
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic acetylacetone-based carbon dots†
Sergei A.
Cherevkov‡
a,
Evgeniia A.
Stepanidenko‡
a,
Mikhail D.
Miruschenko
 a,
Andrei M.
Zverkov
a,
Alexander M.
Mitroshin
ab,
Igor V.
Margaryan
a,
Igor G.
Spiridonov
a,
Denis V.
Danilov
c,
Aleksandra V.
Koroleva
c,
Evgeniy V.
Zhizhin
c,
Marina V.
Baidakova
d,
Roman V.
Sokolov
d,
Maria A.
Sandzhieva
e,
Elena V.
Ushakova
*af and
Andrey L.
Rogach
fg
a,
Andrei M.
Zverkov
a,
Alexander M.
Mitroshin
ab,
Igor V.
Margaryan
a,
Igor G.
Spiridonov
a,
Denis V.
Danilov
c,
Aleksandra V.
Koroleva
c,
Evgeniy V.
Zhizhin
c,
Marina V.
Baidakova
d,
Roman V.
Sokolov
d,
Maria A.
Sandzhieva
e,
Elena V.
Ushakova
*af and
Andrey L.
Rogach
fg
aInternational Research and Education Centre for Physics of Nanostructures, ITMO University, Saint Petersburg 197101, Russia. E-mail: elena.ushakova@itmo.ru
bInstitute of Macromolecular Compounds Russian Academy of Sciences, Saint Petersburg 199004, Russia
cResearch Park, Saint Petersburg State University, Saint Petersburg 199034, Russia
dIoffe Institute, Saint Petersburg 194021, Russia
eSchool of Physics and Engineering, ITMO University, Saint Petersburg 197101, Russia
fDepartment of Materials Science and Engineering, and Centre for Functional Photonics (CFP), City University of Hong Kong, Hong Kong SAR 999077, P. R. China
gIT4Innovations, VSB – Technical University of Ostrava, 17. listopadu 2172/15, Ostrava-Poruba 70800, Czech Republic
First published on 9th February 2024
Abstract
The ongoing development of carbon dots (CDs) for different applications calls for researching novel methods for their synthesis and surface functionalization. For the fabrication of photonic devices, apart from the obvious requirement of bright luminescence, CDs also should be soluble in the non-polar solvents used for the ink-printing of their functional layers. Herein, we introduce amphiphilic CDs synthesized from a mixture of benzoic acid and ethylenediamine in acetylacetone, which satisfy both of the abovementioned requirements. These CDs are quasi-spherical nanoparticles that are 20–50 nm in size, with aliphatic, carbonyl, amide, imine, and carbamate groups at the surface. This wide spectrum of surface groups renders them amphiphilic and soluble in a variety of substances, such as toluene, chloroform, alcohol, and water, with relative polarity ranging from 0.002 to 1. By variation of the molar ratio of benzoic acid and ethylenediamine, the highest quantum yield reported so far of 36% in isopropanol is achieved for the amphiphilic CDs. As a demonstration of the use of developed amphiphilic CDs in LEDs, green-emitting charge-injection devices were fabricated with a broad emission band centered at 515 nm, maximal luminance of 1716 cd m−2, and CCT of 5627 K. These LEDs are the first ones based on amphiphilic CDs. Furthermore, these CDs can be used as luminescent inks and as an active material for solar concentrators.
Introduction
Recently, carbon dots (CDs) have been recognized as an important class of light-emitting nanomaterials due to the ease of preparation, bright photoluminescence (PL), high colloidal stability, and low/negligible toxicity.1–3 Using different chemical precursors in the bottom-up synthesis of CDs makes it possible to functionalize their surface for different purposes, ranging from biomedical to energy/lighting applications.4 For the latter direction, which includes light-emitting devices (LEDs), solar cells, composite catalysts, the energy level structure and existence of electron-withdrawing or -donating groups at the surface of CDs play a crucial role.5–8 In many cases, however, the surface functionality of CDs is somewhat limited by the fact that their synthesis and further solubilization are traditionally performed in polar solvents. Moreover, the application of CDs in devices as a part of the composite emissive/charge-transport layers demands their compatibility with a variety of polymers and other materials, which are mostly soluble in non-polar solvents.9Despite this demand, studies devoted to the development of CDs soluble in nonpolar organic solvents and amphiphilic nanoparticles are still rare. Currently, the non-polar based CDs have been produced by hot injection,10 microwave-assisted,11,12 solvothermal methods,13–15 and by refluxing.16 In most of the studies where hydrophobic CDs were produced, combinations of aliphatic compounds (amines, aldehydes, acids) with hydrophobic molecules, including organic dyes, were used as precursors. For example, hydrophobic CDs were prepared by hot-injection method from melted Paraplast granules injected in toluene and stabilized with dodecanethiol.10 In another study,17 hydrophobic CDs were prepared from rhodamine B and then functionalized with polyethylene glycol (PEG) to increase their solubility in polar media. By refluxing dodecylamine in chlorobenzene, hydrophobic blue-emissive CDs with the PL quantum yield (QY) reaching 10% were made;16 these CDs could be easily mixed with polymers such as poly(methyl methacrylate) and polydimethylsiloxane. In another study, dodecylamine-based CDs were used as surfactants to stabilize multi-walled carbon nanotubes.18 Moreover, methods for fabricating bright amphiphilic CDs are still in their infancy.19 The amphiphilicity of CDs can be introduced via post-synthetic surface modification with long hydrophobic chain epoxides or acrylamides, as was recently shown in a study by Chen et al.20 Another example is the synthesis of CDs in acetone, which mixes well with both polar and non-polar solvents.14 For most of the reported hydrophobic or amphiphilic CDs,10,16,17,20–25 the PL bands experienced shifts with the change in polarity of the medium. This is commonly referred to as solvatochromism, and their PL QYs did not exceed the value of 33%.22 Another class of CDs that can be used for composite fabrication and further implementation as phosphors in down-conversion LEDs are silane-functionalized CDs.26–29 A summary of the synthetic methods and optical properties of hydrophobic and amphiphilic CDs is provided in Table S1 (ESI†).
In this work, we aimed to develop a simple one-pot synthesis of amphiphilic CDs with increased PL QY in a range of solvents. For that, we selected acetylacetone as a solvent and benzoic acid (BA) and ethylenediamine (EDA) as precursors of CDs. The synthesized CDs demonstrate amphiphilic properties, remarkable stability in a variety of solvent media, as well as chemical yield and PL QY up to 36%, which is the highest value reported so far for amphiphilic CDs. Importantly, these CDs are easily mixed with glycerin, a toluene solution of polystyrene, and the common charge transport polymer, polyvinylcarbazole (PVK). We used the amphiphilic CDs to fabricate charge-injection green LEDs with a broad emission band and maximal luminance of 1716 cd cm−2.
Results and discussion
Synthesis of amphiphilic CDs
To synthesize amphiphilic CDs in acetylacetone, a combination of BA and EDA was used (Fig. 1(a)). This synthesis was inspired by a ‘classical’ synthetic pathway towards bright CDs using citric acid and EDA,30,31 which also allowed researchers to produce anisotropic hydrophobic32 and red-emitting33–35 CDs. In this work, citric acid was replaced by BA which served as a hydrophobic component. BA derivatives were previously shown to increase the amount of carbonyl groups at the CD surface, which induced phosphorescence for those CDs in a powder.36 Acetylacetone is a high-boiling solvent (140 °C), which is easily mixed with both polar and non-polar solvents. Thus, it is an ideal candidate as a reactive medium, facilitating the formation of amphiphilic groups at the CD's surface via keto–enol reaction. However, it was previously used only as a carbon source in microwave-assisted syntheses37,38 and Aldol reactions under mild conditions, resulting in the formation of hydrophilic CDs.39 | ||
| Fig. 1 (a) Scheme of the synthesis of amphiphilic CDs from benzoic acid and ethylenediamine in acetylacetone. (b) Photographs of amphiphilic CDs in different solvents, taken under sunlight (left panel) and UV light (right panel). | ||
We envisage three possible mechanisms for the formation of amphiphilic CDs through interaction of acetylacetone with EDA only, BA only, and both precursors together, as demonstrated in Fig. S2–S4 (ESI†), respectively. When EDA reacts with acetylacetone as its carbonyl derivative, bis(acetylacetone)ethylenediamine (Acacen) is formed.40 Subsequently, Acacen in an excess of acetylacetone forms CDs enriched with imine groups at the surface (Fig. S2, ESI†). When BA reacts with acetylacetone, a denser CD core is formed. This is based mostly on acetylacetone covered with hydrophobic benzene rings (Fig. S3, ESI†). The reaction of acetylacetone with BA and EDA at the same time increases the hydrophobicity of the formed CDs and accelerates the formation of Acacen such that esterification inside CDs and 1,4-addition reactions are possible (Fig. S4, ESI†). The proposed mechanisms of efficient interaction between ethylenediamine, benzoic acid, and acetylacetone can be the reason for the rather high mass ratio of the synthesized CDs in relation to the amount of initial precursors (up to 10%). We expect that the CDs formed by the latter reaction would possess a variety of functional groups at the surface which create an amphiphilic shell (Fig. 1(a)), thus rendering those nanoparticles soluble in both polar and nonpolar media. Indeed, we have found that the produced CDs can be easily dispersed in a broad set of polar and nonpolar solvents such as water, methanol, ethanol, isopropanol, acetonitrile, dimethyl sulfoxide (DMSO), acetone, chloroform, toluene, and tetrachloromethane (TCM), as shown by the photographs in Fig. 1(b) obtained under sunlight and UV excitation. In the following, we will elaborate in detail on the morphology, surface chemistry, and optical properties of the produced amphiphilic CDs. In the forthcoming discussion, CD samples produced using EDA or BA as the only precursors are denoted as CDEDA and CDBA, respectively. For the CDs produced from the mixtures of BA and EDA in acetylacetone while changing the molar ratio of these two precursors, samples are designated as CDXX, where XX is the BA-to-EDA molar ratio. The details of the synthesis are provided in the Experimental section.
Morphology and surface functionalities of CDs
The study of the CDs’ morphology performed by a combination of microscopy and spectroscopy methods has confirmed the assumption of their core–shell structure, as shown by TEM images in Fig. 2(a)–(c) and outlined in the forthcoming discussion. | ||
| Fig. 2 Morphology and size of amphiphilic CDs. (a)–(c) TEM images (with HRTEM images as insets) of CDEDA (a), CD0.22 (b), and CDBA (c). (d) AFM image of CD0.22; inset shows a height profile along 2 particles highlighted on the AFM image. (e) Mean size values determined from TEM images and AFM height analysis for CDEDA, CD0.22, and CDBA. (f) XRD pattern of CD0.22. | ||
As can be seen from the TEM image in Fig. 2(a) and Fig. S5(a) (ESI†), CDEDA consisted of a carbonized core of 7.8 ± 1.5 nm, and a rather thick polymer shell (20–25 nm). CDBA consisted of a carbonized core of up to 40 nm in size, whose interplanar distance of 0.34 nm (inset in Fig. 2(c)) corresponds to the distance between the sp2 planes in graphite,41 surrounded by a thicker polymer shell (Fig. 2(c) and Fig. S5(c), ESI†). The CD0.22 sample comprises particles with a 5–7 nm core and interplane distance of 0.25 nm, which can be ascribed to doped sp2-carbon domains as was previously reported for S,N-doped CDs42 or N,Ca-doped CDs.43 These domains are surrounded by a polymer shell so that the overall size of the particles becomes 15.1 ± 4.9 nm (Fig. 2(b) and Fig. S5(b), (c), ESI†). From the analysis of the AFM images of CDEDA, CDBA, and CD0.22 shown in Fig. S6(a) and (b) (ESI†) and Fig. 2(d), respectively, one can derive their height to be less than 20 nm, with a rather broad distribution (Fig. S6(c)–(e), ESI†). The average height was determined as 6.0 ± 3.2, 12.0 ± 8.0, and 10.4 ± 4.0 for CDEDA, CD0.22, and CDBA, respectively. The sizes of the three CD samples estimated from TEM and AFM images are summarized in Fig. 2(e); one can see that they increase with the increase of the BA amount. These CDs are quasi-spherical, as seen in Fig. S5(c) (ESI†) with the example of a single CD0.22 particle. The difference in the TEM and AFM sizes for CD0.22 and CDBA can be attributed to an increased thickness of their polymer shell, as can be seen for CDBA in Fig. 2(c) and Fig. S5(d)–(g) (ESI†), and for CD0.22 in Fig. S5(c) (ESI†).
In the XRD pattern of CD0.22 shown in Fig. 2(f), wide peaks at 16°, 24°, and 42° 2θ are observed; they are attributed to the (004), (002), and (100) planes of the defective planar structures similar to those reported for doped CDs43,44 and graphitic carbon nitride.45 The broadening of the XRD peaks can be related both to the amorphous structure (more than 95% estimated amorphous content) and small size of crystallites (less than 2 nm).
As shown in Fig. S7(a) (ESI†), amphiphilic CDs are composed of carbon, oxygen, and nitrogen (except for the case of CDBA where no nitrogen was present). The contents of these three elements were determined as 67%, 77%, and 65% for carbon, 29%, 16%, and 32% for oxygen, and 1%, 4%, and 0% for nitrogen in CDEDA, CD0.22, and CDBA, respectively. This experimentally determined chemical composition agrees well with the reaction schemes provided in Fig. S2–S4 (ESI†), adjusted to the BA/EDA molar ratio and an excess of acetylacetone in the syntheses. The highest amount of carbon and nitrogen alongside with the lowest amount of oxygen was observed for CD0.22 synthesized from the mixture of BA and EDA in acetylacetone, which confirms that BA facilitates the reaction of EDA with acetylacetone, as illustrated in Fig. S3 (ESI†). From the high-resolution XPS data shown in Fig. S8 (ESI†), it is seen that CDBA are carbonized particles and their surface is rich with hydroxyl and carbonyl groups. For CDEDA, only a small amount of nitrogen atoms is incorporated into the CD structure as pyridinic N. For CDs produced from both EDA and BA, pyridinic and graphitic N are present as well (Fig. S8(e) and (f), ESI†).
The Raman spectrum of CD0.22 (Fig. S7(b), ESI†) has 5 distinct bands with peaks at 1245, 1315, 1415, 1570, and 1655 cm−1, which are attributed to the D*, D, D′′, G, and D′ bands typical for doped graphene and graphene oxide structures46 with relative intensities of 15%, 38%, 5%, 20%, and 27%, respectively. The D* band can be attributed to the sp3-hybridized carbon observed in polyene47 and disordered graphite.48 In addition, the signal at 1130–1200 cm−1 can be attributed to trans-polyacetylene chains.49 The D and G bands are the breathing and stretching modes of the sp2-hybridized carbon domains, respectively.50 The D′′ band originates from the amorphous carbon phase,51 and the D′ band corresponds to an intra-valley resonance with the G band, and undergoes a splitting due to the presence of impurities.52 Thus, we infer that the CDs synthesized here are carbon polymorphs with N and O atoms incorporated into sp2-domains. They demonstrate a high degree of presence of the amorphous/polymer phase (as manifested through the presence of the D′′ and D′ bands and a large width of the D and G bands) and a large amount of surface polymer chains such as trans-polyacetylene (manifested through the presence of the D* band).
From the FTIR spectra of the CDs provided in Fig. S7(c) (ESI†), one can see that those spectra are different from the FTIR spectrum represented by a sum of the precursors (shown by a grey line). The FTIR spectra in the range of 4000–2500 cm−1 of CDs formed from a mixture of BA and EDA have peaks at 3300, 3060, 2974, 2924, and 2886 cm−1, which can be attributed to the stretching vibrations of OH-bonding, Carom-H, asymmetric CH3, asymmetric CH2, and symmetric CH3, respectively. For the 2500–1300 cm−1 range, strong peaks at 1680 and 1610 cm−1, and medium-intensity peaks at 1560, 1530, 1490, 1450, 1380, and 1360 cm−1 are observed. The former two peaks can be attributed to the stretching vibrations of the -CO–CH2–CO- bond in acetylacetone-related structures and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups;53 the peak at 1490 cm−1 originates from the arene stretching; and the peaks at 1300–1500 cm−1 can be attributed to the bonds of the aromatic carbon with nitrogen and/or oxygen atoms. A gradual change in the peaks’ intensities and their positions with an increase of EDA and decrease of the BA content in the precursor mixture is observed: the amount of aromatic phase decreases with an increase of N-doping, which manifests itself as the increase of intensity in the wavenumber regions attributed to the C–N groups vibrations. From the above chemical composition analysis, it can be inferred that amphiphilic CDs are O,N-doped carbon particles with an excess of aliphatic and aromatic groups at the surface. Moreover, they possess –CO–CH2–CO– groups from acetylacetone, which should make them highly soluble in a variety of solvents.
C groups;53 the peak at 1490 cm−1 originates from the arene stretching; and the peaks at 1300–1500 cm−1 can be attributed to the bonds of the aromatic carbon with nitrogen and/or oxygen atoms. A gradual change in the peaks’ intensities and their positions with an increase of EDA and decrease of the BA content in the precursor mixture is observed: the amount of aromatic phase decreases with an increase of N-doping, which manifests itself as the increase of intensity in the wavenumber regions attributed to the C–N groups vibrations. From the above chemical composition analysis, it can be inferred that amphiphilic CDs are O,N-doped carbon particles with an excess of aliphatic and aromatic groups at the surface. Moreover, they possess –CO–CH2–CO– groups from acetylacetone, which should make them highly soluble in a variety of solvents.
Optical properties of amphiphilic CDs
Optical properties were monitored for amphiphilic CDs dissolved in three different solvents–water, isopropanol, and chloroform (Fig. 3 and Fig. S9–S15, ESI†). The peak positions for absorption, PL excitation and PL spectra of these samples are summarized in Table S3 (ESI†). The absorption spectra of all CDs show peaks at 220, 260, and 300–315 nm. Upon addition of EDA, several other peaks emerge at 390, 465, and 490 nm (Fig. 3(a)–(c)). While comparing the PLE-PL maps for CDBA with other samples (Fig. S15(c)–(e), ESI†), the following observations can be made: CDBA in isopropanol has a PL band at 480 nm excited over a wide spectral range (240–400 nm), whereas the EDA-based CDs in isopropanol have a PL band at 350 nm efficiently excited at 240 and 290 nm and PL band at 460 nm efficiently excited at 240, 300, and 390 nm. | ||
| Fig. 3 Optical properties of CDs in water, isopropanol, and chloroform: CDEDA (violet), CD0.15 (blue), CD0.22 (cyan), CD0.33 (green), CD1 (yellow), CD2 (orange), and CDBA (red). (a)–(c) Absorption and (e)–(g) PL spectra excited at 400 nm. (d) PL QY of CDs in water (black squares), isopropanol (red circles), and chloroform (blue triangles), depending on the BA/EDA molar ratio in the synthesis. (h) Radiative (Γ, solid symbols) and nonradiative (knr, open symbols) relaxation constants of CDs in water (black squares), isopropanol (red circles), and chloroform (blue triangles), as a function of the BA/EDA molar ratio used in the synthesis. | ||
The most intense emission among CDs dispersed in different solvents is observed for those dissolved in isopropanol, with a maximal PL QY of 36% for CD0.22 (Fig. 3(d)). The least intense emission in general is observed for CDBA, meaning that optical centers formed under participation of EDA and BA result in CDs with a higher brightness. From the PL decays and estimated PL lifetimes shown in Fig. S16 (ESI†) and summarized in Table S4 (ESI†), radiative (Γ) and nonradiative (knr) relaxation rates were calculated. The radiative rates follow the trend of the PL QY as shown in Fig. 3(h), varying from 0.003 to 0.09 × 109 s−1, whereas nonradiative rates fluctuate around the mean value of 0.2 × 109 s−1. From the spectra, the position of the absorption and PL of optical centers formed in amphiphilic CDs are only shifted slightly upon changing the solvent, indicating that they are not significantly affected by the medium and are well protected within the CD structure.
Optical properties for CD0.22 of identical weights were measured in different solvents with varying relative polarity given in brackets: tetrachloromethane (0.052), toluene (0.099), chloroform (0.259), acetone (0.355), dimethyl sulfoxide (0.444), acetonitrile (0.46), isopropanol (0.617), ethanol (0.654), methanol (0.762), and water (1.0), as summarized in Fig. 4. The CD concentration was estimated as ∼0.5 mg mL−1 for all of these samples. With the increase of the relative polarity from 0.052 to 1.0, the absorption peaks slightly blue-shifted from 402 to 385 nm (Fig. 4(a) and (b)), while the PL spectra blue-shifted from 465 to 457 nm (Fig. 4(e) and (f)). In comparison to reported hydrophobic CDs,13,54 where the absorption and PL bands experienced much larger spectral shifts of up to 160 nm, the observed changes in the absorption and PL peak positions are rather minor. A similar behavior was observed for amphiphilic CDs synthesized from (formylmethyl)triphenylphosphonium chloride in acetone, whose PL peak shifted from 595 and 620 nm by changing the solvent from water to chloroform.14 The optical density of the amphiphilic CDs increases with the relative polarity of the solvent (Fig. 4(c)), while PL QYs reach the maximal value of 36% in the solvent with the relative polarity of 0.617 (isopropanol) and are lower in both more polar and less polar solvents, going down to 3% only for CD0.22 in tetrachloromethane (Fig. 4(g)). The change in PL QY can be explained by the change in either radiative or nonradiative decay rate depending on the type of emission centers. Both radiative and nonradiative decay rates are affected by the interaction of the excited dipole with solvent molecules. However, no single model can explain the diverse spectral properties displayed by fluorophores in various environments.55
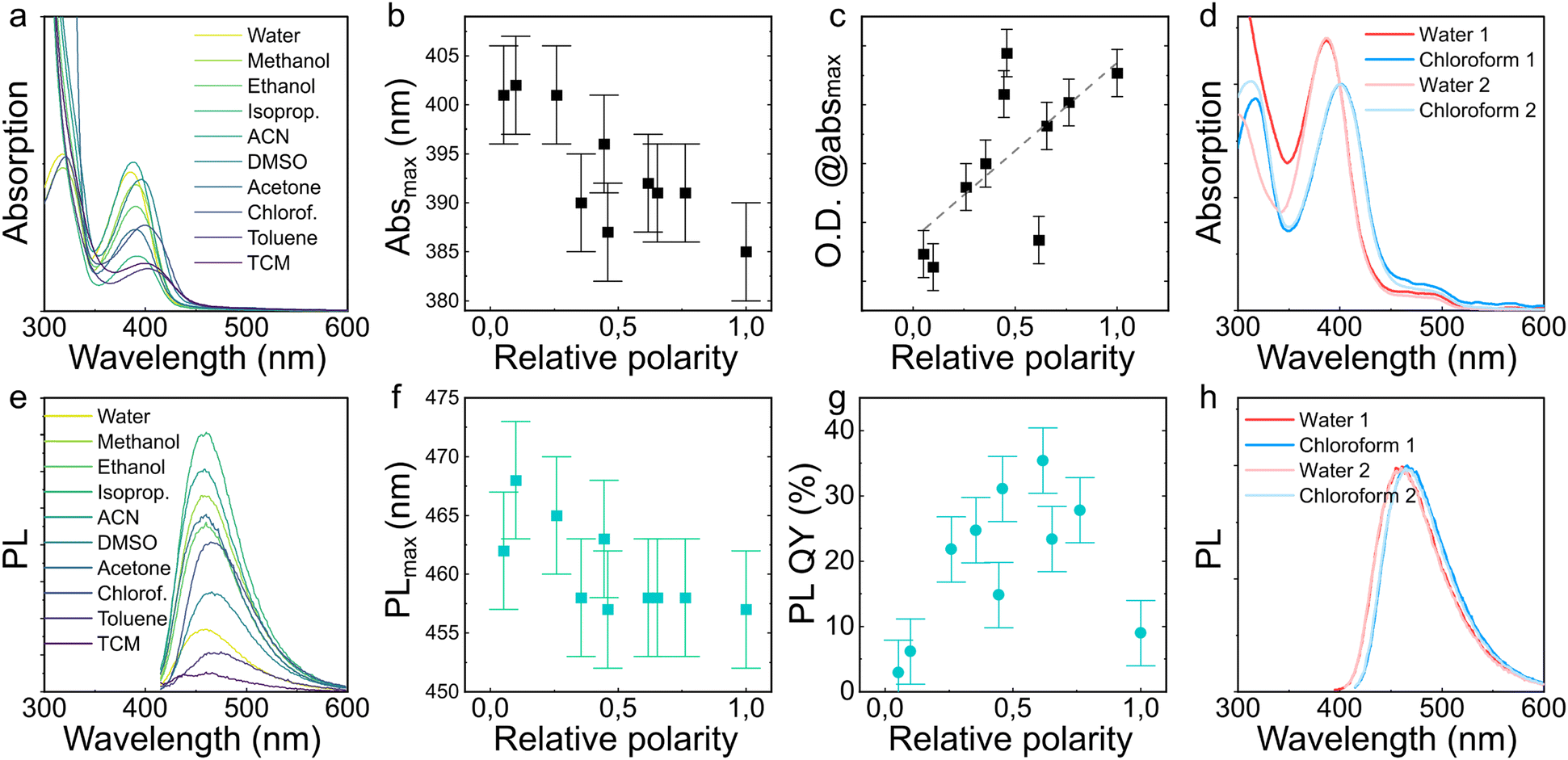 | ||
| Fig. 4 Optical properties of CD0.22 dispersed in a set of solvents with varying relative polarity: (a) absorption spectra, (b) long-wavelength absorption peak position and (c) optical density at its maximum depending on the relative polarity of the solvent; (e) PL spectra, (f) PL peak position excited at 400 nm, and (g) PL QY as a function of the relative polarity of the solvent; (d) absorption and (h) normalized PL spectra excited at 400 nm recorded while changing the solvents from water to chloroform in 2 cycles (1 and 2). | ||
To demonstrate the amphiphilicity of CDs, their optical spectra during several cycles of dissolution in water and chloroform were measured, and are shown in Fig. 4(d) and (h). It is seen that both absorption and PL spectra are the same in water/chloroform after the 1st and 2nd dispersion cycles of CD0.22, in terms of the positions of the respective maxima, indicating that the produced CDs are indeed amphiphilic, as was shown in Fig. 1(b).
Energy level structure of CDs, and their application in LEDs and luminescent inks
The amphiphilic property of the CDs opens a possibility to fabricate composite materials in combination with non-polar conductive polymers, such as polyvinyl carbazole (PVK), for both optoelectronic and photovoltaic devices. For this purpose, we firstly determined the energy structures of CDEDA, CDBA, and CD0.22 by UPS measurements,56 with the UPS spectra presented in Fig. S17 (ESI†). The derived energy structure schemes are shown in Fig. 5(a) for CD0.22 and in Fig. S18 (ESI†) for CDEDA and CDBA. The depth of the highest occupied molecular orbital (HOMO) was estimated from the UPS spectra (Fig. S17, ESI†) from the width of the He I UPS spectrum according to EHomo = (Ecut-off − Ef) − hλ, where hλ is the energy of the incident photons of 21.22 eV; Ecut-off – the energy of the secondary electron cut-off regions (right side edge of the spectrum shown in Fig. S17(a), ESI†), EF – the left-side edge of the spectrum as shown in Fig. S17(a) (ESI†). The ground state S0 corresponding to the HOMO energy was plotted from the vacuum level with Evac = 0 eV, and the position of the lowest unoccupied molecular orbital (LUMO) was estimated according to ELUMO = EHOMO + Eg, where Eg was estimated as the edge of the absorption spectrum (Fig. S17b, ESI†). The excited states S1, S2, and S3 were plotted according to the positions of ELUMO, and absorption bands Abs-1, Abs-2, and Abs-3, respectively, and the radiative transitions were shown as colored arrows corresponding to emissive centers determined from PLE-PL maps. The values of all energy level positions are summarized in Table S5 (ESI†). After absorption of light with the wavelength of 260 nm (black arrow in Fig. 5(a) and S18, ESI†), the excited state S3 related to the Abs-3 band becomes deactivated through the nonradiative transition to the lowest excited states S1 and S2, from which radiative transitions PL-1 and PL-2 (green arrows in Fig. 5(a) and Fig. S18, ESI†) to the ground state S0 can occur. The S1 and S2 states can also be populated by a direct absorption of light with wavelengths of 315 and 395 nm, respectively (black arrows in Fig. 5(a)), and be deactivated through both radiative (green arrows in Fig. 5(a) and Fig. S18, ESI†) and nonradiative transitions to the ground state S0. The radiative relaxation of photoexcited charge carriers for CD0.22 are observed as emission bands at 350 and 460 nm, which can be excited at 260, 315, and 395 nm (Fig. S11(d) and Table S5, ESI†). | ||
| Fig. 5 (a) Energy level structure of CD0.22. HOMO and LUMO energy levels are shown by thick black lines. Upon excitation, the charge carriers move from the ground state (S0) to higher energy states S1, S2, or S3 corresponding to the experimentally observed absorption bands (thin black lines). From these states, the charge carriers can relax radiatively, which is represented by colored arrows (the length of the arrows corresponds to the energy of the emission), or non-radiatively, which is represented by wavy lines. (b) Energy level diagram of the charge-injection CD-LED: the levels for PVK are within the cyan box, and those for the CD0.22 are within the empty box. (c) CIE coordinates for the green light emitted by the fabricated CD-LEDs; the inset shows a photograph of the operating LED. (d) PL spectra of the CD0.22 solution in isopropanol (dashed black line) and CD0.22@PVK excited at 405 nm (orange line); EL spectrum of CD-LED is shown by green line. (e) Current density–luminance–voltage (J–L–V) characteristic curve and (f) luminance–current density (L–J) curve of the CD-LED. | ||
For the CD0.22, their highest occupied molecular orbital (HOMO) lies at −7.2 eV, while the lowest unoccupied molecular orbital (LUMO) is at −4.4 eV. From the optical data discussed above, one can identify excited states of S1, S2, and S3 located at −4.0, −3.3, and −2.4 eV, which correspond to the absorption peaks Abs-1, Abs-2, and Abs-3 in Table S3 (ESI†), respectively; those levels are presented in Fig. 5(a) as thin black lines. The radiative transitions of the two emissive centers corresponding to PL-1 and PL-2 in Table S3 (ESI†) are shown by colored arrows from the S1 and S2 energy levels, respectively. CD0.22 has a Stokes shift, which is shown as a difference between the ground state (S0 level) and the energy level. The charge carriers from this level radiatively relax (dotted line), as shown in Fig. 5(a). A very similar energy structure could be derived for CDEDA, as shown in Fig. S18 (ESI†). However, for the CDBA, HOMO and LUMO levels lie at −6.5 and −3.1 eV, respectively; and there are only two excited states, S1 and S2, with one radiative transition from S2. It is worth noting that the energy level engineering of CDs is important, since they can be then further used as auxiliary components to improve the charge transporting layer/active materials interfaces.57 In a recent review, the positions of the HOMO and LUMO energy levels of different materials were summarized for the electron transport layers applicable in perovskite solar cells: the LUMO energy level varied from −3.3 to −4.5 eV.58 The similar values of LUMO levels achieved for the CDs developed in this work, combined with the possibility to mix them with organic polymers, open an opportunity to use amphiphilic CDs in perovskite solar cells, including those on flexible substrates.59
Considering that the highest PLQY of 36% was achieved for CD0.22, these particles were used for the fabrication of CD-LEDs. A very common conductive polymer PVK was chosen as a polymer host for the CDs. CD0.22 was mixed with PVK in chlorobenzene with a final concentration of 25 wt%; the detailed procedure for the CD-LED fabrication is provided in Supporting Information. The multilayer structure and the band structure for the fabricated CD-LED is shown in Fig. 5(b). CD0.22 has a lower HOMO level (−7.2 eV) as compared to PVK (−5.8 eV). This helps to facilitate the charge carrier accumulation within the CD-PVK layer. The Commission Internationale de L'Eclairage (CIE) coordinates of the EL spectrum is (0.332, 0.430), and the correlated color temperature (CCT) is 5627 K (Fig. 5(c)). Fig. 5(d) shows the electroluminescence (EL) spectrum of the operating CD-LED in comparison with the PL spectra of the same device excited at 405 nm from the ITO side, and of the reference sample of CD0.22 embedded in PVK (CD0.22@PVK). Both EL and PL bands of the LED device and the CD0.22@PVK film are redshifted to 490–530 nm as compared to the PL band of CD0.22 in isopropanol (shown by the dashed line in Fig. 5(d) and in the PL-PLE map in Fig. S19, ESI†), along with the emergence of emission in the 600–700 nm spectral region. A similar redshift was observed in ref. 60, where it was attributed to an activation of emission from surface states with a lower bandgap as compared to the CD's core states caused by injection of charge carriers. In our case, the similarity of the PL spectrum of CD0.22@PVK with the EL from CD-LED points to the possibility of activation of the radiative transitions related to the absorption band of CDs at 510 nm, as well as the formation of additional emissive centers arising from the interaction between CDs and PVK. Further investigations will be needed to address those two possibilities.
From Fig. 5(e), the turn-on voltage (Von), which is defined as the bias voltage to be applied to a CD-LED to reach the luminance of 1 cd m−2, is equal to 5.2 V. The threshold voltage for the current density, which is estimated from the current density slope in Fig. 5(e), is 9.7 V. The current efficiency of the devices was estimated as 0.2 cd A−1 at the current density of 31 mA cm−2 (Fig. 5(f)). The average brightness of the CD-LEDs exceeds 1000 cd cm−2, and the maximal brightness reaches 1716 cd cm−2. The latter value is comparable to or better than those for the reported CD-based LEDs listed in Table S6 (ESI†) for devices employing CDs with a PL QY near 40%. The CD-LEDs kept their emission after at least 50 on/off cycles.
The amphiphilicity of the developed CDs opens up an opportunity to easily mix them with different kinds of polymer solutions. As an example, CD0.22 was mixed with 5 wt% of polystyrene (PS) in toluene, with a resulting concentration of 0.5 mg mL−1 (CD0.22/PS-1) and 0.25 mg mL−1 (CD0.22/PS-2). The appearance of the CD0.22/PS-1 sample under UV light is shown in Fig. 6(a). The mixtures were deposited on glass slides, forming highly transparent thin films with a transmission of 88.7% for both CD0.22/PS-1 and CD0.22/PS-2 at 365 nm (Fig. 6(b) and Fig. S20, ESI†). From Fig. 6(c), one can see that the CD films demonstrate bright emission at the substrate edge under UV light (365 nm) as compared to clear glass, thus providing a simple solar concentrator. From PLE-PL maps, the emission band slightly blueshifts from 460 nm to 440 nm and broadens compared to that of CD0.22 in isopropanol solution with excitation bands observed at 220, 315, and 390 nm (Fig. S11, ESI†). We also checked the solubility of CD0.22 in glycerin and glycerin/isopropanol media for obtaining luminescent inks (CD0.22/Gl), as shown in Fig. 6(a). The obtained mixture can be easily transferred into a fountain pen for writing, and the resulting inscription “Nano” shows emission similar to that in the isopropanol solution, as shown in Fig. 6(d) and (e). We also noticed that apart from the demonstration of amphiphilic CDs produced in this work as active material for LEDs, solar concentrators, and luminescent inks, they can also be used as auxiliary components of perovskite-based solar cells.61
 | ||
| Fig. 6 (a) Photographs of C0.22 dispersed in polystyrene/toluene (CD0.22/PS, left) and glycerin/isopropanol (CD0.22/Gl, right) solutions, taken under 365 nm UV lamp. (b) and (c) Photographs of thin films of CD0.22 (CD0.22/PS-1 and CD0.22/PS-2 samples) on glass substrates under white light (b) and under 365 nm UV lamp (c), compared to bare glass slide. (d) and (e) Photographs of a “Nano” inscription made with a CD0.22 ink on a filter paper, taken under white light (d) and under 365 nm UV lamp (e). | ||
Conclusions
To conclude, we introduced the synthesis of amphiphilic CDs from a mixture of benzoic acid and ethylenediamine in acetylacetone, which are easily dispersible in a variety of solvents of different polarity, such as water, methanol, ethanol, isopropanol, acetonitrile, dimethyl sulfoxide, acetone, chloroform, toluene, and tetrachloromethane without any significant changes of the absorption and emission peak positions. The high PL QY reaching 36% makes these amphiphilic CDs suitable as an active material for different applications, including charge-injection CD-LEDs, where they can be used in combination with a common polymer PVK. The fabricated LEDs show the EL peak at 515 nm with a 200 nm width, maximal luminance of 1716 cd cm−2, CIE coordinates of X = 0.332, Y = 0.430 and CCT of 5627 K. Mixing amphiphilic CDs with glycerin and polystyrene makes it possible to fabricate luminescent inks and thin films with optical responses similar to that for CDs in solution. The developed synthesis of amphiphilic CDs opens up an opportunity to conveniently fabricate different optoelectronic and photovoltaic devices with improved characteristics.Experimental
Synthesis of CDs
BA was dissolved in 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mL of acetylacetone under sonication, and different amounts of EDA were added to the mixture; the masses of BA and the volumes of EDA used to produce 7 different CD samples are provided in Table S2 (ESI†). The mixture was transferred into a 40mL Teflon reactor and maintained at 190 °C for 8h in an oven. After the solvothermal treatment, the CD solution was dialyzed through a membrane with a molecular weight cut-off of 7 kDa against deionized water for two days to remove any residual raw materials. Details of the purification procedure are given in ESI.† After the dialysis, the CDs were redispersed in isopropanol and stored at 5 °C.
mL of acetylacetone under sonication, and different amounts of EDA were added to the mixture; the masses of BA and the volumes of EDA used to produce 7 different CD samples are provided in Table S2 (ESI†). The mixture was transferred into a 40mL Teflon reactor and maintained at 190 °C for 8h in an oven. After the solvothermal treatment, the CD solution was dialyzed through a membrane with a molecular weight cut-off of 7 kDa against deionized water for two days to remove any residual raw materials. Details of the purification procedure are given in ESI.† After the dialysis, the CDs were redispersed in isopropanol and stored at 5 °C.
CD-LED fabrication
A layer of ITO pre-etched with hydrochloric acid on glass substrates was used as a transparent anode. The substrates were cleaned by ultrasonic treatment in soap solution, distilled water, deionized water, acetone, and isopropanol for 10 min, and dried by an air flow. Next, the substrates were treated with UV and oxygen plasma for 20 and 10 min, respectively. A layer of PEDOT-PSS was deposited on the clean ITO surface by spin-coating (2000 rpm, 1 min), and dried at 140 °C for 10 min. A layer of PolyTPD was deposited from its chlorobenzene solution by spin-coating (2000 rpm, 1 min) in a nitrogen atmosphere, and dried at 120 °C for 10 min. Next, the mixture of CD0.22 (10 mg mL−1) and PVK (10 mg mL−1) in chlorobenzene was deposited by spin-coating (2000 rpm, 1 min) and dried at 80 °C for 10 min. The final concentration of CDs in PVK was estimated as 25 wt%. Finally, layers of TPBI, LiF, and Al were deposited by spraying in a vacuum spraying chamber at a pressure of 1–2 × 10−6 mbar. The film prepared using the same procedure from the mixture of CD0.22 (10 mg mL−1) and PVK (10 mg mL−1) in chlorobenzene was used as a reference sample; it is designated hereafter as CD0.22@PVK. The figures-of-merit of CD-LEDs were measured at room temperature inside a nitrogen-filled glovebox without additional cooling. Under these conditions, the CD-LEDs maintained their functionality for up to 30 min of continuous operation.CD-based solar concentrators
A toluene solution containing 5 wt% polystyrene was mixed with different amounts of stock solution of CD0.22 in isopropanol to obtain two mixtures with CD concentrations of 0.5 mg mL−1 (CD0.22/PS-1) and 0.25 mg mL−1 (CD0.22/PS-2). A 100-μL volume of the obtained mixtures was spin-casted (3000 rpm, 30 s) onto clean glass slides.CDs ink fabrication
A 10-μL volume of CD0.22 stock solution in isopropanol with a concentration of 13 mg mL−1 was added to 1 mL of glycerin and glycerin/isopropanol (1/1 volume mixture). The obtained mixture was used as ink for writing on filter paper.The information on the materials used and characterization is given in the ESI.†
Author contributions
Conceptualization – S. A. C. and E. V. U., data curation – S. A. C. and E. A. S., formal analysis – S. A. C. and E. A. S., funding acquisition – E. V. U. and A. L. R., investigation – S. A. C., E. A. S., M. D. M., A. M. Z., A. M. M., I. V. M., I. G. S., D. V. D., A. V. K., E. V. Z., M. V. B., R. V. S., M. A. S., project administration and supervision – E. V. U., writing (original draft) – S. A. C. and E. V. U., writing (review & editing) – S. A. C. and A. L. R.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Priority 2030 Federal Academic Leadership Program, RSF 22-13-00294, and by the project “Experimental and theoretical studies of near-infrared-emitting and chiral carbon dot luminophores project” from Moravian-Silesian Region, contract no. 00734/2023/RRC. TEM studies were performed on the equipment of the Interdisciplinary Resource Centre for Nanotechnology of the Scientific Park of St. Petersburg State University. XPS studies were performed using the equipment of the Resource Center “Physical methods of surface investigation” of the Scientific Park of St. Petersburg State University. The authors express their gratitude to the ITMO University Core Facility Center “Nanotechnologies”.Notes and references
- G. Ragazzon, A. Cadranel, E. V. Ushakova, Y. Wang, D. M. Guldi, A. L. Rogach, N. A. Kotov and M. Prato, Chem, 2021, 7, 606–628 CAS.
- B. Wang, H. Cai, G. I. N. Waterhouse, X. Qu, B. Yang and S. Lu, Small Sci., 2022, 2, 2200012 CrossRef CAS.
- B. Wang and S. Lu, Matter, 2022, 5, 110–149 CrossRef CAS.
- L. Dorđević, F. Arcudi, M. Cacioppo and M. Prato, Nat. Nanotechnol., 2022, 17, 112–130 CrossRef PubMed.
- A. P. Litvin, X. Zhang, E. V. Ushakova and A. L. Rogach, Adv. Funct. Mater., 2021, 2010768 CrossRef CAS.
- Y. Zhai, B. Zhang, R. Shi, S. Zhang, Y. Liu, B. Wang, K. Zhang, G. I. N. Waterhouse, T. Zhang and S. Lu, Adv. Energy Mater., 2022, 12, 2103426 CrossRef CAS.
- S. Mandal and P. Das, Appl. Mater. Today, 2022, 26, 101331 CrossRef.
- T. Feng, S. Tao, D. Yue, Q. Zeng, W. Chen and B. Yang, Small, 2020, 16, 2001295 CrossRef CAS PubMed.
- Q. Xu, H. Cai, W. Li, M. Wu, Y. Wu and X. Gong, J. Mater. Chem. A, 2022, 10, 14709–14731 RSC.
- A. Talib, S. Pandey, M. Thakur and H. F. Wu, Mater. Sci. Eng., C, 2015, 48, 700–703 CrossRef CAS PubMed.
- S. Mitra, S. Chandra, T. Kundu, R. Banerjee, P. Pramanik and A. Goswami, RSC Adv., 2012, 2, 12129–12131 RSC.
- M. Liu, X. Li, Y. Zheng, Y. Zhu, T. Li, Z. He, C. Zhang and K. Zhang, J. Mater. Sci., 2023, 58, 902–910 CrossRef CAS.
- H. Yang, Y. Liu, Z. Guo, B. Lei, J. Zhuang, X. Zhang, Z. Liu and C. Hu, Nat. Commun., 2019, 10, 1789 CrossRef PubMed.
- S. Rajendran and S. K. Bhunia, Colloids Surf., A, 2023, 661, 130882 CrossRef CAS.
- S. Pagidi, H. K. Sadhanala, K. Sharma and A. Gedanken, Adv. Electron. Mater., 2022, 8, 2100969 CrossRef CAS.
- K. Yin, D. Lu, L. Wang, Q. Zhang, J. Hao, G. Li and H. Li, J. Phys. Chem. C, 2019, 123, 22447–22456 CrossRef CAS.
- X. Shi, X. Wang, S. Zhang, Z. Zhang, X. Meng, H. Liu, Y. Qian, Y. Lin, Y. Yu, W. Lin and H. Wang, Langmuir, 2023, 39, 5056–5064 CrossRef CAS PubMed.
- Y. Yang, M. Zhao and L. Lai, Carbon, 2023, 202, 398–413 CrossRef CAS.
- P. Zhao and L. Zhu, Chem. Commun., 2018, 54, 5401–5406 RSC.
- P. Chen, X. He, Y. Hu, X. L. Tian, X. Q. Yu and J. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 19937–19950 CrossRef CAS PubMed.
- P. Zhao, X. Li, G. Baryshnikov, B. Wu, H. Ågren, J. Zhang and L. Zhu, Chem. Sci., 2018, 9, 1323–1329 RSC.
- Y. Liu, D. Sun, Z. Zhang, L. Zhang, S. Nie, J. Xiao, Y. S. Chung, H. Choi, S. Kim and C. Liu, Part. Part. Syst. Charact., 2020, 37, 2000146 CrossRef CAS.
- S. Prakash, S. Sahu, B. Patra and A. K. Mishra, Spectrochim. Acta, Part A, 2023, 290, 122257 CrossRef CAS PubMed.
- Y. Choi, S. Jo, A. Chae, Y. K. Kim, J. E. Park, D. Lim, S. Y. Park and I. In, ACS Appl. Mater. Interfaces, 2017, 9, 27883–27893 CrossRef CAS PubMed.
- A. Mikhraliieva, O. Tkachenko, R. Freire, V. Zaitsev, Y. Xing, A. Panteleimonov, M. Strømme and T. M. Budnyak, ACS Appl. Nano Mater., 2022, 10962–10972 CrossRef CAS.
- Z. Xie, Z. Yin, Y. Wu, C. Liu, X. Hao, Q. Du and X. Xu, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- Z. Xie, F. Wang, C.-Y. Liu, Z. Xie, F. Wang and C.-Y. Liu, Adv. Mater., 2012, 24, 1716–1721 CrossRef CAS PubMed.
- X. Dong, Y. Wang, R. Guan, J. Ren, Z. Xie, X. Z. Dong, Y. Wang, J. Ren, Z. Xie and R. F. Guan, Small, 2021, 17, 2105273 CrossRef CAS PubMed.
- Z. Xie, Q. Du, Y. Wu, X. Hao and C. Liu, J. Mater. Chem. C, 2016, 4, 9879–9886 RSC.
- E. A. Stepanidenko, I. A. Arefina, P. D. Khavlyuk, A. Dubavik, K. V. Bogdanov, D. P. Bondarenko, S. A. Cherevkov, E. V. Kundelev, A. V. Fedorov, A. V. Baranov, V. G. Maslov, E. V. Ushakova and A. L. Rogach, Nanoscale, 2020, 12, 602–609 RSC.
- C. J. Reckmeier, Y. Wang, R. Zboril and A. L. Rogach, J. Phys. Chem. C, 2016, 120, 10591–10604 CrossRef CAS.
- Y. Xiong, X. Zhang, A. F. Richter, Y. Li, A. Döring, P. Kasák, A. Popelka, J. Schneider, S. V. Kershaw, S. J. Yoo, J. G. Kim, W. Zhang, W. Zheng, E. V. Ushakova, J. Feldmann and A. L. Rogach, ACS Nano, 2019, 13, 12024–12031 CrossRef CAS PubMed.
- X. Miao, D. Qu, D. Yang, B. Nie, Y. Zhao, H. Fan, Z. Sun, X. Miao, B. Nie, Z. Sun, H. Fan, Y. Zhao and D. Yang, Adv. Mater., 2018, 30, 1704740 CrossRef PubMed.
- X. Bao, Y. Yuan, J. Chen, B. Zhang, D. Li, D. Zhou, P. Jing, G. Xu, Y. Wang, K. Holá, D. Shen, C. Wu, L. Song, C. Liu, R. Zbořil and S. Qu, Light: Sci. Appl., 2018, 7, 1–11 CrossRef CAS PubMed.
- H. Ding, J.-S. Wei, N. Zhong, Q.-Y. Gao and H.-M. Xiong, Langmuir, 2017, 33, 12635–12642 CrossRef CAS PubMed.
- X. Sun, W. He and B. Liu, J. Phys. Chem. C, 2022, 126, 3540–3548 CrossRef CAS.
- S. Umrao, M. H. Jang, J. H. Oh, G. Kim, S. Sahoo, Y. H. Cho, A. Srivastva and I. K. Oh, Carbon, 2015, 81, 514–524 CrossRef CAS.
- G. He, M. Shu, Z. Yang, Y. Ma, D. Huang, S. Xu, Y. Wang, N. Hu, Y. Zhang and L. Xu, Appl. Surf. Sci., 2017, 422, 257–265 CrossRef CAS.
- H. G. Ye, X. Lu, R. Cheng, J. Guo, H. Li, C. F. Wang and S. Chen, J. Lumin., 2021, 238, 118311 CrossRef CAS.
- S. Özkar, D. Ülkü, L. T. Yildirim, N. Biricik and B. Gümgüm, J. Mol. Struct., 2004, 688, 207–211 CrossRef.
- I. Kovalenko, D. G. Bucknall and G. Yushin, Adv. Funct. Mater., 2010, 20, 3979–3986 CrossRef CAS.
- S. Do, W. Kwon, Y.-H. H. Kim, S. R. Kang, T. T.-W. W. Lee, T. T.-W. W. Lee and S.-W. W. Rhee, Adv. Opt. Mater., 2016, 4, 276–284 CrossRef CAS.
- V. Dutt Sharma, V. Kansay, G. Chandan, A. Bhatia, N. Kumar, S. Chakrabarti and M. K. Bera, Carbon, 2023, 201, 972–983 CrossRef.
- J. Bai, N. Xiao, Y. Wang, H. Li, C. Liu, J. Xiao, Y. Wei, Z. Guo and J. Qiu, Carbon, 2021, 174, 750–756 CrossRef CAS.
- K. J. Mintz, M. Bartoli, M. Rovere, Y. Zhou, S. D. Hettiarachchi, S. Paudyal, J. Chen, J. B. Domena, P. Y. Liyanage, R. Sampson, D. Khadka, R. R. Pandey, S. Huang, C. C. Chusuei, A. Tagliaferro and R. M. Leblanc, Carbon, 2021, 173, 433–447 CrossRef CAS.
- A. Y. Lee, K. Yang, N. D. Anh, C. Park, S. M. Lee, T. G. Lee and M. S. Jeong, Appl. Surf. Sci., 2021, 536, 147990 CrossRef CAS.
- B. Dippel, H. Jander and J. Heintzenberg, Phys. Chem. Chem. Phys., 1999, 1, 4707–4712 RSC.
- R. Al-Jishi and G. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 26, 4514 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 121405 CrossRef.
- A. C. Ferrari, S. E. Rodil, J. Robertson, S. E. Rodil and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 155306 CrossRef.
- S. Vollebregt, R. Ishihara, F. D. Tichelaar, Y. Hou and C. I. M. Beenakker, Carbon, 2012, 50, 3542–3554 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- N. Tajaddini, M. Talebizadeh and M. Anary-Abbasinejad, Tetrahedron Lett., 2019, 60, 366–370 CrossRef CAS.
- D. Gao, Y. Zhang, K. Wu, H. Min, D. Wei, J. Sun, H. Yang and H. Fan, Biosens. Bioelectron., 2022, 200, 113928 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer S., Baltimore, USA, 2006 Search PubMed.
- K. D. Kosolapova, A. V. Koroleva, I. A. Arefina, M. D. Miruschenko, S. A. Cherevkov, I. G. Spiridonov, E. V. Zhizhin, E. V. Ushakova and A. L. Rogach, Nanoscale, 2023, 15, 8845–8853 RSC.
- A. P. Litvin, X. Zhang, E. V. Ushakova and A. L. Rogach, Adv. Funct. Mater., 2021, 31, 2010768 CrossRef CAS.
- H. Wang, H. Li, W. Cai, P. Zhang, S. Cao, Z. Chen and Z. Zang, Nanoscale, 2020, 12, 14369–14404 RSC.
- Z. Xu, Q. Zhuang, Y. Zhou, S. Lu, X. Wang, W. Cai and Z. Zang, Small Struct., 2023, 4, 2200338 CrossRef CAS.
- N. Urushihara, T. Hirai, A. Dager, Y. Nakamura, Y. Nishi, K. Inoue, R. Suzuki, M. Tanimura, K. Shinozaki and M. Tachibana, ACS Appl. Nano Mater., 2021, 4, 12472–12480 CrossRef CAS.
- I. V. Margaryan, A. A. Vedernikova, P. S. Parfenov, M. A. Baranov, D. V. Danilov, A. V. Koroleva, E. V. Zhizhin, S. A. Cherevkov, X. Zhang, E. V. Ushakova and A. P. Litvin, Photonics, 2023, 10, 379 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc04675c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |