantially higher k(r)ISC values of 4-TPA-DBPzCN in zeonex can be explained by the presence of its unique T1 (LE) state. The LE nature of its triplet state enables faster (r)ISC through more efficient SOC with the CT S1 state, in accordance to the El Sayed rule.58 Interestingly, 2-TPA-DBPzCN seems to express better TADF than 3-TPA-DBPzCN, despite their seemingly identical molecular conformations, with the 3-TPA-DBPzCN exhibiting a faster PF and an oxygen invariant film PLQY more reminiscent of purely fluorescent chromophores. Since 4-TPA-DBPzCN also shows the largest increase in PLQY from air to inert atmosphere in zeonex, it clearly stands out as the best TADF emitter among the investigated regio-isomer series. Considering the rare utilisation of this substitution position in existing reports of TADF materials using this and similar acceptors, we envisage that the discovery of superior TADF in 4-TPA-DBPzCN compared to its regio-isomers will stimulate further investigation of similarly substituted materials in future. A final comparison of the main emission characteristics for the four isomer materials in zeonex can be found in Fig. 8.
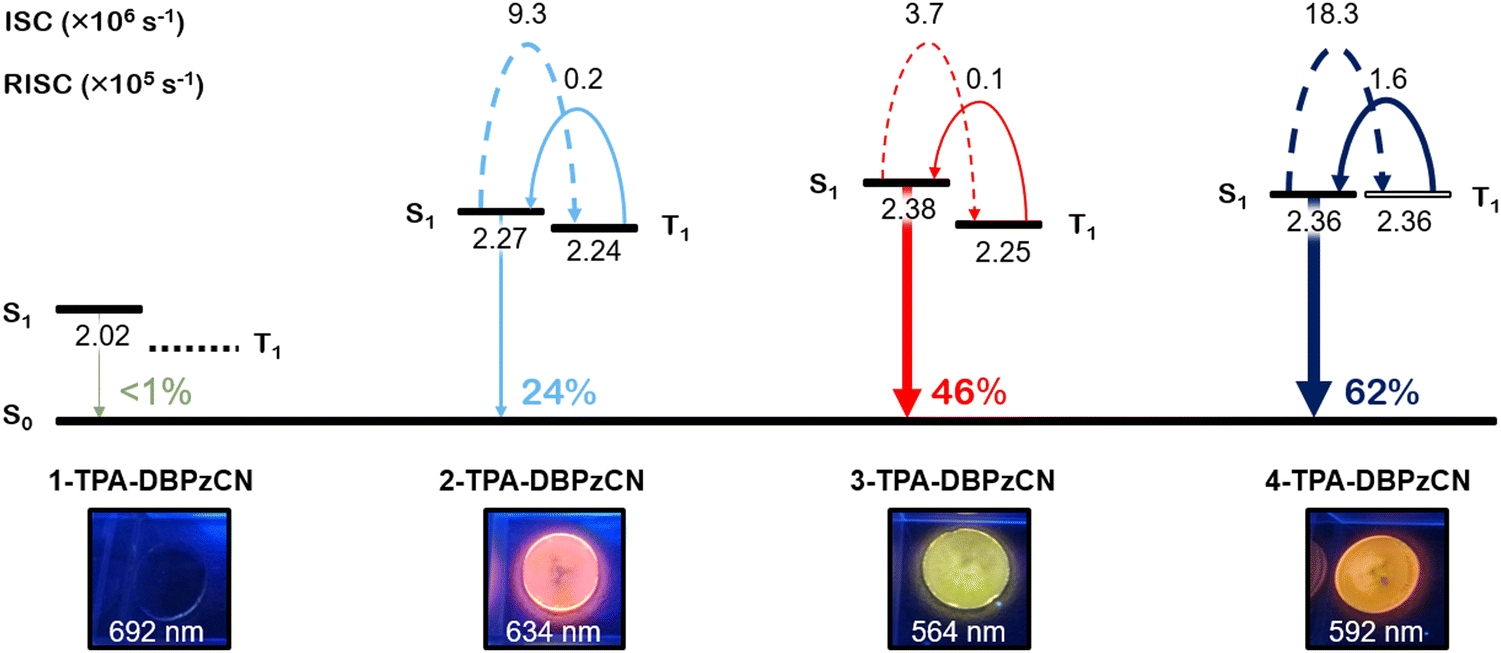 | ||
| Fig. 8 Schematic overview of the main emission characteristics of 1-TPA-DBPzCN (green), 2-TPA-DBPzCN (blue), 3-TPA-DBPzCN (red), and 4-TPA-DBPzCN (black) in 0.1 w/w% zeonex films. The relative positions of the first excited singlet and triplet states are shown according to their experimentally determined energies (in eV), with the exception of the triplet state of 1-TPA-DBPzCN. The corresponding CT (black) or LE (white) character of the excited state is also indicated. The photoluminescence quantum yields are expressed in percentages. | ||
3. Conclusions
In this work, the influence of the regio-isomeric position of a triphenylamine donor on the standard and delayed emission properties of TPA-DBPzCN was investigated. Changes in relative orientation between the donor and acceptor subunits have a profound effect on the emission wavelength, photoluminescence quantum yield, and delayed fluorescence rate and efficiency. The twisted conformation and larger dihedral angles in 1-TPA-DBPzCN and 4-TPA-DBPzCN play an important role in changing their emission characteristics. Notwithstanding, different behaviour is even observed between 2-TPA-DBPzCN and 3-TPA-DBPzCN, which exhibit the same dihedral angle according to the TDDFT calculations. While 1-TPA-DBPzCN shows the desired strongly red-shifted emission in different host materials, its emission is largely quenched. This is mainly attributed to the unfavourable molecular “cis” conformation and extreme twisting of the donor unit. Additionally, this conformation seems to give rise to a low-energy triplet trap state with large spin–orbit coupling to the ground state. 2-TPA-DBPzCN shows surprisingly decent TADF characteristics despite its limited dihedral angle, possibly explained by a suitable alignment of both theoretical and experimental triplet and singlet energy states. Meanwhile, 3-TPA-DBPzCN shows noticeably different emission behaviour. A relatively high fluorescence quantum yield is attributed to the favorable molecular “trans” conformation and larger oscillator strength. Unfortunately, the emission red-shift and TADF characteristics are rather poor in zeonex film, in which it acts more similarly to typical fluorescent dyes. Finally, 4-TPA-DBPzCN is revealed as the overall best TADF emitter in this regio-isomer series. In zeonex, it exhibits the fastest (r)ISC rates and highest (increase in) quantum yield through improved S1–T1 spin–orbit coupling as a result of its near-zero ΔEST and unique 3LE first excited triplet state. This series therefore showcases the importance of investigating underexplored structural motifs and substitution positions in progressing the design and properties of TADF emitters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Research Foundation – Flanders (FWO Vlaanderen) for financial support through projects G087718N, G0D1521N, I006320N, GOH3816NAUHL, the Scientific Research Community ‘Supramolecular Chemistry and Materials’ (W000620N), postdoctoral fellowship 1284623N (T. Cardeynaels), and PhD scholarship 1SC8621N (S. Brebels). The calculations were performed on the computers of the ‘Consortium des équipements de Calcul Intensif (CÉCI)’ (https://www.ceci-hpc.be), including those of the ‘UNamur Technological Platform of High-Performance Computing (PTCI)’ (https://www.ptci.unamur.be), for which we gratefully acknowledge financial support from the FNRS-FRFC, the Walloon Region, and the University of Namur (Conventions No. GEQ U.G006.15, U.G018.19, U.G011.22, RW/GEQ2016, RW1610468, and RW2110213). S. Kuila and A. P. Monkman are supported by EPSRC grant EP/T02240X/1. We also thank Dr Dmitry S. Yufit of the Durham crystallography service for XRD measurements and technical support.References
- E.-L. Hsiang, Z. Yang, Q. Yang, Y.-F. Lan and S.-T. Wu, J. Soc. Inf. Disp., 2021, 29, 446–465 CrossRef.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- M. Vasilopoulou, A. Fakharuddin, F. P. García de Arquer, D. G. Georgiadou, H. Kim, A. R. B. Mohd Yusoff, F. Gao, M. K. Nazeeruddin, H. J. Bolink and E. H. Sargent, Nat. Photonics, 2021, 15, 656–669 CrossRef CAS.
- A. Minotto, P. A. Haigh, G. Łukasiewicz, E. Lunedei, D. T. Gryko, I. Darwazeh and F. Cacialli, Light: Sci. Appl., 2020, 9, 70 CrossRef PubMed.
- Y. Khan, D. Han, A. Pierre, J. Ting, X. Wang, C. M. Lochner, G. Bovo, N. Yaacobi-Gross, C. Newsome, R. Wilson and A. C. Arias, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E11015–e11024 CAS.
- C.-H. Lin, K. S. Karim and Y.-H. Tai, SID Symposium Digest of Technical Papers, 2020, 51, pp. 1855–1858.
- C. Lian, M. Piksa, K. Yoshida, S. Persheyev, K. J. Pawlik, K. Matczyszyn and I. D. W. Samuel, npj Flexible Electron., 2019, 3, 18 CrossRef.
- F. Zhang and B. Z. Tang, Chem. Sci., 2021, 12, 3377–3378 RSC.
- S. Qi, S. Kim, V.-N. Nguyen, Y. Kim, G. Niu, G. Kim, S.-J. Kim, S. Park and J. Yoon, ACS Appl. Mater. Interfaces, 2020, 12, 51293–51301 CrossRef CAS PubMed.
- F. Ni, N. Li, L. Zhan and C. Yang, Adv. Opt. Mater., 2020, 8, 1902187 CrossRef CAS.
- Y. Yamaguchi, Y. Matsubara, T. Ochi, T. Wakamiya and Z.-I. Yoshida, J. Am. Chem. Soc., 2008, 130, 16442 CrossRef CAS.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- C. Adachi, Jpn. J. Appl. Phys., 2014, 53, 060101 CrossRef.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- X.-K. Chen, CCS Chem., 2020, 2, 1256–1267 CrossRef CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- M. K. Etherington, F. Franchello, J. Gibson, T. Northey, J. Santos, J. S. Ward, H. F. Higginbotham, P. Data, A. Kurowska, P. L. Dos Santos, D. R. Graves, A. S. Batsanov, F. B. Dias, M. R. Bryce, T. J. Penfold and A. P. Monkman, Nat. Commun., 2017, 8, 14987 CrossRef CAS PubMed.
- T. Yang, Z. Cheng, Z. Li, J. Liang, Y. Xu, C. Li and Y. Wang, Adv. Funct. Mater., 2020, 30, 2002681 CrossRef CAS.
- T. Yang, J. Liang, Y. Cui, Z. Li, X. Peng, S.-J. Su, Y. Wang and C. Li, Adv. Opt. Mater., 2023, 11, 2201191 CrossRef CAS.
- C. Zhou, Y. Liu, Z. Sun, H. Liu, L. Xu, D. Hu and J. Hu, Dyes Pigm., 2022, 205, 110488 CrossRef CAS.
- A. Shang, T. Lu, H. Liu, C. Du, F. Liu, D. Jiang, J. Min, H. Zhang and P. Lu, J. Mater. Chem. C, 2021, 9, 7392–7399 RSC.
- K. Zhang, J. Fan, C.-K. Wang and L. Lin, Phys. Chem. Chem. Phys., 2021, 23, 21883–21892 RSC.
- S. Kothavale, W. J. Chung and J. Y. Lee, J. Mater. Chem. C, 2022, 10, 6043–6049 RSC.
- Z. Yang, X. Ge, W. Li, Z. Mao, X. Chen, C. Xu, F. Long Gu, Y. Zhang, J. Zhao and Z. Chi, Chem. Eng. J., 2022, 442, 136219 CrossRef CAS.
- W. Xie, M. Li, X. Peng, W. Qiu, Y. Gan, Z. Chen, Y. He, W. Li, K. Liu, L. Wang, Q. Gu and S.-J. Su, Chem. Eng. J., 2021, 425, 131510 CrossRef CAS.
- B. Liu, W.-C. Chen, R. Zhang, Q. Liu, H. Wei, W.-L. Wu, L. Xing, R. Wang, Y. Liu, S. Ji, H.-L. Zhang and Y. Huo, Dyes Pigm., 2023, 216, 111314 CrossRef CAS.
- N. A. Kukhta, H. F. Higginbotham, T. Matulaitis, A. Danos, A. N. Bismillah, N. Haase, M. K. Etherington, D. S. Yufit, P. R. McGonigal, J. V. Gražulevičius and A. P. Monkman, J. Mater. Chem. C, 2019, 7, 9184–9194 RSC.
- A. Danos, D. Gudeika, N. A. Kukhta, R. Lygaitis, M. Colella, H. F. Higginbotham, A. N. Bismillah, P. R. McGonigal, J. V. Grazulevicius and A. P. Monkman, J. Mater. Chem. C, 2022, 10, 4737–4747 RSC.
- Y. Xiao, H. Wang, Z. Xie, M. Shen, R. Huang, Y. Miao, G. Liu, T. Yu and W. Huang, Chem. Sci., 2022, 13, 8906–8923 RSC.
- S. Kothavale, J. Lim and J. Yeob Lee, Chem. Eng. J., 2022, 431, 134216 CrossRef CAS.
- H. Ye, J. Yang, K. Stavrou, M. Li, F. Liu, F. Li, S.-J. Su and A. P. Monkman, Dyes Pigm., 2023, 219, 111568 CrossRef CAS.
- K. Rayappa Naveen, K. Prabhu CP, R. Braveenth and J. Hyuk Kwon, Chem. – Eur. J., 2022, 28, e202103532 CrossRef CAS PubMed.
- S. Wang, Y. Miao, X. Yan, K. Ye and Y. Wang, J. Mater. Chem. C, 2018, 6, 6698–6704 RSC.
- U. Balijapalli, R. Nagata, N. Yamada, H. Nakanotani, M. Tanaka, A. D'Aléo, V. Placide, M. Mamada, Y. Tsuchiya and C. Adachi, Angew. Chem., Int. Ed., 2021, 60, 8477–8482 CrossRef CAS PubMed.
- T. Yang, B. Liang, Z. Cheng, C. Li, G. Lu and Y. Wang, J. Phys. Chem. C, 2019, 123, 18585–18592 CrossRef CAS.
- Y.-Y. Wang, Y.-L. Zhang, K. Tong, L. Ding, J. Fan and L.-S. Liao, J. Mater. Chem. C, 2019, 7, 15301–15307 RSC.
- T. Cardeynaels, S. Paredis, A. Danos, D. Vanderzande, A. P. Monkman, B. Champagne and W. Maes, Dyes Pigm., 2021, 186, 109022 CrossRef CAS.
- S. Kothavale, W. J. Chung and J. Y. Lee, J. Mater. Chem. C, 2020, 8, 7059–7066 RSC.
- F.-M. Xie, H.-Z. Li, G.-L. Dai, Y.-Q. Li, T. Cheng, M. Xie, J.-X. Tang and X. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 26144–26151 CrossRef CAS PubMed.
- Y.-L. Zhang, Q. Ran, Q. Wang, Y. Liu, C. Hänisch, S. Reineke, J. Fan and L. S. Liao, Adv. Mater., 2019, 31, 1902368 CrossRef CAS PubMed.
- J.-L. He, F.-C. Kong, B. Sun, X.-J. Wang, Q.-S. Tian, J. Fan and L.-S. Liao, Chem. Eng. J., 2021, 424, 130470 CrossRef CAS.
- H. Wang, J. X. Chen, X. C. Fan, Y. C. Cheng, L. Zhou, X. Zhang, J. Yu, K. Wang and X. H. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 1685–1692 CrossRef CAS PubMed.
- F.-M. Xie, X.-Y. Zeng, J.-X. Zhou, Z.-D. An, W. Wang, Y.-Q. Li, X.-H. Zhang and J.-X. Tang, J. Mater. Chem. C, 2020, 8, 15728–15734 RSC.
- R. Furue, K. Matsuo, Y. Ashikari, H. Ooka, N. Amanokura and T. Yasuda, Adv. Opt. Mater., 2018, 6, 1701147 CrossRef.
- A. Urbano, A. M. del Hoyo, A. Martínez-Carrión and M. C. Carreño, Org. Lett., 2019, 21, 4623–4627 CrossRef CAS PubMed.
- T. Matsushima, S. Kobayashi and S. Watanabe, J. Org. Chem., 2016, 81, 7799–7806 CrossRef CAS PubMed.
- T. Cardeynaels, S. Paredis, J. Deckers, S. Brebels, D. Vanderzande, W. Maes and B. Champagne, Phys. Chem. Chem. Phys., 2020, 22, 16387–16399 RSC.
- T. J. Penfold, J. Phys. Chem. C, 2015, 119, 13535–13544 CrossRef CAS.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 Revision A03, Gaussian, Inc., Wallingford CT, 2016.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed.
- X. Gao, S. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, J. Chem. Theory Comput., 2017, 13, 515–524 CrossRef CAS PubMed.
- W. Shipan, Z. Cheng, X. Song, X. Yan, K. Ye, Y. Liu, G. Yang and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 9892–9901 CrossRef PubMed.
- H. S. Kim, S. H. Lee, S. Yoo and C. Adachi, Nat. Commun., 2024, 15, 2267 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 2004, 38, 2834–2838 CrossRef.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, Chem. Phys. Chem., 2016, 17, 2956–2961 CrossRef CAS PubMed.
- S. Paredis, T. Cardeynaels, S. Brebels, J. Deckers, S. Kuila, A. Lathouwers, M. Van Landeghem, K. Vandewal, A. Danos, A. P. Monkman, B. Champagne and W. Maes, Phys. Chem. Chem. Phys., 2023, 25, 29842–29849 RSC.
- J. Gibson and T. J. Penfold, Phys. Chem. Chem. Phys., 2017, 19, 8428–8434 RSC.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- N. Notsuka, H. Nakanotani, H. Noda, K. Goushi and C. Adachi, J. Phys. Chem. Lett., 2020, 11, 562–566 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, C. Xu, X. Chen, J. Zhao, Z. Yang, Y. Zhang, W. Wu, S. Jiao, Y. Liu, M. P. Aldred and Z. Chi, Chem. Sci., 2019, 10, 8129–8134 RSC.
- K. Stavrou, L. G. Franca and A. P. Monkman, ACS Appl. Electron. Mater., 2020, 2, 2868–2881 CrossRef CAS PubMed.
- M. A. Baldo and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 10958–10966 CrossRef CAS.
- Q. Zhang, T. Komino, S. Huang, S. Matsunami, K. Goushi and C. Adachi, Adv. Funct. Mater., 2012, 22, 2327–2336 CrossRef CAS.
- S. Bagnich, A. Rudnick, P. Schroegel, P. Strohriegl and A. Köhler, Philos. Trans. R. Soc. London, Ser. A, 2015, 373 CrossRef CAS PubMed.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- D. Kelly, L. G. Franca, K. Stavrou, A. Danos and A. P. Monkman, J. Phys. Chem. Lett., 2022, 13, 6981–6986 CrossRef CAS PubMed.
- N. Haase, A. Danos, C. Pflumm, A. Morherr, P. Stachelek, A. Mekic, W. Brütting and A. P. Monkman, J. Phys. Chem. C, 2018, 122, 29173–29179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2354499. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc01214c |
| This journal is © The Royal Society of Chemistry 2024 |