
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanofluorochromism and self-recovery of alkylsilylpyrene-1-carboxamides†
Yuichi
Hirai
 *a,
Anna
Wrona-Piotrowicz
b,
Janusz
Zakrzewski
b,
Magdalena
Ciechańska
b,
Takahito
Ohmura
d,
Takashi
Takeda
a,
Takayuki
Nakanishi
a,
Rémi
Métivier
*c and
Clémence
Allain
*c
*a,
Anna
Wrona-Piotrowicz
b,
Janusz
Zakrzewski
b,
Magdalena
Ciechańska
b,
Takahito
Ohmura
d,
Takashi
Takeda
a,
Takayuki
Nakanishi
a,
Rémi
Métivier
*c and
Clémence
Allain
*c
aNational Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, 3050044, Japan. E-mail: hirai.yuichi@nims.go.jp
bDepartment of Organic Chemistry, Faculty of Chemistry, University of Łódź, Tamka 12, 91-403 Łódź, Poland
cUniversité Paris-Saclay, ENS Paris-Saclay, CNRS, PPSM, 4 Avenue des Sciences, 91190, Gif-sur-Yvette, France. E-mail: remi.metivier@ens-paris-saclay.fr; clemence.allain@ens-paris-saclay.fr
dNational Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, 3050047, Japan
First published on 19th January 2024
Abstract
A family of (alkylsilyl)pyrene-1-carboxamides exhibits similar mechanofluorochromic responses upon grinding. However, their spontaneous fluorescence recovery processes are distinct despite their similarity in chemical structures and crystal packings. Fluorescence spectroscopy, crystallography, and nanomechanical tests revealed that the chromic direction is dominated by the packing motif, while the fluorescence recovery is driven by the intermolecular interactions and the reversibility of deformation.
Introduction
Mechanofluorochromism (MFC), the phenomenon where organic and coordination compounds exhibit reversible changes in their emission properties under mechanical stimuli, has emerged as a captivating area of research in materials science, chemistry, and photonics.1–3 Thus, it has garnered considerable attention owing to its potential applications in sensors to visualise the location/magnitude of the stress and information encryption systems for anticounterfeiting technologies.4,5The ability to modulate the emission properties of compounds through mechanical deformation can be applied to the design of smart materials with tuneable and responsive luminescent behaviour. When subjected to external mechanical stimuli, such as compression, shear, grinding, or stretching, MFC materials experience changes in discrete molecular conformations, intermolecular interactions, and corresponding electronic and vibrational properties, which in turn manifest as shifts in their emission wavelength, intensity, and decay dynamics.6,7 One of the primary drivers of MFC is the reversible breaking and formation of non-covalent interactions, such as π–π stacking, hydrogen bonding, or metallophilic interactions, which play a crucial role in the molecular packing and electronic communication within the material.8–10 In addition to the disruption or re-establishment of these interactions under mechanical stress, the presence of twisted intramolecular charge transfer states and aggregation-induced emission (AIE) units can also contribute to the MFC response, thereby enhancing the emission efficiency and spectral shifts upon deformation.11–13 Thus, understanding the fundamental principles governing MFC and elucidating structure–property relationships are crucial for designing and optimising materials with tailored MFC behaviour.
To investigate these relationships, we focused on a series of pyrene-derived compounds by introducing different substituents to realise diverse steric and electronic structures.14–16 These structural modifications induce changes in the intermolecular interactions and molecular flexibility/rigidity, which influenced the mechanical response and consequent luminescent properties. While relevant research primarily focuses on large-wavelength shifts and multi-functionality,17–20 there have been rare comprehensive demonstrations of the intricate interplay between molecular structures and their dynamic mechanical/photophysical responses in a bulk solid state, including spontaneous recovery processes.21
In this study, we focused on a family of pyrene-derived carboxamides with alkylsilyl groups22 because of their reported monomer-like purple fluorescence in the solid state and the characteristic “half-stacking” arrangement, which allows realising MFC by increasing or decreasing the effective π-orbitals overlaps (Fig. 1). We investigated the fluorescence responsivity from a broad perspective using crystallography, spectroscopy, and nanomechanics to guide the design and development of novel materials with enhanced MFC properties.
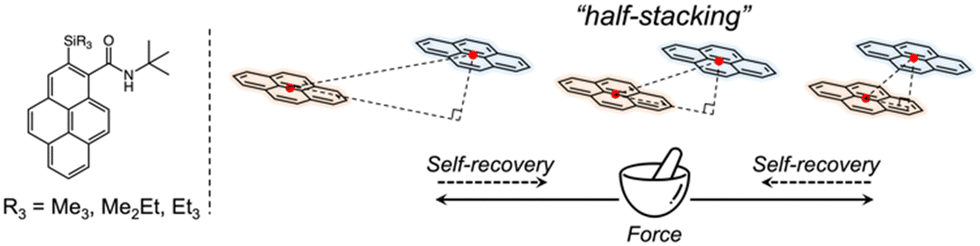 | ||
| Fig. 1 Chemical structures of pyrene-derived carboxamides with alkylsilyl groups, and a schematic of the stacking model. | ||
Results and discussions
N-tert-butyl-2-(alkylsilyl)pyrene-1-carboxamides (alkyl = (CH3)3, (CH3)2(C2H5) and (C2H5)3) were synthesised following a reported method22 and denoted as Me3, Me2Et, and Et3, respectively. The pristine powders exhibited purple fluorescence, and the recorded profiles (Fig. 2, black lines) were the same as those reported previously; λem2 was in the range of 405–410 nm, and a slightly broader spectrum was observed in the case of Et3 where the vibrational structure is attenuated. As mentioned already, the “half-stacking” arrangement of the π-orbitals of Me3 contributed to the monomeric emission bands, even in the solid state. Thus, the same stacking is expected for Me2Et, and the less-dominant monomeric emission bands in Et3 indicates the larger overlapping of π-orbitals. Upon mechanical grinding using a pestle and a mortar, a purple-to-blue bathochromic MFC was observed for all compounds, which indicates the formation of aggregated conformations with new intermolecular interactions. MFC spectroscopy was performed following the reported procedures to determine the mechanical responsivity including spontaneous self-recovery processes in the solid state (Fig. S1, ESI†).16 An increased broad emission band (λem3 ∼ 460 nm) and accompanying near-ultraviolet (UV) shoulder bands (λem1 ∼ 388 nm) with different degrees of contribution were observed (Fig. 2, blue lines). The relative intensity of the near-UV bands was minimal immediately after grinding and increased over time, particularly for Me2Et and Et3 (Fig. S2, ESI†). The shoulder bands should stem from the residues of the pristine and/or the ground forms after undergoing fast spontaneous molecular rearrangements under ambient conditions. In this study, the self-recovery rates were compared qualitatively by the spectral intensity ratios of the ground forms at λem2 and λem3, which resulted in the order of Me3 < Me2Et ≪ Et3. The spectral feature was almost preserved for Me3 after 30 min at room temperature, whereas the fluorescence spectra of Me2Et and Et3 showed a remarkably diminished emission band at 460 nm and concomitant increase in the UV emission for the same duration (Fig. 2, red lines). All compounds exhibited pristine-like spectra with an additional peak at approximately 388 nm after heating (Fig. 2, green lines), which are denoted as “recovered” in the remainder of the text. This indicates the mobility of the compounds in the solid state for reverting back to the pristine form and beyond without the aid of organic vapours or solvents. Differential scanning calorimetry (DSC) of pristine powders also indicated the lack of phase transitions under the condition of the MFC recovery processes (Fig. S3, ESI†).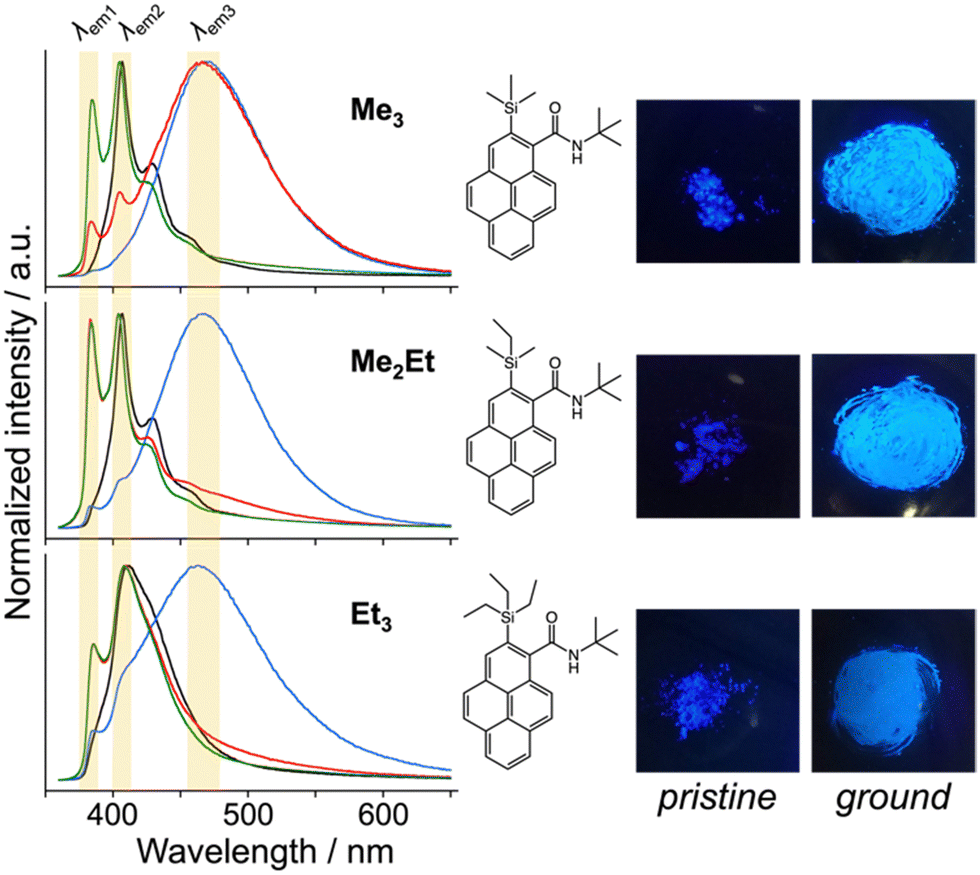 | ||
| Fig. 2 Normalised emission spectra of Me3, Me2Et, and Et3 at room temperature (RT) (black: pristine, blue: ground, red: 30 min at RT after grinding, green: 30 min at RT and 30 min heating at 50 °C after grinding, λex = 340 nm, in the solid state), and chemical structures and photographs under UV irradiation (left: pristine, right: ground, λex = 365 nm). Emission bands at approximately 388, 405–410 and 460 nm are highlighted by yellow shadows. | ||
To correlate the molecular arrangement and MFC response, the crystal structures of all compounds were determined using single-crystal X-ray diffraction (XRD) measurements and those of Me3 were re-determined. The space group was determined to be P21/c with half-stacking features, and the coincidence of simulated and experimental powder XRD (PXRD) patterns confirmed the phase purity of the pristine powders (Fig. S4, ESI†). The packing manner was categorised as a herringbone motif, which has atomic contact ratios %C⋯H/%C⋯C > 4.5, via the Hirshfeld surface analysis23–25 for all molecules (Fig. S5, ESI†). This corroborates the purple-to-blue bathochromic MFC behaviours of Me3, Me2Et, and Et3, where the monomer-like conformation shifts to a dimer-/aggregated-like conformation under mechanical stimuli.16 However, the similar values of atomic contacts between adjacent molecular surfaces and packing motifs still do not explain the distinct kinetic gap in the self-recovery processes that spontaneously occur after unloading. An energy framework analysis was also performed using CrystalExplorer to quantitatively visualise the topology of the intermolecular interactions in these crystals.26–29 The three-dimensional (3D) topologies illustrated the strongest interaction energy (EInterMol of approximately −70 kJ mol−1) between head-to-head pairs that involve NH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonding in all cases, which dominates the crystal packing manner (Fig. 3). In addition to the 30–39% larger EInterMol between half-stacked pairs compared with others, Et3 formed another interaction (−27.5 kJ mol−1) between its head-to-head pairs without NH⋯OC hydrogen bonding. Therefore, relatively stronger and multidimensional interactions between molecules could retain fewer monomeric fluorescence features of Et3 and drive the faster reconstruction of molecular packing in the ground form.
C hydrogen bonding in all cases, which dominates the crystal packing manner (Fig. 3). In addition to the 30–39% larger EInterMol between half-stacked pairs compared with others, Et3 formed another interaction (−27.5 kJ mol−1) between its head-to-head pairs without NH⋯OC hydrogen bonding. Therefore, relatively stronger and multidimensional interactions between molecules could retain fewer monomeric fluorescence features of Et3 and drive the faster reconstruction of molecular packing in the ground form.
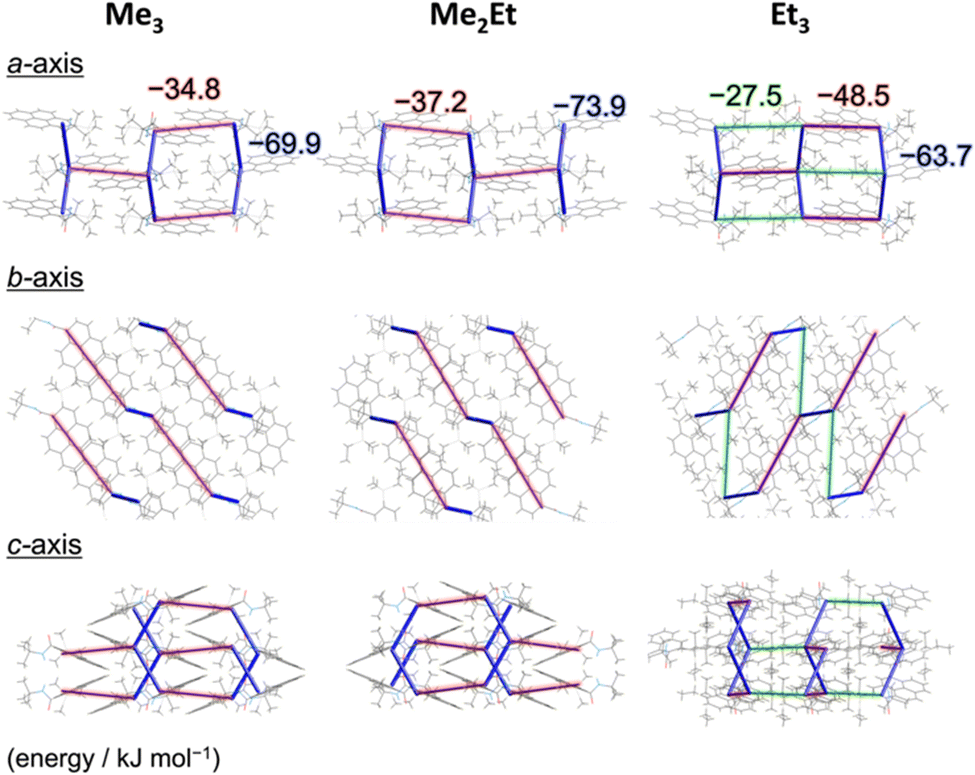 | ||
| Fig. 3 CE-B3LYP energy frameworks of Me3, Me2Et, and Et3 from different perspectives representing the net interaction energy EInterMol (cut-off: 25.0 kJ mol−1) at 123 K. The blue cylinders connect the centres of mass of adjacent molecules, and the diameter of cylinders are proportional to the magnitutdes of EInterMol. Opaque red and green shadows highlight the interactions between parallel half-stacked pairs and non-parallel head-to-head pairs without NH⋯OC hydrogen bonding, respectively. | ||
Following those crystallographic characteristics, the mechanical properties of the crystals were also investigated using nanoindentation experiments (see Fig. S6 in ESI† for testing procedures). The load–displacement (p–h) curves exhibited similar responses without sudden bursts (so-called “pop-ins”),30 which is common in brittle molecular crystals (Fig. 4a). The reduced Young's modulus (Er) and hardness (H) were estimated from more than 300 data sets for each molecule using the Oliver–Phar method (Fig. 4b and c, left).31 The estimated values indicated that Me3 was the softest among the studied compounds, as it exhibited relatively low Er and H values (3.9 and 0.26 GPa, respectively). The slow recovery nature of softer compounds was also supported by the pioneering study on MFC using polymorphic difluoroboron avobenzone crystals with distinct plasticity.21 In this study, additionally, the elasticity index (H/E) and geometric mean ((EH)1/2) were introduced to reflect the type of deformation and include both stiffness and hardness (Fig. 4c, right).32,33 Thus, the indentation hardness of Me3 was dominated by a more irreversible (plastic) character than the reversible (elastic) ones present in Me2Et and Et3. Although no general mathematical relationships yet exists between stiffness and density, highly dense materials are more prone to show higher Er values.34 In this case, the higher void volume of Me3 (Vvoid = 285, 266, and 267 Å3 for Me3, Me2Et, and Et3, respectively) possibly resulted in softer and more irreversible characteristics than in the other compounds because Me3 and Me2Et show similar EInterMol distributions and all compounds have similar crystal densities. Therefore, microscopic mechanical parameters may govern some of the self-recovery trends after unloading, which explains the relatively slow fluorescence recovery in Me3.
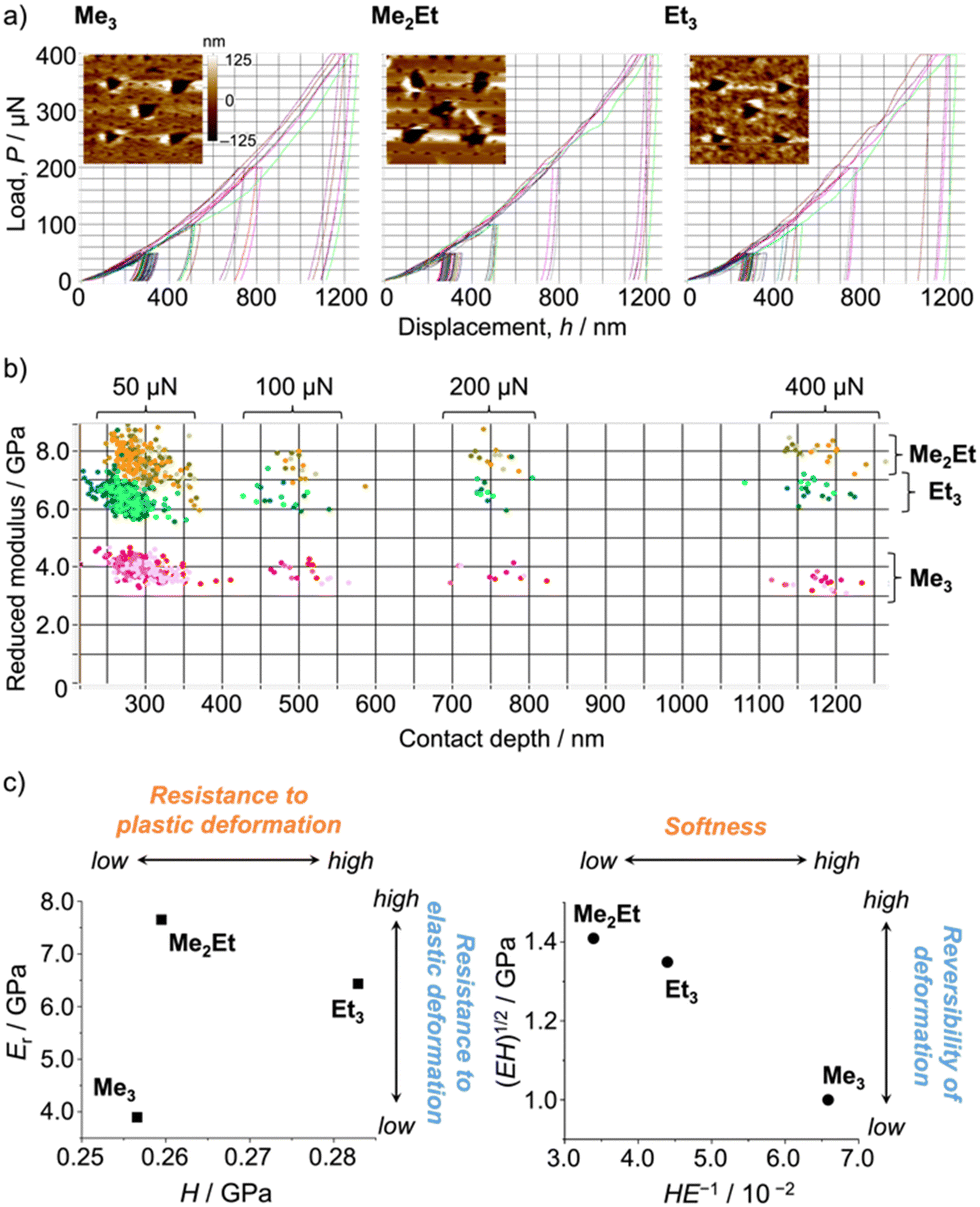 | ||
| Fig. 4 (a) Selected load–displacement curves and in situ scanning probe microscopy two-dimensional images after indentations (20 μm × 20 μm). (b) Distribution map of Young's modulus and the contact depth (>300 indentations for each molecule). (c) Young's modulus versus the hardness plot (left) and the geometric mean versus the elasticity index plot (right). | ||
Finally, we performed in situ temperature-variable spectroscopy on the recovered solids and pristine powders and crystallography on the single crystals in the range of 123–393 K to consider the solid-state mobility of the molecules in terms of fluorescence signals and absolute molecular configurations. The temperature-dependent emission spectra of the recovered samples revealed nearly identical profiles and thermal responses for Me3 and Me2Et (Fig. 5a, top). In addition to the small overlap of effective π-orbitals in the pristine forms, mechanical grinding further dislocated the half-stacked pairs, thereby resulting in the typically structured fluorescence of monomeric pyrenes at low temperatures. By contrast, the recovered form of Et3 showed ratiometric spectral changes between the VIS (λem ∼ 410–419 nm) and UV (λem ∼ 388 nm) bands. The intensity ratios (IVIS/IUV) decreased by 15-folds upon heating, accompanying a hypsochromic shift of 9 nm of the VIS peak (Fig. S7, ESI†). Pristine Et3 also showed a similar response with a smaller contribution of the UV band (Fig. 5a bottom), which indicated the existence of effective stacking in both the pristine and recovered forms. Such a temperature-dependent fluorescence response based on the interchromophoric interactions in a bulk solid material was also reported in a pyrene-based metal–organic framework.35
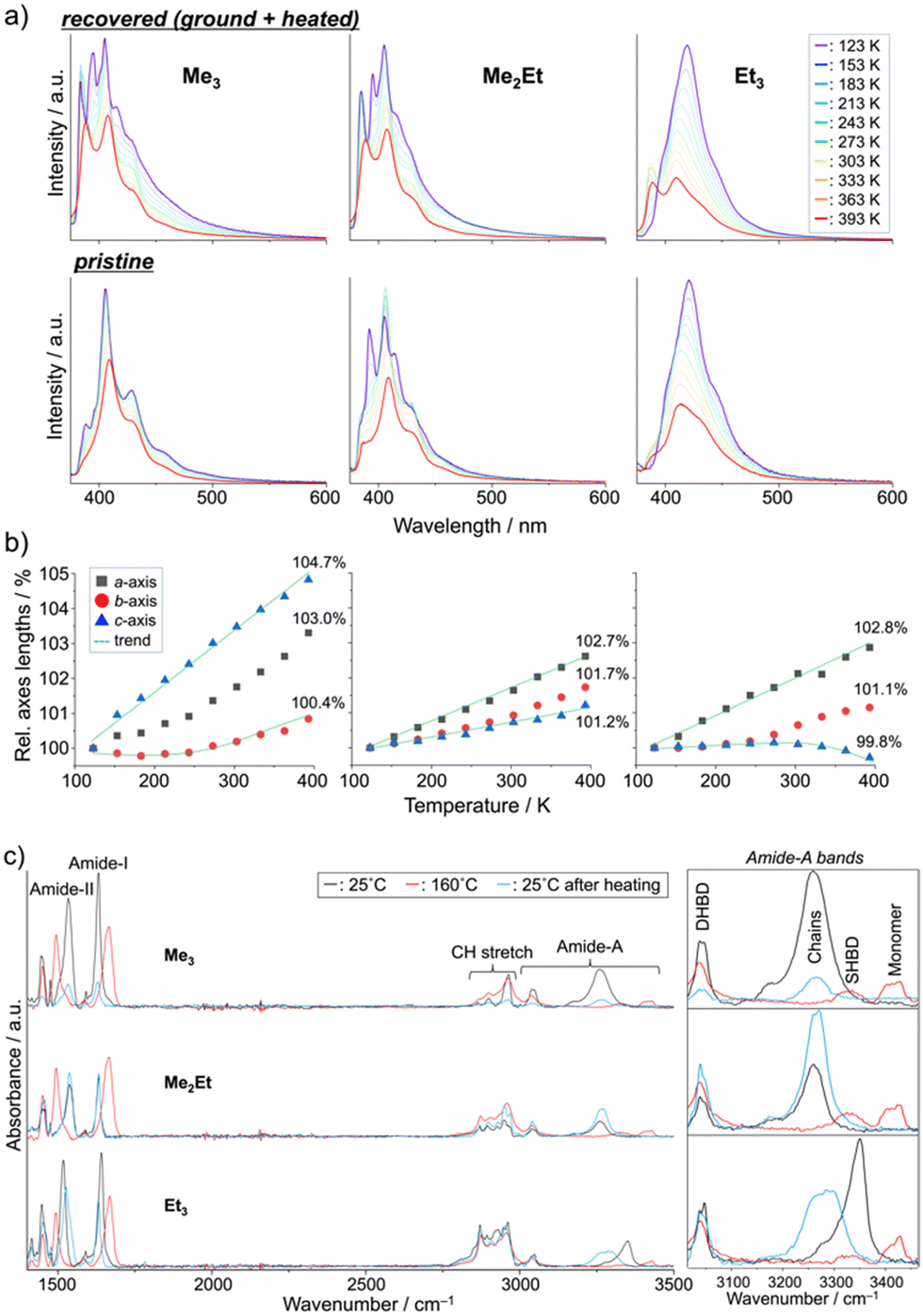 | ||
| Fig. 5 Temperature-dependent (a) fluorescence spectra (top: recovered, bottom: pristine), (b) relative unit-cell axes lengths (values at 393 K are indicated), and (c) ATR-FTIR spectra (DHBD: doubly hydrogen-bonded dimers, SHBD: single hydrogen-bonded dimers) of Me3, Me2Et, and Et3. | ||
Single-crystal XRD (SCXRD) experiments at different temperatures demonstrated the evolution of the unit-cell parameters upon heating (Fig. 5b and Tables S1–S3, ESI†). The contraction/expansion of crystals is generally observed under a controlled pressure using a diamond anvil cell (DAC), and the magnitude of cell expansion in this study (3–5% in the Me3c-axis, Me2Eta-axis, and Et3a-axis at 393 K relative to 123 K) is in the same order as that reported for pyrene-1-carbaldehyde (PA) at several GPa in DAC (relative to the atmospheric pressure).36 As previously mentioned for the emission of PA at a higher pressure, the higher emission intensity at lower temperature in this study may be explained by the suppression of vibrational relaxation processes or AIE due to the higher crystal density.37 The blue-shifted emission of Et3 at higher temperatures also coincides with the shorter emission wavelengths of PA under lower pressures. The intramolecular torsion angles (θtorsion) and energy frameworks (Fig. S8 and S9, ESI†) correlate the similar spectral shapes of Me3 at 123 K and 393 K with a slight change in EInterMol (Δ = 0.4%) and relatively large θtorsion relaxation. In addition to the approximately 5% decrease in EInterMol for half-stacked pairs and head-to-head pairs with NH⋯OC hydrogen bonding in Me2Et and Et3, respectively, the EInterMol between head-to-head pairs without NH⋯OC interaction in Et3 decreased by 25% after heating, which drove the anisotropic expansion of the cell and the appearance of a monomeric emission peak at a higher temperature. Therefore, the intra- and inter-molecular arrangements may change in either ordered or disordered solids depending on the temperature, thus resulting in characteristic emission spectral shifts in accordance with the overlap of the stacking.
These dynamic molecular conformation changes were also confirmed by the temperature-variable FT-IR spectra (Fig. 5c). Amide-I (ca. 1630 cm−1, CO stretch), amide-II (ca. 1530 cm−1, N–H bend/C–N stretch), and amide-A (ca. 3000–3500 cm−1, N–H stretch) bands were identified,38–40 and the shifts in amide-I and II bands (Δ ∼ +30 and −40 cm−1, respectively) and the appearance of the monomer's amide-A bands (ca. 3420 cm−1) upon heating above their melting points indicated the breakage of hydrogen bonding, which is in accordance with the spectroscopic and crystallographic results.
Experimental
Synthesis
N-tert-butyl-2-(alkylsilyl)pyrene-1-carboxamides (alkyl = (CH3)3: Me3, (CH3)2(C2H5): Me2Et and (C2H5)3: Et3) were synthesized following the reported methods.22 See ESI† for 1H- and 13C-NMR, FTIR, and DSC data. ATR-FTIR spectra were performed with a Nicolet 6700 spectrometer equipped with a deuterated triglycine sulphate detector. The spectra were obtained by adding together 64 scans at a resolution of 2 cm−1. DSC analyses were performed using a DSC 2500 Discovery, TA Instruments, under a nitrogen atmosphere for all samples at a heating and cooling rate of 10 °C min−1.Room-temperature MFC spectroscopy
Emission and excitation spectra of pristine powder samples were recorded on a HORIBA Jobin-Yvon Fluorolog FL3-221 spectrometer using a short path length optical quartz cell (20/C/Q/0.2, Starna), and the spectra were corrected for the response of the detector system. The pristine powder (<1 mg) was ground using an agate mortar, and the resulting amorphous solid adhered on a pestle surface was immediately transferred to a quartz plate by smearing to record the spectra within ca. 1 min and minimize the effect of self-recovery at room temperature. Due to the small amount of sample and the short time of measurements for the optimized experiments, emission decay lifetimes and quantum yields of the ground forms were not discussed in this study.In situ temperature-dependent fluorescence spectroscopy
Pristine powders and recovered solids were placed in a THMS600 temperature control stage equipped with a LNP-96 liquid nitrogen pump and a T96-S controller (Linkam Scientific Instruments Ltd). Emission spectra were recorded using an array spectrometer MCPD-9800 and a quantum efficiency measurement system QE-2100 (Otsuka Co., Ltd) in the range between 123 K and 393 K (150 K min−1).SCXRD experiments
Single crystals were mounted on a glass capillary using Araldite® epoxy adhesive. The measurements were performed on a Rigaku Synergy Custom system with a Hybrid Pixel Array Detector HyPix-Arc 150 using graphite monochromated Mo-Kα radiation. Correction for decay and Lorentz-polarization effects were made using empirical absorption correction, solved by direct methods, and expanded using Fourier techniques. Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model except for the ones on the amide donor nitrogen atoms which were refined freely. All calculations were performed using the Rigaku CrysAlis(Pro) crystallographic software package. CIF data was confirmed by checkCIF/PLATON service. CCDC data (2285534–2285543: Me3, 2285544–2285553: Me2Et, 2285523–2285532: Et3).†PXRD experiments
PXRD patterns were recorded on a Rigaku SmartLab X-ray Diffractometer using a single-crystal Si specimen holder to confirm the crystallinity of the pristine powders (Fig. S4, ESI†).Energy framework analysis
The intermolecular interaction energy was calculated at the B3LYP/6-31+G(d,p) level of theory using CrystalExplorer (version 21.5).27 The interaction energy was calculated for a target single molecule with all molecules having any atom within 3.8 Å.Nanoindentation tests
The target crystals were glued on a glass plate with a Crystalbond™ 509 mounting adhesive using a micromanipulator (Fig. S6, ESI†). Nanoindentations were carried out on a Triboindenter system (Hysitron Inc. Triboindenter TI950) equipped with a 60° 3-sided pyramidal tip on fused quartz (Hysitron Inc. Ti-0038). All experiments were performed in the load-controlled mode using a loading/unloading time of 10 s and a hold time of 10 s at peak load (50–400 μN). More than 4 sets of 81 indentations (9 × 9 array) were performed on each crystal. Young's modulus and hardness were estimated from the unloading curves using the reported eq. 1 and eq. 2 in Fig. S6 (ESI†).31In situ SPM images were taken using the same probe after indentations.Conclusions
In this work, we elucidated the structure–property relationships governing the MFC behaviour using pyrene-derived carboxamide with alkylsilyl and tert-butyl groups. All compounds exhibited similar purple-to-blue bathochromic MFC upon grinding. However, the spontaneous fluorescence recovery rates were significantly different, despite their slight differences in the chemical components and molecular packing motifs. Single-crystal XRD experiments and Hirshfeld and energy framework analyses indicated that the chromic direction was determined by the packing motif, whereas the self-recovery process was not simply dominated by the steric hinderance but by the distribution of intermolecular interaction energies. Nanomechanical tests also revealed the reversibility of deformation after unloading, and the “mobility” of molecular solids was demonstrated by temperature-dependent spectroscopy and crystallography either in crystalline or ground/annealed samples. The outcomes of our study contribute to a broader and deeper understanding of the MFC behaviour, including spontaneous fluorescence recovery processes, and inspire further exploration of innovative materials and devices based on simple organic systems.Author contributions
YH: conceptualisation, methodology, crystallography, spectroscopy, funding acquisition, writing; AW-P: organic synthesis, characterization; JZ: organic synthesis, characterization; MC: organic synthesis, characterization; TO: nanoindentation experiments; TT: single-crystal and powder XRD experiments; TN: in situ temperature-dependent fluorescence spectroscopy; RM: conceptualisation, review and editing; CA: conceptualisation, funding acquisition, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Grant Number 22K14661 and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 715757 MECHANO-FLUO to C. A.). Y. H.* gratefully acknowledges Dr Shiro Funahashi (National Institute for Materials Science, Japan) for single crystal X-ray analyses and Dr Bartłomiej Kost (Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Poland) for DSC and FTIR measurements.References
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- K. Muthamma, D. Sunil and P. Shetty, Mater. Today Chem., 2020, 18, 100361 CrossRef CAS.
- P. Shi, D. Deng, C. He, L. Ji, Y. Duan, T. Han, B. Suo and W. Zou, Dyes Pigm., 2020, 173, 107884 CrossRef CAS.
- D. Avobenzone, G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef PubMed.
- M. Raisch, W. Maftuhin, M. Walter and M. Sommer, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- Y. Sagara, H. Traeger, J. Li, Y. Okado, S. Schrettl, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2021, 143, 5519–5525 CrossRef CAS PubMed.
- H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044–10045 CrossRef CAS PubMed.
- T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252–6260 CrossRef CAS PubMed.
- Y. Sun, Z. Lei and H. Ma, J. Mater. Chem. C, 2022, 10, 14834–14867 RSC.
- J. Chen, S. Tan, Y. Yu, S. Zhang, W. Li, Q. Song, Y. Dong, C. Zhang and W. Y. Wong, J. Lumin., 2021, 237, 118179 CrossRef CAS.
- A. Pucci, Sensors, 2019, 19, 4969 CrossRef CAS PubMed.
- Y. Hirai, A. Wrona-Piotrowicz, J. Zakrzewski, A. Brosseau, R. Guillot, R. Métivier and C. Allain, Photochem. Photobiol. Sci., 2020, 19, 229–234 CrossRef CAS PubMed.
- Y. Hirai, A. Wrona-Piotrowicz, J. Zakrzewski, A. Brosseau, R. Métivier and C. Allain, J. Photochem. Photobiol., A, 2021, 405, 112972 CrossRef CAS.
- Y. Hirai, L. Laize-Générat, A. Wrona-Piotrowicz, J. Zakrzewski, A. Makal, A. Brosseau, L. Michely, D. L. Versace, C. Allain and R. Métivier, ChemPhysChem, 2021, 22, 1638–1644 CrossRef CAS PubMed.
- X. Zeng, T. Zhou, J. Liu, K. Wu, S. Li, X. Xiao, Y. Zhang, S. Gong, G. Xie and C. Yang, Adv. Opt. Mater., 2018, 6, 1801071 CrossRef.
- S. Takahashi, S. Nagai, M. Asami and S. Ito, Mater. Adv., 2020, 1, 708–719 RSC.
- S. Ito, R. Sekine, M. Munakata, M. Yamashita and T. Tachikawa, Chem. – Eur. J., 2021, 27, 13982–13990 CrossRef CAS PubMed.
- Z. Wen, T. Yang, D. Zhang, Z. Wang, S. Dong, H. Xu, Y. Miao, B. Zhao and H. Wang, J. Mater. Chem. C, 2022, 10, 3396–3403 RSC.
- G. R. Krishna, M. S. R. N. Kiran, C. L. Fraser, U. Ramamurty and C. M. Reddy, Adv. Funct. Mater., 2013, 23, 1422–1430 CrossRef CAS.
- A. Wrona-Piotrowicz, M. Ciechańska, J. Zakrzewski, R. Métivier, A. Brosseau and A. Makal, Dyes Pigm., 2016, 125, 331–338 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, University of Western Australia, 2012.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735–3738 RSC.
- M. J. Turner, S. Grabowsky, D. Jayatilaka and M. A. Spackman, J. Phys. Chem. Lett., 2014, 5, 4249–4255 CrossRef CAS PubMed.
- B. P. A. Gabriele, C. J. Williams, M. E. Lauer, B. Derby and A. J. Cruz-Cabeza, Cryst. Growth Des., 2020, 20, 5956–5966 CrossRef CAS PubMed.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7, 1564–1583 CrossRef CAS.
- D. Labonte, A. K. Lenz and M. L. Oyen, Acta Biomater., 2017, 57, 373–383 CrossRef CAS PubMed.
- M. F. Ashby, Materials Selection in Mechanical Design, Elsevier Science, 2016 Search PubMed.
- D. P. Karothu, J. Mahmoud Halabi, E. Ahmed, R. Ferreira, P. R. Spackman, M. A. Spackman and P. Naumov, Angew. Chem., Int. Ed., 2022, 61, 1–12 CrossRef PubMed.
- A. Gładysiak, T. N. Nguyen, R. Bounds, A. Zacharia, G. Itskos, J. A. Reimer and K. C. Stylianou, Chem. Sci., 2019, 10, 6140–6148 RSC.
- D. Tchoń and A. Makal, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 343–353 CrossRef PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- A. Warshel, M. Levitt and S. Lifson, J. Mol. Spectrosc., 1970, 33, 84–99 CrossRef CAS.
- L. P. Deflores, Z. Ganim, R. A. Nicodemus and A. Tokmakoff, J. Am. Chem. Soc., 2009, 131, 3385–3391 CrossRef CAS PubMed.
- K. S. Maiti, A. Samsonyuk, C. Scheurer and T. Steinel, Phys. Chem. Chem. Phys., 2012, 14, 16294–16300 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2285534–2285543: Me3, 2285544–2285553: Me2Et and 2285523–2285532: Et3. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc03968d |
| This journal is © The Royal Society of Chemistry 2024 |