
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Humidity and temperature driven transformations in ferroelectric quinuclidine-based chlorocobaltate(II) complex salt: bulk and thin films with preferred orientation†
Mia
Mesić
 ,
Marko
Dunatov
,
Andreas
Puškarić
,
Zoran
Štefanić
and
Lidija
Androš Dubraja
*
,
Marko
Dunatov
,
Andreas
Puškarić
,
Zoran
Štefanić
and
Lidija
Androš Dubraja
*
Ruđer Bošković Institute, Bijenička cesta 54, 10000 Zagreb, Croatia. E-mail: lidija.andros@irb.hr; Tel: +385 1 4561184
First published on 3rd July 2024
Abstract
Ferroelectricity remains a fascinating phenomenon in materials science due to the constant uncovering of novel ferroelectric hybrid materials and new features of the phenomenon, related to its potential applications for the development of new technologies. Herein, preparation of a new inorganic–organic hybrid material composed of 3-quinuclidinone cations and a tetrachlorocobaltate(II) anion is reported, by utilization of a green chemistry approach without the use of solvents through a mechanochemically driven reaction. Two forms of the same compound; anhydrous and hydrate, can be prepared depending on the reactants used in the synthesis. Alternately two compounds can be transformed into one another by exposure to humid conditions or a dry atmosphere. In addition to humidity-driven compositional transformation, the prepared material undergoes multiple temperature-induced phase transformations. For the anhydrous phase, low-, room- and high-temperature phases were detected, belonging to the ferroelectric space group. Multiple transformations that occur in this material are an indication of the soft crystal packing driven by non-covalent interactions. Due to structural flexibility, this system is an ideal candidate for deposition on a substrate, for which the solution-based dip-coating method was used. The obtained metal–organic thin films grow in a specific crystal direction on Si(100) and ITO-coated glass substrates, forming crystalline and uniform films. The self-assembly between cations and anions is discussed, along with the energetic driving forces that lead to a particular thin film structure. Ferroelectric testing through remanent polarization experiments under ambient conditions confirms the uniformity and purity of this metal–organic thin-film system.
Introduction
Ferroelectric polarization and various resistive switching effects are among the key features that can advance modern technologies, such as in data storage devices, sensors and other semiconductor related applications.1–3 Due to their remarkable piezoelectric properties, inorganic ferroelectric ceramics have long dominated research and almost exclusively technology development.4,5 However, conventional ferroelectric ceramics are being challenged by the growing demand for flexible and thin-film integrated devices, as they lack mechanical flexibility and require high processing temperatures. In this sense, molecular ferroelectrics represent a promising platform for ferroelectric compounds that can be designed, fabricated and structurally tuned under environmentally friendly and economically viable conditions.6,7 In particular, hybrid metal–organic complex salts composed of organic amine cations and inorganic halide anions have attracted great interest as materials with prospects for applications in data storage, signal processing, environmental monitoring and energy harvesting.8–10 Organic amine molecules, which have a spherical polar structure and low rotational dynamics in the solid state, have attracted considerable interest in the field of molecular ferroelectrics because realignment of polar molecules in response to an applied electric field can induce ferroelectric polarization.11–14 Simple organic molecules from the family of 1,4-diazabicyclo[2.2.2]octane (C6H12N2, DABCO) and 1-azabicyclo[2.2.2]octane (C7H13N, ABCO) are particularly suitable for this purpose due to their availability and structural tunability.12–14 Thermally induced dynamic movement of the unsymmetrical molecules, such as 1-azabicyclo[2.2.2]octan-3-one [C7H11NO, (O)ABCO], can give rise to excellent thermistor sensitivity in some systems.15 In order to understand the principles underlying the stimulus-structure-response property of molecule-based dielectrics and ferroelectrics at the molecular level, it is imperative to prepare materials with high crystallinity and a well-defined chemical composition and crystal structure.16,17In our recent work, we have pointed out the important role that water molecules play in crystal formation and transformations in the solid-state driven by processes occurring on a solid–gas interface.18,19 Apart from changing the structural arrangement of the constituent cations and anions in the crystal packing, humidity-driven transformations can induce specific properties, such as ferroelectricity, proton conductivity or dielectric switching.18 In some cases, the unit cell of a compound can be reversibly increased/decreased remarkably by 103 Å3 without damaging the crystallinity and functionality.20 Another advantage over conventional inorganic dielectrics and ferroelectrics is that metal–organic complexes and complex salts can be prepared by a simple and environmentally friendly mechanochemical synthesis at room temperature, which we have demonstrated on a series of complex salts of DABCO- and ABCO-based cations and bis(oxalato)chromium(III) anions.18 In addition to the rotating DABCO- and ABCO-based cations, we have now introduced simpler inorganic anions with more rotational freedom, e.g. the tetrachlorocobaltate(II) anion, to produce new and better performing molecular ferroelectric materials. This work presents the formation of an inorganic–organic hybrid material consisting of 3-quinuclidinone cations and tetrachlorocobaltate(II) anions synthesized by an environmentally friendly, solvent-free, and mechanochemically driven reaction. This green chemistry approach is innovative in the context of synthesizing ferroelectric materials and represents a sustainable alternative to conventional methods. In addition to the bulk crystals, thin films of the investigated inorganic–organic material were prepared by dip-coating in order to perform ferroelectric polarization experiments. The possibility of depositing the material as thin films on substrates such as silicon and ITO-coated glass using a solution-based dip-coating method is of great importance. Among the various fabrication methods, dip-coating stands out for its simplicity and effectiveness. In this work, it has been shown that soft materials, where crystal packing is determined by non-covalent interactions, are ideal candidates for creating crystalline and uniform films, which are crucial for potential technological applications.
Experimental
Materials
3-Quinuclidinone hydrochloride (99%), [H-(O)ABCO]Cl, and CoCl2·6H2O (98%) were purchased from Alfa Aesar. Anhydrous CoCl2 and CoCl2·2H2O were prepared by thermal treatment of CoCl2·6H2O at 393 K and 373 K, respectively. Si(100) substrates were purchased from Siegert wafer GmbH and indium-doped tin oxide (ITO) coated glass substrates from Ossila BV. Methanol (99.8%) and acetonitrile (99.9%) used to prepare the dip-coating solutions were purchased from Fisher Scientific.Synthesis of [H–(O)ABCO]2(CoCl4)·H2O and [H–(O)ABCO]2(CoCl4)
Both compounds were prepared by mechanochemical synthesis using a Retsch MM 400 mixer mill at a frequency of 25 Hz, in 5 mL agate grinding jars using one 7 mm agate ball. [H–(O)ABCO]Cl (64 mg, 0.4 mmol) and CoCl2·2H2O (33 mg, 0.2 mmol) were mixed and milled for 30 minutes, giving the phase I [H–(O)ABCO]2(CoCl4)·H2O compound in stoichiometric yield. Performing the mechanochemical reaction with anhydrous CoCl2 instead of CoCl2·2H2O yields the phase II [H–(O)ABCO]2(CoCl4). Blue prism-like single crystals of phase I can be obtained from water solutions by recrystallization. Phase II can be also obtained from ethanol solution of starting reactants with the addition of anti-solvent diethyl ether. Anal. calcd % for phase I C14H26N2O3CoCl4 (found, %): C, 35.69 (35.91); H, 5.56 (5.87); N, 5.94 (5.91); Co, 12.50 (12.29). Anal. calcd % for phase II C14H24N2O2CoCl4 (found, %): C, 37.11 (37.17); H, 5.33 (5.40); N, 6.18 (6.22); Co, 13.07 (13.09).Thin film preparation
For the fabrication of thin films, [H–(O)ABCO]2(CoCl4) (27 mg) was dissolved in a solvent mixture of methanol (3 mL) and acetonitrile (3 mL). Thin films were prepared by dip-coating (50 mm s−1 withdrawal rate) on Si(100) and ITO-coated glass substrates in a humidity-controlled chamber set to 29% relative humidity at 293 K. After the deposition cycle was finished, the as-made films were transferred on a hot plate heated at 343 K to complete the drying and condensation process.Spectroscopic measurements
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were recorded in the 4000–400 cm−1 range using a PerkinElmer FT-IR Frontier spectrometer. Humidity-dependent ATR-FTIR measurements were recorded in situ in the chamber in which the relative humidity was adjusted with a flow of dry or wet gas.Differential scanning calorimetry
Differential scanning calorimetry (DSC) thermograms were recorded using the Netzsch DSC 214 Polyma calorimeter. The DSC signal was measured during heating and cooling cycles in the temperature range from 293 to 453 K in an extra pure nitrogen environment at a rate of 10 K min−1.Surface morphology
Scanning electron microscopy imaging (SEM) and energy dispersive X-ray spectroscopy (EDX) were performed on a field emission scanning electron microscope (model JSM-7000F) operated at 10 keV.Electrical measurements
Ferroelectric tests were performed using a TF Analyzer 1000 at room temperature in the frequency range of 10–125 Hz. A positive-up-negative-down (PUND) test was applied consisting of five triangular voltage pulses of 2.2 V amplitude and 1 ms rise time and a delay time between pulses of 1 s.Crystallographic measurements
The single-crystal X-ray diffraction data were collected by ω-scans using Cu-Kα radiation (λ = 1.54179 Å, microfocus tube, mirror monochromator) on a Rigaku XtaLAB Synergy diffractometer at 100 K and 293 K. The crystal data, experimental conditions and final refinement parameters are summarized in Table S1 in the ESI.† Data reduction, including the multiscan absorption correction, was performed with the CrysAlisPRO software package. Molecular and crystal structures were solved by direct methods using the program SIR201921 and refined by the full-matrix least-squares method based on F2 with anisotropic displacement parameters for all non-hydrogen atoms (SHELXL-2014/7).22 Both programs were operated under the WinGX program package.23 The positions of the hydrogen atoms attached to the carbon and nitrogen of the [H–(O)ABCO]+ cation were found in the electron density map, but were placed in idealized positions. The hydrogen atoms of the water molecule in phase I were also identified based on the difference Fourier map [O–H distances were restrained to a target value of 0.85(2) Å, and the H–O–H angle to 104°]. Geometrical calculations were carried out with PLATON24 and the figures were generated using the CCDC-mercury program.25The powder X-ray diffraction (PXRD) data were collected in reflection mode with Cu-Kα1 radiation (λ = 1.54060 Å) on a Malvern Panalytical Empyrean diffractometer using a step size of 0.013° in the 2θ range between 5° and 50°. Rietveld refinement of the PXRD patterns of mechanochemically prepared phases I and II, as well as of phase III, was carried out using the TOPAS software (details given in Table S2 in the ESI†). For in situ PXRD measurements, a high-temperature camera was used in the temperature range of 293–453 K.
Results and discussion
Synthesis, spectroscopic and thermal characterisation
The compound [H–(O)ABCO]2(CoCl4)·H2O (phase I) shown in Scheme 1 can be prepared in a mechanochemical reaction without solvent, or from aqueous solutions of the precursors in appropriate ratios. The compound [H–(O)ABCO]2(CoCl4) (phase II) is an anhydrous phase of the phase I, and can be prepared directly by mechanochemical synthesis or from the ethanol/diethyl ether solutions of the starting precursors.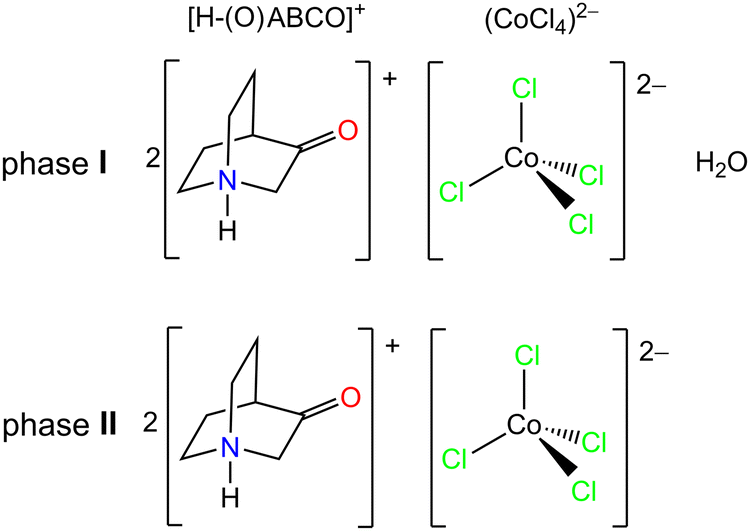 | ||
| Scheme 1 Schematic representation of 3-quinuclidinone cations and the tetrachlorocobaltate(II) anion. | ||
Phases I and II are related by a solid state transformation that can be triggered by changes in relative humidity. This was confirmed by different techniques, i.e. in situ FTIR-ATR spectroscopy, DSC and in situ PXRD measurements. Fig. 1a shows the FTIR-ATR spectrum of phase I (hydrate phase) together with the time-resolved spectra after the introduction of dry nitrogen into the system. It is obvious that transformation of phase I into phase II occurs already after 1 minute of exposure to dry conditions (RH below 5%). The broad bands at 3500 and 3412 cm−1 related to the ν(O–H) stretching vibrations in the water molecule decrease during drying and disappear completely after 10 minutes of nitrogen exposure.19 Other bands in the spectra are not significantly affected by the loss of water molecules. For example, the most intense band in the spectra associated with the stretching vibration of the carbonyl group centred at 1745 cm−1 shifts only slightly to 1743 cm−1 upon drying.26 The transformation from I to II is reversible, and phase I recovers when higher relative humidity levels are restored.
 | ||
| Fig. 1 (a) Time-resolved FTIR-ATR spectra during exposure of phase I (blue line) to dry nitrogen giving the phase II (brown line); (b) DSC-thermogram in the 10 K min−1 heating cycle showing thermally induced processes from the initial phase I. | ||
According to the DSC analysis (Fig. 1b), the loss of water molecules from the phase I in a dynamic 10 K min−1 heating cycle starts at 320 K and ends at 333 K, resulting in anhydrous phase II. DSC shows two additional endothermic processes beyond the mentioned water loss. The first appears to consist of three consecutive sub-processes centred at 360, 366 and 381 K and ending with phase III. Thereafter, another endothermic peak at 433 K indicates a phase transformation from III to IV. The values of the associated latent heat (ΔH) and entropy change (ΔS), obtained from the peak integration of the heat flow curve, are given in Table 1. The latent heat and entropy change values are similar in magnitude to those reported for compounds [H–(O)ABCO](FeCl4)27 and [H-ABCO]2(CoCl4).28
| Phase transitions | Temperature range | Enthalpy change, ΔH (J g−1) | Entropy change, ΔS (J K−1 kg−1) |
|---|---|---|---|
| I → II | 320–333 | 23.5 | 72.1 |
| II → III | 350–395 | 65.4 | 181.4 |
| III → IV | 413–436 | 22.1 | 51.2 |
Further insights into the structure of these high-temperature phases were obtained from PXRD analyses carried out in situ during heating (see Fig. 2). Heating phase I to 333 K leads to the loss of water molecules from the structure and the formation of phase II. Phase II is further stable until 393 K (Fig. S5, ESI†), when, according to PXRD, another phase is formed, referred as III. Phase III crystalizes in a polar space group, Pc (Table 2 and Fig. S3, Table S2, ESI†).
 | ||
| Fig. 2 Temperature modulated PXRD experiments in the [H–(O)ABCO]2(CoCl4)·nH2O (n = 0, 1) compound. | ||
| Phase | I | II-LT | II | III |
|---|---|---|---|---|
| T/K | 293 | 100 | 293 | 393 |
| M r/g mol−1 | 471.11 | 453.09 | 453.09 | 453.09 |
| Space group | Pc21n | P21 | P21 | Pc |
| a/Å | 14.2651(2) | 6.9382(3) | 6.5717(5) | 18.519(2) |
| b/Å | 9.0392(2) | 12.3036(7) | 13.5034(7) | 12.227(1) |
| c/Å | 15.7057(3) | 11.5132(7) | 21.0441(12) | 18.429(3) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 90 | 105.807(6) | 90.322(6) | 120.508(9) |
| γ/° | 90 | 90 | 90 | 90 |
| Z | 4 | 2 | 2 | 2 |
| V/Å3 | 2025.17(7) | 945.66(9) | 1867.43(2) | 3595.66(2) |
A further structural transformation before thermal decomposition of the material is observed at 438 K and is referred as phase IV, for which a unit cell determination from PXRD data was unsuccessful due to severe peak overlap. Further heating to 453 K leads to irreversible thermal decomposition of the material. The experimental PXRD patterns at room temperature for phases I and II agree with the patterns calculated from the single-crystal structures at 293 K and give a satisfactory fit by Rietveld refinement (see Fig. S1 and S2 in ESI†).
Based on DSC, FTIR-ATR and PXRD measurements, the system consisting of two [H–(O)ABCO]+ cations, a (CoCl4)2− anion and a variable water molecule is shown to undergo three different transformations: I → II hydrate phase undergoes a reversible phase transition at 333 K to an anhydrous phase; II → III anhydrous phase undergoes a reversible phase transition at 393 K to an anhydrous high-temperature phase III; and III → IV additional crystalline high-temperature phase forms upon further thermal stimulation. Based on the PXRD patterns measured after thermal treatment at 393 and 438 K, the structure transforms into an anhydrous phase II after cooling to room temperature. The transformation I → II can be triggered either by an increase in temperature or by exposure to low relative humidity (in a dry nitrogen atmosphere). According to PXRD (Fig. S4 in the ESI†), the transformation is reversible and the hydrate phase returns when the relative humidity increases.
Crystal structure of phases I and II
The compound [H–(O)ABCO]2(CoCl4)·H2O phase I; crystalizes in the orthorhombic system, non-centrosymmetric polar space group Pc21n. The parameters of the unit cell are given in Table 2. The crystal structure is free of disorder at room temperature. The asymmetric unit consists of two protonated [H–(O)ABCO]+ molecules, a tetrahedral (CoCl4)2− anion and a crystal water molecule (Fig. 3a). The deviation from the tetrahedral geometry in the (CoCl4)2− anion is considerable with Co–Cl bond lengths in the range of 2.084(1)–2.497(1) Å and Cl–Co–Cl bond angles in the range of 103.83(6)–113.88(5)°. Crystal water molecules form hydrogen bonds with two surrounding (CoCl4)2− anions and this type of contact extends along the c-axis.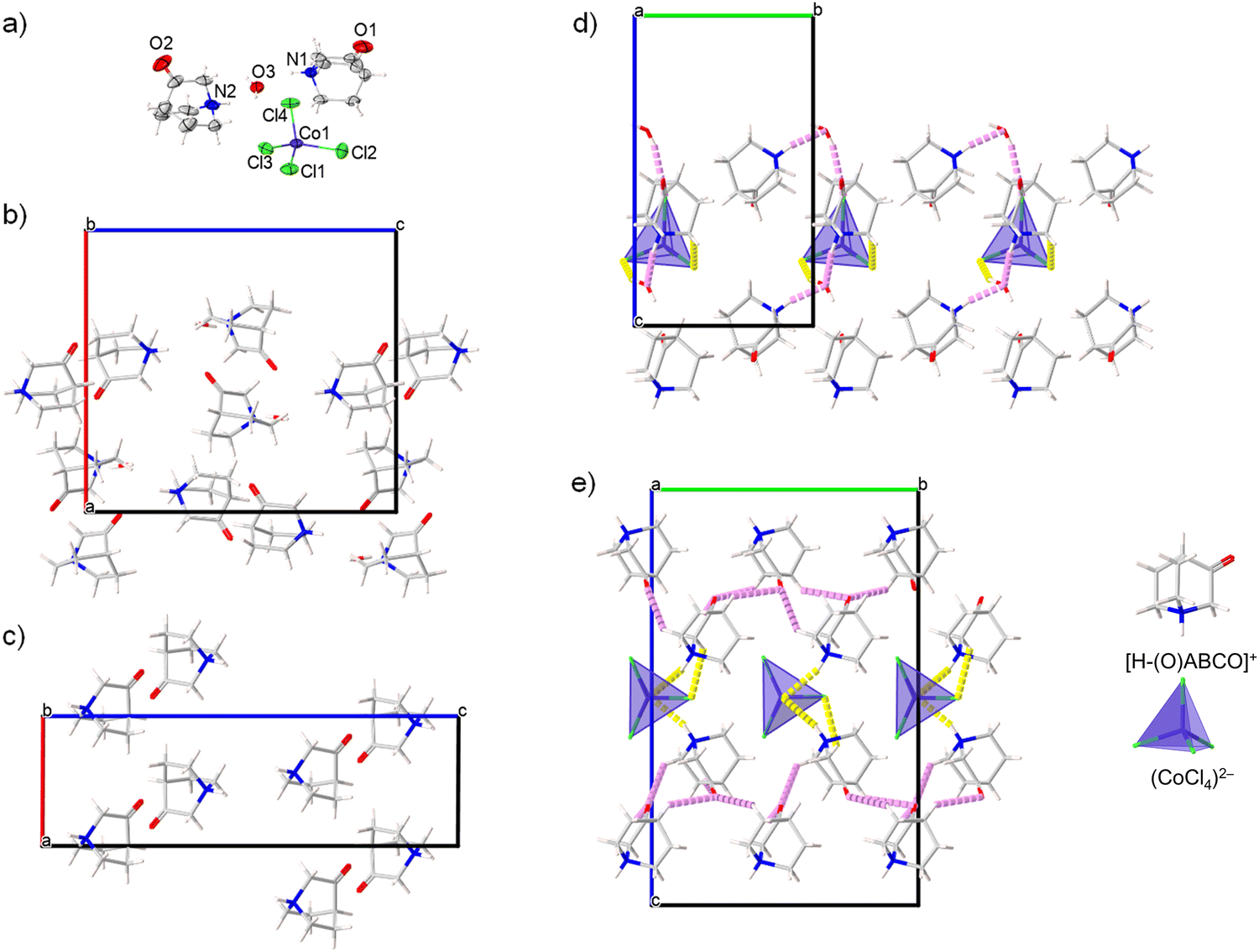 | ||
| Fig. 3 (a) Molecular structure of [H–(O)ABCO]2(CoCl4)·H2O, phase I; (b) and (c) crystal packing of cations in phases I and II; (d) and (e) hydrogen bonding in phases I and II, yellow colour is used to mark the contacts involving anions, and violet colour for contacts formed exclusively between cations. | ||
In addition, the water molecule also acts as a proton acceptor for the protons coming from two crystalographically independent [H–(O)ABCO]+ molecules, forming the O3⋯H1N–N1 and O3⋯H2N–N2 contacts. Two crystallographically independent [H–(O)ABCO]+ molecules are also connected by hydrogen bonds via O2⋯H1N–N1, O1⋯H12A–C12 and O1⋯H13A–C13 contacts (Fig. 3b). The crystal packing achieved by hydrogen bonding is three-dimensional. The distance between the nearest cation and anion (their centre of gravity) in this structural arrangement is 5.485 Å, between two cations 6.370 Å and between two anions 8.364 Å.
Compound [H–(O)ABCO]2(CoCl4)·H2O phase I undergoes symmetry breaking transformation to an anhydrous [H–(O)ABCO]2(CoCl4) phase II by heating to 333 K. The anhydrous phase crystallizes in the monoclinic system, in a non-centrosymmetric polar space group P21. The parameters of the unit cell (Table 2) show a reduction of the a-axis by half, while the b- and c-axes increase. The new phase contains four [H–(O)ABCO]+ molecules and two tetrahedral (CoCl4)2− anions in the asymmetric unit. Compared to phase I, the Co–Cl bond distances in the anions are more uniform and range from 2.239(1)–2.331(7) Å, but the angles deviate from 103.5(3) to 125.5(4)°. Each inorganic anion forms two hydrogen contacts Cl⋯H–N which affect the above-mentioned elongation of the Co–Cl bond, and additionally three Cl⋯H–C with three surrounding [H–(O)ABCO]+ cations. Hydrogen bonds exist between oxo-oxygen atoms acting as proton acceptors to hydrogen atoms bonded to sp3-carbon atoms, with five contacts of this type for each [H–(O)ABCO]+ cation. The crystal packing achieved in this way is altogether three-dimensional. The distance between the nearest cation and anion (their centre of gravity) in this structural arrangement is 5.017 Å, between two cations 6.066 Å and between two anions 6.572 Å. In the low temperature phase II-LT (100 K), which also crystalizes in the monoclinic space group P21 and whose c-axis is almost half as large as that of phase II, the thermal movement of the rotor molecules is damped, which leads to a shortening of the distance between the nearest cation and anion to 4.910 Å and between two cations to 5.879 Å, while the two anions are much more separated from each other with a distance of 8.228 Å, similar to hydrate phase I. Comparing the hydrate (phase I) and the anhydrous (phase II) structures at room temperature, the greatest difference in the crystal packing is due to the geometry of the hydrogen bonds. While in phase I the tetrachlorocobaltate anions act as acceptors for the hydrogen atoms of the water molecules and form an infinite hydrogen bond chain along the c-axis (Fig. 3d), in phase II the anions and cations rotate slightly to achieve closer contact between them and form multiple hydrogen bonds (Fig. 3e). Between two consecutive cation⋯anion hydrogen bond contacts in the crystal packing of phase II, there is one cation⋯cation hydrogen bond contact formed by an oxo-oxygen atom and a hydrogen attached to the sp3-carbon of the [H–(O)ABCO]+ molecule (Fig. 3c).
Thin film synthesis and characterisation
Thin films of phase II were deposited on Si(100) and ITO-coated glass substrates by the dip-coating method. The dip-coating process usually allows the coating of large surfaces with different types of materials, either inorganic oxides, organic or metal–organic compounds, which makes this method very interesting for the fabrication of thin films for various applications, especially for enhancing miniaturization and integration.29,30 The dip-coating process is solution-based and the films are formed as a result of an evaporation-induced self-assembly process. A number of parameters must be optimized in order to obtain crystalline and uniform thin films. The first step is to prepare the solution by adjusting the concentration of precursors, the pH and the combination of solvents to ensure a suitable viscosity and density of the solution for the coating process.31 In addition to these parameters, the type of substrate is another important factor, along with the withdrawal rate of the substrate from the solution, relative humidity, temperature and drying time.32,33 In the production of metal–organic thin films, dip-coating is a simple and efficient process that can result in lightweight, flexible, non-toxic and economically favourable products.34Fig. 4a and b show top view electron microscopy images of the film produced from methanol/acetonitrile solution of compound [H–(O)ABCO]2(CoCl4). The films formed are continuous, dense and homogeneous, but some cracks or pinholes can be seen when viewed over large areas (Fig. 4a), which is due to the solution-based preparation method. In the high magnification images (Fig. 4b), the films are more uniform without pinholes or defects, which is important for potential electronic, optical, and optoelectronic applications. The qualitative elemental composition was determined by energy dispersive X-ray analysis (Fig. 4c) to verify the purity of the produced materials. The films show a very high intensity peak related to the substrate (Si wafer) and elements present in the [H–(O)ABCO]2(CoCl4) compound, namely Co, Cl, N, O, and C, which cannot be reliably determined by EDX analysis.
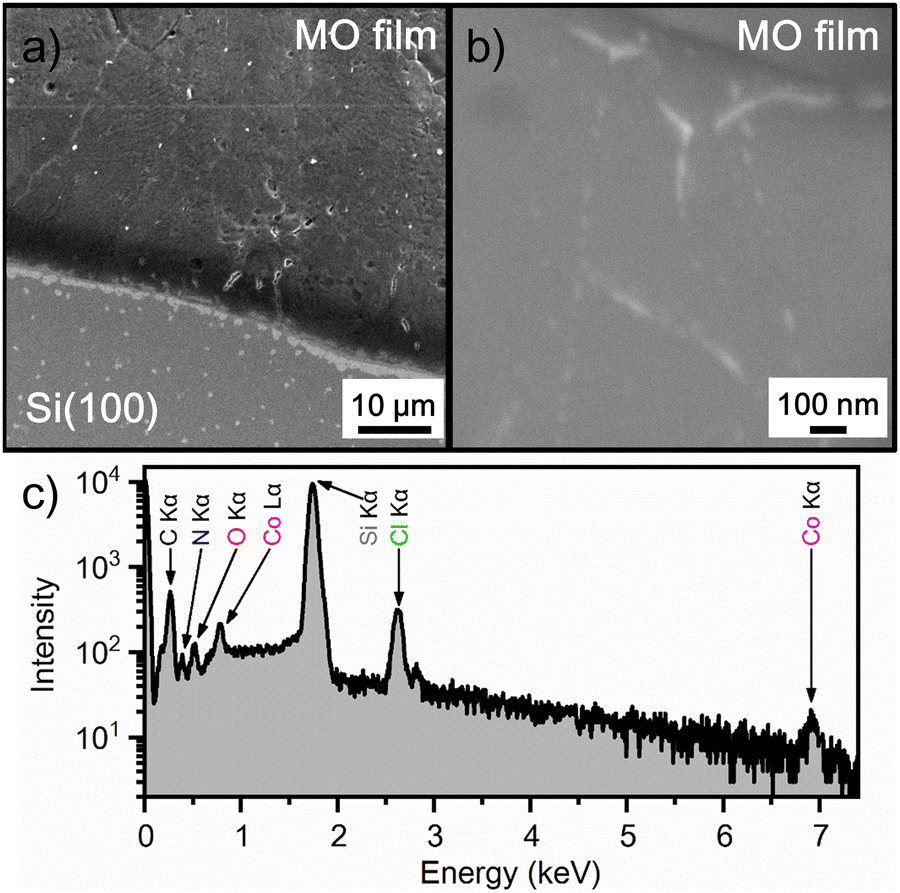 | ||
| Fig. 4 (a) Low and (b) high magnification top view SEM images for the metal–organic (MO) [H–(O)ABCO]2(CoCl4) thin film deposited on the Si(100) substrate; (c) EDX spectra of the corresponding thin film. | ||
The crystal structure of the films deposited on Si(100) was confirmed by powder X-ray diffraction measured in a Bragg–Brentano geometry. The thin films are crystalline regardless of whether they were deposited on a silicon wafer or an ITO/glass substrate. The weakening of most of the diffraction peaks indicates the growth of the films with preferred orientation (Fig. 5a). From the PXRD pattern, it can be seen that only peaks corresponding to the 00l reflections of [H–(O)ABCO]2(CoCl4) (phase II) are observed up to the seventh order. In addition to these peaks, the XRD pattern shows peaks originating from the Si(100) substrate (2θ peaks at 33° and 70°)35 and the SiO2 layer (2θ peak at 61.8°),36 with no additional phase evident.
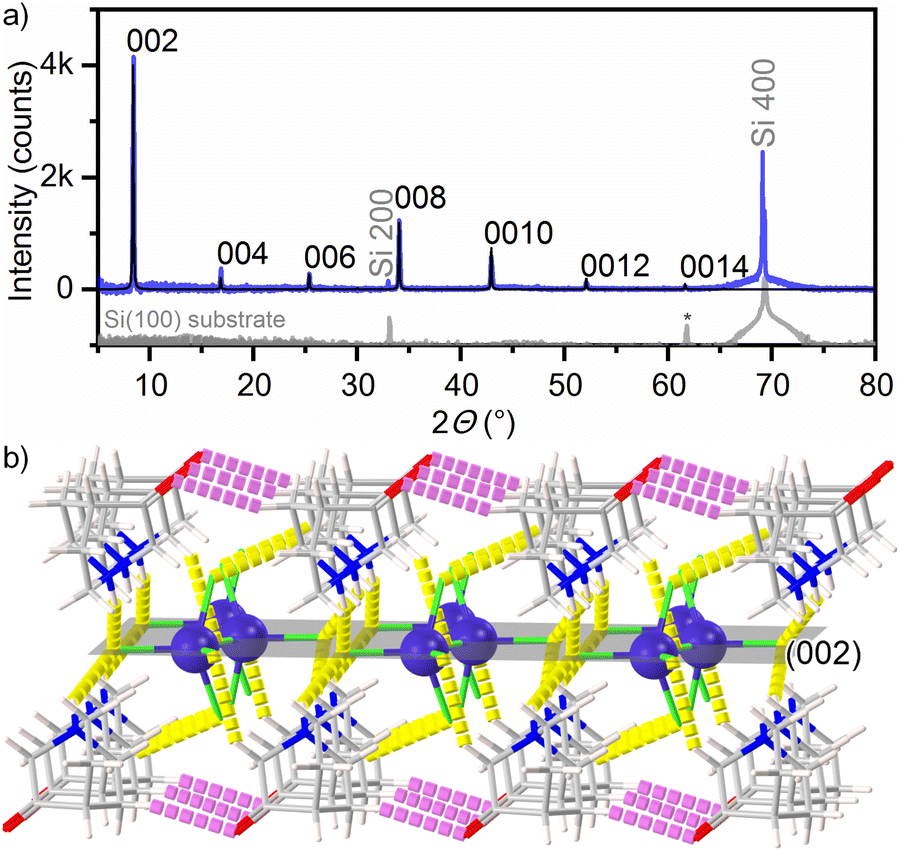 | ||
| Fig. 5 (a) PXRD pattern of the [H–(O)ABCO]2(CoCl4) thin film (phase II) deposited on the Si(100) substrate (blue line). PXRD pattern of the Si(100) substrate is shown as a grey line and on a logarithmic scale for comparison. Calculated PXRD pattern from single-crystal data is shown as a black line. Peak marked with an asterisk comes from the SiO2 layer; (b) crystal packing in the (002) plane of the [H–(O)ABCO]2(CoCl4) phase II. Cobalt atoms are shown as violet balls and chlorine, nitrogen and carbon atoms as green, blue and grey sticks, respectively. | ||
Considering that these thin films were prepared by a dip-coating process in an evaporation-induced self-assembly process, it is rather surprising that such a high degree of the preferred orientation in the [001] direction was achieved. A closer examination of the crystal packing shows that the cobalt(II) ions are located in the (002) plane (Fig. 5b). At the top and bottom of the (002) plane organic cations are located that form hydrogen bonds with (CoCl4)2− anions (shown as yellow sticks in Fig. 5b). In addition, there are hydrogen bonds between the two organic cations (shown as purple sticks in Fig. 5b). This indicates that in addition to the electrostatic interaction between cations and anions, hydrogen bonds also seem to be a driving force for the self-assembly of cations and anions during the evaporation of the solvent and the formation of the film.
Ferroelectric properties
A very convenient approach to maximize polarization along the switching direction is to grow epitaxial ferroelectric thin films along the polar axis on appropriately selected conductive substrates.37 Since phase II forms thin films that grow along the direction of the two-fold rotation axis, it could be expected that they will exhibit out-of-plane polarization in electric field. For the purpose of ferroelectric testing, the 200 nm thick metal–organic films were deposited on conductive ITO/glass substrates, which served as the bottom electrode, and gold was thermally evaporated on the top of the metal–organic film onto an area of 0.1 mm2, which served as the top electrode. The ferroelectric properties of the as-made films investigated by polarization-electric field (P–E) measurements at room temperature (295 K) are shown in Fig. 6. To exclude the non-switching contributions (dielectric capacitance and leakage current) in this metal–organic thin film sample, a positive-up-negative-down (PUND) method with a double triangular voltage pulses was used.20,33,38 | ||
| Fig. 6 Room temperature ferroelectric polarization and the corresponding current response of the [H–(O)ABCO]2(CoCl4) thin film (phase II) on the ITO:glass substrate with Au top electrodes for 100 Hz switching frequency. | ||
The spontaneous polarization (Ps) of the [H–(O)ABCO]2(CoCl4) thin film (phase II) obtained from PUND measurements corresponds approximately to the remanent polarization, and reaches a value of 2.75 μC cm−2 at a coercive field (Ec) of about 40 kV cm−1. If these values are compared with the ferroelectric properties of related materials with rotating polar globular ionic molecules of the DABCO and ABCO family, it is noticeable that the ferroelectric properties are strongly dependent on the capacitor architecture, in addition to the chemical composition and crystal structure. For example, single crystals of the complex salt [(CH3)2-DABCO](CuCl4), which crystalize in the space group Pca21 give a Ps value that is an order of magnitude smaller, but a comparable coercive field (Ps = 0.53 μC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−2, Ec = 45 kV cm−1)39 to that of phase II. Thin films of complex salt with the same organic cation and perchlorate anion, [H–(O)ABCO](ClO4) (space group Pna21), showed a spontaneous polarization almost three times high (Ps = 6.7 μC cm−2).40 Since this material was deposited on a conductive ITO surface and contains GaIn eutectic top electrodes, it is comparable to the capacitor architecture used herein. Other comparable materials are thin films also deposited on ITO-coated substrates, such as [H-ABCO](IO4) (space group Pmn21, Ps = 7.71 μCcm−2, and Ec =255 kV cm−1)13 and [H–(F)ABCO](ReO4) (space group Pn, Ps = 11.2 μCcm−2, and Ec = 20 kV cm−1)41 for which theoretical calculations have confirmed the experimentally obtained value of spontaneous polarizations. In some materials containing ABCO cations, the presence of parasitic contributions, especially leakage currents, limits the detection of macroscopic ferroelectricity, despite the polar crystal structure and the ferroelectric response confirmed by piezoresponse force microscopy, which was the case for thin films of [H-(OH)ABCO](FeCl4) (space group P1).42 Nevertheless, our results show that ABCO- and DABCO-based cations are superior and thermally quite stable organic molecules for the design and fabrication of next-generation ferroelectric thin film capacitors.
cm−2, Ec = 45 kV cm−1)39 to that of phase II. Thin films of complex salt with the same organic cation and perchlorate anion, [H–(O)ABCO](ClO4) (space group Pna21), showed a spontaneous polarization almost three times high (Ps = 6.7 μC cm−2).40 Since this material was deposited on a conductive ITO surface and contains GaIn eutectic top electrodes, it is comparable to the capacitor architecture used herein. Other comparable materials are thin films also deposited on ITO-coated substrates, such as [H-ABCO](IO4) (space group Pmn21, Ps = 7.71 μCcm−2, and Ec =255 kV cm−1)13 and [H–(F)ABCO](ReO4) (space group Pn, Ps = 11.2 μCcm−2, and Ec = 20 kV cm−1)41 for which theoretical calculations have confirmed the experimentally obtained value of spontaneous polarizations. In some materials containing ABCO cations, the presence of parasitic contributions, especially leakage currents, limits the detection of macroscopic ferroelectricity, despite the polar crystal structure and the ferroelectric response confirmed by piezoresponse force microscopy, which was the case for thin films of [H-(OH)ABCO](FeCl4) (space group P1).42 Nevertheless, our results show that ABCO- and DABCO-based cations are superior and thermally quite stable organic molecules for the design and fabrication of next-generation ferroelectric thin film capacitors.
Conclusion
Herein, we report an environmentally friendly mechanochemical approach for the preparation of new ferroelectric complex salts consisting of 3-quinuclidinone cations and tetrachlorocobaltate(II) anions. Depending on the reactants used for the mechanochemical synthesis, a hydrate or anhydrous phase can be formed. Structural and spectroscopic measurements have shown that the hydrate (or anhydrous) phase can also be formed post-synthetically, by exposure to humid (or dry) conditions. The hydrate phase belongs to a polar orthorhombic Pc21n structure, and the loss of water from the structure leads to a symmetry-breaking to the monoclinic P21 anhydrous phase. In addition to humidity-driven compositional transformation, the anhydrous phase undergoes several temperature-induced phase transformations, exhibiting different low- and high-temperature phases in addition to the room-temperature phase.Metal–organic thin films of anhydrous phase were successfully prepared by solution-phase co-assembly of 3-quinuclidinone cations and tetrachlorocobaltate(II) anions using the dip-coating method. The structural characterization confirmed both the polar ferroelectric structure of the room temperature anhydrous phase with a strongly preferred 00l orientation and the single phase nature of the films. The reason for preferential crystal growth in the thin film morphology is directly related to the hydrogen bonding between cations and anions. More importantly, the films were not only structurally but also compositionally uniform throughout, making them good model materials for studying ferroelectric properties. Room temperature polarization switching experiments revealed a stable switching polarization of about 2.75 μC cm−2 with a coercive field of 40 kV cm−1, which represents a good ferroelectric performance for a molecular ferroelectric material and might lead to potential applications in soft piezoelectrics, thin film memory devices and flexible electronics.
Data availability
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Croatian Science Foundation (UIP-2019-04-7433) is gratefully acknowledged.Notes and references
- S. Li, Y. Wang, M. Yang, J. Miao, K. Lin, Q. Li, X. Chen, J. Deng and X. Xing, Mater. Adv., 2022, 3, 5735–5752 RSC.
- J. Liao, W. Wen, J. Wu, Y. Zhou, S. Hussain, H. Hu, J. Li, A. Liaqat, H. Zhu, L. Jiao, Q. Zheng and L. Xie, ACS Nano, 2023, 17(6), 6095–6102 CrossRef CAS.
- K. Yao, S. Chen, S. Cheng Lai and Y. M. Yousry, Adv. Sci., 2022, 9, 2103842 CrossRef CAS PubMed.
- T. Mikolajick, S. Slesazeck, H. Mulaosmanovic, M. Park, S. Fichtner, P. Lomenzo, M. Hoffmann and U. Schroeder, J. Appl. Phys., 2021, 129, 100901 CrossRef CAS.
- N. Setter, D. Damjanovic, L. Eng, G. Fox, S. Gevorgian, S. Hong, A. Kingon, H. Kohlstedt, N. Y. Park, G. B. Stephenson, I. Stolitchnov, A. K. Taganstev, D. V. Taylor, T. Yamada and S. Streiffer, J. Appl. Phys., 2006, 100, 051606 CrossRef.
- S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357 CrossRef CAS PubMed.
- X. Qian, X. Chen, L. Zhu and Q. M. Zhang, Science, 2023, 380, 596 CrossRef.
- H. Xu, F. Sun, W. Guo, S. Han, Y. Liu, Q. Fan, L. Tang, W. Liu, J. Luo and Z. Sun, Angew. Chem., Int. Ed., 2023, e202309416 CAS.
- F.-X. Wang, Y.-Z. Tang, Y.-H. Tan, Y.-R. Zhao, J.-Y. Wang, Y.-K. Li, H. Zhang, T.-T. Ying and M.-Y. Wan, Inorg. Chem. Front., 2023, 10, 4881–4888 RSC.
- Y. Huang, J. L. Gottfried, A. Sarkar, G. Zhang, H. Lin and S. Ren, Nat. Commun., 2023, 14, 5041 CrossRef CAS.
- L. Catalano, S. Perez-Estrada, H.-H. Wang, A. J.-L. Ayitou, S. I. Khan, G. Terraneo, P. Metrangolo, S. Brown and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2017, 139(2), 843–848 CrossRef CAS.
- J. Harada, T. Shimojo, H. Oyamaguchi, H. Hasegawa, Y. Takahashi, K. Satomi, Y. Suzuki, J. Kawamata and T. Inabe, Nat. Chem., 2016, 8, 946–952 CrossRef CAS.
- Y.-M. You, Y.-Y. Tang, P.-F. Li, H.-Y. Zhang, W.-Y. Zhang, Y. Zhang, H.-Y. Ye, T. Nakamura and R.-G. Xiong, Nat. Commun., 2017, 8, 14934 CrossRef CAS.
- H. Seob Choi, S. Li, I.-H. Park, W. Heng Liew, Z. Zhu, K. Chang Kwon, L. Wang, I.-H. Oh, S. Zheng, C. Su, Q.-H. Xu, K. Yao, F. Pan and K. P. Loh, Nat. Commun., 2022, 13, 794 CrossRef.
- T.-T. Ying, M.-Y. Wan, F.-X. Wang, Y. Zhang, Y.-Z. Tang, Y.-H. Tan, J. Liao and L.-J. Wang, Chem. – Eur. J., 2023, 29, e202203893 CrossRef CAS.
- A. Cizman, D. Kowalska, M. Trzebiatowska, W. Medycki, M. Krupiński, P. Staniorowski and R. Poprawski, Dalton Trans., 2020, 49, 10394–10401 RSC.
- X.-M. Zhao, D. Li, H.-X. Zhao, Y.-P. Ren, L.-S. Long and L.-S. Zheng, Inorg. Chem., 2020, 59, 5475–5482 CrossRef CAS PubMed.
- M. Dunatov, A. Puškarić, L. Pavić, Z. Štefanić and L. Androš Dubraja, J. Mater. Chem. C, 2022, 10, 8024–8033 RSC.
- M. Dunatov, K. Molčanov, Z. Štefanić, R. Kruk and L. Androš Dubraja, Inorg. Chem., 2024, 63, 163–172 CrossRef CAS.
- M. Dunatov, A. Puškarić and L. Androš Dubraja, J. Mater. Chem. C, 2023, 11, 2880–2888 RSC.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct, Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef.
- F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley, New York, 6th edn, 2009 Search PubMed.
- P. González-Izquierdo, I. de Pedro, L. Cañadillas-Delgado, G. Beobide, O. Vallcorba, M. Sánchez-Andújar, M. T. Fernández-Díaz, J. Rodríguez Fernández and O. Fabelo, CrystEngComm, 2023, 25, 579–592 RSC.
- C. Belloso-Casuso, I. de Pedro, L. Canadillas-Delgado, G. Beobide, M. Sánchez-Andújar, J. Gárcia Ben, J. Walker, P. González Izquierdo, I. Cano, J. Rodríguez Fernández and O. Fabelo, CrystEngComm, 2024, 26, 439–451 RSC.
- C. Reitz, P. M. Leufke, H. Hahn and T. Brezesinski, Chem. Mater., 2014, 26, 2195–2202 CrossRef CAS.
- I.-G. Bae and B. Park, Sustainable Energy Fuels, 2020, 4, 3115–3128 RSC.
- D. Grosso, J. Mater. Chem., 2011, 21, 17033–17038 RSC.
- J. Hwang, N. Shoji, A. Endo and H. Daiguji, Langmuir, 2014, 30, 15550–15559 CrossRef CAS.
- L. Androš Dubraja, R. Kruk and T. Brezesinski, Adv. Electron. Mater., 2019, 5, 1800287 CrossRef.
- J. E. Ellis, S. E. Crawford and K.-J. Kim, Mater. Adv., 2021, 2, 6169–6196 RSC.
- P. Zaumseil, J. Appl. Cryst., 2015, 48, 528–532 CrossRef CAS.
- S. Neaosuphap, J. Prabket, N. Chanlek and C. Kobdaj, AIP Conf. Proc., 2020, 2279, 140001 CrossRef.
- A. Roy, R. Prasad, S. Auluck and A. Garg, Appl. Phys. Lett., 2013, 102, 182901 CrossRef.
- J. F. Scott, L. Kammerdiner, M. Parris, S. Traynor, V. Ottenbacher, A. Shawabkeh and W. F. Oliver, J. Appl. Phys., 1988, 64, 787–792 CrossRef CAS.
- J.-C. Liu, W.-Q. Liao, P.-F. Li, Y.-Y. Tang, X.-G. Chen, X.-J. Song, H.-Y. Zhang, Y. Zhang, Y.-M. You and R.-G. Xiong, Angew. Chem., Int. Ed., 2020, 59, 3495–3499 CrossRef CAS.
- C.-K. Yang, W.-N. Chen, Y.-T. Ding, J. Wang, Y. Rao, W.-Q. Liao, Y. Xie, W. Zou and R.-G. Xiong, J. Am. Chem. Soc., 2019, 141, 1781–1787 CrossRef CAS.
- Y.-Y. Tang, Y. Xie, Y. Ai, W.-Q. Liao, P.-F. Li, T. Nakamura and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 21932–21937 CrossRef CAS PubMed.
- P. González-Izquierdo, O. Fabelo, L. Canãdillas-Delgado, G. Beobide, O. Vallcorba, J. Salgado-Beceiro, M. Sánchez-Andújar, C. Martin, J. Ruiz-Fuentes, J. Eduardo García, M. T. Fernández-Díaz and I. de Pedro, J. Mater. Chem. C, 2021, 9, 4453–4465 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Single-crystal XRD data (Table S1) and PXRD data (Table S2 and Fig. S1–S5). CCDC: 2342113–2342115. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc01150c |
| This journal is © The Royal Society of Chemistry 2024 |