
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bay-substituted octaazaperopyrenedioxides as solid-state emitters for strong light-matter coupling†
Manuel
Hertzog‡
 a,
Robert
Eichelmann‡
b,
Pierre
Jeudy
b,
Tobias
Wesp
b,
Joachim
Ballmann
b,
Simon
Settele
a,
Finn L.
Sebastian
a,
Andreas
Mischok
c,
Florian
Le Roux
c,
Francisco
Tenopala-Carmona
c,
Malte C.
Gather
c,
Lutz H.
Gade
*b and
Jana
Zaumseil
*a
a,
Robert
Eichelmann‡
b,
Pierre
Jeudy
b,
Tobias
Wesp
b,
Joachim
Ballmann
b,
Simon
Settele
a,
Finn L.
Sebastian
a,
Andreas
Mischok
c,
Florian
Le Roux
c,
Francisco
Tenopala-Carmona
c,
Malte C.
Gather
c,
Lutz H.
Gade
*b and
Jana
Zaumseil
*a
aInstitute for Physical Chemistry, Universität Heidelberg, 69120 Heidelberg, Germany. E-mail: zaumseil@uni-heidelberg.de
bInstitute of Inorganic Chemistry, Universität Heidelberg, 69120 Heidelberg, Germany. E-mail: lutz.gade@uni-heidelberg.de
cHumboldt Centre for Nano- and Biophotonics, Department of Chemistry, University of Cologne, 50939 Köln, Germany
First published on 24th January 2024
Abstract
Molecules based on the octaazaperopyrenedioxide (OAPPDO) motif represent a new class of organic emitters with the ability to form amorphous films when processed from solution while retaining their emissive properties. Here, we report the synthesis of bay-functionalized OAPPDO derivatives with thioether-substituents, which are structurally characterized by their twisted π-core. The combination of this structural motif with suitable substituents at the chromophore facilitates the deposition of neat and smooth films from solution, in which the molecules remain electronically decoupled, thus drastically reducing non-radiative decay pathways. An analysis of absorption and emission spectra of solutions and spincoated thin films is provided along with details of their structural morphology and thermodynamics. The amorphous thin films display spectroscopic characteristics similar to those of the isolated molecules in solution and exhibit photoluminescence quantum yields up to 17%. The high oscillator strength of smooth films of these OAPPDO derivatives enables their integration in Fabry–Pérot microcavities with high quality factors and the observation of strong light-matter coupling with clear exciton–polariton modes and large coupling strength.
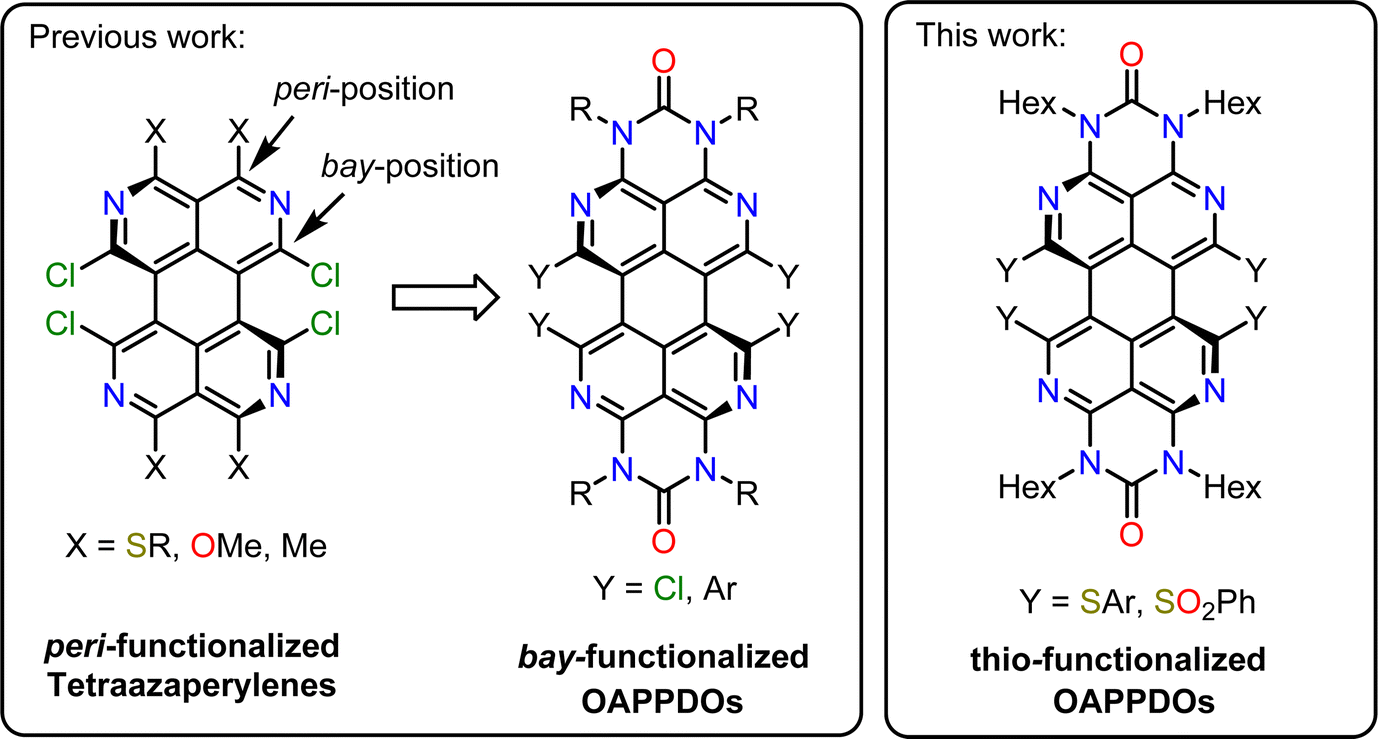
1. Introduction
Strong interactions between dye molecules in crystalline or amorphous thin films are beneficial for charge transport but pose a significant challenge for the development of highly emissive thin films for applications such as organic light-emitting diodes (OLEDs),1,2 luminescent solar concentrators,3,4 or laser gain media.5,6 Various nonradiative decay pathways reduce the photoluminescence quantum yield (PLQY) of densely packed and electronically coupled molecules, such as concentration quenching, singlet fission, or the formation of non-emissive aggregates.7,8 Methods to electronically decouple luminescent molecules, and thus maintain their emissive properties in the solid state, include dilution within a polymer or small molecule matrix,9,10 integration in nanoporous frameworks,11,12 or the protection of the chromophore core with bulky substituents.13–15 Unfortunately, these strategies often lead to a lower density of emitters per volume or poor film formation.One large area of application for molecular solid-state emitters is exciton–polaritonics, i.e., strong light-matter coupling in optical microcavities. Excitonic materials enclosed in a resonant Fabry–Pérot (or similar) cavity can reach the so-called strong-coupling regime when the exchange of energy between the cavity photons and excitons is faster than the decay of either component.16–18 New mixed light-matter states are formed (lower and upper polariton) that enable the precise control of light absorption and emission in these microcavities while keeping the emitters the same. Optoelectronic devices, such as OLEDs and photodiodes can be tuned in the strong coupling regime through adjusting the coupling strength (via the concentration of emitters) and detuning of the cavity (via cavity thickness).19–22 An example of such tuning is the angle-independent emission wavelength of ultrastrongly coupled OLEDs.23,24 Furthermore, due to the high exciton binding energy of their excitons, organic emitters can facilitate exciton–polariton condensation and polariton lasing under suitable conditions even at room temperature.25–30 To reach the strong or ultrastrong coupling regime, the density and oscillator strength of the molecules in the cavity must be high enough. Furthermore, the lifetime of the excitons must be sufficiently long and the quality factor of the produced cavity should be as high as possible. The latter is often limited by the roughness of the emitter film. To reach polariton lasing the photoluminescence quantum yield of the emitter also has to be high. Although there have been several examples of polariton lasing, the field is still limited by the availability of organic emitters with high oscillator strength, which retain their absorption and emission properties in solution-processed neat and smooth thin films.
To address this limitation, we apply an alternative strategy for the formation of smooth, amorphous and aggregate-free thin films with a high density of emissive small molecules. To suppress aggregation of π-conjugated molecules we make use of non-planar π-systems instead of bulky sidegroups. Based on initial studies with peri-substituted tetraazaperylenes,31 we have recently begun to investigate the suitability of octaazaperopyrenedioxides (OAPPDOs)32,33 (see Fig. 1) as emitters at high concentrations in solution as well as in condensed thin films and their application as fluorophores in two-photon microprinted 3D emissive structures.34
 | ||
| Fig. 1 Previously investigated fluorophores based on a tetraazaperylene core and their functionalization with sulfur-based groups at bay-positions (this work). | ||
Formally, the OAPPDO compounds may be viewed as tetraazaperylenes that are decorated with N-substituted urea units in both opposite peri-positions. In particular, we found that the facile modification of these fluorophores in the bay-positions enables efficient tuning of their luminescent properties. Moreover, the bay-substituents of the tetraazaperylene core induce a twist of the polycyclic aromatic core which may reduce the degree of π–π interaction. This twist therefore potentially favours retention of the photophysical properties of isolated chromophores in condensed amorphous or crystalline thin films.
Here, we investigate the impact of introducing sulfur-based groups in substituents in the bay positions of OAPPDOs (see Fig. 1) on their photophysical properties. This structural modification barely alters their absorption and photoluminescence properties, both in solution and in solution-processed thin films. Moreover, we found that these compounds have the tendency to form smooth and amorphous neat films with good PLQY. We further demonstrate the ability of thin films of OAPPDO derivatives to enter the strong coupling regime in suitable Fabry–Pérot microcavities with nearly angle-independent emission, hence expanding the library of potential organic molecules for polaritonic devices.
2. Results and discussion
2.1. Synthesis of OAPPDO thioethers and sulfones and their crystal structures
The previously described33bay-chlorinated OAPPDO 1 enabled the facile preparation of thioether-functionalized derivatives 2a–fvia nucleophilic substitution in high yields (see Scheme 1). The corresponding thiolates were prepared in situ or in the case of the perfluoro derivative 2e the copper thiolate was reacted with 1. Both are common approaches for the sulfur-functionalization of perylene dyes.35 Thioethers 2a, b and f were oxidized to the corresponding sulfones 3a–c with excess meta-chloroperbenzoic acid (m-CPBA) in good yields (for a detailed description of the synthesis, see ESI†). To investigate the packing of the thioether-functionalized OAPPDOs in the solid state, single crystals of 2a–c, 2e and 3a were grown by slow evaporation of concentrated solutions of each compound and crystallographic structure analysis was performed (see Fig. 2 and Tables S1–S3, ESI†). The tetraazaperylene core of all OAPPDO fluorophores was found to be highly twisted (twist angles for 2a: 28.99(4)° and 30.43(5)° for two independent molecules in the cell, 2b: 30.83(4)°, 2c: 29.92(5)°, 2e: 30.32(8)°, 3a: 31.16(4)°) due to the large van der Waals radii of the sulfur atoms of the thioether or sulfone functionalities and the resulting steric repulsion in the bay area. Consistent with the general structural characteristics of rylenes, the individual naphthyridine moieties within the tetraazaperylene core were found to be “fused” through long C–C bonds of around 1.46 Å. The principal structural data are in the typical range of previously reported bay-functionalized tetraazaperylene dyes.31–33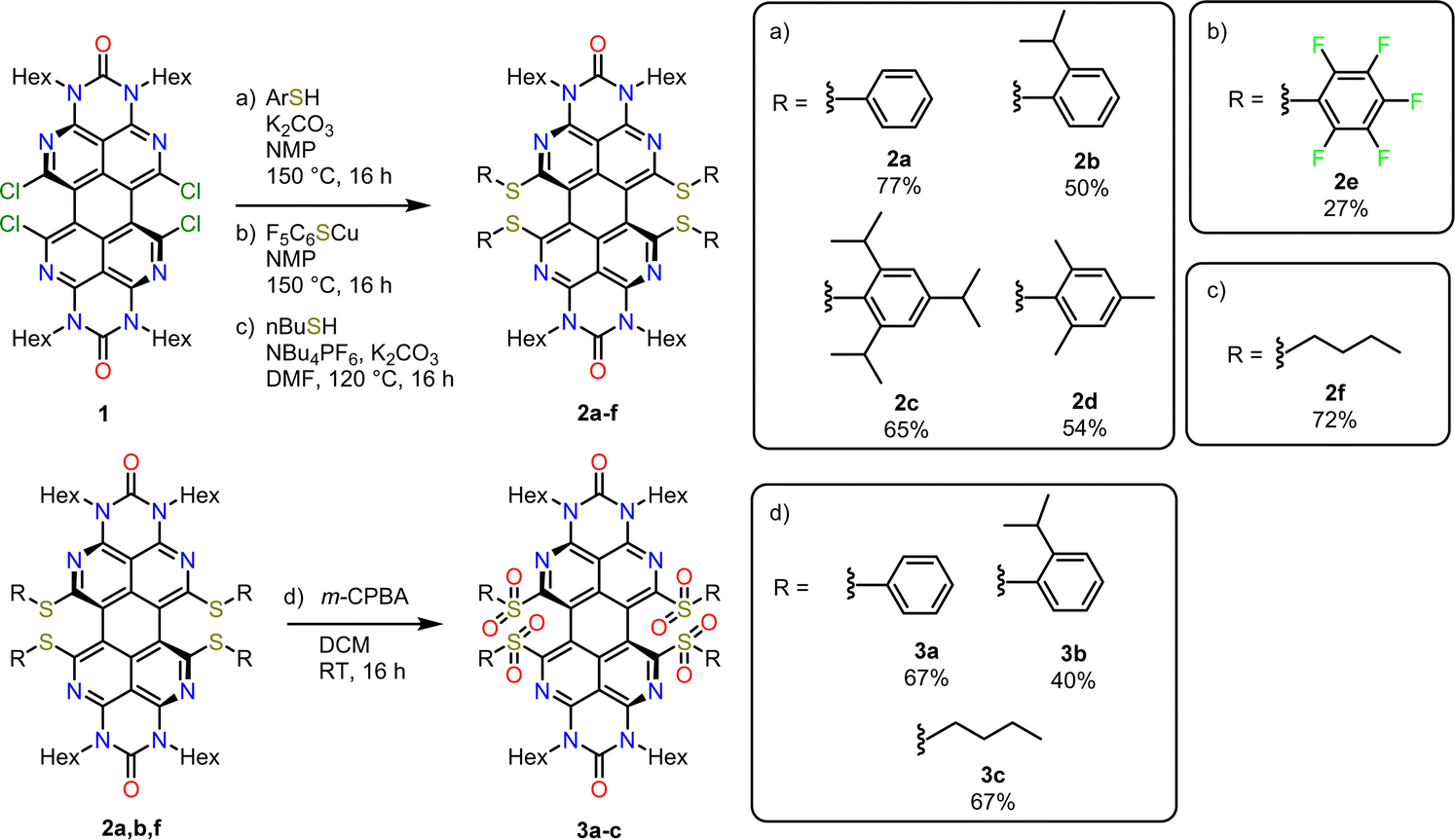 | ||
| Scheme 1 Synthesis of thioether- and sulfone-functionalized OAPPDOs. Reaction conditions: (a) ArSH, K2CO3, NMP, 150 °C, 16 h; (b) F5C6SCu, NMP, 150 C, 16 h; (c) n-BuSH, NBu4PF6, K2CO3, DMF, 120 °C, 16 h; (d) m-CPBA, DCM, room temperature, 16 h. | ||
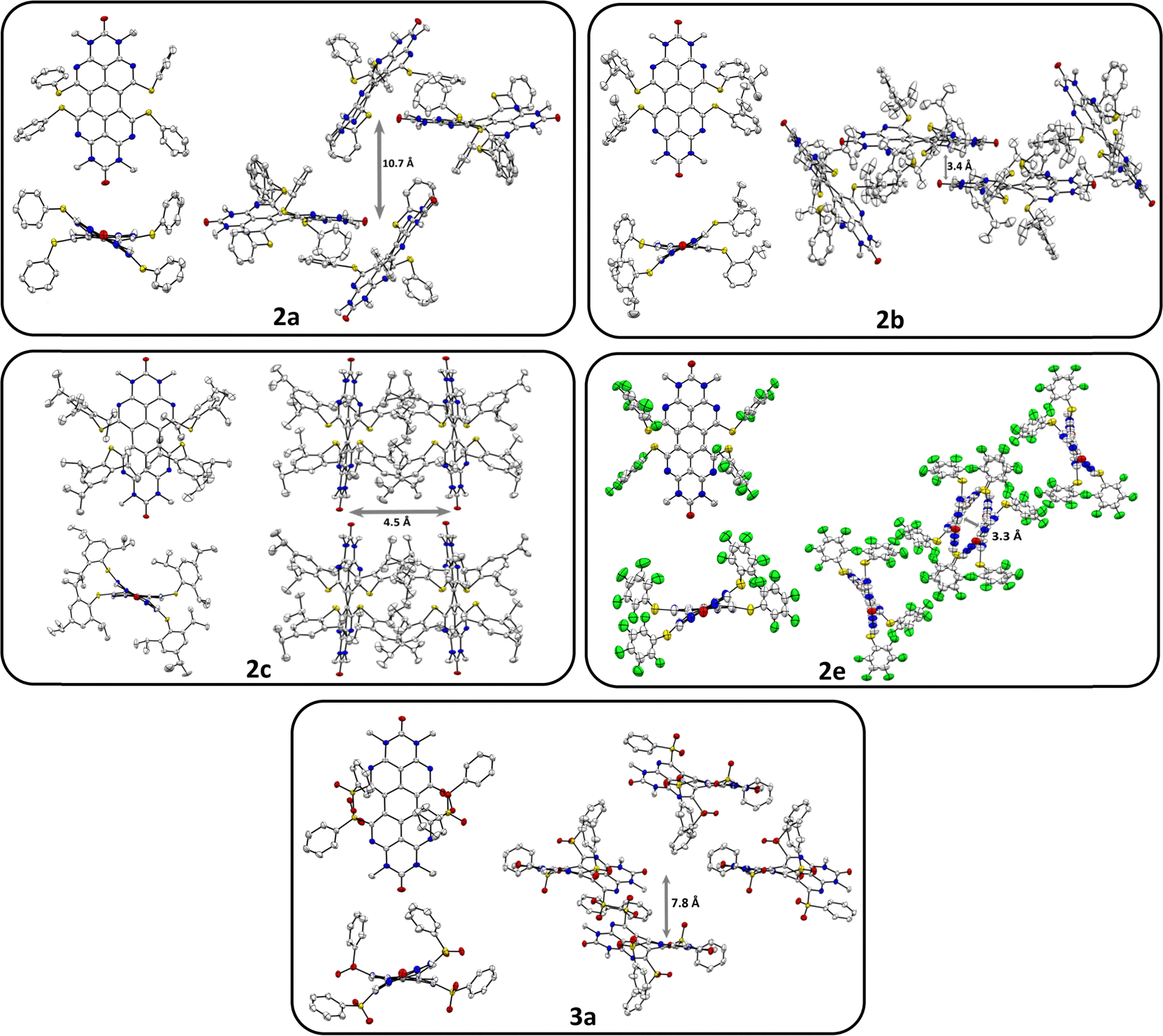 | ||
| Fig. 2 Solid state structures and molecular packing motifs of thioether-functionalized OAPPDOs 2a (view along [111]), 2b (view along a axis), 2c (view along c axis), 2e (view along a axis), and sulfone-functionalized OAPPDO 3a (view along a axis). N-hexyl substituents, hydrogen atoms and disorder are omitted for clarity. Thermal displacement ellipsoids at 50% probability level. | ||
In all compounds, the steric demand of the bay- and N-hexyl-substituents at the periphery of the fluorophore suppresses significant π–π-stacking of the perylene cores. Additionally, the variation of the bay-substituents leads to distinctly different patterns of molecular packing. In compounds 2a, 2b and 2e the molecules are stacked to give a herringbone (2a) and sandwich herringbone (2b and 2e) packing motif with distances of approximately 10.7 Å (2a), 3.4 Å (2b) and 3.3 Å (2e) between the polycyclic aromatic cores. The latter two suggest weak π–π-interactions at the peri region of the perylene units while the central pericyclic cores appear to be ‘decoupled’. In contrast to that, compounds 2c and 3a exhibit both an offset stacked packing pattern with inter-layer distances of approximately 4.5 Å and 7.8 Å, respectively. Such inhibition of π–π-interactions by twisting the π-core system has been studied for numerous perylenediimide (PDI) derivatives36–40 and can lead to dramatic changes in the photophysical properties of thin PDI films. For example, successive brominations in the bay position of a PDI derivative and concomitant opening of the twist angle of the perylenic core drastically increased the yield of triplet formation.41 Moreover, a recent study on singlet fission in amorphous PDI thin films has shown that the twisted polycyclic aromatic cores suppressed the formation of competing excimer states.42 The following sections will therefore first focus on the optical properties of these new OAPPDO dyes in solution and, subsequently, in neat thin films.
2.2. Absorption and emission properties in solution
All OAPPDO derivatives are highly soluble in polar organic solvents, while the thioether-functionalized derivatives also display good solubility in nonpolar solvents such as n-hexane. The absorption spectra of the compounds exhibit intense bands in the visible spectral region with large extinction coefficients (ε) and a characteristic vibronic progression (see Fig. 3 and Table 1 and Fig. S1, S2 (ESI†) for spectra of the other derivatives), indicating coupling between the electronic transitions and the vibrational modes of the tetraazaperylene cores.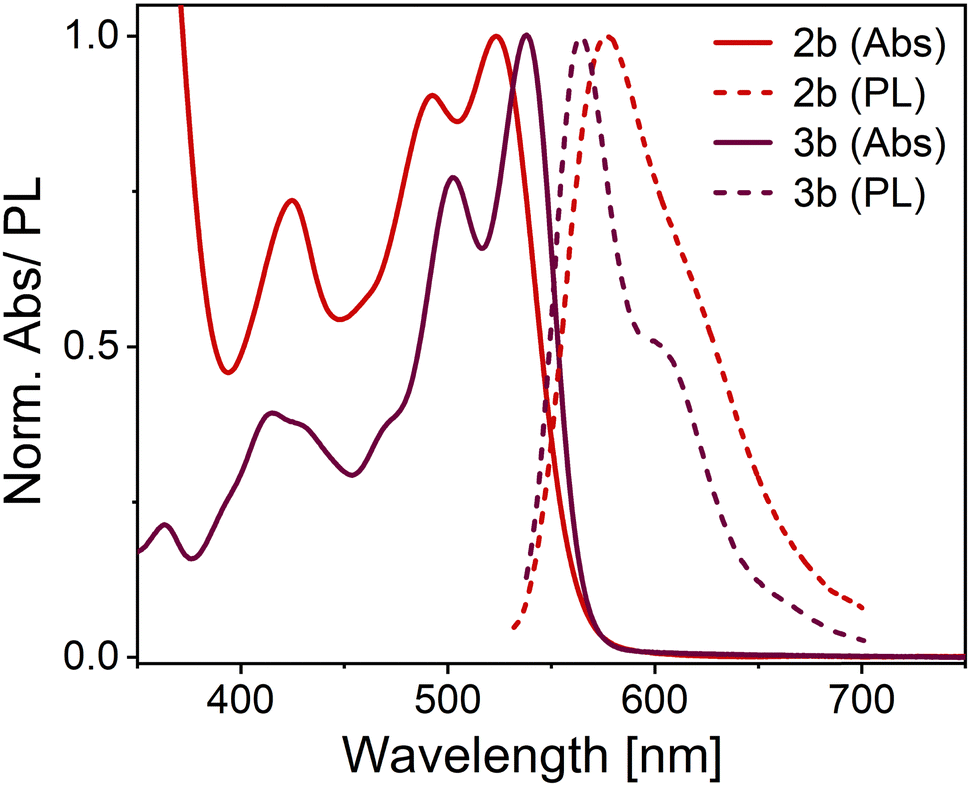 | ||
| Fig. 3 Normalized absorption (Abs) and emission (PL) spectra of compounds 2b and 3b in dichloromethane (concentration ca. 10−5 M). | ||
λ
abs,max [nm] (log10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) ε) |
λ em,max [nm] (PLQY)a (%) | Stokes shift [cm−1] | τ [ns] | k r [107 s−1] | k nr [107 s−1] | |
|---|---|---|---|---|---|---|
| a Excitation wavelength: λabs,max – 5 nm. PLQY measured with an Ulbricht sphere (absorbance < 0.1). | ||||||
| 2a | 525 (4.33) | 577 (68) | 1716 | 7.4 | 9.2 | 4.3 |
| 2b | 532 (4.37) | 576 (67) | 1408 | 6.9 | 9.7 | 4.8 |
| 2c | 533 (4.47) | 575 (73) | 1370 | 6.4 | 11.4 | 4.2 |
| 2d | 527 (4.52) | 569 (55) | 1401 | 5.9 | 9.3 | 7.6 |
| 2e | 521 (4.36) | 563 (77) | 1431 | 6.1 | 12.6 | 3.8 |
| 2f | 530 (4.31) | 573 (70) | 1416 | 8.0 | 8.8 | 3.7 |
| 3a | 536 (4.42) | 558 (82) | 736 | 6.1 | 13.4 | 2.9 |
| 3b | 537 (4.57) | 553 (84) | 891 | 6.6 | 12.7 | 2.4 |
| 3c | 528 (4.42) | 551 (81) | 791 | 5.7 | 14.2 | 3.3 |
The absorption maxima of the thioether-functionalized OAPPDOs 2a–f range from 521 to 533 nm with molar absorption coefficients of around 22400 M−1 cm−1. Although all derivatives are non-planar, the vibronic structure for the S0 → S1 (ground state to first excited state) transition remained similar for all OAPPDOs, indicating the pronounced rigidity of the tetraazaperylene core, consistent with previous reports for PDI derivatives.43 Time-dependent density-functional theory (TD-DFT) calculations (basis set: B3LYP-GD3(BJ)/Def2-TZVPP)44–49 of 2a, 2e, 2f, 3a and 3c confirmed that the maxima around 500 nm correspond to the first singlet transition (S0 → S1) (see Fig. S3–S7 and Tables S4–S8, ESI†). Additionally an intense band between 400 nm and 450 nm is observed and can be attributed to a S0 → S2 transition, which is enhanced due to the core twist of the OAPPDOs.37
All the OAPPDO derivatives are fluorescent and their photoluminescence (PL) spectra are approximatively the mirror image of their respective S0 → S1 transition (see Fig. 3 and Fig. S1, S2 for PL spectra of the other derivatives, ESI†). Notably, this type of mirror image emission pattern was found to be even more pronounced for the sulfone-derivatized compounds 3a–c, indicating a highly rigid tetraazaperylene core. This increased core rigidity for the sulfone-functionalized OAPPDOs is also reflected in the emission Stokes shifts (see Table 1), which are between 1400 cm−1 and 1700 cm−1 for bay-sulfido derivatives 2a-f but only 736–891 cm−1 for the sulfone-derivatives 3a–c. The observed photophysics reflect the key structural modification upon oxidation of the sulfide-functions in the bay-positions, namely the transformation of the twofold substituted sulfur atoms in the thioethers to the fourfold coordinated S-atoms in the sulfones. This significant increase in steric bulk in the immediate proximity of the bay-area rigidifies the chromophore with regard to the (activated) libration around the long molecular axis. Concomitantly, the increased oxidation state of the sulfur leads to a slight contraction and thus stabilization of the C–S bonds. The latter is a well-established pattern in organo-element chemistry.
PLQYs of up to 84% in solution were found for the compounds listed in Table 1. Notably, the oxidation of the bay-thioether units to the corresponding sulfones did not change the absorption behaviour significantly and only led to a small bathochromic shift of the maxima of less than 10 nm. However, the PLQY increased by about 10% upon oxidation. The fluorescence lifetimes (τ) of the OAPPDOs in dichloromethane (DCM) solution are listed in Table 1. All compounds showed a simple mono-exponential decay with lifetimes values between 5.7 ns and 8.0 ns, as typically expected for good fluorescent dyes. The corresponding rate constants for radiative (kr) and non-radiative (knr) decay were also estimated from the measured PLQY and fluorescence lifetime. However, they were all fairly similar, except for slightly higher kr and lower knr for the sulfones compared to the thioethers, which is in agreement with their higher PLQYs.
2.3. Thin films of OAPPDO derivatives
Neat thin films of compounds 2b–e and 3a were prepared by spin-coating from o-xylene or dichloromethane solution (for parameters see Table S9, ESI†). The compounds and solvents were mainly chosen to maximize the concentration of dissolved molecules while also ensuring smooth film formation during spincoating. Dichloromethane was required for the sulfone compounds due to their higher polarity. The thicknesses of the spin-coated neat films ranged from 120 nm to 200 nm. All films were annealed at 80 °C for two hours to remove residual solvents and to avoid thermal cracking during the atomic layer deposition of aluminium oxide (AlOx) for encapsulation. All five OAPPDO compounds studied in this context (2b–e, 3a) formed homogeneous and smooth thin films that displayed no visible signs of crystallization or birefringence in cross-polarized optical micrographs (see Fig. S8–S12, ESI†). All thin films were found to be amorphous at room temperature. Atomic force microscopy (AFM) images of the films of compounds 2d and 3a (see Fig. 4) also showed no signs of crystallization on the nanometer scale and were extremely smooth. The same was observed for the other OAPPDO derivatives (see Fig. S13, ESI†). The root mean square (RMS) roughness determined from AFM images was less than 0.5 nm (see Table 2), highlighting the amorphous nature and the excellent smoothness of these films, which is necessary for the fabrication of high quality optical cavities.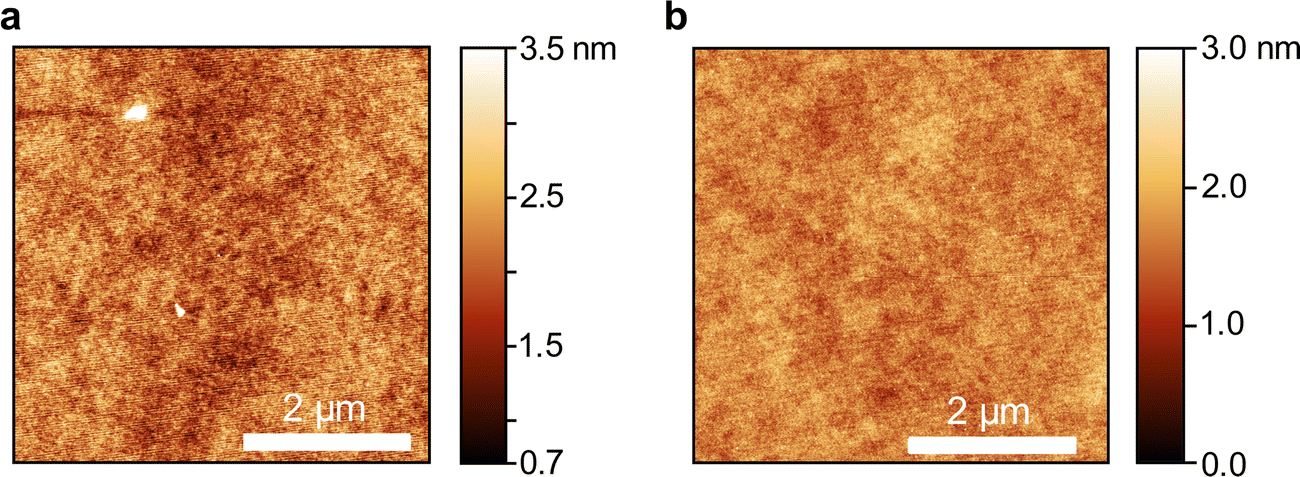 | ||
| Fig. 4 Atomic force microscopy images of a neat film of (a) compound 2d and (b) compound 3a. The scale bar is 2 μm. | ||
| 2b | 2c | 2d | 2e | 3a | |
|---|---|---|---|---|---|
| RMS [nm] | 0.40 | 0.40 | 0.40 | 0.40 | 0.30 |
| Stokes shift [cm−1] | 1230 | 985 | 930 | 1495 | 850 |
| PLQY (%) | 2.2 | 4.5 | 2.8 | 17 | 3.8 |
| τ 1 [ns] | 0.23 | 0.28 | 0.56 | 4.20 | 0.14 |
| τ 2 [ns] | 1.88 | 2.46 | 3.08 | 1.00 | 3.93 |
| τ 3 [ns] | — | 0.66 | 0.20 | — | 0.70 |
| τ avg [ns] | 0.36 | 0.37 | 0.37 | 2.04 | 0.23 |
| k r [107 s−1] | 6.1 | 12.2 | 7.6 | 8.3 | 16.5 |
| k nr [107 s−1] | 271.7 | 258.1 | 262.7 | 40.7 | 418.3 |
To further confirm the amorphous nature of the compounds in the solid state, their thermal behaviour was studied. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) of all compounds were performed under suitable heating/cooling conditions (see Fig. S14–S31, ESI†). Of all derivatives, only compound 2d displayed exothermic crystallization, setting in at 175 °C (max. at 189 °C) with an enthalpy of 16.1 J g−1, followed by an endothermic melting process beginning at 211 °C (see Fig. 5). Interestingly, compounds 2f, 3b and 3c feature one or two glass transitions at 55 °C, 54 °C (and 119 °C) and 45 °C, respectively. Compounds 2a and 2b only showed an endothermic melting process starting at 147 °C and 138 °C, respectively, while compounds 2c–d did not show any observable thermodynamic processes between 0 °C and 275 °C. In summary, except for compound 2d, the investigated OAPPDO derivatives generated no crystalline phases under the conditions of film processing and annealing.
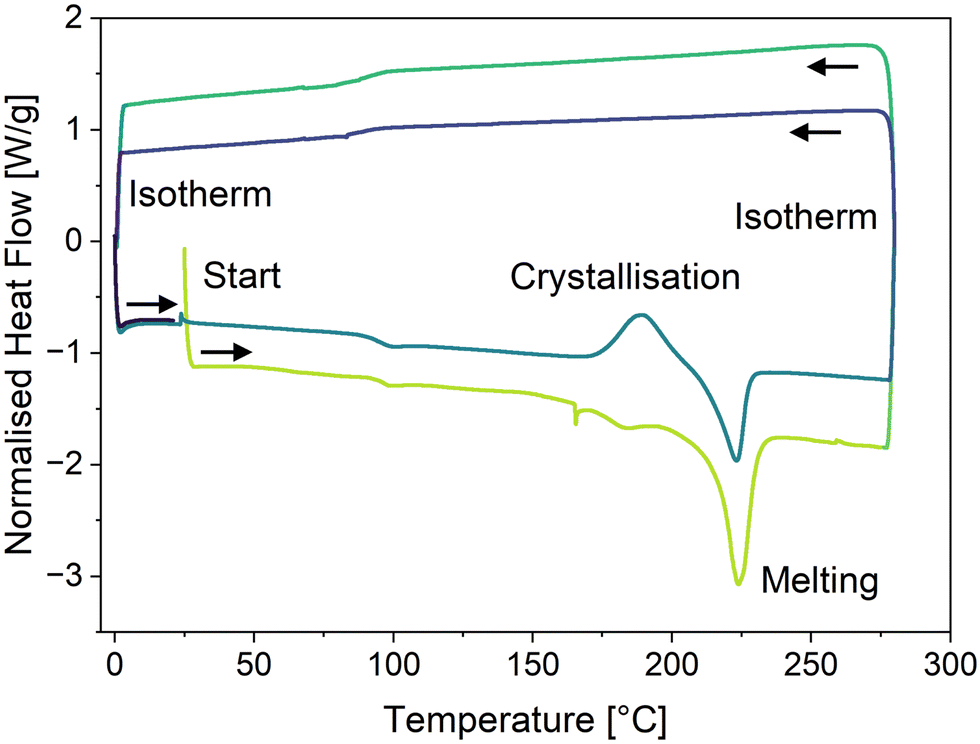 | ||
| Fig. 5 Differential scanning calorimetry thermogram of compound 2d showing an exothermic crystallisation process at ca. 180 °C and an endothermic melting process at ca. 220 °C (positive heat flow: exotherm, negative heat flow: endotherm). The first cycle is indicated in green and the second cycle in blue. | ||
2.4. Absorption and PL of OAPPDO thin films
The absorption and photoluminescence spectra of neat thin films of the five OAPPDO derivatives, 2a–2e and 3a were recorded. As shown in Fig. 6, all of these films display a well-defined vibronic progression of the S0 → S1 transition between 500 nm and 550 nm. The absorption spectra are very similar in terms of shape and spectral position to the absorption spectra in solution (see Fig. S32, ESI†). The principal absorption band in the visible exhibits only a minor shift compared to the spectrum in solution (from 0 nm to 10 nm), which can be attributed solely to the change in the dielectric environment. There is no sign of aggregate formation or excitonic coupling, excluding any formation of J- or H-aggregates.50 The absence of such spectral changes further corroborates the extremely weak π–π and Coulombic interactions between OAPPDO molecules in the solid state as already indicated in the molecular packing (see Fig. 2).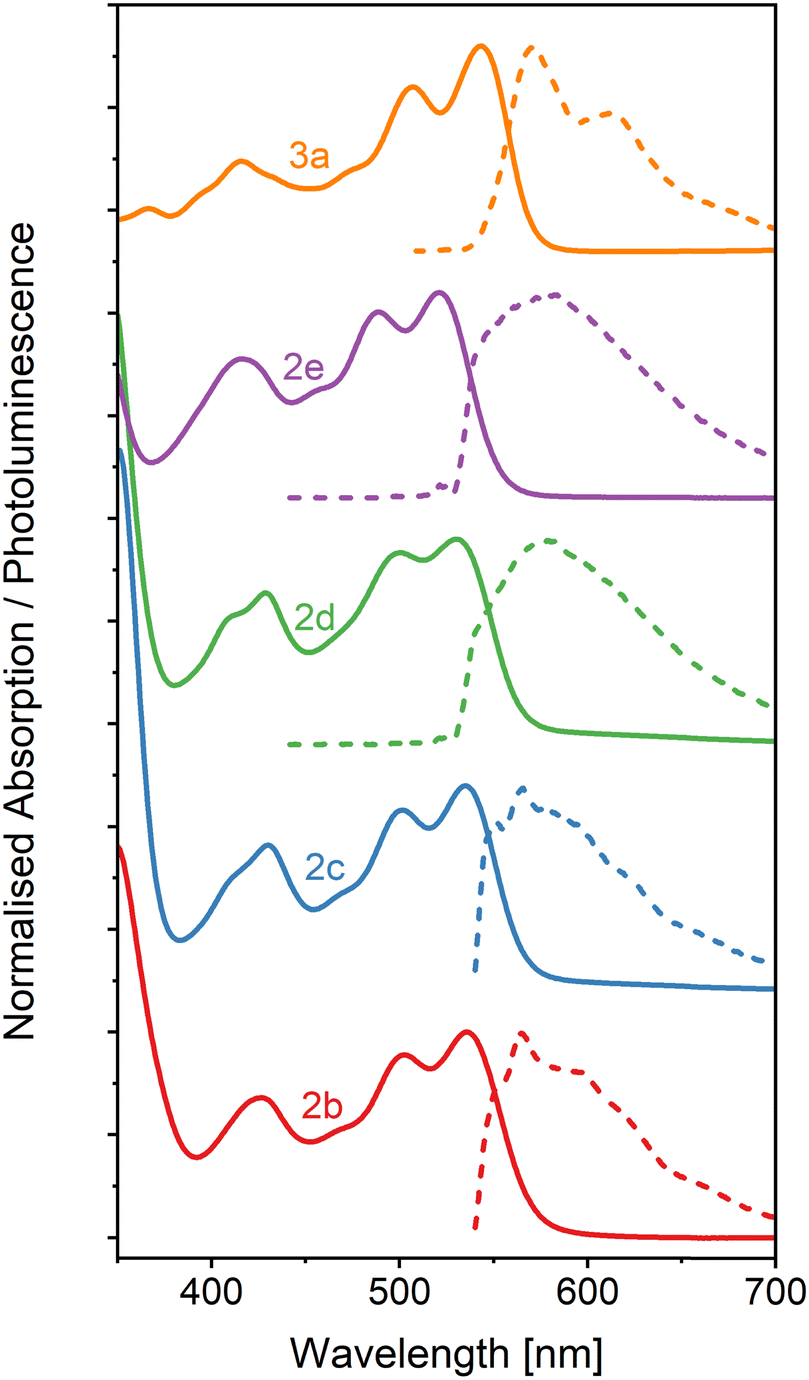 | ||
| Fig. 6 Absorption (solid lines) and PL (dashed lines) spectra of neat OAPPDO thin films. All films were excited at 532 nm. | ||
The PL spectra of thin films of compounds 2b–e display an emission band around 550–600 nm with Stokes shifts of about 1230 cm−1, 985 cm−1, 930 cm−1 and 1495 cm−1, respectively (see Table 2) which, except for compound 2e, is even smaller than the values observed in solution. Interestingly, the solid-state PL of compound 3a retains its well-resolved vibronic structure (see Fig. 7), which is a rare feature for small organic molecules. It was previously only observed for highly twisted41 or shielded43 PDIs. It further corroborates the lack of electronic coupling between the molecules. The well-resolved absorption and emission peaks in the neat film make compound 3a a prime candidate for strong light-matter coupling.
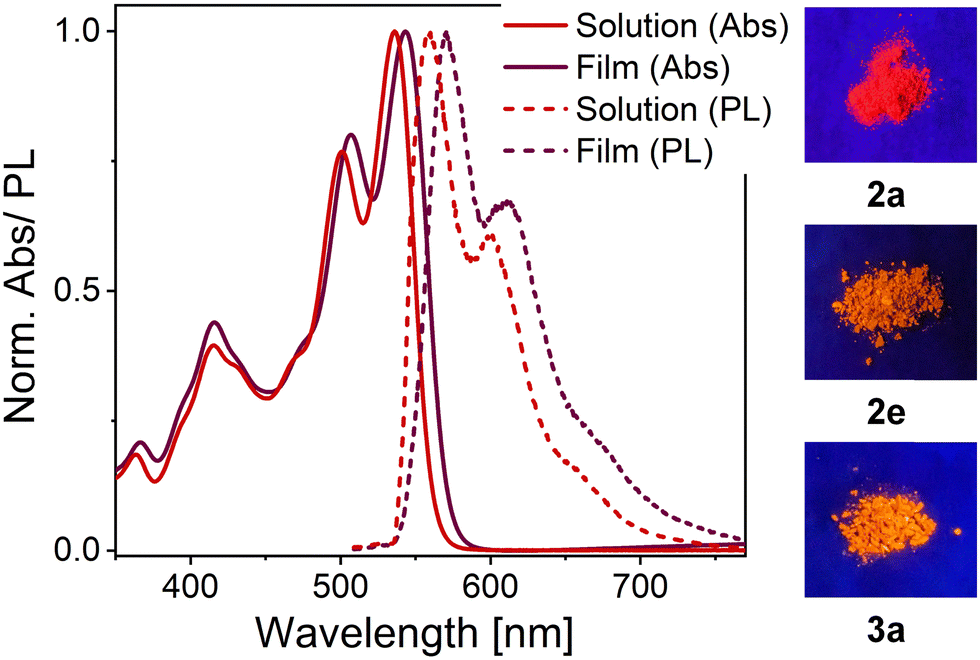 | ||
| Fig. 7 Absorption (Abs, solid lines) and emission (PL, dashed lines) of compound 3a in dichloromethane (red) and in neat thin film (dark red). The clear vibronic progression is retained for both absorption and emission spectra of the neat film (left). Photographs of OAPPDOs 2a, 2e and 3a as powders under UV-illumination (λexc = 365 nm, right). | ||
The PLQYs (see Table 2) measured for thin films of compounds 2b–d and 3a were significantly reduced (to ca. 3%) compared to their PLQY in solution (for excitation wavelengths see Tables S10 and S11, ESI†). The films also showed multiexponential PL decays that could be characterized by three lifetimes (τ1–τ3) or one amplitude-averaged lifetime (τavg) of a few hundred picoseconds (see Table 2 and Fig. S33–S35 (ESI†), for excitation wavelengths and film formation parameters see Table S12, ESI†). This fast and multiexponential decay indicates the presence of efficient non-radiative decay channels (quenching) despite the negligible electronic coupling of the molecules. Interestingly, the PLQY of compound 2e remained fairly high (17%) even for a neat film. This value is comparable to PLQYs reported for neat PDI films.9,15,51 The corresponding rate constants for radiative and non-radiative decay were estimated from the measured PLQYs and amplitude-averaged lifetimes (see Table 2). Despite the increased uncertainty of the measured values for thin films compared to solutions, the rate constants for radiative decay remained within the same range as for the solutions. However, the non-radiative decay increased by almost two orders of magnitude for all films except for compound 2e, for which it only increased by a factor of ten. The amplitude-averaged fluorescence lifetime for thin films of compound 2e stayed in the nanosecond range and thus similar to that in solution. All of these properties indicate that the chromophores of compound 2e interact even less than the other OAPPDO compounds. This might be a result of partial shielding of the polycyclic aromatic cores by perfluorophenyl groups on the thioether units in the bay-positions. Fluorinated groups are also known to efficiently limit water and oxygen diffusion in molecular thin films, which may also reduce the related quenching.
Furthermore, the photostability of the thin films was investigated under UV light exposure (see Experimental section) in air. Films made of compounds 2b–e were photobleached within seconds, probably due to the weak photostability of the thioether C–S bonds. However, compound 3a, with the oxidized sulfone units, showed much higher photostability, with only a 10% reduction of the PL intensity after UV exposure (see Table S13, ESI†). It is noteworthy, that encapsulation of the OAPPDO thin films with 10 nm of aluminium oxide resulted in much higher photostability (i.e., less the 5% PL reduction after the same exposure), suggesting that the photobleaching is largely caused by reactive oxygen species. High PLQY but also high photostability of the neat films are basic requirements for any possible observation of polariton lasing in strongly coupled microcavities.
2.5. Strong light-matter coupling in Fabry–Pérot cavities
Given the high density of emitters and the excellent smoothness of neat OAPPDO films, these new fluorophores should be suitable for achieving strong light-matter coupling in cavities. To this end, the films were integrated in Fabry–Pérot cavities (see Fig. 8a). This was accomplished by spincoating neat OAPPDO layers (100–200 nm thickness) on a highly reflective silver mirror (100 nm Ag) and thermal evaporation of 30 nm of silver as a semi-transparent top mirror.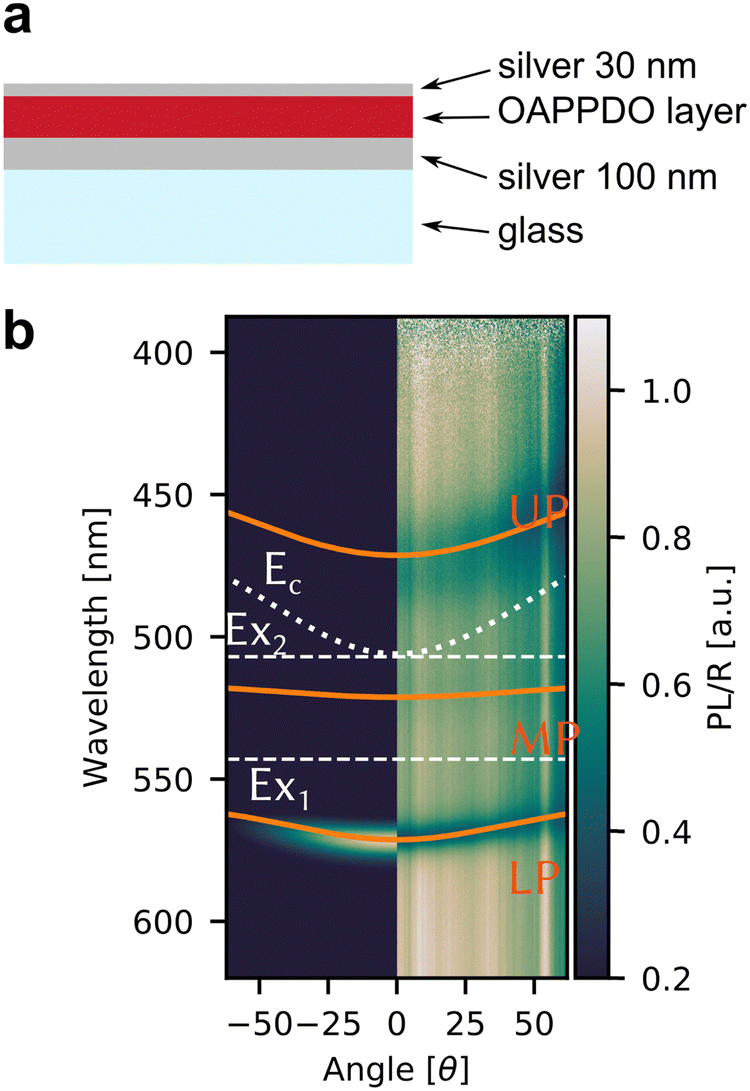 | ||
| Fig. 8 (a) Schematic cross section of a semi-transparent Fabry–Pérot cavity with a neat OAPPDO layer between two silver mirrors. (b) Angle-resolved PL (left) and reflectivity (right) spectra of a Fabry–Pérot cavity containing compound 3a. The solid orange lines are the fitted upper (UP), middle (MP) and lower (LP) polaritonic branches, the grey dotted line is the bare cavity mode (Ec). The horizontal grey dashed lines indicate the excitonic transitions (EX1, EX2) of compound 3a. | ||
Fig. 8b displays the angle resolved reflectivity and photoluminescence spectra of the highly photostable compound 3a embedded in a Fabry–Pérot cavity. They clearly show that the bare excitonic transitions (EX1, EX2 of the vibronic progression of the first singlet transition) and the cavity mode (Ec) are replaced by an upper (UP), middle (MP) and lower (LP) polariton branch resulting from hybridization as shown previously for molecules with prominent vibronic progression.17 The avoided crossing of the polariton modes and the excitonic transitions is a strong indication that the cavity is indeed in the strong coupling regime. PL only occurs from the LP mode and hence fitting of the three polariton modes was performed based on the reflectance data. The same observations were made for cavities with compounds 2b–e (see Fig. S36, ESI†). The linewidth of the polariton modes enabled the extraction of the quality factors of the cavities, which range between 20 and 64. These values are very good for simple silver mirrors and are a direct result of the excellent smoothness of the OAPPDO thin films.
From the angle-resolved reflectivity spectrum of the cavity (right panel of Fig. 8b), the Rabi splitting ħΩR can be extracted by fitting the UP, MP and LP modes with a coupled harmonic oscillator model, taking into account the two excitonic transitions and the cavity mode (see Experimental section). The Rabi splitting for a cavity with compound 3a reached 304 meV for the first excitonic transition and 290 meV for the second excitonic transition. The data for cavities with all other OAPPDO derivatives are summarised in Table 3. The extracted values for ħΩR are significantly larger than the linewidths of both the bare excitonic transitions and the bare cavity mode, hence confirming again that the cavities are in the strong coupling regime.
| Compound | 2b | 2c | 2d | 2e | 3a |
|---|---|---|---|---|---|
| ħΩ 1 [meV] | 270 | 240 | 242 | 280 | 304 |
| ħΩ 2 [meV] | 190 | 220 | 238 | 200 | 290 |
| E c [eV] | 2.45 | 2.33 | 2.30 | 2.40 | 2.45 |
| Q | 48 | 37 | 48 | 21 | 64 |
A comparison of the Rabi splittings (304 meV and 290 meV) for compound 3a with its excitonic transition energies (2.17 eV and 2.03 eV) indicates that the cavities are approaching the so-called ultrastrong coupling regime, i.e., the coupling strength becomes comparable (>10–20%) to the molecular transition energy.52 It is difficult to reach the ultrastrong coupling regime in a cavity with diluted emitters in a matrix, hence, it is usually overserved for neat films of emitters with high oscillator strength.53,54 Such strong coupling also leads to a very flat dispersion of the lower polariton modes and hence nearly angle-independent emission wavelengths, which is very useful for light-emitting devices.24 These observations emphasize again the excellent film quality and high molecular density of neat OAPPDO films as well as their strong light absorbing and emitting properties.
3. Conclusion
In summary, we have presented a new class of amorphous solid-state emitters, which are derivatives of octaazaperopyrenedioxide (OAPPDO), an N-heteropolycyclic dye with a non-planar azaperylene-derived π-system at its core. The introduction of sulfur-based groups at the bay-positions of the tetraazaperylene core leads to a torsional twist in the π-core of about 30° that results in highly soluble compounds that exhibit high molar extinction coefficients and photoluminescence quantum yields in solution. Their tailored molecular properties allow the OAPPDOs to be solution-processed into high density neat films with monomer-like absorption and emission properties and photoluminescence quantum yields up to 17%. Those thin films are amorphous at room temperature, highly homogeneous and exhibit exceptional surface smoothness. Moreover, OAPPDO films are photostable against UV irradiation when encapsulated while the sulfone derivatives were found to be photostable even without any encapsulation. Finally, we demonstrated strong light-matter coupling of thin films of OAPPDOs in simple metal-clad microcavities. We believe that this new class of amorphous solid-state emitters provides promising candidates for optoelectronic and photonic applications where amorphous and highly emissive layers are required.4. Experimental section
4.1. Synthesis
The detailed synthetic protocol for all compounds is described in the ESI.†4.2. Thermal characterization of materials
TGA (thermal gravimetric analysis) measurements were performed with a Mettler Toledo TGA 2 device. DSC (differential scanning calorimetry) was performed using a TA Instruments Discovery DSC 250 calorimeter (2b, 2c, 2d, 2e) or a Mettler Toledo DSC821 (2a, 2f, 3a, 3b, 3c).4.3. Thin film and preparation
Cleaned, alkali-free glass substrates (AF32eco, Schott, 20 × 25 × 0.5 mm3) and EagleXG glass substrates were used for all thin film measurements. Compounds 2b–e were dissolved in o-xylene and compound 3a in DCM, respectively. Neat films were prepared by spincoating directly from solution on cleaned glass substrates (for parameters see Table S9, ESI†). Atomic force microscopy images were recorded with a Bruker Dimension Icon atomic force microscope (AFM) in ScanAsyst® mode. The root mean square surface roughness was extracted using Gwyddion 2.59 software. For encapsulation, a 10 nm AlOx layer was deposited on top of the neat OAPPDO layer by atomic layer deposition (ALD) (Ultratech, Savannah S100, precursor trimethyl-aluminium, Strem Chemicals, Inc.) at low temperature (80 °C) to avoid thermal degradation of the molecules. For Fabry–Pérot cavities, a reflective bottom silver mirror (100 nm) was deposited by thermal evaporation onto a cleaned glass substrate. The OAPPDO layer was spincoated on top and a 30 nm silver mirror completed the cavity.4.4. Optical characterization
Absorption spectra were acquired with an Agilent Cary 6000i or Cary 5000i UV-Vis-NIR absorption spectrometer. Photoluminescence spectra in solution were recorded with a Varian Cary Eclipse fluorescence spectrophotometer and photoluminescence quantum yields (PLQY) were measured on a JASCO spectrofluorometer FP-8500 equipped with an ILF-835j100 mm integrating sphere. PL spectra of thin films were obtained by excitation with a 532 nm laser diode (OBIS, cw) and collection of the emitted light with a 60× objective (Olympus, NA = 0.9). Scattered laser light was blocked with a 550 long pass filter. The emission was imaged onto the entrance slit of a Princeton Instruments IsoPlane SCT 320 spectrometer and spectra were acquired with a PIXIS:400 BR camera (Princeton Instruments).For PLQY values of thin films, the emitter materials were spin-coated onto EagleXG glass substrates as described above. They were subsequently characterized in a PLQY measurement system (Hamamatsu Photonics) equipped with an integrating sphere, xenon lamp (L9799-01), monochromator (A10080-01), and multi-channel analyser (C10027). The samples were excited at different wavelengths (see Table S10, ESI†). All data were analyzed using the Hamamatsu U6039-06 software. All PLQY measurements were performed in dry nitrogen.
All optical images of thin films were acquired using an Olympus BX51 microscope. The optical constants and thickness of the thin films (on EagleXG glass) were determined from variable angle spectroscopic ellipsometry (M2000, J.A. Woollam), transmission spectra, and subsequent modeling (CompleteEase software, J.A. Woollam) using a general oscillator model (see Fig. S37, ESI†).
4.5. Angle-resolved measurements
Angle-resolved reflectivity spectra of the Fabry–Pérot cavities were measured in a home-built Fourier microscopy setup55,56 using a halogen light source (Ocean Optics, HL-2000-HP) focused onto the sample via a 60× objective (Olympus, MPLAPON60X) with a numerical aperture of 0.9. For photoluminescence measurements, a laser diode (OBIS, CW 532 nm) was used as an excitation source, and the scattered laser light was blocked by a long-pass filter (550 nm cut-off). The reflected/emitted light was imaged from the back focal plane of the objective onto the entrance slit of the spectrometer (Princeton Instruments IsoPlane SCT 320). A linear polarizer was placed in front of the spectrometer to select TE polarization. Angle-resolved spectra were recorded with a Si CCD camera (Princeton Instruments, PIXIS:400).4.6. Fluorescence lifetime measurements
For fluorescence lifetime measurements by time-correlated single-photon counting (TCSPC), dilute solutions and thin films of compounds 2b and 2e were excited with the spectrally filtered output of a picosecond-pulsed super-continuum laser source. A dichroic long-pass filter blocked scattered laser light (550 nm cut-off). The collected emission was focused onto a silicon avalanche photodiode (Micro Photon Devices) and a time-correlated single-photon counting module (Picoharp 300, PicoQuant) recorded the PL decay traces, which were fitted as a mono-exponential or multi-exponential decay. Time-resolved PL decays of dilute solutions and thin films of compounds 2c–d and 3a were measured with a time-correlated single photon counting setup (FluoTime 250, TimeHarp 260, PMA Hybrid detector, PicoQuant) at room temperature at an excitation wavelength of 373 nm. The employed excitation wavelengths and solvents are given in Tables S11 and S12 (ESI†).4.7. Photostability measurements
The photostability of thin films of all compounds was estimated using the following procedure. A fluorescence image was recorded with an Olympus BX51 fluorescence microscope under UV excitation using a 10× objective. Samples were then exposed to the same UV light using a 20× objective for 5 minutes. Finally, a fluorescence image was recorded using the same parameters as the first image. The resulting photostability is reported as the decrease of fluorescence intensity before and after UV exposure or as ‘bleached’ when no more fluorescence could be recorded from the exposed area.4.8. Coupled harmonic oscillator (CHO) fitting procedure
The reflectivity dispersion plots were fitted using a 3 × 3 coupled harmonic oscillator (CHO) model to extract couplings strengths. Solving the following Hamiltonian leads to the polariton energy as eigenvalues:where E1 and E2 are the exciton energies, V1 and V2 are the coupling strengths for the corresponding transitions. The cavity energy Ec as a function of angle θ is given by:
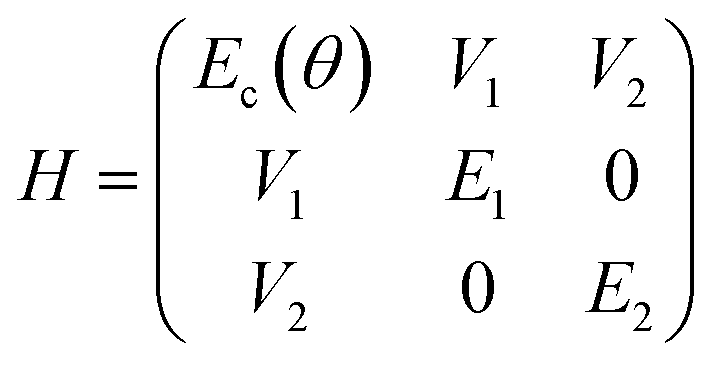
with neff the effective refractive index.
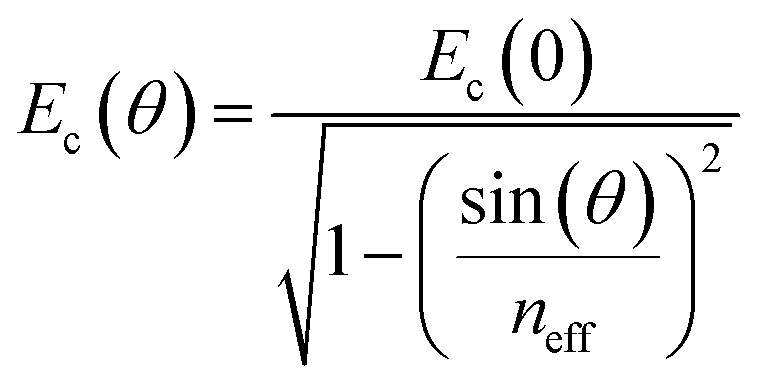
The quality factors (Q) of the cavities were determined using:
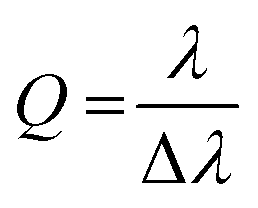
Author contributions
M. H. performed optical experiments if not otherwise noted and analyzed the data. R. E. and P. J. carried out all syntheses and chemical characterizations. T. W. proofread the manuscript. J. B. collected and analyzed the crystallographic data. S. S. and S. L. F. performed and analyzed the TCSPC measurements of compound 2a-b, 2f and 3b-c. A. M., F. L. R. and F. T. C. carried out the ellipsometry measurements, PLQY and lifetime measurements of thin films of compounds 2c-d and 3a under the supervision of M. C. G.; J. Z. and L. H. G. conceived and supervised the project. J. Z., L. H. G., R. E., and M. H. co-wrote the manuscript with input from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Deutsche Forschungsgemeinschaft (DFG) via SFB 1249 (projects A02 and C09). The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through no. INST40/467-1FUGG (Justus cluster). M. C. G. and F. T. C. acknowledge support from the Alexander von Humboldt Stiftung through the Humboldt-Professorship. A. M. acknowledges funding from the European Union's Horizon 2020 research and innovation program under Marie Skłodowska-Curie grant agreement no. 101023743 (PolDev). F. L. R. acknowledges funding from the Alexander von Humboldt Foundation through a Humboldt Fellowship.References
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234–238 CrossRef CAS PubMed.
- J. Roncali, Adv. Energy Mater., 2020, 10, 2001907 CrossRef CAS.
- B. Zhang, P. Zhao, L. J. Wilson, J. Subbiah, H. Yang, P. Mulvaney, D. J. Jones, K. P. Ghiggino and W. W. H. Wong, ACS Energy Lett., 2019, 4, 1839–1844 CrossRef CAS.
- A. J. C. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS PubMed.
- Y. Jiang, Y.-Y. Liu, X. Liu, H. Lin, K. Gao, W.-Y. Lai and W. Huang, Chem. Soc. Rev., 2020, 49, 5885–5944 RSC.
- A. Huber, J. Dubbert, T. D. Scherz and J. Voskuhl, Chem. – Eur. J., 2023, 29, e202202481 CrossRef CAS PubMed.
- J. Gierschner, J. Shi, B. Milián-Medina, D. Roca-Sanjuán, S. Varghese and S. Park, Adv. Opt. Mater., 2021, 9, 2002251 CrossRef CAS.
- B. Zhang, H. Soleimaninejad, D. J. Jones, J. M. White, K. P. Ghiggino, T. A. Smith and W. W. H. Wong, Chem. Mater., 2017, 29, 8395–8403 CrossRef CAS.
- M. G. Ramírez, S. Pla, P. G. Boj, J. M. Villalvilla, J. A. Quintana, M. A. Díaz-García, F. Fernández-Lázaro and Á. Sastre-Santos, Adv. Opt. Mater., 2013, 1, 933–938 CrossRef.
- Y. Wen, T. Sheng, X. Zhu, C. Zhuo, S. Su, H. Li, S. Hu, Q.-L. Zhu and X. Wu, Adv. Mater., 2017, 29, 1700778 CrossRef PubMed.
- J. Yu, Y. Cui, H. Xu, Y. Yang, Z. Wang, B. Chen and G. Qian, Nat. Commun., 2013, 4, 2719 CrossRef PubMed.
- M. J. Hollamby and T. Nakanishi, J. Mater. Chem. C, 2013, 1, 6178–6183 RSC.
- D. Schmidt, M. Stolte, J. Süβ, A. Liess, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2019, 58, 13385–13389 CrossRef CAS PubMed.
- M. Stolte, T. Schembri, J. Süβ, D. Schmidt, A.-M. Krause, M. O. Vysotsky and F. Würthner, Chem. Mater., 2020, 32, 6222–6236 CrossRef CAS.
- D. G. Lidzey, D. D. C. Bradley, M. S. Skolnick, T. Virgili, S. Walker and D. M. Whittaker, Nature, 1998, 395, 53–55 CrossRef CAS.
- R. J. Holmes and S. R. Forrest, Phys. Rev. Lett., 2004, 93, 186404 CrossRef CAS PubMed.
- A. Graf, L. Tropf, J. Zaumseil and M. C. Gather, in Handbook of Organic Materials for Electronic and Photonic Devices, ed. O. Ostroverkhova, Woodhead Publishing, 2nd edn, 2019, ch. 9, pp. 281–307 Search PubMed.
- J. R. Tischler, M. S. Bradley, V. Bulović, J. H. Song and A. Nurmikko, Phys. Rev. Lett., 2005, 95, 036401 CrossRef PubMed.
- B. Siegmund, A. Mischok, J. Benduhn, O. Zeika, S. Ullbrich, F. Nehm, M. Böhm, D. Spoltore, H. Fröb, C. Körner, K. Leo and K. Vandewal, Nat. Commun., 2017, 8, 15421 CrossRef CAS PubMed.
- A. Mischok, J. Lüttgens, F. Berger, S. Hillebrandt, F. Tenopala-Carmona, S. Kwon, C. Murawski, B. Siegmund, J. Zaumseil and M. C. Gather, J. Chem. Phys., 2020, 153, 201104 CrossRef CAS PubMed.
- E. Eizner, J. Brodeur, F. Barachati, A. Sridharan and S. Kéna-Cohen, ACS Photonics, 2018, 5, 2921–2927 CrossRef CAS.
- S. Gambino, M. Mazzeo, A. Genco, O. Di Stefano, S. Savasta, S. Patanè, D. Ballarini, F. Mangione, G. Lerario, D. Sanvitto and G. Gigli, ACS Photonics, 2014, 1, 1042–1048 CrossRef CAS.
- A. Mischok, S. Hillebrandt, S. Kwon and M. C. Gather, Nat. Photonics, 2023, 17, 393–400 CrossRef CAS.
- S. Kena-Cohen and S. R. Forrest, Nat. Photonics, 2010, 4, 371–375 CrossRef CAS.
- J. D. Plumhof, T. Stöferle, L. Mai, U. Scherf and R. F. Mahrt, Nat. Mater., 2014, 13, 247–252 CrossRef CAS PubMed.
- T. Cookson, K. Georgiou, A. Zasedatelev, R. T. Grant, T. Virgili, M. Cavazzini, F. Galeotti, C. Clark, N. G. Berloff, D. G. Lidzey and P. G. Lagoudakis, Adv. Opt. Mater., 2017, 5, 1700203 CrossRef.
- C. P. Dietrich, A. Steude, L. Tropf, M. Schubert, N. M. Kronenberg, K. Ostermann, S. Höfling and M. C. Gather, Sci. Adv., 2016, 2, e1600666 CrossRef PubMed.
- S. K. Rajendran, M. Wei, H. Ohadi, A. Ruseckas, G. A. Turnbull and I. D. W. Samuel, Adv. Opt. Mater., 2019, 7, 1801791 CrossRef.
- T. Ishii, K. Miyata, M. Mamada, F. Bencheikh, F. Mathevet, K. Onda, S. Kéna-Cohen and C. Adachi, Adv. Opt. Mater., 2022, 10, 2102034 CrossRef CAS.
- T. Wesp, P. Valsalan, A. Kochan, M. Hertzog, H. Wadepohl, J. Zaumseil and L. H. Gade, Chem. – Eur. J., 2022, 28, e202202661 CrossRef CAS PubMed.
- T. Wesp, T. Bruckhoff, J. Petry, H. Wadepohl and L. H. Gade, Chem. – Eur. J., 2022, 28, e202200129 CrossRef CAS PubMed.
- T. Wesp, T. Bruckhoff, H. Wadepohl and L. H. Gade, Chem. – Eur. J., 2022, 28, e202201706 CrossRef CAS PubMed.
- R. Eichelmann, J. Monti, L.-Y. Hsu, F. Kröger, J. Ballmann, E. Blasco and L. H. Gade, Mol. Syst. Des. Eng., 2023, 8, 1470–1476 RSC.
- Y. Zhou, B. Xue, C. Wu, S. Chen, H. Liu, T. Jiu, Z. Li and Y. Zhao, Chem. Commun., 2019, 55, 13570–13573 RSC.
- Z. Chen, M. G. Debije, T. Debaerdemaeker, P. Osswald and F. Würthner, ChemPhysChem, 2004, 5, 137–140 CrossRef CAS PubMed.
- Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem. – Eur. J., 2007, 13, 450–465 CrossRef CAS PubMed.
- T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541–5544 CrossRef CAS PubMed.
- T. E. Kaiser, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6719–6732 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- K. Nagarajan, A. R. Mallia, V. S. Reddy and M. Hariharan, J. Phys. Chem. C, 2016, 120, 8443–8450 CrossRef CAS.
- K. M. Felter, R. K. Dubey and F. C. Grozema, J. Chem. Phys., 2019, 151, 094301 CrossRef PubMed.
- Á. J. Jiménez, M.-J. Lin, C. Burschka, J. Becker, V. Settels, B. Engels and F. Würthner, Chem. Sci., 2014, 5, 608–619 RSC.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- M.-J. Lin, Á. J. Jiménez, C. Burschka and F. Würthner, Chem. Commun., 2012, 48, 12050–12052 RSC.
- P. Forn-Díaz, L. Lamata, E. Rico, J. Kono and E. Solano, Rev. Mod. Phys., 2019, 91, 025005 CrossRef.
- S. Kéna-Cohen, S. A. Maier and D. D. C. Bradley, Adv. Opt. Mater., 2013, 1, 827–833 CrossRef.
- M. Held, A. Graf, Y. Zakharko, P. Chao, L. Tropf, M. C. Gather and J. Zaumseil, Adv. Opt. Mater., 2018, 6, 1700962 CrossRef.
- A. Graf, L. Tropf, Y. Zakharko, J. Zaumseil and M. C. Gather, Nat. Commun., 2016, 7, 13078 CrossRef CAS PubMed.
- J. M. Lüttgens, F. J. Berger and J. Zaumseil, ACS Photonics, 2021, 8, 182–193 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2310448–2310452. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc04653b |
| ‡ Both authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |