
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-terminalized titanium carbide MXene coupled with graphene oxide toward optimized temporal domain-dependent nonlinear absorption†
Naying
Shan
a,
Zihao
Guan
a,
Zhiyuan
Wei
a,
Lulu
Fu
a,
Yang
Zhao
a,
Fang
Liu
a,
Lu
Chen
 a,
Yanyan
Xue
b,
Zhipeng
Huang
a,
Mark G.
Humphrey
c,
Jun
Xu
b and
Chi
Zhang
*a
a,
Yanyan
Xue
b,
Zhipeng
Huang
a,
Mark G.
Humphrey
c,
Jun
Xu
b and
Chi
Zhang
*a
aChina-Australia Joint Research Center for Functional Molecular Materials, School of Chemical Science and Engineering, Tongji University, Shanghai 200092, China. E-mail: chizhang@tongji.edu.cn
bSchool of Physical Science and Engineering, Tongji University, Shanghai 200092, China
cResearch School of Chemistry, Australian National University, Canberra, Australian Capital Territory 2601, Australia
First published on 20th March 2024
Abstract
A stepwise approach was implemented in this research to synthesize the covalent linkage between graphene oxide (GO) and MXene for the first time. The preparatory procedure involves the exchange of surface functional groups on MXene, followed by the covalent modification of N-terminalized MXene with GO, making a pioneering advancement in the domain of nonlinear optics. The resulting nanohybrids, labeled as f-MXene-GO, underwent fully comprehensive characterization by crystallographic, spectroscopic, and microscopic techniques. Z-scan measurements revealed that f-MXene-GO features an optimized nonlinear optical (NLO) response in both nanosecond (ns) regimes at 532 nm and femtosecond (fs) regimes at 800 nm across the visible and near-infrared spectra, surpassing its precursor and physically blended samples. The transmittance of f-MXene-GO gradually diminishes with increasing incident energy, displaying enhanced reverse saturable absorption (RSA) and optical limiting effects upon ns pulse excitation. Conversely, f-MXene-GO demonstrates promoted saturable absorption (SA) under fs laser illumination. The effective electron transfer process from MXene to the grafted GO, validated by theoretical calculations, stimulates superior NLO absorption. These findings not only elucidate the intricate relationship between the structure and the NLO response across wide spectral regions and time scales but also provide invaluable insights into the untapped potential of MXene-based materials for NLO applications.
Introduction
Nonlinear optical (NLO) materials are essential in various optical applications, including optical limiting, optical switching, optical communication, and so forth.1–3 Materials with extended π-conjugation have been elaborately designed and explored to pursue remarkable NLO performance.4–6 The successful delamination of graphene7–9 has prompted heightened interest in numerous two-dimensional (2D) materials within the realm of nonlinear optics, which encompass graphene derivatives, transition metal sulfides,10–16 black phosphorus (BP),17,18 and MXene.19–24 However, distinct drawbacks hinder the potential of individual materials: graphene's inherent zero bandgap and limited modulation depth hamper its advancement in optoelectronic devices; the large bandgap of MoS2 along with the narrow tunable bandgap range poses challenges in future devices; the air-instability of BP and perovskites is not ideal in practical developments. To address these limitations and fulfill the expanding technical requirements of optical nonlinearity, the incorporation of varied 2D materials in constructing heterostructures emerges as an appealing approach. Harnessing the collective benefits of diverse materials presents a formidable task. Consequently, the synergy of advantages from 2D materials within heterostructures is anticipated as the incident light passes through the heterojunction boundary, yielding the optimized NLO performance.MXene materials, denoted as Mn+1XnTx (n = 1–3), boast a distinctive molecular composition where M corresponds to a transition metal (e.g., Ti, V, or Nb), X encompasses carbon and/or nitrogen, and T represents a surface termination. Experimental findings have proved the outstanding NLO properties of the MXene family, emphasizing their immense potential in optical limiting, mode-locking pulse generation, and Q-switching pulse generation.25–27 The reported inaugural MXene, Ti3C2Tx, has undergone extensive scrutiny, particularly regarding its NLO response.28–31 The enhanced NLO performance of Ti3C2Tx has been achieved through successful integration, facilitated by van der Waals forces, involving functional materials such as MoS2,32 BP,19 TiO2,33 Fe3O4,22,34 and Ag.35 In contrast to the aforementioned non-covalent functionalization methods, covalent functionalization strategies hinge on the establishment of robust covalent bonds between the anchoring groups and the terminal surface atoms, which not only capitalize on the advantages of both components, but also enable charge transfer at the intimate contact interface. The discerning deployment of these covalent functionalization strategies holds a significant importance in crafting high-performance NLO nanohybrids. Indeed, a variety of ligands have been utilized to covalently functionalize Ti3C2Tx MXene via diazonium,36,37 catechol,38 isocyanate,39 phosphonic acid,40 esterification,41 and silanization.42 These reactions enhance the versatility and performance of the resulting nanohybrids, rendering them more applicable in the fields of optics, electronics, and mechanics. Notably, studies on the covalently functionalized Ti3C2Tx-based materials for the NLO response remain limited.23
The forefront technique for synthesizing Ti3C2Tx involves a hydrofluoric acid-based solution etching process, leading to surface terminations (Tx) consisting of oxo (–O or ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), hydroxyl (–OH), and fluoro (–F) terminations distributed across both edges and basal planes.43 Fluorine forms ionic connections with metal atoms, allowing for chemical modifications.44 Additionally, distinct surface functional group terminations on Ti3C2Tx exhibit varying work functions,45,46 profoundly influencing its optical and electronic properties, which provides an effective avenue to enhance the optical nonlinearity of Ti3C2Tx nanosheets.47,48 Recent research from our group has unveiled that switching the NLO performance of Ti3C2Tx nanosheets is linked to the intrinsic surface terminals via electrical modulation, oscillating between the –OH-rich and O-rich states.24 Inspired by this concept, we introduce a novel approach to design the NLO-active Ti3C2Tx nanosheets, involving the exchange of surface functional groups followed by the covalent modification, for the first time. In essence, the design of its structure encompasses the following three methods: expanding the layer spacing, optimizing the surface groups, and modifying the functional complexes. Importantly, the combined strategy has rarely been reported. Amino-functionalized Ti3C2Tx nanosheets have been commonly employed in energy storage, detector, and adsorption applications.42,49–51 When the N group binds to the Ti atom, the promoted electron transfer causes the surface charge of MXene to be redistributed, significantly enhancing its conductivity.52 Due to the adjustment of surface functional groups, the subsequent covalent functionalization significantly departs from the existing methods.41,53,54 Within the domain of nonlinear optics, this innovative strategy combines the alteration of surface functional groups with subsequent covalent modification, representing a pioneering step in the covalent functionalization of MXene-based materials.55,56
O), hydroxyl (–OH), and fluoro (–F) terminations distributed across both edges and basal planes.43 Fluorine forms ionic connections with metal atoms, allowing for chemical modifications.44 Additionally, distinct surface functional group terminations on Ti3C2Tx exhibit varying work functions,45,46 profoundly influencing its optical and electronic properties, which provides an effective avenue to enhance the optical nonlinearity of Ti3C2Tx nanosheets.47,48 Recent research from our group has unveiled that switching the NLO performance of Ti3C2Tx nanosheets is linked to the intrinsic surface terminals via electrical modulation, oscillating between the –OH-rich and O-rich states.24 Inspired by this concept, we introduce a novel approach to design the NLO-active Ti3C2Tx nanosheets, involving the exchange of surface functional groups followed by the covalent modification, for the first time. In essence, the design of its structure encompasses the following three methods: expanding the layer spacing, optimizing the surface groups, and modifying the functional complexes. Importantly, the combined strategy has rarely been reported. Amino-functionalized Ti3C2Tx nanosheets have been commonly employed in energy storage, detector, and adsorption applications.42,49–51 When the N group binds to the Ti atom, the promoted electron transfer causes the surface charge of MXene to be redistributed, significantly enhancing its conductivity.52 Due to the adjustment of surface functional groups, the subsequent covalent functionalization significantly departs from the existing methods.41,53,54 Within the domain of nonlinear optics, this innovative strategy combines the alteration of surface functional groups with subsequent covalent modification, representing a pioneering step in the covalent functionalization of MXene-based materials.55,56
In this work, we introduce a novel covalently linked composite, f-MXene-GO, comprising graphene oxide (GO) and amino-functionalized MXene, characterized by the extensive 2D π-electron conjugation systems. The carboxyl groups on the surface of GO undergo covalent reactions with the amino groups on the surface of Ti3C2Tx nanosheets, resulting in the formation of amide bonds. To the best of our knowledge, the hybrids assembled by the amino-functionalized MXene and GO have not been explored within the realm of nonlinear optics. Upon excitation by nanosecond (ns) pulses, the normalized transmittance curves of f-MXene-GO deepened as the incident energy increased, indicating an obvious reverse saturable absorption (RSA). When exposed to femtosecond (fs) laser illumination, the resulting material f-MXene-GO exhibits a different NLO performance, that is, saturable absorption (SA). Our study delineates a clear correlation between the structure of MXene-based materials and their NLO responses under the different durations of the laser pulse. These electron–donor/electron–acceptor interactions optimize the NLO effect, augmenting RSA under ns irradiation as well as SA under fs irradiation. This suggests that the incorporation of NLO-active species stands as an effective strategy to bolster the NLO response across the visible and near-infrared regions.57 In conclusion, our investigations present a promising strategy involving the exchange of terminal surface groups and covalent functionalization of MXene materials. This approach not only contributes to the post-synthesis of MXene materials but also offers a new perspective on MXene-based materials in the field of nonlinear optics.
Experimental section
1. Reagents
Ti3AlC2 MAX powder was obtained from 11 Technology Co. Ltd (Jilin, China). Ammonium fluoride (NH4F), tetramethylammonium hydroxide (TMAOH), and N,N-dimethylformamide (DMF) were purchased from Energy Chemical Corporation (Shanghai, China). 1-(3-Dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (C8H17N3·HCl), N,N-diisopropylethylamine (C8H19N), and 1-hydroxybenzotriazole hydrate (C6H5N3O·H2O) were purchased from Aladdin Biochemical Technology Co. Ltd (Shanghai, China). Hydrofluoric acid (HF, 40 wt%) was obtained from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). All reagents were of analytical grade and used without further purification. Reactions that required water-free and oxygen-free conditions were carried out under a N2 atmosphere using standard Schlenk techniques.2. Preparation of amino-functionalized f-MXene
MXene was prepared through the selective etching of aluminum from the MAX phase. Initially, Ti3AlC2 MAX powder (1 g, 200 mesh) was added into 20 mL of HF etchant solution and stirred for 48 hours at room temperature under a N2 atmosphere. Subsequently, the dispersion was subjected centrifugation and washed with ethanol and deionized water until the pH of the supernatant reached approximately 6. After discarding the supernatant, the clay-like precipitates were immersed in 10 mL of TMAOH and stirred for 24 hours at room temperature. The delaminated-Ti3C2Tx MXene was freeze-dried under vacuum overnight. In the next step, NH4F (4.44 g) was added to 5 mL of the 5 mg mL−1 delaminated-Ti3C2Tx MXene dispersion. The combined solution was stirred at 75 °C for 24 hours under a N2 atmosphere. Finally, the amino-modified Ti3C2Tx MXene, denoted as f-MXene, was obtained by freeze-drying the resulting precipitate.3. Preparation of f-MXene-GO
Graphene oxide (GO) was synthesized from graphite powder via the modified Hummers' method (ESI†). GO (150 mg) and f-MXene (150 mg) were dispersed in 150 mL of DMF through ultrasonication for 30 minutes to obtain a uniform suspension. Deoxygenation was carried out by N2 bubbling for 20 minutes. C8H17N3·HCl (80 mg), C8H19N (80 mg), and C6H5N3O·H2O (1.2 mL) were added to the suspension, and a reaction was initiated that spanned for 24 hours. It should be noted that the final product must undergo sequential washing with DMF and deionized water. The layered structured product, denoted as f-MXene-GO, was collected after freeze-drying.4. Material characterization
Scanning electron microscopy (SEM) images using a Hitachi S-4800 instrument and transmission electron microscopy (TEM) images using a JEOL-2100 instrument were obtained. The X-ray photoelectron spectra were obtained using an X-ray photoelectron spectroscopy (XPS) system with Al Kα line excitation on a Kratos Axis Ultra DLD. The structures of the sample were characterized by X-ray powder diffraction (XRD) using a Bruker D8 Advance instrument with Cu Kα radiation (λ = 0.154178 nm). Ultraviolet-visible (UV-Vis) absorption spectra were recorded with an Agilent Cary 5000 spectrophotometer. Fourier transform infrared spectra (FTIR) were recorded using a Thermo Scientific Nicolet iS10 spectrometer in the 4000–400 cm−1 region. The Raman spectra were obtained using an inVia confocal Raman microscope (Renishaw, U.K.) with an excitation laser of 514 nm. For thermal stability analysis, thermal gravimetric analysis (TGA) was carried out with a thermal analyzer STA 409, where the samples were heated to 600 °C at a rate of 15 °C min−1 under a dry N2 flow (20 sccm). Brunauer–Emmett–Teller (BET) surface area measurements were conducted using a TRISTAR 3020 gas adsorption equipment.5. Z-Scan measurements
The nonlinear optical (NLO) characteristics of the samples in the nanosecond (ns) regime were investigated using the open-aperture Z-scan technique. A linearly polarized 12 ns pulsed 532 nm laser at a repetition rate of 10 Hz was generated from a ns Q-switched YAG:Nd3+ laser system (LS-2145-OPO-PC). For the femtosecond (fs) timescale, an 800 nm pulsed laser with a pulse duration of 34 fs and a repetition rate of was generated by Solstice Ace (Spectra-Physics). The Gaussian laser was focused using a converging lens with a focal length of 25 cm. The beam waist values were approximately 60 μm for ns pluses and 30 μm for fs pulses. The input fluence and transmitted fluence of the laser pulses were checked using two energy detectors. In the nonlinear scattering (NLS) experiments, an additional energy detector was positioned at an approximate horizontal angle of 30° from the incident fluence to capture a portion of the scattering signals. DMF dispersions of the samples were tested in a 1 mm thick quartz cell at room temperature.Results and discussion
1. Synthesis of f-MXene–GO
The delaminated-Ti3C2Tx was derived from the Ti3AlC2 powder by a two-step process including HF etching and TMAOH intercalation reactions. Subsequently, the delaminated-Ti3C2Tx was further modified with NH4F, referred to as f-MXene, which then reacted with graphene oxide (GO). The synthesis scheme of f-MXene–GO is illustrated in Fig. 1. The GO is successfully incorporated into amino-functionalized f-MXene through the amide bonds via the carbodiimide reaction, leading to the covalent linkage between f-MXene and GO and the formation of f-MXene–GO.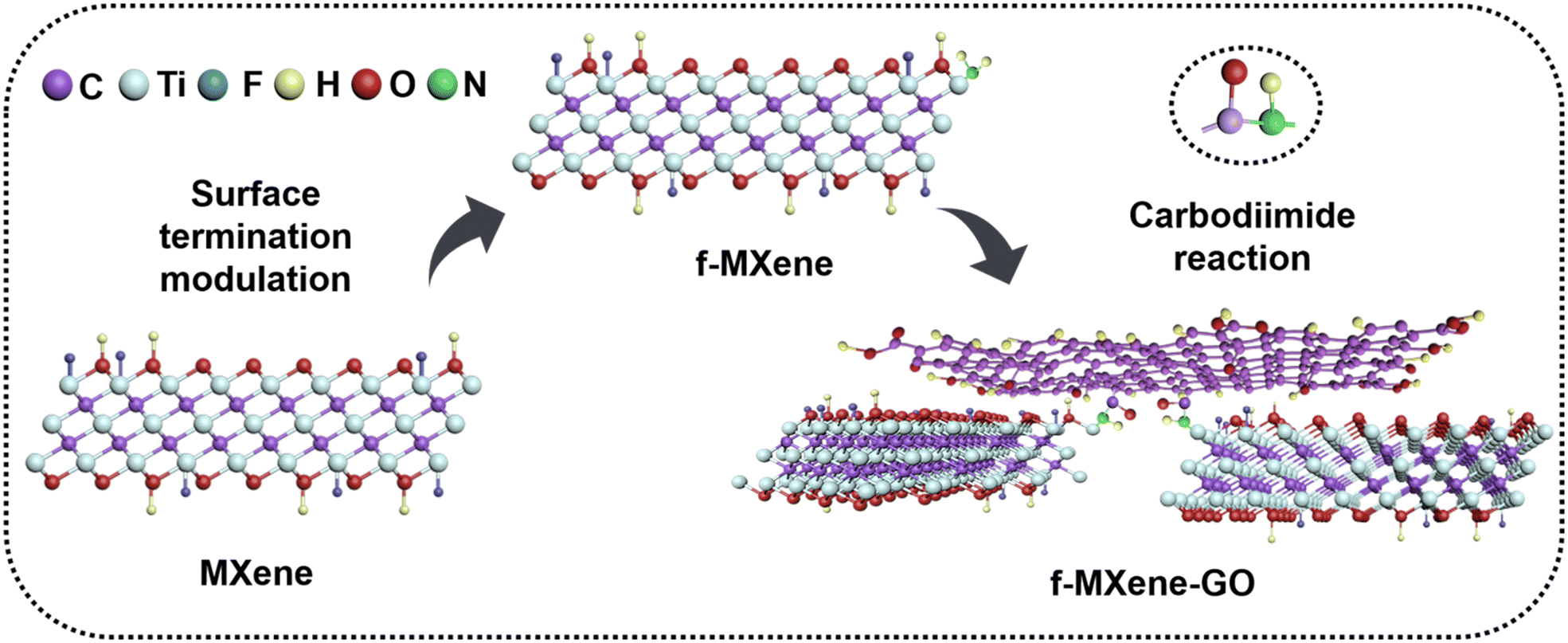 | ||
| Fig. 1 Preparation of f-MXene-GO. | ||
2. Characterization methods
The morphological and microstructural characteristics of GO, f-MXene, and f-MXene–GO were investigated using a scanning electron microscope (SEM) and a transmission electron microscope (TEM), as depicted in Fig. 2. GO manifested extensive sheet-like structures, and MXene (Fig. S1, ESI†) displayed a slender sheet with a morphology distinct from the accordion-like structure, signifying the success of the HF etching and TMAOH intercalation reactions. The f-MXene presented the morphological characteristics similar to MXene. The elemental surface distribution image of f-MXene–GO revealed even dispersion of Ti, C, N, and O elements on the surface. Moreover, the distribution areas of N elements and the regions of Ti and C elements overlapped, demonstrating the uniform amine modification on the Ti3C2Tx MXene surface. The N elements may be traced back to the amide bond (HN–CO), which requires further characterization. The HN–CO bonds were attributed to the dehydration condensation reaction, inducing the spontaneous arrangement of Ti3C2Tx MXene and GO. This process resulted in the formation of the covalently functionalized material f-MXene–GO with an adaptive layered structure.58 The TEM analysis revealed distinctive features for pure Ti3C2Tx MXene (Fig. S2, ESI†) and f-MXene, characterized by the dark wrinkles in the edge region indicative of the self-stacking between layers. In Fig. 2d and f, similar characteristics were observed for GO and f-MXene–GO, while GO displayed a thin layer structure attributed to the harmonic coupling between the bending and stretching modes.59 In the high-resolution transmission electron microscopy (HRTEM) images of f-MXene–GO, parallel fringes were evident in the MXene regions, with a lattice spacing of approximately 1.3 nm. This spacing indicated that the covalent linkage reaction between Ti3C2Tx MXene and GO molecules increased the interlayer spacing. In contrast, the lack of overlapping layers in the GO regions resulted in unclear parallel fringes, presenting as a white area with a water ripple pattern.58,60 The layered structure of f-MXene–GO exhibited non-uniform dark folds, suggesting that GO with Ti3C2Tx MXene linked together still possessed flexible mechanical properties. Furthermore, the self-adaptive layered structure of f-MXene–GO induced by GO maintained the intrinsic properties and original appearance of the Ti3C2Tx MXene.
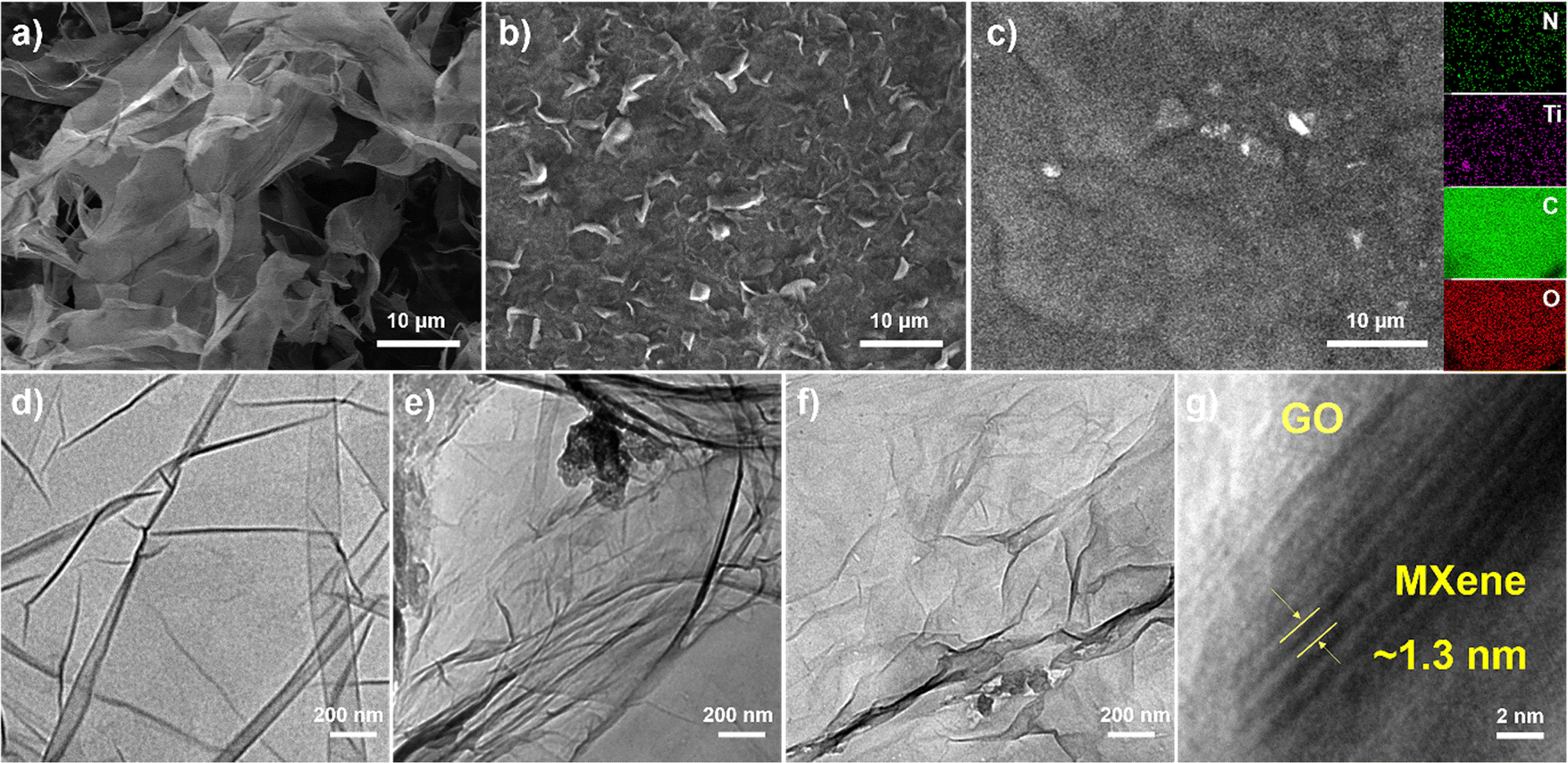 | ||
| Fig. 2 SEM images of (a) GO, (b) f-MXene, and (c) f-MXene–GO (inset: the corresponding elemental mapping images). TEM images of (d) GO, (e) f-MXene, and (f) f-MXene–GO. (g) High-resolution TEM image of f-MXene–GO. | ||
To investigate the amide bonds between MXene and GO, Fourier transform infrared spectroscopy (FTIR) analysis was conducted as shown in Fig. 3a. In Ti3C2Tx MXene, peaks located at 550, 1440, and 1630 cm−1 were attributed to the Ti–O vibration, C–O stretching vibration, and CC stretching vibration, respectively. The resulting f-MXene exhibited a new peak at 3300 cm−1 compared to the precursor, which represents N–H bonds after the reaction with NH4F, confirming the presence of amino groups on the surface. When GO was covalently bonded to f-MXene, the stretching peak of carboxyl groups (–COOH) at 1730 cm−1 almost vanished in the case of f-MXene–GO. Notably, the stretching peak of the amide bond (–CONH) at 1670 cm−1 appeared, identifying the highly efficient formation of the amide bond between the amine group of MXene and the carboxyl group of GO. The information on the crystal structure was obtained from the X-ray powder diffraction (XRD) patterns of GO, MXene, f-MXene, and f-MXene–GO. In Fig. 3b, compared with Ti3C2Tx MXene, the (002) plane of f-MXene was shifted to 7.07°, indicating that an increased d-spacing was around 1.25 nm. The (002) plane of f-MXene was shifted to the lower angle after HF etching, TMAOH intercalation process, and NH4F modification, as reported previously.23 For f-MXene–GO, the (002) plane of MXene was shifted to 6.90° with an enlarged interlayer spacing (∼1.28 nm), whereas the (001) plane of GO was shifted from 10.53° to 8.78°. The further expansion in the interlayer pacing of both MXene and GO was observed due to the carbodiimide reactions, consistent with the TEM results.60 Compared with MXene, f-MXene–GO possessed an increase in interlayer spacing, which was beneficial for the enhanced surface areas in Fig. S3 (ESI†). Raman spectroscopy, an important technique for characterizing two-dimensional materials, was employed (Fig. 3c). D and G peaks were observed in pristine GO at 1343 and 1590 cm−1, respectively, with an intensity ratio of D and G peaks (ID/IG) of 0.735. The carbon atom defect is the source of the D peaks, which are distinctive peaks of carbon atoms. The in-plane stretching vibration of sp2 hybridization is represented by the G peaks. Furthermore, the unbroadened G peak indicates that the edges are the source of D peaks rather than structural defects.59 After f-MXene was covalently attached to GO, the ID/IG of f-MXene–GO was increased to 0.937, suggesting a reduction in the in-plane sp2 hybridization of GO58 as a result of the carbodiimide reactions.
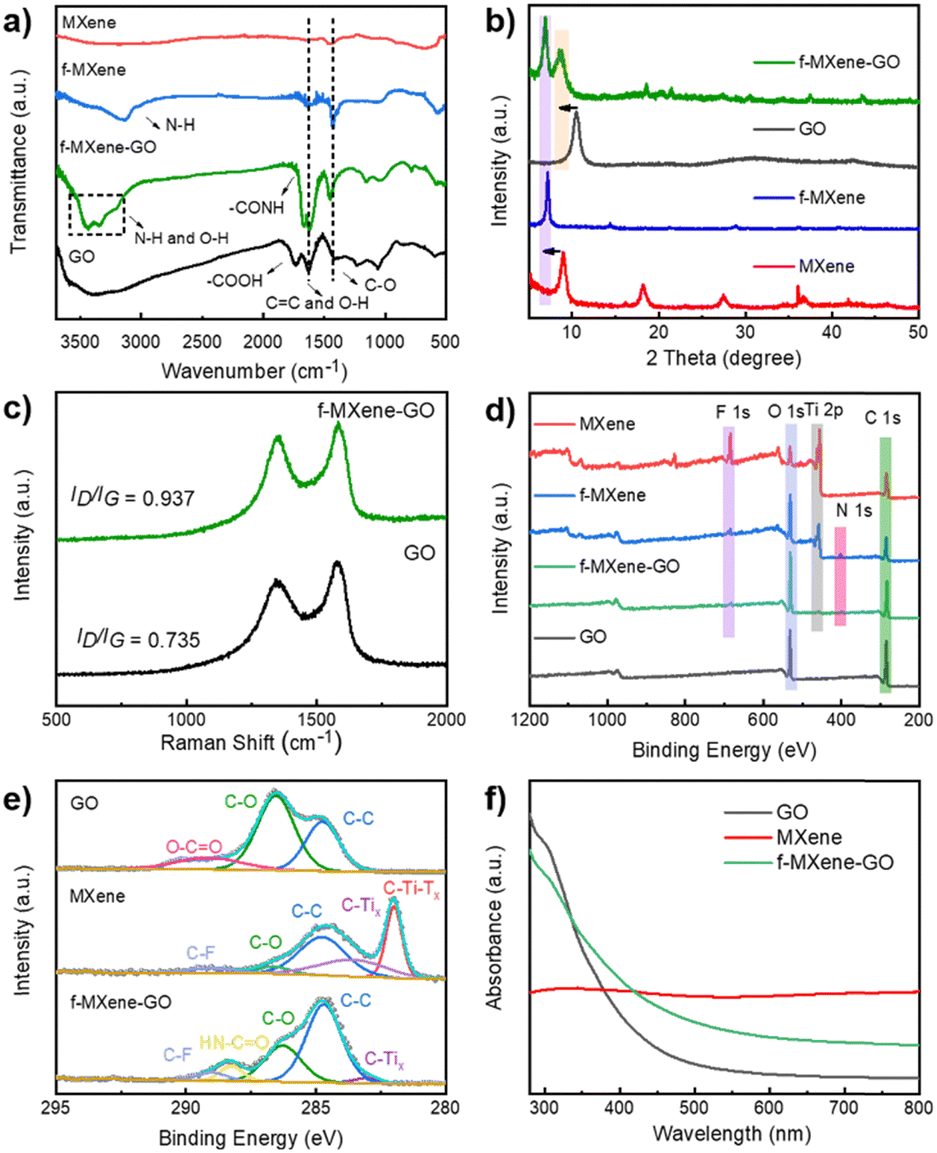 | ||
| Fig. 3 (a) FTIR spectra and (b) XRD patterns of GO, MXene, f-MXene, and f-MXene–GO. (c) Raman spectra of GO and f-MXene–GO. (d) Survey XPS spectra of GO, MXene, f-MXene, and f-MXene–GO. (e) C 1s spectra of GO, MXene, and f-MXene–GO. (f) UV-Vis spectra of GO, MXene, and f-MXene–GO. | ||
The X-ray photoelectron spectra (XPS) of the as-prepared samples were recorded in Fig. 3d. The atomic percentages of GO, MXene, f-MXene, and f-MXene–GO were listed in Table S1 (ESI†). There were C elements (∼284 eV), Ti elements (∼455 eV), O elements (∼532 eV), and F elements (∼684 eV) in MXene. A new peak appeared for f-MXene with a binding energy of 401.7 eV, corresponding to N 1s. After f-MXene reacted with GO to form f-MXene–GO, the intensities of the C 1s and O 1s peaks further increased, whereas the intensities of the Ti 2p, F 1s, and N 1s peaks decreased. To further understand the bond formation and structural changes in detail, the deconvoluted C 1s spectrum of GO exhibited three characteristic peaks in Fig. 3e, including 284.7 eV (C–C), 286.8 eV (C–O), and 288.2 eV (O–CO). The high-resolution C 1s spectrum of MXene can be fitted into five components attributed to 281.7 eV (C–Ti–Tx), 282.8 eV (C–Tix), 284.7 eV (C–C), 285.8 eV (C–O), and 288.7 eV (C–F), which confirmed the modification of F species on the surface. After covalent attachment with GO, a new peak appeared at 287.6 eV, representing the HN–CO bond, while the peak corresponding to O–CO disappeared. In contrast, the high-resolution C 1s spectra of the physical mixture MXene/GO (Fig. S4, ESI†) showed the characteristic peaks of GO and MXene without the peak (HN–CO). Consistent with the FTIR analysis, the results illuminated that GO was covalently linked to f-MXene via the amide bond. The high-resolution Ti 2p spectra of the sample can be deconvoluted into two distinct peaks as shown in Fig. S5 (ESI†), which were ascribed to Ti 2p1/2 and Ti 2p3/2, respectively. Moreover, the peaks of TiO2 (∼458.4 eV) were not apparent, evidencing the avoidable oxidation of Ti atoms in f-MXene and f-MXene–GO. The carbodiimide reaction did not significantly affect the surface composition and structure of Ti atoms. Thermal gravimetric analysis (TGA) was carried out to further investigate the functionalization reaction, as illustrated in Fig. S6 (ESI†). The weight loss for GO was 62 wt% at ∼600 °C, resulting from the decomposition of the surface functional groups to produce carbon dioxide and water. Notably, the weight losses of MXene and f-MXene were much lower. In the case of f-MXene–GO, the weight loss was only 26 wt%, either due to the loss or transformation of some oxygen-containing groups or the reaction with amines.58
3. Linear optical properties
As shown in Fig. 3f, the ultraviolet-visible (UV-Vis) absorption spectra of GO exhibited a broad linear absorption that includes both infrared and ultraviolet bands. The absorption increased rapidly near the ultraviolet band, with a small broad peak at ∼300 nm, corresponding to the n–π* transition of the CO bond. The position of the peak was related to the content of the CO bond in GO, which depended on the degree of oxidation.61 There was no obvious absorption edge in the spectrum, and the absorbance decreased slowly in the visible light wavelength region. With broadband absorption, the ultraviolet absorption spectra of Ti3C2Tx MXene were closely related to the surface functional groups. Two characteristic absorptions were centered at 330 and 760 nm, which represent the interband transition and the surface transversal plasmon mode.62 The intrinsic interband transitions were directly visualized and correlated with the size and thickness of Ti3C2Tx MXene.63 The transversal surface plasmon mode of MXene appeared with increasing concentrations in Fig. S7 (ESI†), which strongly depends on the free carrier density on the nanomaterial surface. The resulting f-MXene–GO improved the absorption in the visible light region. In particular, due to the covalent functionalization, the hybrid f-MXene–GO behaved differently from Ti3C2Tx MXene, which had implications for the linear optical absorption of the transversal surface plasmon mode.
4. NLO properties
Due to the ease of data interpretation, the Z-scan technique is the most commonly used method to evaluate the nonlinear optical (NLO) properties of all as-prepared samples. In the open-aperture Z-scan tests, the transmittance is recorded at various positions and plotted as a function of the Z position, with the focus of the laser being the zero position. According to the fitting formula (ESI†), the theoretical curve shows good agreement with these experimental scattering point data of the NLO responses. The absorptions of all samples show a proportional relationship with different concentrations at 532 and 800 nm as shown in Fig. S7 (ESI†). GO, MXene, MXene/GO with a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 based on the synthesis, and f-MXene–GO were tested in DMF solution with linear transmittance values of 88.7%, 60.0%, 58.0%, and 84.0% at 532 nm, as well as 97.0%, 56.0%, 82.0%, and 95.5% at 800 nm, respectively. The blank solvent DMF has no absorption in the absorption spectra at 532 and 800 nm, which excludes the influence of its linear absorption. The purpose is to provide a theoretical basis for the subsequent NLO measurement. In Fig. S8 (ESI†), the NLO response of the DMF at 532 and 800 nm is almost negligible. Therefore, any NLO response observed in the as-prepared samples is solely due to their properties.
:1 based on the synthesis, and f-MXene–GO were tested in DMF solution with linear transmittance values of 88.7%, 60.0%, 58.0%, and 84.0% at 532 nm, as well as 97.0%, 56.0%, 82.0%, and 95.5% at 800 nm, respectively. The blank solvent DMF has no absorption in the absorption spectra at 532 and 800 nm, which excludes the influence of its linear absorption. The purpose is to provide a theoretical basis for the subsequent NLO measurement. In Fig. S8 (ESI†), the NLO response of the DMF at 532 and 800 nm is almost negligible. Therefore, any NLO response observed in the as-prepared samples is solely due to their properties.
The examination of the NLO properties was conducted at 532 nm using the open-aperture Z-scan technique with a 12 nanosecond (ns) laser excitation at 10 Hz. The contour map serves to visualize two-dimensional Z-scan data and avoids cluttering the plot with excessive data points when the color bar represents the normalized transmittance. As depicted in Fig. 4a–c, the Z-scan results of GO, Ti3C2Tx MXene, and f-MXene–GO indicate energy-dependent reverse saturable absorption (RSA). The transmittance curves exhibit a valley-shaped profile from non-focus to on-focus. Furthermore, the depth of the valley intensifies with the increasing pulse energy from 50 to 125 μJ. In Fig. S9 (ESI†), the minimum normalized transmittance (Tmin) values decrease with higher pulse energy of all materials. The Tmin values of GO at Z = 0 are 0.95 (50 μJ), 0.91 (75 μJ), 0.88 (100 μJ), and 0.85 (125 μJ), while the Tmin values of Ti3C2Tx MXene at Z = 0 are 0.76 (50 μJ), 0.73 (75 μJ), 0.67 (100 μJ), and 0.65 (125 μJ). Similarly, the Tmin values of f-MXene–GO at Z = 0 are 0.66 (50 μJ), 0.53 (75 μJ), 0.41 (100 μJ), and 0.30 (125 μJ). Fig. 4d presents a more comprehensive comparison through the Z-scan results of the precursor materials (GO and Ti3C2Tx MXene), the physically mixed samples (MXene/GO), and the resulting products (f-MXene–GO). It is noteworthy that the nonlinear absorption coefficient (βeff) value of f-MXene–GO is 393.13 cm GW−1, surpassing other materials in Table S2 (ESI†). The optical limiting threshold (FOL) is defined as the fluence at which the transmittance diminishes to half of the linear transmittance. Additionally, the FOL for f-MXene–GO is low at 0.49 J cm−2, rendering it an exemplary candidate in the ns regime and significantly outperforming other materials listed in Table S3 (ESI†). Fig. 4e elucidates a comparison between the βeff and the third-order nonlinear susceptibility (Imχ(3)). Table S4 (ESI†) details the values for βeff, Imχ(3), and the figure of merit (FOM) for GO, MXene, MXene/GO, and f-MXene–GO, utilizing ns pulses centered at 532 nm with an input intensity of 125 μJ. FOM, specifically defined to mitigate the impact of linear absorption on third-order optical nonlinearity, is a key parameter. In terms of the third-order NLO absorption coefficient, the covalently functionalized material f-MXene–GO exceeds that of both the precursor and the physically mixed material. This observation strongly emphasizes the superiority conferred by the covalent bond.
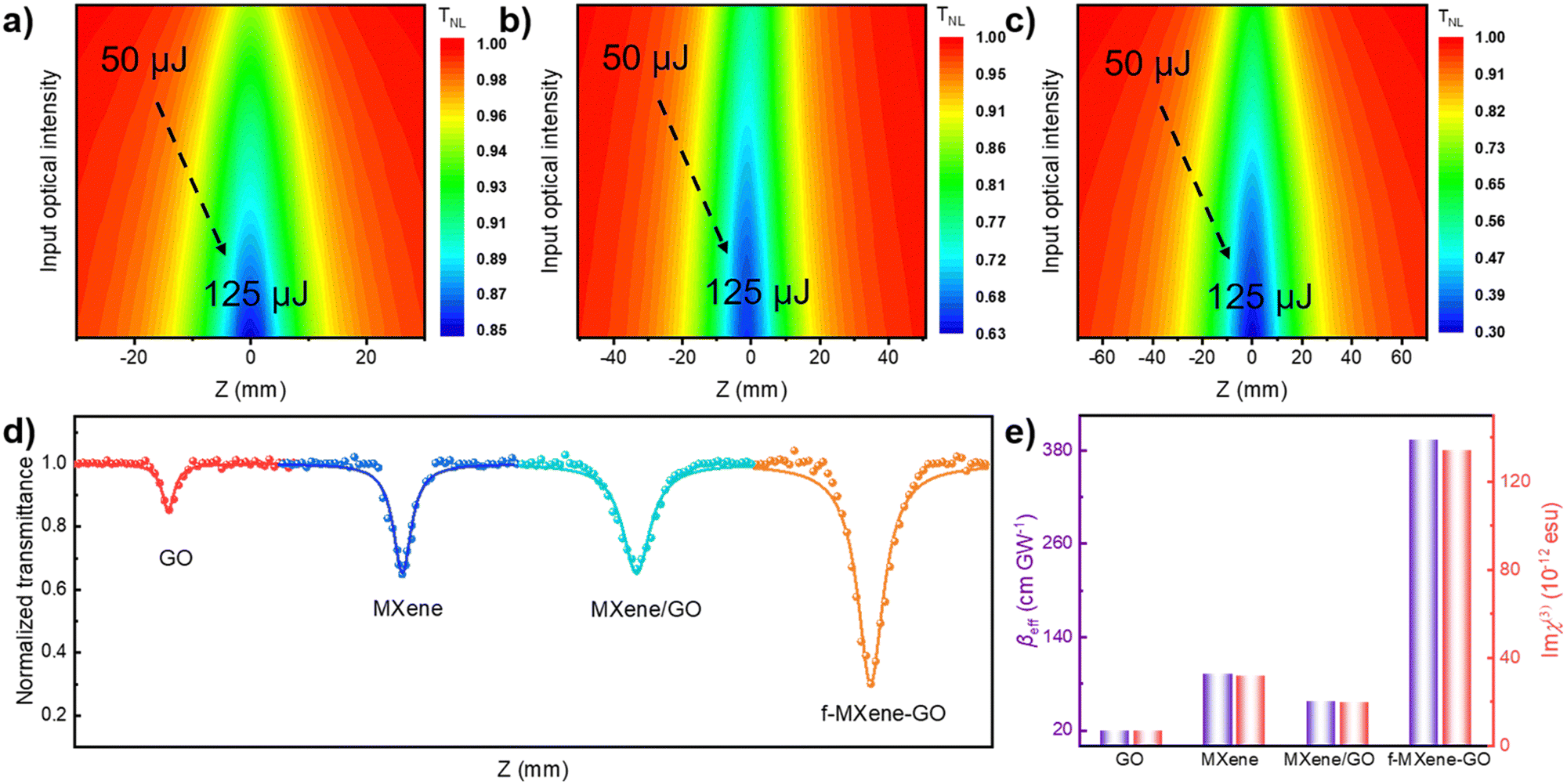 | ||
| Fig. 4 Normalized transmittance distributions of (a) GO, (b) MXene, (c) f-MXene–GO with the excitation of a 12 ns and 10 Hz laser centered at 532 nm. The characteristic RSA responses of samples are displayed in figures (a–c). (d) The open-aperture Z-scan NLO data of GO, MXene, MXene/GO, and f-MXene–GO at a same incident pulse energy of 125 μJ in the ns regime of 532 nm. (e) The comparison of βeff and Imχ(3) of GO, MXene, MXene/GO, and f-MXene–GO. | ||
The combination of both nonlinear absorption (NLA) and nonlinear scattering (NLS) effects collectively contributes to the RSA response in nanoparticle dispersions exposed to a high-intensity ns pulse. The NLS process involves solvent evaporation from the laser-irradiate nanoparticles, triggering the formation of microbubbles and/or microplasmas. These serve as scatterers that disperse the intense laser beam, consequently enhancing the NLS effect. The positioning of an additional photodetector at an approximately 30° horizontal angle to the incident laser beam allows for the analysis of the NLS contribution to the RSA response. As the dispersion samples went through the on-focus point, the symmetric peak-shaped curve in transmittance exhibited augmentation with increasing energy, enabling the detection of scattering signals in the dispersions. The Z-scan measurements, as illustrated in Fig. S10 (ESI†), delineate the scattering data for functionalized nanohybrids. Remarkably, f-MXene–GO shows no active NLS signals, indicating that NLA alone is responsible for the observed RSA response.
Furthermore, an in-depth exploration of the NLO properties was extended to the femtosecond (fs) regime using the open-aperture Z-scan technique with 34 fs at 800 nm and a repetition rate of 1 kHz. In contrast to the ns timescale, Fig. 5a–c depict the Z-scan outcomes in the fs regime. MXene demonstrates RSA, while GO and f-MXene–GO exhibit saturable absorption (SA). SA is characterized by a deepening peak-shaped transmittance curve with increasing pulse energy, indicating the dominance of single-photon absorption in the nonlinear absorption process. In Fig. S11 (ESI†), the maximum normalized transmittance (Tmax) values of GO at Z = 0 are 1.02 (40 nJ), 1.03 (60 nJ), and 1.05 (80 nJ). Correspondingly, the Tmin values of Ti3C2Tx MXene at Z = 0 are 0.97 (40 nJ), 0.94 (60 nJ), and 0.84 (80 nJ). In the case of f-MXene–GO, the Tmax values at Z = 0 are 1.03 (40 nJ), 1.05 (60 nJ), and 1.09 (80 nJ), respectively. Fig. 5d visually elucidates the NLO behavior, emphasizing the substantial difference in the βeff values between MXene/GO (7.03 × 10−2 cm GW−1) and f-MXene–GO (−8.12 × 10−3 cm GW−1). This highlights the advantages of covalent bonding in the case of f-MXene–GO. Fig. 5e presents a comparison of the as-prepared samples in terms of βeff and Imχ(3) with an input intensity of 80 nJ in the fs regime.
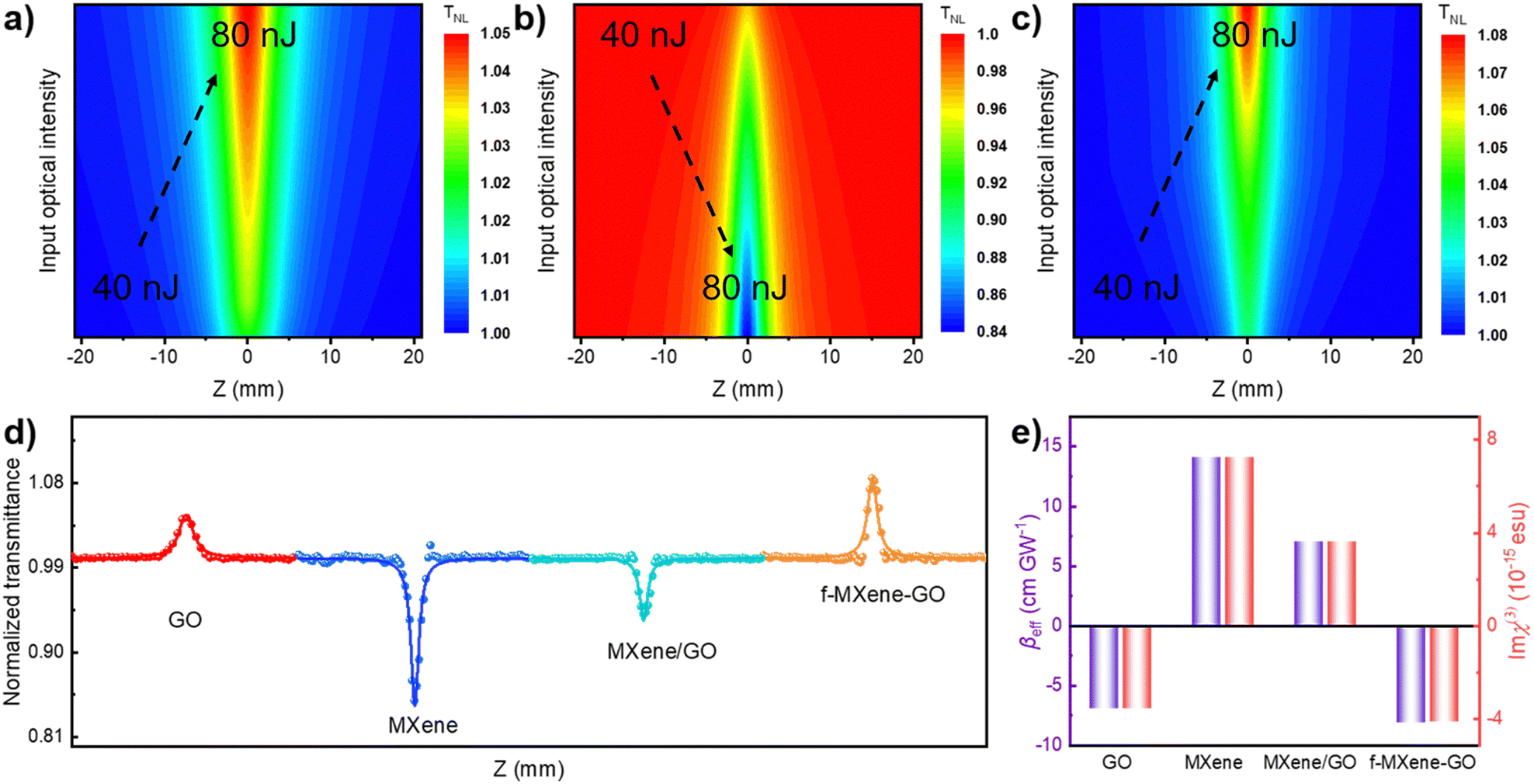 | ||
| Fig. 5 Normalized transmittance distributions of (a) GO, (b) MXene, (c) f-MXene–GO with the excitation of a 34 fs and 1 kHz laser centered at 800 nm. The characteristic SA responses of samples are displayed in figure (a) and (c), while the characteristic RSA responses of samples are presented in figure (b). (d) The open-aperture Z-scan NLO data of GO, MXene, MXene/GO, and f-MXene–GO at the same incident pulse energy of 80 nJ in the fs regime of 800 nm. (e) The comparison of βeff and Imχ(3) of GO, MXene, MXene/GO, and f-MXene–GO. | ||
The comparison of NLO parameters in the ns regime of 532 nm and the fs regime of 800 nm with other typical materials is presented in Fig. 6. Moreover, the stability of the NLO response over time was extensively explored. Fig. S12 (ESI†) illustrates that the NLO performance of f-MXene–GO materials remained unchanged after 30 and 60 days. In stark contrast, the XRD patterns and Raman spectra of the structural analysis revealed the oxidation of MXene after the 3-month storage, while f-MXene–GO exhibited anti-oxidation (Fig. S13, ESI†). This highlights the protective effect of covalently linking GO to the MXene surface against external factors such as oxygen and water, consistent with the antioxidant capacity reported in the literature.64 This superiority can be attributed to the implementation of covalent functionalization, reinforcing its environmental stability.
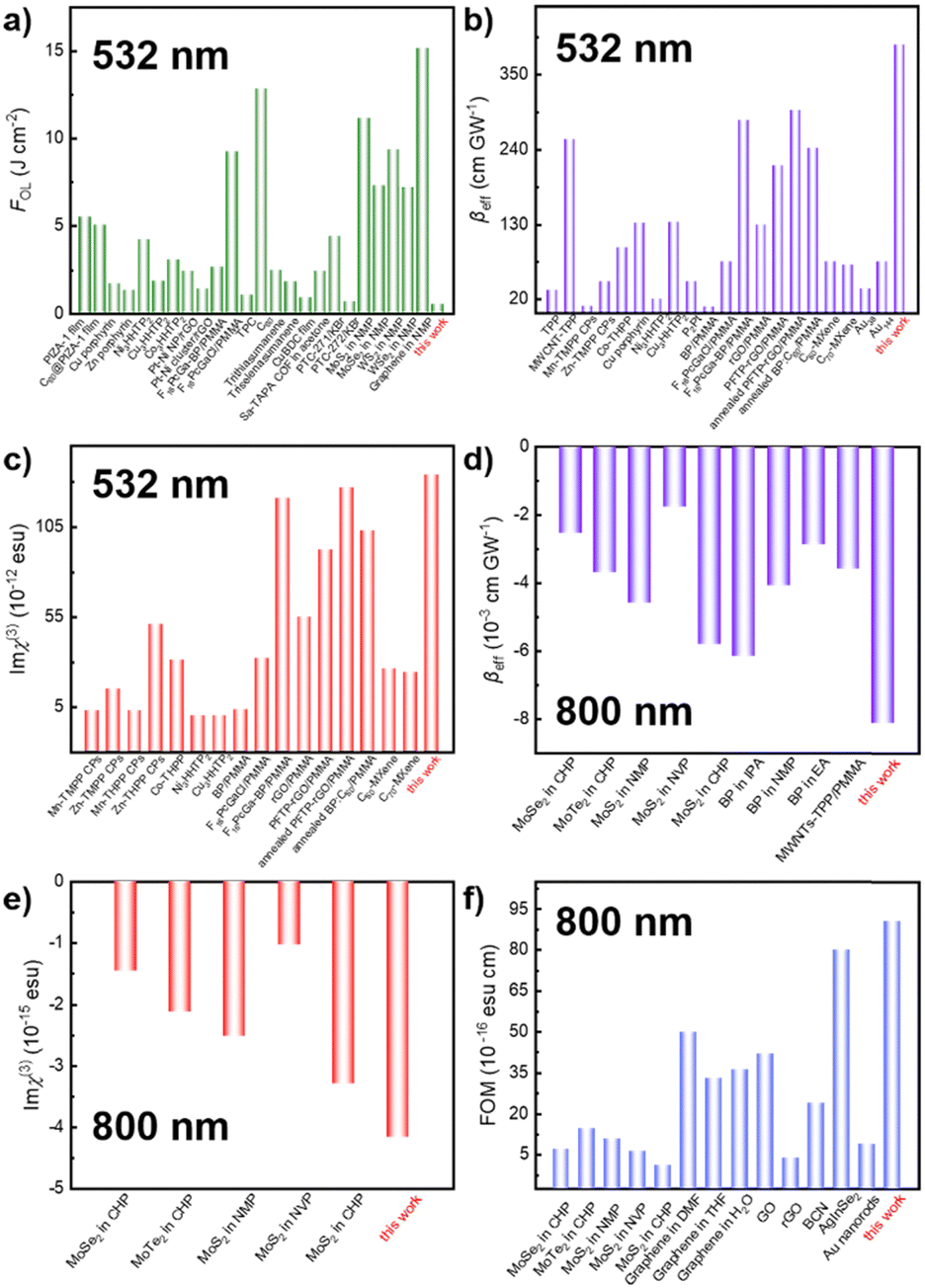 | ||
| Fig. 6 Comparison of (a) FOL, (b) βeff, and (c) Imχ(3) of f-MXene–GO in the ns regime of 532 nm. The comparison of (d) βeff, (e) Imχ(3), and (f) FOM of f-MXene–GO in the fs regime of 800 nm. | ||
Graphene has garnered widespread attention among physicists owing to its unique electronic structure. The Pauli blocking effect makes graphene an ideal ultra-broadband and fast saturable absorber for applications in ultrafast laser technology. GO, a derivative of graphene, possesses chemically active oxygen functional groups, allowing for covalent linking with other nonlinear optically active molecules. The strong electron transfer behavior between electron acceptors and electron donors results in the hybrids demonstrating excellent optical limiting effects in the field of nonlinear optics, as exemplified by nanohybrids of GO and porphyrin.65,66 In the realm of GO materials, the sp2 moiety induces SA behavior due to the Pauli blocking effect while the sp3 fraction introduces two-photon absorption (2PA) and excited state absorption (ESA), leading to RSA behavior. The NLO response can be finely tailored by manipulating the oxidation degree of GO nanomaterials, that is, by adjusting the ratio of sp2 and sp3 hybridization. In the case of the ns pulsed laser, RSA can be attributed to synergistic mechanisms, including NLA such as 2PA and ESA, as well as NLS.67 Unlike pure graphene, GO possesses a limited band gap due to surface functionalization, allowing for the possibility of NLA.68 GO shows RSA, which results from 2PA or ESA under radiation at 1064 nm67 and is dominated under ESA radiation at 532 nm.69,70 Therefore, in the study, under the ns radiation of 532 nm, the prepared GO exhibits RSA behavior primarily governed by ESA, with the result of NLS synergy (Fig. S14, ESI†).67,71–73 With a fs pulse width, the ground state of GO absorbs the laser light and transitions to the excited state. The electronic transition and relaxation between the ground state and the excited state reach a steady state, limiting the further absorption of single photon due to the Pauli blocking effect, inducing SA behavior. Simultaneously, in contrast to short pulses with high pulse repetition rates, the solvothermal-induced NLS phenomenon is significantly weakened. Furthermore, the 2PA of the sp3 fraction in the structure of GO induces RSA while the SA is caused by the band-filling effect induced by the sp2 fraction.61,69,74 However, it has been proved that pulsed laser radiation at 800 nm is unfavorable to the RSA of GO in the fs regime. Therefore, under 800 nm fs pulsed laser irradiation, the NLO response of GO is dominated by sp2-induced single-electron absorption because of the band-filling effect.
On the other hand, recent research has generated considerable interest in attributing the RSA of Ti3C2Tx MXene to mechanisms such as 2PA, bandgap renormalization, and ESA of surface functional groups.20,25,31,48 Under the irradiation at 532 nm in the ns regime and at 800 nm in the fs regime, the log(ΔT0)–log(I) diagram was used to investigate the relationship between the NLO absorption (ΔT0, the depth of the valley bottom) and the laser pulse energy density (I) in Ti3C2Tx MXene. The slope is correlated with the order of the nonlinear optics, corresponding to the slope of n, which is a nominal (n + 1) photon absorption process. In order to validate the above statement, Fig. S15 (ESI†) demonstrates that the slope of the MXene material is 0.81, which is close to 1. Consequently, it is reasonable to infer that the RSA behavior of MXene is derived from the 2PA at 532 nm in the ns regime. Similarly, the fitting data yields slopes at 1.02 for MXene at 800 nm in the fs regime. Additionally, if the 2PA induces the NLO behavior, the βeff of the MXene remains essentially unchanged at different energies, as shown in Fig. S16 and S17 (ESI†).
Consequently, the high Fermi energy level of metallic MXene, the interlayer coupling reaction, and the electron transfer play pivotal roles in the NLO process.75–77 Under 532 nm ns laser irradiation (Fig. 7a), the proposed mechanism involves the few-layer MXene materials continuously transitioning electrons to the excited state through 2PA. In the covalently functionalized f-MXene–GO, the electron transfer from the excited state of MXene to the excited state (S1) of GO induces electrons of GO to move to a higher state (Sn) due to the electron accumulation in the S1 state, thereby enhancing ESA. Moreover, the promotion of 2PA of MXene occurs as the excited state electrons are transferred to GO. Therefore, in f-MXene–GO, the synergistic effects of 2PA in MXene and the ESA in GO collectively enhance the RSA. Turning attention to the fs excitation wavelength at 800 nm, Fig. 7b explains the NLO behavior of f-MXene–GO, where electrons are transferred from the excited state of MXene to the S1 state of GO, increasing the electron accumulation of the S1 state of GO. Benefiting from the band-filling effect, the GO component of the f-MXene–GO nanohybrid mainly contributes to the NLO signal at 800 nm. The NLO response of MXene/GO at 800 nm is thus more likely to be a result of counteracting SA from GO with RSA from MXene. Also, the SA performance exhibited by f-MXene–GO is better than that of pure GO due to the electron transfer.
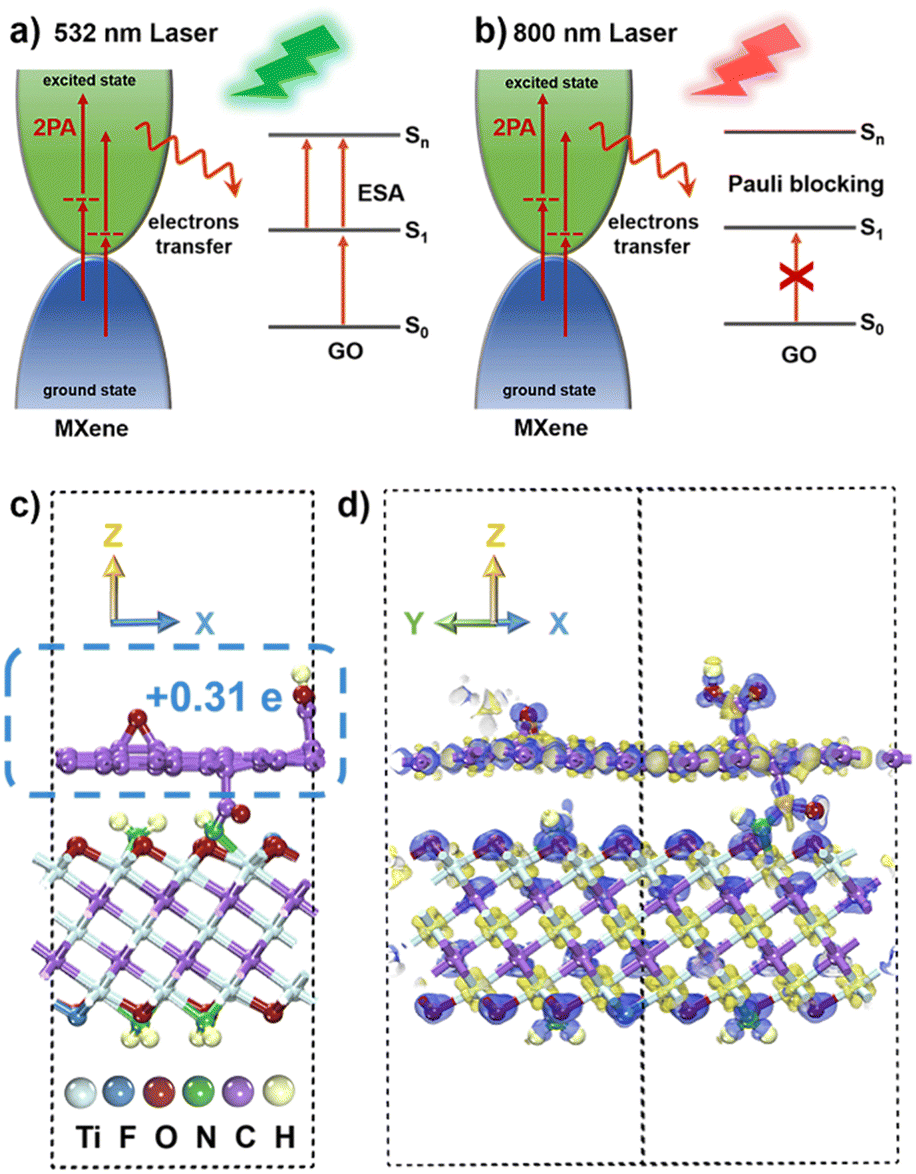 | ||
| Fig. 7 Proposed mechanisms of the NLO response of f-MXene–GO (a) in the ns regime of 532 nm and (b) in the fs regime of 800 nm. (c) The model for f-MXene–GO. (d) Charge density difference diagram of f-MXene–GO. | ||
5. Theoretical calculations
Theoretical calculations utilizing the density functional theory (DFT) method were performed to provide a comprehensive understanding of alterations in the NLO properties. For the MXene model construction, three O atoms on the surface of MXene were replaced by one F atom and two N atoms, respectively, with the N atoms saturated by H atoms. The GO model was constructed by adding an epoxy group and a carboxyl group to each of the upper and lower surfaces of the monolayer graphene. The heterogeneous model of f-MXene–GO was created from a (4 × 4) supercell for MXene and a (5 × 5) supercell for GO in order to minimize the lattice mismatch. In the final model for f-MXene–GO (Fig. 7c), the dashed box represents the GO segment with a transferred charge value of 0.31 e, based on the Mulliken population analysis.The charge density difference diagram in Fig. 7d vividly depicts the electron density distribution within the f-MXene–GO. The light yellow part represents the charge accumulation area, while the light purple area denotes the region of charge reduction, providing a clear visualization of the electron density distribution in f-MXene–GO. The electron transfer from MXene to GO is consistent with the proposed mechanisms in NLO analysis. The covalently functionalized construction of f-MXene–GO facilitates more efficient charge transport, thus enabling significantly enhanced NLO response under both the ns and fs laser irradiation.
Conclusions
The successful synthesis of f-MXene–GO through the amidation method, along with its extensive characterization using various techniques, has paved the way for evaluating the NLO properties. The resulting f-MXene–GO demonstrates significant promise in nonlinear optics, demonstrating the improved optical limiting effect under ns laser irradiation and enhanced SA under fs laser irradiation. The enhancement was attributed to the synergistic effects of multiple mechanisms including the NLO behavior of GO, the RSA of MXene, and the photoinduced electron/energy transfer from MXene to GO. This research provides valuable insights into the intricate NLO properties and associated mechanisms of f-MXene–GO under different laser pulse widths. Overall, f-MXene–GO is anticipated to emerge as a high-performance material within the domain of nonlinear optics.Author contributions
Naying Shan: conceptualization, project administration, and writing and editing. Zihao Guan and Zhiyuan Wei: investigation. Yang Zhao, Fang Liu, and Lu Chen: formal analysis and software. Yanyan Xue: methodology. Zhipeng Huang: resources and validation. Mark G. Humphrey, Jun Xu, and Lulu Fu: writing – review and editing. Chi Zhang: conceptualization, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51432006), the Ministry of Education and the State Administration of Foreign Experts Affairs for the 111 Project (B13025), the Ministry of Education of China for the Changjiang Innovation Research Team (IRT14R23), and the Innovation Program of Shanghai Municipal Education Commission are gratefully acknowledged. M. G. Humphrey thanks the Australian Research Council (DP170100411) for support.Notes and references
- Q. Guo, Y. Yao, Z. Luo, Z. Qin, G. Xie, M. Liu, J. Kang, S. Zhang, G. Bi, X. Liu and J. Qiu, ACS Nano, 2016, 10, 9463–9469 CrossRef CAS PubMed.
- R. Thakur, A. VahidMohammadi, J. Smith, M. Hoffman, J. Moncada, M. Beidaghi and C. A. Carrero, ACS Catal., 2020, 10, 5124–5134 CrossRef CAS.
- J. M. Hales, J. Matichak, S. Barlow, S. Ohira, K. Yesudas, J. L. Bredas, J. W. Perry and S. R. Marder, Science, 2010, 327, 1485–1488 CrossRef CAS PubMed.
- J. Sun, Z. Liu, C. Yan, X. Sun, Z. Xie, G. Zhang, X. Shao, D. Zhang and S. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 2944–2951 CrossRef CAS PubMed.
- Y. Song, J. Sun, X. He, M. Liao, J. Zhao, W. Zeng, S. Zhou and H. Chen, Angew. Chem., Int. Ed., 2023, 62, e202306418 CrossRef CAS PubMed.
- J. Wang, J. Zhao, S. Han, Y. Li, W. Zeng, J. Sun and S. Zhou, Macromolecules, 2024, 57, 1030–1037 CrossRef CAS.
- A. Ganesan, A. Husain, M. Sebastian and S. Makhseed, Dyes Pigm., 2021, 196, 109794 CrossRef CAS.
- T. Ahmed, K. Roy, S. Kakkar, A. Pradhan and A. Ghosh, 2D Mater., 2020, 7, 025043 CrossRef CAS.
- A. Wang, L. Long, W. Zhao, Y. Song, M. G. Humphrey, M. P. Cifuentes, X. Wu, Y. Fu, D. Zhang, X. Li and C. Zhang, Carbon, 2013, 53, 327–338 CrossRef CAS.
- Z. Wei, Y. Fang, H. Li, Z. Guan, N. Shan, F. Liu, Y. Zhao, L. Fu, Z. Huang, M. G. Humphrey and C. Zhang, J. Mater. Chem. C, 2023, 11, 5190–5198 RSC.
- Y. Fang, H. Li, Z. Wei, Z. Guan, N. Shan, L. Fu, Z. Huang, M. G. Humphrey and C. Zhang, J. Mater. Chem. C, 2022, 10, 10876–10887 RSC.
- Z. Wei, Z. Guan, N. Shan, H. Li, Y. Fang, Y. Zhao, L. Fu, Z. Huang, M. G. Humphrey and C. Zhang, J. Alloys Compd., 2023, 934, 167902 CrossRef CAS.
- M. Diao, H. Li, R. Hou, Y. Liang, J. Wang, Z. Luo, Z. Huang and C. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 7423–7431 CrossRef CAS PubMed.
- Y. Li, N. Dong, S. Zhang, X. Zhang, Y. Feng, K. Wang, L. Zhang and J. Wang, Laser Photonics Rev., 2015, 9, 427–434 CrossRef CAS.
- X. Sun, B. Zhang, Y. Li, X. Luo, G. Li, Y. Chen, C. Zhang and J. He, ACS Nano, 2018, 12, 11376–11385 CrossRef CAS PubMed.
- M. Diao, H. Li, X. Gao, R. Hou, Q. Cheng, Z. Yu, Z. Huang and C. Zhang, Adv. Funct. Mater., 2021, 31, 2106930 CrossRef CAS.
- M. Shi, S. Huang, N. Dong, Z. Liu, F. Gan, J. Wang and Y. Chen, Chem. Commun., 2018, 54, 366–369 RSC.
- Z. Liu, B. Zhang, N. Dong, J. Wang and Y. Chen, J. Mater. Chem. C, 2020, 8, 10197–10203 RSC.
- X. Xia, C. Ma, H. Chen, K. Khan, A. K. Tateen and Q. Xiao, Opt. Mater., 2021, 112, 110809 CrossRef CAS.
- G. Wang, D. Bennett, C. Zhang, C. O. Coileain, M. Liang, N. McEvoy, J. J. Wang, J. Wang, K. Wang, V. Nicolosi and W. J. Blau, Adv. Opt. Mater., 2020, 8, 1902021 CrossRef CAS.
- N. Xu, H. Li, Y. Gan, H. Chen, W. Li, F. Zhang, X. Jiang, Y. Shi, J. Liu, Q. Wen and H. Zhang, Adv. Sci., 2020, 7, 2002209 CrossRef CAS PubMed.
- Y. Hu, H. Chu, X. Ma, Y. Li, S. Zhao and D. Li, Mater. Today Phys., 2021, 21, 100482 CrossRef CAS.
- Y. Fang, Z. Wei, Z. Guan, N. Shan, Y. Zhao, F. Liu, L. Fu, Z. Huang, M. G. Humphrey and C. Zhang, J. Mater. Chem. C, 2023, 11, 7331–7344 RSC.
- H. Li, S. Chen, D. W. Boukhvalov, Z. Yu, M. G. Humphrey, Z. Huang and C. Zhang, ACS Nano, 2022, 16, 394–404 CrossRef CAS.
- G. Liu, J. Yuan, Z. Wu, P. Yu, Z. Wang and X. Xu, J. Mater. Chem. C, 2023, 11, 2355–2363 RSC.
- B. Guo, Q.-l Xiao, S.-h Wang and H. Zhang, Laser Photonics Rev., 2019, 13, 1800327 CrossRef CAS.
- X. Jiang, S. Liu, W. Liang, S. Luo, Z. He, Y. Ge, H. Wang, R. Cao, F. Zhang, Q. Wen, J. Li, Q. Bao, D. Fan and H. Zhang, Laser Photonics Rev., 2018, 12, 1700229 CrossRef.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS.
- Y. Wang, Y. Wang and J. He, Laser Photonics Rev., 2023, 17, 2200733 CrossRef CAS.
- Y. Dong, S. Chertopalov, K. Maleski, B. Anasori, L. Hu, S. Bhattacharya, A. M. Rao, Y. Gogotsi, V. N. Mochalin and R. Podila, Adv. Mater., 2018, 30, 1705714 CrossRef PubMed.
- H. Chen, L. Gao, O. A. Al-Hartomy, F. Zhang, A. Al-Ghamdi, J. Guo, Y. Song, Z. Wang, H. Algarni, C. Wang, S. Wageh, S. Xu and H. Zhang, Nanoscale, 2021, 13, 15891–15898 RSC.
- L. Guo, J. Lei, Y. Zhang, X. Duan, X. Wang, Z. Zhang, Z. Wei, L. Li, Q. Guo, X. Liu, R. Ning, J. Wang, W. Hu and W. Wu, Nanotechnology, 2022, 33, 315701 CrossRef CAS PubMed.
- J. Liu, S. Chen, J. He, L. Tao and Y. Zhao, Opt. Mater., 2023, 136, 113417 CrossRef CAS.
- H. Pan, Y. Hu, X. Ma, H. Chu, Y. Li, Z. Pan, J. Zhao, S. Zhao and D. Li, Mater. Today Nano, 2023, 23, 100356 CrossRef CAS.
- J. Sun, H. Cheng, L. Xu, B. Fu, X. Liu and H. Zhang, ACS Photonics, 2023, 10, 3133–3142 CrossRef CAS.
- H. Y. Jing, H. Yeo, B. Z. Lyu, J. Ryou, S. Choi, J. H. Park, B. H. Lee, Y. H. Kim and S. Lee, ACS Nano, 2021, 15, 1388–1396 CrossRef CAS PubMed.
- P. Zhang, L. Wang, K. Du, S. Wang, Z. Huang, L. Yuan, Z. Li, H. Wang, L. Zheng, Z. Chai and W. Shi, J. Hazard. Mater., 2020, 396, 122731 CrossRef CAS PubMed.
- J. E. Heckler, G. R. Neher, F. Mehmood, D. B. Lioi, R. Pachter, R. Vaia, W. J. Kennedy and D. Nepal, Langmuir, 2021, 37, 5447–5456 CrossRef CAS PubMed.
- R. M. McDaniel, M. S. Carey, O. R. Wilson, M. W. Barsoum and A. J. D. Magenau, Chem. Mater., 2021, 33, 1648–1656 CrossRef CAS.
- D. Kim, T. Y. Ko, H. Kim, G. H. Lee, S. Cho and C. M. Koo, ACS Nano, 2019, 13, 13818–13828 CrossRef CAS PubMed.
- T. Zhou, C. Wu, Y. Wang, A. P. Tomsia, M. Li, E. Saiz, S. Fang, R. H. Baughman, L. Jiang and Q. Cheng, Nat. Commun., 2020, 11, 2077 CrossRef CAS PubMed.
- G. Zhang, T. Wang, Z. Xu, M. Liu, C. Shen and Q. Meng, Chem. Commun., 2020, 56, 11283–11286 RSC.
- Z. Li, L. Wang, D. Sun, Y. Zhang, B. Liu, Q. Hu and A. Zhou, Mater. Sci. Eng., B, 2015, 191, 33–40 CrossRef CAS.
- H. Qiu, F. Li, C. Jin, Z. Yang, J. Li, S. Pan and M. Mutailipu, Angew. Chem., Int. Ed., 2024, 63, e202316194 CrossRef CAS PubMed.
- T. Schultz, N. C. Frey, K. Hantanasirisakul, S. Park, S. J. May, V. B. Shenoy, Y. Gogotsi and N. Koch, Chem. Mater., 2019, 31, 6590–6597 CrossRef CAS.
- A. Di Vito, A. Pecchia, M. Auf der Maur and A. Di Carlo, Adv. Funct. Mater., 2020, 30, 1909028 CrossRef CAS.
- Y. Li, J. Si, L. Yan, Z. Fan, Z. Liang and X. Hou, J. Chem. Phys., 2022, 157, 164704 CrossRef CAS PubMed.
- J. Liu, S. Chen, J. He, R. Huang, L. Tao, Y. Zhao and Y. Yang, Nanomaterials, 2022, 12, 2043 CrossRef CAS PubMed.
- L. Wang, N. Zhang, Y. Li, W. Kong, J. Gou, Y. Zhang, L.-N. Wang, G. Yu, P. Zhang, H. Cheng and L. Qu, ACS Appl. Mater. Interfaces, 2021, 13, 42442–42450 CrossRef CAS PubMed.
- F. Ai, C. Fu, G. Cheng, H. Zhang, Y. Feng, X. Yan and X. Zheng, ACS Appl. Nano Mater., 2021, 4, 8192–8199 CrossRef CAS.
- M.-C. Liu, B.-M. Zhang, Y.-S. Zhang and Y.-X. Hu, ACS Sustainable Chem. Eng., 2021, 9, 12930–12939 CrossRef CAS.
- P. Cai, L. Ding, Z. Chen, D. Wang, H. Peng, C. Yuan, C. Hu, L. Sun, Y. N. Luponosov, F. Huang and Q. Xue, Adv. Funct. Mater., 2023, 33, 2300113 CrossRef CAS.
- M. Mozafari and M. Soroush, Mater. Adv., 2021, 2, 7277–7307 RSC.
- V. Neubertova, O. Guselnikova, Y. Yamauchi, A. Olshtrem, S. Rimpelova, E. Cizmar, M. Orendac, J. Duchon, L. Volfova, J. Lancok, V. Herynek, P. Fitl, P. Ulbrich, L. Jelinek, P. Schneider, J. Kosek, P. Postnikov, Z. Kolska, V. Svorcik, S. C. Hertopalov and O. Lyutakov, Chem. Eng. J., 2022, 446, 136939 CrossRef CAS.
- A. Hazan, B. Ratzker, D. Zhang, A. Katiyi, M. Sokol, Y. Gogotsi and A. Karabchevsky, Adv. Mater., 2023, 35, 2210216 CrossRef CAS PubMed.
- Z. Yang, W. Tan, T. Zhang, C. Chen, Z. Wang, Y. Mao, C. Ma, Q. Lin, W. Bi, F. Yu, B. Yan and J. Wang, Adv. Opt. Mater., 2022, 10, 2200714 CrossRef CAS.
- Y. Liu, Y. Xu, C. Xu, J. Chen, H. Liu, H. Zhang, L. Jin, J. Fan, Y. Zou and X. Ma, Adv. Opt. Mater., 2024, 12, 2301439 CrossRef CAS.
- O. B. Seo, S. Saha, N. H. Kim and J. H. Lee, J. Membr. Sci., 2021, 640, 119839 CrossRef CAS.
- J. Wang, Y. Hernandez, M. Lotya, J. N. Coleman and W. J. Blau, Adv. Mater., 2009, 21, 2430–2435 CrossRef CAS.
- X.-P. Li, X. Li, H. Li, Y. Zhao, J. Wu, S. Yan and Z.-Z. Yu, Adv. Funct. Mater., 2022, 32, 2110636 CrossRef CAS.
- X. F. Jiang, L. Polavarapu, S. T. Neo, T. Venkatesan and Q. H. Xu, J. Phys. Chem. Lett., 2012, 3, 785–790 CrossRef CAS PubMed.
- G. Valurouthu, K. Maleski, N. Kurra, M. Han, K. Hantanasirisakul, A. Sarycheva and Y. Gogotsi, Nanoscale, 2020, 12, 14204–14212 RSC.
- D. B. Lioi, P. R. Stevenson, B. T. Seymour, G. Neher, R. D. Schaller, D. J. Gosztola, R. A. Vaia, J. P. Vernon and W. J. Kennedy, ACS Appl. Nano Mater., 2020, 3, 9604–9609 CrossRef CAS.
- X. Tang, J. Luo, Z. Hu, S. Lu, X. Liu, S. Li, X. Zhao, Z. Zhang, Q. Lan, P. Ma, Z. Wang and T. Liu, Nano Res., 2023, 16, 1755–1763 CrossRef CAS.
- L. Fu, M. G. Humphrey and C. Zhang, Nano Res., 2023, 16, 25–32 CrossRef CAS.
- M. Bala Murali Krishna, N. Venkatramaiah, R. Venkatesan and D. Narayana Rao, J. Mater. Chem., 2012, 22, 3059–3068 RSC.
- J. Zhu, Y. Li, Y. Chen, J. Wang, B. Zhang, J. Zhang and W. J. Blau, Carbon, 2011, 49, 1900–1905 CrossRef CAS.
- D. W. Boukhvalov and M. I. Katsnelson, J. Am. Chem. Soc., 2008, 130, 10697–10701 CrossRef CAS PubMed.
- Z. Liu, Y. Wang, X. Zhang, Y. Xu, Y. Chen and J. Tian, Appl. Phys. Lett., 2009, 94, 021902 CrossRef.
- M. K. Kavitha, H. John, P. Gopinath and R. Philip, J. Mater. Chem. C, 2013, 1, 3669–3676 RSC.
- Y.-X. Li, J. Zhu, Y. Chen, J. Zhang, J. Wang, B. Zhang, Y. He and W. J. Blau, Nanotechnology, 2011, 22, 205704 CrossRef PubMed.
- Y. Du, N. Dong, M. Zhang, K. Zhu, R. Na, S. Zhang, N. Sun, G. Wang and J. Wang, Phys. Chem. Chem. Phys., 2017, 19, 2252–2260 RSC.
- T. Bai, C.-Q. Li, J. Sun, Y. Song, J. Wang, W. J. Blau, B. Zhang and Y. Chen, Chem. – Eur. J., 2015, 21, 4622–4627 CrossRef CAS PubMed.
- M. Saravanan, T. C. Sabari Girisun, G. Vinitha and S. Venugopal Rao, RSC Adv., 2016, 6, 91083–91092 RSC.
- J. S. D. Rodriguez, T. Ohigashi, C.-C. Lee, M.-H. Tsai, C.-C. Yang, C.-H. Wang, C. Chen, W.-F. Pong, H.-C. Chiu and C.-H. Chuang, Carbon, 2021, 185, 410–418 CrossRef CAS.
- J. K. El-Demellawi, A. E. Mansour, A. M. El-Zohry, M. N. Hedhili, J. Yin, A.-H. M. Emwas, P. Maity, X. Xu, O. M. Bakr, O. F. Mohammed and H. N. Alshareef, ACS Mater. Lett., 2022, 4, 2480–2490 CrossRef CAS.
- M. Mariano, O. Mashtalir, F. Q. Antonio, W.-H. Ryu, B. Deng, F. Xia, Y. Gogotsi and A. D. Taylor, Nanoscale, 2016, 8, 16371–16378 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc00366g |
| This journal is © The Royal Society of Chemistry 2024 |