
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the relationship between molecular and order-dependent photostability of ITIC derivatives for the production of photochemically stable blends†
Y. A.
Quiroz Avalos
 a,
Q.
Eynaud
a,
P.
Perkhun
a,
A.
Rivaton
b,
W.
Köntges
c,
R. R.
Schröder
c,
T.
Koganezawa
d,
N.
Yoshimoto
e,
A. Kumar
Bharwal
f,
D.
Duché
f,
C. M.
Ruiz
f,
O.
Margeat
a,
C.
Aumaître
g,
R.
Demadrille
*g,
C.
Videlot-Ackermann
*a and
J.
Ackermann
*a
a,
Q.
Eynaud
a,
P.
Perkhun
a,
A.
Rivaton
b,
W.
Köntges
c,
R. R.
Schröder
c,
T.
Koganezawa
d,
N.
Yoshimoto
e,
A. Kumar
Bharwal
f,
D.
Duché
f,
C. M.
Ruiz
f,
O.
Margeat
a,
C.
Aumaître
g,
R.
Demadrille
*g,
C.
Videlot-Ackermann
*a and
J.
Ackermann
*a
aAix Marseille Univ., CNRS, CINAM, Marseille, France. E-mail: jorg.ackermann@univ-amu.fr; christine.ackermann@cnrs.fr
bUniv. Clermont Auvergne, CNRS, SIGMA Clermont, Institut de Chimie de Clermont-Ferrand, Clermont-Ferrand F-63000, France
cUniversität Heidelberg, CAM, Centre for Advanced Materials, Heidelberg, Germany
dIndustrial Application Division, Japan Synchrotron Radiation Research Institute (JASRI), Sayo, Hyogo 679-5198, Japan
eDepartment of Physical Science and Materials Engineering, Iwate University, Ueda Morioka 020 8551, Japan
fAix Marseille Univ., Univ. de Toulon, UMR CNRS 7334, IM2NP, Marseille, France
gUniv. Grenoble Alpes, CEA, CNRS, INP, IRIG, SyMMES, Grenoble 38000, France. E-mail: renaud.demadrille@cea.fr
First published on 19th February 2024
Abstract
We present a systematic study of the intrinsic photostability of ITIC derivative acceptors, namely ITIC, ITIC-Th and ITIC-4F, in solution, in layers and in blends with donor polymers such as PBDB-T (also known as PCE12) or its fluorinated derivative PBDB-T-2F (known as PM6). We followed the evolution of the absorption spectra of ITIC derivative acceptors and blends under constant irradiation using different light sources (AM1.5 solar simulator, SUNTest or LED). Any interaction with oxygen under illumination was avoided by placing the solutions and thin films under inert conditions, i.e. nitrogen or vacuum. While donor polymers are highly photostable, all three NFAs show a common photodegradation process with the formation of photoproducts and molecular structure modifications under light exposure. Compared to the solutions where the degradation kinetics are very fast, the degradation in films is significantly slowed down. The corresponding photodegradation process including a cis–trans isomerisation of end groups is found to be common for all ITIC derivatives and independent of the light source. In blends, photodegradation is directly attributable to the acceptor, but varies according to the derivative. In fact, ITIC-4F based blends will benefit from a stabilizing mechanism due to a favorable molecular packing inside the blend. Thereafter, we have studied the photostability of blend layers post-annealed at different temperatures, and we show that the thermally induced packing and crystallinity of the ITIC derivatives dominate the photostability of the blends. We finally investigate the photostability of organic solar cells based on thermally annealed PM6:ITIC-4F blend layers.
Introduction
In recent years, polymer solar cells (PSCs) have shown tremendous increases in performance. They reached power conversion efficiencies (PCE) of over 19% under simulated sunlight1,2 and over 36% under indoor light.3–5 This brings PSCs into the realm of industrially relevant emerging photovoltaic (PV) technologies since they can be produced at low-cost and with a low environmental impact. They also show appealing features such as flexibility, light weight and colour tuning.1,6 The recent improvement of the performances in PSCs is mainly due to the development of novel non-fullerene acceptors (NFA),7,8 the improvement of donor polymers9–11 and the strategy of using multiple components in blends.12 Today, it still seems realistic to increase the efficiency of PSCs further beyond 20% by chemically engineering the materials10,11 but current performance levels already make it possible to envisage innovative photovoltaic applications such as energy collection systems for powering connected objects or applications such as glazing in buildings and agrivoltaic sectors.13 Motivated by these perspectives, research has started to address key industrial requirements such as large-scale processability, cost reduction in material production, and long-term stability of the technology.14 In this context, the photostability of the materials and some highly efficient PSCs has been studied in more detail,15 leading to the identification of degradation mechanisms of NFAs under different stress conditions.16–23 Among NFA materials, the photostability of indacenodithienothiophene (IDTT)-based electron acceptors, such as ITIC and its derivatives ITIC-4F and ITIC-Th, has been intensively studied in recent years with encouraging work on the photostability of solar cells highlighting a very high stability depending on the chemical structure of ITIC derivatives. Indeed, solar cells using ITIC-4F and ITIC-Th as acceptors in blend with a donor polymer PBDB-T show device stabilities with an extrapolated T80 up of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h, while ITIC based solar cells degrade much faster suggesting a molecular-dependent photodegradation of the polymer blend.17Fig. 1(a) shows the chemical structures of ITIC, ITIC-4F, ITIC-Th together with the donor polymer PBDB-T named PCE12 and its fluorinated derivative PBDB-T-2F named PM6 (see the ESI† for chemical names). However, recent studies on the photodegradation mechanism of ITIC derivatives in solution have shown that ITIC is significantly more stable than ITIC-4F24 showing an opposite trend to the photostability of the corresponding PSCs.17 Interestingly, photostability studies of ITIC derivatives carried out in films, in air, have shown that there is a strong effect of crystalline order, and more specifically polymorphism, on the photooxidation rates of NFAs.25 Thus, these studies suggest that there is a rather complex relationship between molecular structure, crystalline order and intrinsic photostability of ITIC derivatives, which requires further studies to complete the picture of the photodegradation of these NFAs.
000 h, while ITIC based solar cells degrade much faster suggesting a molecular-dependent photodegradation of the polymer blend.17Fig. 1(a) shows the chemical structures of ITIC, ITIC-4F, ITIC-Th together with the donor polymer PBDB-T named PCE12 and its fluorinated derivative PBDB-T-2F named PM6 (see the ESI† for chemical names). However, recent studies on the photodegradation mechanism of ITIC derivatives in solution have shown that ITIC is significantly more stable than ITIC-4F24 showing an opposite trend to the photostability of the corresponding PSCs.17 Interestingly, photostability studies of ITIC derivatives carried out in films, in air, have shown that there is a strong effect of crystalline order, and more specifically polymorphism, on the photooxidation rates of NFAs.25 Thus, these studies suggest that there is a rather complex relationship between molecular structure, crystalline order and intrinsic photostability of ITIC derivatives, which requires further studies to complete the picture of the photodegradation of these NFAs.
 | ||
| Fig. 1 (a) Molecular structure of acceptor molecules ITIC, ITIC-Th and ITIC-4F and donor polymers PCE12 (PBDB-T) and PM6 (PBDB-T-2F). (b) Normalized absorption spectra of thin films based on organic molecules (right Y axis) compared to different light sources, AM1.5 for the standard solar spectrum emitted by the sun with an integrated power of 1000 W m−2 (100 mW cm−2), energy spectrum of the SUNTest and LED illumination. The hatched area indicates UV light below 400 nm cut by an UV-filter. | ||
In this work, we have studied the intrinsic photostability of ITIC derivatives, namely ITIC, ITIC-Th and ITIC-4F in solution, in single layers and in blends with polymers i.e. PBDB-T (PCE12) or its fluorinated derivative PBDB-T-2F (PM6) (Fig. 1(a)). We focused on the potential changes in photodegradation of the NFA polymer blend due to donor–acceptor interactions and ordering. For this purpose, we applied a setup of advanced methods to study the film morphology including GIWAXS (Grazing-Incidence Wide-Angle X-ray Scattering), analytical TEM (ATEM) and Raman spectroscopy. Our results reveal that the intrinsic photostability of all three molecules is very similar in solution, but shows strong variation as a function of molecule structure when studied in films. Interestingly, when the NFAs are mixed with a donor polymer, we again observe changes in the photodegradation rates between the three ITIC derivatives, suggesting that there are indeed molecular independent parameters that directly influence the degradation pathways. A detailed analysis of the crystalline order in pure films and in polymer blends allowed us to attribute the variation in photodegradation mainly to changes in the crystalline order. Importantly, further enhancement of the crystalline order by thermal annealing shows that the photostability of all three NFAs is mainly determined by the polymorphs, while the subtle changes in the chemical structure of the ITIC derivatives studied have only a small effect. We also show that there is a common photodegradation process for all molecules, including a cis–trans isomerisation of the 2-(3-oxo-2,3-dihydroinden-1-ylidene)malononitrile (INCN) end groups. This process is independent of the light source used, i.e. AM1.5 or LED (Fig. 1(b)). Overall, the enhancement of crystalline order within the acceptor phase is found to be essential to suppress or slow down the cis–trans isomerisation, allowing the photostability of all ITIC derivatives to be improved by over an order of magnitude.
Results and discussion
Photostability of NFAs in solution
As a first photodegradation study, we performed a stability test of the three ITIC molecules in chlorobenzene solutions. Fig. 2(a) shows the evolution of the absorption spectra for the three NFAs upon constant irradiation using an AM1.5 light source (25 °C, 1000 W m−2). The initial absorption spectrum of each molecule is characterized by a main band located between 550 and 750 nm, composed of a shoulder at 600 nm and a maximum around 670 nm corresponding to the A0–1 and A0–0 transitions, respectively.25 During illumination, the A0–0 band shows a rapid decrease in intensity, whereas the A0–1 band shows a less pronounced decrease in absorption. This behaviour is identical for all three molecules. After 24 h, we observe an almost complete disappearance of the A0–0 transition. In parallel, a new band appears between 450 and 550 nm, which increases in intensity as the A0–1 transition decreases. These spectral features, which are identical for the three molecules, suggest the formation of photoproducts and molecular structure modifications under light exposure. Recent work by Perepichka et al.24 analysed the photodegradation of ITIC and ITIC-4F in solution and showed that two isomeric photoproducts P1 and P2 are formed upon exposure to light (see Fig. S1(a), ESI†). The degradation mechanism was considered to be a cis–trans isomerisation of the terminal groups, followed by an electrocyclic reaction between the INCN moiety and the thiophene ring, followed by a 1,5-sigmatropic hydride shift (see Fig. S1(b), ESI†). The photoproduct P1 was identified as an asymmetric isomer of ITIC-4F with a subsequent 1,5-hydride shift, while P2 is the fully fused photoproduct after the isomerization of the second INCN unit. In a more recent article from the same group, this mechanism was confirmed and a Diels–Alder cycloaddition of singlet oxygen was evidenced when the thiophene was replaced by a furan.26 In their initial work, Perepishka et al. showed that the absorption of the photoproduct P2 is found blue shifted with respect to the main absorption peak at 510 nm. Our observations are fully consistent with the ones of Perepishka et al. and thus we attributed the absorption band between 450 and 550 nm to the formation of P2. We found that all three molecules have similar degradation rates over the first 10 hours, while the rate of degradation of ITIC accelerates over the last 14 hours. However, the general degradation mechanism related to the photoinduced isomerization can be considered as common for all three ITIC derivatives on the basis of the variation of their absorption spectra in solution and the conclusion of previous studies. | ||
| Fig. 2 Evolution of absorption spectra of ITIC, ITIC-Th and ITIC-4F: (a) in solution under constant UV filtered AM1.5 illumination and (b) in layers under constant UV filtered SUNTest illumination. (c) Corresponding FTIR spectra of initial layers and after 500 h under constant UV filtered AM1.5 illumination. | ||
Photostability of NFAs and donor polymers in thin films
To further investigate the degradation of the NFA molecules and donor polymers in the solid state, the photostability of pure films under UV-filtered SUNTest illumination was investigated. The films of pure NFAs and polymers were deposited in a glove box atmosphere on inert substrates (glass and KBr) and thermally annealed at 100 °C for solvent evaporation. Prior to light soaking, the films were vacuum sealed in the absence of oxygen and water. The photodegradation of acceptor and donor pure films was studied over exposure times up to 500 h using UV-Vis absorption and FTIR spectroscopy. The evolution of the absorption and FTIR spectra as a function of time exposure is shown in Fig. 2(b) and (c), respectively. Films based on ITIC derivatives show very similar absorption spectra, consisting mainly of a large band in the visible region extending to the near infrared with two peaks corresponding to the A0–1 and A0–0 transitions. The maximum absorption of A0–0 is observed at 705 nm for ITIC, 698 nm for ITIC-Th and 720 nm for ITIC-4F. Within the first 100 hours of exposure, the A0–0 band degrades faster than the A0–1 band, whereas with longer exposure, both A0–0 and A0–1 are degraded almost equally. In parallel, we observe the formation of a band between 450 and 550 nm that can be attributed to the formation of P2. After 500 h under illumination, the two initial bands have almost completely disappeared for ITIC-4F and ITIC-Th, and although they can still be distinguished for ITIC, the absorption band corresponding to P2, with a maximum around 520 nm, is dominant in all the three dyes.FTIR analysis was used to study in detail the modification of the structure of the NFAs upon irradiation (Fig. 2(c)). For the ITIC films, a peak at 1702 cm−1 can be observed, corresponding to the unsaturated carbonyl stretching vibrations (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the INCN units. This peak is common to all ITIC derivatives and decreases in intensity with exposure time. The signal at 1702 cm−1 progressively shifts towards higher wavenumbers and a new signal at 1716 cm−1 is clearly distinguishable after 500 hours under light. This suggests that the carbonyl group is not degraded during light soaking, but rather that there is a change in its chemical environment. A shift in the carbonyl signal towards higher wavenumbers was also observed by Park et al.19 for ITIC layers deposited on ZnO and exposed to UV light. In this case, a more significant shift at 1732 cm−1 was found. This shift was attributed to the saturation of the vinyl group connected to the INCN after the reaction with hydroxyl radicals. The hydroxyl radicals were formed by degradation of water at the interface with ZnO under UV light. Perepichka et al. stated that the vinylene bridge is not the immediate reaction centre.24 Therefore, the shift of the carbonyl group observed in the IR spectra, in the absence of water and oxygen, can be attributed to an electrocyclic reaction involving the vinyl bridge and the INCN moiety. In addition, we note that the CN peak at 2220 cm−1 decreases upon illumination, which also correlates with the cyano group being affected by the degradation process. These results suggest an identical transformation of the three molecules in films, with cis–trans isomerisation of the terminal groups as the starting point for the phototransformation. Compared to the solutions where the degradation kinetics are very fast, the degradation in thin films is significantly slowed down. The complete photo-transformation of the molecules occurs on a time scale of 500 hours.
O) of the INCN units. This peak is common to all ITIC derivatives and decreases in intensity with exposure time. The signal at 1702 cm−1 progressively shifts towards higher wavenumbers and a new signal at 1716 cm−1 is clearly distinguishable after 500 hours under light. This suggests that the carbonyl group is not degraded during light soaking, but rather that there is a change in its chemical environment. A shift in the carbonyl signal towards higher wavenumbers was also observed by Park et al.19 for ITIC layers deposited on ZnO and exposed to UV light. In this case, a more significant shift at 1732 cm−1 was found. This shift was attributed to the saturation of the vinyl group connected to the INCN after the reaction with hydroxyl radicals. The hydroxyl radicals were formed by degradation of water at the interface with ZnO under UV light. Perepichka et al. stated that the vinylene bridge is not the immediate reaction centre.24 Therefore, the shift of the carbonyl group observed in the IR spectra, in the absence of water and oxygen, can be attributed to an electrocyclic reaction involving the vinyl bridge and the INCN moiety. In addition, we note that the CN peak at 2220 cm−1 decreases upon illumination, which also correlates with the cyano group being affected by the degradation process. These results suggest an identical transformation of the three molecules in films, with cis–trans isomerisation of the terminal groups as the starting point for the phototransformation. Compared to the solutions where the degradation kinetics are very fast, the degradation in thin films is significantly slowed down. The complete photo-transformation of the molecules occurs on a time scale of 500 hours.
Previous studies have claimed the remarkable stability of ITIC derivatives under LED light. In fact, the strong absorption dips in the absorption spectra of pure films of the three ITIC derivatives have never been seen before in photostability tests using LED light.17,23 To confirm that the degradation mechanism observed here is not dependent on the light source, the stability of the ITIC layers was investigated under LED illumination (see Fig. 1(b) for the light source spectrum). As shown in Fig. S2 (see ESI†), the A0–0 band of ITIC decreases continuously over the exposure time, and the shoulder between 450 nm and 550 nm corresponding to the photoproducts starts to form. The degradation mechanism is identical under LED light and therefore independent of the light source, although LED light leads to a much slower process compared to SUNTest illumination which can be mainly attributed to the reduced amount of photons range absorbed by the NFAs under these conditions (see Fig. 1(b)).
Following this study, it seemed important to study the intrinsic stability of donor polymers in order to gain a better understanding of the degradation phenomena that can occur in blends. As a next step, we investigated the photostability of the donor by studying the evolution of the absorption and IR spectra of the donor polymer films under SUNTest illumination as shown in Fig. S3 (see ESI†). Importantly, the absorption and FTIR spectra of both polymers are almost unchanged after 400 h, demonstrating excellent photostability of the polymers under illumination in the absence of oxygen. This is consistent with recent results on ageing under LED light17,23 and under UV light.19 There is therefore a marked difference in photostability between donor and acceptor materials. Fig. 3(a) summarizes the degradation kinetics of NFAs and polymers based on absorption by plotting the evolution of the low energy band for each material as a function of illumination time. The results were confirmed by plotting the diminution in the intensity of the peak at 2220 cm−1 related to the C–N vibrational signal of the corresponding FTIR spectra (see Fig. S4(a), in the ESI†). Interestingly, the degradation rates of ITIC and ITIC-4F films are comparable, while ITIC-Th shows the highest degradation rate. Furthermore, the stability in pure films for each of the three NFAs does not match that found in the solution, suggesting that another parameter leads to the variation in the intrinsic photostability.
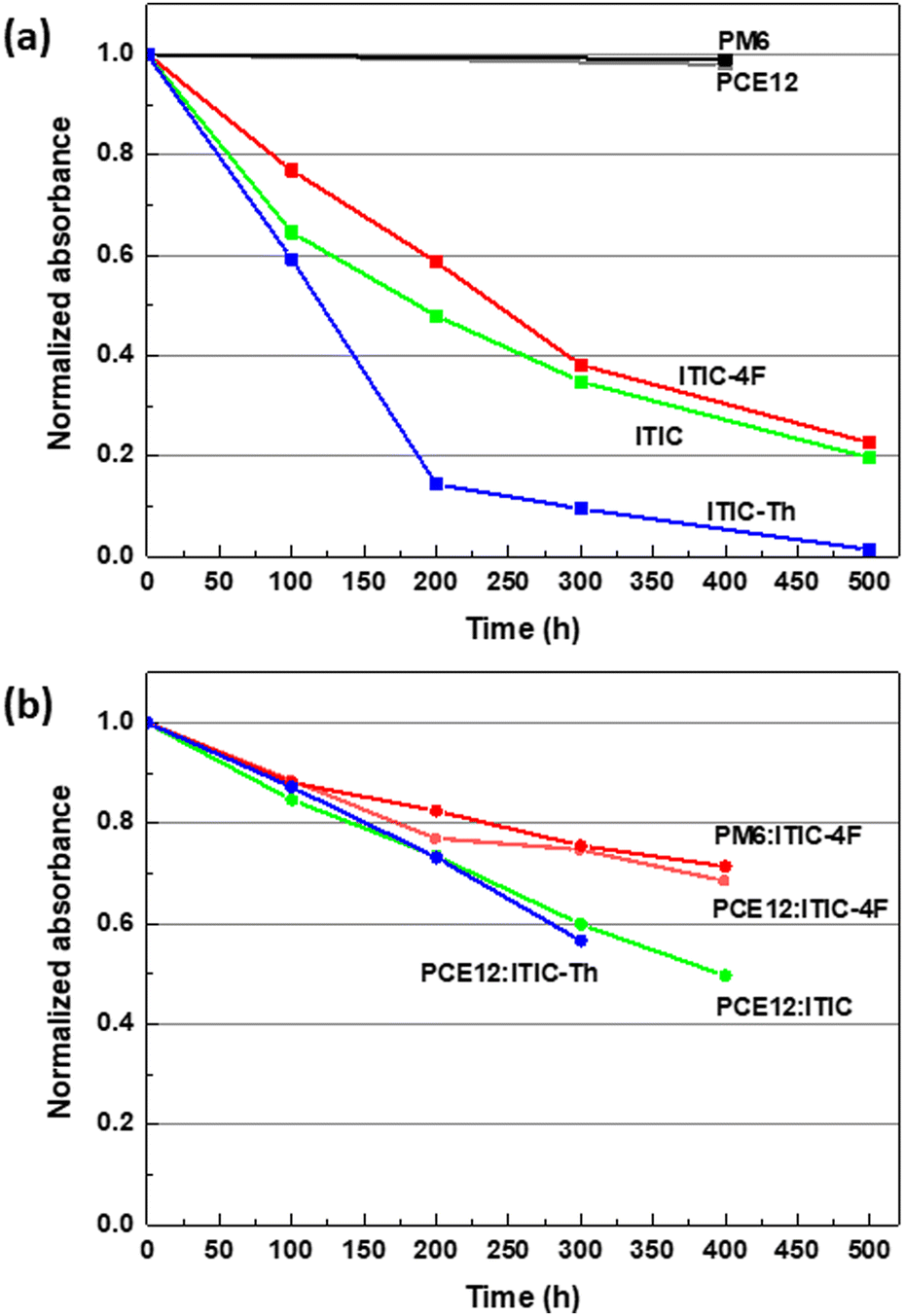 | ||
| Fig. 3 Time evolution of absorption spectra of pure material films (a) and PM6:ITIC-4F, PCE12:ITIC-4F, PCE12:ITIC and PCE12:ITIC-Th blends (b). The values were taken from the A0–0 peak corresponding to the degradation of the NFA under constant UV filtered SUNTest illumination in the absence of oxygen. | ||
Photostability of blend films
We have already seen that the degradation kinetics of donor and acceptor materials are quite different when studied in pure films under identical conditions. Similar degradation experiments were then carried out on the polymer blends typically used to make high-efficiency organic solar cells. Blends composed of PCE12:ITIC, PCE12:ITIC-Th, PCE12:ITIC-4F and PM6:ITIC-4F were processed with a D:A ratio of 1:1. Compared to pure films, the interpretation of the absorption spectra of blends is more complex as they are composed of both donor and acceptor absorption bands. As an example, Fig. S5 (in ESI†) shows the spectrum of the PCE12:ITIC blend, with three peaks in the visible range compared to the spectrum of PCE12 and ITIC. In the mixture, only the low energy band located at around 700 nm can be attributed to the A0–0 absorption of ITIC, while the maximum absorption peak located at around 640 nm is the result of the addition of the absorption bands of the two materials (A0–1 of the acceptor and ICT of the donor). Fig. S6 (see ESI†) shows the time evolution of the absorption spectra of all blends during light soaking under constant UV-filtered SUNTest illumination. The light-induced changes in the NFA absorption range are much less pronounced for all blends compared to pure NFA films. This direct observation can be attributed to a filtering or screening effect of the polymer which reduces the number of absorbed photons in the spectral region of common absorption. The inner-filter effect is similar for all blends because PM6 and PCE12 have identical molar extinction coefficient spectra as their chemical structure differing by only two fluor atoms. To further clarify whether the polymer retains the same high stability within the blend, IR spectra at 1600–1700 cm−1 of blends and pure donor polymer films were compared (see Fig. S7, in ESI†). The vibrational signal at 1648 cm−1 is only observable in the spectra of the two polymers, PCE12 and PM6, and corresponds to the vibration of the two carbonyl groups of the dithiophene dione unit. This signal remained unchanged under light exposure for the polymer mixtures. We can therefore conclude that both polymers, in the blends, have an unchanged high photostability.
Therefore, we suggest that any changes in the absorption spectra of the blends can only be attributed to the degradation of the NFAs during the interval time of this study. With respect to the A0–0 absorption peaks in the spectra shown in Fig. S6 (ESI†), the blend samples behave similarly, showing a loss of absorption and a shoulder formation in the 450–550 nm region. In the case of ITIC-Th which is the less stable material when studied in film, we can clearly see a better stability in blends. Reporting the evolution of the absorption (Fig. 3(b)) highlights that the PM6:ITIC-4F blend is clearly the most stable, as expressed by a lower overall degradation of the A0–0 absorption band of the NFA. In this case, we cannot see the spectral signature of the formation of photoproducts P2 after 400 h. To investigate whether the higher stability of ITIC-4F in PM6:ITIC-4F blends is only related to the mixing with PM6, we also prepared PCE12:ITIC-4F blends and studied their photodegradation as shown in Fig. S6 (see the ESI†). Again, the stability of ITIC-4F in blends with PCE12 was found to be significantly better than that of the film alone. This proves that the mechanism of stabilization of ITIC-4F in blends is independent of the polymer. Fig. 3(b) compares the degradation rate of the four blends. During the first 100 h, all blends show a similar degradation rate, whereas after 200 h, the ITIC-4F based blends show lower losses compared to the ITIC and ITIC-Th based blends. The same tendency has been observed by plotting the diminution in the intensity of the peak at 2216 cm−1 of the corresponding FTIR spectra (see Fig. S4(b), in the ESI†).
To understand the origin of the difference in photostability of the three NFA based blends, it is important to remember that the photodegradation of the three ITIC derivatives starts with a photoinduced cis–trans isomerisation of the end groups involving a twist of the INCN unit on the vinyl bridge. Crystallinity plays an important role in determining molecular packing and it has been shown that molecules with higher crystallinity also have shorter π–π stacking distances.27,28 At shorter π–π stacking distances, molecular motions are restricted by intermolecular steric hindrance and thus photoinduced isomerisation of the molecule is less favourable.29 It is therefore possible that changes in the photostability of the three NFAs are related to differences in molecular packing inside the blend.
Morphological analysis of blend films
In order to obtain more information about the crystallinity and ordering of the ITIC derivatives, a series of experiments using advanced analytical techniques, including GIWAXS, Raman spectroscopy and analytical TEM analysis, were carried out on ITIC and ITIC-4F pure materials in film and in blends. The 1D in-plane profiles of these films are shown in Fig. 4(a). In the case of pure films, ITIC shows a (100) lamellar diffraction peak, indicating a crystalline organisation, whereas ITIC-4F is amorphous. It is important to note that the films were deposited on glass substrates or on a ZnO buffer layer and post-annealed at a relatively low temperature (100 °C). Under these conditions, ITIC-4F films were found to be amorphous, whereas an ordering was observed for films deposited under other conditions30 and annealed at higher temperatures. For pure polymer films, PCE12 and PM6 show a strong (100) lamellar diffraction peak at qz = 0.345 Å−1 (d = 1.82 nm) and qz = 0.323 Å−1 (d = 1.94 nm) in the out-of-plane direction, respectively (see Fig. S8(a), ESI†). It is attributed to an edge-on lamellar stacking of PCE12 and PM6. For PCE12, with a marked (100) peak in the in-plane direction, the polymer chains are also oriented with a face-on orientation. In the case of polymer blends, the overlap of the π–π scattering reflections of PCE12 and PM6 with those of ITIC and ITIC-4F, respectively, makes it difficult to distinguish the contribution of polymer and NFA in the (100) lamellar diffraction peak. However, a Gaussian fit highlights the presence of a shoulder in the (100) peak of both blends that can be associated with the diffraction peak of the NFAs as shown in the deconvolution of Fig. 4(b). Table S1 (ESI†) gives the peak position (qxy), the peak area and the full width at half maximum (FWMH) values of each contribution of each material resulting from the Gaussian fit. The d-spacing and the crystal coherence length (CCL) of pure film materials and blend layers are calculated from these data. In PCE12:ITIC blend (after 100 °C thermal annealing), the d-spacing value of each material increases with respect to the pure materials. However, for the PM6:ITIC-4F blend, the d-spacing value of PM6 remains constant with the addition of a contribution of ordered ITIC-4F crystallites. In the case of ITIC-4F, the amorphous state in pure film materials is transformed into a crystalline order with d = 1.93 nm and CCl = 0.63 nm in the PM6:ITIC-4F blend.These results show that mixing the NFAs with the polymers changes their crystalline order, which can affect the photodegradation rate, as previously shown in photo-oxidation studies of pure film materials.25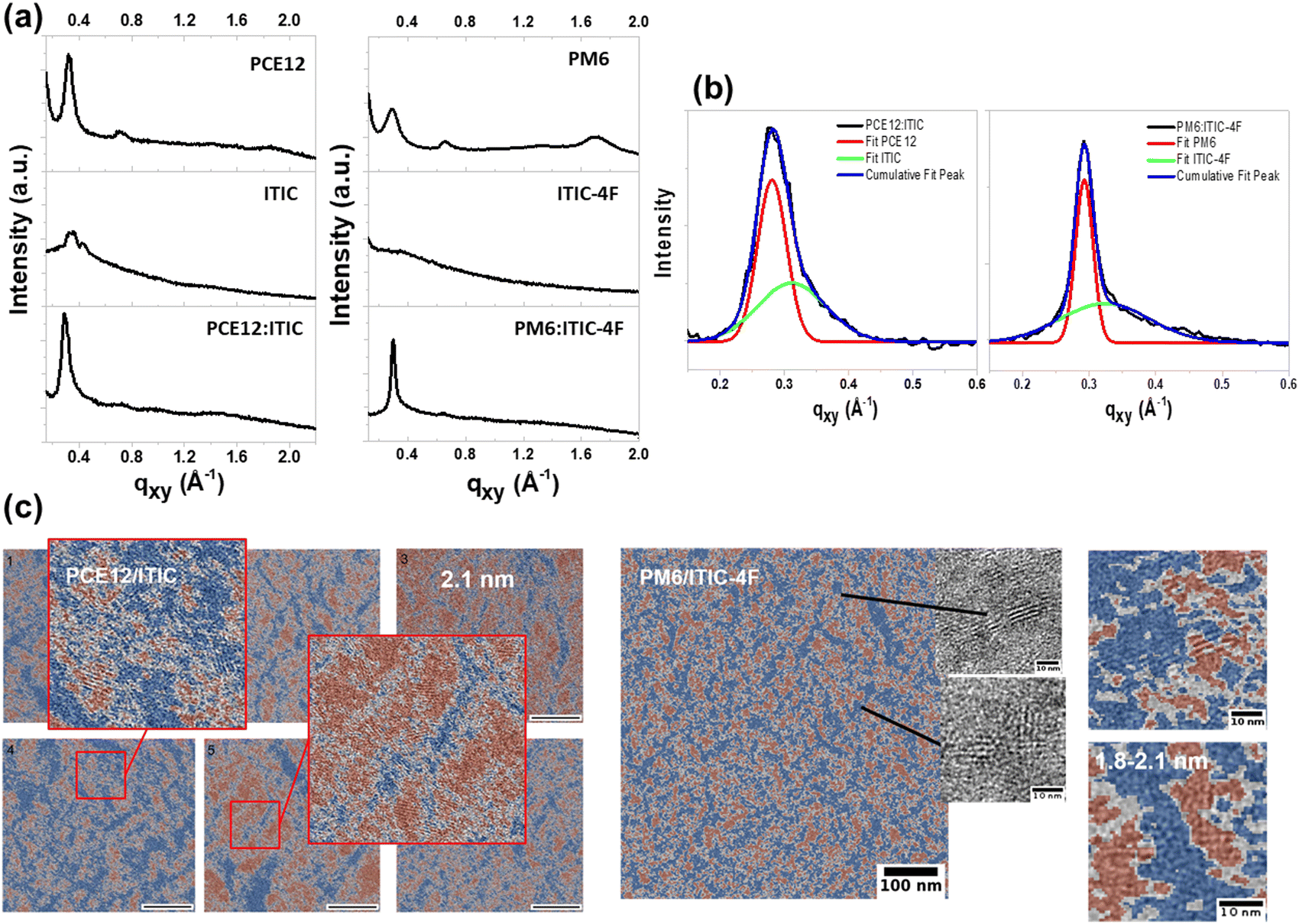 | ||
| Fig. 4 (a) 2D-GIXRD profiles of pure material and blend layers at 100 °C. (b) Gaussian fitting of the IP (100) peak of the blend layers. (c) Material distribution map of blend layers at 100 °C obtained by ATEM from bright-field TEM images of corresponding areas (in blue: polymers and in red: acceptors). | ||
To gain deeper insight into the blend morphology, we performed analytical TEM (ATEM), which has been previously applied to study ITIC polymorphism within polymer blends with PCE12.31 Details on processing and analyzing data by ATEM, electron energy spectra of pure polymer and NFAs can be found in previous works.31,32 In the case of ITIC, a high degree of crystallinity was found in both pure materials and blend films like in a recent work.33 ATEM analysis also reveals differences in the crystalline organisation of ITIC when comparing pure film and blended films. Indeed, there is a distribution of lamellar spacing with a d-spacing ranging from 1.9 nm to 2.1 nm in films of pure materials, which changes to a dominant d-spacing of 2.1 nm when ITIC is blended with PCE12 (Fig. 4(c)). In the case of ITIC-4F, which is amorphous in pure films, ATEM images of PM6:ITIC-4F films show the formation of a crystalline organisation, but on rather short length scales. In comparison to ITIC, there is no dominant lamellar spacing in the PM6:ITIC-4F blend but rather a large distribution from 1.8 nm to 2.1 nm signals. From these analyses, some conclusions can be drawn about the relationship between photostability and crystallinity of ITIC derivatives. Considering that the photostability of ITIC was similar in pure films and polymer blends, the results suggest that the observed rather small increase in the degree of crystallinity and uniformity of d-spacing does not change the photodegradation rate of this molecule. In contrast, ITIC-4F undergoes a clear transition from an amorphous state in pure films to ordered domains when mixed with the polymer. The polymer-induced increase in the crystalline order of ITIC-4F was also recently demonstrated when blended with PM6 so the observed photostability enhancement of ITIC-4F in PM6:ITIC-4F blend may also originate from crystal order.34 Interestingly, Ciammaruchi et al. recently reported a large set of different polymorphisms in ITIC and ITIC-4F layers for specific thermal annealing treatments that modified the photostability of the molecules in air.25
Therefore, in the next part of our study, we first subjected the PM6:ITIC-4F blend and the pure PM6 and ITIC-4F layers to thermal annealing at different temperatures to see whether the crystalline order affects the photostability under the same conditions as for untreated layers.
Morphology of highly ordered ITIC-4F films and PM6:ITIC-4F polymer blends
For this purpose, temperatures between 100 °C and 200 °C were applied to the layers after deposition. Fig. S9 (in ESI†) shows the absorption spectra of PM6 and ITIC-4F films as well as the corresponding blend as a function of thermal annealing temperature. While the absorption spectra of PM6 are not affected by thermal annealing, a strong red shift of about 41 nm of the low energy band is observed for both the pure ITIC-4F film and the PM6:ITIC-4F blend annealed at 200 °C. The red-shifted and narrower ITIC-4F peaks indicate an increase in thermal-induced aggregation and thus better organization of molecules, where predominance J-like aggregates in ITIC-4F films have been reported.30,35To further analyse the thermally induced aggregation of ITIC-4F, AFM, 2D-GIXD and Raman spectroscopy analyses were performed on the annealed layers. The AFM images in Fig. S10 (in ESI†) show the clear difference between the as-cast, 100 °C and 150 °C annealed layers compared with layers annealed at 200 °C. We found that thermal annealing up to 150 °C has no effect on the surface morphology, since at all three temperatures (as-cast, 100 °C and 150 °C), all layers have the same roughness of about 1.56 nm and are composed of similar “fibril” structures. In contrast, annealing at 200 °C induces larger round aggregates and an increase in roughness to 2.58 nm.
Additional 2D-GIXD measurements of the same layers help us to better understand the evolution of ITIC-4F aggregation with temperature in blends (Fig. 5). We compare the in-plane and out-of-plane intensity profiles of 100 °C, 150 °C and 200 °C annealed layers in Fig. S8(b) (in ESI†).
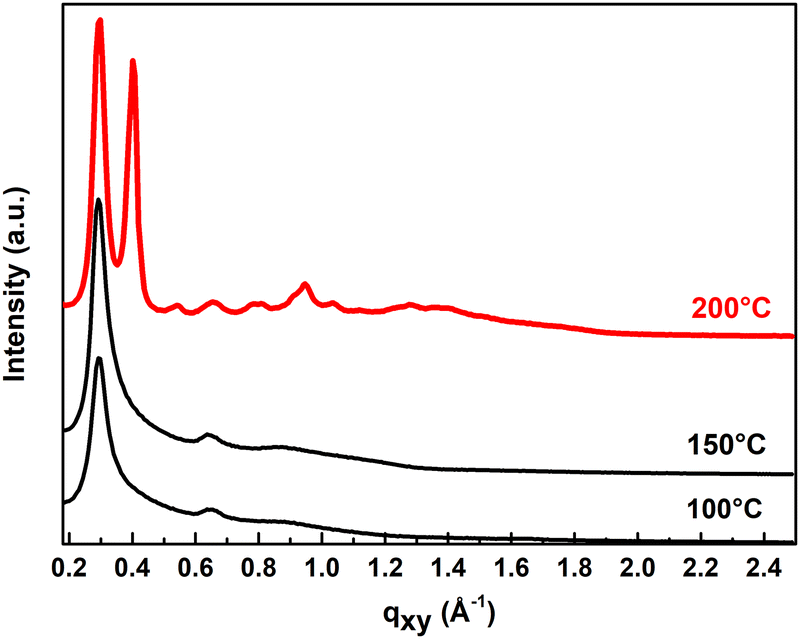 | ||
| Fig. 5 2D-GIXRD profiles of PM6:ITIC-4F films as function of thermal annealing temperature. | ||
From UV-Vis and AFM, there is no evidence of increased crystallinity at temperatures below 200 °C, but the intensity profiles from 2D-GIXD show the evolution of the aggregation of ITIC-4F at each temperature and reveal that thermal annealing strongly enhances the crystallinity of the films, expressed by much brighter and sharper peaks. The bottom of Table S1 (ESI†) shows the values of the Gaussian fit of the (100) in-plane peaks for PM6:ITIC-4F blends annealed at 150 °C and 200 °C. The contribution of ITIC-4F is even evident at 200 °C by the appearance of two separate peaks (see Fig. 5). While the d-spacing of the PM6 polymer chain is constant over temperature at a value of 2.12–2.15 nm, the most significant change is observed for ITIC-4F at 200 °C with a reduced d-spacing of 1.57 nm. In addition, a CCL of 2.38 nm highlights the formation of crystallites three times larger than at 100 °C or 150 °C (CCL = 0.63–0.75 nm). These two observations indicate that annealing the blend at 200 °C induces the formation of larger and denser ITIC-4F crystallites. Nevertheless, the typical diffraction peak (100) assigned to ITIC-4F is as intense and narrow in the out-of-plane as in the in-plane profiles at 200 °C, suggesting edge-on and face-on lamellar orientation of the ITIC-4F crystallites. The face-on orientation of the crystallites at 200 °C is also highlighted by an intense (010) diffraction peak (qz = 1.86 Å−1) in the out-of-plane direction corresponding to the short π–π stacking distance of the closely packed ITIC-4F (d = 0.33 nm (Fig. S8(b), ESI†).
In order to gain a deeper insight into the ordering of polymer and acceptors, Raman analysis was additionally performed on mixed layers at the three temperatures and on pure PM6 and ITIC-4F layers as references. Raman can probe molecular conformation as well as chemical structure as it is sensitive to the polarisable electron density, e.g. pi- conjugated systems, providing important information on structural and morphological changes in semi-crystalline materials25 The Raman spectra of ITIC-4F and PM6 materials as well as blends are shown in Fig. S11 (in the ESI†) and Fig. 6, respectively. In Fig. S11 (ESI†), it can be seen that PM6 shows specific peak structures that can be attributed to the formation of crystalline order in the polymer independently of the thermal treatment. In contrast, the pure ITIC-4F films only show highly crystalline structures at 200 °C. At 200 °C, both PM6 and ITIC-4F films exhibit highly crystalline structures in agreement with the results obtained using GIWAXS. In particular, crystalline ITIC-4F films show the signals of the C–C intra-ring stretching mode at 1248 cm−1 and the fused phenyl ring at 1600 cm−1. In addition, peaks at 970 cm−1, 1138 cm−1, 1285 cm−1 and 1340 cm−1 are assigned to ITIC-4F, while peaks at 1075 cm−1 and 1230 cm−1 can be attributed to the PM6 polymer alone. Finally, peaks in the 1400–1550 cm−1 region are a contribution from both materials. These identifications allow us to assign the different peaks in the spectrum of the blend to PM6 or ITIC-4F. The spectra of the PM6:ITIC-4F blend as a function of annealing temperature are shown in Fig. 6. Interestingly, we find several Raman peaks already in the as-cast layers, indicating the formation of a crystalline order in the mixture at room temperature. Increasing the annealing temperature causes small changes, only the intensity of two signals at 1248 cm−1 and 970 cm−1 (indicated by red arrows) increases with temperature. These results suggest that there is a blend induced increase in ordering, i.e. an increase in crystallinity of both PM6 and ITIC-4F, not only due to temperature but also due to interactions of ITIC-4F and PM6 within the blend. Together with the GIWAXS results presented above, we show here that the thermal annealing of the pure films and blend layers, together with the interaction with the polymer PM6, has resulted in an increased crystallinity of ITIC-4F.
 | ||
| Fig. 6 Raman spectra of as-cast PM6:ITIC-4F blends and annealed at 100 °C, 150 °C and 200 °C. | ||
Photostability of highly ordered ITIC-4F films and PM6:ITIC-4F polymer blends
After confirming the presence of ITIC-4F crystallites in the pure layer at 200 °C and in the blends, the layers were subjected to photodegradation studies to analyse the effect of the enhanced crystallinity on the stability of ITIC-4F. The evolution of the absorption spectra of the degraded layers under light exposure is shown in Fig. 7(a) and Fig. S12 (in the ESI†). It can be clearly seen in Fig. S12 (ESI†) that the stability of the pure ITIC-4F annealed at 200 °C is greatly improved, as only 30% of the loss of the A0–0 peak is observed after more than 700 h compared to the ITIC-4F layer processed at 100 °C as shown in Fig. 2, which shows the total degradation of the initial absorbance and the formation of P2 after only 500 h of illumination. The stability is confirmed by the IR spectra, with almost no change in the CO vibration peak at 1702 cm−1. This signal was shifted to 1716 cm−1 for ITIC-4F processed at 100 °C. We then investigated the stability of ITIC-4F within the PM6:ITIC-4F blends by varying the annealing temperature from 100 °C to 200 °C. As shown in Fig. 7(b), the stability of the PM6:ITIC-4F blends, and therefore of ITIC-4F, is progressively improved with increasing annealing temperature. All these results indicate the importance of crystalline order in improving the photostability of ITIC-4F, both in pure films and in blends.
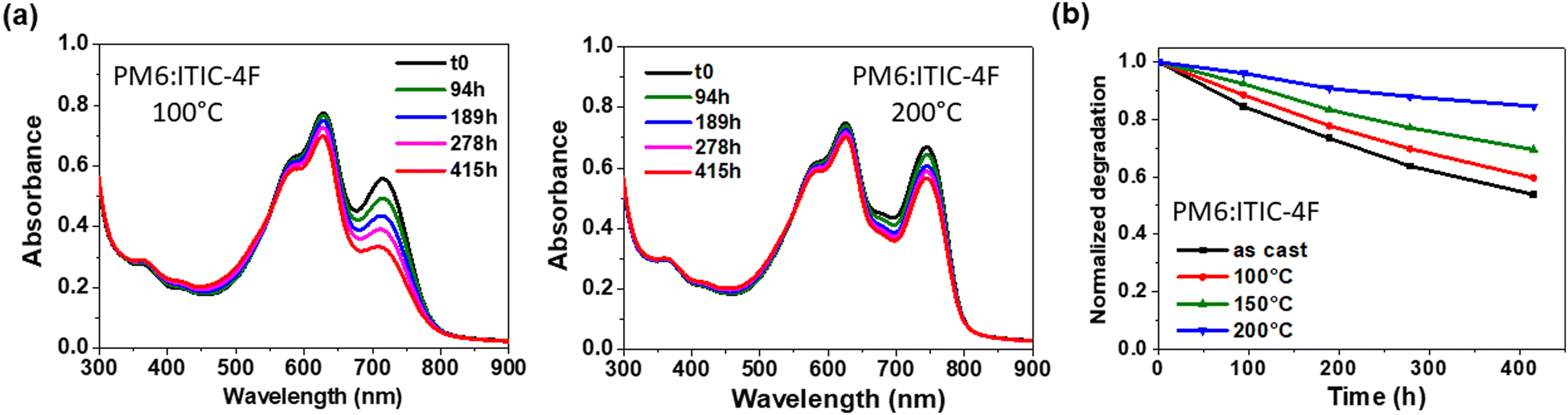 | ||
| Fig. 7 UV-visible absorption spectra of PM6:ITIC-4F blends at 100 °C and 200 °C under constant UV filtered SUNTest illumination (a) and time evolution of absorption spectra of PM6:ITIC-4F blends as a function of thermal annealing temperature (b). In (b), the values were taken from the A0–0 peak corresponding to the degradation of the NFA under constant UV filtered SUNTest illumination. | ||
Photostability polymer blends based on ITIC and ITIC-Th
We then applied the same approach, i.e. inducing improved crystallinity by thermal annealing, to ITIC and ITIC-Th and their respective blends with the PCE12 polymer to see if their stability was also improved by crystalline order. Based on previously reported study on charge transport properties in ITIC derivatives, optimized annealing temperatures for ITIC and ITIC-Th were 240 °C and 200 °C, respectively.36 The absorption spectra of PCE12 polymer, ITIC and ITIC-Th and PCE12:ITIC and PCE12:ITIC-Th blends as a function of annealing temperature are shown in Fig. S13 (in the ESI†). A red shift of the A0–0 band and an increase in the ratio between the A0–0 and A0–1 peaks at high annealing temperatures are observed for ITIC and ITIC-Th, as previously observed for ITIC-4F and reported in the section above. ITIC-Th layers annealed at 150 °C show increased absorbance and a narrow A0–0 peak, while the layer annealed at 200 °C shows decreased intensity. In the case of ITIC annealed at 240 °C, the intensity of the absorption spectra decreases but the ratio between A0–0 and A0–1 increases. As observed for ITIC-4F layers annealed at 200 °C, a redshifted and narrower peak is a clear indication of more ordered ITIC aggregates. The evolution of the absorption spectra under constant SUNTest illumination of annealed ITIC and ITIC-Th layers is presented in Fig. S14 (in ESI†). Importantly, ITIC layers annealed at 240 °C retained 74% of the initial absorbance after 590 h, while ITIC annealed at 100 °C was completely converted in P2 after 500 h as shown before. The same stability enhancement is observed for thermally annealed ITIC-Th layers, which maintain 90% of the initial absorption upon 590 h illumination compared to a complete photo-transformation after 500 h when annealed at 100 °C as shown in Fig. 2. FTIR spectra of ITIC annealed at 240 °C and ITIC-Th at 200 °C confirm the improved stability as the intensity of most peaks is less reduced and the shift of the CO vibration peak at 1702 cm−1 is suppressed.
In the case of the blended layers, the evolution of the absorption spectra of both PCE12:ITIC and PCE12:ITIC-Th blends is shown in Fig. S15 (in ESI†). The PCE12:ITIC blend shows a loss of about 15% of the initial absorption spectrum of the A0–0 peak after 286 h, but after this period no changes are observed up to 700 h. This result suggests that the initial degradation that is observed for 286 h is essentially related to the degradation of ITIC in the residual amorphous regions within the films. By suppressing photoinduced isomerisation in the ordered domains, the mixture is strongly stabilised and no significant degradation occurs after 286 h. The FTIR spectra of PCE12:ITIC blends annealed at 240 °C support the previous observations and confirm the improved stability of the blend. Fig. S.16 (in the ESI†) shows the degradation kinetics of PCE12:ITIC and PCE12:ITIC-Th films annealed at 100 °C, 150 °C, 200 °C and 240 °C, showing greatly improved stability after high temperature annealing. Our results highlight the general relationship between the crystallinity of NFAs in the ITIC family and the improved photochemical stability in pure material films and, more importantly, in blends.
Photovoltaics properties of polymer blends based on ITIC-4F
Films casted from o-xylene were implemented as the active layer in an inverted device structure (glass/ITO/ZnO/PM6:ITIC-4F/MoO3/Ag) to evaluate the performance and phtostability of the solar cells as a function of annealing temperature. Blend layers were annealed at 100 °C, 150 °C or 200 °C before the thermal evaporation of the interfacial layer and the top electrode. A reference device based on a complete device post-annealed at 100 °C during 10 minutes was realized also. The devices were encapsulated before being placed in a SUNTest for photodegradation experiments. The initial performances (PCE, Jsc, Voc and FF) of four systems are listed in Table S2 (ESI†). Compared to the reference device, the thermal annealing at high temperatures of the active layer critically affects the performances. Voc decreases continuously from 0.88 V to 0.68 V for the annealing from 100 °C to 200 °C, while Jsc is slightly increased, except for the highest temperature. The efficiency value (PCE) decreases strongly to 6.6% at 200 °C due to a very low FF and Voc. This drastic decline may be a consequence of the strong aggregation and larger domains formed by ITIC-4F at such temperature, which increases charge recombination. Furthermore, the high temperature annealing may degrade the interface between the ETL ZnO and the blend. Interestingly, annealing at 100 °C has different effects depending on the step in which it is applied. Annealing at 100 °C of the complete device typically increases the FF and delivers a PCE of more than 11%, while 100 °C annealing applied only to the ITO/ZnO/Active layer delivers only 10% efficiency. These results suggest a possible different interaction with the interfacial layers during the annealing step.Performance evolution of PM6:ITIC-4F based solar cells at each annealing temperature is presented in Fig. 8. First, we notice a different behavior of the devices that have been post-annealed at 100 °C, which corresponds to optimal process conditions, compared to cells processed with annealing only of the active layer at 100 °C. The latter exhibit better stability with only 10% losses in PCE after 150 h of constant illumination, while the annealing of the complete device leads to solar cells that degrade much faster with performance losses of 30% of the initial efficiency. Devices with the active layer annealed at 150 °C exhibit the same evolution than devices using annealing of the complete device, while the device using the active layer annealed at 200 °C degrades faster. Importantly, all the devices exhibit very stable Jsc while major losses are caused by losses in FF and Voc (Fig. S17, ESI†). The cause of the faster degradation of 200 °C annealed active layer based solar cells is not clear but it may arise from interfacial reaction with ZnO at high annealing temperature or due to the increased roughness of the active layer which may affect the quality of the subsequent MoO3/Ag electrode and thus device stability.
 | ||
| Fig. 8 PCE as a function of illumination time for encapsulated solar cells based on PM6:ITIC-4F blends annealed at different temperatures. Post-annealing at 100 °C indicates an annealing treatment to the full devices during 10 minutes. Illumination was carried out in a SUNTest where UV light below 400 nm was cut off with an industrial UV filter. | ||
Conclusion
In previous studies, it has been shown that the photostability of ITIC-based OSCs depends on the nature of the side chains and end groups, and changes in the microstructure of the blends have also been identified as strongly influencing the photostability of ITIC-4F-based solar cells.17,37 In this work, we were able to show that the intrinsic photostability of three NFAs from the ITIC family strongly varies between solution, pure material and blend films. In particular, we have annealed blends at temperatures of up to 200 °C, which has never been done before. Thanks to our results, we have obtained a better understanding of the relationships that exist between the molecular structure and the crystalline order to the intrinsic stability of NFAs. We find that the low photostability of the three ITIC derivatives in pure films is comparable whatever the difference between the molecules. However, when these NFA molecules are studied in the presence of a donor polymer, we observe significant differences, especially in the case of ITIC-4F, which exhibits greatly enhanced stability. Our results indicated polymer-induced crystallisation of ITIC-4F within the blend, which was confirmed by several characterisation techniques.We found that the stability of the ITIC derivatives within the blends or in layers of pure material is completely dominated by the crystalline order of the NFA. It has been shown that high temperature annealing (typically above 150 °C) can increase the packing and crystallinity of ITIC derivatives in pure films and blends, resulting in much better photostability of the films.
Our results confirm the observations of Perepichka et al.24 who showed that the photodegradation process of ITIC derivatives is based on a cis–trans isomerisation step of the INCN end groups and a rearrangement. By increasing the molecular order in the acceptor phase, the cis–trans isomerisation of the INCN group is slowed or even inhibited due to strong intermolecular interactions and steric hindrance in the crystalline phase. Given that INCN end groups are used in a wide range of NFAs, including the Y-NFA family, our results indicate the importance of crystal order for the production of highly stable NFA PSCs.
These results reveal that enhancing crystallinity in ITIC derivatives renders these NFA materials more photostable in polymer blends. However, the organic solar cells using more photostable blends are surprisingly less stable under light soaking than those that have faster intrinsic degradation. This result shows that the photostability of organic solar cells is complex and degradation of the devices in our study is not primarily governed by the photostability of the blends but rather driven by other degradation paths. Thus, the intrinsic photostability of active materials is an important key function but not necessarily sufficient to gain stable solar cells.
Experimental
Solutions and films preparation
ITIC, ITIC-4F, ITIC-Th and PBDB-T (PCE12), PBDB-T-2F (PM6) were purchased from 1-Materials, while o-xylene and chlorobenzene were acquired from Sigma-Aldrich. To prepare pure material solutions, a quantity of 10 mg ml−1 for polymer donors and 20 mg ml−1 for acceptors were prepared in chlorobenzene and stirred overnight at room temperature or 60 °C depending on the molecule. For blend solutions, a mixture in a weight ratio of 1:1 was prepared at a total concentration of 20 mg ml−1. For PCE12:ITIC and PCE12:ITIC-Th, the blends were prepared in chlorobenzene and stirred at 60 °C. The PM6:ITIC-4F blend was prepared in o-xylene and stirred at room temperature, while the PCE12:ITIC-4F blend was prepared in chlorobenzene and stirred at room temperature. All blend solutions were stirred overnight. For UV-Vis and FTIR samples, glass and KBr substrates were used, respectively. The solutions were then spin-coated on top of cleaned substrates under an inert atmosphere in a glove box, films were subsequently annealed at 100 °C for 10 min to eliminate residual solvent.
Photodegradation in solution
The solutions for the stability test were prepared inside a nitrogen-filled glove-box using anhydrous and high purity solvents. The total concentration of the solutions was fixed to 0.05 mg ml−1 (5% vv/v from a mother solution of 1 mg ml−1) for all ITIC derivatives. The solutions were then transferred to transparent glass vials and illuminated from the bottom using a solar simulator at 1 Sun intensity AM1.5G light. UV light below 400 nm was cut off with an industrial UV filter placed between the source of light and the solutions to avoid UV-induced degradation of the molecules and solvents. The absorption spectra of solutions were measured before illumination (named 0 h) and at different time periods using a UV-Vis-near infrared Spectrophotometer Cary 5000. For measurements, the solutions were taken out of the glove box to be transferred to a quartz cuvette and measured their UV-Vis spectra, then the solution was again transferred to the glass vial to continue degradation inside the glove box. Here it is important to note that, after each measurement and before continuing the light exposure, the vials were filled again with nitrogen to avoid interactions with oxygen during illumination.Photodegradation in pure material films and polymer blends
All films were placed in a sample holder that allowed measuring the same spot in the film during characterization. Films were sealed under inert conditions in a glass tube under a vacuum. Photodegradation experiments were performed using a SUNTest CPS/XLS Atlas device designed to simulate the AM1.5 spectrum. UV light below 400 nm was cut off with an industrial UV filter. The black standard temperature was set at 60 °C, which corresponds to a chamber temperature of around 35 °C. Irradiation provided by a xenon lamp from Atlas (NXE1700) was set at 1000 W m−2 in the UV-visible domain. Photodegradation experiments were also performed using white LED light from a homemade set-up. The films were characterized before degradation and at different time periods during illumination. UV-visible spectra were obtained using a Shimadzu UV-2600 spectrophotometer equipped with an integrating sphere. The absorbance of the samples was measured and integrated with the 200–1000 nm range. Infrared transmission spectra were recorded using a Thermo Scientific Nicolet 6700 spectrophotometer purged with dry air (32 acquisitions summation with 4 cm−1 resolution). The sample holder used for the films allowed measuring the same spot in the film. An average of 2 to 3 films have been tested.Characterization of organic films
The surface morphology of the blend layers was investigated by AFM (NTEGRA from NT-MIDT) in semi-contact mode using the silicon tips (MikroMash) with a theoretical resonant frequency of 150 kHz and a force constant of 5.4 N m−1 at room temperature. Thin films were further analyzed with high-brightness synchrotron radiation at BL19B2 in SPring-8 (Japan). GIWAXS (Grazing-Incidence Wide-Angle X-ray Scattering) measurements were performed using a high-sensitive 2D X-ray detector (PILATUS 300K). The incident angle and wavelength of X-rays were 0.13° and 0.100 nm, respectively. The crystal coherence length (CCL) values were extracted by the Scherer equation:38 | (1) |
Solar cell fabrication and I–V measurement
PM6:ITIC-4F ink was prepared in o-xylene (96.5% oxylene + 3.5% tetralin) at a total concentration of 20 mg ml−1 in ratio 1:1 and stirred at 50 °C overnight. ITO-coated glass (sheet resistance 10–15 Ω per square) purchased from LUMTEC were sequentially cleaned with deionized water, acetone and isopropanol under sonication for 15 min each, dried with argon and then treated in a UV-ozone oven for 15 min at 80 °C. All the devices were fabricated in inverted structure ITO/ZnO/Active Layer/MoO3/Ag. The znO layer was spin-coated in air from a 1% v/v solution in isopropanol at 5000 rpm followed by thermal annealing at 120 °C for 10 min. The substrates were then transferred to an N2-filled glovebox where a blend film of ∼100 nm thickness was spin-coated at 2200 rpm for 2 min on top of ZnO films. At this stage of solar cell fabrication, thermal annealing was applied at different temperatures (100, 150 or 200 °C) for 10 min. Finally, 5 nm of MoO3 and 100 nm of Ag were deposited by thermal evaporation under a mask to define an active area of 0.27 cm2. The current density–voltage characterization of the devices under the AM1.5G light solar simulator (Newport Sol3A Class AAA) was recorded using a Keithley 238 source meter unit inside the glove box. The illumination intensity of the light source was calibrated to 100 mW cm−2 using a standard silicon solar cell (Newport Company, Oriel no. 94043A) calibrated by the National Renewable Energy Laboratory (NREL). A reference device based on a complete device post-annealed at 100 °C for 10 minutes was realized. Before solar cell stability, the devices were encapsulated using DELO epoxy glue KATIOBOND LP655 and a glass substrate. The glue was cured using AM1.5 light containing UV. After encapsulation, the cells are kept one day inside the glovebox to dry before air exposure. For the stability test, the devices were protected individually with a 400 nm UV filter. The photodegration experiments were performed in a SUNTest XXL+ Atlas where the performances (PCE, Jsc, Voc and FF) were constantly measured using a Keithley 238 Source Measure Unit.
Author contributions
Y. A. A. Q., A. R. and J. A. designed and performed the photo-degradation study. Y. A. A. Q., Q. E. and P. P. prepared pure material films and blend films. Y. A. A. Q., A. R., D. D. and J. A. analyzed the absorption data. Y. A. A. Q. prepared and characterized the solar cells. T. K. and N. Y. have performed the acquisition of GIWAXS patterns; N. Y. and C. V. A. have analyzed the data. W. K. and R. R. S. have performed and analyzed the ATEM data. A. K. B. and C. M. R. have performed and analyzed the IR data. C. V. A., J. A. and O. M. supervised the work. R. D., C. A., C. V. A. and J. A. provided an interpretation of experimental data. R. D., C. V. A. and J. A. wrote the final manuscript including the contributions of all coauthors. All the authors contributed to the preparation of the manuscript and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project received funding from the French National Research Agency (ANR) under projects ANR-17-CE05-0020-01 named NFA-15 and ANR-18-CE04-0007-04 named BELUGA. The synchrotron radiation experiments were performed at BL19B2 in SPring-8 with the approval of Japan Synchrotron Radiation Research Institute (JASRI) (Proposal Nos. 2021B1947, 2022A2069 and 2022A2063).Notes and references
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 1–8 Search PubMed.
- J. Fu, P. W. K. Fong, H. Liu, C.-S. Huang, X. Lu, S. Lu, M. Abdelsamie, T. Kodalle, C. M. Sutter-Fella, Y. Yang and G. Li, Nat. Commun., 2023, 14, 1760 CrossRef CAS PubMed.
- H. K. H. Lee, J. Wu, J. Barbé, S. M. Jain, S. Wood, E. M. Speller, Z. Li, F. A. Castro, J. R. Durrant and W. C. Tsoi, J. Mater. Chem. A, 2018, 6, 5618–5626 RSC.
- T. H. Kim, N. W. Park, M. A. Saeed, S. Y. Jeong, H. Y. Woo, J. Park and J. W. Shim, Nano Energy, 2023, 112, 108429 CrossRef CAS.
- C. Lee, J.-H. Lee, H. H. Lee, M. Nam and D.-H. Ko, Adv. Energy Mater., 2022, 12, 2200275 CrossRef CAS.
- Y. Li, G. Xu, C. Cui and Y. Li, Adv. Energy Mater., 2018, 8, 1701791 CrossRef.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Q.-Q. Pan, S.-B. Li, Y. Wu, Y. Geng, M. Zhang and Z.-M. Su, Org. Electron., 2018, 53, 308–314 CrossRef CAS.
- A. Karki, A. J. Gillett, R. H. Friend and T.-Q. Nguyen, Adv. Energy Mater., 2021, 11, 2003441 CrossRef CAS.
- Y. Firdaus, V. M. Le Corre, J. I. Khan, Z. Kan, F. Laquai, P. M. Beaujuge and T. D. Anthopoulos, Adv. Sci., 2019, 6, 1802028 CrossRef PubMed.
- S. H. K. Paleti, S. Hultmark, J. Han, Y. Wen, H. Xu, S. Chen, E. Järsvall, I. Jalan, D. R. Villalva, A. Sharma, J. I. Khan, E. Moons, R. Li, L. Yu, J. Gorenflot, F. Laquai, C. Müller and D. Baran, Nat. Commun., 2023, 14, 4608 CrossRef CAS PubMed.
- R. Meitzner, U. S. Schubert and H. Hoppe, Adv. Energy Mater., 2021, 11, 2002551 CrossRef CAS.
- E. Y. Ko, G. E. Park, J. H. Lee, H. J. Kim, D. H. Lee, H. Ahn, M. A. Uddin, H. Y. Woo, M. J. Cho and D. H. Choi, ACS Appl. Mater. Interfaces, 2017, 9, 8838–8847 CrossRef CAS PubMed.
- Y. Wang, J. Luke, A. Privitera, N. Rolland, C. Labanti, G. Londi, V. Lemaur, D. T. W. Toolan, A. J. Sneyd, S. Jeong, D. Qian, Y. Olivier, L. Sorace, J.-S. Kim, D. Beljonne, Z. Li and A. J. Gillett, Joule, 2023, 7, 810–829 CrossRef CAS.
- N. Y. Doumon, M. V. Dryzhov, F. V. Houard, V. M. Le Corre, A. Rahimi Chatri, P. Christodoulis and L. J. A. Koster, ACS Appl. Mater. Interfaces, 2019, 11, 8310–8318 CrossRef CAS PubMed.
- X. Du, T. Heumueller, W. Gruber, A. Classen, T. Unruh, N. Li and C. J. Brabec, Joule, 2019, 3, 215–226 CrossRef CAS.
- X. Du, T. Heumueller, W. Gruber, O. Almora, A. Classen, J. Qu, F. He, T. Unruh, N. Li and C. J. Brabec, Adv. Mater., 2020, 32, 1908305 CrossRef CAS PubMed.
- S. Park and H. J. Son, J. Mater. Chem. A, 2019, 7, 25830–25837 RSC.
- A. Classen, T. Heumueller, I. Wabra, J. Gerner, Y. He, L. Einsiedler, N. Li, G. J. Matt, A. Osvet, X. Du, A. Hirsch and C. J. Brabec, Adv. Energy Mater., 2019, 9, 1902124 CrossRef CAS.
- L. Duan, H. Yi, Y. Zhang, F. Haque, C. Xu and A. Uddin, Sustainable Energy Fuels, 2019, 3, 723–735 RSC.
- J. Luke, E. M. Speller, A. Wadsworth, M. F. Wyatt, S. Dimitrov, H. K. H. Lee, Z. Li, W. C. Tsoi, I. McCulloch, D. Bagnis, J. R. Durrant and J.-S. Kim, Adv. Energy Mater., 2019, 9, 1803755 CrossRef.
- A. J. Clarke, J. Luke, R. Meitzner, J. Wu, Y. Wang, H. K. H. Lee, E. M. Speller, H. Bristow, H. Cha, M. J. Newman, K. Hooper, A. Evans, F. Gao, H. Hoppe, I. McCulloch, U. S. Schubert, T. M. Watson, J. R. Durrant, W. C. Tsoi, J.-S. Kim and Z. Li, Cell Rep. Phys. Sci., 2021, 2, 100498 CrossRef CAS.
- Y. Che, M. R. Niazi, R. Izquierdo and D. F. Perepichka, Angew. Chem., Int. Ed., 2021, 60, 24833–24837 CrossRef CAS PubMed.
- L. Ciammaruchi, O. Zapata-Arteaga, E. Gutiérrez-Fernández, J. Martin and M. Campoy-Quiles, Mater. Adv., 2020, 1, 2846–2861 RSC.
- Y. Che, M. R. Niazi, Q. Chan, P. Ghamazi, T. Yu, C. Ruchlin, H. Yu, H. Yan, D. Ma, S. S. Xiao, R. Izquierdo and D. F. Perepichka, Angew. Chem., Int. Ed., 2023, 62, e202309003 CrossRef CAS PubMed.
- M. Jung, Y. Yoon, J. H. Park, W. Cha, A. Kim, J. Kang, S. Gautam, D. Seo, J. H. Cho, H. Kim, J. Y. Choi, K. H. Chae, K. Kwak, H. J. Son, M. J. Ko, H. Kim, D.-K. Lee, J. Y. Kim, D. H. Choi and B. Kim, ACS Nano, 2014, 8, 5988–6003 CrossRef CAS PubMed.
- H. Cha and J. Wu, Joule, 2021, 5, 1322–1325 CrossRef.
- A. Gonzalez, E. S. Kengmana, M. V. Fonseca and G. G. D. Han, Mater. Today Adv., 2020, 6, 100058 CrossRef.
- Q. Eynaud, Y. A. A. Quiroz, T. Koganezawa, R. Sato, N. Yoshimoto, O. Margeat, C. M. Ruiz, J. Ackermann and C. Videlot-Ackermann, J. Mater. Chem. C, 2023, 11, 9657–9669 RSC.
- W. Köntges, P. Perkhun, J. Kammerer, R. Alkarsifi, U. Würfel, O. Margeat, C. Videlot-Ackermann, J.-J. Simon, R. R. Schröder, J. Ackermann and M. Pfannmöller, Energy Environ. Sci., 2020, 13, 1259–1268 RSC.
- P. Perkhun, W. Köntges, F. Pourcin, D. Esteoulle, E. Barulina, N. Yoshimoto, P. Pierron, O. Margeat, C. Videlot-Ackermann, A. K. Bharwal, D. Duché, C. R. Herrero, C. Gonzales, A. Guerrero, J. Bisquert, R. R. Schröder, M. Pfannmöller, S. Ben Dkhil, J.-J. Simon and J. Ackermann, Adv. Energy Sustainability Res., 2021, 2, 2000086 CrossRef CAS.
- J. Xin, X. Meng, X. Xu, Q. Zhu, H. B. Naveed and W. Ma, Matter, 2019, 1, 1316–1330 CrossRef.
- L. Zhang, H. Zhao, B. Lin, J. Yuan, X. Xu, J. Wu, K. Zhou, X. Guo, M. Zhang and W. Ma, J. Mater. Chem. A, 2019, 7, 22265–22273 RSC.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- Y. A. Avalos-Quiroz, T. Koganezawa, P. Perkhun, E. Barulina, C. M. Ruiz, J. Ackermann, N. Yoshimoto and C. Videlot-Ackermann, Adv. Electon. Mater., 2022, 8, 2100743 CrossRef CAS.
- N. Y. Doumon, F. V. Houard, J. Dong, H. Yao, G. Portale, J. Hou and L. J. A. Koster, Org. Electron., 2019, 69, 255–262 CrossRef CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc04614a |
| This journal is © The Royal Society of Chemistry 2024 |