DOI:
10.1039/D3TC04292H
(Paper)
J. Mater. Chem. C, 2024,
12, 4273-4286
Evaluation of acenes as potential acceptors in thermally activated delayed fluorescence emitters and the promise of a phenoxazine–naphthalene emitter for OLEDs†‡
Received
21st November 2023
, Accepted 16th January 2024
First published on 18th January 2024
Abstract
Thermally activated delayed fluorescence (TADF) is one of the most promising technologies for harvesting triplet excitons in all-organic emitters, a property that is essential for achieving high efficiency in devices. Compounds that operate via this mechanism for emission typically rely on a combination of electron donating and accepting moieties separated by an aromatic bridge. Here we demonstrate that although naphthalene is underutilised as an acceptor, it can nonetheless be used in a donor–acceptor TADF emitter when coupled to two phenoxazines in the 1- and 4-positions. The compound 1,4-PXZ-Nap-PXZ emits at 508 nm, has a photoluminescence quantum yield of 48% and a delayed lifetime of 22.7 ms in a 20 wt% doped film in 1,3-bis(N-carbazolyl)benzene (mCP). An organic light-emitting diode (OLED) using this emitter showed a maximum external quantum efficiency (EQEmax) of 11% and green emission at λEL of 505 nm, demonstrating for the first time the potential of naphthalene-acceptor based emitters for devices. Finally, we have demonstrated by way of a density functional theory (DFT) study why naphthalene alone amongst linear acenes is suitable for this role.
Introduction
Of the various competing display technologies used in the modern world, that of the organic light-emitting diode (OLED) is rapidly becoming one of the most widely adopted. This is because OLEDs offer numerous advantages over other display technologies, including high efficiencies, the capacity to view the display at wide angle without loss of image quality, improved contrast, faster refresh rate and an access to a multitude of form factors. In particular, OLEDs can be integrated into flexible and foldable displays and lighting panels, something that is not possible with other display technologies.1 In an OLED, light is generated by the radiative decay of an exciton that forms following electrical excitation of an emitting molecule, which can be, for instance, an organic (or organometallic) small-molecule or polymer that is located centrally in a stack of organic semiconducting layers sandwiched between two electrodes.1,2 The overall efficiency of an OLED is quantified in terms of its external quantum efficiency (EQE), which relates to the ratio of supplied electrons to observed photons.EQE is the product of four terms (eqn (1)),3 the most important of which for emitter design is the exciton utilization efficiency (nST), the ratio of generated excitons that can undergo allowed radiative transitions to the ground state. This is because the charge carrier balance (γ) and photoluminescence quantum yield (PLQY, ΦPL) can both equal unity in an optimally functioning device,1 while the outcoupling efficiency (nout) is typically limited to only 20% as the transition dipole moments of the emitter molecules are distributed isotropically.4 Meanwhile, nST depends on the mechanism of emission, because under electrical excitation excitons are generated through hole and electron recombination in a 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 ratio of triplets to singlets.1,3 Thus if a material is only capable of productively harvesting singlet excitons to produce light, as is the case for purely fluorescent emitters, its internal quantum efficiency (IQE; the product of γ, nST and ΦPL) will be limited to 25%, and its maximum EQE will be limited to around 5%. By contrast, phosphorescent emitters can harvest both singlet and triplet excitons, owing to strong spin–orbit coupling between the excited states which permits singlet to triplet interconversion via intersystem crossing (ISC) and reverse intersystem crossing (RISC). These triplet excitons can then radiatively decay via phosphorescence, thereby allowing 100% of the generated excitons to emit light. High spin–orbit coupling is normally induced by the inclusion of a heavy metal atom (or atoms) in the emitter, as is the case with organometallic complexes. However, the heavy metals, most commonly Pt and Ir, that these so-called 2nd-generation emitters rely upon have a number of inherent drawbacks, including: uneven distribution across the Earth's crust, low natural abundance compared to the atoms of all-organic emitters and, to a degree, relatively high cost.5,6 As such, there is still significant demand for materials that emit via alternative triplet harvesting mechanisms, and of these the most promising is that of thermally activated delayed fluorescence (TADF). In this mechanism the singlet–triplet splitting energy (ΔEST) is sufficiently small to permit endothermic upconversion of triplet excitons into singlets via RISC.1,7 If the gap is sufficiently small, this can be achieved even with the modest spin–orbit coupling present in all-organic emitters. These upconverted triplet excitons can then emit from the singlet excited state at the same emission wavelength as the initially generated singlet excitons, but with a longer overall excited state lifetime. This mechanism likewise allows 100% of generated excitons to emit light but without necessitating the inclusion of a heavy metal.
:25 ratio of triplets to singlets.1,3 Thus if a material is only capable of productively harvesting singlet excitons to produce light, as is the case for purely fluorescent emitters, its internal quantum efficiency (IQE; the product of γ, nST and ΦPL) will be limited to 25%, and its maximum EQE will be limited to around 5%. By contrast, phosphorescent emitters can harvest both singlet and triplet excitons, owing to strong spin–orbit coupling between the excited states which permits singlet to triplet interconversion via intersystem crossing (ISC) and reverse intersystem crossing (RISC). These triplet excitons can then radiatively decay via phosphorescence, thereby allowing 100% of the generated excitons to emit light. High spin–orbit coupling is normally induced by the inclusion of a heavy metal atom (or atoms) in the emitter, as is the case with organometallic complexes. However, the heavy metals, most commonly Pt and Ir, that these so-called 2nd-generation emitters rely upon have a number of inherent drawbacks, including: uneven distribution across the Earth's crust, low natural abundance compared to the atoms of all-organic emitters and, to a degree, relatively high cost.5,6 As such, there is still significant demand for materials that emit via alternative triplet harvesting mechanisms, and of these the most promising is that of thermally activated delayed fluorescence (TADF). In this mechanism the singlet–triplet splitting energy (ΔEST) is sufficiently small to permit endothermic upconversion of triplet excitons into singlets via RISC.1,7 If the gap is sufficiently small, this can be achieved even with the modest spin–orbit coupling present in all-organic emitters. These upconverted triplet excitons can then emit from the singlet excited state at the same emission wavelength as the initially generated singlet excitons, but with a longer overall excited state lifetime. This mechanism likewise allows 100% of generated excitons to emit light but without necessitating the inclusion of a heavy metal.
Modern emitter design is frequently aided by in silico modelling. Density functional theory (DFT) can predict the energies of both singlet and triplet excited states via the time-dependent (TD-)DFT formalism. These calculations are able to predict real-world trends among a series of compounds.8 Accurate prediction of absolute energies is also possible, but is more challenging to achieve since it requires the proper inclusion of multiple effects such as solvent stabilization, changes in the excited-state geometry, vibronic effects, as well as conformational disorder. In addition to energies, TD-DFT can also calculate the oscillator strength and the nature of the excited sate. The oscillator strength closely relates to the radiative decay rate and thus the ΦPL, and larger values correspond to a more probable transition. Meanwhile, the ‘nature’ of the excited state indicates which molecular orbitals contribute to the transition, and from the localization of these orbitals around the molecule it is possible to estimate the degree of charge-transfer (CT) or locally-excited (LE) character of the excited state, and which molecular units of the molecule are involved in the electronic transition. DFT can also calculate the energies of the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO respectively), which can be used to quantify the strength of the donor and acceptor moieties of the emitter. A more stabilised LUMO and more destabilised HOMO correspond to a stronger acceptor and stronger donor, respectively. Finally, it is also possible to calculate the spin–orbit coupling (SOC, HSO) between two excited states of different spin multiplicities, which in the case of TADF involves a singlet and a triplet excited state. This property indicates the degree of coupling between the two states which thus opens both intersystem and reverse-intersystem crossing pathways, both of prime importance when discussing the TADF mechanism.1
TADF emitter design typically targets minimising ΔEST by localising the hole and the electron densities (for a two-state electronic transition, these usually correspond to the HOMO and LUMO, respectively) of the emitter to a (number of) donor and acceptor moieties. These moieties are then typically linked via an aromatic bridge7 to ensure sufficient overlap between the HOMO and LUMO, which is necessary to maintain a large oscillator strength and thus efficient fluorescence. The most commonly used aromatic bridge is benzene, yet its larger homolog, naphthalene, has been used only rarely.9–12 This is curious, both because naphthalene is bulkier than benzene and ought to induce larger torsion angles between the donor/acceptor and the central bridge, which is beneficial for reducing ΔEST, and also because the larger π-system possesses a more stabilised LUMO, opening the possibility of using naphthalene itself as the electron-accepting moiety.
Indeed, of the plethora of OLED emitters, only three notable reports can be found of compounds containing naphthalene (Fig. 1). These are: (1) 1,4-naphthyl-CF3 and 1,5-Naphthyl-CF3, two fluorescent blue (λEL = 453 and 446 nm, respectively) donor–bridge–acceptor emitters where naphthalene acts as a bridge9 that show moderate ΦPL (67 and 36%, respectively) in toluene solution. OLEDs with these emitters show a maximum external quantum efficiency (EQEmax) of 2.3% in both cases; (2) a donor–acceptor–donor emitter, DPTZN, in which naphthalene acts as the acceptor and that emits via room-temperature phosphorescence (RTP).10 This molecule shows a remarkable enhancement of the RTP when blended with TRZ-BIM at 10 wt% doping, where the ΦPL is 38% compared to only 3% in the neat film. A device based on this blend showed yellow emission (λEL = 570 nm) with an EQEmax of 11.5%; notably, the mechanism underpinning the RTP enhancement was not elucidated by the authors; (3) finally, a donor–bridge–acceptor TADF emitter, BTaz-Na-PXZ,11 which is to the best of our knowledge the first known example of a naphthalene-containing compound showing TADF. This compound exhibits weak, blue emission (λPL = 473 nm) as the ΦPL = 18% as a 1 wt% doped film in Zeonex; no devices were fabricated. In addition, two further reports document the use of naphthalene-containing compounds as materials used in OLEDs: (1) a fluorescent emitter with the naphthalene moiety attached to the periphery of the structure that likely is not involved in the electronic transition;13 and (2) two naphthalene-containing molecules used as the charge recombination layer in white OLEDs (WOLEDs), which likewise are not involved directly in the emission.14
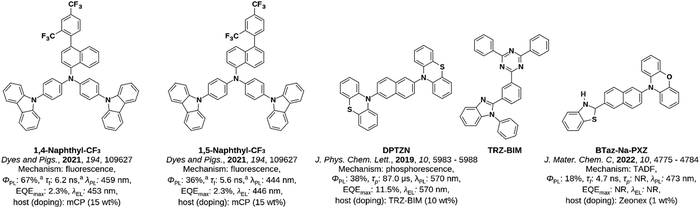 |
| | Fig. 1 Structures and photophysical properties of previously reported examples of naphthalene-based emitters. Results are shown for doped films (or doped devices) with the host and doping concentration shown, unless otherwise specified. aIn degassed toluene solution. NR: not recorded. | |
Here, we demonstrate the viability of naphthalene-based compounds as emitters in OLEDs where the naphthalene acts as the electron-accepting moiety in a donor–acceptor–donor compound. We additionally demonstrate using density-functional theory (DFT) computations that naphthalene, of all the linear acenes up to hexacene, is ideally suited to act as an acceptor in TADF emitter design.
Computational screen
We initiated a DFT computational screen of 36 naphthalene-based compounds containing one of six different donor moieties with a range of donating strengths and sizes to simultaneously probe the effect of both electronics and sterics. These donors were diphenylamine (DPA), carbazole (Cz), 3,6-di-tert-butylcarbazole (DTBCz), 1,8-dimethylcarbazole (DMCz), 9,9-dimethyldihydroacridine (DMAC) and phenoxazine (PXZ). These donors decorate the naphthalene in either the 1-, 1,4-, 2- or 2,6-configurations where in the doubly substituted compounds, the second substituent was either another equivalent of the same donor or alternatively 2,4-diphenyl-1,3,5-triazine (TRZ) (Fig. 2), the latter being a stronger acceptor than naphthalene and serving to further stabilise the LUMO of the emitter.
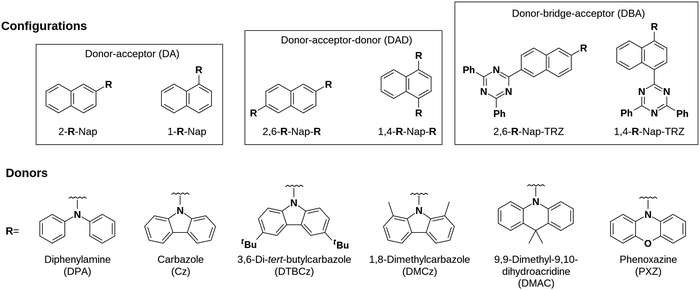 |
| | Fig. 2 General structures of the 36 compounds investigated computationally. | |
The full results from this investigation are available in the ESI† (Table S1). In summary, four of the compounds, 1-PXZ-Nap, 1,4-PXZ-Nap-PXZ, 2-PXZ-Nap and 2,6-PXZ-Nap-PXZ were identified as having the most promising traits to show TADF. Crucially, each of these four compounds were computed to have a ΔEST of below 0.1 eV. The analogous structures 1,4-PXZ-Nap-TRZ and 2,6-PXZ-Nap-TRZ have similarly small ΔEST, but the stronger TRZ acceptor further stabilises the S1 energy to 2.20 and 2.34 eV, respectively. As blue-emitting OLEDs are typically in higher demand than green or yellow emitting, we chose to target the higher energy compounds. A marginally higher ΔEST was calculated for 2-PXZ-Nap and 2,6-PXZ-Nap-PXZ of 0.06 and 0.04 eV, respectively, compared to 0.01 eV for each of 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ. However, in the compounds with a larger ΔEST, the HSO was also larger, at 0.24 and 0.15 cm−1 for 2-PXZ-Nap, 2,6-PXZ-Nap-PXZ, respectively, compared to 0.03 and <0.01 cm−1 for 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ, respectively. This competing effect between diminishing ΔEST and maintaining SOC has been described previously in the literature.16 Hence, three of the four emitters have an effectively equivalent first-order mixing coefficient of around 10−4, with only 1,4-PXZ-Nap-PXZ having a significantly smaller mixing coefficient of around 10−6 due to the much smaller HSO in this emitter. This highlights that the S1 and T1 excited states remain largely pure spin states. In all cases, the small ΔEST values can be attributed to excellent confinement of the HOMO and LUMO onto the PXZ donor and naphthalene acceptor moieties, respectively, as evidenced from the orbital distributions of the DFT-optimised structures (Fig. 3). In turn, this localization is the result of the large torsion angles between the donor(s) and acceptor, which is close to perpendicular in all cases, and, to a lesser extent, the high donating strength of the PXZ moiety, as reflected by the computed HOMO energies. The four molecules have a predicted S1 excitation energy ranging from 433 nm for 2-PXZ-Nap to 479 nm for 1,4-PXZ-Nap-PXZ. This trend is naturally mirrored in the calculated HOMO–LUMO gap (ΔEHOMO–LUMO), being largest in 2-PXZ-Nap (3.66 eV) and decreasing to 3.46 eV in 1,4-PXZ-Nap-PXZ since the S1 excited states of all molecules are dominated by a HOMO to LUMO transition. As might be expected, the largest change in both the S1 energy and ΔEHOMO–LUMO was observed between the singly and doubly donor-substituted examples; however, this change was driven primarily by a stabilisation of the LUMO level as opposed to a destabilisation of the HOMO, despite the addition of a second donor unit. Finally, in all cases the predicted oscillator strength for the S0 → S1 transition is negligible. However, this is typical in charge-transfer type emitters such as those investigated here, in which the HOMO–LUMO overlap in the ground state-optimised structure is intentionally minimised and the probability of the transition becomes small.17 These emitters can still possess high efficiencies in the real world so long as emission of the S1 is coupled to vibrational modes inducing either: (1) a reduced donor–acceptor torsion and hence greater HOMO–LUMO overlap or (2) contributions from electronic transitions between deeper occupied molecular orbitals and higher-lying unoccupied molecular orbitals.18
 |
| | Fig. 3 Computed HOMO & LUMO energies with associated densities (isovalue = 0.02) as computed at the PBE0-D3(BJ)/6-31G(d,p) level of theory and excited state energies, calculated at the TDA-PBE0-D3(BJ)/6-31G(d,p) level of theory in the gas phase. | |
Synthesis
Based on the theoretical study, we next targeted the synthesis of the four compounds. 1-PXZ-Nap, 2-PXZ-Nap and 1,4-PXZ-Nap-PXZ were each obtained following a Pd-catalysed Buchwald–Hartwig cross-coupling reaction of the relevant mono or di-halogenated naphthalene and phenoxazine in 34%, 56% and 31% yield, respectively (Schemes S3–S5, ESI†). Surprisingly, 2,6-PXZ-Nap-PXZ could not be obtained by this route and thus this target was abandoned at this stage for further investigation. We also note that the mono-substituted product from the reaction of 1,4-dibromonaphthalene and phenoxazine, 1-bromo-4-phenoxylnaphthalene, could not be isolated, regardless of the stoichiometry of phenoxazine used. The synthesised emitters were fully characterised by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, gas-chromatography/mass-spectrometry (GCMS), high-pressure liquid chromatography (HPLC), melting point analysis, and structure elucidation by single-crystal X-ray diffraction. Full synthetic details are available in the ESI.†
Crystal structures
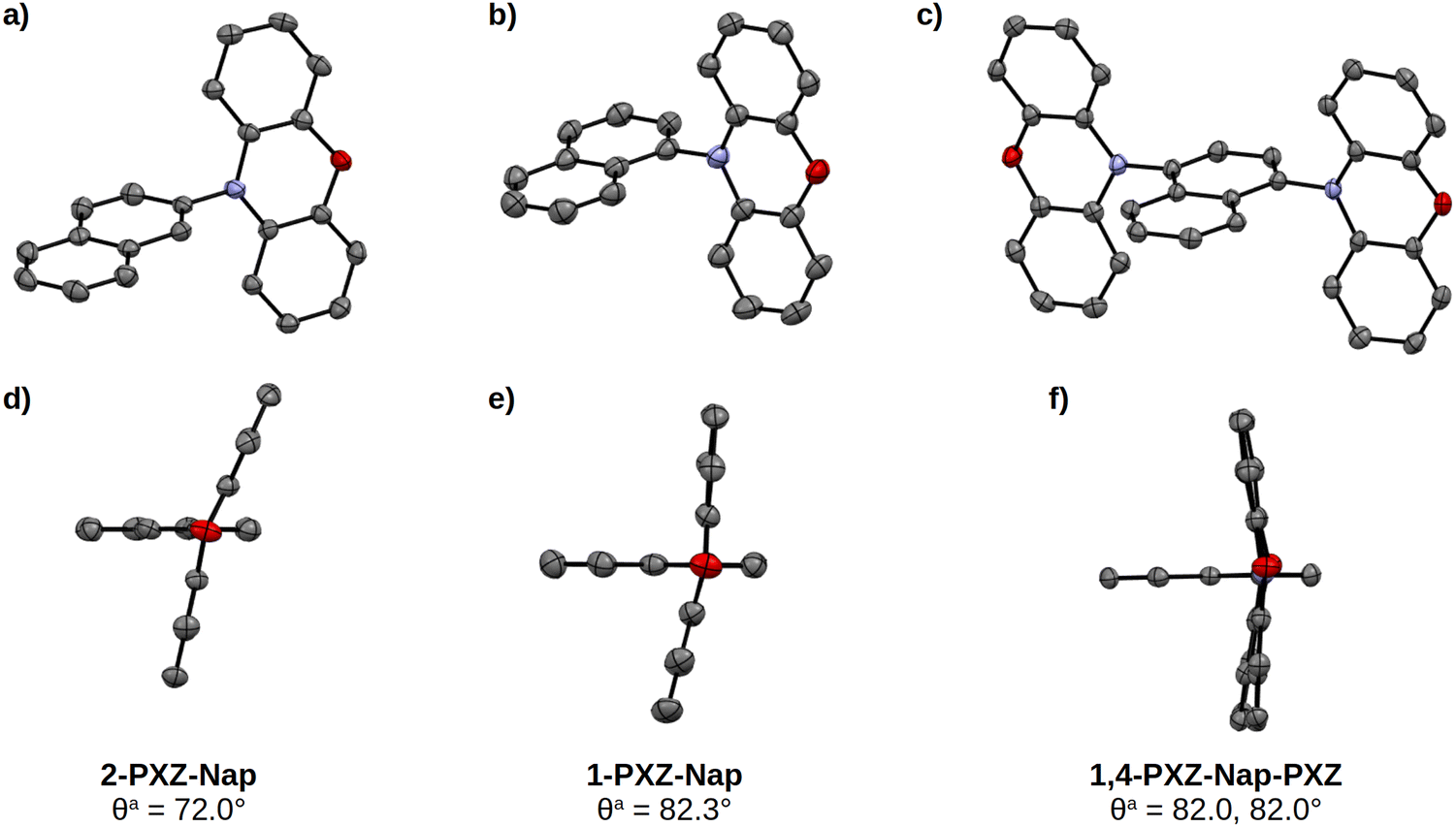
The structures of each of the three emitters were determined by single crystal X-ray crystallography. Each of the crystals was grown by vapour diffusion of hexane into toluene. These structures are shown in Fig. 4; in all three compounds the central naphthalene is planar while the phenoxazine donor is puckered around the connecting nitrogen atom. In the related 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ structures, there is a close to perpendicular dihedral angle between the phenoxazine and naphthalene groups of 82.3° and 82.0°, respectively, while the less sterically hindered 2-PXZ-Nap has a more relaxed dihedral angle of 72.0°. The difference in geometry between the 1-PXZ-Nap/1,4-PXZ-Nap-PXZ and 2-PXZ-Nap agrees with the trend predicted by DFT (Table 1). A complete analysis of the obtained crystal structures is available in the ESI† (S6).
 |
| | Fig. 4 Structures of 2-PXZ-Nap (a) and (d), 1-PXZ-Nap (b) and (e) and 1,4-PXZ-Nap-PXZ (c) and (f) derived from single-crystal X-ray crystallography. Atoms are shown as thermal ellipsoids at the 50% probability level. Hydrogen atoms have been omitted for clarity. aDihedral angle between donor and acceptor moieties, measured as the average of the four absolute dihedral angles between the two groups where angles >180° have been normalised according to the formula θnorm = θ − 180. | |
Table 1 Computational results computed at the TDA-PBE0-D3(BJ)/6-31G(d,p) level of theory
| Structure |
E
S1
/eV (nm) |
f
S1
|
ΔESTc/eV |
H
SO
/cm−1 |
H
SO/ΔESTe |
E
HOMO/eV |
E
LUMO/eV |
θ
/° |
|
Energy of the first singlet excited state.
Oscillator strength of the S0 → S1 transition.
S1–T1 energy gap.
Spin–orbit coupling between S1 and T1 at the ground state geometry, calculated using the PySOC program.15
First-order mixing coefficient between S1 and T1.
Dihedral angle between the donor and acceptor moieties, measured as the average of the four absolute dihedral angles between the two groups where angles >180° have been normalised according to the formula θnorm = θ − 180.
|
|
2-PXZ-Nap
|
2.87 (433) |
<0.01 |
0.06 |
0.24 |
5.27 × 10−4 |
−4.90 |
−1.25 |
80.7 |
|
1-PXZ-Nap
|
2.78 (447) |
<0.01 |
0.01 |
0.03 |
2.83 × 10−4 |
−4.91 |
−1.26 |
83.2 |
|
2,6-PXZ-Nap-PXZ
|
2.70 (459) |
<0.01 |
0.04 |
0.15 |
4.79 × 10−4 |
−5.03 |
−1.56 |
80.4, 80.4 |
|
1,4-PXZ-Nap-PXZ
|
2.59 (479) |
<0.01 |
0.01 |
<0.01 |
9.76 × 10−7 |
−5.05 |
−1.58 |
82.8, 82.8 |
Electrochemistry
Cyclic (CV) and differential pulse (DPV) voltammograms for each of the three synthesised emitters were recorded in acetonitrile and are shown in Fig. 5 and the electrochemical data are collated in Table 2. Clear and reversible oxidation and reduction waves are observed for each of 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ, corresponding to estimated HOMO/LUMO values of −5.08/−3.52, −5.12/−3.53 and −5.13/−3.52 eV, respectively. The LUMO energy shows essentially no variance with emitter structure, which is to be expected considering how electronically decoupled the donor PXZ groups are with the naphthalene acceptor. There is also little variation in the HOMO energy, even between 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ. The corresponding HOMO–LUMO gaps are 1.56, 1.59 and 1.61 eV for 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ, respectively. Compared to the naphthalene-based emitters that have been previously reported,9,10 the HOMO energies of these three emitters are generally more destabilised while the LUMO energies are notably more stabilised. As such, the HOMO–LUMO gaps are smaller at ca. 1.6 eV, compared to ca. 3.0, 3.0 and 2.7 eV for 1,4-naphthyl-CF3,91,5-naphthyl-CF3,9 and DPTZN,10 respectively (Table 2). Finally, the trend in the measured HOMO/LUMO energies matches that predicted by DFT, although the absolute energies of the LUMO are more stabilised than the predictions.
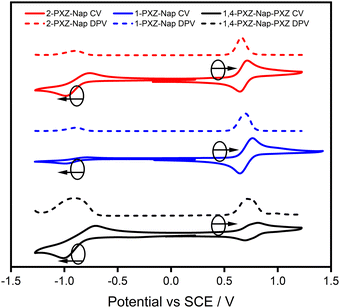 |
| | Fig. 5 Cyclic and differential pulse voltammograms for the emitters 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ, measured in acetonitrile and referenced to a saturated calomel electrode (0.45 V vs. SCE19) with a scan rate of 0.1 V s−1. | |
Table 2 Summary of electrochemical data of the new emitters in this work and literature examples
| Emitter |
E
ox
/V |
E
red
/V |
E
HOMO
/eV |
E
LUMO
/eV |
ΔEHOMO–LUMO/eV |
|
Oxidation/reduction potentials obtained from the peak of the DPV using Fc/Fc+ as the internal standard and referenced to SCE.19
Measured from the onset of oxidation/reduction peak.
Calculated by the authors as ELUMO = EHOMO + Eopt, where Eopt is the optical gap estimated from UV-vis absorption spectra.
Calculated from reported EHOMO/ELUMO, according to Eox = EHOMO − 4.8 and Ered = 4.8 − ELUMO.11NR: not recorded. |
|
2-PXZ-Nap
|
0.28 |
−1.28 |
−5.08 |
−3.52 |
1.56 |
|
1-PXZ-Nap
|
0.32 |
−1.27 |
−5.12 |
−3.53 |
1.59 |
|
1,4-PXZ-Nap-PXZ
|
0.33 |
−1.28 |
−5.13 |
−3.52 |
1.61 |
|
1,4-naphthyl-CF3
9
|
0.46 |
NR |
−5.24 |
−2.21c |
3.03 |
|
1,5-naphthyl-CF3
9
|
0.45 |
NR |
−5.23 |
−2.18c |
3.05 |
|
DPTZN
10
|
0.55d |
−2.1d |
−5.35 |
−2.70 |
2.65 |
|
BTaz-Ph-PXZ
11
|
NR |
NR |
NR |
NR |
NR |
Absorption and photoluminescence
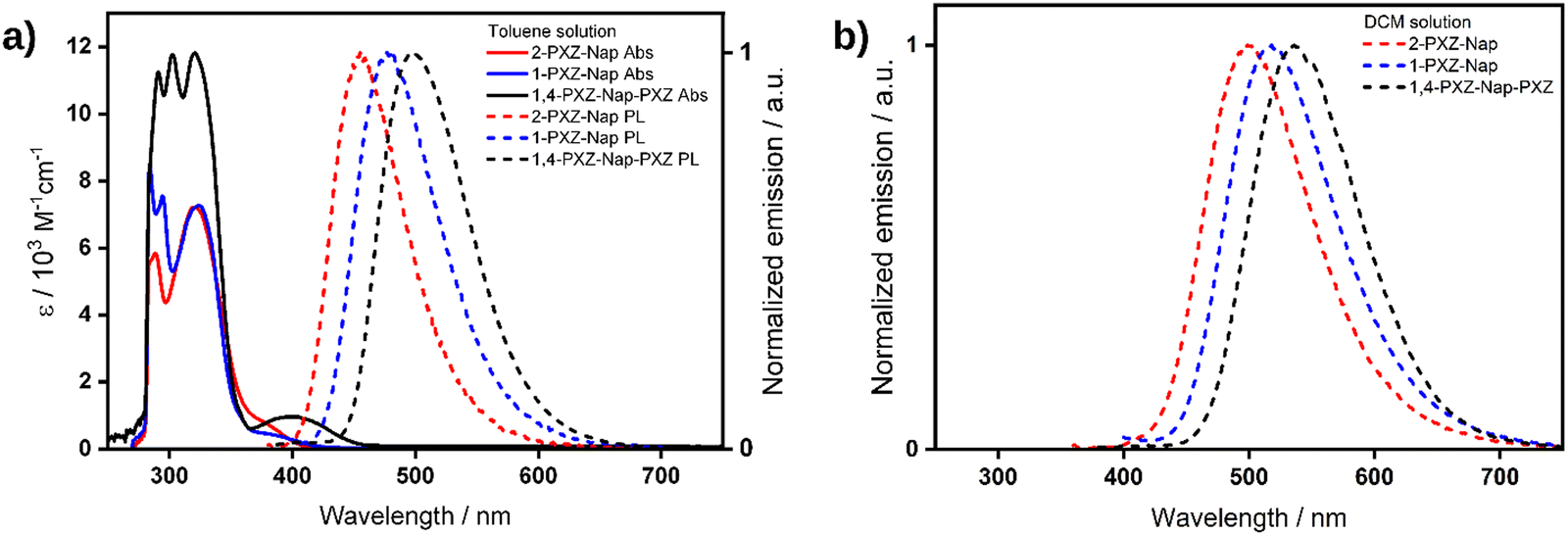
Absorption (Abs) and photoluminescence (PL) spectra for each of the three emitters were recorded in dilute toluene solution (Fig. 6a). Each emitter shows an intense and structured absorption band centred around ca. 300 nm corresponding to the overlap of peaks from the local excitation (LE) of both the central naphthalene moiety20 and the terminal phenoxazine donor.21 The contribution of the phenoxazine moiety is clearly demonstrated by the increase in molar absorptivity of this LE band in the doubly substituted 1,4-PXZ-Nap-PXZ compared to either of the singly substituted emitters. In addition, an unstructured and less intense band can be observed that is red-shifted relative to the LE band in all three compounds, appearing as a shoulder in 2-PXZ-Nap and 1-PXZ-Nap at ca. 375 nm and as a distinct band in 1,4-PXZ-Nap-PXZ at 402 nm. This lower energy band is assigned to the charge transfer (CT) transition between the phenoxazine donor and naphthalene acceptor and is noticeably more intense in the doubly substituted 1,4-PXZ-Nap-PXZ. This arises because the second equivalent of the phenoxazine donor moiety increases the overlap between the HOMO and LUMO, thus increasing the probability of transition. As well, the CT absorption band is red-shifted in 1,4-PXZ-Nap-PXZ compared to the singly substituted emitters as the HOMO is destabilized due to the presence of the second electron-donating PXZ. The observed trend in the CT band absorption energies mirrors the S1 energies predicted by TDA-DFT; however, the molar absorptivities do not, where the predicted oscillator strength, f, of the S0 → S1 transition at the optimised geometries for each of the emitters is ca. 0, as at this geometry there is a near orthogonal conformation between the donor and acceptor groups. The PL spectrum of each emitter shows a single, unstructured band peaking at λPL = 455, 476 and 496 nm for 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ, respectively, reflecting emission from a CT state. The corresponding Stokes shifts are 80, 101 and 94 nm (4687, 5614 and 4714 cm−1), respectively, relative to the CT absorption band. In contrast to the absorption spectra, which show little variation with structure, a clear trend can be seen with the λPL values, with 2-PXZ-Nap emitting in the deep blue, followed by 1-PXZ-Nap and then 1,4-PXZ-Nap-PXZ emitting in the sky-blue regime. In particular, the difference between the two singly substituted structures demonstrates that the emission energy of this class of donor-naphthalene type emitters is influenced not only by the number of donors but also their substitution pattern around the naphthalene ring. We hypothesise that the apparent difference in behaviour of the CT absorption band and the CT emission band in relation to molecular structure is caused by the CT absorption band being partially masked by the much larger LE absorption band, which makes its peak position difficult to ascertain accurately. TDA-DFT calculations at the relaxed excited state of all three emitters show an equivalent stabilisation of ca. 0.56 eV (Table S3, ESI†) compared to the vertical excited state. We thus conclude that there is no increased stabilisation of the conformation of 2-PXZ-Nap in the relaxed excited state that is responsible for its apparent reduced Stokes shift compared to 1-PXZ-Nap.
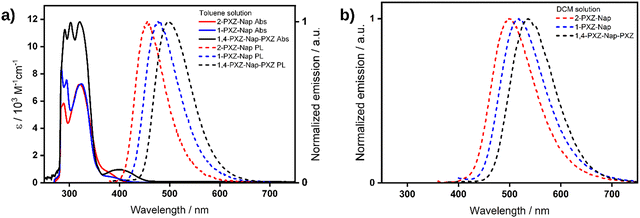 |
| | Fig. 6 (a) UV-vis absorption (Abs, solid lines) and photoluminescence (PL, dashed lines) of dilute toluene solutions of 2-PXZ-Nap (red), 1-PXZ-Nap (blue) and 1,4-PXZ-Nap-PXZ (black), λexc = 322, 340, 325 nm, respectively. (b) PL spectra of dilute dichloromethane solutions of the same, λexc = 326, 326, 400 nm, respectively. | |
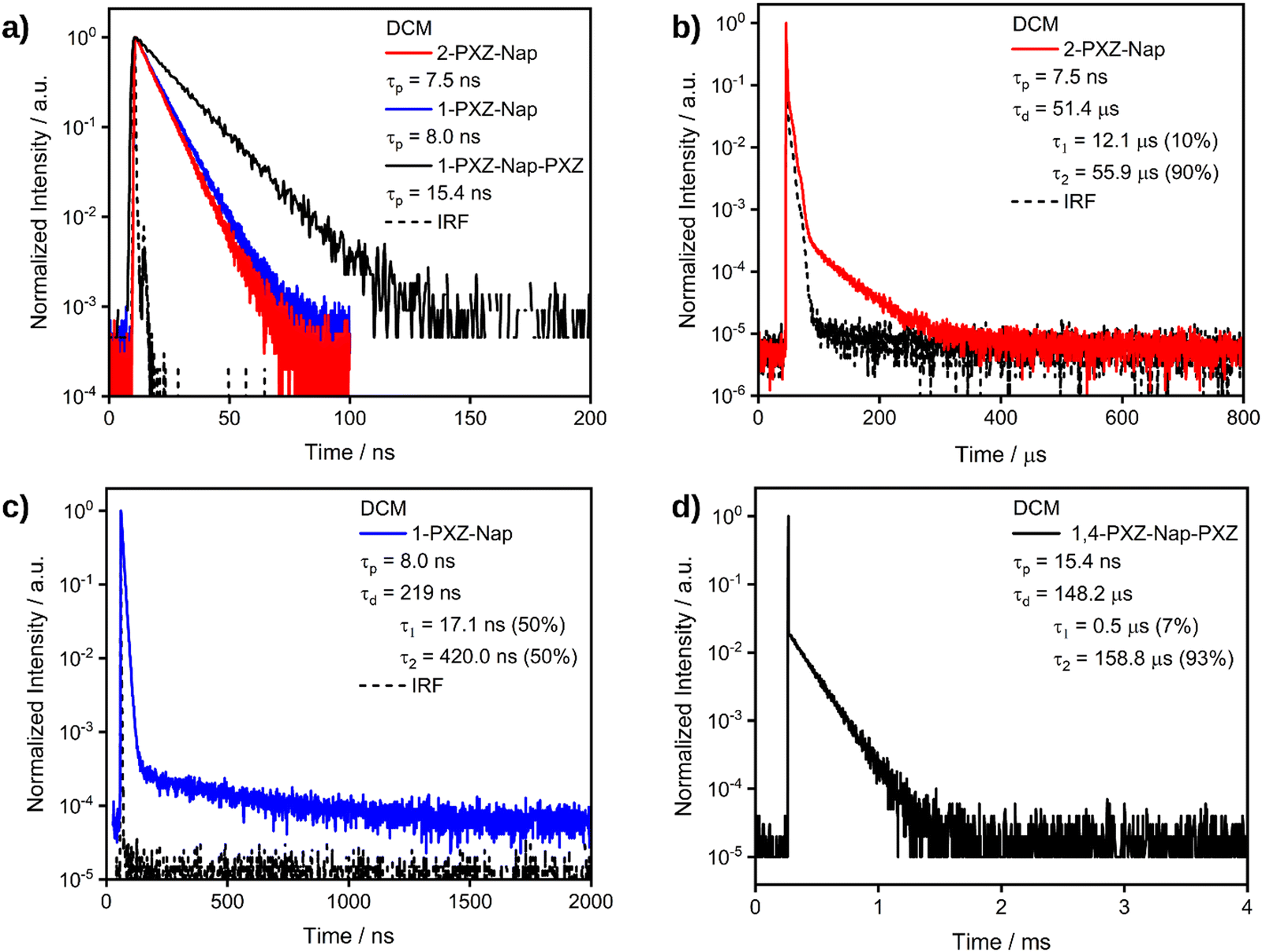
We then proceeded to investigate the photophysical properties of each of the emitters in greater detail in solution (Table 3). In toluene, the time-resolved PL decays of both of the singly substituted emitters exhibited a single decay process with comparable lifetimes, τp, of 2.8 and 3.1 ns for 2-PXZ-Nap and 1-PXZ-Nap, respectively, indicating that the origin of the emission is fluorescence. By contrast, the τp of 1,4-PXZ-Nap-PXZ is approximately double at 6.4 ns. No delayed lifetime was observed for any of the three compounds in toluene. In dichloromethane, the λPL of each emitter was red-shifted relative to toluene (Fig. 6b) to λPL = 502, 518 and 536 nm for 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ, respectively. These values correspond to a red-shift of 40–50 nm compared to toluene, and originate from the increased stabilisation of the singlet excited state of the emitters in the more polar DCM. In comparison to toluene, each emitter in DCM exhibited two distinct decay processes with the same emission maxima (Fig. 7), with prompt lifetimes τp, of 7.5, 8.0 and 15.4 ns, and delayed lifetimes, τd, of 51.4, 0.2 and 148.2 μs, for 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ respectively. The prompt lifetimes retain the same pattern as was observed in toluene, with the singly substituted emitters decaying roughly twice as quickly as the doubly substituted. However, all three decays were slower in DCM than in toluene which is consistent with an overall increase of the CT character of the first singlet excited state in the more polar solvent. By contrast, the delayed lifetimes show a much greater dependence on substitution pattern, spanning four orders of magnitude from 1-PXZ-Nap (219 ns) to 1,4-PXZ-Nap-PXZ (148 μs), although the decays of the two singly substituted emitters remain faster than that of the doubly substituted, as was observed in toluene. In addition to having a shorter lifetime, the delayed fluorescence of 1-PXZ-Nap and 2-PXZ-Nap was also significantly weaker than that of 1,4-PXZ-Nap-PXZ, as evidenced by their respective decay curves (Fig. 7). The delayed emission in all three emitters is biexponential, indicating contribution from multiple emissive states, and the ratio of the two delayed processes is smaller in 1-PXZ-Nap and 2-PXZ-Nap than in 1,4-PXZ-Nap-PXZ (τ1:τ2 = 50:50, 10:90 and 7:93, respectively). Considering the large dihedral angle between the phenoxazine and naphthalene, and the puckering of the phenoxazine moiety observed in the crystal structure (Fig. 4), we hypothesize that the multiple lifetimes originate from different conformers. The ΦPL of 1,4-PXZ-Nap-PXZ is 40% in degassed DCM solution, compared to only 22% in aerated DCM, indicating a significant contribution from triplet excited states. Owing to the poor ΦPL recorded in the film state (see below), the ΦPL of 2-PXZ-Nap and 1-PXZ-Nap were not measured in DCM.
Table 3 Summary of the photophysical properties of the three emitters in solution
| Emitter |
Toluene |
Dichloromethane |
2MeTHF |
|
λ
abs
/nm (ε/103 M−1 cm−1) |
λ
PL
/nm |
τ
p
/ns |
λ
PL
/nm |
Φ
PL
/% |
τ
p
/ns |
τ
d
/μs |
ΔEST/eV |
|
Estimated from the peak of the measured spectrum.
Photoluminescence quantum yield in toluene relative to quinine sulfate in 1 M H2SO4 (ΦPL = 54.6%), measured in degassed solution.22 Value in parentheses is in air.
Prompt (τp) and delayed (τd) lifetime. NR: not recorded. No delayed emission was observed in toluene. Steady-state and time-gated spectra in 2-MeTHF are available in the ESI (Fig. S15). λexc = 322, 340, 325 nm (toluene λPL, 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ, respectively), 375 nm (toluene τp), 326, 326, 400 nm (DCM λPL, 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ, respectively), 360 nm (DCM ΦPL), 326, 326, 375 nm (DCM, τp and τd, 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ, respectively), and 310 nm (2-MeTHF ΔEST).
|
|
2-PXZ-Nap
|
375 (0.81) |
455 |
2.8 |
502 |
NR |
7.5 |
51.4 |
0.52 |
|
1-PXZ-Nap
|
375 (0.48) |
476 |
3.1 |
518 |
NR |
8.0 |
0.2 |
0.40 |
|
1,4-PXZ-Nap-PXZ
|
402 (0.83) |
496 |
6.4 |
536 |
40 (22) |
15.4 |
148.2 |
0.24 |
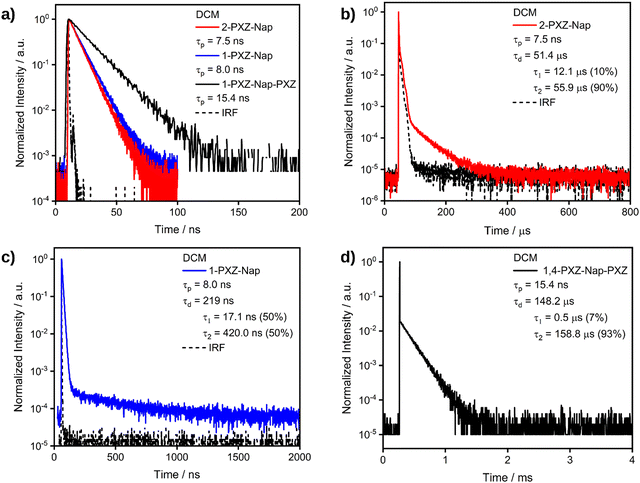 |
| | Fig. 7 (a) TRPL of the prompt decay process of 2-PXZ-Nap-PXZ, 1-PXZ-Nap-PXZ and 1,4-PXZ-Nap-PXZ. (b) TRPL of the delayed decay process of 2-PXZ-Nap. (c) TRPL of the delayed decay process of 1-PXZ-Nap. (d) TRPL of the delayed decay process of 1,4-PXZ-Nap-PXZ. Measured in dilute DCM solutions with λexc: 375 nm in all cases. | |
In 2-methyltetrahydrofuran (2-MeTHF), large ΔEST of 0.52 and 0.40 eV were recorded for 2-PXZ-Nap and 1-PXZ-Nap, respectively. These large singlet–triplet gaps result in very sluggish triplet to singlet up-conversion and are the cause of the low delayed emission intensity observed in Fig. 7b and c. By comparison, 1,4-PXZ-Nap-PXZ has a significantly smaller ΔEST of 0.24 eV, which results in more efficient RISC, and as such exhibits a greater proportion of delayed emission (Fig. 7d). None of the experimentally obtained ΔEST values correspond well to the DFT predicted values, which all approach zero. This likely suggests that the geometrically relaxed structures that have approximately orthogonally linked donor and acceptor groups, as observed both computationally and in the crystal structures, are not the predominantly emissive species in 2-MeTHF glass. This is to be expected, considering the ca. 0 oscillator strength predicted for the orthogonal structures. Further, DFT computations reveal an equally small ΔEST and approximately equivalent HSO in both DCM and toluene (Table S4, ESI†), and so differences in the RISC rate of these structures are unlikely to explain the switching on of the delayed emission in DCM only. We instead suggest that non-radiative decay from T1 is relatively much more suppressed in DCM.
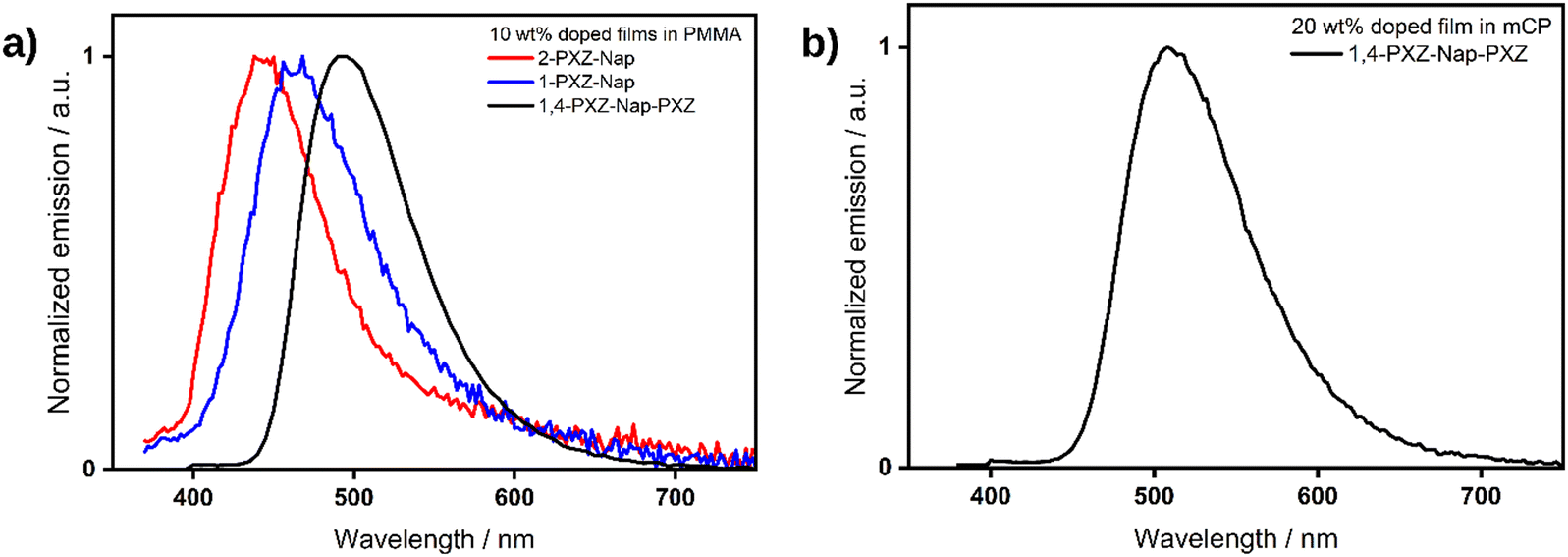
We next investigated the photophysical properties in the solid state. In spin-coated 10 wt% doped films using polymethylmethacrylate (PMMA) as a host, the PL is slightly blue-shifted compared to that in toluene at λPL of 441, 462 and 491 nm for 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ, respectively (Table 4). The trend in λPL between the three emitters, however, remained the same as in toluene. The photoluminescence quantum yields, ΦPL, are very low for the two singly substituted emitters at ca 2% but is notably higher in 1,4-PXZ-Nap-PXZ at 22%, which decreases to 18% upon exposure to air. Time-resolved PL of the films revealed the same pattern as was observed in toluene (Fig. 8), with a rapid and broadly equivalent τp of 2.9 and 3.6 ns for 2-PXZ-Nap and 1-PXZ-Nap, respectively, and a longer τp of 11.7 ns for 1,4-PXZ-Nap-PXZ. The τd of 1,4-PXZ-Nap-PXZ film is 8.0 μs, significantly faster than the 148.2 μs observed in dilute DCM solution, and retains the biexponential character. Compared to DCM, the ratio of the two delayed lifetimes is much small at 33:67, indicating a greater contribution to the emission from multiple emissive states. This is typical of emission from doped glassy films, in which the emitter is partially frozen in multiple different conformations, resulting in varying emission lifetimes.23–25 Neither of the singly substituted emitters demonstrated any delayed emission in the film state.
Table 4 Summary of the photophysical properties of the three emitters in doped films
| Emitter |
10 wt% in PMMA |
10 wt% in mCP |
20 wt% in mCP |
|
λ
PL
/nm |
Φ
PL
/% |
τ
p
/ns |
τ
d
/μs |
Φ
PL/% |
λ
PL
/nm |
Φ
PL/% |
τ
p
/ns |
τ
d
/μs |
|
Estimated from the peak of the measured spectrum.
Measured under vacuum. Values in parentheses are in air. ΦPL values measured using an integrating sphere.
Not observed. NR: not recorded. PMMA: polymethylmethacrylate, mCP: 1,3-bis(N-carbazolyl)benzene. λexc = 340, 340, 360 nm (PMMA λPL and ΦPL, 10 wt% mCP; 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ respectively), 375 nm (PMMA τp and τd) and 340 nm (20 wt% mCP).
|
|
2-PXZ-Nap
|
441 |
<1 (2) |
2.9 |
—c |
7 (2) |
NR |
NR |
NR |
NR |
|
1-PXZ-Nap
|
462 |
2 (1) |
3.6 |
—c |
7 (2) |
NR |
NR |
NR |
NR |
|
1,4-PXZ-Nap-PXZ
|
491 |
22 (18) |
11.7 |
8.0 |
32 (7) |
508 |
48 (7) |
88.2 |
22.7 × 103 |
 |
| | Fig. 8 (a) TRPL of 2-PXZ-Nap and 1-PXZ-Nap in 10 wt% doped films in PMMA measured by time-correlated single photon counting (TCSPC), λexc: 375 nm. (b) TRPL of 1,4-PXZ-Nap-PXZ in 10 wt% doped films in PMMA measured by time-correlated single photon counting (TCSPC), in λexc: 375 nm. The data points at ca. 25 and 30 μs are an artefact of the measurement process. | |
The PL spectra were then measured in an OLED relevant host of suitably high triplet energy, 1,3-bis(N-carbazolyl)benzene (mCP) at 10 wt% doping concentration. The ΦPL increased to 7% for each of 2-PXZ-Nap, 1-PXZ-Nap and 32% for 1,4-PXZ-Nap-PXZ. There is also a clear decrease in the ΦPL in air, implying that available triplet excited states are being quenched by oxygen, down to 7% for 1,4-PXZ-Nap-PXZ and 2% for both 2-PXZ-Nap and 1-PXZ-Nap, although in the latter case the change in ΦPL is close to the measurement error of ±5%.22 Upon increasing the doping concentration to 20 wt%, the ΦPL of the 1,4-PXZ-Nap-PXZ film increased to 48%, which is the highest ΦPL we recorded for this emitter. A full doping screen for the three emitters is available in the ESI† (Table S5). In the 20 wt% mCP film, the λPL red-shifted by 17 nm to 508 nm (Fig. 9) compared to the 10 wt% PMMA, likely owing to increased contribution to the emission from aggregates at the higher doping concentration. Temperature-dependent time-resolved PL decay measurements of the 20 wt% mCP film are shown in Fig. 10. As the temperature increases, the initial proportion of delayed emission also increases (Fig. 10b), which is consistent with a thermally activated pathway in which there is more energy available to upconvert triplets to singlets. The emission lifetime of 1,4-PXZ-Nap-PXZ also decreases at higher temperature, as evidenced from the steeper slope (Fig. 10a), indicating a faster decay process. This is caused by increased rates of both RISC and non-radiative decay. Finally, the total proportion of delayed emission appears to decrease at elevated temperature. This is caused by the increase in non-radiative decay, which competes with triplet up-conversion and results in reduced delayed emission at long emission times. In considering 2-PXZ-Nap and 1-PXZ-Nap, we note that: (1) TDA-DFT calculations predicted a similarly small ΔEST for the two singly-substituted emitters as the doubly substituted emitter; (2) the measured ΦPL of the films was extremely low, even under vacuum; (3) the delayed lifetime in solution was extremely short. We therefore contend that the absence of TADF in the film state of 2PXZ-Nap and 1-PXZ-Nap is largely driven by quenching of the triplet states through non-radiative decay pathways, which simultaneously explains the low ΦPL and short/absent delayed emission.
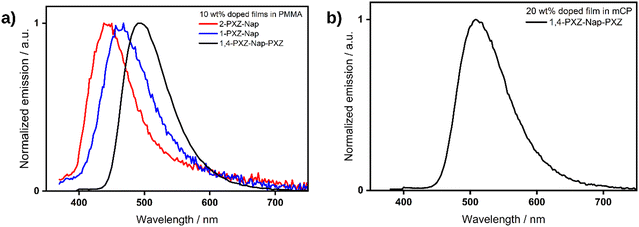 |
| | Fig. 9 (a) PL spectra of 10 wt% doped PMMA films of 2-PXZ-Nap (red), 1-PXZ-Nap (blue) and 1,4-PXZ-Nap-PXZ (black), λexc = 340, 340, 360 nm, respectively. (b) PL of 1,4-PXZ-Nap-PXZ in 20 wt% mCP doped film. λexc: 340 nm. | |
 |
| | Fig. 10 (a) Temperature-dependent TRPL of the 20 wt% film of 1,4-PXZ-Nap-PXZ in mCP. (b) Inset of the same. λexc: 375 nm. | |
Electroluminescent (EL) device
As only 1,4-PXZ-Nap-PXZ was identified as showing TADF in the film state, we only assessed the potential of this compound as an emitter in an OLED. The optimized device structure used was ITO/N,N′-di(1-naphthyl)-N,N′-diphenyl-(1,1′-biphenyl)-4,4′-diamine (NPB) (30 nm)/tris(4-carbazoyl-9-ylphenyl)amine (TCTA) (20 nm)/20 wt% 1,4-PXZ-Nap-PXZ in mCP (20 nm)/2,8-bis(diphenyl-phosphoryl)-dibenzo[b,d]thiophene (PPT) (10 nm)/1,3,5-tris(3-pyridyl-3-phenyl)benzene (TmPyPB) (40 nm)/LiF (1 nm)/Al (100 nm). The electroluminescence (EL) spectrum of this device along with the recorded efficiency data are presented in Fig. 11. The EL peak, λEL, at 505 nm is blue-shifted by 3 nm compared to the PL of the 20 wt% mCP film, corresponding to Commission Internationale de l'Éclairage (CIE) coordinates of (0.23, 0.47). The peak external quantum efficiency (EQEmax) is 11.2%, clearly more than the 5% theoretical limit for fluorescent OLEDs. Assuming Lambertian emission and values of γ = 1.0, nout = 0.2 and using the measured ΦPL of 48%, this EQEmax is larger than the theoretical maximum of 9.6%, assuming isotropic alignment of the transition dipole moment (TDM). This suggests that there is enhanced outcoupling efficiency of the device,4 implying a degree of preferential horizontal alignment of the TDM of the emitter in the film state. The EQE decreases relatively severely at higher luminance, approaching 1% at 100 cd m−2. Considering the long-delayed lifetime of 22.7 ms and the high doping concentration of the 20 wt% doped film in mCP, this efficiency roll-off is likely caused by the accumulation of triplets leading to emission degradation via triplet–triplet annihilation and/or triplet–polaron quenching. Future devices may be able to mitigate this issue through optimisation, particularly by either using a host that promotes a faster delayed emission or that uses a lower emitter doping concentration. To the best of our knowledge, this is the first demonstrated TADF OLED utilising a naphthalene-based emitter. The turn-on voltage (Von) was modest at 3.2 V and the peak power efficiency (PEmax) was 28.7 lm W−1. Although the peak performance of the device establishes the naphthalene-acceptor based design as an effective strategy for designing efficient OLEDs, the severe efficiency roll-off at higher luminance values indicates that there remains significant effort to improve the device performance further.
 |
| | Fig. 11 Electroluminescence spectrum and efficiency data of the OLED. | |
Theoretical investigation of larger acenes
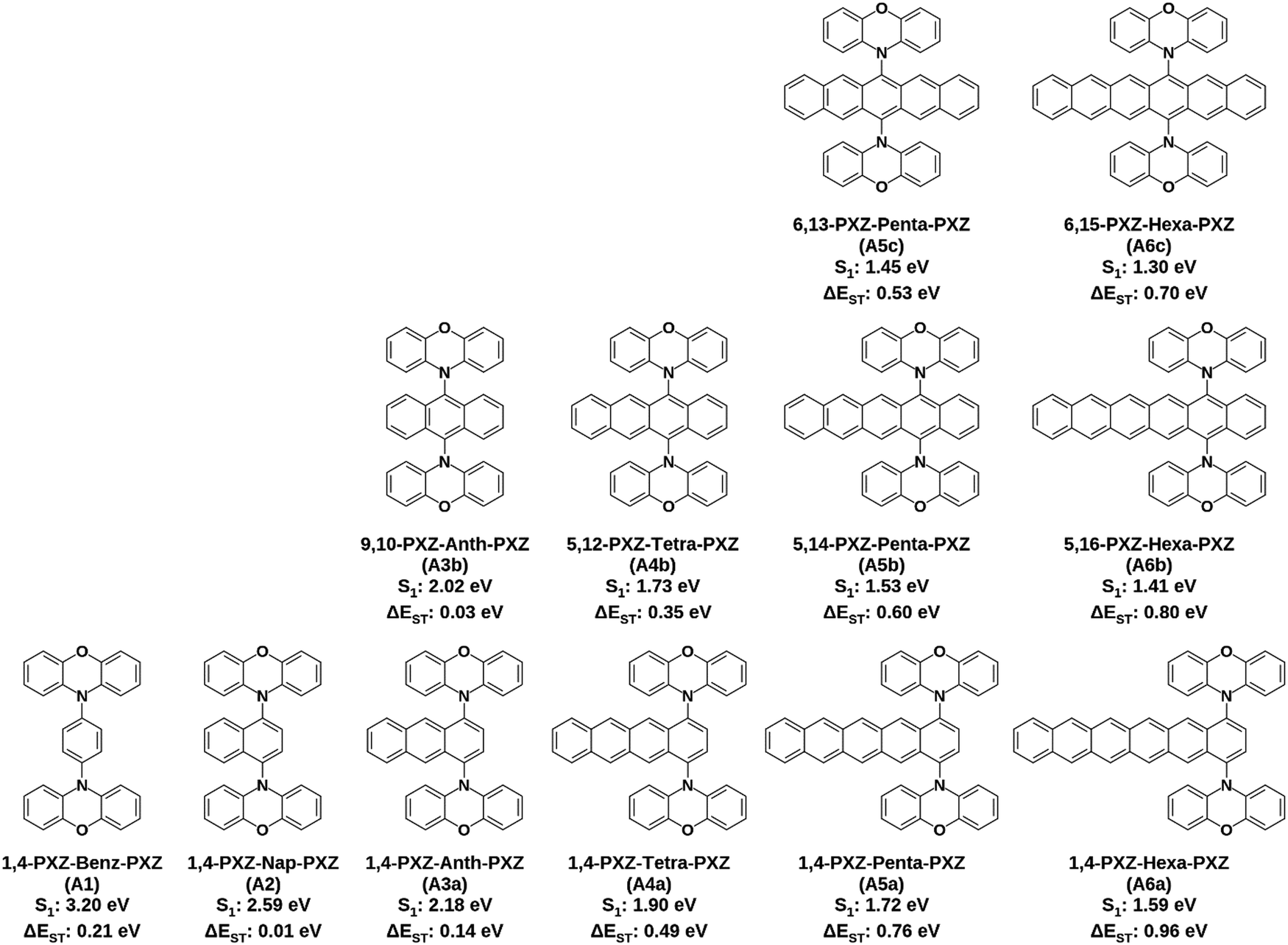
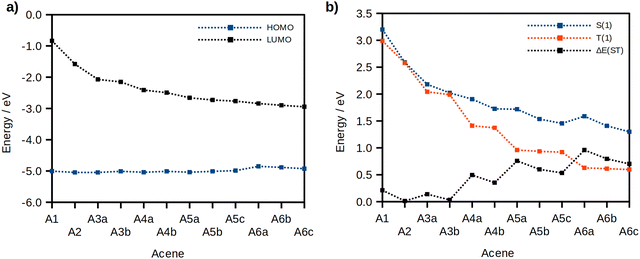
Considering the results obtained from the device based on 1,4-PXZ-Nap-PXZ we next chose to investigate in silico whether an even larger acene acting as an electron-acceptor would also yield compounds likely to show TADF. To this end we designed an additional 11 molecules based on the PXZ-acene-PXZ structure where acene is one of either benzene, anthracene, tetracene, pentacene or hexacene and the two donors were placed in all symmetrical combinations on either side of the ring system (Fig. 12). The results from this computational study are shown in Fig. 13 and 14. As the size of the acene part of the compound is increased, the energy of the LUMO is substantially stabilized because the LUMO is localized to the acene moiety (Fig. 13). Meanwhile, the HOMO is almost unchanged since it remains largely localized to the phenoxazine units, except in the hexacene derivatives where the HOMO is instead localized to the acene (the HOMO−1 is now localized on the phenoxazine unit), which is accompanied by a minor destabilization effect. Between the different substitution patterns, there is a simultaneous stabilization of the LUMO and a slight destabilization of the HOMO as the phenoxazines are placed closer to the centre, but the change in the LUMO is greater, resulting in a decrease of the electronic band gap. Hexacene again acts as an exception, with both the HOMO and LUMO stabilized simultaneously as the donors retreat from the periphery of the acene. In all cases, the stabilization of the LUMO is associated with a stronger electronic interaction between the LUMO of the phenoxazine and the acene when the donors are placed near the centre of the acene core. A small coefficient of the LUMO density is therefore located on the phenoxazine donor (Fig. S1, ESI†) and this coefficient is greater in the centrally substituted case, thus leading to a stronger stabilization of the LUMO. This stronger electronic interaction is a consequence of the larger LUMO density near the centre of the acene core as opposed to the outer rings.
 |
| | Fig. 12 Structures, names, S1 energies and ΔEST of the 12 emitters investigated, calculated at the PBE0-D3(BJ)/6-31G(d,p) level of theory in the gas phase. | |
 |
| | Fig. 13 (a) Computed HOMO and LUMO energies and (b) S1 and T1 energies calculated at the PBE0-D3(BJ)/6-31G(d,p) level of theory in the gas phase. | |
 |
| | Fig. 14 (a) The computed lowest energy locally excited singlet (1LE), locally excited triplet (3LE), charge-transfer singlet (1CT) and charge-transfer triplet (3CT), and (b) the computed S1 and T1 energies (as shown in Fig. 13b) of the centrally substituted acenes only, calculated at the PBE0-D3(BJ)/6-31G(d,p) level of theory in the gas phase. | |
Across the entire series, the S1 state retains a large CT character and is mostly described by a HOMO to LUMO transition, except in the hexacene derivatives where it is best described by a HOMO−1 to LUMO transition (Table 5). Correspondingly, a significant red-shift in S1 is observed as the acene size increases due to the increasing stabilization of the LUMO (Fig. 13b), with 1,4-PXZ-Hexa-PXZ (S1 = 1.59 eV) and 6,15-PXZ-Hexa-PXZ (S1 = 1.30 eV) exhibiting the smallest S1 energies for their respective substitution patterns. We also observed that the magnitude of the S1 energy red-shift is larger when the donors are placed closer to the centre of the acene core, consistent with the trend in the electronic band gap, and this effect is more prominent in the larger acenes. As such, the S1 of the terminally substituted hexacene derivative (1,4-PXZ-Hexa-PXZ, A6a, S1 = 1.59 eV) is higher in energy than that of the centrally substituted pentacene derivative (6,13-PXZ-Pent-PXZ, A5a, S1 = 1.45).
Table 5 Principal transitions and resulting nature of the S1 and T1 excited states
| Emitter |
S1 principal transitions (probability/%) |
S1 naturea |
T1 principal transitions (probability/%) |
T1 naturea |
|
CT = charge-transfer, HLCT = hybrid local and charge transfer, LE = local excitation.
|
|
A1
|
HOMO → LUMO (99) |
CT |
HOMO−1 → LUMO+2 (37) |
HLCT |
| HOMO → LUMO+3 (25) |
| HOMO → LUMO+1 (30) |
|
A2
|
HOMO → LUMO (99) |
CT |
HOMO → LUMO (99) |
CT |
|
A3a
|
HOMO → LUMO (99) |
CT |
HOMO−2 → LUMO (94) |
LE |
|
A3b
|
HOMO → LUMO (99) |
CT |
HOMO−2 → LUMO (95) |
LE |
|
A4a
|
HOMO → LUMO (98) |
CT |
HOMO−2 → LUMO (94) |
LE |
|
A4b
|
HOMO → LUMO (99) |
CT |
HOMO−2 → LUMO (95) |
LE |
|
A5a
|
HOMO → LUMO (98) |
CT |
HOMO−2 → LUMO (94) |
LE |
|
A5b
|
HOMO → LUMO (99) |
CT |
HOMO−2 → LUMO (95) |
LE |
|
A5c
|
HOMO → LUMO (99) |
CT |
HOMO−2 → LUMO (95) |
LE |
|
A6a
|
HOMO−1 → LUMO (97) |
CT |
HOMO → LUMO (94) |
LE |
|
A6b
|
HOMO−1 → LUMO (98) |
CT |
HOMO → LUMO (94) |
LE |
|
A6c
|
HOMO−1 → LUMO (99) |
CT |
HOMO → LUMO (95) |
LE |
The evolution of the T1 energy differs significantly from that of the S1 because of the competing contributions from states possessing locally excited (3LE) and charge-transfer (3CT) character (Fig. 14a). For the phenyl and naphthalene derivatives, the T1 state is predominantly CT in character (Table 5). However, starting from the anthracene derivative, the locally excited triplet state localized on the acene core becomes more stabilised than the 3CT. This arises because the energy ordering of the triplet states of donor–acceptor compounds is ruled by the interplay between the magnitude of the electronic interaction between the electron-donating and the electron-accepting units, which tends to stabilize the 3CT, and the exchange interaction which favours the 3LE as the lowest triplet energy state. As a result, the energy of the T1 state decreases significantly with increasing ring size but is essentially invariant across different substitution patterns where the acene acceptor remains constant since the T1 transition is essentially localized on the acene bridge. The corresponding locally-excited singlet state (1LE), meanwhile, remains higher in energy than the charge-transfer state (1CT) because the exchange interaction energy does not affect singlets.
As a direct consequence, we observed a sizeable decrease in ΔEST from 0.21 to 0.01 eV for the phenyl and naphthalene derivatives respectively, explained by the larger delocalization of the LUMO, which further strengthens the CT character of both S1 and T1. However, starting from the anthracene derivatives, the disproportionate stabilisation of the locally excited triplet compared to the charge-transfer singlet results in a steep increase in ΔEST (Fig. 14b), up to a maximum of 0.96 eV in 1,4-PXZ-Nap-PXZ (A6a). Conversely, there is a stabilization of the 1CT as the donor position is moved from the terminus of the acene towards the centre, while the 3LE remains constant, resulting in a sizeable decrease in ΔEST, and as such the centrally substituted hexacene derivative (6,15-PXZ-Hex-PXZ, A6c) has a noticeably smaller ΔEST of 0.70 eV.
With these results in hand, it becomes clear that of all the acenes evaluated naphthalene is the most ideally suited to act as an acceptor for developing TADF emitters. The ΔEST of the centrally substituted anthracene derivative was also minimized, but the inversion of the 3LE and 3CT states means this is not guaranteed for other donors, and any blue-shifting of the emission is likely to be accompanied by an increase in ΔEST. In particular, acenes larger than anthracene will likely possess a ΔEST that is too large for TADF, unless either the S1 energy is substantially stabilized and/or the T1 state is sufficiently destabilised. This observation is corroborated by recent reports in the literature of donor–acceptor emitters containing larger acenes such as anthracene, pyrene and perylene (Fig. 15). The emitters DPAAnCN and CzAnCN have been previously reported by our group26 and consist of a cyanoanthracene core centrally substituted with diphenylamine and carbazole, respectively. Similarly, the emitters Pery-2PXZ and Pery-2PTZ, reported by Li et al.,27 contain a perylene core substituted with either phenoxazine or phenothiazine as donor groups, respectively. Each of these four emitters has a large ΔEST of >0.6 eV (in DPAAnCN and CzAnCN this ΔEST is reported from TD-DFT,26 while in Pery-2PXZ and Pery-2PTZ it is calculated by the authors from the difference in energy of the TD-DFT-calculated T1 energy and the measured onset of fluorescence27), which is too large for efficient TADF. Instead, these emitters operate via alternative triplet harvesting mechanisms. In the case of DPAAnCN and CzAnCN, the exciton harvesting mechanism was hypothesised to employ a ‘hot-exciton’ channel,26 while the perylene derivatives Pery-2PXZ and Pery-2PTZ were shown to operate via triplet–triplet-annihilation (TTA).27 Devices based on these hot-exciton emitters achieved appreciable EQEmax of 6.0% and 4.0% for the OLEDs with DPAAnCN and CzAnCN, respectively,26 while no devices were fabricated based on the TTA emitters. Finally, a family of pyrene derived emitters were reported by Park et al.,28 exemplified by 3Me-1Bu-TPPDA, which possesses a very large ΔEST of 0.84 eV and was reported to be fluorescent. However, it should be noted that an OLED based on this emitter achieved an EQEmax of 9.3%, in excess of the 5% limit for fluorescent devices. This strongly suggests that a triplet-harvesting mechanism is operational in the device.
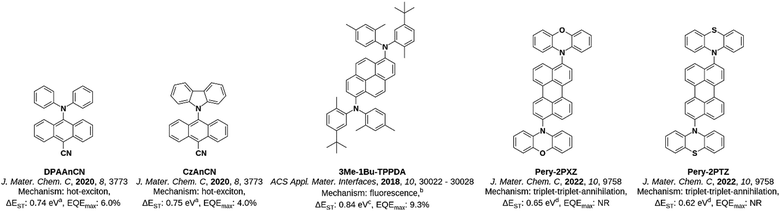 |
| | Fig. 15 The structure, emission mechanism and ΔEST of emitters based on larger acenes reported in the literature. aComputed at the TDA-PBE0-D3(BJ)/6-31G(d,p) level. bAs stated by the authors.28cThe difference in energy of the fluorescence peak measured in toluene at room temperature and the phosphorescence peak measured in toluene at 77 K with a 30 ms delay. dThe difference in energy of the onset of fluorescence measured in DCM at room temperature and the T1 state calculated at the TD-DFT-B3LYP/6-31G level. | |
Conclusions
Herein, we have conducted a computational screen of 36 naphthalene-containing emitters to evaluate their promise as TADF emitters. Three of the compounds, 2-PXZ-Nap, 1-PXZ-Nap and 1,4-PXZ-Nap-PXZ, were then identified as being the most promising and were subsequently synthesised. The photophysical properties of these emitters in toluene and DCM solution and doped films in PMMA and mCP were measured. A high photoluminescence quantum yield of 48% and evidence of TADF were recorded for 1,4-PXZ-Nap-PXZ as a 20 wt% doped film in mCP. An OLED employing this emitter demonstrated an EQEmax = 10.5%, PEmax = 28.7 lm W−1 with green emission (λEL = 505 nm). This is, to the best of our knowledge, the first reported OLED utilising a TADF emitter in which naphthalene alone acts as the acceptor moiety. This initial study clearly demonstrates the potential of the acene acceptor design for the development of TADF emitters. Finally, a comprehensive computational study has been presented to demonstrate why naphthalene alone of the linear acenes is suitable for constructing donor-acene-donor TADF emitters of this type.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Some computations were performed using resources kindly provided by the University of Mons (UMONS), supported by the Belgian National Fund for Scientific Research (FRS-FNRS). These computational resources were provided by the Consortium des Équipements de Calcul Intensif (CÉCI) funded by F. R. S.-FNRS under Grant 2.5020.11. Y. O. acknowledges funding by the Fonds de la Recherche Scientifique-FNRS under Grant no. F.4534.21 (MIS-IMAGINE).
References
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- S. Reineke and M. A. Baldo, Phys. Status Solidi A, 2012, 209, 2341–2353 CrossRef CAS.
- T. D. Schmidt, M. Flämmich, B. J. Scholz, D. Michaelis, C. Mayr, N. Danz and W. Brütting, Org. Photonics V, 2012, 8435, 843513 Search PubMed.
- F. Tenopala-Carmona, O. S. Lee, E. Crovini, A. M. Neferu, C. Murawski, Y. Olivier, E. Zysman-Colman and M. C. Gather, Adv. Mater., 2021, 33, 2100677 CrossRef CAS PubMed.
- P. Nuss and M. J. Eckelman, PLoS One, 2014, 9, e101298 CrossRef PubMed.
- G. M. Mudd, Ore Geol. Rev., 2012, 46, 106–117 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- D. Hall, J. C. Sancho-García, A. Pershin, D. Beljonne, E. Zysman-Colman and Y. Olivier, J. Phys. Chem. A, 2023, 127, 4743–4757 CrossRef CAS PubMed.
- L. Zhu, H. Zhang, X. Peng, M. Zhang, F. Zhou, S. Chen, J. Song, J. Qu and W. Y. Wong, Dyes Pigm., 2021, 194, 109627 CrossRef CAS.
- J. Wang, J. Liang, Y. Xu, B. Liang, J. Wei, C. Li, X. Mu, K. Ye and Y. Wang, J. Phys. Chem. Lett., 2019, 10, 5983–5988 CrossRef CAS PubMed.
- S. Paredis, T. Cardeynaels, J. Deckers, A. Danos, D. Vanderzande, A. P. Monkman, B. R. Champagne and W. Maes, J. Mater. Chem. C, 2022, 10, 4775–4784 RSC.
- Y. Li, J.-Y. Liu, Y.-D. Zhao and Y.-C. Cao, Mater. Today, 2017, 20, 258–266 CrossRef.
- H. Park, J. Lee, I. Kang, H. Y. Chu, J. I. Lee, S. K. Kwon and Y. H. Kim, J. Mater. Chem., 2012, 22, 2695–2700 RSC.
- M. Mahmoudi, J. Keruckas, D. Volyniuk, V. Andrulevičienė, R. Keruckienė, E. Narbutaitis, Y. C. Chao, M. Rutkis and J. V. Grazulevicius, Dyes Pigm., 2022, 197, 109868 CrossRef CAS.
- Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui, J. J. Eriksen, Y. Gao, S. Guo, J. Hermann, M. R. Hermes, K. Koh, P. Koval, S. Lehtola, Z. Li, J. Liu, N. Mardirossian, J. D. McClain, M. Motta, B. Mussard, H. Q. Pham, A. Pulkin, W. Purwanto, P. J. Robinson, E. Ronca, E. R. Sayfutyarova, M. Scheurer, H. F. Schurkus, J. E. T. Smith, C. Sun, S.-N. Sun, S. Upadhyay, L. K. Wagner, X. Wang, A. White, J. D. Whitfield, M. J. Williamson, S. Wouters, J. Yang, J. M. Yu, T. Zhu, T. C. Berkelbach, S. Sharma, A. Y. Sokolov and G. K.-L. Chan, J. Chem. Phys., 2020, 153, 024109 CrossRef CAS PubMed.
- Y. Olivier, B. Yurash, L. Muccioli, G. D’Avino, O. Mikhnenko, J. C. Sancho-García, C. Adachi, T.-Q. Nguyen and D. Beljonne, Phys. Rev. Mater., 2017, 1, 075602 CrossRef.
- W. Zeng, S. Gong, C. Zhong and C. Yang, J. Phys. Chem. C, 2019, 123, 10081–10086 CrossRef CAS.
- Y. Olivier, M. Moral, L. Muccioli and J.-C. Sancho-García, J. Mater. Chem. C, 2017, 5, 5718–5729 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- G. Grabner, K. Rechthaler, B. Mayer, G. Köhler and K. Rotkiewicz, J. Phys. Chem. A, 2000, 104, 1365–1376 CrossRef CAS.
- B. Sk, S. Sharma, A. James, S. Kundu and A. Patra, J. Mater. Chem. C, 2020, 8, 12943–12950 RSC.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- T.-Y. Hwang, Y. Choi, Y. Song, N. S. A. Eom, S. Kim, H.-B. Cho, N. V. Myung and Y.-H. Choa, J. Mater. Chem. C, 2018, 6, 972–979 RSC.
- P. Sudhakar, A. Kumar Gupta, D. B. Cordes and E. Zysman-Colman, Chem. Sci., 2024, 15, 545–554 RSC.
- C. Si, Y.-N. Hu, D. Sun, K. Wang, X.-H. Zhang and E. Zysman-Colman, J. Mater. Chem. C, 2023, 11, 12174–12184 RSC.
- N. Sharma, M. Y. Wong, D. Hall, E. Spuling, F. Tenopala-Carmona, A. Privitera, G. Copley, D. B. Cordes, A. M. Z. Slawin, C. Murawski, M. C. Gather, D. Beljonne, Y. Olivier, I. D. W. Samuel and E. Zysman-Colman, J. Mater. Chem. C, 2020, 8, 3773–3783 RSC.
- M. Imran, J. Pang, J. Zhao and M.-D. Li, J. Mater. Chem. C, 2022, 10, 9758–9772 RSC.
- H. Jung, S. Kang, H. Lee, Y. J. Yu, J. H. Jeong, J. Song, Y. Jeon and J. Park, ACS Appl. Mater. Interfaces, 2018, 10, 30022–30028 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Nidhi
Sharma
ab,
Tomas
Matulaitis
ab,
Nidhi
Sharma
ab,
Tomas
Matulaitis