
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rationalizing the carborane versus phenyl-driven luminescence in related dicarboxylic ligands and their antenna effect for their Eu3+ and Tb3+ metal–organic frameworks: a combined experimental and computational study†
Zhen
Li
ab,
Claudio
Roscini
 c,
Rosario
Núñez
a,
Francesc
Teixidor
a,
Clara
Viñas
a,
Eliseo
Ruiz
*d and
José Giner
Planas
*a
c,
Rosario
Núñez
a,
Francesc
Teixidor
a,
Clara
Viñas
a,
Eliseo
Ruiz
*d and
José Giner
Planas
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Bellaterra, Spain. E-mail: jginerplanas@icmab.es
bShandong Provincial Key Laboratory of Monocrystalline Silicon Semiconductor Materials and Technology, College of Chemistry and Chemical Engineering, Dezhou University, Dezhou 253023, China
cCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC, and The Barcelona Institute of Science and Technology (BIST), Campus UAB, Bellaterra, Barcelona 08193, Spain
dDepartament de Química Inorgànica i Orgànica and Institut de Recerca de Química Teòrica i Computacional, Universitat de Barcelona, Diagonal 645, 08028 Barcelona, Spain. E-mail: eliseo.ruiz@qi.ub.es
First published on 4th January 2024
Abstract
Replacement of a phenyl moiety by a carborane in 1,3-di(4-carboxyphenyl) derivatives has a pronounced effect on the photophysical properties of the compounds themselves and their corresponding Eu3+ and Tb3+ metal organic frameworks. Herein, we demonstrate that while the luminescence of the carborane-derivative 1,7-di(4-carboxyphenyl)-1,7-dicarba-closo-dodecaborane (mCB-L) is negligible in the solid state (0.3%), the corresponding phenyl-derivative [1,1′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3′,1′′-terphenyl]-4,4′′-dicarboxylic] (TDCA) is highly luminescent (quantum yield of 47.8%). However, the latter one is a much worse antenna for sensitizing Eu3+ and Tb3+ cations than the former one. Based on our previous studies where mCB-L behaves as a good antenna for these two lanthanides, we have now comparatively studied the optical properties of mCB-L and TDCA and their efficiency as sensitizers for Eu and Tb cations, in the solid state at room temperature. STEOM-DLPNO-CCSD calculations revealed a larger energy difference between the singlet S1 and the lowest conical intersection (CI) points for TDCA (33.0 kcal mol−1) than that for mCB-L (18.4 kcal mol−1), explaining the observed different luminescence properties of these two compounds. Contrary to the carborane derivative, TDCA exhibits a more distorted structure at the CI point. TDCA-Eu and TDCA-Tb MOFs have been synthesized and characterized by infrared spectroscopy (IR) and powder X-ray diffraction (PXRD). The optical properties of TDCA-Eu and TDCA-Tb MOFs were investigated, and both MOFs displayed luminescence in the visible region with characteristic emission bands attributed to Eu3+ and Tb3+ transitions, respectively. However, the quantum yields for TDCA-Ln (Eu, 11.1%; Tb, 4.8%) were found to be much lower than those of carborane-based MOFs (Eu, 20.5%; Tb, 49.8%), indicating that the TDCA ligand is a less efficient light-absorbing sensitizer for Eu3+ and Tb3+ ions compared to the carborane-based ligand and may favor undesired back-energy transfer, as suggested by their faster decay times in comparison with the carborane counterparts. STEOM-DLPNO-CCSD calculations are used to analyze the changes in electron densities on photo-excitation from S0 to S1 and de-excitation from T1 to S0, as well as the differences in the excited state energies for mCB-L, TDCA, and the hypothetical longer derivatives (mCB-L2 and QDCA). Our research, which combines experimental and computational methods, offers valuable data for optimizing the singlet and triplet energies, as well as their differences (gaps), by choosing between phenyl and carborane as the building scaffold for 1,3-di(4-carboxyphenyl) compounds. This demonstrates that using phenyl or m-carborane as the building scaffold leads to distinct luminescence properties.
:3′,1′′-terphenyl]-4,4′′-dicarboxylic] (TDCA) is highly luminescent (quantum yield of 47.8%). However, the latter one is a much worse antenna for sensitizing Eu3+ and Tb3+ cations than the former one. Based on our previous studies where mCB-L behaves as a good antenna for these two lanthanides, we have now comparatively studied the optical properties of mCB-L and TDCA and their efficiency as sensitizers for Eu and Tb cations, in the solid state at room temperature. STEOM-DLPNO-CCSD calculations revealed a larger energy difference between the singlet S1 and the lowest conical intersection (CI) points for TDCA (33.0 kcal mol−1) than that for mCB-L (18.4 kcal mol−1), explaining the observed different luminescence properties of these two compounds. Contrary to the carborane derivative, TDCA exhibits a more distorted structure at the CI point. TDCA-Eu and TDCA-Tb MOFs have been synthesized and characterized by infrared spectroscopy (IR) and powder X-ray diffraction (PXRD). The optical properties of TDCA-Eu and TDCA-Tb MOFs were investigated, and both MOFs displayed luminescence in the visible region with characteristic emission bands attributed to Eu3+ and Tb3+ transitions, respectively. However, the quantum yields for TDCA-Ln (Eu, 11.1%; Tb, 4.8%) were found to be much lower than those of carborane-based MOFs (Eu, 20.5%; Tb, 49.8%), indicating that the TDCA ligand is a less efficient light-absorbing sensitizer for Eu3+ and Tb3+ ions compared to the carborane-based ligand and may favor undesired back-energy transfer, as suggested by their faster decay times in comparison with the carborane counterparts. STEOM-DLPNO-CCSD calculations are used to analyze the changes in electron densities on photo-excitation from S0 to S1 and de-excitation from T1 to S0, as well as the differences in the excited state energies for mCB-L, TDCA, and the hypothetical longer derivatives (mCB-L2 and QDCA). Our research, which combines experimental and computational methods, offers valuable data for optimizing the singlet and triplet energies, as well as their differences (gaps), by choosing between phenyl and carborane as the building scaffold for 1,3-di(4-carboxyphenyl) compounds. This demonstrates that using phenyl or m-carborane as the building scaffold leads to distinct luminescence properties.
Introduction
Luminescent materials have attracted significant attention due to their wide-ranging applications in fields such as optoelectronics, photonics, and bioimaging.1–5 The ability to control and manipulate the luminescent properties of materials is of utmost importance for designing advanced functional materials with tailored properties. In this context, the choice of molecular scaffolds as ligands plays a crucial role in determining the luminescent behavior of a material. To date, a predominant approach for designing organic fluorophores has been through conjugated systems, wherein diverse emitting units are interconnected by means of double bonds, triple bonds, and aromatic rings.6In recent years, icosahedral closo-carboranes 1,n-C2B10H12 (n = 2, 7 or 12), which are molecular carbon–boron polyhedral clusters, have emerged as promising building blocks for a wide variety of applications.7–13 Carboranes, as boron-rich clusters with three-dimensional aromatic structures,14–16 exhibit exceptional stability and possess advantageous material properties such as robust thermal and chemical stability, as well as notable hydrophobicity.7,10,11,17–21 We and others have demonstrated that carboranes are excellent scaffolds in luminescent materials both in solution9,22–32 and, more recently, in the solid state.33–38 Regarding the latter, in recent years significant progress has been made in the development of carborane-based active luminophores,39 which exhibit properties that align with solid-state luminescence and stimuli-responsiveness. This indicates that the luminescent features, including the color, can be adjusted by manipulating the molecular morphology in the solid and aggregate states. By utilizing these adjustable luminescent properties, it is possible to create unique stimuli-responsive luminescent materials using carborane based molecules. Our group has recently contributed to this area of research through the development of highly emissive water dispersible nanoparticles,40 fluorescent metal complexes with unique structures41 and luminescent lanthanide metal–organic frameworks (Ln-MOFs) that are printable from water inks for potential applications in anticounterfeiting and bar-coding.42 However, the vast majority of luminescent materials in aggregate or solid state based on carboranes are associated with the ortho–closo-carborane (1,2-C2B10H12) isomer or its nido-carborane derivatives (C2B9H12−),36,43 while only a limited number of examples have been reported for the meta-carborane (1,7-C2B10H12; Chart 1) isomer, mostly from our group.41,42,44–48 Carboranes are often viewed as three-dimensional analogues to benzene (Chart 1, top).41,48 The former are significantly larger (∼40%) than the benzene ring rotation envelope10 and behave as strong electron-withdrawing groups (similar to a fluorinated aryl) on a substituent at one of the cluster carbons.10,49–51 The replacement of a phenyl moiety by a carborane in a molecule can have a significant impact on the vertical excitation processes, including the electronic structure, optical properties, and excited-state behavior of the molecule. Given the distinctive features of carboranes and phenyl rings as molecular scaffolds, a comparative study of their luminescence and vertical excitation properties would provide valuable insights into their potential as ligands for designing luminescent materials.
 | ||
| Chart 1 A representation of m-carborane and benzene (top) and dicarboxylic acids studied in this work (bottom). | ||
Here, we present a comparative study of luminescence and vertical excitation properties of carborane-based and phenyl-based ligands themselves, and their corresponding Eu3+ and Tb3+ metal organic frameworks. In this study, two 1,3-di(4-carboxyphenyl) derivatives were experimentally examined (1,7-di(4-carboxyphenyl)-1,7-dicarba-closo-dodecaborane (mCB-L) and [1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic (TDCA); Chart 1). These two compounds differ in their central moiety, with one having a 2D aromatic phenyl ring (TDCA), and the other having a 3D aromatic carboranyl moiety (mCB-L). These materials exhibit differences in their photoluminescence quantum yields and their ability to act as antennas for Eu3+ and Tb3+ cations in the solid state. To prove this, we have synthesized and characterized the corresponding TDCA-Eu and TDCA-Tb MOFs. We have combined experimental approaches and a recently developed similarity transformed equation-of-motion domain-based local pair natural orbital coupled-cluster singles and doubles (STEOM-DLPNO-CCSD) theoretical method (see Computational details section), and determined that the molecular structures play a crucial role in the corresponding energy levels of the excited states and therefore in the photophysical properties of these carboxyphenyl and carboxycarboranyl derivatives and their corresponding lanthanide metal organic frameworks. extended mCB-L2 and QDCA compounds have also been studied by computational methods.
Results and discussion
We have investigated the optical properties of TDCA by UV-vis absorption and emission spectroscopies in the solid-state at room temperature. The free TDCA compound exhibits a broad absorption band centered around 300 nm (Fig. S1, ESI†), which is attributed to the π → π* transitions. The luminescence spectrum for TDCA upon continuous-wave irradiation (λex = 290 nm) shows a strong emission at λem = 390 nm and an overall quantum yield (Φ) of 47.8% (Fig. S2, ESI†). As summarized in Table 1, the observed Φ for TDCA is much larger than that for the carborane derivative mCB-L (0.3%).42 In order to understand the photophysical properties of these two related derivatives, we have calculated their ground state electronic structures and vertical excitation energies. We have performed the calculations by using the recently developed coupled cluster method STEOM-DLPNO-CCSD. This is a wave function-based quantum chemistry approach based on coupled cluster calculation,52 which gives accurate results for the prediction of excited energy values.53 The calculated values of singlet (S) and triplet (T) levels for mCB-L and TDCA are summarized in Table 1. Similar results regarding the energy differences S0–S1 and T1–S0 for TDCA and mCB-L1 ligands were obtained at both B3LYP-TDDFT42 and STEOM-DLPNO-CCSD level of theory (Table S1, ESI†). We have computed the vertical and adiabatic excitation energies.54 For mCB-L, the vertical S1 excitation energy of 272 nm is in agreement with the experimental value determined for this compound in the solid state with a λabsmax ∼ 289 nm. The lowest energy of the S1 vertical excitation than the adiabatic value is due to differences between the potential surfaces between DFT and the coupled cluster method. For TDCA, the S1 excitation energy values are 280 and 294 nm (vertical and adiabatic values, respectively, see Table S2, ESI†), also very close to the experimental broad absorption band centered around 300 nm.| Experimental | Calculated | ||||
|---|---|---|---|---|---|
| Compounds | λ ex (nm) | λ em (nm) | Φ (%) | S0–S1 nm (cm−1) | T1–S0 nm (cm−1) |
| TDCA | 290 | 390 | 47.8 |
280 (35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 727) 727)
|
624 (16033)
|
294 (34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 065) 065)
|
476 (20989)
|
||||
| mCB-L | 280 | 312 | 0.3 |
272 (36806)
|
526 (19006)
|
|
241 (41437)
|
424 (23593)
|
||||
To better comprehend the contrasting luminescent behavior in the solid state exhibited by TDCA and mCB-L, we conducted a comprehensive investigation into the deexcitation process via conical intersections (CI), which denotes points where the potential energy surfaces of two electronic states intersect (Fig. 1).55–58 The kinetics of non-radiative decay crucially relies on the presence of CI. Hence, the accessibility of CI directly impacts the rates of non-radiative decay and, consequently, the fluorescence quantum yield. Fig. 1 portrays schematic representations of two distinct potential energy profiles: one indicating the dominance of non-radiative decay (Fig. 1a), and the other highlighting the predominance of radiative decay with fluorescence (Fig. 1b). In Fig. 1a, a minimum energy CI is observed in a lower energy region relative to the Franck–Condon (FC) point, thereby facilitating rapid, non-radiative decay through the corresponding CI region. Conversely, in Fig. 1b, the lowest energy CI is situated significantly higher in energy compared to the FC point, resulting in infrequent access of the molecule to the CI region and consequently slower non-radiative decay. By comparing the energy differences between the singlet S1 states and the optimized CI structures of the investigated molecules, one can elucidate the relative significance of radiative versus non-radiative decay. We have therefore explored the minimum energy CI structures of TDCA and mCB-L. The geometries corresponding to the conical intersections were also optimized at the TDDFT-B3LYP level (Fig. 1c, d and Experimental section). The energy differences between the singlet S1 and the optimized conical intersection structures were then calculated at DLPNO-CCSD level. A much larger difference was found for TDCA (33.0 kcal mol−1) than for mCB-L (18.4 kcal mol−1). Consequently, the energy barrier between the singlet S1 and the lowest CI point decreases when replacing a phenyl ring by a carborane moiety as the central core of 1,3-di(4-carboxyphenyl) derivatives. Analysis of the structures at the conical intersection points reveals notable differences in geometries between the two systems, as shown in Fig. 1c and d. The TDCA ligand exhibits a quite distorted structure that deviates from planarity, whereas the mCB-L compound does not show any distortion. These findings confirm that the m-carborane-based system, with a smaller energy difference at the CI point, may facilitate a more accessible non-radiative process compared to the phenyl-based system.
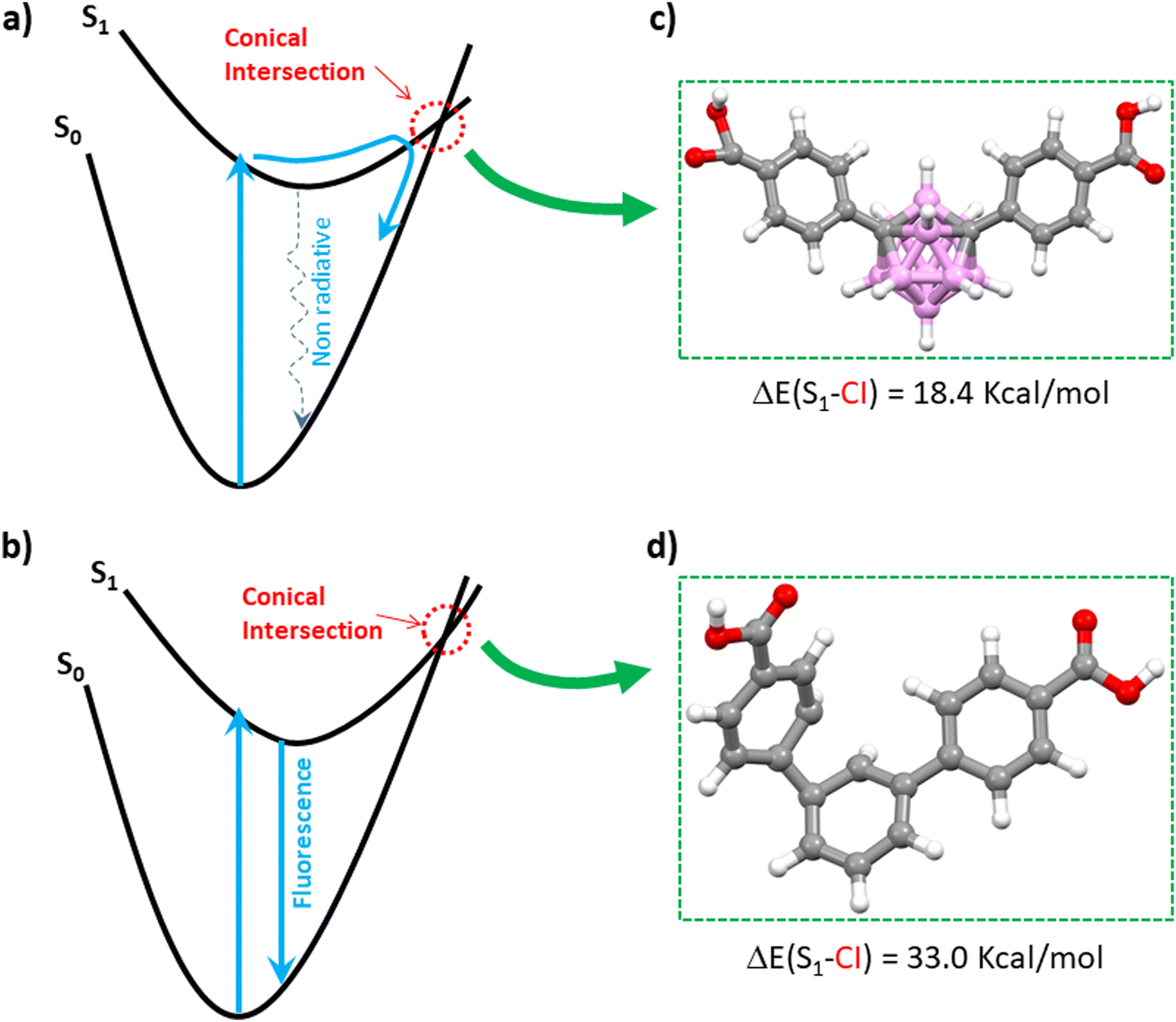 | ||
| Fig. 1 Schematic representation of the potential energy surfaces for two types of photo-excitations and geometries of the studied molecules optimized at the conical intersection points. (a) The case in which an ultrafast non-radiate decay via the conical intersection (CI) region (see dotted blue arrow) is dominant. (b) The case in which the decay via a fluorescence process (solid blue arrow) is dominant. Views of mCB-L (c) and TDCA (d) optimized molecular structures at their CI points. Fig. 1a and b is adapted from ref. 46 with permission from the Royal Society of Chemistry. Color codes for atoms in the structures: C, gray; H, white; O, red; B, pink. | ||
We also wondered how the replacement of carborane by a phenyl moiety might affect the vertical excitation processes in these 1,3-di(4-carboxyphenyl) derivatives. Fig. 2 shows a representation of the change of electron densities on photo-excitation from S0 to S1 and Fig. S3 (ESI†) represents the de-excitation from T1 to S0 obtained from the STEOM-DLPNO-CCSD calculations. Lobes in Fig. 2 and Fig. S3 should not be confused with the common HOMO–LUMO orbitals. Lobes in these figures represent electron density differences between the initial and the final state, and the color relates to loss (purple) or a gain (pale brown) of electron density through the excitation or de-excitation processes. Thus, during the vertical S0 → S1 photo-excitation of mCB-L, there is a net electron density transfer from the phenyl rings toward the carboxylic acids (Fig. 2). The absence of a lobe in the carborane moiety means that its electron density change on excitation is zero, although the carborane orbitals do participate.42 In the case of the TDCA, there is an involvement of the three aromatic rings, indicating that on excitation, the transfer occurs from the external aromatic rings to the central one, being the electron density concentrated in the central phenyl group. The different electron density transfer between these two closely related molecules is probably due to their different symmetry, being mCB-L the less symmetrical. The analysis of the net electron density transfer on vertical S0 → S1 excitation of the longer molecules (mCB-L2 and QDCA; Chart 1) revealed clear differences between them and with their shorter counterparts. mCB-L2 shows a net electron density transfer from the two consecutive phenyl rings from one side of the molecule toward the rings in the other side. As in mCB-L, the carborane moiety shows an imperceptible electron density change. The QDCA shows an electron density transfer between the three central phenyl rings towards the phenyl-carboxylic moieties at the extremes of the molecule.
 | ||
| Fig. 2 Schematic diagrams for the energy absorption on photoexcitation from singlet states (S0 to S1). Purple (negative values) and pale brown (positive values) lobes represent the electron density differences between the initial and final state calculated at STEOM-DLPNO-CCSD level. | ||
Although mCB-L shows negligible fluorescence in the solid state, it was shown to behave as a good antenna ligand for Eu3+ and Tb3+.42 Such lanthanide cations are highly adaptable for use in lighting and sensing applications, but their effectiveness depends on the presence of an efficient antenna due to their low absorption capability.59,60 We previously reported a strong luminescence of mCB-Eu and mCB-Tb MOFs,42 which indicates that in the presence of any of these two lanthanide cations, excitation of mCB-L is followed by intersystem crossing (ICS) from its singlet state (S1) to the triplet excited state (T1) and subsequent energy transfer (ET) from T1 of mCB-L to the emissive levels of the metals, thus originating the observed metal-centered emissions.42 Even though this is the more accepted intramolecular ET process (also known as antenna effect) for lanthanide complexes and MOFs, a number of reports have shown that the ET can also be originated from the singlet state S1 (or charge transfer (CT) state) to the emissive levels of the metals.61–63 Nevertheless, the lowest STEOM-DLPNO-CCSD calculated excited states energies for TDCA and mCB-L (Table S2, ESI†) show, in agreement with our previous prediction based on time-dependent density functional theory (TDDFT) methods (Table S1, ESI†), that the energies of the S1 and T1 states for TDCA are lower than those for mCB-L. The latter indicates that energy transfer is expected to be much less efficient in the case of the TDCA ligand, as the energy of its triplet state is slightly lower than that of Eu3+ and much lower than that of Tb3+ (vide infra). In order to verify this prediction, we have synthesized a series of lanthanide MOFs based on the TDCA ligand and studied their photophysical properties.
Needle-like {[Ln2(TDCA)3(DMF)]n·Solv} (TDCA-Ln, where Ln = Eu, Tb) crystals were prepared by a solvothermal reaction between the TDCA ligand and the corresponding lanthanide nitrate in a mixture of DMF/HNO3 at 175 °C for 3 days (Fig. S5, ESI†). Infrared spectra (IR) and powder X-ray diffraction (PXRD) for TDCA-Eu and TDCA-Tb match well with the reported for TDCA-Ce64 and proved to be isostructural (Fig. S5 and S6, ESI†, respectively). Although the 3D structures of the latter differ to those for the carborane based MOF family, they all consist of 1D rod-shaped LnOx polyhedral, with carboxylate linkers coordinating in a bridging mode.64 The optical properties of TDCA-Ln (Ln = Eu, Tb) were investigated by measuring their UV-vis absorption (Fig. S1, ESI†) and emission spectra (Fig. 3 and Table 2) in the solid state at room temperature. Both MOFs display similar broad absorption bands to that of the free ligand, with the peak maximum slightly blue shifted to 290 nm (Fig. S1, ESI†). The luminescence spectra for TDCA-Eu (λex = 290 nm) displays red luminescence with well-resolved emission bands in the visible region at 591, 614, 650 and 699 nm (Fig. 1a), attributed to the characteristic Eu3+ transitions (5D0 → 7FJ; J = 1–4). These emission bands were similar to those of carborane-based analogue mCB-Eu.42 No emission bands were observed in the UV region, indicating a good quenching of the luminescence of the ligand. Contrary to the TDCA-Eu MOF, the related TDCA-Tb compound exhibits the simultaneous luminescence of the TDCA ligand and the Tb3+ ions upon excitation at 290 nm (Fig. 3b). The emission spectrum of TDCA-Tb shows a weak blue emission band centered at 377 nm, ascribed to the π → π* transitions of the ligand, and sharp bands at 489, 541, 582, and 621 nm, typical of the Tb3+ transitions (5D4 → 7FJ (J = 6–3). The lifetimes (τ) and Φ for TDCA-Eu, TDCA-Tb are summarized in Table 2. As can be seen in Table 2, Φ for TDCA-Eu and TDCA-Tb are 11.1% and 4.8%, respectively. Slight variations of the excitation wavelength (280–310 nm) show rather consistent Φ values and emission spectra (Fig. S7, ESI†). Mono-exponential fitting of the slower part of the decay curves was carried out to obtain the longest luminescence lifetimes, while avoiding possible background interferences. The obtained lifetimes for the TDCA MOFs resulted quite shorter than those of the corresponding mCB-Ln MOFs, as shown in the Table 2.
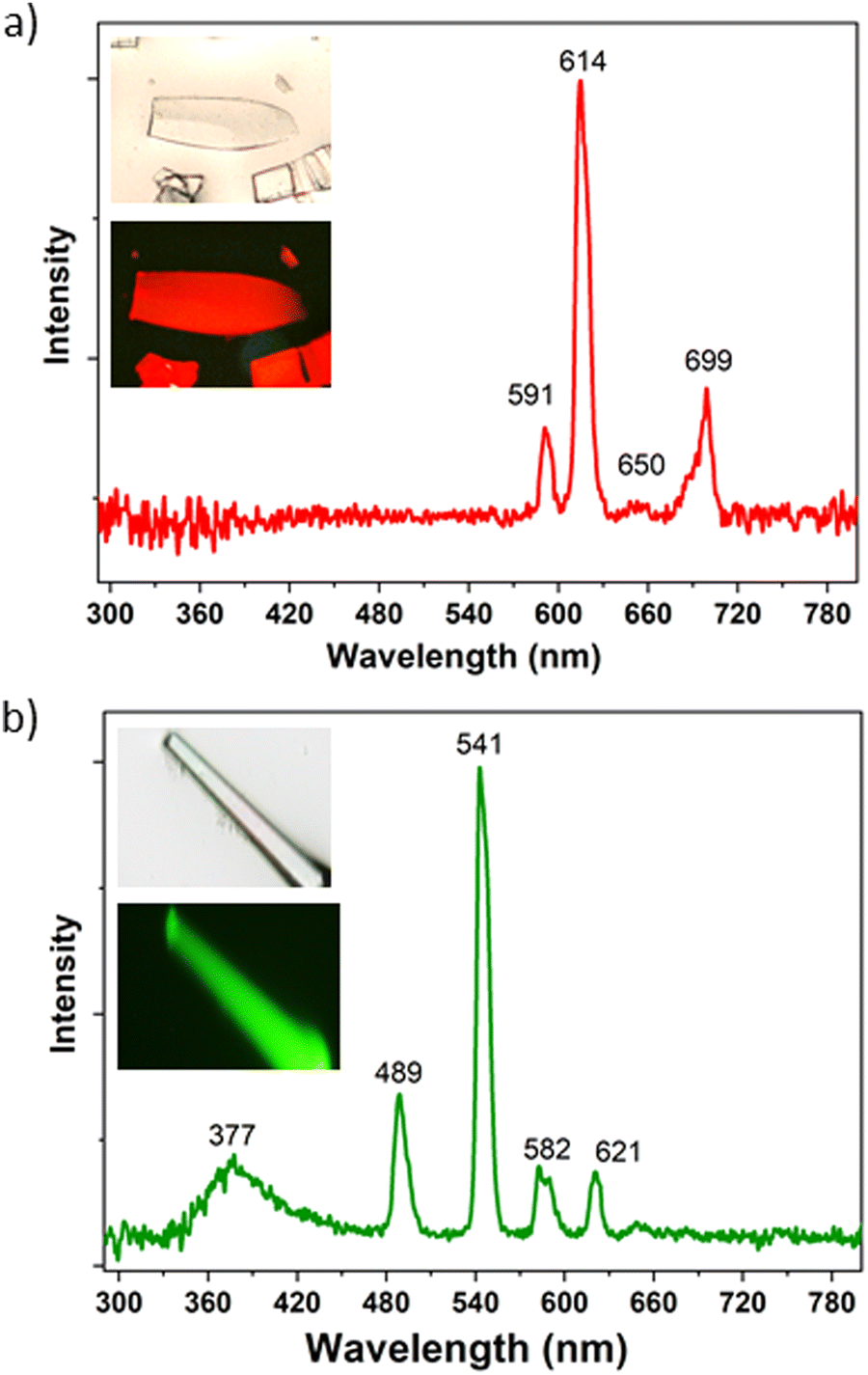 | ||
| Fig. 3 Solid-state emission spectra of TDCA-Eu (a) and TDCA-Tb (b) under continuous-wave irradiation (λex = 290 nm) at room temperature. Insets: Optical microscopy images of the corresponding crystals under white and the corresponding luminescence images). | ||
The shorter lifetimes of the TDCA-Ln MOFs suggest the presence of quenching effects of the sensitized Ln luminescence (Fig. S8, ESI†). Since both types of MOFs (TDCA-Ln MOFs and mCB-Ln MOFs) are measured in the same conditions and in the solid state, external causes for quenching (e.g. solvent) should be excluded. Thus, a plausible explanation could be related to the back-energy transfer from the Ln to the ligand, mainly occurring in the TDCA-based MOFs. This is also supported by the lower quantum yield values of these MOFs, compared to the mCB-based MOFs. If part of the energy of the Ln is quenched by the back-energy transfer, the amount of energy released through radiative decay is reduced, decreasing the value of the luminescence quantum yield. Nevertheless, the lower Φ values for the phenyl based TDCA-Ln than the related carborane based mCB-Ln MOFs could be also ascribed to less efficient direct energy transfer from the TDCA ligand to the Ln (for sensitizing both Ln3+ cations), compared to the mCB-L ligand. The observed broad blue emission for TDCA-Tb indicates that for this particular ion, the energy absorbed by the TDCA ligand is not completely transferred to the Tb3+ cations in the solid state. The absence of such blue emission for TDCA ligand in TDCA-Eu indicates a better energy transfer to the metal ion.
The above experimental data clearly corroborates our computational predictions. That is, the TDCA ligand is much worse antennae than the carborane based ligand mCB-L. Fig. 4 summarizes the STEOM-DLPNO-CCSD calculated S1, T1 energy values and gaps (ΔES–T) for mCB-L, TDCA and two hypothetical ligands mCB-L2 and mQDCA which are the result of adding two additional aromatic rings to the previous ones (Chart 1). Efficient lanthanide-centered luminescence of MOFs is fulfilled by the use of antenna linkers with the lowest triplet state of the organic linkers located at least 1850 cm−1 above the lowest emitting excited states of the lanthanide ions.65 Regarding the family of isostructural lanthanide (Eu3+ and Tb3+) MOFs containing mCB-L, the T1 energy level of the carborane ligand was calculated to be 23593 cm−1 and thus capable of sensitizing both cations (Eu3+, 17300 cm−1; Tb3+, 20500 cm−1; Fig. 4). In the case of the TDCA ligand, its T1 energy level is calculated to be 20989 cm−1, that is, 489 cm−1 above that for the Tb3+ excited state and 3689 cm−1 above that for Eu3+. The very much lower T1 energy of TDCA than that for mCB-L explains the more ineffective ET and the experimental observation of the free ligand emission and very weak Tb-centered emission (Fig. 3 and Table 2). In other words, the higher energy of T1 for mCB-L than that for TDCA allows to sensitize the emissive state of the Tb3+ cations, explaining the 10 times-fold quantum yield for the former one. The quantum yield difference between mCB-Eu and TDCA-Eu is less pronounced as the corresponding ligands T1 energies are well above the resonance level of the Eu ions in these cases. In the case of Ln-MOF formation of the two hypothetical ligands (mCB-L2 or QDCA, Chart 1), their very much lower T1 energies (Table S2, ESI† and Fig. 4) suggest that the QDCA ligand would be able to sensitize only Eu3+, while the mCB-L2 would not be acting as a good antenna for any of these two cations.
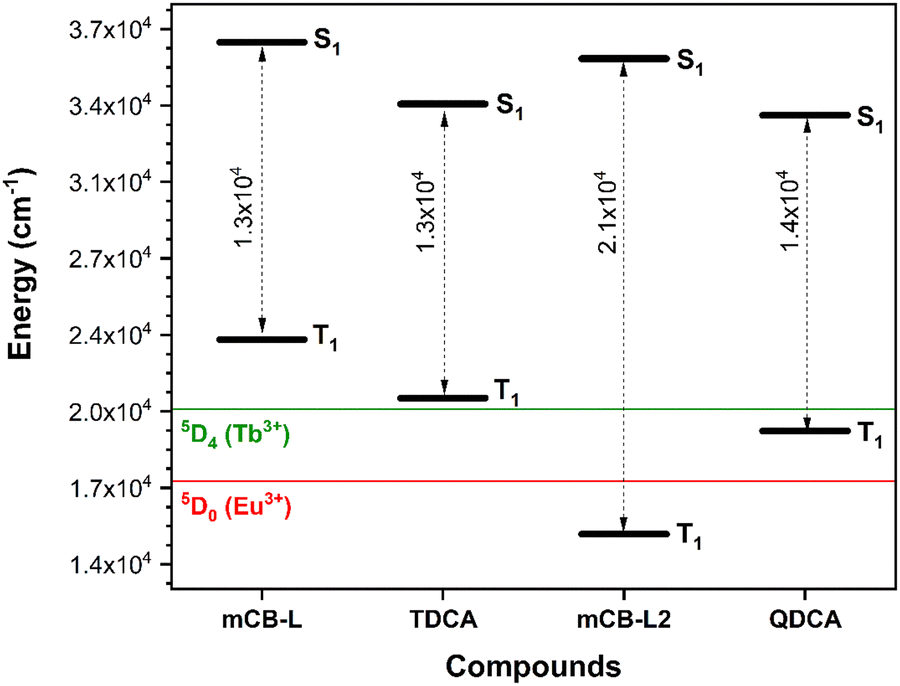 | ||
| Fig. 4 Dependence of the STEOM-DLPNO-CCSD calculated S1, T1 and energy gap (dashed grey arrows) values as a function of the molecular structures. Energies of the lowest emitting excited states of Tb3+ and Eu3+ are indicated as green and red lines, respectively. | ||
With regards to the STEOM-DLPNO-CCSD calculated minimum excited state energies for the extended compounds mCB-L2 and QDCA (Fig. 4 and Tables S1–S2, ESI†), the results indicate that the replacement of a central phenyl moiety by a 3-D m-carborane cluster increases the S1 energies (Fig. 4). Those energies are however rather uniform within the same series of ligands (mCB-L, 36806 cm−1/mCB-L2, 36080 cm−1versus TDCA, 34065 cm−1/QDCA, 33565 cm−1). We found however a higher variability in the calculated minimum T1 excited energies, as can be seen in Fig. 4. There is a decrease of the T1 energies of the compounds in the order mCB-L > TDCA > QDCA > mCB-L2. Thus, one could “modulate” the energy gaps (ΔES1–T1) by selecting the appropriate molecular structure of the ligand. Controlling the energy gaps between the S and T states is essential in the development of energy-efficient light-emitting diodes, various solar energy conversion methods, and photocatalytic transformations.66–68 Our data reveals that the shorter mCB-L and TDCA ligands show the smallest energy gaps (13213 and 13076 cm−1, respectively; Table S2, ESI† and Fig. 4), than the longer ligands QDCA (14034 cm−1) and mCB-L2 (21148 cm−1). The smaller gap of mCB-L, in addition to the high energy of its T1 state, explains the much better efficiency of this ligand as good antennae for the studied cations. It has been demonstrated that the intersystem crossing (ISC) efficiency can be improved by reducing the energy gap between S1 and T1 (ΔES1–T1).69,70 However, we cannot exclude a simultaneous ET process from S1 to the emitting states of the lanthanide ions.62 In such scenario (simultaneous S1 and T1 ET to Lanthanide cations), the observed changes in electron densities on vertical singlet excitations between mCB-L and TDCA (Fig. 2 left) are worth commenting as those may have an impact on the observed lanthanide based luminescence. On excitation, the electron density transfer takes places from the phenyl rings in mCB-L to the carboxylic acids, so that such electron density could be further transfer to the lanthanide cations on coordination. Such electron density transfer would not happen, or at least not preferentially, in the case of TDCA. All these might also explain why the carborane based linker is a much better antenna for Eu3+ and Tb3+ than the corresponding phenyl based linker.
We highlight the use of STEOM-DLPNO-CCSD calculations in conjunction with experimental findings to showcase how the replacement of a planar phenyl group with a 3D carborane fragment produces a significant impact in the photophysical properties of both the ligands and their lanthanide metal–organic frameworks (MOFs).
Conclusions
In conclusion, the TDCA ligand exhibited strong fluorescence with a high quantum yield in the solid state at room temperature, which is basically quenched when replacing the central phenyl moiety in TDCA by a carborane moiety (mCB-L). Quantum chemistry calculations (STEOM-DLPNO-CCSD and B3LYP-TDDFT) provided insights into the electronic structures and excited state energies of TDCA and mCB-L, indicating that the m-carborane-based mCB-L ligand may have a more accessible non-radiative decay process compared to the phenyl-based TDCA ligand. It was found that structural deformation in the minimal energy conical intersection (CI) was localized on a single phenyl ring of the TDCA, that was not taking place in the corresponding carborane derivative mCB-L. The findings highlight the importance of the molecular structure and symmetry in determining the photophysical properties of 1,3-di(4-carboxyphenyl) derivatives in the solid state.TDCA-Ln (Ln = Eu, Tb) MOFs were successfully synthesized and their optical properties were investigated. TDCA-Eu displayed red emission with sharp and well-resolved emission bands at 591, 614, 650, and 699 nm, characteristic of Eu3+ transitions, while TDCA-Tb exhibited simultaneous luminescence of the TDCA ligand and Tb3+ ions with emission bands at 489, 541, 582, and 621 nm, typical of Tb3+ transitions. However, the quantum yields for TDCA-Eu and TDCA-Tb were found to be much lower than those of carborane-based MOFs, indicating that the TDCA ligand is a less efficient light-absorbing antenna chromophore for sensitizing Eu3+ and Tb3+ ions compared to the carborane-based ligand. However, the faster and multiexponential decays of the TDCA MOFs indicate the presence of back energy-transfer, which could also contribute to the lower quantum yield values. The observed broad blue emission at 377 nm for TDCA-Tb suggests that the energy absorbed by the TDCA ligand is not completely transferred to the Tb3+ ions in the solid state. Computational calculations also supported the experimental results, showing that the TDCA ligand has a lower triplet energy level compared to the carborane-based ligand, which could result in the lower quantum yields observed for TDCA-Ln compounds.
This study provides insights into how the replacement of carborane by a phenyl moiety affects the vertical excitation processes in 1,3-di(4-carboxyphenyl) derivatives. The analysis of electron density changes on photo-excitation and de-excitation using STEOM-DLPNO-CCSD calculations reveals differences in electron density transfer between the carborane-based and phenyl-based derivatives. The findings suggest that the electron density transfer from the phenyl rings to the carboxylic acids upon excitation may play a role in the observed luminescence properties of lanthanide-based metal–organic frameworks (MOFs) containing carborane-based linkers. The use of wave function-based quantum chemistry calculations in conjunction with experimental results provides valuable insights into the photophysical properties of these compounds and their potential applications in luminescent materials.
Experimental section
All chemicals were of reagent-grade quality. They were purchased from commercial sources and used as received.Synthesis of {[(Ln)2(TDCA)3(DMF)x]n·Solv} (TDCA-Ln, where TDCA = [1,1′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :3′,1′′-terphenyl]-4,4′′-dicarboxylic])
:3′,1′′-terphenyl]-4,4′′-dicarboxylic])
A solvothermal method was adopted to prepare the TDCA-Ln crystal. Briefly, 0.02 mmol of Ln(NO3)3 (Ln = Ce, Eu or Tb) and 0.03 mmol of TDCA ligand were dissolved in 0.5 mL of DMF with the help of sonication. 7 μL of concentrated HNO3 was added into the above mixture. The above solution was further transferred to an 8-dram vial and heated at 175 °C in an oven for 72 h. The colorless crystals were obtained after washing with DMF and methanol for several times (Yield based on the Ln3+ cation: 42.7% for TDCA-Ce, 36.2% for TDCA-Eu and 33.7% for TDCA-Tb). Elemental analysis (%): calculated for {Ce2(TPDC)3(DMF)5]n·18H2O}: C 46.91, H 5.58, N 3.65; found: C 47.00, H, 4.16, N 3.64; calculated for {Eu2(TPDC)3(DMF)4]n·5H2O·4MeOH}: C 51.55, H 5.09, N 3.16; found: C 51.41, H, 3.94, N 3.05; calculated for {Tb2(TPDC)3(DMF)3]n·8H2O·2MeOH}: C 50.53, H 4.80, N 2.49; found: C 50.20, H, 3.82, N 2.45.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by MICINN (PID2019-106832RB-I00, PID2021-122464NB-100, PID2022-136892NB-I00), and through the Severo Ochoa Program for Centers of Excellence for the FUNFUTURE (EX2019-000917-S project), and by the Generalitat de Catalunya (2021/SGR/00286, 2021/SGR/00442). E. R. thanks Generalitat de Catalunya by an ICREA Academia award, Spanish Ministry Science for a Maria de Maeztu excellence grant (CEX2021-001202-M) and computer resources, technical expertise and assistance provided by the CSUC. Zhen Li acknowledges the China Scholarship Council (CSC) for his PhD grant (201808310071) and Scientific Research Foundation of Dezhou University (2023xjrc210). The present publication is dedicated to Prof. John D. Kennedy on the occasion of his 80th Birthday. Yin-yang symbol via Flickr (CC BY 2.0 licence).References
- Y.-C. Wei, K.-H. Kuo, Y. Chi and P.-T. Chou, Acc. Chem. Res., 2023, 56, 689–699 CrossRef CAS PubMed.
- T. Mehmood and J. P. Reddy, in Advances in aggregation induced emission materials in biosensing and imaging for biomedical applications, PT B, ed. R. S. Bhosale and V. Singh, 2021, vol. 185, pp. 179–198 Search PubMed.
- C. Chiatti, C. Fabiani and A. L. Pisello, in Annual review of materials research, ed. D. R. Clarke, 2021, vol. 51, pp. 409–433 Search PubMed.
- S. Gao, Z. Cui and F. Li, Chem. Soc. Rev., 2023, 52, 2875–2885 RSC.
- J. Liu, J. Xue, G.-P. Yang, L.-L. Dang, L.-F. Ma, D.-S. Li and Y.-Y. Wang, Coord. Chem. Rev., 2022, 463, 214521 CrossRef CAS.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, Soft Matter, 2013, 9, 4564–4579 RSC.
- R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC.
- A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed.
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed.
- M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed.
- F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed.
- A. M. Spokoyny, Pure Appl. Chem., 2013, 85, 903–919 CrossRef CAS PubMed.
- S. P. Fisher, A. W. Tomich, S. O. Lovera, J. F. Kleinsasser, J. Guo, M. J. Asay, H. M. Nelson and V. Lavallo, Chem. Rev., 2019, 119, 8262–8290 CrossRef CAS PubMed.
- J. Poater, C. Viñas, I. Bennour, S. Escayola, M. Solà and F. Teixidor, J. Am. Chem. Soc., 2020, 142, 9396–9407 CrossRef CAS PubMed.
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed.
- J. Poater, C. Viñas, M. Solà and F. Teixidor, Nat. Commun., 2022, 13, 3844 CrossRef CAS PubMed.
- J. Plesek, Chem. Rev., 1992, 92, 269–278 CrossRef CAS.
- R. N. Grimes, Carboranes, Academic Press, 2016 Search PubMed.
- F. Teixidor and D. E. Kaufmann, Science of Synthesis: Houben-Weyl Methods of Molecular Transformations, Georg Thieme Verlag, Stuttgart, 5th edn, 2015, vol. 6 Search PubMed.
- S. Fujii, MedChemComm, 2016, 7, 1082–1092 RSC.
- J. F. Valliant, K. J. Guenther, A. S. King, P. Morel, P. Schaffer, O. O. Sogbein and K. A. Stephenson, Coord. Chem. Rev., 2002, 232, 173–230 CrossRef CAS.
- P. Labra-Vázquez, R. Flores-Cruz, A. Galindo-Hernández, J. Cabrera-González, C. Guzmán-Cedillo, A. Jiménez-Sánchez, P. G. Lacroix, R. Santillan, N. Farfán and R. Núñez, Chem. – Eur. J., 2020, 26, 16530–16540 CrossRef PubMed.
- X. Wu, J. Guo, Y. Quan, W. Jia, D. Jia, Y. Chen and Z. Xie, J. Mater. Chem. C, 2018, 6, 4140–4149 RSC.
- S.-Y. Kim, J.-D. Lee, Y.-J. Cho, M. R. Son, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2018, 20, 17458–17463 RSC.
- K. Nishino, H. Yamamoto, J. Ochi, K. Tanaka and Y. Chujo, Chem. – Asian J., 2019, 14, 1577–1581 CrossRef CAS PubMed.
- D. K. You, H. So, C. H. Ryu, M. Kim and K. M. Lee, Chem. Sci., 2021, 12, 8411–8423 RSC.
- C. Bellomo, D. Zanetti, F. Cardano, S. Sinha, M. Chaari, A. Fin, A. Maranzana, R. Núñez, M. Blangetti and C. Prandi, Dyes Pigm., 2021, 194, 109644 CrossRef CAS.
- L. Ji, S. Riese, A. Schmiedel, M. Holzapfel, M. Fest, J. Nitsch, B. F. E. Curchod, A. Friedrich, L. Wu, H. H. Al Mamari, S. Hammer, J. Pflaum, M. A. Fox, D. J. Tozer, M. Finze, C. Lambert and T. B. Marder, Chem. Sci., 2022, 13, 5205–5219 RSC.
- J. Ochi, K. Tanaka and Y. Chujo, Dalton Trans., 2021, 50, 1025–1033 RSC.
- K. L. Martin, J. N. Smith, E. R. Young and K. R. Carter, Macromolecules, 2019, 52, 7951–7960 CrossRef CAS.
- D. Tu, S. Cai, C. Fernandez, H. Ma, X. Wang, H. Wang, C. Ma, H. Yan, C. Lu and Z. An, Angew. Chem., Int. Ed., 2019, 58, 9129–9133 CrossRef CAS PubMed.
- X. Wu, J. Guo, Y. Cao, J. Zhao, W. Jia, Y. Chen and D. Jia, Chem. Sci., 2018, 9, 5270–5277 RSC.
- J. Ochi, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2020, 59, 9841–9855 CrossRef CAS PubMed.
- Z. Wang, J. Zhao, M. Muddassir, R. Guan and S. Tao, Inorg. Chem., 2021, 60, 4705–4716 CrossRef CAS PubMed.
- K. Yuhara, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2022, 6, 1414–1420 RSC.
- F. Sun, S. Tan, H.-J. Cao, J. Xu, V. I. Bregadze, D. Tu, C. Lu and H. Yan, Angew. Chem., Int. Ed., 2022, 61, e202207125 CrossRef CAS PubMed.
- J. Tong, Y. Cao, Y.-W. Zhang, P. Wang, P. Wang, X.-J. Liao, W. Zhang, Y. Wang, Y.-X. Zheng, J.-J. Zhu and Y. Pan, Angew. Chem., Int. Ed., 2022, 61, e202209438 CrossRef CAS PubMed.
- Z. Wang, B. Chen, H. Zhang, H. Yang, S. Tao and R. Guan, Mater. Chem. Front., 2022, 6, 783–792 RSC.
- K. Tanaka, M. Gon, S. Ito, J. Ochi and Y. Chujo, Coord. Chem. Rev., 2022, 472, 214779 CrossRef CAS.
- S. Sinha, Z. Kelemen, E. Hümpfner, I. Ratera, J.-P. Malval, J. P. Jurado, C. Viñas, F. Teixidor and R. Núñez, Chem. Commun., 2022, 58, 4016–4019 RSC.
- J. Soldevila-Sanmartín, E. Ruiz, D. Choquesillo-Lazarte, M. E. Light, C. Viñas, F. Teixidor, R. Núñez, J. Pons and J. G. Planas, J. Mater. Chem. C, 2021, 9, 7643–7657 RSC.
- Z. Li, R. Núñez, M. E. Light, E. Ruiz, F. Teixidor, C. Viñas, D. Ruiz-Molina, C. Roscini and J. G. Planas, Chem. Mater., 2022, 34, 4795–4808 CrossRef CAS PubMed.
- D. Tu, J. Li, F. Sun, H. Yan, J. Poater and M. Solà, JACS Au, 2021, 1, 2047–2057 CrossRef CAS PubMed.
- A. Jana, M. Jash, W. A. Dar, J. Roy, P. Chakraborty, G. Paramasivam, S. Lebedkin, K. Kirakci, S. Manna, S. Antharjanam, J. Machacek, M. Kucerakova, S. Ghosh, K. Lang, M. M. Kappes, T. Base and T. Pradeep, Chem. Sci., 2023, 14, 1613–1626 RSC.
- J. Cabrera-González, A. Ferrer-Ugalde, S. Bhattacharyya, M. Chaari, F. Teixidor, J. Gierschner and R. Núñez, J. Mater. Chem. C, 2017, 5, 10211–10219 RSC.
- A. Ferrer-Ugalde, J. Cabrera-González, E. J. Juárez-Pérez, F. Teixidor, E. Pérez-Inestrosa, J. M. Montenegro, R. Sillanpää, M. Haukka and R. Núñez, Dalton Trans., 2017, 46, 2091–2104 RSC.
- M. Chaari, Z. Kelemen, J. G. Planas, F. Teixidor, D. Choquesillo-Lazarte, A. Ben Salah, C. Viñas and R. Núñez, J. Mater. Chem. C, 2018, 6, 11336–11347 RSC.
- M. Chaari, Z. Kelemen, D. Choquesillo-Lazarte, N. Gaztelumendi, F. Teixidor, C. Viñas, C. Nogués and R. Núñez, Biomater. Sci., 2019, 7, 5324–5337 RSC.
- L. Schwartz, L. Eriksson, R. Lomoth, F. Teixidor, C. Viñas and S. Ott, Dalton Trans., 2008, 2379–2381 RSC.
- F. Teixidor, R. Núñez, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2000, 39, 4290–4292 CrossRef CAS PubMed.
- R. Núñez, P. Farràs, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2006, 45, 1270–1272 CrossRef PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- J. Bae, M. Sakai, Y. Tsuchiya, N. Ando, X. K. Chen, T. B. Nguyen, C. Y. Chan, Y. T. Lee, M. Auffray, H. Nakanotani, S. Yamaguchi and C. Adachi, Front. Chem., 2022, 10 DOI:10.3389/fchem.2022.990918.
- C. Fang, B. Oruganti and B. Durbeej, J. Phys. Chem. A, 2014, 118, 4157–4171 CrossRef CAS PubMed.
- P. Rybczyński, M. H. E. Bousquet, A. Kaczmarek-Kędziera, B. Jędrzejewska, D. Jacquemin and B. Ośmiałowski, Chem. Sci., 2022, 13, 13347–13360 RSC.
- K. Ikemoto, T. Tokuhira, A. Uetani, Y. Harabuchi, S. Sato, S. Maeda and H. Isobe, J. Org. Chem., 2020, 85, 150–157 CrossRef CAS PubMed.
- Y. Harabuchi, M. Hatanaka and S. Maeda, Chem. Phys. Lett., 2019, 737, 100007 CrossRef.
- Y. Harabuchi, T. Taketsugu and S. Maeda, Phys. Chem. Chem. Phys., 2015, 17, 22561–22565 RSC.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- T. Gorai, W. Schmitt and T. Gunnlaugsson, Dalton Trans., 2021, 50, 770–784 RSC.
- P. A. Tanner, W. Thor, Y. Zhang and K.-L. Wong, J. Phys. Chem. A, 2022, 126, 7418–7431 CrossRef CAS PubMed.
- E. Kasprzycka, V. A. Trush, V. M. Amirkhanov, L. Jerzykiewicz, O. L. Malta, J. Legendziewicz and P. Gawryszewska, Chem. – Eur. J., 2017, 23, 1318–1330 CrossRef CAS PubMed.
- C. Yang, L.-M. Fu, Y. Wang, J.-P. Zhang, W.-T. Wong, X.-C. Ai, Y.-F. Qiao, B.-S. Zou and L.-L. Gui, Angew. Chem., Int. Ed., 2004, 43, 5010–5013 CrossRef CAS PubMed.
- Z. Li, X.-B. Li, M. E. Light, A. E. Carrillo, A. Arauzo, M. Valvidares, C. Roscini, F. Teixidor, C. Viñas, F. Gándara, E. Bartolomé and J. G. Planas, Adv. Funct. Mater., 2023, 33, 2307369 CrossRef CAS.
- L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487–505 CrossRef CAS.
- E. J. Peterson, J. Rawson, D. N. Beratan, P. Zhang and M. J. Therien, J. Am. Chem. Soc., 2022, 144, 15457–15461 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- T. Chen, L. Zheng, J. Yuan, Z. An, R. Chen, Y. Tao, H. Li, X. Xie and W. Huang, Sci. Rep., 2015, 5, 10923 CrossRef PubMed.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- S. Xu, Y. Yuan, X. Cai, C.-J. Zhang, F. Hu, J. Liang, G. Zhang, D. Zhang and B. Liu, Chem. Sci., 2015, 6, 5824–5830 RSC.
- F. Neese, WIREs Comput. Mol. Sci., 2018, 8, e1327 CrossRef.
- M. Nooijen and R. J. Bartlett, J. Chem. Phys., 1997, 106, 6441–6448 CrossRef CAS.
- M. Nooijen and R. J. Bartlett, J. Chem. Phys., 1997, 107, 6812–6830 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Sous, P. Goel and M. Nooijen, Mol. Phys., 2014, 112, 616–638 CrossRef CAS.
- A. K. Dutta, F. Neese and R. Izsák, J. Chem. Phys., 2016, 145, 34102 CrossRef PubMed.
- A. K. Dutta, M. Nooijen, F. Neese and R. Izsák, J. Chem. Phys., 2017, 146, 74103 CrossRef PubMed.
- A. K. Dutta, M. Nooijen, F. Neese and R. Izsák, J. Chem. Theory Comput., 2018, 14, 72–91 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc04018f |
| This journal is © The Royal Society of Chemistry 2024 |