
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmart emissive hybrid dynamer and nanocomposite made of complementary organic and inorganic emitters combined via a supramolecular Janus synthon†
Ilya V.
Kashnik
ab,
Jeanne
Rebours
a,
Noée
Dumait
a,
Konstantin A.
Brylev
 b and
Yann
Molard
*a
b and
Yann
Molard
*a
aUniversité de Rennes, CNRS, ISCR – UMR 6226, SnMAT – UAR 2025, Rennes F-35000, France. E-mail: yann.molard@univ-rennes
bNikolaev Institute of Inorganic Chemistry SB RAS, 3 Acad. Lavrentiev ave., 630090 Novosibirsk, Russian Federation
First published on 14th May 2024
Abstract
Monotopic and ditopic blue-emitting anthracene derivatives bearing one or two thymine moieties, respectively, are associated via hydrogen bonds to a red-emitting [Mo6I8(OCOC2F5)6]2− anion, whose charge is counter-balanced by a Janus-type organic synthon made of an imidazolium head bearing an organic chain terminated with a complementary diamidopyridine group. The generated supramolecular hydrogen-bonded complex and dynamers show blue emission in solution and red emission in the solid state. Once embedded in the PMMA film, the dynamer emission colour is controlled by the irradiation time and power in the UV-2A region. This dynamic luminescent behaviour allows for evanescent optical writing on the film.
Introduction
Organic–inorganic materials are versatile substances whose abilities depend on the intrinsic properties of the isolated components and cooperative effects induced by their interactions. Depending on their functionalities, they show great potential in a tremendous number of fields such as housing, health and diagnosis, electronics, energy, and optics.1,2 In particular, emissive responsive materials able to react to one or several external stimuli such thermal treatment, electrical or mechanical stress, chemical or vapor exposure, and light irradiation,3–7 have been used in applications such as sensors, lighting, optoelectronics, data recording and storage, security and anti-counterfeiting.8–10 In this regard, using supramolecular self-assembly to construct hybrid materials facilitates the structural control of molecular functionalities via a bottom-up process. Such a strategy, which enters in the paradigm of supramolecular material nanoarchitectonics,11,12 can be used to design hybrid dynamers. Hybrid dynamers are supramolecular polymers, generated by the association of organic and inorganic monomers bearing complementary binding groups able to connect through reversible non-covalent interactions.13,14In this work, we describe the generation of a supramolecular polymer via the recognition-controlled polyassociation of ditopic heterocomplementary blue- and red-emitting monomers.
While the blue emitter contains a well-known anthracene central core functionalized with two thymine moieties, the red emitter is a dianionic octahedral molybdenum nanocluster. The dynamic assembly between both emitters relies on a Janus-type organic linker made of a diamidopyridine moiety on one side and an imidazolium cationic head on the other.
Anthracene is an attractive building block owing to its ease of functionalization and efficient deep-blue emission with a high photoluminescence quantum efficiency.15–18 It has been widely used in the design of photocyclizable supramolecular receptors for organic and inorganic guests,19–22 blue organic light-emitting diodes (OLEDs),23 and emissive moisture sensors24 and was combined with various inorganic phosphors, among which CsCu2I325 or CsPb(Br/I)326 nanocrystals are used to design white light-emitting diodes. However, in the solid state, π–π stacking between two anthracene units generally leads to a red shift and a strong quenching of its fluorescence due to excimer formation. This problem can be partially resolved by inhibiting π–π stacking through functionalization.
Octahedral molybdenum cluster complexes with the general formula [Mo6Li8La6]2− (Li = inner ligand and La = apical ligand, see Scheme 1) are nanoscale inorganic building blocks obtained first through a high-temperature solid state chemistry route. The metallic Mo6 scaffold is maintained by direct metal–metal bonds, covalently bonded to eight face-capping ligands (μ3-Li) and stabilized by six apical ligands (La).27,28 Depending on the nature of inner and apical ligands, octahedral molybdenum cluster compounds can be highly red-NIR phosphorescent29–31 with a very large Stokes shift32 and show excellent photostability.33,34 So far, they have been integrated in soft nanocomposites without the modification of their emission abilities,35–38 thanks to their mainly metal-centred emission properties. In this work, we decided to use the Cs2[Mo6I8(OCOC2F5)6] compound33 as the starting material because it represents one of the most emissive and stable cluster complex described to date in the literature.39–41
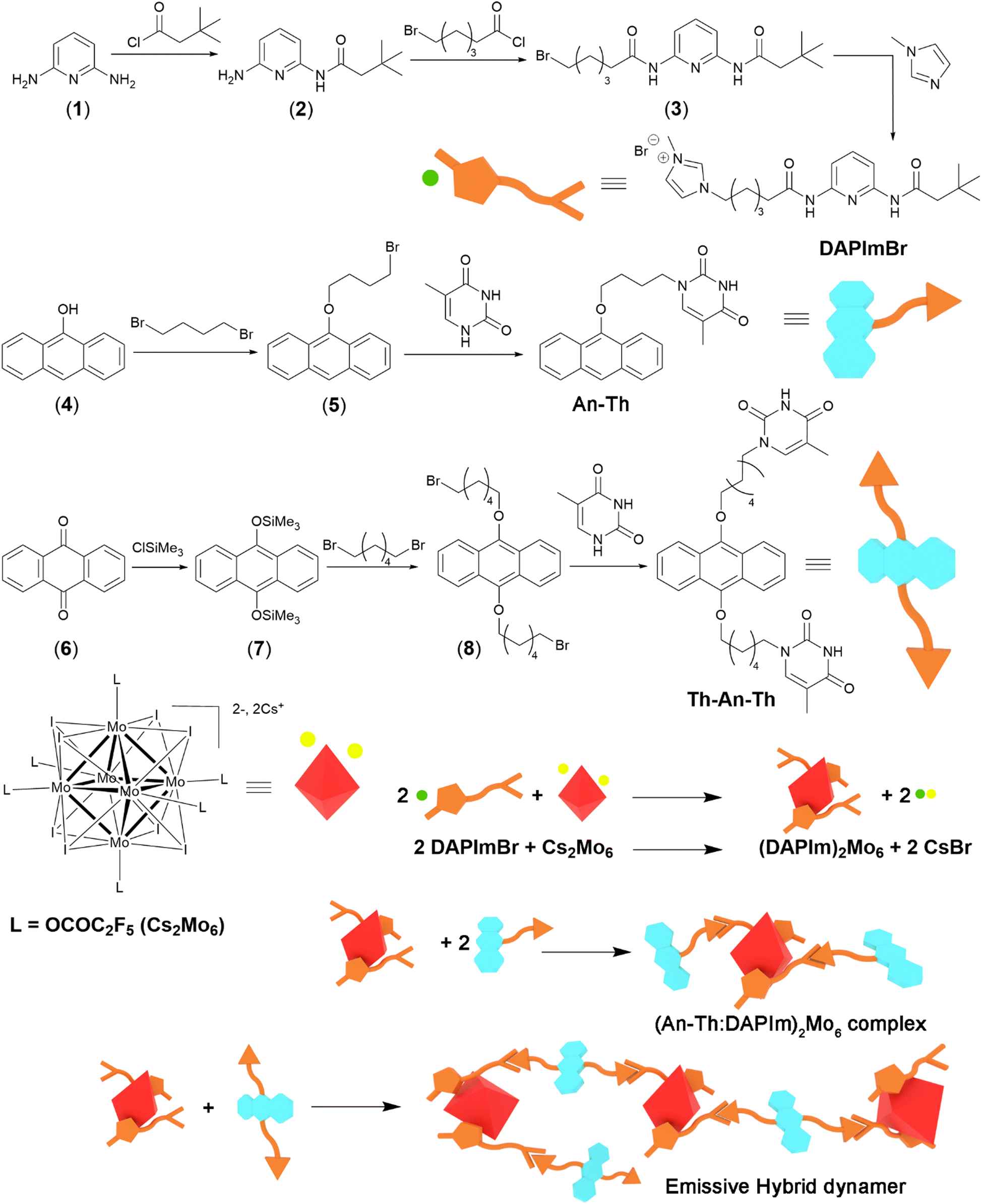 | ||
| Scheme 1 Synthetic scheme of DAPImBr salt, monotopic Th-An and ditopic Th-An-Th ligands, functionalised cluster complex (DAPIm)2Mo6, emissive complex and hybrid dynamer containing complementary emitters. | ||
Usually, dual-emissive materials are able to efficiently deliver a tailored emission that depends on the concentration and ratio of dyes. However, the degree of interactions between the emitters should also be considered to adjust the emission response toward the applied stimulus. As demonstrated herein, such an adjustment can be performed by introducing a hybrid dynamer in a host matrix, leading to a smart emissive nanocomposite whose emission colour is dependent on the irradiation time and power.
Results and discussion
Synthesis
As depicted in Scheme 1, the DAPImBr salt is a Janus-type organic linker. It bears an imidazolium head that will interact electrostatically with the dianionic cluster complex [Mo6I8(OCOC2F5)6]2− (Mo6) on one side, and on the other side, with a diamidopyridine moiety that possesses three complementary hydrogen-bond sites of thymine derivatives. It additionally bears a tert-butyl group to enhance its solubility in non-protic solvents.DAPImBr was obtained in three steps starting from 2,6-diaminopyridine (1) by amide bond formation using first 3,3-dimethylbutyryl chloride to afford compound 2 and then 6-bromohexanoyl chloride to give compound 3. Such reactions were performed in dry THF and in the presence of triethylamine. The intermediates were purified by column chromatography on silica gel. The reaction of 3 with N-methyl imidazole in CHCl3 and a catalytic amount of I2 led to the formation of the desired organic salt DAPImBr. The monotopic An-Th compound was mainly chosen as a model compound to study the supramolecular interactions between the thymine derivatives and DAPImBr or (DAPIm)2Mo6. It was synthesized in two steps starting from commercially available anthrone (4) that reacted with a large excess of 1,4-dibromobutane to afford compound 5. The alkylation of thymine was then performed with 5 in DMSO at 25 °C under argon. The ditopic Th-An-Th was obtained in several steps: first, the reduction of 9,10-anthraquinone (6) with magnesium powder at reflux in THF under argon and in the presence of chlorotrimethylsilane, giving 7, which was required prior to the reaction with 1,6-dibromohexane. 7 was isolated and crystallized as 9,10-dihydroxyanthracene.22Th-An-Th was then obtained by reacting 8 with a large excess of thymine. This compound shows a relatively poor solubility in non-polar solvents.
Targeted organic compounds were characterized by usual techniques such as 1H and 13C NMR (see ESI† Fig. S1–S13), mass spectrometry and elemental analysis. The Mo6 cluster compound was functionalized with DAPIm+ cation quantitatively via a cationic metathesis to give (DAPIm)2[Mo6I8(OCOC2F5)6] (further labelled as (DAPIm)2Mo6). The purity of this hybrid building block was assessed by EDS (no traces of Br or Cs) and by elemental analysis. The 1H NMR studies in CDCl3 indicate that associating DAPIm+ with the Mo6 anion induces an increase in the electron density on the imidazolium ring, which is reduced by an upfield shift of the imidazolium protons signals from 10.26, 7.46 and 7.32 ppm down to 9.18, 7.35 and 7.26 ppm (see ESI† Fig. S16).
Complexes were obtained in solution by mixing (DAPIm)2Mo6 with An-Th or Th-An-Th in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:1 stoichiometric ratio, respectively, followed by solvent evaporation. Due to the weakness and plurality of soft interactions, no single crystal suitable for structural determination could be obtained. Hence, spectroscopic methods were used to get insights into the assembling behaviour.
:1 or 1:1 stoichiometric ratio, respectively, followed by solvent evaporation. Due to the weakness and plurality of soft interactions, no single crystal suitable for structural determination could be obtained. Hence, spectroscopic methods were used to get insights into the assembling behaviour.
Binding studies
DAPIm+ and thymine-containing anthracene derivatives possess hydrogen bond binding sites, allowing the formation of triply hydrogen-bonded complementary pairs that should lead for Th-An-Th to the formation of supramolecular polymers known as dynamers. The association of such units is well described in the literature,42–46 and the calculated binding constant are generally lower than 1000 M−1,21 which makes 1H NMR spectroscopy the most suited spectroscopic method to evaluate them.47 Hence, we first investigated the behaviour of DAPImBr in CDCl3 upon the addition of aliquots of a CDCl3 solution containing An-Th, and, to get insight into the influence of the Mo6 cluster, we performed the same study with (DAPIm)2Mo6. The 1H NMR spectra area of interest are reproduced in Fig. 1a for the titration involving the bromide salt, and Fig. 1b presents the plots of amide proton signal positions for DAPImBr and (DAPIm)2Mo6 against the number of An-Th equivalents added to the deuterated solutions (see ESI† Tables S1, S2 and Fig. S17 for all other titration data). In both cases, the sequential addition of An-Th solution aliquots leads to a downfield shift of the two amide proton signals belonging to DAPIm+ from 8.32 and 8.98 ppm to 9.77 and 9.98 ppm, respectively. Meanwhile, the NH proton signal of thymine shows an upfield shift from 8.77 to 8.57 ppm. These results are a clear indication of the formation of the complementary hydrogen-bonded pairs in solution. From these titration curves, binding constant values of Ka = 261 ± 2 M−1 and Ka = 420 ± 3 M−1 were deduced for An-Th:DAPImBr and (An-Th:DAPIm)2Mo6 complexes, respectively.47 Such values are in the expected range for supramolecular systems based on triple hydrogen-bonded pairs and demonstrate a poor influence of the cluster core on the hydrogen-bonded assemblies. This is also expected considering the size of the linker between the imidazolium head and the diamido pyridine moiety. | ||
| Fig. 1 (a) Evolution of the 1H NMR spectrum of DAPImBr in CDCl3 ([DAPImBr] = 9.86 × 10−4 M) upon the addition of An-Th; (b) follow up of the chemical shift of amide protons vs. [An-Th]/[DAPIm+] in CDCl3 (black triangles for titration with DAPImBr; red squares for titration with (DAPIm)2Mo6). | ||
The dynamer formation between Th-An-Th and (DAPIm)2Mo6 could also be evidenced by looking at the upfield shift from 8.88 down to 8.46 ppm of the amine proton signals belonging to the thymine moieties upon increasing the Th-An-Th concentration in a solution of (DAPIm)2Mo6 (see ESI† Fig. S18). However, considering that Th-An-Th started to crystallize at concentration higher than about 10−4 M in aprotic deuterated solvent, its poor solubility prohibited the calculation of the binding constant. Considering that the monotopic and ditopic ligands are made of the same building blocks, we can reasonably assume that interactions between Th-An-Th and DAPIm+ should be of the same strength than with the previous system.
Absorption and emission studies
The absorption spectra of An-Th and Th-An-Th recorded in CH3CN show the typical signature of the anthracene 1La absorption band between 300 and 420 nm (Fig. 2a), while the (DAPIm)2Mo6 spectrum presents the typical absorption band of the {Mo6I8}4+ cluster core in the UV area and up to 500 nm, together with the absorption band of diamido pyridine centred at about 293 nm. | ||
| Fig. 2 (a) Absorption spectra (plain lines) and emission spectra (λexc = 375 nm, dashed lines) in CH3CN of An-Th (black), Th-An-Th (red) and (DAPIm)2Mo6 (blue); (b) emission spectra (λexc = 375 nm) of (An-Th:DAPIm)2Mo6 (black) and Th-An-Th:(DAPIm)2Mo6 (red) in CHCl3 (plain lines) and in the solid state (dashed lines). | ||
At this stage, we wish to state that thymine derivatives are well known to undergo a reversible photocyclization under UV-irradiation at their absorption maximum wavelength, which stands at about 280 nm.48–50 In our case, the excitation wavelength will always be at a much lower energy (375–450 nm), which precludes the formation of thymine photodimers.
The addition of 1 equivalent of An-Th in a CH3CN DAPImBr or (DAPIm)2Mo6 solution does not induce a shift of the diamido pyridine absorption band, which is a typical behaviour for host:guest systems with low binding constant values.20,21 At higher An-Th concentration, the overlap between the thymine and the diamidopyridine absorption bands precludes any clear conclusion about a shift appearance (see ESI† Fig. S19 and S20).
Steady state and time-dependent emission studies were realized in solution and in the solid state. Fig. 2 presents the emission spectra observed for An-Th, Th-An-Th, (DAPIm)2Mo6, and the corresponding mixtures, while all the recorded photophysical data are gathered in Table 1 (see ESI† Fig. S21–S31 for emission decay and fits). An-Th and Th-An-Th exhibit the typical cyan broad emission of anthracene in solution in its monomeric form with emission maxima at 422 and 443 nm, respectively. Emission lifetime decay could be fitted with one component, confirming the monomeric nature of the anthracene emission (no interactions between anthracene moieties in solution) with lifetime values of 5.7 and 13.4 ns for An-Th and Th-An-Th, respectively. Their emission in the solid state differs largely: while the same emission envelope is observed for An-Th, Th-An-Th shows a red-shifted and broader emission envelope that is attributed to anthracene excimer formation, as observed previously in the literature (see ESI† Fig. S32).21 The emission signal of the hexamolybdenum cluster anion is not sensitive to the nature of its counter cation nor to the complex formation with anthracene-based compounds. A broad emission centred at 670 nm is always observed either in deaerated solution (as all phosphorescent species, Mo6 emissive derivatives are subject to O2 quenching) or in the solid state (see ESI† Fig. S33).
| Compound | λmax, nm | τ 1 (a1, %) | τ 2 (a2, %) | τ 3 (a3, %) | τ av | Φ, % |
|---|---|---|---|---|---|---|
a In solution.
b In the solid state.
c Lifetime measured in vacuum.
d After 1 minute under UV-2A irradiation. Average lifetime is calculated as  . .
|
||||||
| An-Th | 422 | 5.7 ns | — | — | 5.7 ns | 44 |
| An-Th | 422 | 8.5 ns (1) | 1.5 ns (14) | 0.4 ns (85) | 1.7 ns | 3 |
| Th-An-Th | 443 | 13.4 ns | — | — | 13.4 ns | 60 |
| Th-An-Th | 455 | 15 ns (6) | 5.8 ns (18) | 1.0 ns (76) | 7.5 ns | 17 |
| (DAPIm)2Mo6 | 670 | 63 μs (69) | 36 μs (31) | — | 57 μs | 32 |
| (DAPIm)2Mo6 | 670 | 39 μs (49) | 18 μs (51) | — | 32 μs | 12 |
| (An-Th:DAPIm)2Mo6 | 668 | 70 μs (42) | 33 μs (58) | — | 55 μs | 6 |
| Th-An-Th:(DAPIm)2Mo6 | 668 | 158 μs (9) | 69 μs (36) | 17 μs (55) | 84 μs | 1 |
| PolAn | 440 | 17 ns | — | — | 17 ns | 97 |
| PolMo | 668 | 111 μs (45) | 53 μs (55) | — | 90 μs | 12 |
| Poldyn | 440 | 11 ns (63) | 3.4 ns (37) | — | 9.8 ns | 10 47d |
| 668 | 124 μs (7) | 38 μs (93) | — | 55 μs | ||
| Poldyn | 440 | 10 ns (48) | 3.4 ns (52) | — | 8.2 ns | — |
| 668 | 243 μs (73) | 123 μs (27) | — | 224 μs | — | |
The overlap between the anthracene emission band and the Mo6 cluster anion absorption band leads to a very efficient energy transfer. Indeed, the calculated Förster distance for this system is 5.85 nm (see ESI† Fig. S34). The efficiency of this energy transfer was verified in a CHCl3 solution by a titration experiment involving the stepwise increase of (DAPIm)2Mo6 concentration while keeping the An-Th concentration constant. Each addition of the cluster complex led to the partial quenching of An-Th, whose emission disappeared completely after the addition of 14 equivalents of (DAPIm)2Mo6 (see ESI† Table S4 and Fig. S35). The nature of this energy transfer seems to be of trivial type (resonance energy transfer), as confirmed by evaluating the anthracene moiety excited state lifetime value that remained unchanged (around 6 ns) upon the addition of up to 3 equivalents of (DAPIm)2Mo6 (see ESI† Table S3 and Fig. S36–S39).
Fig. 2b presents the emission spectra recorded in solution and in the solid state for hybrid supramolecular complexes (An-Th:DAPIm)2Mo6 and Th-An-Th:(DAPIm)2Mo6. They were prepared by mixing stoichiometric amounts of DAP2Mo and An-Th or Th-An-Th in CHCl3 in 1:2 or 1:1 ratio, respectively, and then evaporating to dryness. In aerated solution, the anthracene emission band is more intense, while deaerating the solution leads to the increase of the cluster emission band. In the solid state, however, only the red emission of the cluster component is observed with an enhancement of its average lifetime compared to DAP2Mo from 32 μs to 55 μs. These facts actually indicate the efficient energy transfer from the anthracene to the cluster complex.
The dynamer was also let under constant irradiation at 375 nm to check its stability, and we observed an enhancement of the cluster emission band by about 14% after the first 20 seconds of irradiation, which was followed by a slight decrease.
Design of a smart emissive nanocomposite
Considering that in the solid state only the emission band of (DAPIm)2Mo6 is observed in the overall emission signal envelope of pure Th-An-Th:(DAPIm)2Mo6 dynamer, we decided to lower the supramolecular interactions strength between the emitters by embedding the dynamer in a poly(methylmetacrylate) (PMMA) matrix. We hypothesized that such an embedment would affect the inter-emitters distance and lead to the enhancement of the anthracene emission band by lowering the energy transfer efficiency to the cluster compound. Considering that PMMA has a high degree of rigidity compared to other host matrices and a triplet energy of 3.1 eV,51 this is a suitable wide band gap host matrix and a material of choice to observe luminescence from our emitters.52Hence, a homogeneous 2.3 wt%-doped PMMA film was obtained by the slow evaporation of the CHCl3/MeOH (8:1) mixture containing commercial PMMA and the dynamer (Poldyn). For the sake of comparison, PMMA samples containing either (DAPIm)2Mo6 (PolMo) or Th-An-Th (PolAn) at the same concentration than in Poldyn were also prepared. The samples are presented in Fig. 3a under day light and UV-2A irradiation, while the emission spectra at the beginning of the irradiation are presented in Fig. 3b. According to its emission spectrum and calculated excited state lifetime, Th-An-Th behaves in the PMMA film like in solution. Only the bifunctionalized anthracene monomer emission is recorded with an excitation decay that could be fitted with a 17 ns single component. In the presence of (DAPIm)2Mo6, the Th-An-Th emission decay was fitted with two components with an average lifetime value of 9.8 or 8.2 ns in air or in vacuum, respectively. Such a decrease is attributed to the remaining energy transfer from the organic emitter to the inorganic one.
 | ||
| Fig. 3 (a) Doped PMMA films under daylight and UV-2A irradiation immediately after turning on the UV lamp and after 30 s (left: PolMo, center: PolAn, right: Poldyn); (b) emission spectra recorded for PolAn (blue), PolMo (red) and Poldyn (black); (c) evolution of the emission envelop of Poldyn film under 1 min irradiation at 375 nm in air (inset: follow up of the emission maxima intensity ratio with the irradiation time); (d) 1931 CIE coordinates diagram for 1 min irradiation at 375 nm; (e) Poldyn film exposed under UV-2A after the optical writing up of “Hi!” with a 405 nm laser pointer. | ||
As presented in Fig. 3c, a prolonged excitation of Poldyn at 375 nm, with the pulsed laser diode used for lifetime studies, leads to the enhancement of the cluster compound emission band and a slight lowering of the anthracene one. The ratio value between the two emission maxima intensity I668/I440 passes from 0.9 to 2.8 after 10 s and stabilizes around 3 after 1 min of irradiation. Therefore, the emission colour of the dynamer containing film depends on the irradiation time and power and passes from blue (CIE coordinates: x = 0.25, y = 0.14) to red (CIE coordinates: x = 0.38, y = 0.19) upon irradiation (Fig. 3d). Moreover, the irradiation also affects the absolute quantum yield, which goes from 0.10 to 0.47 after 1 min under a 4 W UV-2A lamp. Keeping the sample in the dark leads to the recovery of the blue emission colour. Such behaviour allowed us to use Pdyn as a support for optical writing with a 5 mW 405 nm laser pointer, as depicted in Fig. 3e (see also ESI† Video S1 for the writing process). The mechanism of the colour changes, and thus the writing process, is attributed to the increase of the cluster compound emission efficiency induced by a depletion of the local O2 concentration within the PMMA film upon irradiation. We already observed such a phenomenon when [Mo6I8(OCOC2F5)6]2− was associated with oxindole53 or tetraphenylethylene54 derivatives. In fact, the O2 concentration lowering is induced by the reaction of the fundamental O2 (3Σg−) triplet state with the cluster compound excited triplet state. The O2 consumption rate is related to the cluster compound excited state generation, which explains why irradiation time and power are deeply related to stabilize the cluster emission. Indeed, when the dynamer-doped PMMA film is placed under high vacuum (1.9 × 10−6 hPa), the intensity of the blue and red components does not vary with a I668/I440 ratio value of 10.3 upon constant irradiation even after 300 s (see ESI† Fig. S40).
Experimental
Starting materials were purchased from Alfa Aesar or Aldrich and used without any further purification. All solvents were distilled and dried before use. Cs2[Mo6I8(OCOC2F5)6] precursor was obtained by the reported procedures.33 NMR experiments were realized at 298 K on a Bruker AV III 400 MHz spectrometer fitted with a BBFO probe and Bruker AV III HD 500 MHz spectrometer fitted with a BBFO probe. All peaks were referenced to the methyl signals of TMS at δ = 0 ppm. Mass spectrometry and elemental analysis were performed with a Bruker MaXis 4G spectrometer and a Thermo Fisher Flash EA1112 microanalyzer, respectively, at the Centre Régional de Mesures Physiques de l’Ouest (CRMPO). UV-vis absorption spectra and excitation vs. emission maps were recorded with a Horiba Duetta spectrophotometer. The absolute quantum yields were measured with a C9920-03 Hamamatsu system. Lifetime measurements and TRPL mapping at 296 K were realized using a picosecond laser diode (Jobin Yvon deltadiode, 375 nm) and a Hamamatsu C10910-25 streak camera mounted with a slow single sweep unit. Signals were integrated on the whole emission decay. Fits were calculated using Hamamatsu software and the goodness of fit judge by the reduced χ2 value and residual plot shape. Irradiation time-dependent emission studies were performed with the picosecond 375 nm laser diode used for lifetime measurements. Spectra were recorded with an Ocean Optics QEPro CCD spectrometer.Synthetic procedures
:1) as eluent). Then, the product was dried at 60 °C overnight under vacuum to give compound 2 as a white solid (3.7 g, 78%). 1H NMR (400 MHz, CDCl3) δ = 7.63 (s, 1H, NH), 7.58 (d, J = 8.0, 1H, CHpy–C–NH), 7.46 (t, J = 7.9, 1H, CH), 6.26 (d, J = 7.9, 1H, CHpy–C–NH2), 4.32 (s, 2H, NH2), 2.21 (s, 2H, CH2), 1.09 (s, 9H, 3CH3). 13C NMR (101 MHz, CDCl3) δ = 170.21 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 157.05 (Cpy), 149.83 (Cpy), 140.12 (Cpy), 104.18 (Cpy), 103.26 (Cpy), 51.72 (CH2), 31.29 (C–(CH3)3), 29.80 (3CH3). MS (ESI, CH3OH/CH2Cl2 90/10): [M+H] C11H18N3O m/z (theoretical) = 208.1444, m/z (found) = 208.1448; [M+Na] C11H17N3ONa m/z (theoretical) = 230.1264, m/z (found) = 230.1266. E.A.: calc. for C11H17N3O: C, 63.74; H, 8.27; N, 20.27; found: C, 63.87; H, 8.18; N, 19.97.
:1) as eluent). Then, the product was dried at 60 °C overnight under vacuum to give compound 3 as a white solid (4.68 g, 72%). 1H NMR (400 MHz, CDCl3) δ = 7.91 (dd, J = 13.7, 8.1 Hz, 2H, 2CHpy–C–NH), 7.71 (t, J = 8.1 Hz, 1H, CHpy), 7.65 (s, 1H, NH), 7.57 (s, 1H, NH), 3.43 (t, J = 6.7 Hz, 2H, CH2–Br), 2.41 (t, J = 7.4 Hz, 2H, CH2–C(O)NH), 2.25 (s, 2H, CH2–C(CH3)3), 1.97–1.86 (m, 2H, CH2), 1.82–1.70 (m, 2H, CH2), 1.60–1.48 (m, 2H, CH2), 1.11 (s, 9H, 3CH3). 13C NMR (101 MHz, CDCl3) δ = 171.05 (CO), 170.25 (CO), 149.42 (Cpy), 149.35 (Cpy), 140.86 (Cpy), 109.44 (Cpy), 109.41 (Cpy), 51.75 (CH2), 37.44 (CH2), 33.46 (CH2), 32.42 (CH2), 31.36 (C–(CH3)3), 29.79 (3CH3), 27.69 (CH2), 24.36 (CH2). MS (ESI, CH3OH/CH2Cl2 90/10): [M+Na] C17H26N3O2BrNa m/z (theoretical) = 406.11006, m/z (found) = 406.1110. [M+H] C17H27N3O2Br m/z (theoretical) = 384.1281, m/z (found) = 384.1283. Traces: [M2+Na] C17H26N3O2ClNa m/z (theoretical) = 362.1606, m/z (found) = 362.1609. E.A.: calc. for C17H26N3O2Br: C, 53.13; H, 6.82; N, 10.93; found: C, 53.51; H, 6.64; N, 10.45.
O), 171.14 (CO), 149.88 (Cpy), 140.45 (Cpy), 137.58 (Cpy), 123.12 (Cimidazole), 122.26 (Cimidazole), 109.40 (Cpy), 109.34 (Cpy), 51.03 (CH2), 49.77 (CH2), 36.72 (Cimidazole), 36.62 (CH2), 31.35 (C–(CH3)3), 29.79 (3CH3), 29.41 (CH2), 25.10 (CH2), 24.23(CH2). MS (ESI, CH3OH/CH2Cl2 90/10): [M+] C21H32N5O2m/z (theoretical) = 386.2551, m/z (found) = 386.2548. E.A.: calc. for C21H32N5O2Br·0.25CHCl3: C, 51.43; H, 6.55; N, 14.11; found: C, 51.30; H, 6.82; N, 14.09. UV-vis in CH3CN λmax, nm (ε, mol−1 dm3 cm−1): 297 (12352), 293 sh (11570), 285 sh (9399), 261 (3548), 254 (3534).
:1) as eluent). Then, the product was dried at 60 °C overnight under vacuum to give compound 5 as a white solid (1.82 g, 37%). 1H NMR (400 MHz, CDCl3) δ = 8.33–8.18 (m, 3H, 3CHanthracene), 8.06–7.99 (m, 2H, 2CHanthracene), 7.56–7.44 (m, 4H, 4CHanthracene), 4.26 (t, J = 6.2 Hz, 2H, O–CH2), 3.64 (t, J = 6.5 Hz, 2H, Br–CH2), 2.36 (dtd, J = 8.9, 6.7, 4.9 Hz, 2H, CH2), 2.29–2.19 (m, 2H, CH2). MS (ESI, CH3OH/CH2Cl2 90/10): [M+H] C18H18OBr m/z (theoretical) = 329.0535, m/z (found) = 329.0534; [M] C18H17OBr m/z (theoretical) = 328.0457, m/z (found) = 328.0449. E.A.: calc. for C18H17OBr: C, 65.67; H, 5.20; found: C, 65.83; H, 4.99.
O), 150.89 (N–CO/Canthracene–O), 140.54 (N–CH), 132.43 (Canthracene), 128.56 (Canthracene), 125.53 (Canthracene), 125.46 (Canthracene), 125.33 (Canthracene), 125.13 (Canthracene), 124.61 (Canthracene), 122.50 (Canthracene), 122.35 (Canthracene), 122.08 (Canthracene), 110.77 (C–CH3), 75.09 (CH2–O), 48.64 (CH2–N), 27.54 (CH2–CH2–O), 26.32 (CH2–CH2–N), 12.37 (CH3). MS (ESI, CH3OH/CH2Cl2 90/10): [M+Na] C23H22N2O3Na m/z (theoretical) = 397.1523, m/z (found) = 397.1525; [M–H+2Na] C23H21N2O3Na2m/z (theoretical) = 419.1342, m/z (found) = 419.1342; [M+K] C23H22N2O3K m/z (theoretical) = 413.1262, m/z (found) = 413.1260. E.A.: calc. for C23H22N2O3: C, 73.78; H, 5.92; N, 7.48; found: C, 73.77; H, 5.74; N, 7.36. UV-vis in CH3CN λmax, nm (ε, mol−1 dm3 cm−1): 390 (6967), 370 (8107), 352 (5375), 335 (2718), 320 sh (1202).
:1) as the eluent). Then, the product was dried at 60 °C overnight under vacuum to give compound 8 as a yellow-greenish solid (1 g, 66%). 1H NMR (300 MHz, CDCl3) δ = 8.33–8.25 (m, 4H, 4CHanthracene), 7.54–7.46 (m, 4H, 4CHanthracene), 4.19 (t, J = 6.5 Hz, 4H, 2CH2–O), 3.50 (t, J = 6.7 Hz, 4H, 2CH2–Br), 2.04 (ddt, J = 24.8, 14.1, 6.8 Hz, 8H, 4CH2), 1.83–1.59 (m, 8H, 4CH2). MS (ESI, CH3OH/CH2Cl2 90/10): [M+H] C26H33O2Br2m/z (theoretical) = 535.08418, m/z (found) = 535.0842; [M] C26H32O2Br2m/z (theoretical) = 534.07635, m/z (found) = 534.0760; [M–HBr+H] C26H32O2Br m/z (theoretical) = 455.15802, m/z (found) = 455.1562. E.A.: calc. for C26H32O2Br2: C, 58.22; H, 6.01; found: C, 58.23; H, 6.03.
O), 151.60 (CO), 147.14, 141.77 (CHthymine), 125.02 (CHanthracene), 122.18 (CHanthracene), 109.72, 75.52 (CH2–O), 47.90 (CH2–N), 30.04 (CH2–CH2–O), 28.55 (CH2), 26.01 (CH2), 25.64 (CH2), 10.74 (CH3). MS (ESI, CH3OH/CH2Cl2 90/10): [M+Na] C36H42N4O6Na m/z (theoretical) = 649.29966, m/z (found) = 649.3002; [M+K] C36H42N4O6K m/z (theoretical) = 665.27359, m/z (found) = 665.2736; [M–H+2Na] C36H41N4O6Na2m/z (theoretical) = 671.2816, m/z (found) = 671.2816; [M–H+Na+K] C36H41N4O6NaK m/z (theoretical) = 687.25554, m/z (found) = 687.2554. E.A.: calc. for C36H42N4O6 + 0.5H2O: C, 68.01; H, 6.81; N, 8.81; found: C, 68.26; H, 6.70; N, 8.62. UV-vis in CHCl3λmax, nm (ε, mol−1 dm3 cm−1): 407 (6670), 385 (7686), 367 (5531), 349 sh (2783), 332 sh (1153).
:I = 6.0:7.8 (no presence of Cs). UV-vis in CH3CN λmax, nm (ε, mol−1 dm3 cm−1): 406 sh (4538), 338 sh (8035), 289 (51036).
O), 157.05 (Cpy), 149.83 (Cpy), 140.12 (Cpy), 104.18 (Cpy), 103.26 (Cpy), 51.72 (CH2), 31.29 (C–(CH3)3), 29.80 (3CH3). MS (ESI, CH3OH/CH2Cl2 90/10): [M+H] C11H18N3O m/z (theoretical) = 208.1444, m/z (found) = 208.1448; [M+Na] C11H17N3ONa m/z (theoretical) = 230.1264, m/z (found) = 230.1266. E.A.: calc. for C11H17N3O: C, 63.74; H, 8.27; N, 20.27; found: C, 63.87; H, 8.18; N, 19.97.
:1) as eluent). Then, the product was dried at 60 °C overnight under vacuum to give compound 3 as a white solid (4.68 g, 72%). 1H NMR (400 MHz, CDCl3) δ = 7.91 (dd, J = 13.7, 8.1 Hz, 2H, 2CHpy–C–NH), 7.71 (t, J = 8.1 Hz, 1H, CHpy), 7.65 (s, 1H, NH), 7.57 (s, 1H, NH), 3.43 (t, J = 6.7 Hz, 2H, CH2–Br), 2.41 (t, J = 7.4 Hz, 2H, CH2–C(O)NH), 2.25 (s, 2H, CH2–C(CH3)3), 1.97–1.86 (m, 2H, CH2), 1.82–1.70 (m, 2H, CH2), 1.60–1.48 (m, 2H, CH2), 1.11 (s, 9H, 3CH3). 13C NMR (101 MHz, CDCl3) δ = 171.05 (CO), 170.25 (CO), 149.42 (Cpy), 149.35 (Cpy), 140.86 (Cpy), 109.44 (Cpy), 109.41 (Cpy), 51.75 (CH2), 37.44 (CH2), 33.46 (CH2), 32.42 (CH2), 31.36 (C–(CH3)3), 29.79 (3CH3), 27.69 (CH2), 24.36 (CH2). MS (ESI, CH3OH/CH2Cl2 90/10): [M+Na] C17H26N3O2BrNa m/z (theoretical) = 406.11006, m/z (found) = 406.1110. [M+H] C17H27N3O2Br m/z (theoretical) = 384.1281, m/z (found) = 384.1283. Traces: [M2+Na] C17H26N3O2ClNa m/z (theoretical) = 362.1606, m/z (found) = 362.1609. E.A.: calc. for C17H26N3O2Br: C, 53.13; H, 6.82; N, 10.93; found: C, 53.51; H, 6.64; N, 10.45.
O), 171.14 (CO), 149.88 (Cpy), 140.45 (Cpy), 137.58 (Cpy), 123.12 (Cimidazole), 122.26 (Cimidazole), 109.40 (Cpy), 109.34 (Cpy), 51.03 (CH2), 49.77 (CH2), 36.72 (Cimidazole), 36.62 (CH2), 31.35 (C–(CH3)3), 29.79 (3CH3), 29.41 (CH2), 25.10 (CH2), 24.23(CH2). MS (ESI, CH3OH/CH2Cl2 90/10): [M+] C21H32N5O2m/z (theoretical) = 386.2551, m/z (found) = 386.2548. E.A.: calc. for C21H32N5O2Br·0.25CHCl3: C, 51.43; H, 6.55; N, 14.11; found: C, 51.30; H, 6.82; N, 14.09. UV-vis in CH3CN λmax, nm (ε, mol−1 dm3 cm−1): 297 (12352), 293 sh (11570), 285 sh (9399), 261 (3548), 254 (3534).
:1) as eluent). Then, the product was dried at 60 °C overnight under vacuum to give compound 5 as a white solid (1.82 g, 37%). 1H NMR (400 MHz, CDCl3) δ = 8.33–8.18 (m, 3H, 3CHanthracene), 8.06–7.99 (m, 2H, 2CHanthracene), 7.56–7.44 (m, 4H, 4CHanthracene), 4.26 (t, J = 6.2 Hz, 2H, O–CH2), 3.64 (t, J = 6.5 Hz, 2H, Br–CH2), 2.36 (dtd, J = 8.9, 6.7, 4.9 Hz, 2H, CH2), 2.29–2.19 (m, 2H, CH2). MS (ESI, CH3OH/CH2Cl2 90/10): [M+H] C18H18OBr m/z (theoretical) = 329.0535, m/z (found) = 329.0534; [M] C18H17OBr m/z (theoretical) = 328.0457, m/z (found) = 328.0449. E.A.: calc. for C18H17OBr: C, 65.67; H, 5.20; found: C, 65.83; H, 4.99.
O), 150.89 (N–CO/Canthracene–O), 140.54 (N–CH), 132.43 (Canthracene), 128.56 (Canthracene), 125.53 (Canthracene), 125.46 (Canthracene), 125.33 (Canthracene), 125.13 (Canthracene), 124.61 (Canthracene), 122.50 (Canthracene), 122.35 (Canthracene), 122.08 (Canthracene), 110.77 (C–CH3), 75.09 (CH2–O), 48.64 (CH2–N), 27.54 (CH2–CH2–O), 26.32 (CH2–CH2–N), 12.37 (CH3). MS (ESI, CH3OH/CH2Cl2 90/10): [M+Na] C23H22N2O3Na m/z (theoretical) = 397.1523, m/z (found) = 397.1525; [M–H+2Na] C23H21N2O3Na2m/z (theoretical) = 419.1342, m/z (found) = 419.1342; [M+K] C23H22N2O3K m/z (theoretical) = 413.1262, m/z (found) = 413.1260. E.A.: calc. for C23H22N2O3: C, 73.78; H, 5.92; N, 7.48; found: C, 73.77; H, 5.74; N, 7.36. UV-vis in CH3CN λmax, nm (ε, mol−1 dm3 cm−1): 390 (6967), 370 (8107), 352 (5375), 335 (2718), 320 sh (1202).
:1) as the eluent). Then, the product was dried at 60 °C overnight under vacuum to give compound 8 as a yellow-greenish solid (1 g, 66%). 1H NMR (300 MHz, CDCl3) δ = 8.33–8.25 (m, 4H, 4CHanthracene), 7.54–7.46 (m, 4H, 4CHanthracene), 4.19 (t, J = 6.5 Hz, 4H, 2CH2–O), 3.50 (t, J = 6.7 Hz, 4H, 2CH2–Br), 2.04 (ddt, J = 24.8, 14.1, 6.8 Hz, 8H, 4CH2), 1.83–1.59 (m, 8H, 4CH2). MS (ESI, CH3OH/CH2Cl2 90/10): [M+H] C26H33O2Br2m/z (theoretical) = 535.08418, m/z (found) = 535.0842; [M] C26H32O2Br2m/z (theoretical) = 534.07635, m/z (found) = 534.0760; [M–HBr+H] C26H32O2Br m/z (theoretical) = 455.15802, m/z (found) = 455.1562. E.A.: calc. for C26H32O2Br2: C, 58.22; H, 6.01; found: C, 58.23; H, 6.03.
O), 151.60 (CO), 147.14, 141.77 (CHthymine), 125.02 (CHanthracene), 122.18 (CHanthracene), 109.72, 75.52 (CH2–O), 47.90 (CH2–N), 30.04 (CH2–CH2–O), 28.55 (CH2), 26.01 (CH2), 25.64 (CH2), 10.74 (CH3). MS (ESI, CH3OH/CH2Cl2 90/10): [M+Na] C36H42N4O6Na m/z (theoretical) = 649.29966, m/z (found) = 649.3002; [M+K] C36H42N4O6K m/z (theoretical) = 665.27359, m/z (found) = 665.2736; [M–H+2Na] C36H41N4O6Na2m/z (theoretical) = 671.2816, m/z (found) = 671.2816; [M–H+Na+K] C36H41N4O6NaK m/z (theoretical) = 687.25554, m/z (found) = 687.2554. E.A.: calc. for C36H42N4O6 + 0.5H2O: C, 68.01; H, 6.81; N, 8.81; found: C, 68.26; H, 6.70; N, 8.62. UV-vis in CHCl3λmax, nm (ε, mol−1 dm3 cm−1): 407 (6670), 385 (7686), 367 (5531), 349 sh (2783), 332 sh (1153).
:I = 6.0:7.8 (no presence of Cs). UV-vis in CH3CN λmax, nm (ε, mol−1 dm3 cm−1): 406 sh (4538), 338 sh (8035), 289 (51036).
Preparation of supramolecular complexes
(An-Th:DAPIm)2Mo6: 50 mg (15 μmol) of (DAPIm)2Mo6 and 11 mg (30 μmol) of compound An-Th were dissolved in 10 mL of CHCl3. The solvent was removed under reduced pressure to yield an orange powder of (An-Th:DAPIm)2Mo6.Th-An-Th:(DAPIm)2Mo6: 50 mg (15 μmol) of (DAPIm)2Mo6 and 18.8 mg (30 μmol) of compound Th-An-Th were dissolved in 10 mL of CHCl3. The solvent was removed under reduced pressure to yield an orange powder Th-An-Th:(DAPIm)2Mo6.
Preparation of doped PMMA films
All films were prepared using commercial PMMA (MW: 120000 g mol−1).
PolMo: 2 g of PMMA and 40 mg of compound (DAPIm)2Mo6 were dissolved in 30 mL of CH2Cl2. The resulting solution was placed in a closed Petri dish and left for evaporation at room conditions to yield a thin transparent orange film.
PolAn: 2 g of PMMA and 7.5 mg of compound Th-An-Th were dissolved in 30 mL of CH2Cl2/MeOH mixture (8:1 v/v). The resulting solution was placed in a closed Petri dish and left for evaporation at room conditions to yield a thin transparent colorless film.
Poldyn: 2 g of PMMA, 40 mg of compound (DAPIm)2Mo6 and 7.5 mg of compound Th-An-Th were dissolved in 30 mL of CH2Cl2/MeOH mixture (8:1 v/v). The resulting solution was placed in a closed Petri dish and left for evaporation at room conditions to yield a thin transparent orange film.
Conclusion
Supramolecular luminescent organic and inorganic building blocks bearing complementary hydrogen-bond binding group were synthesized. While blue-emitting anthracene derivatives were functionalized by one or two thymine groups, yielding the monotopic An-Th and ditopic Th-An-Th compounds, the red-NIR phosphorescent [Mo6I8(OCOC2F5)6]2− cluster dianion was associated with two tailor-made organic Janus-type cations, i.e., bearing an imidazolium head on one side to interact electrostatically with the inorganic anion and a diamidopyridine moiety on the other side to bind with the complementary thymine group of the blue emitter. The association constant deduced for (An-Th:DAPIm)2Mo6 from the NMR titration experiment in CDCl3 showed typical values for supramolecular assemblies based on triple hydrogen-bonded pair. Photophysical studies were first realized in solution and in the solid state and demonstrated an efficient energy transfer from the blue-emitting organic emitter to the red-NIR phosphorescent cluster to such an extent that only the cluster emission is observable. This behaviour was observed for both (An-Th:DAPIm)2Mo6 supramolecular complex and Th-An-Th:(DAPIm)2Mo6 dynamer. To minimize this energy transfer, the dynamer was embedded in an optically transparent PMMA matrix at 2 wt%. Such an integration increases the distance between the emitters by breaking the hydrogen-bonded network, thus leading to a blue-emitting polymer whose emission colour turns red under prolonged irradiation. This reversible emission colour change shows that the hybrid-doped PMMA film could serve as a medium for data encryption and reading, which would be performed using the same UV-2A light stimulus, a particularly efficient and cost-effective system for the end-user.Author contributions
I. V. K: investigation, validation, writing – review – editing; J. R.: resources, writing – review – editing; N. D.: resources; K. A. B.: supervision, funding acquisition, writing – review – editing; Y. M.: conceptualization, investigation, funding acquisition, supervision, project administration, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge Dr S. Cordier for financial support through the CLUSPOM IRP Network. I. V. K. thanks the French Embassy for providing the Vernadski scholarship for the co-tutelle PhD program between France and Russia. I. V. K. and K. A. B. thank Russian science foundation (project 19-73-20196-p). J. R. thanks the EUR LUMOMAT project and the Investments for the Future program ANR-18-EURE-0012 for PhD financial support.References
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 40, 696–753 RSC.
- S. Parola, B. Julián-López, L. D. Carlos and C. Sanchez, Adv. Funct. Mater., 2016, 26, 6506–6544 CrossRef CAS.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- S. Yamane, K. Tanabe, Y. Sagara and T. Kato, Top. Curr. Chem., 2012, 318, 395–406 CrossRef CAS PubMed.
- X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971–1981 CrossRef CAS PubMed.
- M.-K. Tsang, G. Bai and J. Hao, Chem. Soc. Rev., 2015, 44, 1585–1607 RSC.
- E. Cariati, E. Lucenti, C. Botta, U. Giovanella, D. Marinotto and S. Righetto, Coord. Chem. Rev., 2016, 306, 566–614 CrossRef CAS.
- X. Yu, H. Chen, X. Shi, P.-A. Albouy, J. Guo, J. Hu and M.-H. Li, Mater. Chem. Front., 2018, 2, 2245–2253 RSC.
- Y. Zhang, S. Zhou, K. C. Chong, S. Wang and B. Liu, Mater. Chem. Front., 2019, 3, 836–841 RSC.
- P. Alam, N. L. C. Leung, Y. Cheng, H. Zhang, J. Liu, W. Wu, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 4536–4540 CrossRef CAS PubMed.
- K. Ariga, M. Li, G. J. Richards and J. P. Hill, J. Nanosci. Nanotechnol., 2011, 11, 1–13 CrossRef CAS PubMed.
- M. Aono and K. Ariga, Adv. Mater., 2016, 28, 989–992 CrossRef CAS PubMed.
- J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS.
- N. Roy, B. Bruchmann and J.-M. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2014, 43, 1885–1898 RSC.
- G. S. Baviera and P. M. Donate, Beilstein J. Org. Chem., 2021, 17, 2028–2050 CrossRef CAS PubMed.
- H. Bouas-Laurent, A. Castellan, J. P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC.
- H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2001, 30, 248–263 RSC.
- G. McSkimming, J. H. R. Tucker, H. Bouas-Laurent, J. P. Desvergne, S. J. Coles, M. B. Hursthouse and M. E. Light, Chem. – Eur. J., 2002, 8, 3331–3342 CrossRef CAS PubMed.
- Y. Molard, D. M. Bassani, J.-P. Desvergne, P. N. Horton, M. B. Hursthouse and J. H. R. Tucker, Angew. Chem., Int. Ed., 2005, 44, 1072–1075 CrossRef CAS PubMed.
- Y. Molard, D. M. Bassani, J.-P. Desvergne, N. Moran and J. H. R. Tucker, J. Org. Chem., 2006, 71, 8523–8531 CrossRef CAS PubMed.
- D. Marquis, J.-P. Desvergne and H. Bouas-Laurent, J. Org. Chem., 1995, 60, 7984–7996 CrossRef CAS.
- B. Chen, B. Liu, J. Zeng, H. Nie, Y. Xiong, J. Zou, H. Ning, Z. Wang, Z. Zhao and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1803369 CrossRef.
- S. Miho, K. Imato and Y. Ooyama, RSC Adv., 2022, 12, 25687–25696 RSC.
- W. Liu, K. W. Ng, H. Lin, Z. Dai, J. Xu, S. Su, Z. Tang and S. Wang, J. Phys. Chem. C, 2021, 125, 13076–13083 CrossRef CAS.
- X. Shen, C. Sun, X. Bai, X. Zhang, Y. Wang, Y. Wang, H. Song and W. W. Yu, ACS Appl. Mater. Interfaces, 2018, 10, 16768–16775 CrossRef CAS PubMed.
- S. Cordier, F. Grasset, Y. Molard, M. Amela-Cortes, R. Boukherroub, S. Ravaine, M. Mortier, N. Ohashi, N. Saito and H. Haneda, J. Inorg. Organomet. Polym. Mater., 2015, 25, 189–204 CrossRef CAS.
- F. A. Cotton, Inorg. Chem., 1964, 3, 1217–1220 CrossRef CAS.
- K. Kirakci, P. Kubat, M. Dusek, K. Fejfarova, V. Sicha, J. Mosinger and K. Lang, Eur. J. Inorg. Chem., 2012, 3107–3111 CrossRef CAS.
- M. A. Mikhaylov and M. N. Sokolov, Eur. J. Inorg. Chem., 2019, 4181–4197 CrossRef CAS.
- M. N. Sokolov, M. A. Mihailov, E. V. Peresypkina, K. A. Brylev, N. Kitamura and V. P. Fedin, Dalton Trans., 2011, 40, 6375 RSC.
- Y. Zhao and R. R. Lunt, Adv. Energy Mater., 2013, 3, 1143–1148 CrossRef CAS.
- M. Amela-Cortes, Y. Molard, S. Paofai, A. Desert, J.-L. Duvail, N. G. Naumov and S. Cordier, Dalton Trans., 2016, 45, 237–245 RSC.
- J. Choi, D. Nguyen, E. Gi, K. A. Brylev, J. W. Yu, D. Kim, W. B. Lee, D. H. Kim, I. Chung, K. K. Kim and S.-J. Kim, J. Mater. Chem. C, 2022, 10, 4402–4410 RSC.
- Y. Molard, C. Labbe, J. Cardin and S. Cordier, Adv. Funct. Mater., 2013, 23, 4821–4825 CrossRef CAS.
- M. Amela-Cortes, A. Garreau, S. Cordier, E. Faulques, J.-L. Duvail and Y. Molard, J. Mater. Chem. C, 2014, 2, 1545–1552 RSC.
- M. Robin, W. Kuai, M. Amela-Cortes, S. Cordier, Y. Molard, T. Mohammed-Brahim, E. Jacques and M. Harnois, ACS Appl. Mater. Interfaces, 2015, 7, 21975–21984 CrossRef CAS PubMed.
- M. Robin, N. Dumait, M. Amela-Cortes, C. Roiland, M. Harnois, E. Jacques, H. Folliot and Y. Molard, Chem. – Eur. J., 2018, 24, 4825–4829 CrossRef CAS PubMed.
- K. Kirakci, K. Fejfarova, M. Kucerakova and K. Lang, Eur. J. Inorg. Chem., 2014, 2331–2336 CrossRef CAS.
- K. Kirakci, M. A. Shestopalov and K. Lang, Coord. Chem. Rev., 2023, 481, 215048 CrossRef CAS.
- M. A. Mikhailov, K. A. Brylev, P. A. Abramov, E. Sakuda, S. Akagi, A. Ito, N. Kitamura and M. N. Sokolov, Inorg. Chem., 2016, 55, 8437–8445 CrossRef CAS PubMed.
- C. Fouquey, J.-M. Lehn and A.-M. Levelut, Adv. Mater., 1990, 2, 254–257 CrossRef CAS.
- A. Gooch, N. S. Murphy, N. H. Thomson and A. J. Wilson, Macromolecules, 2013, 46, 9634–9641 CrossRef CAS PubMed.
- R. J. Thibault, T. H. Galow, E. J. Turnberg, M. Gray, P. J. Hotchkiss and V. M. Rotello, J. Am. Chem. Soc., 2002, 124, 15249–15254 CrossRef CAS PubMed.
- O. Uzun, A. Sanyal, H. Nakade, R. J. Thibault and V. M. Rotello, J. Am. Chem. Soc., 2004, 126, 14773–14777 CrossRef CAS PubMed.
- H. Xu, T. B. Norsten, O. Uzun, E. Jeoung and V. M. Rotello, Chem. Commun., 2005, 5157–5159 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- C. M. Cheng, M. I. Egbe, J. M. Grasshoff, D. J. Guarrera, R. P. Pai, J. C. Warner and L. D. Taylor, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 2515–2519 CrossRef CAS.
- A. Udagawa, P. Johnston, H. Uekusa, H. Koshima, K. Saito and T. Asahi, ACS Sustainable Chem. Eng., 2016, 4, 6107–6114 CrossRef CAS.
- M. Inada, A. Udagawa, S. Sato, T. Asahi and K. Saito, Photochem. Photobiol. Sci., 2022, 21, 2169–2177 CrossRef CAS PubMed.
- A. A. Avdeenko, T. L. Dobrovolskaya, V. A. Kultchitsky, Y. V. Naboikin and S. N. Pakulov, J. Lumin., 1976, 11, 331–337 CrossRef CAS.
- G. Oster, N. Geacintov and A. U. Khan, Nature, 1962, 196, 1089–1090 CrossRef CAS.
- S. Khlifi, N. Fournier Le Ray, S. Paofai, M. Amela-Cortes, H. Akdas-Kilic, G. Taupier, S. Derien, S. Cordier, M. Achard and Y. Molard, Mater. Today, 2020, 35, 34–41 CrossRef CAS.
- I. V. Kashnik, B. Yang, S. S. Yarovoi, T. S. Sukhikh, M. Cordier, G. Taupier, K. A. Brylev, P.-A. Bouit and Y. Molard, Chem. – Eur. J., 2024, 30, e202400079 CrossRef CAS PubMed.
- A. Dirksen, U. Hahn, F. Schwanke, M. Nieger, J. N. H. Reek, F. Vögtle and L. De Cola, Chem. – Eur. J., 2004, 10, 2036–2047 CrossRef CAS PubMed.
- Y.-X. Wang, Y.-M. Zhang and Y. Liu, J. Am. Chem. Soc., 2015, 137, 4543–4549 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc01043d |
| This journal is © The Royal Society of Chemistry 2024 |