
The structure of Pd-functionalized UiO-67 during CO2 hydrogenation†
Alina A.
Skorynina
 *a,
Andrea
Lazzarini
bc,
Dag Kristian
Sannes
b,
Elizaveta G.
Kozyr
d,
Christian
Ahoba-Sam
b,
Silvia
Bordiga
bd,
Unni
Olsbye
*b and
Aram L.
Bugaev
*e
*a,
Andrea
Lazzarini
bc,
Dag Kristian
Sannes
b,
Elizaveta G.
Kozyr
d,
Christian
Ahoba-Sam
b,
Silvia
Bordiga
bd,
Unni
Olsbye
*b and
Aram L.
Bugaev
*e
aCELLS-ALBA Synchrotron Light Source, Carrer de la Llum, 2, 26, 08290 Cerdanyola del Vallès, Barcelona, Spain. E-mail: askorynina@cells.es
bCentre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, Sem Saelandsvei 26, 0315 Oslo, Norway. E-mail: unni.olsbye@kjemi.uio.no
cDepartment of Physical and Chemical Sciences, University of L’Aquila, Via Vetoio (“A.C. De Meis” building), 67100 L’Aquila, Italy
dDepartment of Chemistry and NIS Centre, University of Turin, Via Pietro Giuria, 7, 10125 Turin, Italy
eSuperXAS beamline, Paul Scherrer Institute, Forschungsstrasse 111, 5232 Villigen, Switzerland. E-mail: aram.bugaev@psi.ch
First published on 14th February 2024
Abstract
The detailed evolution of the local atomic and electronic structure of palladium nanoparticles (NPs) embedded in Zr-based metal–organic frameworks (MOFs) UiO-67 is presented. The samples were employed for CO2 hydrogenation under different temperatures and at total pressures of 1 or 8 bar. The effects of exposure to the reactive atmosphere and reaction products on the structure of MOFs and Pd NPs were monitored in operando mode by simultaneous X-ray diffraction and X-ray absorption spectroscopy measurements. Advanced analysis of large experimental datasets supported by theoretical simulations revealed the formation of mixed carbide and hydride phases of palladium and the adsorption of CO species as reaction intermediates at the surface of Pd NPs under the reaction conditions. According to available information, the observation of the Pd/Zr interface and its association with methanol formation was first obtained in this work for Pd–Zr-MOFs materials.
Introduction
Hydrogenation of CO2 is an important reaction attracting the attention of researchers over decades and becoming more and more crucial in the context of the green and circular economy. The particular challenge is the ability to reduce the main driver of global warming by converting CO2 into useful value-added products such as methanol. Prior to this, the hydrogenation of CO2 to CH4, also called the Sabatier reaction,1 was carried out at high temperatures (>300 °C) to promote the kinetics of the reaction. The most popular catalysts for methanation are supported metal systems.2–7 However, during the hydrogenation reaction in such catalytic systems, the catalyst ages, which is associated with gradual sintering of metal particles and/or adhesion of carbon atoms to nanoparticles (NPs), reducing the number of active sites involved in the reaction.8–12As a solution to the problem of particle sintering, there is a promising way, namely using a stable porous structure as a substrate, inside the pores of which nanoparticles of fixed sizes could be grown. Among all materials possessing such characteristics, we find metal–organic frameworks (MOFs).13–16 MOFs consist of metal/metal oxide nodes connected by organic linkers into three-dimensional porous crystalline structures. Since these nodes are active by themselves only in a limited number of MOF structures, it is proposed to carry out additional functionalization for catalytic purposes.17–21 Some of the most promising MOFs are the Zr-based UiO family, displaying exceptional thermal and chemical stability up to the required reaction temperatures.22–26 Also, in the case of UiO-66/67/68, functionalization has played an important role in enhancing the material potentialities, by insertion of other metals in the inorganic cornerstones27,28 and by functionalization of linkers with additional metals.29–33
Previously, our group has investigated how the CO2 hydrogenation reaction proceeds on Pt NPs grown inside UiO-67 pores, where the process was monitored by operando FT-IR spectroscopy.34,35 Furthermore, the successful synthesis of palladium NPs inside UiO-67 was studied by X-ray powder diffraction (XRPD) and X-ray absorption spectroscopy (XAS),36,37 where Pd structural changes were monitored during ethylene hydrogenation.32,38
In this work, we have studied structural evolution of Pd@UiO-67 during operando carbon dioxide hydrogenation by simultaneous measurements of XRPD, X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopies. The multivariate curve resolution alternating least squares (MCR-ALS) approach39–41 was applied to XANES spectra to extract pure species from the whole dataset during the CO2 hydrogenation reaction under different temperature and pressure conditions. The obtained experimental spectra were complemented by theoretical XANES simulations and density functional theory (DFT)-calculated atomic structures to define surface adsorbed species. XRPD and EXAFS analysis provided values for lattice expansion of the MOFs structure and, respectively, changes in the local environment of Pd atoms. Furthermore, the CO2 conversion was followed by FT-IR spectroscopy. Finally, the conditions used during XAS and FT-IR measurements were replicated in a catalytic test rig to establish structure/composition – performance correlations for the Pd@UiO-67 catalyst.
Results and discussion
A TEM image of the sample after activation is shown in Fig. 1. The formed Pd NPs, which are regarded to be the active species during CO2 hydrogenation, can be easily distinguished within the framework. Average size of the particles is ca. 2.3 nm (Fig. S1, ESI†),32 which is in good agreement with the results of EXAFS analysis (vide infra). The deviation from the normal distribution (see the inset in Fig. 1) may originate from the co-existence of the particles inside the pores and at the outer surface of MOF crystallites.21,32,36 | ||
| Fig. 1 TEM image of the UiO-67-Pd sample after activation. Size distribution analysis shown in the inset was performed by counting 240 particles from different images of the sample. | ||
Fig. 2 shows details of operando FT-IR spectra of UiO-67-Pd, namely the formate region (2800–2700 cm−1) and CO⋯Me interaction region (2150–1910 cm−1) collected during CO2 hydrogenation reaction. Differently from what was observed in our previous research with its Pt-analogue,35 the Pd catalyst operating at 1 bar total pressure did not develop the typical tiny band of the bidentate formate groups located at 2750 cm−1 (Fig. 2a),35,42 which we identified as the one responsible for methanol production. Indeed, catalytic data collected under the same reaction conditions confirmed such evidence. This behavior was also confirmed at different operating temperatures. Another difference from UiO-67-Pt, is the absence of the interaction of produced CO with Pd NPs that were formed during the activation procedure (Fig. 2b), meaning that CO adsorption is not favorable at such temperature and pressure conditions. However, by varying the total pressure to 8 bar, UiO-67-Pd catalyst started developing the typical bidentate formate spectroscopic feature at 2750 cm−1 (Fig. 2c).35 Such signal is constant throughout the whole catalytic test, similarly to the methanol selectivity that is strictly related to the presence of such species. Furthermore, higher pressure began to favor CO adsorption onto Pd NPs, as visible from gas-phase roto-vibrational signal of CO at 240 °C (red curve, Fig. 2d) and from its typical signal related to linear carbonyl species at 2050 cm−1 (brown curve, Fig. 2d).38,43–45
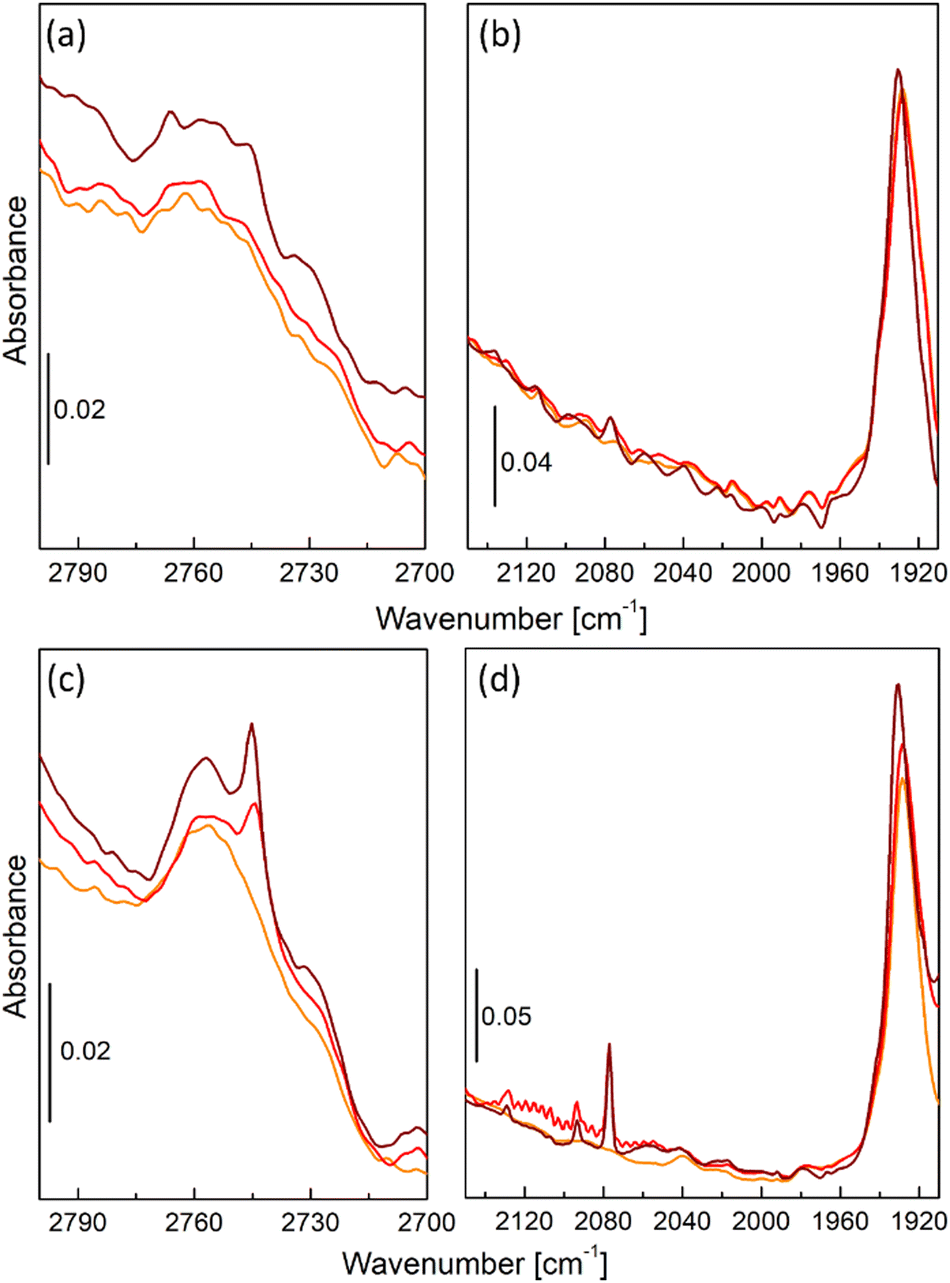 | ||
| Fig. 2 FT-IR spectra in the formate region (parts (a) and (c)) and CO⋯Me interaction region (parts (b) and (d)) of UiO-67-Pd in operando CO2 hydrogenation conditions under different total pressure, namely 1 bar (parts (a) and (b)) or 8 bar (parts (c) and (d)). Orange curves refer to activated catalyst at 240 °C, red curves refer to CO2 hydrogenation at 240 °C, brown curves refer to CO2 hydrogenation at 170 °C. | ||
The local structures of palladium and zirconium, as well the crystal structure of UiO-67 framework was followed in situ starting from the activation of as-prepared UiO-67-Pd under various conditions. Fitting of the EXAFS data collected under each of the experimental conditions provided the evolution of the first-shell Pd–Pd interatomic distances and the averaged first-shell coordination numbers (Fig. 3, parts (a) and (b), respectively). XANES data (Fig. S2, ESI†) confirm the successful reduction of Pd(II) with formation of Pd NPs, in agreement with our previous reports.36,37 The coordination numbers in the activated samples were close to 10 irrespective of the activation temperature (240 and 300 °C). The values correspond to the particle size of about 2.6 nm, which is slightly higher than in TEM due to the stronger influence of larger particles in atom-weighted EXAFS signal. Notably, the cell parameter of the UiO-67 framework decreases together with the formation of Pd NPs (Fig. 4). For the sample activated directly in the reaction mixture, lower values of coordination numbers (N = 9) were observed, which can be explained by the stabilization of the small particles due to the interaction with the reagents (vide infra).
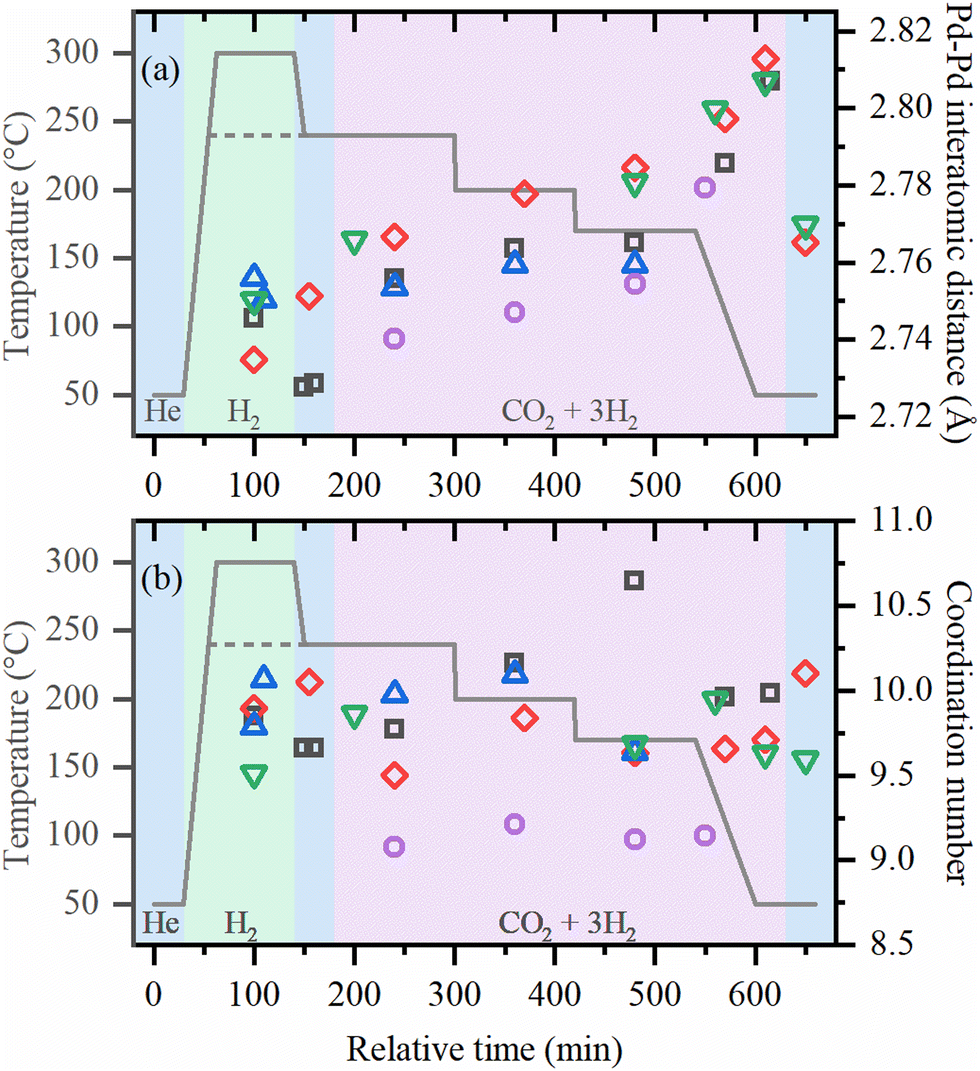 | ||
| Fig. 3 Pd–Pd interatomic distances (a) and coordination numbers (b) determined by first-shell fitting of EXAFS data collected for the samples activated at 300 °C in H2 and used for CO2 hydrogenation under 1 bar and 8 bar (black squares and red diamonds, respectively), activated at 240 °C in H2 and used for CO2 hydrogenation under 1 bar and 8 bar (blue up and green down triangles, respectively) and activated directly in reaction mixture at 1 bar at 240 °C (purple circles). Colored areas of the background correspond to different gas environment: blue – helium flow, green – hydrogen flow, pink – reaction mixture. | ||
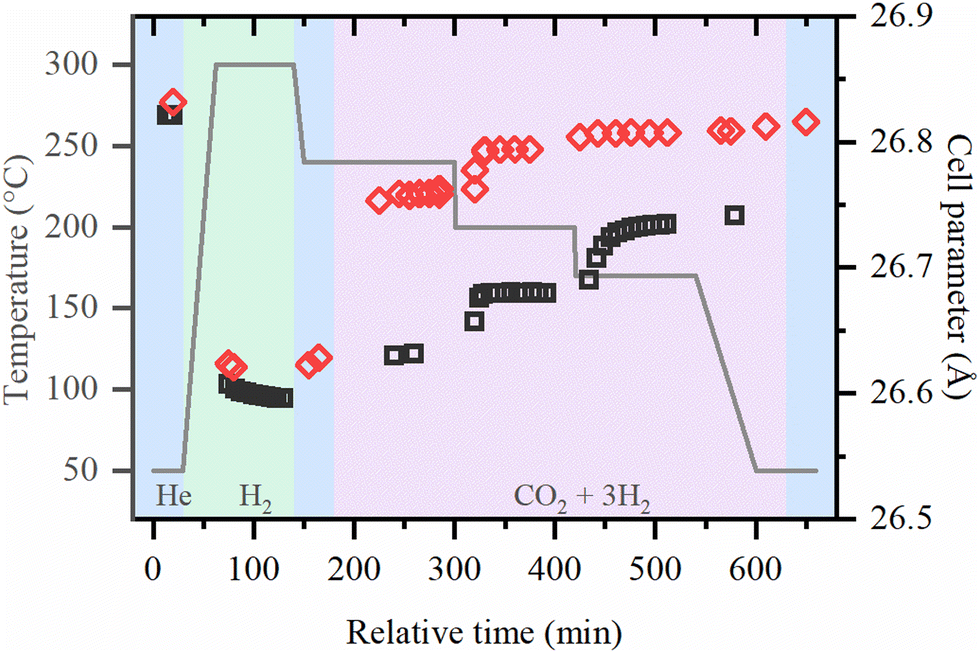 | ||
| Fig. 4 Cell parameter of UiO-67 samples during activation in H2 and reaction under 1 bar (black squares) and 8 bar (red diamonds) obtained by the refinement of XRPD data. | ||
Under reaction conditions, coordination numbers are stable, but interatomic distances systematically increase for all samples, being larger for lower temperatures and high pressures. After cooling down in the reaction mixture, the values of RPd–Pd ∼ 2.81 Å were observed, which is close to those reported for palladium hydrides.46–48 However, flushing with helium, performed for the samples investigated at high pressures, results in only partial decrease of RPd–Pd to ca. 2.77 Å which is considerably higher than RPd–Pd = 2.74 Å expected for metallic palladium, and exceeds the thermal expansion of Pd NPs.49 This indicates that apart from palladium hydride, which is known to form reversibly, irreversible formation of an additional phase occurs. For smaller particles formed after activation in the reaction mixture, shorter RPd–Pd values were observed under reaction conditions. This correlates with the fact that smaller particles have smaller hydrogen (or carbon) storage capacity50 resulting in a less pronounced cell expansion.
The typical XRPD pattern of UiO-67 (Fig. S3, ESI†) is preserved during both activation procedure and CO2 hydrogenation reaction, which proves the extreme stability of MOFs with UiO-67 structure. The evolution of the cell parameter is shown in Fig. 4. Under reaction conditions, the cell parameter is increasing with respect to the activated material in pure He and in H2/He mixture. The increase of the cell parameter is higher at high pressures and lower temperatures, which may indicate the adsorption of reactive molecules inside the pores of UiO-67.
To get further insights into the local structure of Pd sites, we analyzed XANES spectra, which are known to be sensitive to the presence of light atoms, such as H or C, that are not visible in Pd K-edge EXAFS.51–54 Principle component analysis (PCA) analysis revealed the presence of three independent species formed under reaction conditions (Fig. S4 and S5, ESI†). Then, MCR-ALS procedure was applied to represent the whole set of XANES data (Fig. S6, ESI†) by the three representative spectra (Fig. 5a) and their concentration profiles (Fig. 6 and Fig. S7–S9, ESI†). The first component (solid black line) corresponds to the metallic phase of palladium and its concentration is maximal for the fresh samples after activation in H2. The second component (dotted blue line) is characterized by a shift of the second peak towards smaller energies, due to the increased Pd–Pd distances, characteristic for palladium hydride.45,50,54 Furthermore, its concentration decreases when after activation in H2 the sample is flushed with He (Fig. 6 and Fig. S7, ESI†). However, the shape of the first peak is different from that reported for palladium hydrides46 and is slightly broadened toward higher energies, indicative of Pd–C bonds.55 Therefore, this component represents the Pd NPs with increased interatomic distances due to the presence of both H and C impurities in the bulk of the particles (Fig. 5c). The last component (dashed red line) is characterized by smaller interatomic distances, but the carbide-like features in the first peak are even more pronounced than in the second component. To reproduce the observed changes, the theoretical spectra were calculated for the structures with CO2, CO, CHx, H2O, O and OH placed on top, bridge or hollow sites (Fig. S10, ESI†) at of palladium surface. The highest similarity with the experimental data was observed in theoretical spectrum for bridge CO configuration, shown in Fig. 5b by dashed red line together with pure Pd surface (solid black). In agreement with the assignment made, the concentration of the last component (surface adsorbed CO) goes to zero after flushing with He, while the second one (bulk carbide) remains stable. Moreover, the separately measured XAS spectra for Pd@γ-Al2O3 NPs with a particle size of ca. 2.6 nm under CO flow prove the proposed model (Fig. S11, ESI†).
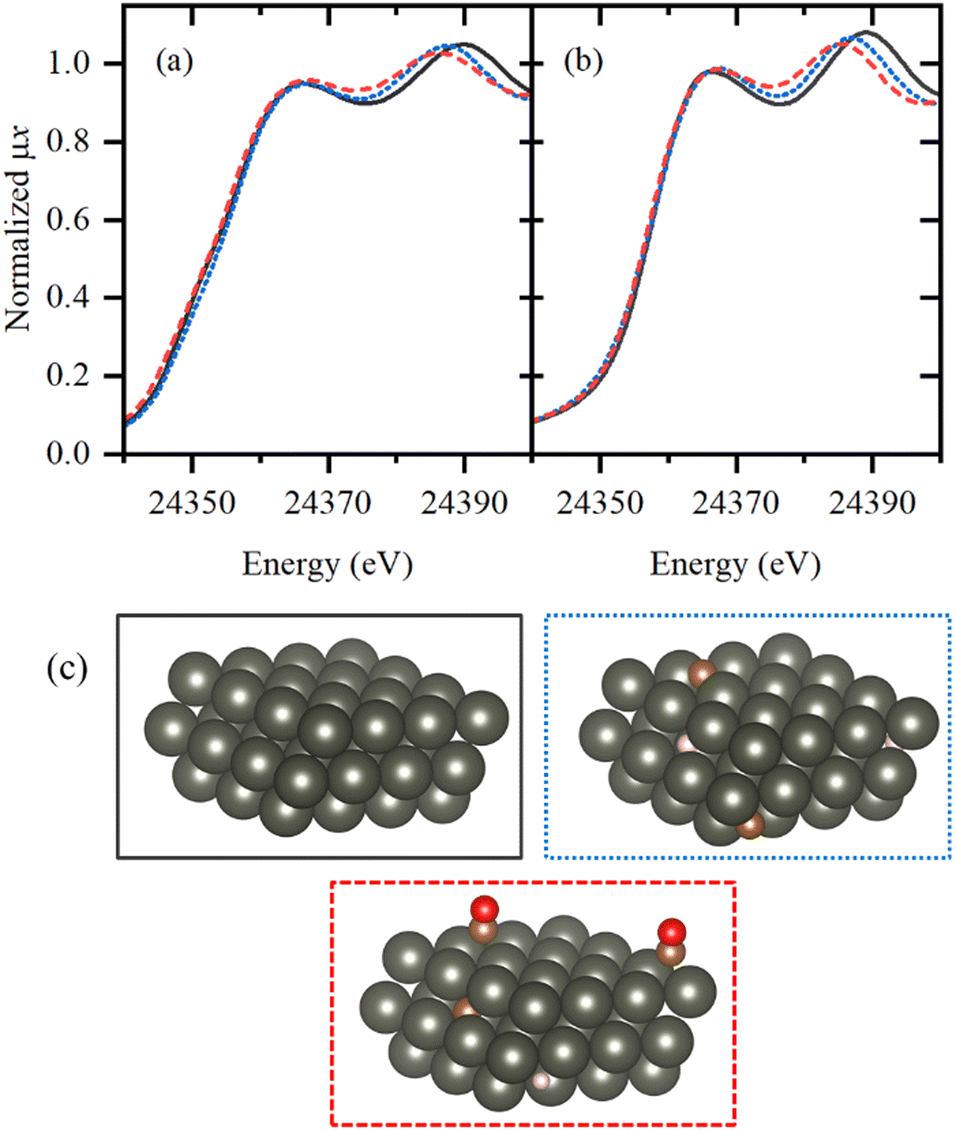 | ||
| Fig. 5 (a) Pd K-edge XANES spectra extracted from the whole experimental dataset by MCR approach. (b) Theoretical spectra calculated for pure Pd surface (solid black), bulk Pd with RPd–Pd = 2.8426 Å absorbed ca. 7% C and ca. 7% H atoms (dotted blue) and Pd surface with bridging CO molecule (dashed red). (c) Schematic illustration of atomic structures of palladium surfaces whose simulated XANES spectra reproduces the features observed in (the frame color is the same as for spectra in part (b)). Atoms color code: Pd – dark grey, C – brown, O – red, H – pink MCR components. | ||
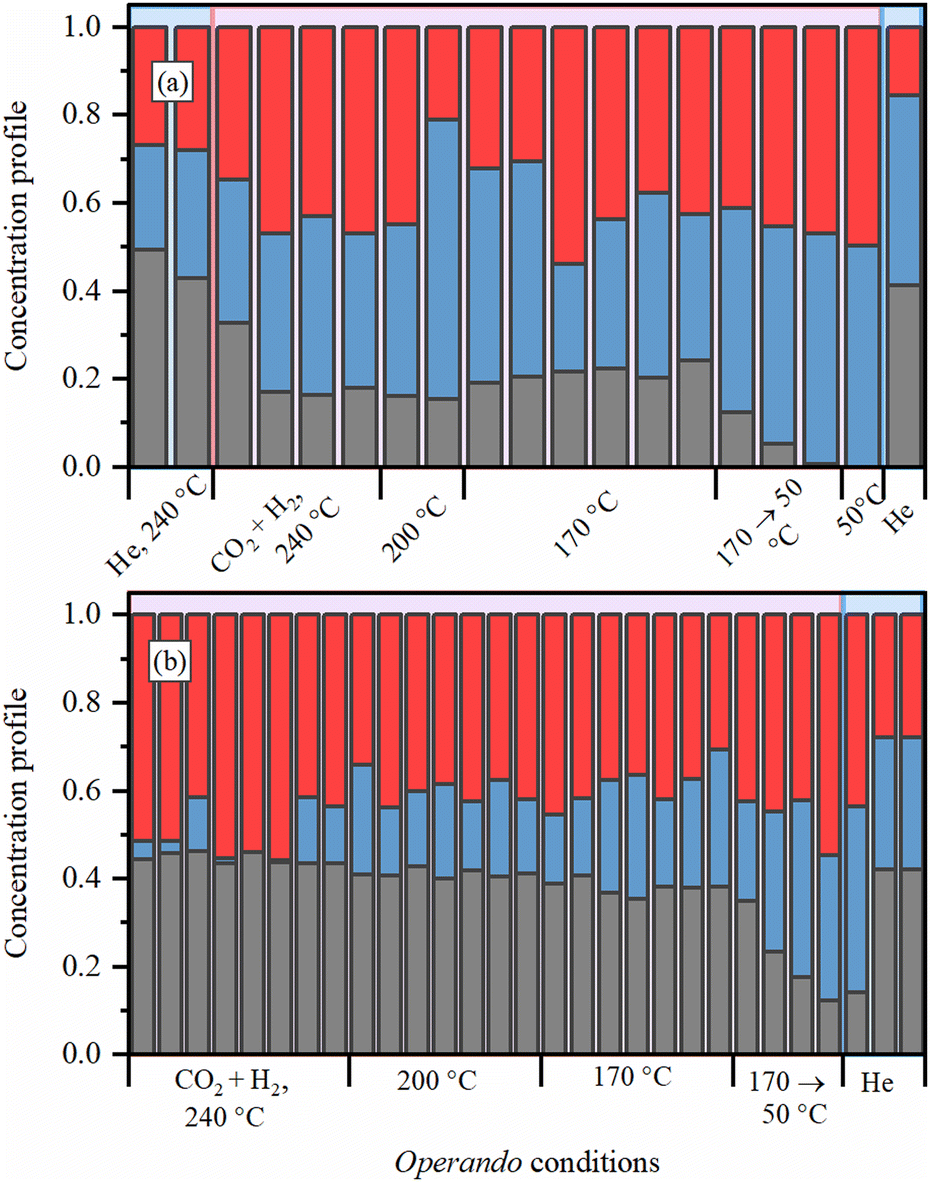 | ||
| Fig. 6 Evolution of MCR components (the color code is the same as for spectra presented in Fig. 5a) for the samples (a) activated in H2 at 300 °C and exposed to reaction mixture under 8 bar and (b) activated directly in the reaction mixture under 1 bar at 240 °C. | ||
Additional insights into the structure of UiO-67-Pd come from the Zr K-edge EXAFS. Due to the complex surroundings of Zr atoms on UiO-67 secondary building units, the fitting of EXAFS data requires a big number of parameters to be taken into consideration. In particular, coordination numbers (N), Debye–Waller factors (σ2) and interatomic distances (R) for single scattering of two Zr–O, Zr–C and Zr–Zr contributions and Zr–O–O, Zr–O–C and Zr–O–C–O multiple scatterings already require 18 parameters with additional energy corrections (ΔE0). To reduce the number of independent fitting parameters, only three scattering paths (Zr–O1, Zr–O2 and Zr–Zr) were used, where two Zr–O contributions depict the interval 1.2–2.6 Å in R-space (phase uncorrected). Furthermore, fitting was performed simultaneously for five EXAFS spectra of the same sample, using shared parameters, and applying several constrains. For instance, interatomic distances for identical paths were used the same for the whole lifespan of the sample, ΔE0 was fixed at zero, and total coordination number of two oxygen scattering paths was set to 8. Thus, the total number of variable parameters of five EXAFS spectra was 20. The fitting results for the sample activated in H2 at 300 °C and exposed to reaction mixture under 8 bar for the most essential parameters are summarized in the Table 1.
| Sample condition/parameter | As such, room temperature, He flow | After activation at 300 °C, reaction mixture under 8 bar at 240 °C | Reaction mixture under 8 bar at 200 °C | Reaction mixture under 8 bar at 170 °C | After reaction, 50 °C, He flow |
|---|---|---|---|---|---|
| S 0 2 × NZr–O1 | 2.77 ± 0.25 | 3.45 ± 0.22 | 3.29 ± 0.24 | 3.12 ± 0.22 | 2.92 ± 0.24 |
| S 0 2 × NZr–O2 | 5.23 ± 0.25 | 4.55 ± 0.22 | 4.71 ± 0.24 | 4.88 ± 0.22 | 5.01 ± 0.24 |
| S 0 2 × NZr–Zr | 4.13 ± 0.07 | 1.26 ± 0.56 | 1.88 ± 0.57 | 2.29 ± 0.57 | 3.32 ± 0.59 |
| R Zr–O1 | 2.12 ± 0.03 | ||||
| R Zr–O2 | 2.27 ± 0.01 | ||||
| R Zr–Zr | 3.52 ± 0.01 | ||||
| σ 2 Zr–O1 | 0.003 ± 0.001 | 0.006 ± 0.001 | 0.005 ± 0.001 | 0.005 ± 0.001 | 0.004 ± 0.001 |
| σ 2 Zr–O2 | 0.004 ± 0.001 | 0.006 ± 0.001 | 0.005 ± 0.001 | 0.005 ± 0.001 | 0.004 ± 0.001 |
| σ 2 Zr–Zr | 0.004 ± 0.001 | 0.006 ± 0.001 | 0.005 ± 0.001 | 0.005 ± 0.001 | 0.004 ± 0.001 |
| R-factor | 0.0238 | 0.0656 | 0.0413 | 0.0379 | 0.0304 |
The Zr–Zr coordination number decreases dramatically upon activation, which is visually seen in Fig. 7, while the increase in the Debye–Waller is consistent with the temperature. Even considering the correlation between these parameters, an additional effect on Zr–Zr coordination beyond thermal disorder is evident. Under reaction conditions, Zr–Zr coordination remains lowered and is only partially restored after cooling down to 50 °C. Therefore, the secondary building units of UiO-67 experience local distortions with a decrease in the coordination of Zr–Zr preserving with the overall stability of the whole UiO-67 framework confirmed by XRPD. FT-EXAFS spectra collected for the sample directly activated in the reaction mixture under 1 bar at 240 °C show the appearance of an additional peak at ca. 3 Å (Fig. 8), attributed to the Zr–Pd contribution. To elucidate this contribution, we have fitted the EXAFS data at 240 °C using two Zr–O paths and one Zr–Pd path with independent variable parameters. The Zr–Pd path was created by substitution of one Zr atom in UiO-67 inorganic cornerstone by Pd. Addition of the Zr–Zr path does not improve the fit quality as shown in ESI† (Fig. S13). The fit results are summarized in the Table 2.
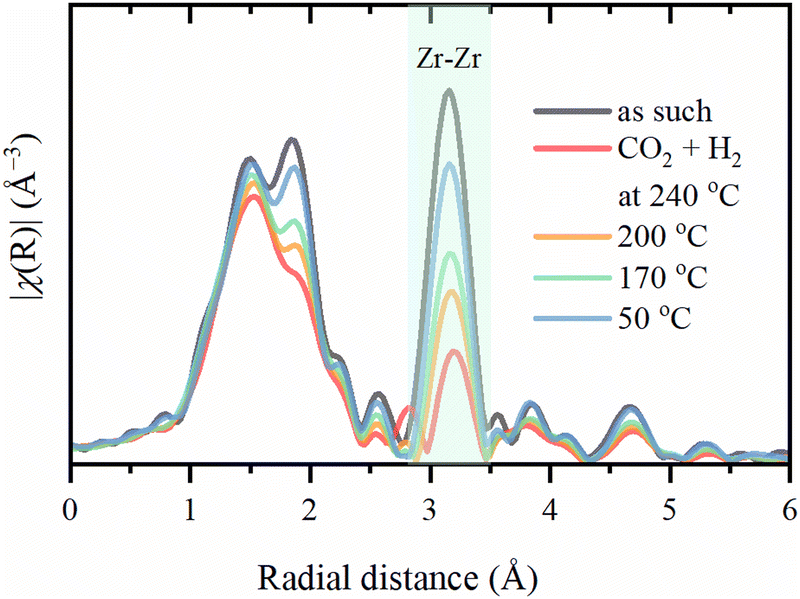 | ||
| Fig. 7 Experimental phase-uncorrected FT of Zr K-edge EXAFS spectra collected for the sample activated in H2 at 300 °C and exposed to reaction mixture under 8 bar. The highlighted area shows the contribution of Zr–Zr. | ||
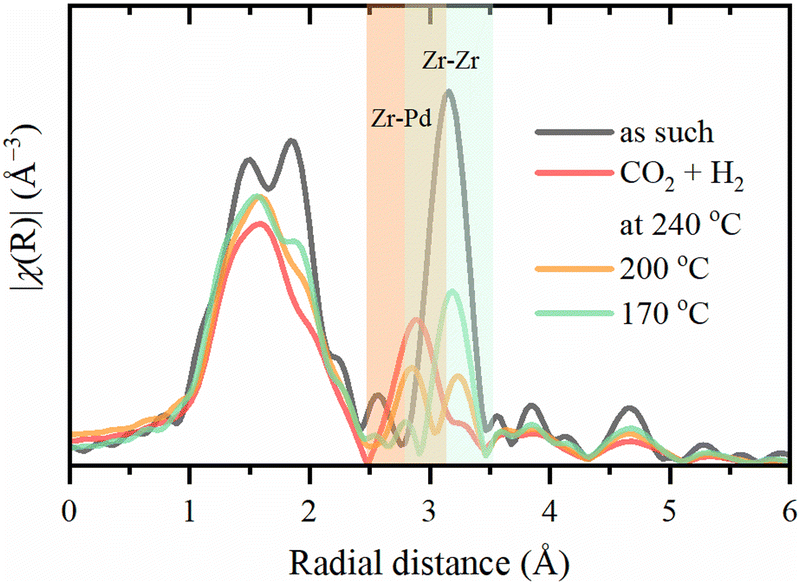 | ||
| Fig. 8 Experimental phase-uncorrected FT of Zr K-edge EXAFS spectra collected for the sample activated directly in the reaction mixture under 1 bar at 240 °C. The orange area shows the contribution of Zr–Pd and the green one – Zr–Zr. | ||
| Sample condition/parameter | Activation and reaction mixture under 1 bar at 240 °C |
|---|---|
| S 0 2 × NZr–O1 | 3.95 ± 8.17 |
| S 0 2 × NZr–O2 | 0.97 ± 6.06 |
| S 0 2 × NZr–Pd | 3.90 ± 2.01 |
| R Zr–O1 | 2.14 ± 0.04 |
| R Zr–O2 | 2.29 ± 0.04 |
| R Zr–Pd | 3.26 ± 0.14 |
| σ 2 Zr–O1 | 0.007 ± 0.018 |
| σ 2 Zr–O2 | 0.002 ± 0.017 |
| σ 2 Zr–Pd | 0.010 ± 0.003 |
| ΔE0 | −3.61 ± 5.39 |
| ΔEZr–Pd | −5.45 ± 4.04 |
| R-factor | 0.0361 |
The CO2 conversion and product distribution obtained over UiO-67-Pd under conditions mimicking those used during FT-IR and operando XAS and XRPD measurements are presented in Fig. 9. The conversion of CO2 was below 2% under all conditions tested. The catalytic results acquired under FT-IR conditions reveal exclusive formation of CO as the sole product at 1 bar. Conversely, at 8 bar, methanol is detected at all temperatures.
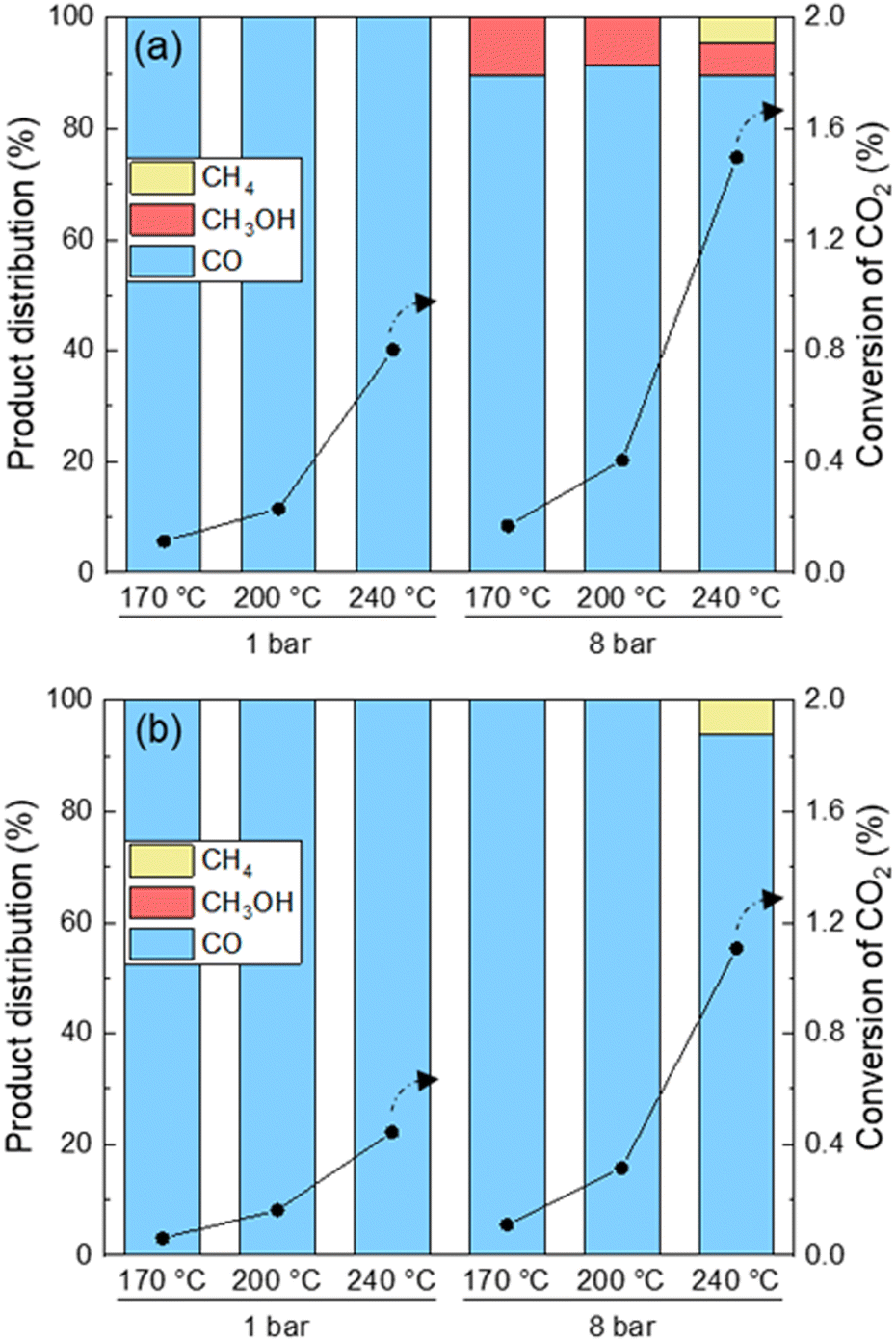 | ||
| Fig. 9 Product distribution (left axes, bars) and CO2 conversion (right axes, circles) at 1 and 8 bar and 170, 200 and 240 °C over UiO-67-Pd. CH4 selectivity (yellow), CH3OH (red) and CO (blue) and CO2 conversion (circles). (a) Reduction: 10% H2 in inert for 4 h at 300 °C. Conditions: H2/CO2 = 6/1, contact time (τ) = 0.01 gcat min ml−1. (b) Reduction: 14% H2 in inert for 0.5 h at 300 °C. Conditions: H2/CO2/inert = 3/1/4, contact time (τ) = 0.01 gcat min ml−1. | ||
This is in agreement with the fact that formate (Fig. 2), a well-known intermediate of methanol, was only observed at 8 bar using FT-IR. Methane is only observed at 8 bar and 240 °C, which is expected due to the limitation of the kinetics of the Sabatier reaction.1
Under XAS/XRPD conditions only CO was observed except at the highest pressure and temperature tested herein, where methane was also formed. The formation of CO, and of methane as a secondary product thereof at high temperature and pressure, is in agreement with literature on Pd nanoparticles in SiO2 and in UiO-66 MOF.1,56 To the best of our knowledge, methanol formation on Pd–Zr-MOFs is unprecedented in the open literature. However, this observation is well supported by prior studies in which a synergistic interaction between open Zr sites on the Zr6O4(OH)4 nodes of UiO-66/-67 and either Pt NPs or Cu NPs has been reported, with the metal NP as the hydrogen activating site, and the metal–Zr interface as the CO2 activation site. In both cases, formate species attached to the Zr-nodes were identified as intermediates to methanol formation.35,57–59
Unfortunately, the overall catalytic performance of Pd NPs embedded in the UiO-67 MOF happened to be quite low for this kind of reaction. Nevertheless, the methodologies developed for the analyses of this material were made available to identify intermediates adsorbed on the surface of the nanoparticles and demonstrated an extreme stability of the Pd-functionalized MOF under reaction conditions.
Materials and methods
Catalyst preparation
The UiO-67-Pd samples were prepared by the following procedure: 135 g of ZrCl4 (1 eq.) was slowly added to a 3L reactor containing 63 ml of distilled water (6 eq.) and 2243 ml of dimethylformamide (DMF) (50 eq.) held at room temperature on a magnetic stirring plate. The solution was heated to 110 °C and 212 g of benzoic acid (3 eq.) was added as a modulator and dissolved rapidly. When the benzoic acid was completely dissolved, 14 g of 2,2′-bipyridine-5,5′-dicarboxylic acid (bpy) was added, followed by 126 g of 1,1′-biphenyl-4,4′-dicarboxylic acid (bpdc). The resulting solution was placed for two days at 120 °C in a jacketed glass reactor (with stirring) equipped with a reflux condenser. The resulting product was isolated by filtration, washed with hot DMF (500 ml) and acetone (1 l) in the filter, and then dried at 150 °C in air overnight. The MOF was then impregnated with PdCl2 (Sigma-Aldrich) in DMF at 100 °C overnight (with stirring).32 This procedure leads to the formation of well-defined BPYDC-PdCl2 sites. The amount of PdCl2 used for the impregnation corresponded to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 bpy:Pd molar ratio and 4.6 wt% Pd in the final material.
:1 bpy:Pd molar ratio and 4.6 wt% Pd in the final material.
Laboratory characterization
Transmission electron microscopy (TEM) and high-resolution TEM (HR-TEM) measurements were performed using a side entry Jeol JEM 3010 (300 kV) microscope equipped with a LaB6 filament and fitted with X-ray EDS analysis by a Link ISIS 200 detector. For the analysis, the powdered samples were deposited on a copper grid, coated with a porous carbon film. All digital micrographs were acquired using an Ultrascan 1000 camera and the images were processed by Gatan digital micrograph. A statistically representative number of particles was counted to obtain the Pd particle size distribution with the average value 2.3 nm. The mean particle diameter (dm) was calculated as follows: dm = Σdini/Σni, where ni was the number of particles of diameter di. Measurements were carried out on UiO-67-Pd after activation at 4 hours at 300 °C in 20 ml (min 0.1gcat)−1 10% H2 in Ar. It is worth noting that the UiO-67-Pd samples proved to be stable to prolonged exposition under the electron beam of the instrument, in terms of UiO-67 crystallinity as well as Pd NPs size (no metal coalescence).Operando FT-IR technique was performed by means of a Bruker Vertex70 instrument, equipped with a liquid nitrogen-cooled MCT detector. Each spectrum was acquired by averaging 64 acquisitions with a spectral resolution of 2 cm−1. The undiluted sample was pressed to form a pellet of approximately 4 mg and was mounted inside a low free-volume cell from AABSPEC (model #CXX). Such cell allowed pressure, temperature, and gas flow control. The sample was activated at 300 °C (5 °C min−1 ramp) in 10% H2/He (10 ml min−1) for 4 h and then cooled to 240 °C in 10 ml min−1 He. Afterwards, the sample was kept under atmospheric pressure and CO2 hydrogenation (CO2/H2 = 1/6, 10 ml min−1, τ = 0.0004 gcat min ml−1) reaction conditions for 2 h. Finally, the temperature was varied to 170 °C, keeping constant the feed composition. Such catalytic test was performed with same temperatures and gas composition, but at 8 bar pressure.
Operando XAS and XRPD
XRPD and Pd K-edge XAS data were collected at the BM31 of the ESRF (Grenoble, France). The sample (ca. 10 mg) was loaded inside a 2.5 mm quartz glass capillary and fixed with quartz wool from both sides. The capillary was glued inside a U-shaped sample holder connected to a remotely controlled gas line (see Fig. S14, ESI†). A gas blower was positioned below the sample to control the temperature. The samples were activated in situ by heating in a flow of 1.4 ml min−1 H2 and 8.6 ml min−1 He from room temperature to 300 °C and left at this temperature for 30 min to allow the formation of nanoparticles. Then, the flow was switched to pure He to remove possible PdHx phase. The activated material was cooled down to 240 °C, and exposed to a reaction mixture of H2/CO2/He = 3/1/4 (7.5, 2.5, and 10 ml min−1 of H2, CO2, and He, respectively) with a total flow of 20 ml min−1 (τ = 0.0005 gcat min ml−1). The reaction was carried out at 240, 200, and 170 °C under a total pressure of 1 and 8 bar, for 2 h under each of the above conditions. The resulting gas mixture after the sample was analyzed by online mass spectrometer (Pfeiffer OmniStar GSD 320).Pd K-edge XAS were obtained in the transmission mode by continuous scanning of the double crystal Si(111) monochromator from 24.1 to 25.3 keV for steady state spectra and during reaction, and from 24.2 to 24.8 keV for measurements during activation. To reduce higher harmonics the monochromator was detuned to 80% of the maximal intensity. The palladium foil was measured simultaneously with each spectrum for energy calibration using a third ionization chamber. For the sample activated at 300 °C at 8 bar and the sample directly activated in reaction mixture, Zr K-edge XAS (see Fig. S15, ESI†) were also collected from 17.8 to 19.0 keV alternatively with Pd K-edge XAS.
XRPD was measured using 0.51067 Å radiation, selected by a Si(111) channel-cut monochromator. CMOS-Dexela 2D detector covered the 2θ region up to 35°. The values of the photon wavelength, sample to detector distance and detectors tilts have been optimized by Rietveld refinement of NIST LaB6 and Si samples and kept fixed in the refinement of the Pd/C samples. For better statistics 20 diffraction images and 20 dark images (without X-ray beam) with acquisition time of 5 seconds were collected at each experimental point.
XAS data analysis
Demeter software package60 was used to process and analyze EXAFS spectra. Background subtraction, normalization and energy calibration were performed in the Athena program using standard parameters. For the Pd K-edge, the first shell Fourier analysis of the EXAFS data was performed in the Artemis program using Pd–Pd contribution with four variable parameters: ΔE0, RPd–Pd, σ2Pd–Pd, NPd–Pd. The fit was performed in R-space in 1.0–3.0 Å region for k2-weighted data Fourier transformed (FT) in 3.0–10.0 Å−1 Δk region (2ΔkΔR/π > 8). The S02 value was fixed at 0.77 as determined for Pd foil. For the Zr K-edge the 4.0–14.0 Å−1 Δk and 1.21–3.7 Å ΔR intervals were used for all spectra. One path of Zr–O shell and one path of Zr–Zr were used in the Fourier analysis of the EXAFS data with 8 variable parameters in total.Analysis of experimental XANES spectra was performed using multivariate curve resolution (MCR) approach implemented in pyMCR 0.3.1.61 The whole dataset of normalized experimental spectra was represented by matrix D of size m × n, where m is the number of spectra and n is the number of energy points. This matrix is decomposed as D = CST + E, where C with size m × k is the concentration profiles of k pure components from matrix S (n × k), and E (m × n) is the error matrix. The energy ranges were limited to 24.33–24.41 keV. The optimal number of components was determined from the PCA implemented in PyFitIt.62–64
Theoretical XANES calculation
Theoretical XANES spectra were calculated within a full potential finite difference method of FDMNES code.65,66 The computational sphere radius was chosen as 5.2 Å. The surfaces were represented by unit cell with 32 Pd atoms, adsorbed species (CO2, CO, CH3, H2O, O and OH placed on top, bridge, or hollow sites) and 13 Å of vacuum (see Fig. S10, ESI†). All geometries were initially optimized in VASP 5.3 code67–69 with PBE exchange–correlation potential using a 8 × 4 × 1 Monkhorst Pack grid with the plane-wave basis cutoff at 500 eV.XRPD data analysis
2D XRPD images were averaged in PyFAI code70 to obtain I(2θ) patterns. Whole-pattern fitting by Pawley method was performed in Jana2006 code.71 The used 2θ-range was from 3 to 30°. The initial structure was based on a cubic cell of UiO-67 in Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m symmetry with cell parameter a = 26.8 Å. The variable parameters included cell parameter (a), three parameters of Pseudo-Voight function and ten parameters of the background function.
m symmetry with cell parameter a = 26.8 Å. The variable parameters included cell parameter (a), three parameters of Pseudo-Voight function and ten parameters of the background function.
Catalytic testing
Catalytic tests were performed under conditions mimicking those used in XAS/XRPD and FT-IR studies, using a commercial Microactivity Effi reactor (PID Eng & Tech) with a fixed bed flow setup. The reactor is a stainless-steel reactor (I.D. 6 mm) coated with silicon, connected to an Agilent 8890 gas chromatogram (GC) equipped with two flame ionization detectors (FIDs) and a thermal conductivity detector (TCD). One of the FIDs was coupled to a methanizer to achieve lower detection limits of CO and CO2. The Pd-UiO-67 sample was tested under two different conditions to better compare the activity of the material under similar conditions to those during XAS and FT-IR characterization. To imitate the conditions during XAS and XRPD, 0.2 g catalyst were reduced in 14% H2 in inert for 0.5 h at 300 °C. The catalyst was tested at 240, 200 and 170 °C at 1 and 8 bar for 4 h under a reaction mixture of H2/CO2/Inert = 3/1/4 with a total flow of 20 ml min−1 (τ = 0.01 gcat min ml−1). To replicate the condition used during FT-IR spectroscopy, 0.2 g UiO-67-Pd was reduced in 10% H2 in inert for 4 h at 300 °C before being exposed to a reaction mixture of H2/CO2 = 6/1 with a total flow of 20 ml min−1 (τ = 0.01 gcat min ml−1). The catalyst was tested at 240, 200 and 170 °C at 1 and 8 bar.Conclusions
Summarizing the operando study on UiO-67-Pd MOF, we can conclude that: (i) under reaction conditions, mixed palladium hydride and carbide phases are formed; (ii) the hydride phase is stronger at high pressure and low temperatures, but is removed upon flushing in He, while the carbide one is stable even after flushing; (iii) formation of CO adsorbed species is observed under reaction conditions, and can act as stabilizing factor for smaller Pd NPs formed upon activation directly in the reaction mixture; (iv) Zr–O coordination in the inorganic building units of UiO-67 is reduced during CO2 hydrogenation reaction that is the more considerable at the higher temperature, while the overall UiO-67 lattice remains stable under all conditions; (v) a Zr/Pd interface is observed in EXAFS, being responsible for methanol formation in UiO-67-Pd samples.Author contributions
ALB, EGK, AAS, AL, DKS, and CA-S carried out experiments, performed data analysis and theoretical calculations. ALB, AL and UO designed the experiments. AAS and ALB wrote the original draft of the manuscript. ALB, SB and UO: manuscript revising and funding acquisition. All authors discussed and contributed to the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
CA-S, AL, DKS, SB and UO acknowledge the Research Council of Norway for financial support of this research through contracts no. 250795 (CONFINE) and 288331 (CO2LO). Dr Gurpreet Kaur is acknowledged for the synthesis of UiO-67-Pd. We thank ESRF for providing the beamtime and Dragos Stoian and Wouter van Beek for their support during experiment at BM31 beamline. We acknowledge ALBA for the additional measurements on commercial Pd NPs under CO atmosphere.References
- J. Martins, N. Batail, S. Silva, S. Rafik-Clement, A. Karelovic, D. P. Debecker, A. Chaumonnot and D. Uzio, Catal. Commun., 2015, 58, 11–15 CrossRef CAS.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- H. Bahruji, M. Bowker, G. Hutchings, N. Dimitratos, P. Wells, E. Gibson, W. Jones, C. Brookes, D. Morgan and G. Lalev, J. Catal., 2016, 343, 133–146 CrossRef CAS.
- A. Ota, E. L. Kunkes, I. Kasatkin, E. Groppo, D. Ferri, B. Poceiro, R. M. Navarro Yerga and M. Behrens, J. Catal., 2012, 293, 27–38 CrossRef CAS.
- N. Koizumi, X. Jiang, J. Kugai and C. Song, Catal. Today, 2012, 194, 16–24 CrossRef CAS.
- L. Zhang, Y. Zhang and S. Chen, Appl. Catal., A, 2012, 415–416, 118–123 CrossRef CAS.
- A. Ramirez, et al. , JACS Au, 2021, 1(10), 1719–1732 CrossRef CAS PubMed.
- R. Raudaskoski, M. V. Niemelä and R. L. Keiski, Top. Catal., 2007, 45, 57–60 CrossRef CAS.
- A. Prašnikar, A. Pavlišič, F. Ruiz-Zepeda, J. Kovač and B. Likozar, Ind. Eng. Chem. Res., 2019, 58, 13021–13029 CrossRef.
- N. Rui, Z. Wang, K. Sun, J. Ye, Q. Ge and C.-J. Liu, Appl. Catal., B, 2017, 218, 488–497 CrossRef CAS.
- A. A. Skorynina, et al. , Radiat. Phys. Chem., 2020, 175, 108079 CrossRef CAS.
- W. Stuyck, et al. , J. Catal., 2022, 408, 88–97 CrossRef CAS.
- V. V. Butova, M. A. Soldatov, A. A. Guda, K. A. Lomachenko and C. Lamberti, Russ. Chem. Rev., 2016, 85, 280–307 CrossRef CAS.
- T. A. Goetjen, J. Liu, Y. Wu, J. Sui, X. Zhang, J. T. Hupp and O. K. Farha, Chem. Commun., 2020, 56, 10409–10418 RSC.
- A. Yu. Pnevskaya, et al. , J. Phys. Chem. C, 2021, 125(40), 22295–22300 CrossRef CAS.
- A. Yu. Pnevskaya and A. L. Bugaev, Results Chem., 2023, 5, 100831 CrossRef CAS.
- D. Ma, B. Li, K. Liu, X. Zhang, W. Zou, Y. Yang, G. Li, Z. Shi and S. Feng, J. Mater. Chem. A, 2015, 3, 23136–23142 RSC.
- L. Wang, P. Jin, S. Duan, J. Huang, H. She, Q. Wang and T. An, Environ. Sci.: Nano, 2019, 6, 2652–2661 RSC.
- Y. Dou, H. Zhang, A. Zhou, F. Yang, L. Shu, Y. She and J.-R. Li, Ind. Eng. Chem. Res., 2018, 57, 8388–8395 CrossRef CAS.
- B. Gole, U. Sanyal, R. Banerjee and P. S. Mukherjee, Inorg. Chem., 2016, 55, 2345–2354 CrossRef CAS PubMed.
- A. A. Tereshchenko, et al. , Inorg. Chem., 2022, 61(9), 3875–3885 CrossRef CAS.
- W. Dong, C. Feng, L. Zhang, N. Shang, S. Gao, C. Wang and Z. Wang, Catal. Lett., 2015, 146, 117–125 CrossRef.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- K. S. Vetlitsyna-Novikova, V. V. Butova, I. A. Pankin, V. V. Shapovalov and A. V. Soldatov, J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech., 2019, 13, 787–792 CrossRef CAS.
- V. V. Butova, O. A. Burachevskaya, I. V. Ozhogin, G. S. Borodkin, A. G. Starikov, S. Bordiga, A. Damin, K. P. Lillerud and A. V. Soldatov, Microporous Mesoporous Mater., 2020, 305, 110324 CrossRef CAS.
- V. V. Butova, K. S. Vetlitsyna-Novikova, I. A. Pankin and A. V. Soldatov, J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech., 2020, 14, 318–323 CrossRef CAS.
- K. E. deKrafft, W. S. Boyle, L. M. Burk, O. Z. Zhou and W. Lin, J. Mater. Chem., 2012, 22, 18139–18144 RSC.
- R. Dalapati, B. Sakthivel, A. Dhakshinamoorthy, A. Buragohain, A. Bhunia, C. Janiak and S. Biswas, CrystEngComm, 2016, 18, 7855–7864 RSC.
- E. Borfecchia, S. Øien, S. Svelle, L. Mino, L. Braglia, G. Agostini, E. Gallo, K. A. Lomachenko, S. Bordiga, A. A. Guda, M. A. Soldatov, A. V. Soldatov, U. Olsbye, K. P. Lillerud and C. Lamberti, J. Phys.: Conf. Ser., 2016, 712, 012125 CrossRef.
- L. Braglia, E. Borfecchia, K. A. Lomachenko, A. L. Bugaev, A. A. Guda, A. V. Soldatov, B. T. L. Bleken, S. Øien-Ødegaard, U. Olsbye, K. P. Lillerud, S. Bordiga, G. Agostini, M. Manzoli and C. Lamberti, Faraday Discuss., 2017, 201, 265–286 RSC.
- L. Braglia, E. Borfecchia, L. Maddalena, S. Øien, K. A. Lomachenko, A. L. Bugaev, S. Bordiga, A. V. Soldatov, K. P. Lillerud and C. Lamberti, Catal. Today, 2017, 283, 89–103 CrossRef CAS.
- A. L. Bugaev, A. A. Guda, K. A. Lomachenko, E. G. Kamyshova, M. A. Soldatov, G. Kaur, S. Øien-Ødegaard, L. Braglia, A. Lazzarini, M. Manzoli, S. Bordiga, U. Olsbye, K. P. Lillerud, A. V. Soldatov and C. Lamberti, Faraday Discuss., 2018, 208, 287–306 RSC.
- C. Wang, A. Wang, Z. Yu, Y. Wang, Z. Sun, V. M. Kogan and Y.-Y. Liu, Catal. Commun., 2021, 148, 106178 CrossRef CAS.
- E. S. Gutterød, S. Øien-Ødegaard, K. Bossers, A.-E. Nieuwelink, M. Manzoli, L. Braglia, A. Lazzarini, E. Borfecchia, S. Ahmadigoltapeh, B. Bouchevreau, B. T. Lønstad-Bleken, R. Henry, C. Lamberti, S. Bordiga, B. M. Weckhuysen, K. P. Lillerud and U. Olsbye, Ind. Eng. Chem. Res., 2017, 56, 13206–13218 CrossRef.
- E. S. Gutterod, A. Lazzarini, T. Fjermestad, G. Kaur, M. Manzoli, S. Bordiga, S. Svelle, K. P. Lillerud, E. Skulason, S. Oien-Odegaard, A. Nova and U. Olsbye, J. Am. Chem. Soc., 2020, 142, 999–1009 CrossRef.
- A. L. Bugaev, A. A. Skorynina, L. Braglia, K. A. Lomachenko, A. Guda, A. Lazzarini, S. Bordiga, U. Olsbye, K. P. Lillerud, A. V. Soldatov and C. Lamberti, Catal. Today, 2019, 336, 33–39 CrossRef CAS.
- E. G. Kamyshova, A. A. Skorynina, A. L. Bugaev, C. Lamberti and A. V. Soldatov, Radiat. Phys. Chem., 2020, 175, 108144 CrossRef CAS.
- O.A. Usoltsev, et al. , Nanomaterials, 2020, 10(9), 1643 CrossRef CAS.
- J. Jaumot, R. Gargallo, A. de Juan and R. Tauler, Chemom. Intell. Lab. Syst., 2005, 76, 101–110 CrossRef CAS.
- R. Tauler, Chemom. Intell. Lab. Syst., 1995, 30, 133–146 CrossRef CAS.
- J. Jaumot, A. de Juan and R. Tauler, Chemom. Intell. Lab. Syst., 2015, 140, 1–12 CrossRef CAS.
- D. Yang, M. A. Ortuno, V. Bernales, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2018, 140, 3751–3759 CrossRef CAS PubMed.
- A. Lazzarini, E. Groppo, G. Agostini, E. Borfecchia, F. Giannici, G. Portale, A. Longo, R. Pellegrini and C. Lamberti, Catal. Today, 2017, 283, 144–150 CrossRef CAS.
- E. Groppo, W. Liu, O. Zavorotynska, G. Agostini, G. Spoto, S. Bordiga, C. Lamberti and A. Zecchina, Chem. Mater., 2010, 22, 2297–2308 CrossRef CAS.
- O. Usoltsev, et al. , Small Methods, 2024, 2301397 CrossRef.
- A. L. Bugaev, A. A. Guda, K. A. Lomachenko, A. Lazzarini, V. V. Srabionyan, J. G. Vitillo, A. Piovano, E. Groppo, L. A. Bugaev, A. V. Soldatov, V. P. Dmitriev, R. Pellegrini, J. A. van Bokhoven and C. Lamberti, J. Phys.: Conf. Ser., 2016, 712, 012032 CrossRef.
- A. L. Bugaev, A. A. Guda, K. A. Lomachenko, V. V. Shapovalov, A. Lazzarini, J. G. Vitillo, L. A. Bugaev, E. Groppo, R. Pellegrini, A. V. Soldatov, J. A. van Bokhoven and C. Lamberti, J. Phys. Chem. C, 2017, 121, 18202–18213 CrossRef CAS.
- Aram L. Bugaev, O. A. Usoltsev, A. Lazzarini, K. A. Lomachenko, A. A. Guda, R. Pellegrini, M. Carosso, J. G. Vitillo, E. Groppo, J. A. van Bokhoven, A. V. Soldatov and C. Lamberti, Faraday Discuss., 2018, 208, 187–205 RSC.
- O. A. Usoltsev, A. A. Skorynina, B. O. Protsenko, V. Martin-Diaconescu, R. Pellegrini, A. V. Soldatov, J. van Bokhoven and A. L. Bugaev, Appl. Surf. Sci., 2023, 614, 156171 CrossRef CAS.
- W. Liu, et al. , J. Mater. Chem. A, 2021, 9(16), 10354–10363 RSC.
- A. L. Bugaev, A. A. Guda, I. A. Pankin, E. Groppo, R. Pellegrini, A. Longo, A. V. Soldatov and C. Lamberti, Catal. Today, 2019, 336, 40–44 CrossRef CAS.
- A. L. Bugaev, V. V. Srabionyan, A. V. Soldatov, L. A. Bugaev and J. A. V. Bokhoven, J. Phys.: Conf. Ser., 2013, 430, 012028 CrossRef CAS.
- O. A. Usoltsev, et al. , Topics in Catalysis, 2020, 63, 58–65 CrossRef CAS.
- O. A. Usoltsev, et al. , J. Phys. Chem. C, 2022, 126(10), 4921–4928 CrossRef CAS.
- A. L. Bugaev, A. A. Guda, A. Lazzarini, K. A. Lomachenko, E. Groppo, R. Pellegrini, A. Piovano, H. Emerich, A. V. Soldatov, L. A. Bugaev, V. P. Dmitriev, J. A. van Bokhoven and C. Lamberti, Catal. Today, 2017, 283, 119–126 CrossRef CAS.
- H. Jiang, Q. Gao, S. Wang, Y. Chen and M. Zhang, J. CO2 Util., 2019, 31, 167–172 CrossRef CAS.
- B. Rungtaweevoranit, J. Baek, J. R. Araujo, B. S. Archanjo, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2016, 16, 7645–7649 CrossRef CAS PubMed.
- Y. Zhu, J. Zheng, J. Ye, Y. Cui, K. Koh, L. Kovarik, D. M. Camaioni, J. L. Fulton, D. G. Truhlar and M. Neurock, Nat. Commun., 2020, 11, 5849 CrossRef CAS PubMed.
- E. S. Gutterød, S. H. Pulumati, G. Kaur, A. Lazzarini, B. G. Solemsli, A. E. Gunnæs, C. Ahoba-Sam, M. E. Kalyva, J. A. Sannes and S. Svelle, J. Am. Chem. Soc., 2020, 142, 17105–17118 CrossRef PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- C. H. Camp Jr., J. Res. Natl. Inst. Stand. Technol., 2019, 124, 1–10 Search PubMed.
- A. Martini, A. A. Guda, S. A. Guda, A. L. Bugaev, O. V. Safonova and A. V. Soldatov, Phys. Chem. Chem. Phys., 2021, 23, 17873–17887 RSC.
- A. Martini, A. A. Guda, S. A. Guda, A. Dulina, F. Tavani, P. D’Angelo, E. Borfecchia and A. V. Soldatov, Cham, 2021.
- A. Martini, S. A. Guda, A. A. Guda, G. Smolentsev, A. Algasov, O. Usoltsev, M. A. Soldatov, A. Bugaev, Y. Rusalev, C. Lamberti and A. V. Soldatov, Comput. Phys. Commun., 2020, 250, 107064 CrossRef CAS.
- A. A. Guda, S. A. Guda, M. A. Soldatov, K. A. Lomachenko, A. L. Bugaev, C. Lamberti, W. Gawelda, C. Bressler, G. Smolentsev, A. V. Soldatov and Y. Joly, J. Phys.: Conf. Ser., 2016, 712, 012004 CrossRef.
- S. A. Guda, A. A. Guda, M. A. Soldatov, K. A. Lomachenko, A. L. Bugaev, C. Lamberti, W. Gawelda, C. Bressler, G. Smolentsev, A. V. Soldatov and Y. Joly, J. Chem. Theory Comput., 2015, 11, 4512–4521 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- J. Kieffer and J. P. Wright, Powder Diffr., 2013, 28, S339–S350 CrossRef CAS.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr. - Cryst. Mater., 2014, 229, 345–352 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme of the experimental setup, additional XANES and FT-EXAFS spectra, MCR analysis of XANES, atomic models, XRPD data. See DOI: https://doi.org/10.1039/d3tc04175a |
| This journal is © The Royal Society of Chemistry 2024 |