
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Is a small singlet–triplet energy gap a guarantee of TADF performance in MR-TADF compounds? Impact of the triplet manifold energy splitting†
Rasa
Keruckiene
 a,
Aliaksei A.
Vaitusionak
b,
Maksim I.
Hulnik
b,
Ivan A.
Berezianko
b,
Dalius
Gudeika
a,
Simas
Macionis
a,
Malek
Mahmoudi
a,
Dmytro
Volyniuk
a,
Danillo
Valverde
c,
Yoann
Olivier
*c,
Kai Lin
Woon
d,
Sergei V.
Kostjuk
be,
Sebastian
Reineke
f,
Juozas V.
Grazulevicius
*a and
Gjergji
Sini
*g
a,
Aliaksei A.
Vaitusionak
b,
Maksim I.
Hulnik
b,
Ivan A.
Berezianko
b,
Dalius
Gudeika
a,
Simas
Macionis
a,
Malek
Mahmoudi
a,
Dmytro
Volyniuk
a,
Danillo
Valverde
c,
Yoann
Olivier
*c,
Kai Lin
Woon
d,
Sergei V.
Kostjuk
be,
Sebastian
Reineke
f,
Juozas V.
Grazulevicius
*a and
Gjergji
Sini
*g
aDepartment of Polymer Chemistry and Technology, Kaunas University of Technology, K. Barsausko st. 59, LT-50254, Kaunas, Lithuania. E-mail: juozas.grazulevicius@ktu.lt
bResearch Institute for Physical Chemical Problems of the Belarusian State University, 14, Leningradskaya st, 220030 Minsk, Belarus
cLaboratory for Computational Modeling of Functional Materials, Namur Institute of Structured Matter, University of Namur, Rue de Bruxelles, 61, B-5000 Namur, Belgium
dLow Dimensional Material Research Centre, Department of Physics, University Malaya, Kuala Lumpur, Malaysia
eDepartment of Chemistry, Belarusian State University, Leningradskaya st. 14, 220006, Minsk, Belarus
fDresden Integrated Center for Applied Physics and Photonic Materials (IAPP), Technische Universität Dresden, Dresden 01062, Germany
gLaboratoire de Physicochimie des Polymères et des Interfaces, EA 2528, CY Cergy Paris Université, 5 mail Gay-Lussac, 95031 Cergy-Pontoise Cedex, France
First published on 16th February 2024
Abstract
Aiming at obtaining pure-color emitters for organic light-emitting diodes (OLEDs), we designed and synthesized a new organic boron-containing indolocarbazole derivative as a candidate for multiple resonance-induced thermally activated delayed fluorescence (MR-TADF) emitters. The efficiency of the targeted MR and TADF characteristics are analyzed by steady-state and time-resolved spectroscopy at different temperatures, and by different theoretical methods. The molecule exhibits green-yellow photoluminescence and electroluminescence with a full-width at half-maximum (FWHM) of 20 nm and 23 nm for thin films observed from photoluminescence (PL) and electroluminescence spectra. The PL intensity increases by up to 100% for the degassed sonicated toluene solutions; however, no TADF behavior is observed. The increase in PL is predominantly due to the decrease of the singlet quenching by the dissolved oxygen and by the remaining microaggregates in solution, as opposed to the frequently reported quenching of triplet excitons by oxygen. The absence of TADF behavior in toluene solution is attributed to very small spin–orbit coupling between S1 and T1 (0.02 cm−1) and a large T1–T2 gap that inhibits the T1 → T2 upconversion of triplets which likely results in a very low rate of intersystem crossing as compared to the radiative decay rate. However, a weak TADF is observed in guest–host mixtures, e.g., in thin films made with mCP, DPEPO, and Zeonex hosts. This is attributed to the reduced T1–T2 gap, resulting from the conformational distortions of the emitter in the guest–host systems. Even with 100% photoluminescence quantum yields of the toluene solution, the efficiency of the first prototypical OLEDs is limited. Different sources for the low device external quantum efficiency are discussed. Ultimately, our structure–property analysis suggests a design rule allowing tuning of the T1–T2 gap for more efficient TADF.
1. Introduction
The field of thermally activated delayed fluorescence (TADF) has witnessed remarkable advancements, thanks to the synthesis of various TADF molecules incorporating diverse electron and donor moieties within a donor–acceptor (D–A) architecture.1–11 Achieving high efficiency with these molecules necessitates an extremely low exchange energy, leading to a near-zero energy splitting between the first singlet- and triplet excited states (ΔEST) of less than 0.03 eV.12–14 This is one of the crucial parameters that facilitate the induction of reverse intersystem crossing (RISC), a process made possible through the vibronic coupling with torsion mode around the D–A bond.15,16However, the flexibility inherent in the D–A bond, while essential, gives rise to a challenge. This architectural characteristic introduces conformational disorder, significantly broadening the emission spectra and compromising the desired color purity—a stringent requirement for display applications, and efforts to circumvent the shortcomings were met with limited success.17–19 The discovery of multi-resonance TADF (MR-TADF) by Hatakeyama et al. has ushered in a paradigm shift in TADF design.20 By employing a rigid polycyclic aromatic framework, MR-TADF exhibits a narrow emission bandwidth coupled with high oscillator strength compared to conventional D–A TADF architecture, together with a small ΔEST. The small ΔEST is achieved by effectively spatially separating the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) distributions on adjacent neighboring atomic sites thanks to opposite resonant effects while maintaining sufficient overlap to guarantee a high oscillator strength.21–24
First MR-TADF materials (DABNA-1 and DABNA-2) were reported in 2016 by Hatakeyama et al.25 These compounds contain a central boron atom and para-disposed nitrogen atoms that achieve the desired alternating pattern of the electron density distribution. Devices fabricated from DABNA-1 and DABNA-2 showed a maximum EQE of 13.5% and 20.2% with CIE coordinates of (0.13, 0.09) and (0.12, 0.13), respectively. The MR based design approach provided sharp EL spectra with FWHMs of 30 and 28 nm for DABNA-1 and DABNA-2, respectively. However, the low rate of RISC resulted in a severe efficiency roll-off. The MR effect in DABNA-1 was further enhanced by changing its structure either by attaching nitrogen phenyl rings with t-butyl groups26 or by further addition of tert-butyl carbazole27 and di(tert-butylphenyl)amine28 moieties at the para-position of the boron atom. Thus, all TADF properties of the DABNA-1 derivative, TBN-TPA, were significantly improved, resulting in a high EQE of 32.1% and FWHM of 27 nm, except for the 15 nm red-shift of the EL spectrum.27 By fusing two DABNA-1 molecules, ν-DABNA was designed, and it was reported to exhibit a quite sharp EL peak (FWHM = 18 nm) at 469 nm and a high EQE of 34.4%.29 Duan et al.30 developed a new strategy of extending the conjugation of the DABNA-1 core by simply replacing the diphenylamine unit with tert-butylcarbazole (BCz-BN). Thus, the first series of green MR-TADF emitters with EQEs of up to 22.4% and FWHM of up to 29 nm was reported. By introduction of four extra tert-butylphenyl groups on the carbazole moieties Wang et al.31 expanded the conjugation skeleton and therefore reported highly efficient (EQE of 25.5%), stable (high EQE of 20.1% was maintained at a practical high luminance of 1000 cd m−2), narrowband (FWHM of 33 nm) and pure green emitters with CIE coordinates of (0.20, 0.65). Subsequently, a large number of studies have been carried out, aiming to extend the conjugation of the DABNA-1 core and to improve device performance, which resulted in synthesis of blue,32,33 green,30,32,34–37 yellow34,36 and orange-red35 MR-TADF emitters by introducing into the DABNA-1 structure a variety of electron-deficient and electron-rich groups. The main tendency observed is that electron-deficient moieties in DABNA (e.g., CN and F) lead to a bathochromic shift of its emission spectra: a high performance OLED device with an outstanding EQE as high as 33.7% and orange-red luminescence was fabricated by introducing cyano substituents.36
Despite the large number of efforts oriented towards the design and synthesis of MR-TADF emitters, clear design rules for new compounds are still missing but urgently needed. While the detailed RISC mechanism is not yet elucidated in MR-TADF, a small ΔEST between S1 and T1 is often seen as a prerequisite for TADF to take place. If the direct RISC occurs from the first triplet excited state (T1) to the first singlet excited state (S1) as proposed by Lin et al.,38 a decrease in ΔEST is expected to lower the activation energy for RISC to take place. Pershin et al.,39 through computational modeling, have demonstrated that ΔEST can be reduced by increasing the π-conjugation of MR-TADF and thus by increasing the excited state electronic density delocalization. However, several studies have shown that S1 and T1 states in these MR-TADF compounds are of the same nature, which, by virtue of El Sayed's rule, limits a direct transition of triplet excitons from T1 to S1. In this context, it has been shown40–44 that internal upconversion from T1 to some higher but low-lying triplet states from which RISC takes place is an alternative path for the TADF mechanism to be efficient. Accordingly, the efficiency of the TADF contribution to the triplet harvesting of the MR-TADF compounds in solution depends on this T1–Tn energy gap, and the S1(Sn)–Tn spin–orbit couplings (SOC). Designing MR-TADF compounds by solely targeting the smallest ΔES1–T1 may then be insufficient, thus rendering important the presence of a dense manifold of triplet excited states up to the S1 energy.
Here, we address focus on both small ΔES1T1 and small T1–Tn (n > 1) energy splitting. To this aim, we have synthesized a boron-based indolocarbazole derivative, an elongated MR-TADF molecule. This compound has a significantly reduced ΔES1T1 but a large T1–T2 energy splitting, resulting in inefficient triplet harvesting. The analysis of the reasons for this large T1–T2 energy splitting allowed us to suggest a new design rule helping to avoid the problem.
2. Results and discussion
2.1 Synthesis
Boron-based compound DG7 was successfully synthesized using a three step synthetic route starting from commercially available 5,11-dihydro-indolo[2,3-b]carbazole, as presented in Scheme 1. In the first two steps, monosubstituted indolocarbazole (dg7-2) was obtained via an Ullmann amination reaction with iodobenzene followed by nucleophilic substitution catalyzed by potassium tert-butoxide (t-BuOK) affording dg7-1. The final target molecule DG7 was synthesized by a one-pot reaction via a lithium–halogen exchange reaction with n-butyllithium, electrophilic trapping with boron tribromide, and high-temperature tandem electrophilic arene borylation in the presence of diisopropylethylamine. The structure of the target compound was confirmed by X-ray single crystal analysis and 1H NMR spectroscopy (see Experimental in the ESI†).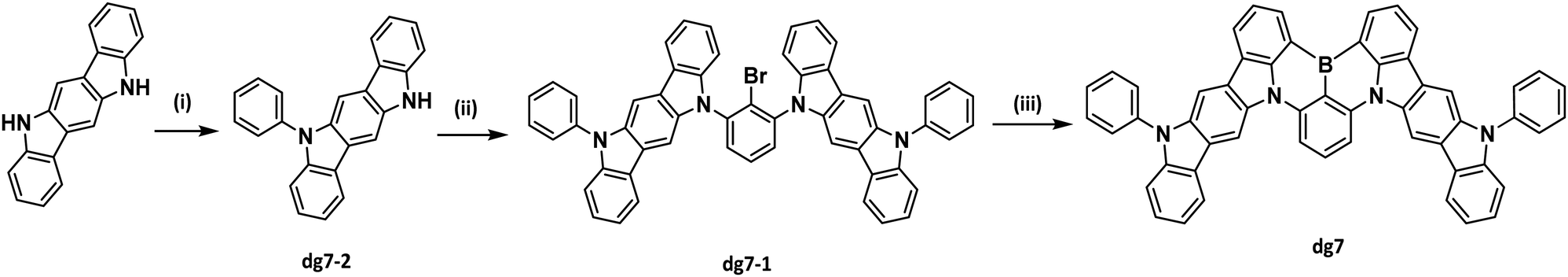 | ||
| Scheme 1 Synthetic route to boron-based compound DG7: (i) iodobenzene, CuI, 1,10-phenanthroline, K2CO3, 18-crown-6, DMF, 20 h at 140 °C, (ii) 1-bromo-2,6-difluorobenzene, t-BuOK, DMF, 48 h at 100 °C, (iii) n-BuLi, tert-butylbenzene, 4 h at r.t.; BBr3, 12 h at r.t., N,N-diisopropylethylamine, 24 h at 170 °C. | ||
 | ||
| Fig. 1 TGA (a), DSC (b) and photoelectron emission (c) curves of compound DG7. | ||
| Property | Sample | DG7 |
|---|---|---|
| a Taken from the 2nd heating scan of DSC measurement. b 5% weight loss, taken from TGA measurement. c Obtained by CELIV measurements at an electric field of 2 × 104 V cm−1. | ||
| T m,a °C | Powder | 357 |
| T g,a °C | 195 | |
| T cr,a °C | 298 | |
| T de-5%,b °C | 517 | |
E
ox![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) vs.Fc/Fc+, V vs.Fc/Fc+, V |
DCM solution with TBAPF6 | 0.75 |
|
E
redvs.Fc/Fc+, V |
−1.47 | |
| IPCV, eV | 5.85 | |
| EACV, eV | 3.63 | |
| IPPE, eV | Film | 5.9 |
| EAPE, eV | 3.7 | |
| μ h,c cm2 (V s)−1 | 3.4 × 10−6 | |
The electrochemical properties of DG7 were investigated by cyclic voltammetry (CV). The voltammograms are presented in Fig. S1a (ESI†), and the derived data are presented in Table 1. DG7 showed quasi-reversible oxidation and reduction in the potential range from −1.8 to 1.8 V. By utilizing the potential oxidation (Eoxvs.Fc/Fc+) of DG7, the quite high ionization potential value (IPCV) of 5.58 eV of the sample implies a not too strong electron donating characteristic of the sample. Nevertheless, the determined electron affinity suggests a well-balanced oxidation–reduction process upon electrical induction of the sample. By utilizing the potential reduction (Eredvs.Fc/Fc+) of DG7, we obtained an electron affinity (EACV) value of 3.63 eV (Table 1).
To investigate the charge-injecting capabilities of DG7, we conducted photoelectron emission (PE) measurements in an air environment. Using data from solid films of DG7, an ionization potential (IPPE) of 5.9 eV and an electron affinity (EAPE) of 3.7 eV were obtained (Table 1) based on PE and absorption spectra. The energy levels of DG7 from CV and PE measurements agree well. The charge-transporting ability of DG7 was investigated using time-of-flight (TOF) and charge extraction by linearly increasing voltage (CELIV) methods (Fig. S1b–d, ESI†). The vacuum-deposited films of DG7 exhibited hole-transporting properties with a hole mobility (μh) of 3.4 × 10−6 cm2 (V s)−1 under a low electric field of 2 × 104 V cm−1 (Table 1). The electroluminescent properties of DG7 were tested using charge-injecting and charge-transporting data (refer to the last section).
2.2 Geometrical and electronic structure characteristics
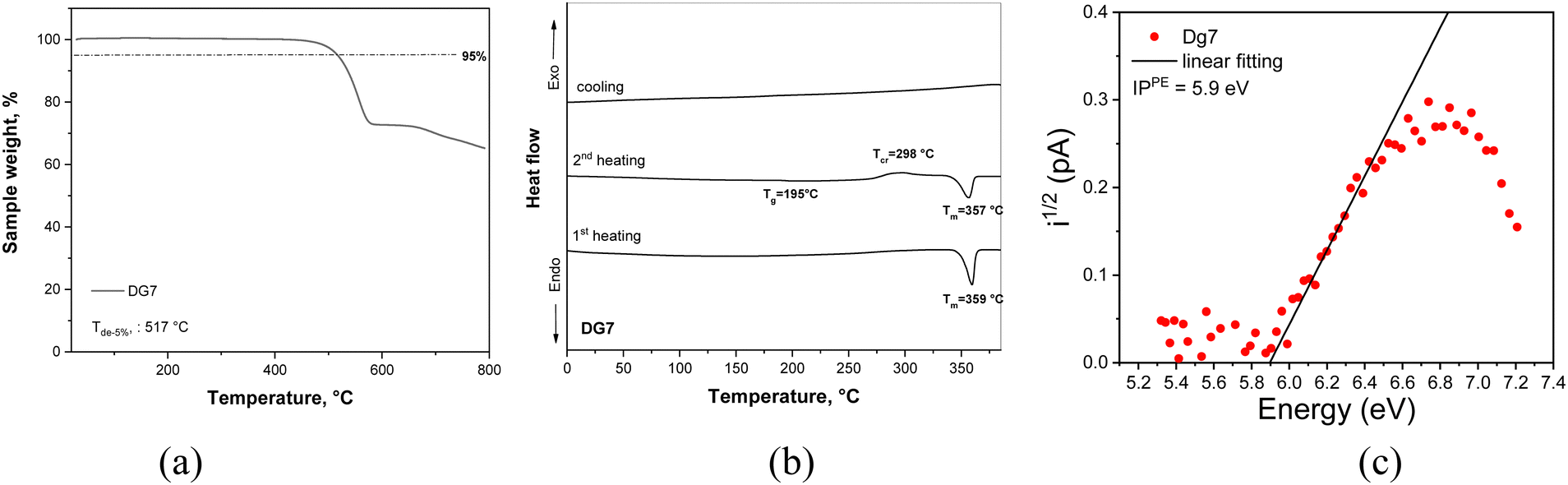
The ground-state optimized geometry of DG7 and some selected molecular orbital (MO) pictures are presented in Fig. 2. The ground-state molecular structure is almost planar, yet the steric hindrance between the closely lying peripheral benzene rings induces some helicoidal-wise deformation of the molecular plane, with a pitch of roughly 19° between their plans, see Fig. 2a. The HOMO and LUMO are both delocalized on the central part of the molecule including contribution from the carbazole inner moieties of the indolocarbazole fragments (Fig. 2b), whereas HOMO−1 and LUMO+1 are localized on the indolocarbazole fragments with no contribution from the boron atom.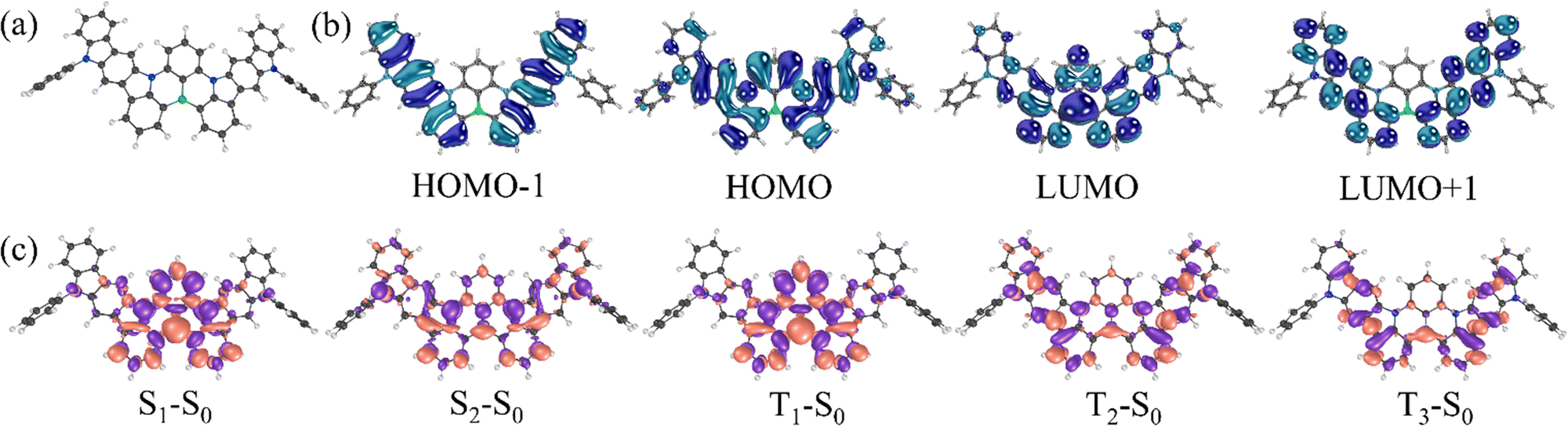 | ||
| Fig. 2 (a) Ground-state molecular structure of DG7 optimized at the PBE0/6-31G(d,p) level. (b) Shape of some selected MOs of DG7, and (c) the difference density plot between the ground-state and the selected excited state (singlet or triplet). | ||
For the sake of better understanding the excited state properties of DG7, we computed vertical excitation energies using the spin-component scaling second-order approximate coupled-cluster (SCS-CC2) together with the cc-pVDZ basis set. This level of theory has been shown to accurately describe the excited states of MR-TADF compounds.46 Our results indicate that the S1 state is predominantly described by a HOMO to LUMO transition (80% weight) with a high oscillator strength (f = 0.701). A similar electronic nature is obtained for the T1 state, as can be seen in the difference-density plot (Fig. 3c). Because of the similar nature of S1 and T1 as testified by their nearly identical electronic density distributions, the SOC value is quite small (0.02 cm−1), in line with the El-Sayed rule. Interestingly, both S1 and T1 states exhibit a short-range CT (SRCT) character, which is an intrinsic feature of MR-TADF compounds. The singlet–triplet energy gap between the S1 and T1 states is predicted to be 0.09 eV, in good agreement with the experimental data (0.19 eV, vide infra). The second singlet state lies 0.48 eV above the S1 states and has almost null oscillator strength (f = 0.001) (see Table S1, ESI†). The T2 and T3 triplet states can be potentially intermediate states for the RISC, since they are nearly degenerate and positioned merely 0.22 eV above the S1 state (see Table S1, ESI†). Additionally, the SOC value between S1 and T3, and between S2 and T2 are considerably higher than that between S1 and T1 (0.50 cm−1 for S2 → T2, and 0.44 cm−1 for S1 → T3) because of the very different electronic-density localization of both T2 and T3 states in comparison to S1.
 | ||
| Fig. 3 (a) UV-vis and (b) photoluminescence spectra of toluene or THF solutions and neat film of DG7. (a) The green-vertical bars (oscillator strengths) correspond to the theoretical absorption spectrum calculated at the hole–hole Tamm–Dancoff approximated density functional theory (hh-tda) using the B3LYP functional with the def2-svp basis set.53 The theoretical excited state energies are blue-shifted by 0.48 eV in order to fit the energy of S1 with the maximum of the experimental low-energy band. (c) and (d) Theoretical absorption and emission spectra obtained at the SCS-CC2/cc-pVDZ level of theory. The simulated spectrum in panel (d) is redshifted by 93 nm to fit with the experimental peak. (d) PL decay curve of the mCP-doped film of DG7. (e) and (f) Emission scans of DG7 in toluene (10−6) from 500 to 700 nm (e) under excitation at 350 nm and (f) under excitation at 530 nm, for 5 different conditions of solution. | ||
Excited-state geometry optimizations of the first lowest singlet and triplet states were carried out at the TD-PBE0/6-31G(d,p) level. Both optimized structures resemble the ground-state minimum structure, as can be seen by superimposing the ground- and excited-state structures (see Fig. S2, ESI†). Due to the very similar excited-state structures for both S1 and T1 the adiabatic ΔEST is close to the one obtained from vertical excitation from the ground state (see Table S2, ESI†).
2.3 Photophysical properties in toluene solution and neat film
The photophysical properties of dilute toluene, tetrahydrofuran (THF) (concentration of 10−5 M) and also neat films of the target compound were studied. UV-vis absorption and steady state PL spectra of the compound are presented in Fig. 3 and the selected spectral data are summarized in Table 1.The absorption spectrum of the THF solution of the compound showed four bands with maxima at 522, 387, 321, and 305 nm, while the solid sample UV spectrum exhibited maxima at 532, 392, 325, and 309 nm which proves a similar absorption profile with a small redshift for neat film in comparison to solution of the compound. As depicted in Fig. 3a, compound DG7 exhibits an intense and sharp absorption band around 530 nm. The longer maximum wavelength as compared to other MR-TADF compounds like DABNA-1 is due to the more extended π-conjugation via the fused indolocarbazole moieties, whereas the sharpness of the absorption band is due to the rigid molecular skeleton hosting the multi resonance-induced charge transfer transition.47
Emission spectra of the DG7 emitter excited at 350 nm show a small, red-shifted maximum upon increasing the solvent polarities from toluene to THF, confirming the SRCT nature of the S1 state (Fig. 3b).48 The green-yellow emission of the solutions of DG7 is still rare and was reported for MR-TADF only once before.49,50 These emission characteristics make DG7 a perfect candidate for hyperfluorescent based OLEDs. The Stokes shifts of diluted toluene solution and neat film PL spectra of DG7 are only 21 nm (0.09 eV) and 34 nm (0.14 eV) with respect to their absorption maxima, indicating that its molecular relaxation is highly restricted. This very small Stokes shift is similar to the ones observed in other MR-TADF materials51,52 and finds support from the theoretical calculations (vide supra).
The vibronically resolved absorption and emission spectra of DG7 were emulated (Fig. 3c) within the displaced undistorted harmonic oscillator model in conjunction with the thermal vibration correlation functional formalism, as implemented in MOMAP/2022A (see the ESI† for further computational details) within the Franck–Condon approximation. Absorption and emission theoretical spectra are shown in Fig. 3a, c and d. Fig. 3a indicates that the lowest-energy absorption band is due to only the 1st excited state (S1), whereas the band peaking around 400 nm includes contributions from the closely lying S2–S4 excited states. A small FWHM value of 29.4 nm is obtained at the theoretical level, as compared to the experimental value of 17.6 nm in toluene solution (Fig. 3c and d). This small broadening is essentially modulated by a low vibrational normal mode (frequency of 25 cm−1 and Huang–Rhys factor of 2.3) characterized by a collective twisting motion of the hydrogen atoms of the phenyl groups (see Fig. S3, ESI†). In Fig. 3d, the simulated emission spectrum is redshifted in relation to the experimental data in toluene in order to match the main experimental peak. The experimental low energy weak emission peak is well reproduced by the theoretical calculations, indicating contribution from the 0–1 vibrational replica in addition to the 0–0 one. According to our theoretical results, the Stokes shift, computed from the absorption and emission peaks, is 7 nm (0.05 eV), confirming the small reorganization energy of the S1 state (see Fig. S4, ESI†).
It is worth noting that possible contribution from the excimer emission in toluene solutions has been previously reported falling in the same region as the theoretical 0–1 vibrational replica of the emission band.54 Interestingly, the sonication of the toluene solutions at different concentrations of DG7 indicates an important increase in the emission intensity (Fig. 3e and f), witnessing the presence of microaggregates and the corresponding aggregation caused quenching (ACQ) in solution. Nevertheless, when varying the concentration from 10−4–10−6 mol L−1, the intensity ratio between the main and the weak-energy peaks remains unchanged (IM/IW = 7, Fig. S5, ESI†), suggesting the absence of excimer emission independently of the presence of microaggregates in toluene solution. This conclusion seems to find support from the time-resolved emission spectra (TRES) measurements: the PL spectra of toluene solutions of DG7 are practically the same at different excitation wavelengths and different delay times after excitation (Fig. 4a and Fig. S6 and S7, ESI†), thus excluding the presence of different emission species in solution. Upon photoexcitation, air-equilibrated and degassed dilute toluene solution of DG7 exhibits the same decent yellowish-green emission with an emission peak maximum λPL at 552 nm and a FWHM of 20 nm (0.09 eV) (Fig. S5, ESI†). Using the integrating sphere, PLQY values of diluted toluene solution of DG7 were measured (Table 2), approaching 100% in the degassed/sonicated toluene solution. When air is present in the dilute toluene solution, the emission intensity of DG7 decreases and the PLQY is reduced to 57%. Because the excitation wavelength of 530 nm is not enough to populate states above S1, and that the S1 radiative decay rate is high and the ISC S1 → T1 is practically El Sayed forbidden, we conclude that the triplets in this case are absent, and that both the sonication and N2 bubbling only help eliminate PL quenching of singlet states by destroying the remaining micro-aggregates of DG7. In the case of an excitation wavelength of 350 nm, only slightly lower PLQY values of 96 and 46% were obtained for the degassed/sonicated and air-containing toluene solutions, respectively (Table 2). Given the similarly important impacts of the sonication on the emission intensities shown in Fig. 3e and f, and the potentially similar type of effects that bubbling the toluene solution with N2 could have on the emission intensity, we conclude that the impact of oxygen on the triplet excitons as shown in Fig. 3e (excitation at 350 nm) is also very small (see Annex I, ESI†).
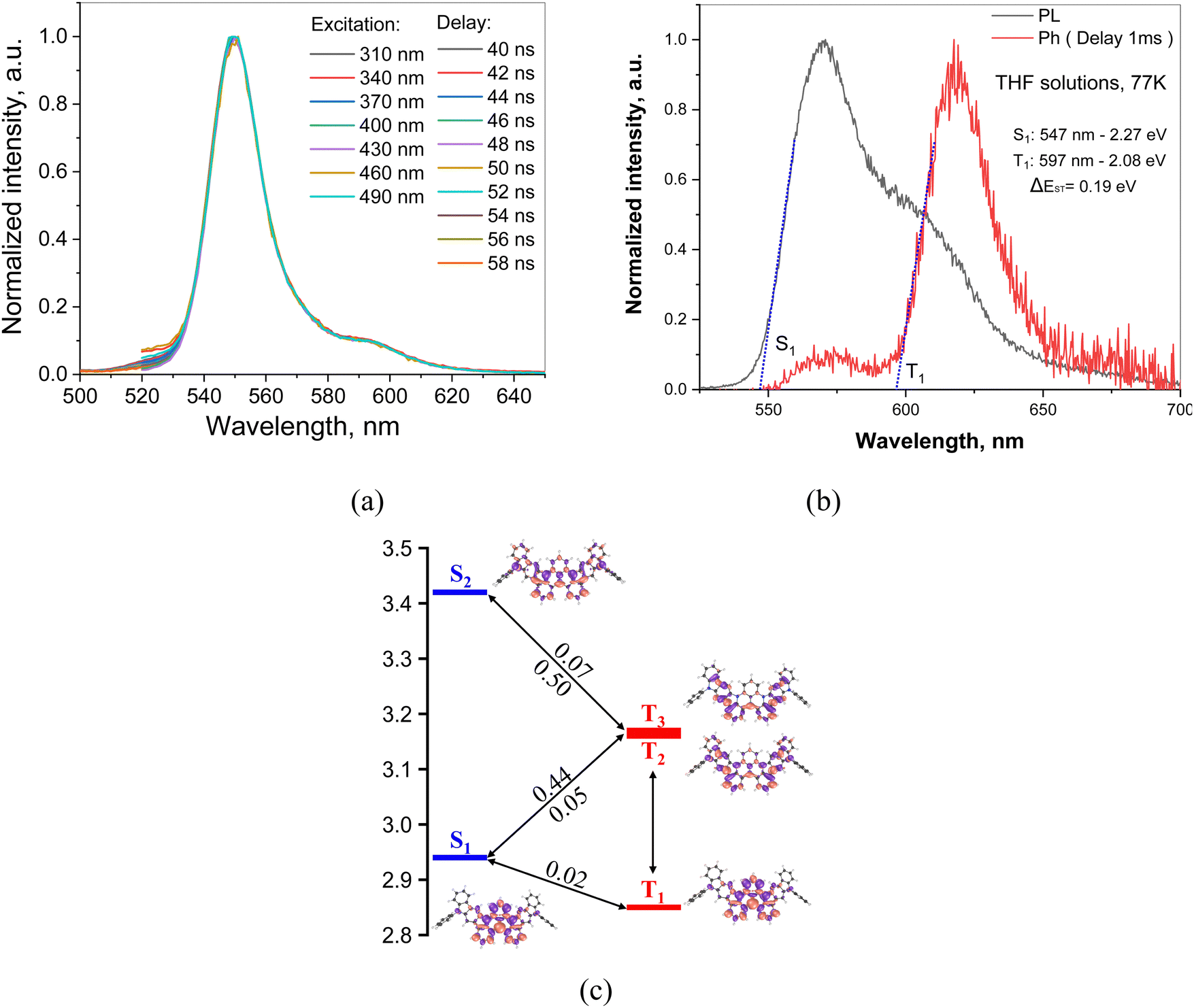 | ||
| Fig. 4 (a) PL spectra at different excitation and delay times after excitation of DG7 in toluene (10−6 M). The PL spectra were plotted from PL decay curves of time-resolved emission spectra (TRES). (b) Fluorescence and phosphorescence spectra of DG7 in THF solution at 77 K (λex = 330 nm). (c) SCS-CC2/cc-PVDZ energy diagram for the low-lying singlet and triplet excited states. The density difference plots with respect to the ground state are also shown for each excited state. A selection of spin–orbit couplings (SOC) between singlet and triplet states is also shown. * In the figure are shown the results obtained at the wB97XD/6-311G(d,p) level. | ||
| Media | Toluene (10−5 M−1) at λex 530/350d nm | THF (10−5 M−1) at 296/77 K | Zeonex (1 wt%) | mCP (5/8 wt%) | TPBi (5/8 wt) | Neat film |
|---|---|---|---|---|---|---|
| a Wavelength of the peak of PL spectrum. b Full-width at half-maximum. c Photoluminescence quantum yield determined using an integrating sphere. d Excitation wavelength. e Values obtained for air-containing toluene solution of DG7. f Obtained in air, recalculated for vacuum. | ||||||
| λ PL,a nm | 552 | 569/570 | 550 | 564/563 | 567/566 | 568 |
| FWHM,b nm | 20 | 51/50 | 30 | 34/34 | 55/34 | 30 |
| PLQY,cf % | 100 (57e)/96 (46e) | — | 68 | 19/12 | 13/11 | 4 |
The absence of contribution from the triplet excitons in the PL emission spectra in toluene solutions is intriguing. Additionally, the experimental results indicate absence of delayed FL in toluene solutions or in the solid film, despite the relatively small ΔE(S1–T1) (0.08–0.09 eV and 0.19 eV obtained theoretically and experimentally, respectively), see Fig. 4b. This characteristic of DG7 is similar to those of other MR-TADF compounds with missing TADF behavior in their dilute solutions, and can be explained by (i) the fast radiative decay timescale of the S1 state and (ii) the very small SOC of 0.02 cm−1 that is found between S1 and T1 (Fig. 4c), in turn being easily explained in the frame of El Sayed's rule:55 S1 and T1 have very similar nature, as can be seen from the density difference plots, see Fig. 4d, making the spin–flip between them practically forbidden. However, while the very small S1–T1 SOC value of 0.02 cm−1 can explain the PL characteristics when the excitation energy is 530 nm, questions remain about the PL characteristics observed through excitation at 350 nm.
Crossing the information extracted so far from Fig. 3a, e, f and Fig. S6 (ESI†), it can be easily concluded that the excitation energy of 3.55 eV (350 nm) allows populating very probably the closely lying excited states S2–S4 of DG7 in toluene solution (Fig. 3a and Fig. S5, ESI†), as compared to the population of only S1 when the excitation energy is 2.34 eV (530 nm, Fig. S8, ESI†) (cf. ESI,† Annex I). The diagram shown in Fig. 5c finally allows proposing a mechanism explaining the absence of delayed emission whatever the excitation wavelength (Fig. 4a): if S2 is populated, the dominant part of the excitons relax to S1 and then to S0, but a small part of them might undergo ISC from S2 to T2 or S1 to T2/3. However, a PLQY of 100% in toluene when excited at 530 nm, and the absence of delayed emission in toluene solution whatever the excitation energy support the hypothesis that the different ISC channels to the triplet manifold of excited states and/or the IC within it are inefficient for DG7 in toluene. This mechanism is expected to quench excitons by populating T1 behaving as a dead-end for the singlet and triplet excitons, which subsequently deexcite non-radiatively to the ground state. Indirect support to this mechanism seems to come from the presence of a weak DF band for DG7 in THF at 77K (Fig. 4b): we hypothesize this DF band stemming from a less endothermic (or possibly exothermic) S1–T2/3 energy gap in THF as compared to toluene, due to the presence of larger CT character for T2/3 as compared to T1 (Fig. 2a), which would increase the S1–T2/3 spin orbit coupling.
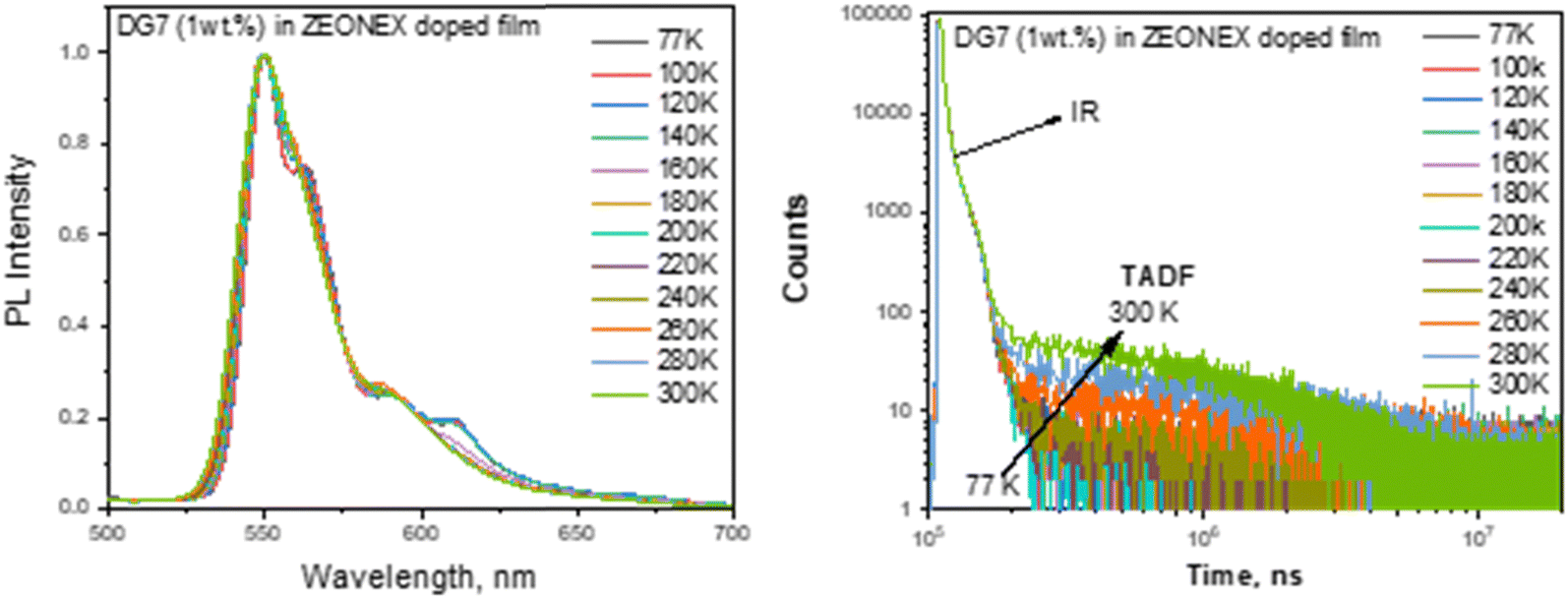 | ||
| Fig. 5 Normalized PL spectra (a) and PL decay curves (b) of the Zeonex doped film of DG7 (1 wt%) recorded at different temperatures. | ||
The observation of DF in the Zeonex and DG7 solid mixture is intriguing, given that Zeonex is an inert non-emissive polymer and hence, no exciplex formation is expected between DG7 and Zeonex that could lead to DF.56 We hypothesize that the solid state polarization and/or distribution of geometrical configurations of DG7 in Zeonex might impact the S1, T1 and/or T2 energies, given their different space localizations in the central part and on the indolocarbazole arms, respectively (see Fig. 2c). Note that the distribution of geometrical configurations considered here comes from a static distribution of the most flexible molecular degrees of freedom (dihedral angles, bending of the DG7 core) leading to a further enhancement of the (non-)planarity of DG7 generally arising in the amorphous solid state, which impacts directly the energy gaps between the different excited states while the dynamic configurational disorder in solution associated with molecular vibrations leaves these gaps unchanged. The geometrical aspect is discussed in Annex II (ESI†) based on the results from molecular dynamics simulations followed by DFT calculations. The results suggest that some geometrical deformations in DG7 induced by the molecular dynamics simulations help in changing the energy gaps between the excited states but it occurs only in few instances (T1–T2 is taken as an example in Fig. S9, ESI†). In addition, because of the weak dielectric constant of Zeonex, it is unlikely that the excited energy gaps would be modulated by solid state polarization effects. A last hypothesis to rationalize the presence of a delayed fluorescence could arise from the damping/reduction of the amplitude of vibrational modes associated with non-radiative decay channels. In the end the large energy gaps between S1, T1 and T2 (Fig. 4c) seem consistent with the DF duration in the millisecond range, which is typically regarded as weak.
Neat films and guest–host mixtures of (8 wt%) DG7 with mCP, mCBP, and with TPBi hosts were additionally characterized theoretically and/or experimentally, see Fig. 6a–e. Recorded spectra in different hosts exhibit intense green fluorescence with sharp emission peaks at 563, 566 and 568 and small full-width at half-maxima (FWHMs) of 34, 34 and 30 nm, in mCP and in TPBi hosts, and in neat film, respectively (Table 2). Fig. 6a exhibits a small shoulder around 625 nm of increasing intensity with increasing temperature; however the relative intensity of this shoulder as compared to the main peak and the temperature impact on its intensity are very small. Importantly, the solid-state PLQY values for mCP- and TPBi-doped samples of 5 wt% DG7 and its neat film were only 19, 13, and 4%, respectively, and were reduced to 12 and 11%, respectively for the 8 wt% DG7–host mixtures (Table 2 and Fig. S10–S13, ESI†). These results indicate that both DG7–mCP and DG7–TPBi mixtures and the neat film of DG7 show very weak delayed fluorescence and TADF efficiencies, which are very different from those reported for other MR-TADF compounds, like DABNA-1, which was attributed to a strong exciplex emission when mixed with mCBP53.
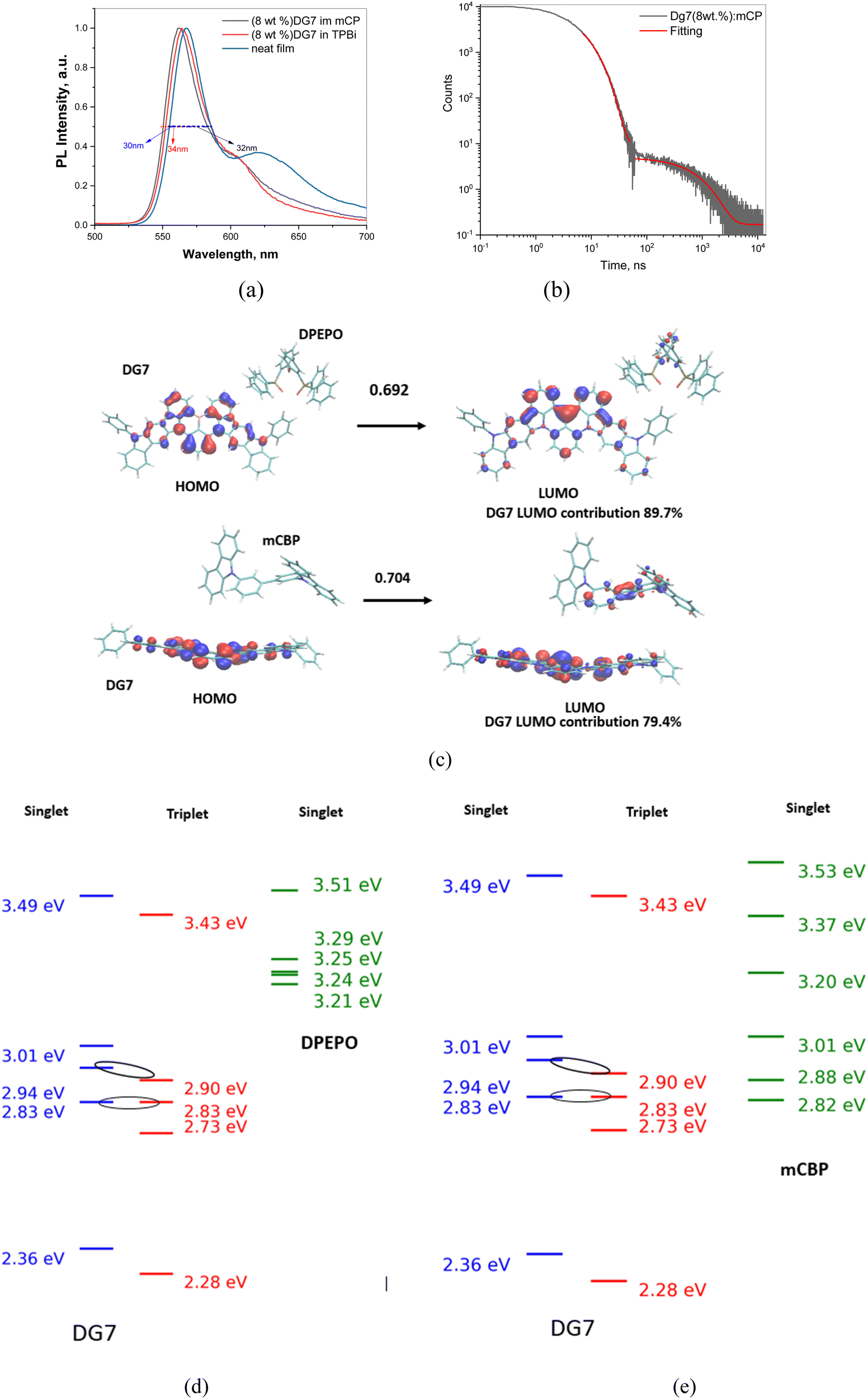 | ||
| Fig. 6 (a) Normalized PL spectra of the DPEPO, mCP, and TPBi doped film of DG7 (λexc = 330 nm). (b) PL decay curve of the mCP doped film of DG7 (λexc = 330 nm). (c) Example of DG7–DPEPO and DG7–mCBP complexes extracted from molecular dynamics simulations. The MO contributions to the first excited state (S1) are also shown, indicating formation of an exciplex with a weak contribution from DPEPO or mCBP. (d) and (e) Comparative energy diagrams between DG7 (Hh-tda/b3lyp/def-2svp −0.45 eV to be in line with experiment) and DPEPO (d) and mCB (e) as reported previously57. | ||
In order to better understand the impact of the DG7–host interactions on the observed DF, from the MD simulation described in Annex II (ESI†) we extracted the molecular configurations of the closest contact between DG7 and the host molecules, and subsequently performed hh-tda/b3lyp/def2-svp calculations on them. While the results shown in Fig. 6c and Fig. S14 (ESI†) indeed indicate examples of DG7–host associations, the very small host contribution (% of LUMO-host in the optical transition) allows excluding the presence of exciplexes. Similar comments apply for the DG7–mCP mixture. Additionally, the DG7–host energy diagrams shown in Fig. 6d and e suggest possible Dexter energy transfer from DG7 to mCP.
Interestingly, the DF lifetime is practically the same for the three solid mixtures of DG7 with Zeonex, mCP, and DPEPO (1.3–1.4 ms), suggesting similar nature for the DF in the three systems. Given the absence of exciplex formation in Zeonex, and the similar conclusion deduced from the results of our MD followed by hh-TDA calculations on DG7–host mixtures (Fig. 6c and Fig. S14, ESI†), we conclude that the main source for the (weak) DF in the case of mCP and DPEPO is not from exciplex formation but from some geometrical configurations exhibiting some intramolecular changes inducing a change in the guest electronic structure (i.e. energy gap between excited states), stemming from different guest–host interactions. Furthermore, the overall impact of DG7 geometrical distortions on the DF efficiency is expected to be small, as confirmed by the theoretical results shown in Fig. S9e and f (ESI†), indicating that the majority of the molecular conformations correspond to large energy gaps between excited states.
We close this section by suggesting two main effects explaining the huge difference between the solution and the solid state PLQY: (i) presence of DG7 microaggregates in DG7–host and DG7–Zeonex mixtures, resulting in important exciton quenching much larger for the DG7–host because of the higher concentration in DG7. This finds support from the strong sonication and bubbling effects observed in toluene solution, and from the smaller PLQY found in DG7–host mixtures with 8 wt% as compared to 5 wt% (Table 2). (ii) Singlet and triplet quenching by oxygen, as supported by the important intensity differences from measurements in air and in degassed and sonicated toluene solutions.
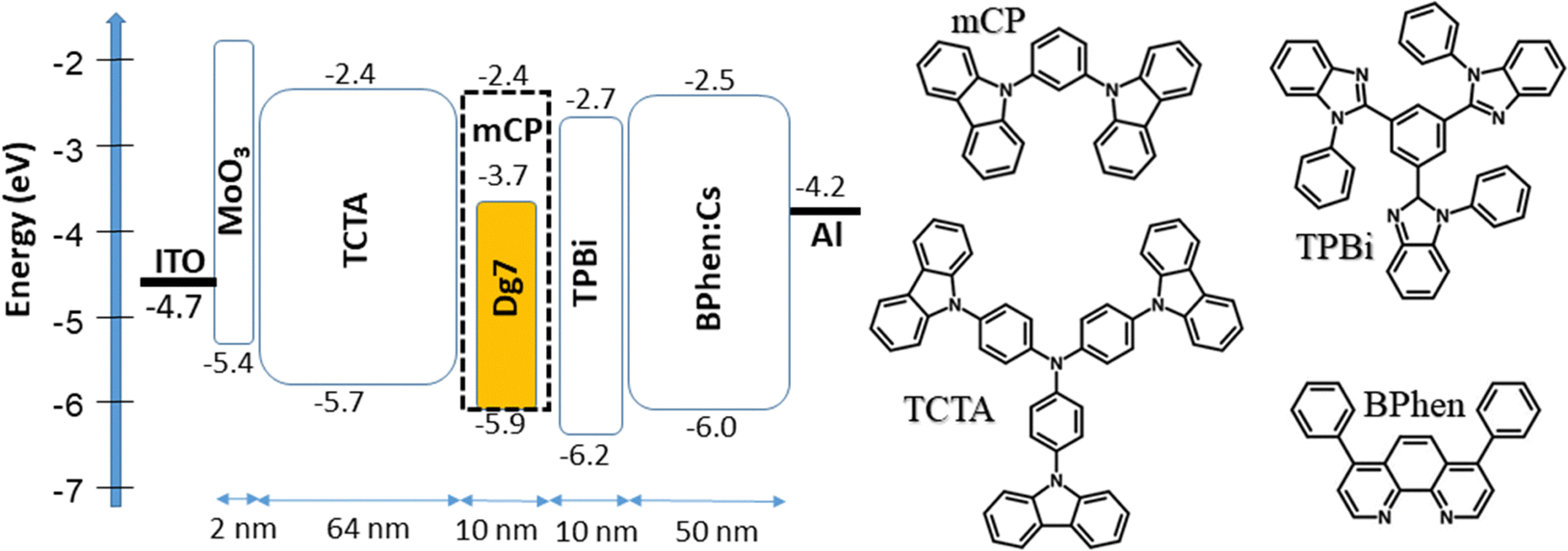 | ||
| Fig. 7 Visualized device structures representing the equilibrium energy diagram and molecular structures of the compounds used in the OLEDs. | ||
The efficient injection of charge carriers from ITO and aluminum electrodes and the following transport to emissive layers of all the fabricated OLEDs are confirmed by their low turn-on voltages (3.1–3.6 V at a luminance of 1 cd m−1) (Fig. 8a and Table 3). With the decreased concentration of DG7 in the hosts from 8 to 5 wt%, devices 1 and 3 showed higher efficiency than devices 2 and 4, respectively (Fig. 8b and Table 3). The highest maximum brightness of 9176 cd m−2 at 10 V, the maximum power efficiency of 7 lm W−1 and the maximum external quantum efficiency of 2.8% were observed for device 3 with 5% of DG7 dispersed in the mCP host as the emitting layer. Devices 1–4 showed low EQE and drastic efficiency roll-off.
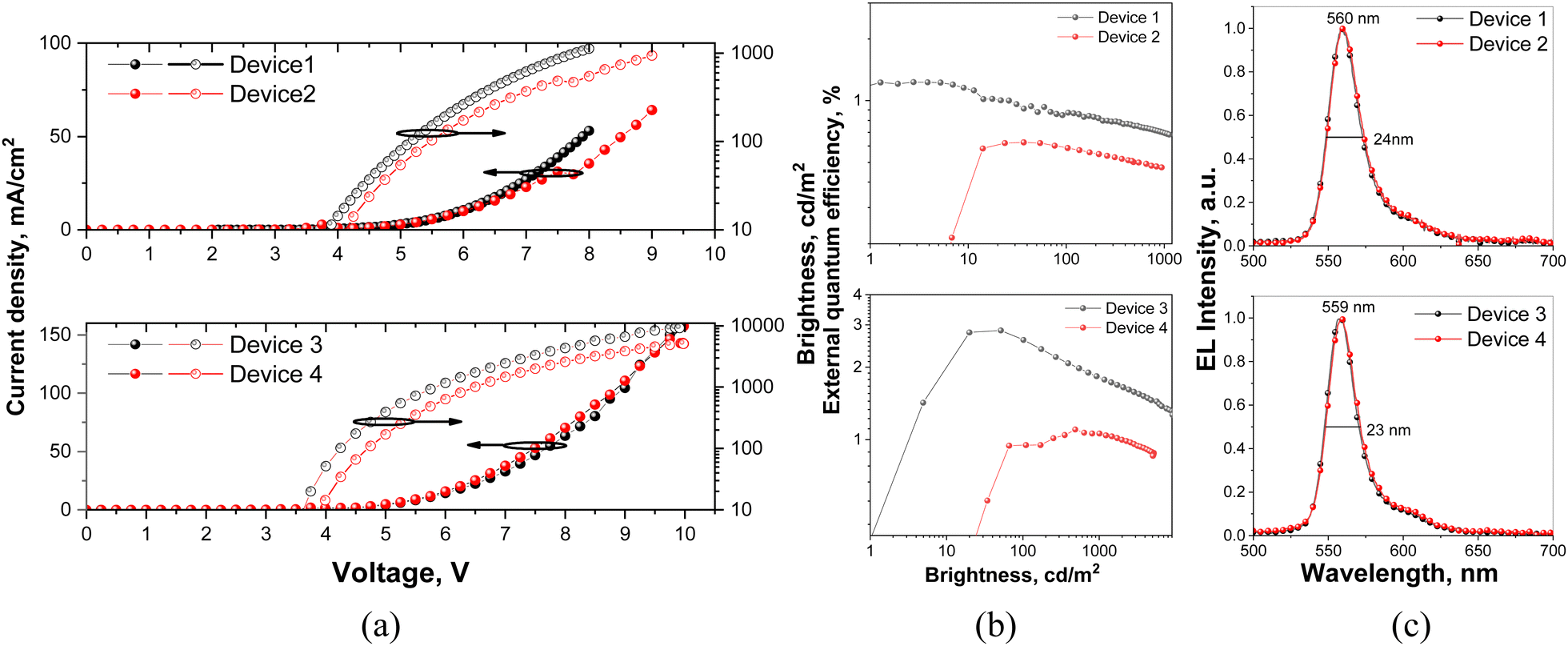 | ||
| Fig. 8 Normalized EL spectra (a), current density/brightness versus applied voltage (b), and external quantum efficiency (c) versus brightness. | ||
| Device | EML | V on,a V | Max brightness,b cd m−2 | PE1000/EQE1000c (lm W−1/%) | PEmax/EQEmaxd (lm W−1/%) | λ,e nm | CIE, x,y | FWHM, nm |
|---|---|---|---|---|---|---|---|---|
| a Turn-on voltage at a luminance of 1 cd m−1. b Maximum brightness. c Power efficiency and external quantum efficiency at 1000 cd m−2. d Maximum power efficiency, and maximum external quantum efficiency. e Maxima of EL (λmax) spectra. | ||||||||
| OLED structures: ITO/MoO3/TCTA/EML/TPBi/BPhen:Cs/Al | ||||||||
| 1 | DG7(5 wt%):TPBi | 3.1 | 1128 | 0.7/0.7 | 2.7/1.2 | 560 | 0.42, 0.54 | 24 |
| 2 | DG7(8 wt%):TPBi | 3.6 | 941 | —/— | 1.1/0.6 | 560 | 0.43, 0.54 | 24 |
| 3 | DG7(5 wt%):mCP | 3.3 | 9176 | 2.9/1.8 | 7/2.8 | 559 | 0.41, 0.55 | 23 |
| 4 | DG7(8 wt%):mCP | 3.5 | 5079 | 1.1/1 | 2.5/1.1 | 559 | 0.41, 0.54 | 23 |
These efficiencies and their trends are coherent with the weak TADF efficiency and small PLQY values of the light-emitting solid layers (Fig. S10–S13, ESI† and Table 2). Thus, severe intermolecular aggregation and exciton loss channels of compound DG7 dispersed in different hosts result in strong ACQ of DG7 emission and the absence of efficient triplet harvesting, which constitute the main reasons for the relatively low maximum EQE value of 2.8% of DG7-based OLEDs.
In spite of these drawbacks, DG7 demonstrated high color purity of electroluminescence (EL). The EL performances of the OLEDs are shown in Fig. 8c and relevant parameters are summarized in Table 3. The shapes of EL spectra of the fabricated OLEDs were in good agreement with the PL profiles. They showed yellowish green emission with peaks at 560 and 559 nm as well as narrow FWHM values of 24 and 23 for devices based on TPBi and mCP hosts, respectively (Fig. 8c and Table 3). The CIE color coordinates of the DG7 devices are collected in Table 3 and shown in Fig. S15b (ESI†). The emission peaks and FHWM values show no change with the increase of doping concentration from 5 wt% to 8 wt%.
Discussion
The indolocarbazole-based MR-TADF emitter presents a unique scenario: while it exhibits outstanding optical properties in solution, reaching a PLQY of 100% in degassed and sonicated toluene, the PLQY in solid mixtures or neat film is reduced to less than 12%, and its EQE of only 2.8% in OLEDs is subpar. Still, these results are quite coherent if one combines the moderate PLQY values of 12–19% in host:guest systems also used in the OLEDs (cf.Table 2) with a typical value of 20% for the outcoupling efficiency, by additionally assuming 100% of singlet–triplet harvesting and 100% of charge balance. The main limitation to higher EQE seems to be the low PLQY of DG7 in the corresponding thin films. Accordingly, there is a first-sight dichotomy between the efficient singlet–triplet harvesting deduced from the electroluminescence measurement in the devices, and the absence of DF deduced from the photoluminescence measurements in toluene solutions. However, the DF timescale of several ms indicates the presence of long-living triplet species, which, in the devices, are expected to take advantage from different slow- or fast channels to undergo rISC. Amongst possible triplet-to-singlet rISC channels present in the devices regardless of their (in)efficiency, the direct S1–T1 channel or a mechanism which would involve higher-lying triplet states such as T2 and T3 could not be excluded in view of the associated energy gaps in the guest–host mixtures. The decrease of the EQE with increased brightness could be associated with annihilation processes such as triplet–triplet annihilation or triplet–polaron annihilation.Accordingly, this huge discrepancy between the toluene solution and the solid-state results seems to stem from a joint contribution from different factors: (i) because of the rigid molecular planar structure, DG7 is prone to strong π–π stacking interactions, leading to the presence of microaggregates even in solution, with a direct impact on ACQ. (ii) The above effect is strongly enhanced in the presence of oxygen. Our study suggests additional significant effects on singlet quenching that could be prevalent in MR-TADF, in addition to common triplet quenching by oxygen. (iii) The energy gaps between the different relevant excited states S1, T1 and T2/3 which limit the DF. The excited state ordering could also be responsible for the very slow device operation.
Consequently, avoiding both aggregation and design of compounds with a small T1–T2 energy gap is crucial for reaching excellent performances with the MR-TADF compounds in general. While the aggregation effect can be overcome by inducing steric hindrance, avoiding a large T1–T2 energy gap is more challenging. Aiming at obtaining insights into the latter parameter, in Fig. 9 we consider five MR-TADF molecules, including two real molecules, DG7 and DABNA-1, along with three model compounds named Model-1 to Model-3. In Fig. 9a we present the HOMO, HOMO−1, and HOMO−2 pictograms of the five molecules and their ΔE(T1–T2) and ΔE(T2–T3) values, whereas in Fig. 9b we show the correlation between ΔE(T1–T2) and ΔE(T2–T3) with ΔE(H–H−1) and ΔE(H−1–H−2), respectively. Based on the good correlations shown in Fig. 9b, in the following we follow the evolution of ΔE(T1–T2) for the five molecules based on the characteristics of their three highest occupied MOs.
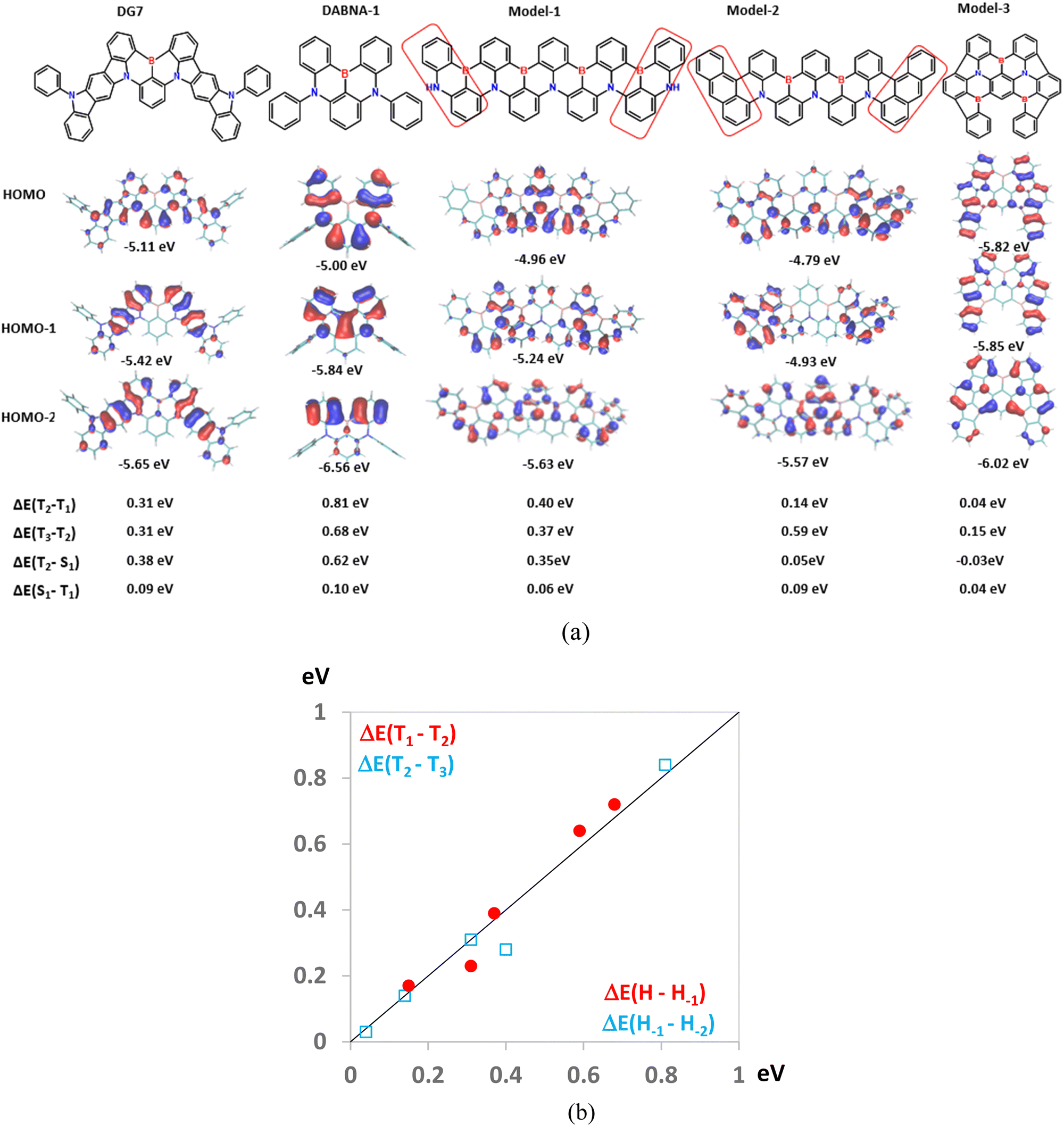 | ||
| Fig. 9 (a) HOMO−2, HOMO−1, and HOMO of the indicated molecules, along with ΔE(T1–T2) and ΔE(T2–T3). (b) Correlation between ΔE(T1–T2) and ΔE(T2–T3) with the energy differences between the HOMO and HOMO−1, reported as ΔE(H–H−1), and between HOMO−1 and HOMO−2, reported as ΔE(H−1–H−2), respectively. | ||
First, we consider DG7. Fig. 2b and Table S1 (ESI†) indicate that T1 corresponds to the HOMO → LUMO transition, both of MOs being localized in the central part of DG7, whereas T2 seems to involve transitions from HOMO−2 localized on the lateral indolocarbazole fragments. Accordingly, the electronic density-differences between ground- and excited states for T1 and for T2 are distributed in different molecular moieties of DG7, in the central part for T1 and in the indolocarbazole arms for T2 (Fig. 9a). These moieties are of a very different nature, thus giving rise to very different triplet energies.
Note that HOMO−1 and HOMO−2 of DG7 correspond to the out-of-phase and in-phase combinations of the two local-indolocarbazole HOMOs (Fig. 9a), with the corresponding energies being obviously lower than the energy of the central-core fragment of DG7. It is tempting to think that, in a new DG7-like molecule, the ΔE(T1–T2) could be much smaller if the constitution of the lateral fragments were to be such that HOMO-DG7 (localized on the molecular central core) becomes HOMO−2 in a new DG7-like molecule. We assume consequently that one possible way to tune ΔE(T1–T2) would be to tune ΔE(H–H−1) by making these MOs delocalized on identical fragments.
To this aim, and for the sake of simplicity, we focus on the B,N containing MR-TADF compounds. Most of them contain a central pseudo-symmetry plane (or axes) orthogonal to (lying on) the average molecular plane; however, the conclusions drawn below hold true independently of the molecular shape. From the chemical-composition standpoint, these molecules either contain a homogenous and symmetrical MR π-conjugated system like DABNA-1 or Model-1, or (ii) like in DG7, they contain a homogenous symmetrical MR-TADF central core, and two identical lateral fragments generally fused to the MR-TADF core.
First, we highlight that designing MR-TADF compounds exhibiting total chemical homogeneity with respect to the central pseudo-symmetry plane seems to be not a good idea. Indeed, the in-phase combination of the two “half-HOMOs” (the local-HOMOs of the two left- and right molecular halves) is expected to strongly overlap between them, thus resulting in large ΔE(H–H−1) and ΔE(T1–T2). This factor is, for instance, the reason why DABNA-1 exhibits huge ΔE(T1–T2) of 0.8 eV (Fig. 9a). Increasing the space delocalization of the π-conjugated system by enlarging the molecular size and keeping the homogeneous chemical constitution as in the case of Model-1 (Fig. 9a) is indeed helpful in reducing the overlap between the two half-HOMOs but cannot prevent them from strongly overlapping in the center of the molecule, in addition to the increased propensity for aggregation. The HOMO of Model-1 is indeed strongly localized in the center of the molecule, whereas HOMO−1 is localized on the lateral parts of the molecule, thus resulting in a very high ΔE(T1–T2) of 0.4 eV.
It is then clear that avoiding strong energy splitting between the HOMO and HOMO−1 would need designing molecules containing lateral groups of different chemical constitution as compared to the central part. Indeed, replacing the lateral parts of Model-1 by anthracene-like groups like in Model-2 is found to localize the half-HOMOs on the lateral parts and reduce their overlap in the central part, thus resulting in strongly reduced ΔE(H–H−1) and in ΔE(T1–T2) of only 0.14 eV.
Finally, in Fig. 9a we show another model molecule, named Model-3, with similar size to DG7, but exhibiting the desired order between the three lowest occupied MOs: HOMO−2 is localized in the central core, and HOMO−1 and HOMO are localized on the lateral fragments. This result indicates a larger donor character for the lateral fragments of Model-3 as compared to the central core. Accordingly, small ΔE(T1–T2) and ΔE(T2–T3) values of 0.04 eV and 0.15 eV were found, respectively. Consequently, the comparison between Model-2 and Model-3 on the one hand, and that between DG7 and DABNA-1 on the other hand support the validity of the idea that ΔE(T1–T2) in MR-TADF compounds can be tuned by judiciously designing lateral groups of higher donor character as compared to the core, whereas the comparison between Model-2 and Model-3 indicates that increasing the size of the molecules is not mandatory for tuning ΔE(T1–T2). Interestingly, this design strategy results in a denser density of triplet states which has been proven to be beneficial for increasing the rate of RISC.58 We speculate that such improved MR-TADF molecules would be able to exhibit intramolecular TADF behavior as opposed to the absence of this desired property for most of the previously reported MR-TADF compounds.
Conclusions
The synthesis, characterization, and comprehensive study of a boron-based MR-TADF compound have demonstrated the intricate relationship between the molecular structure, aggregation, and photophysical properties. DG7 toluene solution exhibits outstanding optical properties, reaching a photoluminescence quantum yield of 100% for the degassed and sonicated sample, and an ultra-pure green-yellow emission with a full width at half maximum (FWHM) of only 20 nm. Despite the high solution PLQY, DG7 experienced a dramatic reduction of PLQY of the solid mixtures. This was attributed to strong π–π stacking interactions, in turn stemming from the rigid planar molecular structure, leading to aggregation-induced quenching. Like other MR-TADF compounds, DG7 exhibits no delayed fluorescence in toluene solution, which is attributed to the large energy gap between the two lowest triplet excited states. Our theoretical analysis on this behavior points to some fundamental aspects in correlation with the chemical constitution. The insights gained from DG7 suggest that optimization of MR-TADF compounds requires a careful balance between molecular design to prevent aggregation and strategic engineering of energy gaps to enhance TADF efficiency.Based on our results, one possible design strategy for new efficient MR-TADF emitters would need (i) avoiding the tendency for aggregation by inducing steric hindrance at some peripheral “strategic” positions, and (ii) combining lateral groups of higher donor character (higher local-HOMO) as compared to the molecular central core, which can be reached by a judicious combination of size and/or chemical constitution of lateral moieties.
Author contributions
Conceptualization – Rasa Keruckiene, Dmytro Volyniuk, Gjergji Sini; methodology and investigation – Aliaksei A. Vaitusionak, Maksim I. Hulnik, Ivan A. Berezianko, Dalius Gudeika, Simas Macionis, Malek Mahmoudi; validation and data curation – Danillo Valverde, Yoann Olivier Kai Lin Woon, Sergei V Kostjuk, Sebastian Reineke; formal analysis and writing – original draft – Yoann Olivier, Kai Lin Woon, Sebastian Reineke, Gjergji Sini; writing – review and editing – Yoann Olivier, Gjergji Sini; supervision and funding acquisition – Juozas. V. Grazulevicius.Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.Acknowledgements
The research was funded by the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement No. 823720. D. V. and Y. O. acknowledge funding by the “Fonds de la Recherche Scientifique-FNRS” under Grant no. F.4534.21 (MIS-IMAGINE). The work in Namur has been funded by the Belgian National Fund for Scientific Research (F. R. S.-FNRS) within the Consortium des Équipements de Calcul Intensif (CÉCI), under Grant No. 2.5020.11. This work is also supported by the University of Malaya Data Intensive Computing Centre. Dr A. Bucinskas is acknowledged for XRDGI measurement.References
- K.-L. Woon, C.-L. Yi, K.-C. Pan, M. K. Etherington, C.-C. Wu, K.-T. Wong and A. P. Monkman, J. Phys. Chem. C, 2019, 123, 12400–12410 CrossRef CAS.
- L. Skhirtladze, K. Lietonas, A. Bucinskas, D. Volyniuk, M. Mahmoudi, O. Mukbaniani, K. L. Woon, A. Ariffin and J. V. Grazulevicius, J. Mater. Chem. C, 2022, 10, 4929–4940 RSC.
- W. Derkowski, D. Kumar, T. Gryber, J. Wagner, M. Morawiak, M. A. Kochman, A. Kubas, P. Data and M. Lindner, Chem. Commun., 2023, 59, 2815–2818 RSC.
- F. Baraket, B. Pedras, É. Torres, M. J. Brites, M. Dammak and M. N. Berberan-Santos, Dyes Pigm., 2020, 175, 108114 CrossRef CAS.
- L. Gan, Z. Xu, Z. Wang, B. Li, W. Li, X. Cai, K. Liu, Q. Liang and S. Su, Adv. Funct. Mater., 2019, 29, 1808088 CrossRef.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- P. Stachelek, J. S. Ward, P. L. dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce and A. P. Monkman, ACS Appl. Mater. Interfaces, 2019, 11, 27125–27133 CrossRef CAS.
- J. Wang, Y. Yang, P. Xu, Z. Cang, C. Yao and X. Zhai, J. Lumin., 2023, 263, 120087 CrossRef CAS.
- J. Wang, X. Zhai, C. Ji, M. Zhang, C. Yao, G. Xie, J. Zhang and X. Xi, Dyes Pigm., 2023, 219, 111586 CrossRef CAS.
- J. Wang, Y. Yang, C. Jiang, M. He, C. Yao and J. Zhang, J. Mater. Chem. C, 2022, 10, 3163–3171 RSC.
- J. Wang, J. Zhang, C. Jiang, C. Yao and X. Xi, ACS Appl. Mater. Interfaces, 2021, 13, 57713–57724 CrossRef CAS PubMed.
- J. Li, R. Zhang, Z. Wang, B. Zhao, J. Xie, F. Zhang, H. Wang and K. Guo, Adv. Opt. Mater., 2018, 6, 1701256 CrossRef.
- X. Liu, Y.-F. Wang, M. Li, Y. Zhu and C.-F. Chen, Org. Electron., 2021, 88, 106017 CrossRef CAS.
- H. Xie, Z. Huang, N. Li, T. Hua, J. Miao and C. Yang, J. Mater. Chem. C, 2022, 10, 11239–11245 RSC.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- T. Serevičius, R. Skaisgiris, J. Dodonova, L. Jagintavičius, J. Bucevičius, K. Kazlauskas, S. Juršėnas and S. Tumkevičius, Chem. Commun., 2019, 55, 1975–1978 RSC.
- T. Serevičius, R. Skaisgiris, J. Dodonova, K. Kazlauskas, S. Juršėnas and S. Tumkevičius, Phys. Chem. Chem. Phys., 2020, 22, 265–272 RSC.
- M. Mahmoudi, D. Gudeika, S. Kutsiy, J. Simokaitiene, R. Butkute, L. Skhirtladze, K. L. Woon, D. Volyniuk and J. V. Grazulevicius, ACS Appl. Mater. Interfaces, 2022, 14, 40158–40172 CrossRef CAS PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- Y. Cheng, X. Fan, F. Huang, X. Xiong, J. Yu, K. Wang, C. Lee and X. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202212575, DOI:10.1002/anie.202212575.
- S. Madayanad Suresh, L. Zhang, D. Hall, C. Si, G. Ricci, T. Matulaitis, A. M. Z. Slawin, S. Warriner, Y. Olivier, I. D. W. Samuel and E. Zysman-Colman, Angew. Chem., Int. Ed., 2023, 62, e202215522, DOI:10.1002/anie.202215522.
- W. Yang, J. Miao, F. Hu, Y. Zou, C. Zhong, S. Gong and C. Yang, Adv. Funct. Mater., 2023, 2213056 CrossRef CAS.
- K. R. Naveen, H. I. Yang and J. H. Kwon, Commun. Chem., 2022, 5, 149 CrossRef CAS PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- S. H. Han, J. H. Jeong, J. W. Yoo and J. Y. Lee, J. Mater. Chem. C, 2019, 7, 3082–3089 RSC.
- X. Liang, Z. P. Yan, H. B. Han, Z. G. Wu, Y. X. Zheng, H. Meng, J. L. Zuo and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 11316–11320 CrossRef CAS PubMed.
- J. H. Kim, W. J. Chung, J. Kim and J. Y. Lee, Mater. Today Energy, 2021, 21, 100792 CrossRef CAS.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu and L. Duan, Angew. Chem., Int. Ed., 2019, 58, 16912–16917 CrossRef CAS.
- Y. Xu, Z. Cheng, Z. Li, B. Liang, J. Wang, J. Wei, Z. Zhang and Y. Wang, Adv. Opt. Mater., 2020, 8, 1902142 CrossRef CAS.
- F. Liu, Z. Cheng, L. Wan, Z. Feng, H. Liu, H. Jin, L. Gao, P. Lu and W. Yang, Small, 2022, 18, 2106462 CrossRef CAS.
- X. Yan, Z. Li, Q. Wang, Y. Qu, Y. Xu and Y. Wang, J. Mater. Chem. C, 2022, 10, 15408–15415 RSC.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468–19472 CrossRef CAS.
- Y. Liu, X. Xiao, Y. Ran, Z. Bin and J. You, Chem. Sci., 2021, 12, 9408–9412, 10.1039/d1sc02042k.
- M. Yang, S. Shikita, H. Min, I. S. Park, H. Shibata, N. Amanokura and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 23142–23147 CrossRef CAS PubMed.
- Y. Xu, C. Li, Z. Li, J. Wang, J. Xue, Q. Wang, X. Cai and Y. Wang, CCS Chem., 2022, 4, 2065–2079 CrossRef CAS.
- L. Lin, J. Fan, L. Cai and C.-K. Wang, Mol. Phys., 2018, 116, 19–28 CrossRef CAS.
- A. Pershin, D. Hall, V. Lemaur, J.-C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- H. Tanaka, S. Oda, G. Ricci, H. Gotoh, K. Tabata, R. Kawasumi, D. Beljonne, Y. Olivier and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 17910–17914 CrossRef CAS PubMed.
- S. Lee, H. Kim and Y. Kim, InfoMat, 2021, 3, 61–81 CrossRef CAS.
- D. Hall, J. C. Sancho-García, A. Pershin, G. Ricci, D. Beljonne, E. Zysman-Colman and Y. Olivier, J. Chem. Theory Comput., 2022, 18, 4903–4918 CrossRef CAS PubMed.
- K. Stavrou, A. Danos, T. Hama, T. Hatakeyama and A. Monkman, ACS Appl. Mater. Interfaces, 2021, 13, 8655 CrossRef PubMed.
- Y. Xu, C. Li, Z. Li, J. Wang, J. Xue, Q. Wang, X. Cai and Y. Wang, CCS Chem., 2022, 4, 2065–2079 CrossRef CAS.
- G. Meng, L. Liu, Z. He, D. Hall, X. Wang, T. Peng, X. Yin, P. Chen, D. Beljonne, Y. Olivier, E. Zysman-Colman, N. Wang and S. Wang, Chem. Sci., 2022, 13, 1665–1674, 10.1039/d1sc05692a.
- Y. Y. Jing, Y. Yang, N. Li, Z. Ye, X. Wang, X. Cao and C. Yang, Luminescence, 2024, 1, e4624, DOI:10.1002/BIO.4624.
- N. Ikeda, S. Oda, R. Matsumoto, M. Yoshioka, D. Fukushima, K. Yoshiura, N. Yasuda, T. Hatakeyama, N. Ikeda, S. Oda, K. Yoshiura, T. Hatakeyama, R. Matsumoto, M. Yoshioka, D. Fukushima and N. Yasuda, Adv. Mater., 2020, 32, 2004072 CrossRef CAS PubMed.
- D. Hall, S. M. Suresh, P. L. dos Santos, E. Duda, S. Bagnich, A. Pershin, P. Rajamalli, D. B. Cordes, A. M. Z. Slawin, D. Beljonne, A. Köhler, I. D. W. Samuel, Y. Olivier and E. Zysman-Colman, Adv. Opt. Mater., 2020, 8, 1901627, DOI:10.1002/ADOM.201901627.
- K. L. Woon, P. A. Nikishau and G. Sini, Adv. Theory Simul., 2022, 5, 2200056 CrossRef CAS.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- B. Wu, H. Su, A. Cheng, X. Zhang, T. Wang and G. Zhang, Cell Rep. Phys. Sci., 2023, 4, 101245 CrossRef CAS.
- X. Wu, B.-K. Su, D.-G. Chen, D. Liu, C.-C. Wu, Z.-X. Huang, T.-C. Lin, C.-H. Wu, M. Zhu, E. Y. Li, W.-Y. Hung, W. Zhu and P.-T. Chou, Nat. Photonics, 2021, 15, 780–786 CrossRef CAS.
- N. Notsuka, R. Kabe, K. Goushi and C. Adachi, Adv. Funct. Mater., 2017, 27, 1703902 CrossRef.
- J. Eng, J. Hagon and T. J. Penfold, J. Mater. Chem. C, 2019, 7, 12942–12952 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc04397e |
| This journal is © The Royal Society of Chemistry 2024 |