
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnexpected p-type thermoelectric transport arising from magnetic Mn substitution in Fe2V1−xMnxAl Heusler compounds†
Rajveer
Jha
a,
Naohito
Tsujii
 *a,
Fabian
Garmroudi
b,
Sergii
Khmelevskyi
c,
Ernst
Bauer
b and
Takao
Mori
*ad
*a,
Fabian
Garmroudi
b,
Sergii
Khmelevskyi
c,
Ernst
Bauer
b and
Takao
Mori
*ad
aResearch Center for Materials Nanoarchitectonics (MANA), Nanomaterials Field, National Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba 305-0047, Japan. E-mail: tsujii.naohito@nims.go.jp; mori.takao@nims.go.jp
bInstitute of Solid State Physics, TU Wien, A-1040 Vienna, Austria
cVienna Scientific Cluster Research Center, TU Wien, A-1040 Vienna, Austria
dGraduate School of Pure and Applied Sciences, University of Tsukuba, Tennodai 1-1-1, Tsukuba 305-8671, Japan
First published on 23rd May 2024
Abstract
p-Type Fe2VAl-based thermoelectrics have been much less investigated compared to their respective n-type counterparts. Thus, it is crucial to identify novel doping strategies to realize enhanced p-type Fe2VAl Heusler compounds. In the current study, the effect of Mn substitution in Fe2V1−xMnxAl is investigated with respect to temperature-dependent electronic transport as well as temperature- and field-dependent magnetic properties. We find an anomalous and unexpected p-type Seebeck coefficient for nominally n-doped Fe2V1−xMnxAl over an extremely large range of concentrations up to x = 0.6. Using density functional theory (DFT) calculations, this is traced back to distinct modifications of the electronic structure, i.e., localized magnetic defect states (m = 2.43μB) at the valence and conduction band edges, and a concomitant pinning of the Fermi level within the pseudogap. Furthermore, we were able to further optimize the thermoelectric properties by co-doping Al antisites in off-stoichiometric Fe2V0.9Mn0.1Al1+y, yielding sizeable values of the power factor, PF = 2.2 mW K−2 m−1 in Fe2V0.9Mn0.1Al1.1 at 350 K, and figure of merit, ZT ∼ 0.1 for highly off-stoichiometric Fe2V0.9Mn0.1Al1.5 at T = 500 K. Our work underlines the prospect of engineering Fe2VAl-based Heusler compounds via magnetic doping to realize enhanced p-type thermoelectris and encourages studies involving other types of co-substitution for Mn-substituted Fe2V1−xMnxAl.
Introduction
Thermoelectric (TE) materials can convert waste heat into electric energy, or vice versa, by utilizing the Seebeck effect, thus representing a viable option for generating eco-friendly energy to address the current energy crisis.1–4 A crucial task, however, lies in further improving the conversion efficiency of thermoelectric materials.5,6 The efficiency of TE materials is determined by a dimensionless figure of merit (ZT) = (S2/ρκ) × T, where S is the Seebeck coefficient, ρ the electrical resistivity, κ = (κelec+ κph) the thermal conductivity, consisting of electron and lattice contributions, and T the temperature. These key properties have tradeoffs that limit progress in enhancing ZT, and it is necessary to develop new enhancement principles.7–9 Among several material classes, the Heusler-type intermetallic compound Fe2VAl is a promising candidate for thermoelectric power generation near room temperature.10,11 This alloy has the cubic L21-ordered crystal structure and exhibits a high thermoelectric power factor PF = S2/ρ near room temperature.12 Fe2VAl shows a semiconductor-like temperature dependence of the electrical resistivity in a wide temperature range up to 1200 K or above.10 The electronic structure of stoichiometric Fe2VAl suggests a deep pseudogap at the Fermi level.13 Stoichiometric Fe2VAl is a nonmagnetic, low carrier, and compensated semimetal.13–18 Recently, it was shown that rational co-substitution can tune the band gap and therefore significantly improve the thermoelectric properties, yielding high power factors up to around 10.3 mW K−2 m−1.19–22 However, the lattice thermal conductivity of Fe2VAl is about one order of magnitude larger than that of other high-performance thermoelectric materials, resulting in much smaller ZT values compared to the best-performing systems at room temperature like Bi2Te3 or Mg3Sb2-based compounds.6,23–28In order to further increase ZT of Fe2VAl-based systems, various strategies have been employed already, including elemental substitution at all lattice sites, tuning the microstructure and grain size as well as thin film deposition.29–50 Particularly, B. Hinterleitner et al. reported an ultrahigh power factor, above 45 mW K−2 m−1, and ZT around 5 at 350 K for Fe2V0.8W0.2Al metastable thin films.50 Also, doping of Fe2VAl p-type Heusler compounds with various elements, such as Ti and Zr, has been studied to understand the effects of different dopants on the thermoelectric performance.40,51,52 However, the performance of p-type Fe2VAl-based thermoelectrics remains still far below those of the best n-type compounds. Therefore, it is crucial to identify novel p-type candidate materials and doping elements.
Furthermore, it has been demonstrated that spin fluctuations can contribute significantly to optimize the power factor (PF = 1.2 mW K−2 m−1 at 400 K) in weakly ferromagnetic Fe2V0.9Cr0.1Al0.9Si0.1 and Fe2.2V0.8Al0.6Si0.4.53 That being said, likewise to spin fluctuations, the usage of magnetism in different forms to enhance the Seebeck coefficient is also rapidly increasing; for example, spin Seebeck effect, anomalous Nernst effect, magnon drag, paramagnon drag, spin entropy, etc.54–61 are most recently studied instances.
Thus, magnetic doping can be a useful strategy in improving thermoelectric properties. In the case of Fe2VAl-based compounds, however, the effect of magnetic element doping with respect to thermoelectric properties, has not been well studied. This motivated us to substitute V/Mn in Fe2V1−xMnxAl. We observed a unusual hole-dominated Seebeck coefficient in nominally n-doped Fe2V1−xMnxAl, which increases with increasing Mn content up to x = 0.2. Employing first principles electronic structure calculations within the framework of the density functional theory62 and the coherent potential approximation,63 used to model the effects of the substitutional atomic disorder, we trace this back to a severe modification of the electronic structure, wherein magnetic impurity states are formed by Mn atoms, leading to a pinning of the Fermi energy near the valence band edge and within the pseudogap.
Moreover, we additionally co-substituted Al antisites at the Fe and V sites in Al-rich Fe2V0.9Mn0.1Al1+y samples to further optimize the thermoelectric performance. Since p-type Fe2VAl Heusler compounds have been reported to a lesser content compared to n-type systems, we systematically studied the effect of combined Al off-stoichiometry and Mn co-substitution in p-type Fe2V1−xMnxAl1+y. As an extra finding, the thermal conductivity is substantially suppressed already in single-substituted Fe2V1−xMnxAl and even further by introducing additional Al off-stoichiometry in Fe2V1−xMnxAl1+y. Moreover, the latter shows a sizeable enhancement of the thermopower and a shift of the maximum towards higher temperatures, together with an unexpectedly low electrical resistivity. It is crucial to focus on and optimize p-type full-Heusler systems suitable for applications in thermoelectric power generation, because the well-studied n-type material must be paired with a p-type counterpart to assemble efficient thermoelectric modules.
Experimental & computational details
High-purity elements of Fe (99.98%), Mn (99.99%), V (99.9%), and Al (99.999%), were weighted in the stoichiometric ratio and were arc-melted in Ar atmosphere on a water-cooled copper hearth. The ingots were turned over on the copper hearth and were re-melted for four times to ensure homogeneity. The obtained ingots were loaded to a tungsten carbide jar in the glove box under an Ar atmosphere and milled to a fine powder in a planetary ball mill at a speed of 200 rpm for 5 hours. The obtained fine powders were loaded in a graphite die and consolidated by spark plasma sintering (SPS, SPS-1080 System, SPS SYNTEX INC) under a pressure of 40 MPa at 1273 K for 10 minutes under static vacuum (2 × 10−2 Pa). We performed post-annealing for all the samples after the SPS sintering the samples were sealed in quartz tubes for high-temperature annealing at 1273 K for 24 hours and then slowly cooled down to 673 K in 48 hours, where the samples were further annealed for 48 hours and finally naturally cooled to room temperature.Powder X-ray diffraction measurements were performed to check the phase purity and lattice parameters of the cubic Heusler compounds using a RINT TTR-3 diffractometer (Rigaku Co., Akishima, Tokyo, Japan) and employing Cu Kα radiation. Experimental data were further analyzed by the Rietveld refinement method using the program RIETAN-FP.64 Finally, bar-shaped samples were cut out of the disk samples and used to measure the Seebeck coefficient and electrical resistivity. Measurements were done with a four-probe method on a commercial system ZEM3 (Advance Riko Inc.). The thermal conductivity κ was calculated by using κ = DCpd, where D is the thermal diffusivity, Cp is heat capacity, and d is density. The thermal diffusivity coefficient (D) and the heat capacity (Cp) of bulk material were concurrently measured for the disk sample on a xenon laser flash system (Netzsch LFA 467, Germany) with a pyro Ceram disk as a reference sample. Magnetization measurements were performed by using a superconducting quantum interference device magnetometer from Quantum Design.
Density functional theory calculations of the electronic structure of Fe2V1−xMnxAl were performed using the coherent potential approximation (CPA) in the framework of the Korringa–Kohn–Rostoker (KKR) method and the atomic sphere approximation (ASA).65,66 In our KKR–ASA calculations the partial wave functions were expanded in a spdf-basis (up to l = 3) and the effects of exchange and correlation are treated within the density functional theory using standard PBE exchange–correlation functionals.62 To model thermal magnetic disorder above the magnetic ordering temperature, we employed the disordered local moments (DLM) formalism.67
Results and discussion
Fig. 1(a) and (c) shows the powder X-ray diffraction patterns for Fe2V1−xMnxAl and Fe2V0.9Mn0.1Al1+y samples, respectively. All the samples crystallize in the Heusler (L21) type structure; no impurity peak or secondary phase has been observed. The (111) peak at around 27° is not very pronounced and is further suppressed with increasing Mn or Al concentration in Fe2V1−xMnxAl or Fe2V0.9Mn0.1Al1+y [see Fig. S1(a) and (b) in the ESI†]. The suppressing of the (111) peak suggests that disorder in the Fe2VAl compound becomes more significant with doping. Because the 111 peak is mainly due to the difference in the scattering factors of the Al and the V-sites, our findings indicate that an inter-atomic B2-type disorder exists between the two sites. The (220) peak at around 44.5° for Fe2V1−xMnxAl and Fe2V0.9Mn0.1Al1+y is shown in Fig. 1(b) and (d), indicating a steady increase in the lattice parameter with increasing Mn/Al concentration. The broadening of (220) peak with increasing Mn concentration indicates that disorder is increasing in Mn-doped Fe2VAl compounds. Also, the grain size reduction is another possibility of broadening of (220) peak. Moreover, the lattice parameter was obtained by Rietveld analysis for all samples. Fig. 1(e) and (f) exhibits the lattice parameters as a function of the concentration x and y for the Mn-doped and Al-rich samples, respectively. The increase in the lattice parameter with Mn doping is basically in line with Vegard's law. Also, we observed a slight deviation from Vegard's law for high Mn concentration, i.e., x = 0.6.68 Furthermore, we found that the lattice parameter for Al-rich Fe2V0.9Mn0.1Al1+y is increasing with increasing Al doping. The increase of the lattice parameter with y in Fe2V0.9Mn0.1Al1+y does not follow Vegard's law. Moreover, the lattice parameter increases rapidly for y = 0.5, which deviates from previously reported results for the Al-rich Fe2VAlx.69 Nonetheless, the overall tendency of an increasing lattice parameter is consistent with that reported on Al-rich Fe2VAlx, although the absolute values are smaller compared to those reported previously. The linear trend observed in the lattice parameter indicates that Mn atoms successfully substitute the V site; the solubility limit of Al in Al-rich Fe2V0.9Mn0.1Al1+y is y = 0.5. In comparison, the solubility limit of Al in pure Fe2VAlx was found to be x = 2 in ref. 64. For Al-rich Fe2V0.9Mn0.1Al1+y, the off-stoichiometry manifests itself through the presence of Al antisites on the Fe and V sites.69 Overall, the crystallographic site occupancies can considerably affect the physical properties of Heusler compounds.70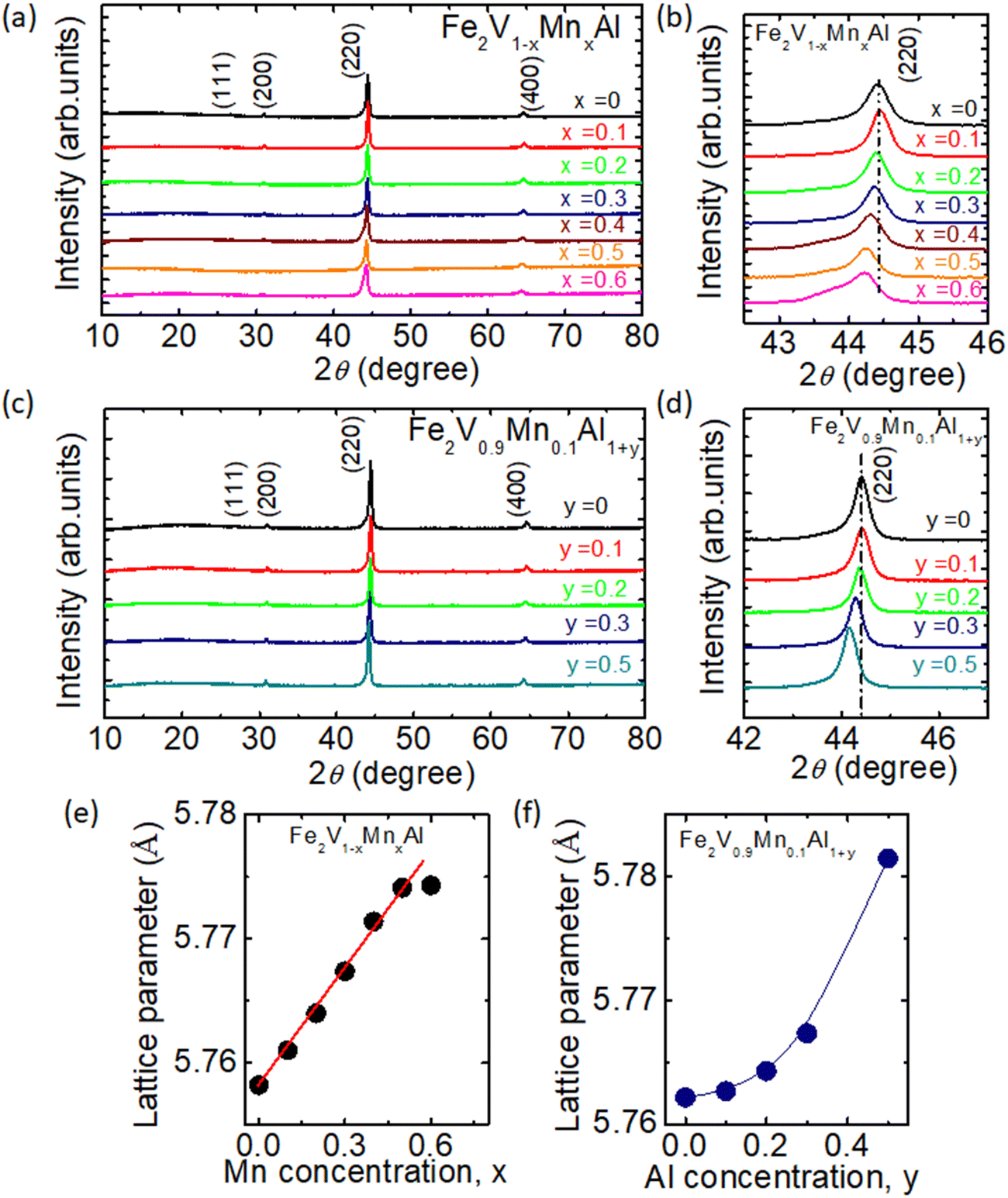 | ||
| Fig. 1 (a) XRD patterns for Fe2V1−xMnxAl sintered samples, with the numbers in the XRD pattern indicating Miller indices. (b) Zoomed view of (220) peak at around 44.5° for Fe2V1−xMnxAl. (c) XRD patterns for Fe2V0.9Mn0.1Al1+y sintered samples. (d) Zoomed view of (220) peak at around 44.5° for Fe2V0.9Mn0.1Al1+y. (e) The lattice parameter was obtained by a refinement using the Rietveld method with RIETAN-FP64 of Fe2V1−xMnxAl as a function of concentration x of Mn, and the solid line represents the linear Vegard's law. (f) Lattice parameter obtained by a refinement of Fe2V0.9Mn0.1Al1+y as a function of y. | ||
Fig. 2 depicts the temperature-dependent thermoelectric properties of Fe2V1−xMnxAl. Firstly, the electrical resistivity at T = 300 K shows a slight increase as x increases up to x = 0.1, then gradually decreases with further increasing the Mn concentration in Fe2V1−xMnxAl [Fig. 2(a)]. We note that the Seebeck coefficient for stoichiometric Fe2VAl (x = 0) is slightly negative in this study, whereas usually sizeable positive values have been reported for samples synthesized via arc or induction melting.45,71 This discrepancy can likely be explained by off-stoichiometry or antisite disorder induced during the synthesis via ball milling and has been found by other groups as well.72,73 However, the Seebeck coefficient becomes p-type upon substituting Mn at the V site in Fe2V1−xMnxAl, even though n-type doping would be expected from a simple estimation of the effective valence electron concentration per atom, indicating non-rigid band doping behavior. The absolute value of the Seebeck coefficient reaches S = 64 μV K−1 at 300 K for Fe2V1−xMnxAl with x = 0.2 [Fig. 2(b)]. For higher Mn-doped samples, the Seebeck coefficient decreases down to S = 16 μV K−1 at 300 K for Fe2V1−xMnxAl (x = 0.6). For non-doped Fe2VAl, the Seebeck coefficient is almost temperature-independent, and the absolute value is near to zero. Again, these results deviate significantly from those obtained in melted samples and suggest the presence of defects such as antisites in the present sample. The highest value of the power factor PF = 0.8 mW K−2 m−1 [Fig. 2(c)] is obtained for Fe2V0.8Mn0.2Al and peaks near room temperature, like all the other samples from Fe2V1−xMnxAl.
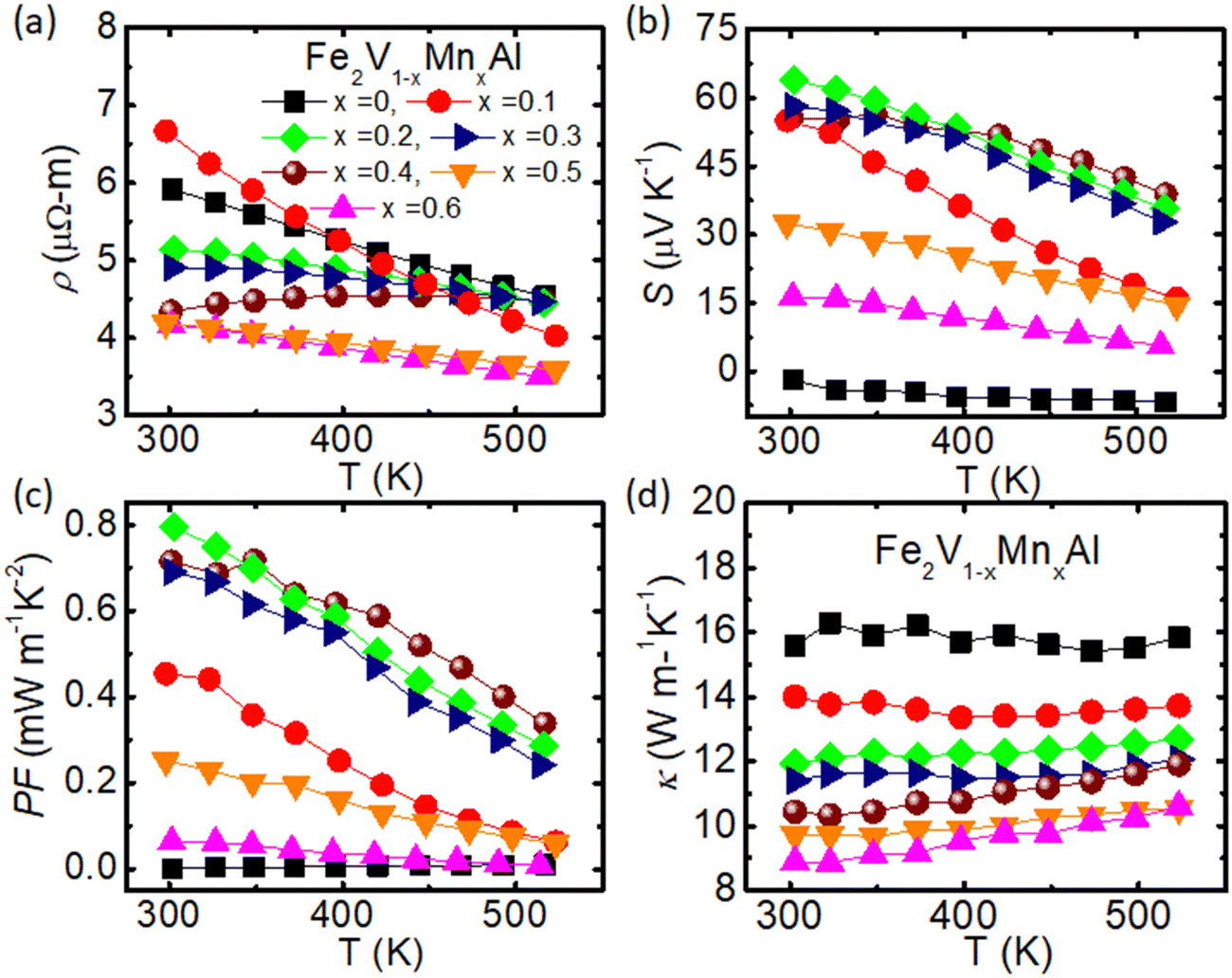 | ||
| Fig. 2 Thermoelectric properties of Fe2V1−xMnxAl (x = 0, 0.1, 0.2, 0.3, 0.4, 0.5 and 0.6) (a)–(d) temperature dependence of electrical resistivity ρ, Seebeck coefficient S, power factor PF, and total thermal conductivity κ, respectively. | ||
The total thermal conductivity of Fe2V1−xMnxAl [Fig. 2(d)], κ, is nearly temperature-independent, and the values decrease substantially upon Mn doping in Fe2V1−xMnxAl. The total thermal conductivity at room temperature for stoichiometric Fe2VAl is 16 W m−1 K−1, which is significantly lower than for melted samples (∼25–30 W m−1 K−1),74,75 again indicating an increased number of defects such as antisite disorder, lattice strain, and grain boundaries that likely arise from the synthesis via ball milling. For Fe2V1−xMnxAl (x = 0.6), the thermal conductivity further decreases down to 9 W m−1 K−1. In general, the total thermal conductivity κ for ordinary metals and semimetals is the sum of electronic and phonon terms, which can be specified as κ = κelec+ κph, where κph is the lattice thermal conductivity and κelec is the electronic contribution, although a bipolar share should be expected when minority carriers actively contribute to transport. Since previous studies on Fe2VAl-based systems have neglected the bipolar contribution, we align our investigation with those works and only distinguish between lattice and electron contributions, where κel is related to the electrical resistivity ρ as given by the Wiedemann–Franz law κel = L0T/ρ, where L0 = 2.45 × 10−8 WΩ K−2 is the Lorenz number, and ρ is the electrical resistivity. The lattice thermal conductivity (κph) can be obtained by subtracting κel from the total thermal conductivity (κ). Fig. 3(a) shows κph as a function of temperature. At room temperature, κph drops from 14 to 7.5 W m−1 K−1 due to the Mn substitution, which is a significant reduction compared to previously reported values for doped Fe2VAl systems,12,30,74,76–78 considering that there are no significant differences in atomic mass and size between Mn and V atoms. Meanwhile, it is obvious that κelec is smaller than κph for all the studied compounds, which signifies that the total thermal conductivity is dominated by lattice phonons rather than charge carriers. This implies that a substantial further enhancement can be achieved by further suppressing heat conduction through phonons, e.g. by tuning the microstructure or via co-substitution as employed in the second part of this study. The dimensionless figure of merit, ZT, was determined and reaches its highest value ZT = 0.025, for Fe2V1−xMnxAl (x = 0.4) at about T = 350 K [Fig. 3(b)].
 | ||
| Fig. 3 (a) Temperature dependence of lattice thermoelectric conductivity for Fe2V1−xMnxAl. (b) The thermoelectric performance, ZT values vs. temperature for Fe2V1−xMnxAl. | ||
In order to further improve the TE properties of Mn-substituted Fe2V1−xMnxAl, additional co-substitution/off-stoichiometry with Al at the Fe and V sites has been performed. This type of off-stoichiometry has been previously shown to not only significantly increase the p-type Seebeck coefficient of Fe2VAl-based thermoelectrics but also greatly reduces the lattice thermal conductivity via substitutions on different sublattices.69Fig. 5 demonstrates the temperature-dependent thermoelectric properties of Al-rich Fe2V0.9Mn0.1Al1+y. Indeed, within this study we have observed that ρ decreases with increasing Al concentration (see Fig. 5(a)). Furthermore, the temperature-dependent behavior of ρ(T) changes from semiconductor-like, dρ/dT < 0, towards metallic-like, dρ/dT > 0. It is interesting to note, however, that ρ reaches its lowest values for Fe2V0.9Mn0.1Al1.3 (y = 0.3) and increases again dramatically as y further rises up to y = 0.5, concomitant with an unexpected decrease of the carrier concentration. This indicates peculiar changes in the electronic structure and clearly shows that such Al off-stoichiometry cannot be regarded as a conventional doping mechanism.
Regarding the Seebeck coefficient, the additional off-stoichiometry clearly has a beneficial effect and not only increases the absolute values of the Seebeck coefficient but also modifies the overall temperature-dependent behavior such that S stays large over a broader temperature interval (see Fig. 5(b)). Upon increasing the amount of Al in Fe2V0.9Mn0.1Al1+y, the maximum of |S| shifts from below room temperature up to almost 500 K for Fe2V0.9Mn0.1Al1.5 (y = 0.5). Moreover, the peak value S = 82 μV K−1 for Fe2V0.9Mn0.1Al1.5 (y = 0.5) is about 20% larger than the maximum value of the Fe2V1−xMnxAl series, S = 61 μV K−1 for x = 0.2 (shown in Fig. 3(b)). Hence, as expected, the Al-excess in terms of antisites on Fe and V sublattices has brought an enhancement in the Seebeck coefficient, consistent with findings in ref. 69. The value S = 82 μV K−1 is comparable to the largest ones in the p-type Fe2VAl-based systems12,19,44–49,69,74 and establishes such type of Al-off stoichiometry and concomitant co-substitution, e.g., with V/Mn, as a desirable route to improve the ZT of p-type full-Heusler systems. The temperature dependence of the Seebeck coefficient shows a maximum at around T = 350 K for y = 0.1 and 0.2, and around T = 450 K for x = 0.3 and 0.5. The maximum for y = 0 is not seen in Fig. 4(b), but is expected below T = 300 K. Thus, the maximum temperature systematically increases with increasing y. This reflects the increase in the hole carrier concentration by extra Al doping and also hints at a possible expansion of the band gap.
 | ||
| Fig. 4 Thermoelectric properties of Fe2V0.9Mn0.1Al1+y (y = 0, 0.1, 0.2, 0.3 and 0.5) (a)–(d) temperature dependence of electrical resistivity ρ, Seebeck coefficient S, power factor PF, and total thermal conductivity κ, respectively. | ||
Compared to the single substitution series Fe2V1−xMnxAl, where the power factor peaks at 0.8 mW K−2 m−1, PF is significantly improved up to 2.0 mW K−2 m−1 for Fe2V0.9Mn0.1Al1.1. However, the power factor decreases with increasing Al concentration in Fe2V0.9Mn0.1Al1+y, hovering around PF = 1.6–1.8 mW K−2 m−1 at 450–500 K for y = 0.3 and 0.5. The thermal conductivity of Fe2V0.9Mn0.1Al1+y shows only weak temperature dependence for the measured temperature range 300–550 K (see Fig. 5(d)). The thermal conductivity at room temperature for stoichiometric Fe2V0.9Mn0.1Al, is 12.1 W m−1 K−1, while it decreases monotonically as a function of y down to 9 W m−1 K−1 for Fe2V0.9Mn0.1Al1+y (y = 0.5). We observed an almost 40% drop in thermal conductivity for Fe2V0.9Mn0.1Al1+y (y = 0.5) compared to the stoichiometric Fe2VAl compound, despite the lack of any heavy element substitution in the investigated samples. The significant decreases in thermal conductivity with increasing Al in Fe2V0.9Mn0.1Al1+y is consistent with the findings in ref. 69 and is assumed to be stemming from the sizeable atomic disorder and point defects brought about by the large number of Al/Fe and Al/V antisites.
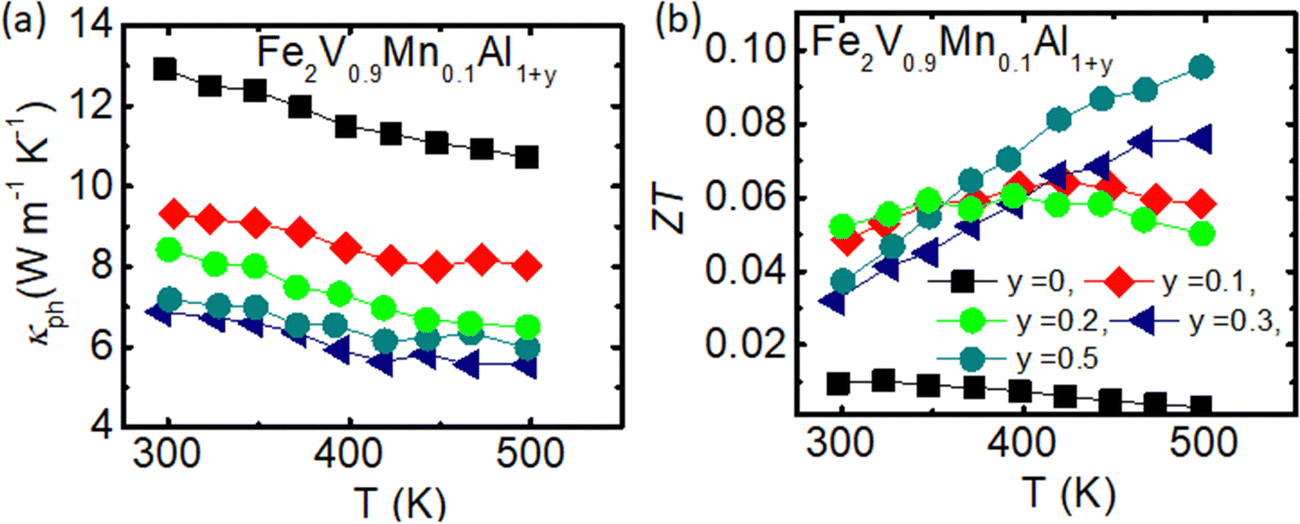 | ||
| Fig. 5 (a) Temperature dependence of lattice thermoelectric conductivity for Fe2V0.9Mn0.1Al1+y. (b) The thermoelectric performance, ZT values vs. temperature for Fe2V0.9Mn0.1Al1+y. | ||
 | ||
| Fig. 6 (a) The Seebeck coefficient as a function of valence electron concentration (VEC) at 300 K. The dotted lines are doped off-stoichiometric Fe2VAl data taken from ref. 78. (b) The Seebeck coefficient as a function of hole carrier concentration p estimated at T = 300 K for Fe2V1−xMnxAl and Fe2V0.9Mn0.1Al1+y with Ioffe–Pisarenko cure. | ||
The temperature dependence of κph is shown in Fig. 5(a), which was again obtained by applying the Wiedemann–Franz law to estimate the electronic contribution from the experimental electrical resistivity. Overall, the thermal conductivity is phonon-driven, while the κelec contribution increases for Al-rich samples. As discussed in the previous section, the electrical resistivity was further decreased, and the Seebeck coefficient enhanced for the Al-rich Fe2V0.9Mn0.1Al1+y samples, Consequently, the power factor has improved, and thermal conductivity decreased for Al-rich Fe2V0.9Mn0.1Al1+y samples. Moderately, the ZT values have been improved for Fe2V0.9Mn0.1Al1+y (y = 0.5), with the highest ZT value 0.1 achieved for Fe2V0.9Mn0.1Al1+y (y = 0.5) at around T = 500 K [Fig. 5(b)].
Furthermore, we have investigated the valence electron concentration (VEC) dependence of the Seebeck coefficient (S) at T = 300 K for Fe2V1−xMnxAl and Fe2V0.9Mn0.1Al1+y. Fe2VAl has 24 valence electrons per formula unit, thereby VEC = 6.40 As aliovalent elements are doped in the Fe2VAl system, the VEC deviates from 6, which affects largely the Seebeck coefficient. For Mn-doped Fe2V1−xMnxAl, the VEC scenario is not in agreement with the behavior observed within the majority of single-element doping studies reported in the literature so far; rather, we have observed a p-type Seebeck coefficient for the VEC > 6.0 (see Fig. 7(a)). The universal curve based on the VEC is only valid for the rigid band case.40 However, substitutions on the Fe- or V-sites with certain transition metals, can cause significant changes in the electronic band structure; thereby, the simple VEC rule is no longer valid. For instance, off-stoichiometric Heusler alloys, Fe2−xV1+x−yTiyAl (p-type Seebeck coefficient), have been investigated using soft X-ray photoelectron spectroscopy.79 The electronic structure near the Fermi level is found to be drastically altered by off-stoichiometry, while doping with Ti has a more moderate effect in a rigid-band-like manner.79 In the case of off-stoichiometric Fe2−xV1+x Al-based Heusler alloys, Soda et al. proposed that the excess V or Fe (antisite) defects may induce impurity states near the valence band edge, resulting in a potential enhancement of the Seebeck coefficient.80 To elucidate the anomalous p-type Seebeck coefficient in nominally n-doped Fe2V1−xMnxAl, we have performed extensive density functional theory calculations of the alloy-averaged densities of states of substituted Fe2V1−xMnxAl, which are summarized in the subsequent section. Our theoretical investigations reveal that the Fermi level remains pinned near the valence band edge inside the pseudogap, owing to the creation of additional electronic states below EF, which balances the chemical potential.
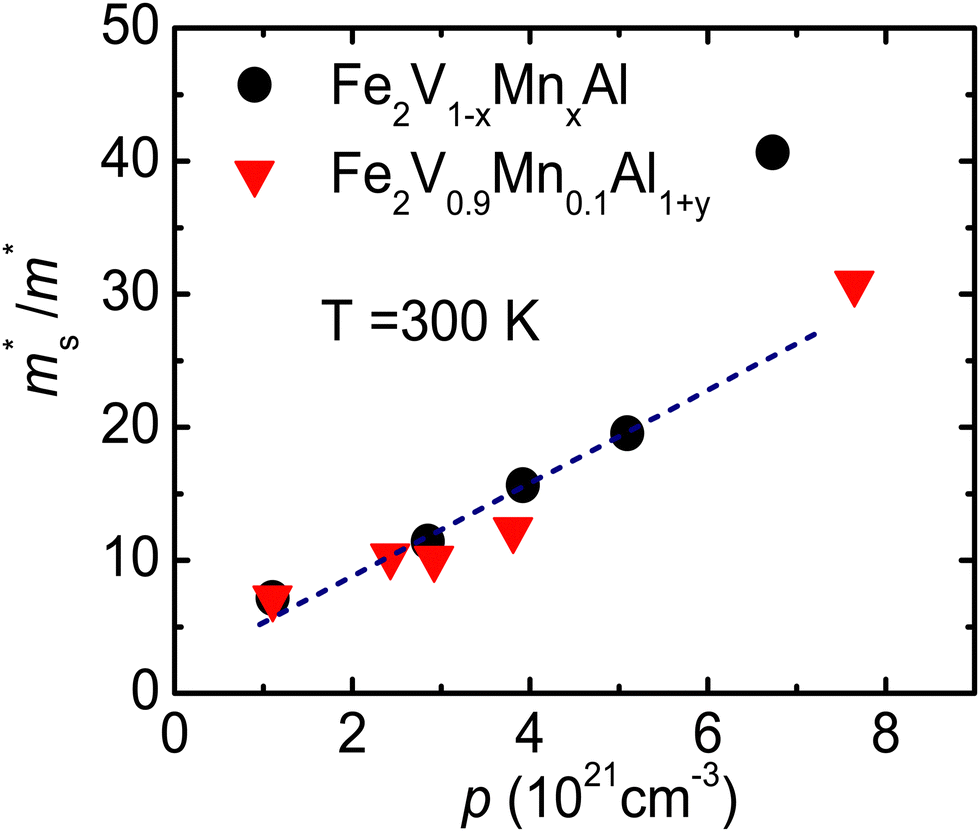 | ||
Fig. 7 The Seebeck effective mass ( ) as a function of hole carrier concentration p estimated at T = 300 K for Fe2V1−xMnxAl and Fe2V0.9Mn0.1Al1+y. The dotted line represents a linear trend in ) as a function of hole carrier concentration p estimated at T = 300 K for Fe2V1−xMnxAl and Fe2V0.9Mn0.1Al1+y. The dotted line represents a linear trend in 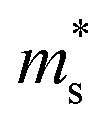 with increasing Mn doping in Fe2V1−xMnxAl. with increasing Mn doping in Fe2V1−xMnxAl. | ||
To further shed light on the anomalous doping dependence of Fe2V1−xMnxAl and Fe2V0.9Mn0.1Al1+y, we have measured the Hall resistivity to estimate the carrier concentration.
The magnetic field-dependent Hall resistivity for Fe2V1−xMnxAl and Fe2V0.9Mn0.1Al1+y at T = 300 K is shown in the ESI† [see Fig. S7]. A linear magnetic field dependence ρxy was observed for all samples. The Hall coefficient RH was then determined from the slope of ρxyversus the applied magnetic field B. The carrier concentration (p) was estimated using the simple equation p = 1/eRH. All the samples (except for x = 0 and y = 0) are p-type, and the hole carrier concentration appears to increase monotonically with doping, except for y = 0.5.
The Seebeck coefficient as a function of carrier concentration at room temperature, known as the Ioffe–Pisarenko plot, is shown in Fig. 7(b). The results here deviate from the Ioffe–Pisarenko curve, especially in the heavily doped region. Although a general trend with a maximum is similar, a clear difference is that the peak is much wider than the normal IP curve, with a shift of maximum to higher carrier concentrations. This is consistent with an increase of the density of states effective mass. Indeed, DFT calculations reveal that rather localized electronic states are formed at both sides of the Fermi level and that the slope of the energy-dependent density of states is enhanced, which is in good agreement with experimental findings. Similar features were reported by Parzer et al. for the Al-rich Fe2VAl system, where they suggested that a shift in a maximum of S vs. carrier concentration occurs due to resonant states located close to the Fermi level.69 Therefore, Mn- and extra Al-doping can be a very fast and effective way to increase the Seebeck coefficient through disorder-related resonant levels in the Heusler compounds. In the present case, the Mn- and Al-doping provoked too much hole carriers, thereby the enhancement in the Seebeck coefficient was just moderate. It should also be stressed that thermally quenched samples of Fe2VAl showed only n-type behavior and an effective strategy for enhancing the p-type Heusler compound has not been reported. The present findings, Mn- and/or excess Al-doping induced disorder may cast a new way for achieving p-type high power factor.
The Hall mobility was derived using μH = ρxy/(ρxxB) [see Table S2, ESI†]. μH decreases by increasing the Mn doping concentration in Fe2V1−xMnxAl or increasing Al in Fe2V0.9Mn0.1Al1+y due to the increasing charge carrier density and/or increasing static disorder in the crystalline unit cell.
Fig. 7 shows the Seebeck effective mass ( ) as a function of hole carrier concentration p estimated at T = 300 K for Fe2V1−xMnxAl and Fe2V0.9Mn0.1Al1+y.
) as a function of hole carrier concentration p estimated at T = 300 K for Fe2V1−xMnxAl and Fe2V0.9Mn0.1Al1+y.  was estimated using eqn (S1) (ESI†), where the
was estimated using eqn (S1) (ESI†), where the 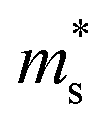 values are directly proportional to the Seebeck coefficient and the carrier concentration (p).81 The formula used was taken from ref. 81 where it is suggested that there are two classes of effective masses; one describes an inertial effective mass that is relevant to a specific electron, and the other one depends more on the density of the electron states (DOS), and is hence called a DOS mass,
values are directly proportional to the Seebeck coefficient and the carrier concentration (p).81 The formula used was taken from ref. 81 where it is suggested that there are two classes of effective masses; one describes an inertial effective mass that is relevant to a specific electron, and the other one depends more on the density of the electron states (DOS), and is hence called a DOS mass,  . Further,
. Further,  can be directly calculated from the Seebeck coefficient employing eqn (S2) (ESI†); thus,
can be directly calculated from the Seebeck coefficient employing eqn (S2) (ESI†); thus,  can be considered as Seebeck effective mass (
can be considered as Seebeck effective mass (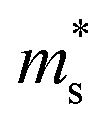 ). We found that with increasing carrier concentration, the Seebeck effective mass
). We found that with increasing carrier concentration, the Seebeck effective mass  increases linearly in the case of Fe2V1−xMnxAl, while for the Fe2V0.9Mn0.1Al1+y, it slightly deviates from linearity. According to literature, the tendencies of the Seebeck effective mass
increases linearly in the case of Fe2V1−xMnxAl, while for the Fe2V0.9Mn0.1Al1+y, it slightly deviates from linearity. According to literature, the tendencies of the Seebeck effective mass  at room temperature and above upon doping are straightforwardly explained by changes in the band structure, e.g., band convergence or changes in the dominant scattering mechanism. In fact, it has been mentioned for Al-rich Fe2VAlx systems that a local distortion of the electronic density of states arises for very large Al concentrations.69 Again, the increase of
at room temperature and above upon doping are straightforwardly explained by changes in the band structure, e.g., band convergence or changes in the dominant scattering mechanism. In fact, it has been mentioned for Al-rich Fe2VAlx systems that a local distortion of the electronic density of states arises for very large Al concentrations.69 Again, the increase of  is fully consistent with first-principles calculations, revealing a modification of the alloy-averaged densities of states, where both conduction and valence band edges become more steep. Thus, the density of states effective mass increases due to the rather localized impurity states by Mn, which spawn at both sides of the Fermi energy.
is fully consistent with first-principles calculations, revealing a modification of the alloy-averaged densities of states, where both conduction and valence band edges become more steep. Thus, the density of states effective mass increases due to the rather localized impurity states by Mn, which spawn at both sides of the Fermi energy.
As can be seen in Fig. 6(a), p-type full Heusler materials have a smaller absolute value of the Seebeck coefficient compared to n-type materials. Additionally, it's challenging to obtain p-type materials due to a more restricted number of doping elements. Mn doping Al-rich Fe2VAl could lead to improved thermoelectric properties in p-type materials near room temperature. The achieved power factor around 2.2 mW K−2 m−1 at T = 350 K, involving a Mn-doped Al-rich Fe2VAl strategy, is larger than Ta-doped or Al-rich Fe2VAl systems, while it is lower than for off-stoichiometric and Ti-doped samples, reaching up to ∼4 mW K−2 m−1.12,52 In the present cases, the highest ZT value of 0.05 was achieved for Fe2V0.9Mn0.1Al1+y (y = 0.1, 0.2) near room temperature. The value of power factor for Mn-doped Al-rich Fe2V0.9Mn0.1Al1+y (y = 0.1) around 2.2 mW K−2 m−1 is a relatively high value for p-type Heusler compound near room temperature. The explored compounds could be useful for applications in thermoelectric power generation.
The magnetic properties of selected samples were also investigated. Fig. 8(a) depicts the temperature-dependent magnetization M under an applied external magnetic field B = 1 T for the Fe2V1−xMnxAl (x = 0, 0.1, 0.2, 0.4) samples in the temperature range 2–300 K. We fitted the inverse of the magnetic susceptibility H/M vs. temperature measured at B = 1 T for x = 0, 0.1, 0.2 and 0.4 in the paramagnetic region above 200 K employing the Curie–Weiss function, H/M = (T − θ)/C, where C and θ are the Curie constant and Weiss temperature, respectively [see Fig. 8(b)]. From least squares fit, the obtained values of parameters C, and θ are summarized in Table 1. Because of the decreasing Curie constants upon an increasing Mn content, the overall susceptibility decreases. The value of θ can be seen as a measure of the inter-site magnetic interaction in the case of a molecular-field approximation. Thus, the samples with x = 0 and 0.1 are weakly ferromagnetic, with a Weiss temperature θ = 35 K and θ = 50 K, respectively, which can most likely be attributed to intrinsic antisite defects involving the Fe sublattices.
 | ||
| Fig. 8 (a) The temperature dependence of magnetization M for Fe2V1−xMnxAl (x = 0, 0.1, 0.2, 0.4), (b) The inverse of magnetic susceptibility H/M vs. T for Fe2V1−xMnxAl (x = 0, 0.1, 0.2, 0.4). The dotted line shows the fitting to the Curie–Weiss function H/M = (T − θ)/C with C = NPeff2/3kB, where Peff, N, and kB are the effective magnetic moment, number of magnetic ions and the Boltzmann constant, respectively. (c) and (d) The magnetization M vs. B curves for Fe2V1−xMnxAl (x = 0, 0.1, 0.2, 0.4) at 2.0 K and 300 K. | ||
| Samples | C (K emu mol−1) | μ eff (μB per atoms) | θ (K) |
|---|---|---|---|
| x = 0 | 5.88 | 3.44 | 35 |
| x = 0.1 | 4.05 | 2.87 | 50 |
| x = 0.2 | 3.59 | 2.69 | −2.5 |
| x = 0.4 | 2.46 | 1.85 | −14.6 |
A further increase of x, however, causes a crossover of θ to negative values (x = 0.4: θ ∼ −16 K), indicating that the growing Mn content triggers eventually antiferromagnetic interactions. This is very much in line with our first-principles calculations of the exchange interactions between Mn atoms shown in the ESI† (Table S7).
The derivative of magnetization dM/dT vs. T shows a peak at around 16 K for undoped Fe2VAl and 30 K for x = 0.1, which suggests a Curie temperature TC ∼ 16 K for x = 0 and TC ∼ 30 K for x = 0.1 [see ESI,† Fig. S6(a)]. The isothermal magnetization at T = 2 K decreases rapidly with increasing Mn concentration in Fe2V1−xMnxAl for x ≥ 0.2. This decrease in magnetization is expected due to the antiferromagnetic correlation between Mn atoms. The higher Mn concentration, for example, the x ≥ 0.2 samples, probably shows a ferrimagnetic nature, which is analogous to the ferrimagnetism reported in the Fe2MnAl compound.82 However, above the Curie point, the inverse magnetic susceptibility is no longer following the Néel hyperbola. This deviation is due to the limitations of magnetization measurements in the paramagnetic state of the alloys without perfect atomic ordering. The effective magnetic moments evaluated from the respective Curie constants C are μeff = 3.44μB per atoms for x = 0, μeff = 2.87μB per atoms for x = 0.1, μeff = 2.69μB per atoms for x = 0.2, and μeff = 1.85μB per atoms for x = 0.4. The μeff values for x ≤ 0.2 are close to those calculated for Fe2V0.9Cr0.1Al and other Fe2VAl-based full-Heusler compounds etc.31,53,83
Beyond that, it is interesting to point out that the Seebeck coefficient of the Mn-doped Fe2V1−xMnxAl exhibits its maximum value of about 60 μV K−1 at 300 K for a wide range of Mn concentrations, x = 0.1 to 0.4, as seen in Fig. 2(b). This plateau-like behavior cannot be explained by a normal doping dependence (see Fig. 6(a)). In addition, the magnetic interaction is also found to be reduced by Mn-doping, as is seen in Fig. 8. The undoped Fe2VAl shows large effective magnetic moment μeff = 3.44μB per atoms, which suggests off-stoichiometry or Fe antisite defects is present in the sample. The values of effective magnetic moments decrease with increasing Mn doping levels in Fe2V1−xMnxAl. We can assume that at low Mn doping level, isolated magnetic ions might act as local perturbations, but their influence on the overall magnetic interaction might be minimal. As the Mn doping level increases, the competing interactions could become more pronounced, potentially leading to a more significant reduction in magnetic interaction. Consequently, the Mn-doped samples need to assume other changes in the electronic structure emerging from the substitution of Mn atoms at the V sublattice that cause an enhancement of the Seebeck coefficient.
The magnetization (M) as a function of applied magnetic field (B) for Fe2V1−xMnxAl (x = 0.1, 0.2, 0.4) is shown in Fig. 8(c) and (d) at 2.0 K and 300 K, respectively. The magnetization curves up to B = 5 T reveal a gradual change, from ferromagnetic-like features (x = 0.1) towards a paramagnetic behaviour for x = 0.4. Most importantly, the magnetization decreases with increasing Mn concentration. It might be possible that introducing Mn magnetic ions at the V site in Fe2VAl could create several competing magnetic interactions. Depending on the specific magnetic ion and its interactions with Fe and Al, these competing interactions might partially cancel out the ferromagnetic order between Fe atoms, leading to a net reduction in the overall magnetic interaction strength. Also, we have studied magnetic properties for Al-rich Fe2V0.9Mn0.1Al1+y compounds [see ESI,† Fig. S5(a)–(d)].
Density functional theory calculations
To further elucidate the anomalous behavior of Mn substitution in Fe2V1−xMnxAl, both with respect to the thermoelectric transport and magnetic properties, we performed extensive density functional theory (DFT) calculations on substituted Fe2V1−xMnxAl Heusler compounds across different concentrations x. Our calculations indicate that Mn atoms, when substituted at the V site, exhibit a large local magnetic moment of around 2.43μB, while they stay non-magnetic, when substituted at the Fe sites.To simulate the true paramagnetic state with thermally disordered magnetic moments at high temperatures, calculations were performed within the framework of the disordered local moment (DLM) approximation.67 Moreover, the substitutional alloy disorder due to Mn atoms randomly replacing V atoms in the unit cell, is fully accounted for in the calculations of the electronic structure by making use of the Coherent Potential Approximation (CPA).63Fig. 9(a) shows the atom-projected and spin-polarized density of states of a single Mn atom in Fe2V0.9Mn0.1Al. A sizeable splitting between majority and minority spin channels is obvious, resulting in a large magnetic moment of around 2.43μB. Interestingly, both minority- and majority-spin contributions exhibit a gap at the Fermi energy. Moreover, rather sharp and localized features in the electronic density of states occur at both sides of the Fermi level for the majority-spin bands. These impurity states contribute to the total DOS of Fe2V1−xMnxAl at the conduction and valence band edge as shown in Fig. 10(b), effectively narrowing the pseudogap of pristine Fe2VAl with increasing Mn substitution.
 | ||
| Fig. 9 (a) Spin-polarized density of states of a single Mn atom in Fe2V0.9Mn0.1Al, showing a sizeable splitting between majority and minority contributions, yielding a large magnetic moment m ∼ 2.43μB for Mn atoms substituted at the V site. (b) Partial atom-projected density of states of Fe2V0.9Mn0.1Al. | ||
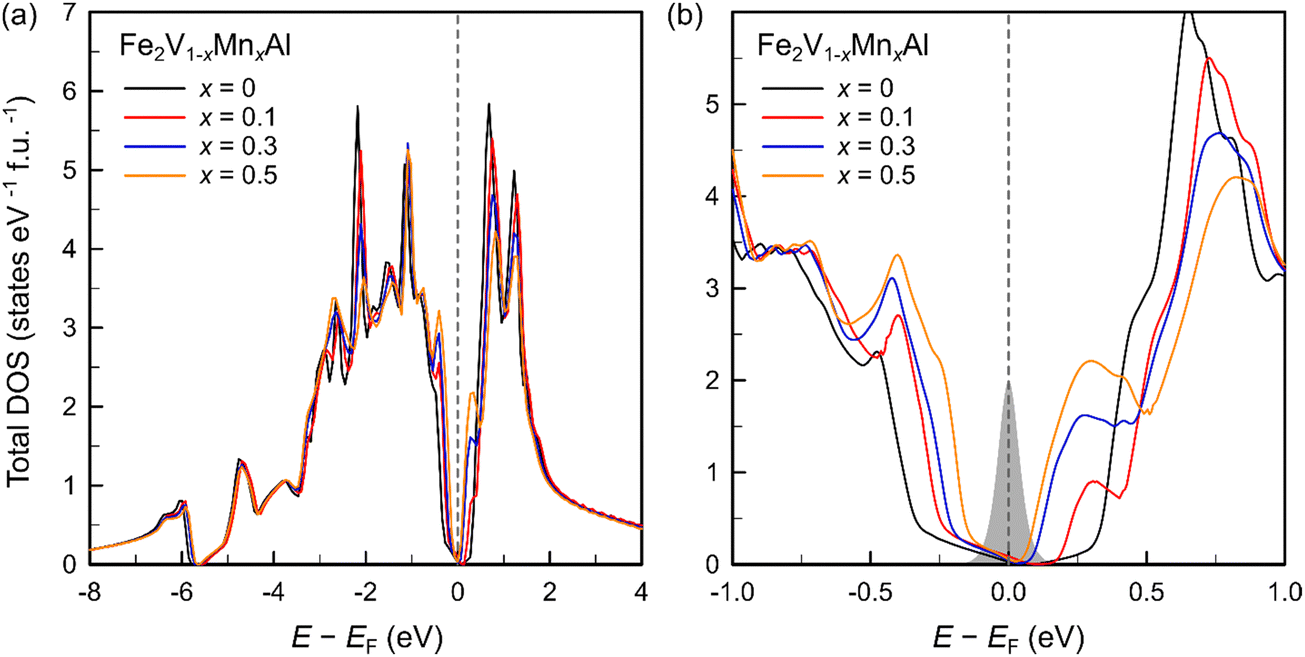 | ||
| Fig. 10 (a) Total density of states (DOS) of Fe2V1−xMnxAl calculated in the disordered local moment (DLM) theory. (b) DOS around the Fermi energy, featuring contributions from rather localized Mn impurity states at the valence and conduction band edges, as well as a pinning of the Fermi level within the pseudogap. To indicate the energy range, relevant for thermoelectric transport, the derivative of the Fermi–Dirac distribution at 300 K is shown in grey. | ||
Fig. 9(b) displays the atom-projected partial DOS of Fe2V0.9Mn0.1Al in the paramagnetic state with the respective contributions from all atoms. We point out that, despite the large electron doping concentration of 0.2 e per formula unit upon substituting 10% Mn at the V site, the Fermi level remains virtually unaffected and stays within the pseudogap. This becomes even more evident in Fig. 10, where the total DOS of Fe2V1−xMnxAl is shown for various Mn concentrations in the paramagnetic state, matching our experimentally investigated samples. Even for Mn substitutions as high as x = 0.5, corresponding to an entire additional electron, the Fermi level remains pinned within the pseudogap and does not get shifted into the conduction band. The anomalous doping behavior can be traced back to Mn forming magnetic impurity states below the Fermi energy (see Fig. 10(a)), which can accommodate the additional electrons doped by Mn in Fe2V1−xMnxAl. Thus, although the valence electron concentration increases, the total number of states below EF increases as well, rendering an unchanged position of EF. This readily explains the unconventional VEC dependence of the Seebeck coefficient in Fig. 7(a), where S retains positive values even for VEC ≫ 6.
We also studied the effect of substituting Mn at the Fe site, i.e., considering that Mn may occupy the Fe site with Fe atoms simultaneously occupying the vacant V positions as antisite defects (Fe1.9Mn0.1V0.9Fe0.1Al), which cannot be ruled out by our standard X-ray diffraction experiments due to the very similar atomic scattering factors of Mn, Fe and V. However, the electronic density of states of Fe1.9Mn0.1V0.9Fe0.1Al (see ESI,† Fig. S8) indicates that this seems very unlikely, since a metallic ground state with a very large DOS at EF emerges, which contradicts the experimental Seebeck coefficient and its VEC dependence.
Conclusion
Overall, p-type Fe2VAl compounds are less developed compared to n-type ones. Here, we performed a systematic experimental and theoretical study of the thermoelectric properties of Mn-doped Fe2V1−xMnxAl Heusler compounds across a wide range of Mn concentrations x = 0 – 0.6. We found an unexpected hole-dominated Seebeck coefficient in nominally n-doped Fe2V1−xMnxAl. Using DFT calculations, the origin for this unconventional doping behavior was found to be rooted in distinct modifications of the electronic density of states upon Mn substitution. More specifically, Mn atoms at the V site form magnetic impurity states with a large local magnetic moment (m ∼ 2.43μB) and antiferromagnetic exchange interactions. The additional electronic states contributed below EF exactly balance the chemical potential, such that EF remains pinned at the valence band edge, within the pseudogap, even for very large Mn concentrations up to x = 0.6. Moreover, rather localized Mn states spawn at both sides of the pseudogap, yielding an increase of the DOS effective mass, albeit a narrowing of the pseudogap is triggered as well. Consequently, relatively large p-type Seebeck coefficients and an anomalous doping/VEC dependence emerges in Fe2V1−xMnxAl.The thermoelectric properties could be further improved by co-substituting Al antisites at the Fe and V sublattices in Al-rich Fe2V0.9Mn0.1Al1+y compounds. Fe2V0.9Mn0.1Al1.1 exhibits the largest Seebeck coefficient (S = 75 μV K−1) and PF = 2.2 mW K−2 m−1 at T = 350 K. Also, the thermal conductivity decreased to 9 W m−1 K−1 for Fe2V0.9Mn0.1Al1.5 (y = 0.5). Consequently, the largest ZT value, ∼0.1, was found for Fe2V0.9Mn0.1Al1.5 (y = 0.5), at T = 500 K. Our study motivates further investigations and optimization studies of Fe2VAl-based Heusler compounds, co-substituted with Mn at the V site, to realize high-performance p-type thermoelectrics, which are relevant for technological applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was mainly supported by JST Mirai Program Grant JPMJMI19A1. N. T. has been supported by JSPS KAKENHI, 22H01761.References
- L. E. Bell, Science, 2008, 321, 1457 CrossRef CAS PubMed.
- J. Heand and T. M. Tritt, Science, 2017, 357, eaak9997 CrossRef PubMed.
- N. Nandihalli, C.-J. Liu and T. Mori, Nano Energy, 2020, 78, 105186 CrossRef CAS.
- I. Petsagkourakis, K. Tybrandt, X. Crispin, I. Ohkubo, N. Satoh and T. Mori, Sci. Technol. Adv. Mater., 2018, 19, 836 CrossRef CAS PubMed.
- T. Zhu, Y. Liu, C. Fu, J. P. Heremans, J. G. Snyder and X. Zhao, Adv. Mater., 2017, 29, 1605884 CrossRef PubMed.
- Z. Liu, N. Sato, W. Gao, K. Yubuta, N. Kawamoto, M. Mitome, K. Kurashima, Y. Owada, K. Nagase, C.-H. Lee, J. Yi, K. Tsuchiya and T. Mori, Joule, 2021, 5, 1196 CrossRef CAS.
- T. Mori, Small, 2017, 13, 1702013 CrossRef PubMed.
- J. Mao, Z. Liu, J. Zhou, H. Zhu, Q. Zhang, G. Chen and Z. Ren, Adv. Phys., 2018, 67, 69–147 CrossRef.
- T. Hendricks, T. Caillat and T. Mori, Energies, 2022, 15, 7307 CrossRef CAS.
- Y. Nishino, M. Kato, S. Asano, K. Soda, M. Hayasaki and U. Mizutani, Phys. Rev. Lett., 1997, 79, 1909 CrossRef CAS.
- H. Kato, M. Kato, Y. Nishino, U. Mizutani and S. Asano, J. Jpn. Inst. Met., 2001, 65, 652 CrossRef CAS.
- Y. Nishino and Y. Tamada, J. Appl. Phys., 2014, 115, 123707 CrossRef.
- G. Y. Guo, G. A. Botton and Y. Nishino, J. Phys.: Condens. Matter, 1998, 10, L119 CrossRef CAS.
- D. J. Singh and I. I. Mazin, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 14352 CrossRef CAS.
- R. Weht and W. E. Pickett, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 6855 CrossRef CAS.
- A. Laila, M. Nanko and M. Takeda, Mater. Trans., 2020, 61, 2216 CrossRef CAS.
- M. Weinert and R. E. Watson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 9732 CrossRef CAS.
- H. Okamura, J. Kawahara, T. Nanba, S. Kimura, K. Soda, U. Mizutani, Y. Nishino, M. Kato, I. Shimoyama, H. Miura, K. Fukui, K. Nakagawa, H. Nakagawa and T. Kinoshita, Phys. Rev. Lett., 2000, 84, 3674 CrossRef CAS PubMed.
- F. Garmroudi, A. Riss, M. Parzer, N. Reumann, H. Muller, E. Bauer, S. Khmelevskyi, R. Podloucky, T. Mori, K. Tobita, Y. Katsura and K. Kimura, Phys. Rev. B, 2021, 103, 085202 CrossRef CAS.
- F. Garmroudi, M. Parzer, A. Riss, A. V. Ruban, S. Khmelevskyi, M. Reticcioli, M. Knopf, H. Michor, A. Pustogow, T. Mori and E. Bauer, Nat. Commun., 2022, 13, 3599 CrossRef CAS PubMed.
- F. Garmroudi, M. Parzer, A. Riss, N. Reumann, B. Hinterleitner, K. Tobita, Y. Katsura, K. Kimura, T. Mori and E. Bauer, Acta Mater., 2021, 212, 116867 CrossRef CAS.
- F. Garmroudi, M. Parzer, A. Riss, S. Beyer, S. Khmelevskyi, T. Mori, M. Reticcioli and E. Bauer, Materials Today, Physics, 2022, 27, 100742 CAS.
- Z. Liu, W. Gao, H. Oshima, K. Nagase, C.-H. Lee and T. Mori, Nat. Commun., 2022, 13, 1120 CrossRef CAS PubMed.
- K. Imasato, S. D. Kang and G. J. Snyder, Energy Environ. Sci., 2019, 12, 965–971 RSC.
- L. Wang, N. Sato, Y. Peng, R. Chetty, N. Kawamoto, D. H. Nguyen and T. Mori, Advanced Energy, Materials, 2023, 13, 2301667 CAS.
- Y. C. Lan, B. Poudel, Y. Ma, D. Z. Wang, M. S. Dresselhaus, G. Chen and Z. F. Ren, Nano Lett., 2009, 9, 1419 CrossRef CAS PubMed.
- Y.-Z. Li, Q. Zhang, K. Liu, Y.-J. Lin, N. Lin, Y. Yu, F. Liu, X.-B. Zhao, B.-H. Ge, O. Cojocaru-Mirédin, C.-G. Fu and T.-J. Zhu, Materials Today, NANO, 2023, 22, 100340 CAS.
- J. Pei, B. Cai, H.-L. Zhuang and J.-F. Li, Natl. Sci. Rev., 2020, 7, 1856–1858 CrossRef PubMed.
- Y. Feng, J. Y. Rhee, T. A. Wiener, D. W. Lynch, B. E. Hubbard, A. J. Sievers, D. L. Schlagel, T. A. Lograsso and L. L. Miller, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 165109 CrossRef.
- K. Soda, H. Murayama, K. Shimba, S. Yagi, J. Yuhara, T. Takeuchi, U. Mizutani, H. Sumi, M. Kato, H. Kato, Y. Nishino, A. Sekiyama, S. Suga, T. Matsushita and Y. Saito, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 245112 CrossRef.
- M. Kato, Y. Nishino, U. Mizutani and S. Asano, J. Phys.: Condens. Matter, 2000, 12, 1769–1779 CrossRef CAS.
- T. Naka, K. Sato, M. Taguchi, T. Nakane, F. Ishikawa, Y. Yamada, Y. Takaesu, T. Nakama and A. Matsushita, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 085130 CrossRef.
- S. Jemima, A. Mani, A. Bharathi, N. Ravindran and Y. Hariharan, J. Alloys Compd., 2001, 326, 183–187 CrossRef CAS.
- E. P. Amaladass, A. T. Satya, S. Sharma, K. Vinod, V. Srinivas, C. S. Sundar and A. Bharathi, J. Alloys Compd., 2015, 648, e34–e38 CrossRef.
- O. Nashima, T. Kanomata, Y. Yamaguchi, S. Abe, T. Harada, T. Suzuki, H. Nishihara, K. Koyama, T. Shishido, K. Watanabe and T. Kaneko, J. Alloys Compd., 2004, 383, 298–301 CrossRef CAS.
- M. Mikami, Y. Kinemuchi, K. Ozaki, Y. Terazawa and T. Takeuchi, J. Appl. Phys., 2012, 111, 093710 CrossRef.
- R. Jha, N. Tsujii, C. Bourgès, W. Gao, E. Bauer and T. Mori, Z. Anorg. Allg. Chem., 2022, 648, e202200058 CrossRef CAS.
- W. Lu, W. Zhang and L. Chen, J. Alloys Compd., 2009, 484, 812–815 CrossRef CAS.
- F. Kobayashi, N. Ide and Y. Nishino, J. Jpn. Inst. Met., 2007, 71, 208 CrossRef CAS.
- Y. Nishino, IOP Conf. Ser.: Mater. Sci. Eng., 2011, 18, 142001 Search PubMed.
- T. Sugiura and Y. Nishino, J. Jpn. Inst. Met., 2009, 73, 846 CrossRef CAS.
- Y. Sandaiji, N. Ide, Y. Nishino, T. Owada, S. Harada and K. Soda, J. Jpn. Soc. Powder Powder Metall., 2010, 57, 207 CrossRef CAS.
- I. Knapp, B. Budinska, D. Milosavljevic, P. Heinrich, S. Khmelevskyi, R. Moser, R. Podloucky, P. Prenninger and E. Bauer, Phys. Rev. B, 2017, 96, 045204 CrossRef.
- K. Iwase, Y. Nishino, H. Miyazaki and K. Soda, J. Jpn. Inst. Met., 2008, 72, 464 CrossRef CAS.
- Y. Nishino, H. Kato, M. Kato and U. Mizutani, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 233303 CrossRef.
- Y. Hanada, R. O. Suzuki and K. Ono, J. Alloys Compd., 2001, 329, 63 CrossRef CAS.
- C. S. Lue and Y.-K. Kuo, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 085121 CrossRef.
- T. Nakama, Y. Takaesu, K. Yagasaki, T. Naka, A. Matsushita, K. Fukuda and Y. Yamada, J. Phys. Soc. Jpn., 2005, 74, 1378 CrossRef CAS.
- Y. Nishino, S. Kamizono, H. Miyazaki and K. Kimura, AIP Adv., 2019, 9, 125003 CrossRef.
- B. Hinterleitner, I. Knapp, M. Poneder, Y. Shi, H. Müller, G. Eguchi, C. Eisenmenger-Sittner, M. Stöger-Pollach, Y. Kakefuda, N. Kawamoto, Q. Guo, T. Baba, T. Mori, S. Ullah, X.-Q. Chen and E. Bauer, Nature, 2019, 576, 85–90 CrossRef CAS PubMed.
- H. Matsuura, Y. Nishino, U. Mizutani and S. Asano, J. Jpn. Inst. Met., 2002, 66, 767 CrossRef CAS.
- H. Miyazaki, S. Tanaka, N. Ide, K. Soda and Y. Nishino, Mater. Res. Express, 2014, 1, 015901 CrossRef CAS.
- N. Tsujii, A. Nishide, J. Hayakawa and T. Mori, Sci. Adv., 2019, 5, eaat5935 CrossRef CAS PubMed.
- R. Ang, A. U. Khan, N. Tsujii, K. Takai, R. Nakamura and T. Mori, Angew. Chem., Int. Ed., 2015, 54, 12909 CrossRef CAS PubMed.
- F. Ahmed, N. Tsujii and T. Mori, J. Mater. Chem. A, 2017, 5, 7545 RSC.
- Y. Zheng, T. Lu, Md. M. H. Polash, M. Rasoulianboroujeni, N. Liu, M. E. Manley, Y. Deng, P. J. Sun, X. L. Chen, R. P. Hermann, D. Vashaee, J. P. Heremans and H. Zhao, Sci. Adv., 2019, 5, eaat9461 CrossRef CAS PubMed.
- J.-B. Vaney, S. Aminorroaya Yamini, H. Takaki, K. Kobayashi, N. Kobayashi and T. Mori, Mater. Today Phys., 2019, 9, 100090 CrossRef.
- S. Hebert, R. Daou, A. Maignan, S. Das, A. Banerjee, C. Bourgès, N. Tsujii and T. Mori, Sci. Technol. Adv. Mater., 2021, 22, 583 CrossRef CAS PubMed.
- W. L. Lee, S. Watauchi, V. L. Miller, R. J. Cava and N. P. Ong, Phys. Rev. Lett., 2004, 93, 226601 CrossRef PubMed.
- R. Ramos, M. H. Aguirre, A. Anadon, J. Blasco, I. Lucas, K. Uchida, P. A. Algarabel, L. Morellón, E. Saitoh and M. R. Ibarra, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 054422 CrossRef CAS.
- H. Matsuura, M. Ogata, T. Mori and E. Bauer, Phys. Rev. B, 2021, 104, 214421 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- B. L. Gyorffy, Phys. Rev. B: Solid State, 1972, 5, 2382 CrossRef.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15 CAS.
- I. A. Abrikosov and H. L. Skriver, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 16532 CrossRef CAS PubMed.
- A. V. Ruban and H. L. Skriver, Comput. Mater. Sci., 1999, 15, 199 CrossRef.
- B. L. Gyorffy, A. J. Pindor, J. Staunton, G. M. Stocks and H. Winter, J. Phys. F: Met. Phys., 1985, 15, 1337 CrossRef CAS.
- A. Denton and N. Ashcroft, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 43, 3161 CrossRef CAS PubMed.
- M. Parzer, F. Garmroudi, A. Riss, S. Khmelevskyi, T. Mori and E. Bauer, Appl. Phys. Lett., 2022, 120, 071901 CrossRef CAS.
- F. Garmroudi, M. Parzer, A. Riss, A. Pustogow, T. Mori and E. Bauer, Phys. Rev. B, 2023, 107, L081108 CrossRef CAS.
- B. Hinterleitner, F. Garmroudi, N. Reumann, T. Mori, E. Bauer and R. Podloucky, J. Mater. Chem. C, 2021, 9, 2073–2085 RSC.
- K. Kurosaki, H. Muta, Y. Endo, A. Charoenphakdee, M. Uno and S. Yamanaka, J. Alloys Compd., 2009, 486, 507–510 CrossRef CAS.
- Y. Takagiwa and Y. Iwasaki, ACS Appl. Energy Mater., 2023, 6(15), 8256–8265 CrossRef CAS.
- Y. Nishino, S. Deguchi and U. Mizutani, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 115115 CrossRef.
- B. Hinterleitner, P. Fuchs, J. Rehak, F. Garmroudi, M. Parzer, M. Waas, R. Svagera, S. Steiner, M. Kishimoto, R. Moser, R. Podloucky and E. Bauer, Phys. Rev. B, 2020, 102, 075117 CrossRef CAS.
- Y. Nishino, H. Sumi and U. Mizutani, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 094425 CrossRef.
- C. S. Lue, C. F. Chen and K. Kuo, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 064204 CrossRef.
- M. Mikami and K. Kobayashi, J. Alloys Compd., 2008, 466, 530–534 CrossRef CAS.
- K. Soda, S. Harada, M. Kato, S. Yagi, M. Inukai, H. Miyazaki, Y. Sandaiji, Y. Tamada, S. Tanaka, T. Sugiura and Y. Nishino, J. Electron Spectrosc. Relat. Phenom., 2011, 184, 236 CrossRef CAS.
- K. Soda, S. Harada, M. Kato, S. Yagi, Y. Sandaiji and Y. Nishino, IOP Conf. Ser.: Mater. Sci. Eng., 2011, 18, 142004 Search PubMed.
- G. J. Snyder, A. Pereyra and R. Gurunathan, Adv. Funct. Mater., 2022, 32, 2112772 CrossRef CAS.
- N. I. Kourov, V. V. Marchenkov, A. V. Korolev, L. A. Stashkova, S. M. Emelyanova and H. W. Weber, Phys. Solid State, 2015, 57, 700–708 CrossRef CAS.
- K. Sato, T. Naka, M. Taguchi, T. Nakane, F. Ishikawa, Y. Yamada, Y. Takaesu, T. Nakama, A. de Visser and A. Matsushita, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 104408 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc00779d |
| This journal is © The Royal Society of Chemistry 2024 |