
Trap engineering through chemical doping for ultralong X-ray persistent luminescence and anti-thermal quenching in Zn2GeO4†
Annu
Balhara
ab,
Santosh K.
Gupta
 *ab,
Malini
Abraham
cd,
Brindaban
Modak
be,
Subrata
Das
cd,
Chandrani
Nayak
f,
Harshini V.
Annadata
g and
Mohit
Tyagi
bh
*ab,
Malini
Abraham
cd,
Brindaban
Modak
be,
Subrata
Das
cd,
Chandrani
Nayak
f,
Harshini V.
Annadata
g and
Mohit
Tyagi
bh
aRadiochemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai-400085, India. E-mail: santoshg@barc.gov.in; santufrnd@gmail.com
bHomi Bhabha National Institute, Anushaktinagar, Mumbai-400094, India
cMaterials Science and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology, Thiruvananthapuram, Kerala 695019, India
dAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
eChemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai-400085, India
fAtomic and Molecular Physics Division, Bhabha Atomic Research Centre, Mumbai-400085, India
gBeamline Development & Application Section, Bhabha Atomic Research Centre, Mumbai 400085, India
hTechnical Physics Division, Bhabha Atomic Research Centre, Trombay, Mumbai-400085, India
First published on 18th December 2023
Abstract
Recently, defect luminescence-based ultralong persistent luminescent (PersL) materials have been increasingly appreciated for advanced applications. However, an in-depth understanding of trap manipulation is a big challenge in controlling trap distribution. In this work, we provide a complete understanding of defect-induced photoluminescence (PL) in Zn2GeO4 and the significant role of different defects and defect complexes. Excitation-dependent tunable emissions from blue to green regions indicated different mechanisms for filling traps for different excitation energies. Time-resolved emission spectroscopy (TRES) revealed time-dependent trap distribution, shifting the PL band from blue to green. The existence of different electron–hole recombination pathways in different time windows shed light on the complex PL of Zn2GeO4. Systematic temperature-dependent PL studies imply different trapping and de-trapping processes. We demonstrate the activation energies for different trapping mechanisms and the role of negative thermal expansion (NTE) of Zn2GeO4 in achieving the negative thermal quenching (NTQ) of PL. Further, the aliovalent doping of Pr3+ was used for trap manipulation and introducing additional intermediate defect states. Density functional theory calculations as well as thermoluminescence and electron paramagnetic resonance studies revealed a reduction in defect formation energies for selective defects, four-fold increase in trap density, and trap re-shuffling to optimum trap depths (0.73 eV) on the doping of Pr3+. The rich trap distribution resulted in a two-fold increase in the quantum yield of green emissions (∼19%) due to Zn interstitial defects. Improved afterglow on UV charging and an increase in the afterglow time from a few minutes in undoped Zn2GeO4 to an intense X-ray activated afterglow for 18 hours was observed in the Pr3+ doped Zn2GeO4 phosphor. Analysis of afterglow decay kinetics revealed the prominent trap-to-trap tunnelling mechanisms for long lifetimes. Impedance studies revealed the widening of electron channels and reduced resistance to electron movement with the incorporation of Pr3+ ions that enhanced PersL. LED fabrication was carried out to demonstrate the potential of the Zn2GeO4 phosphor for solid-state lighting. We believe that such kinetic and thermodynamic interpretation of defect chemistry will be helpful in tailoring the optoelectronic properties of native defect phosphors.
1. Introduction
Native defect phosphors are an ideal candidate for applications such as solid-state lighting, medical diagnosis, optical tagging, optical storage, photocatalysis and anti-counterfeiting.1 Over the past decades, native defect phosphors have attracted scientific attention owing to the wide scope for modulating the emission output, photoluminescence quantum yield (PLQY), afterglow, and thermal quenching (TQ) via defect engineering.2,3 In the past, the trap-tuning strategy was investigated in several native defect phosphors to achieve improved PLQY and afterglow.4 On a broad scale, approaches like doping, synthetic tuning, and band structure engineering have been renowned routes for tuning trap distribution to attain the desired performance for photocatalysis, electrocatalysis, persistent luminescence (PersL), optical information storage and solid-state lighting applications.3–9 Particularly, PersL materials with long-lasting afterglow have aroused scientific interest for broad applications, such as emergency lighting, night surveillance, bioimaging, information storage, optical tagging, and photodynamic therapy.10–12 With the emerging applications of X-ray-activated PersL phosphors in all spheres of mankind, as mentioned above, the quest for understanding the optical phenomenon and afterglow mechanisms stimulated extensive research in this field. However, in-depth investigations need to be conducted on the types of defect states, their behaviour, trapping–detrapping processes, trap distribution, and quenching mechanisms at different excitation wavelengths to develop efficient long PersL materials for targeted applications. Wang et al. in their recently published review article have elaborated on the latest developments in Ce3+/Eu2+-activated phosphors, highlighting the role of correlation between the local structure and photoluminescence (PL) properties to give an overall vision of the composition–structure–property correlations.13 In recent years, phosphors exhibiting defect-mediated negative thermal quenching (NTQ) of luminescence have demonstrated huge potential for lighting, medical, and security applications owing to the improved thermal stability of emissions.14,15 Therefore, extensive studies on materials that exhibit defect-related emissions, NTQ, and long PersL are of great importance for establishing a physical correlation between luminescent properties, charge carrier type and trap distribution.Among the semiconductor oxides, ternary oxide zinc germanate (Zn2GeO4) has been well established for applications such as display and optoelectronic devices, CO2 photoreduction, water splitting, lithium ion batteries, and ultraviolet (UV) photodetectors.15–20 Zn2GeO4 is a wide-band gap oxide (Eg = 4.5 eV) and has received prime attention due to excellent chemical and thermal stability, non-toxicity, self-activated luminescent properties, rich defect density, and high conductivity.17–19 It is one of the defect-rich phosphors with oxygen vacancies (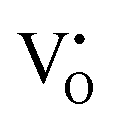 ), zinc interstitials (
), zinc interstitials (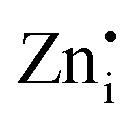 ), Zn and Ge vacancies (VGe, VZn) and antisite defects (ZnGe or GeZn).21–23 The white-bluish or green emission under UV excitation is originated from the donor (
), Zn and Ge vacancies (VGe, VZn) and antisite defects (ZnGe or GeZn).21–23 The white-bluish or green emission under UV excitation is originated from the donor ( )-acceptor (VGe and VZn) pair (DAP) recombination.22 Due to diverse applications of Zn2GeO4, a comprehensive investigation is a prerequisite to correlate the PL and afterglow with the electronic states and intermediate trap levels in Zn2GeO4. Several reports exhibited the visible emission of Zn2GeO4 and investigated the spectral changes on doping of Mn2+, Bi3+, Yb3+ and Li+ ions.5,22–24 In our previous work, a detailed experimental and theoretical investigation of PL due to defect states was carried out to realize the PersL blue-green emission of Zn2GeO4.23 Recently, Yuan et al.15 have also carried out an in-depth study of negative thermal expansion (NTE) mechanism of Zn2GeO4 below room temperature (RT). The NTE will result in increased stiffness of the Zn2GeO4 lattice and can significantly suppress the emission loss due to thermal effects.25,26 Dolado et al.22 investigated the electronic structure changes, optical output, and NTQ of Li-doped Zn2GeO4. Some other prominent works explored the bright green PersL in Mn2+-doped Zn2GeO4 phosphors for applications in fluorescence latent fingerprinting, biosensing, and rewritable information storage.5,27–29 In spite of significant investigations on Zn2GeO4 phosphors for various applications, the discussion about the origin of photoluminescence, trap distribution, afterglow mechanisms, and trapping–detrapping processes is limited to few studies. Unfortunately, reports on complete mechanistic studies of defect-mediated PL, negative thermal quenching of PL in Zn2GeO4 phosphors, role of structure rigidity, and different trapping and recombination processes at different excitation wavelengths and temperatures, are missing in the literature. Short-term afterglow and low PLQY of green emission in Zn2GeO4 phosphors are still the major concerns for real-time applications.
)-acceptor (VGe and VZn) pair (DAP) recombination.22 Due to diverse applications of Zn2GeO4, a comprehensive investigation is a prerequisite to correlate the PL and afterglow with the electronic states and intermediate trap levels in Zn2GeO4. Several reports exhibited the visible emission of Zn2GeO4 and investigated the spectral changes on doping of Mn2+, Bi3+, Yb3+ and Li+ ions.5,22–24 In our previous work, a detailed experimental and theoretical investigation of PL due to defect states was carried out to realize the PersL blue-green emission of Zn2GeO4.23 Recently, Yuan et al.15 have also carried out an in-depth study of negative thermal expansion (NTE) mechanism of Zn2GeO4 below room temperature (RT). The NTE will result in increased stiffness of the Zn2GeO4 lattice and can significantly suppress the emission loss due to thermal effects.25,26 Dolado et al.22 investigated the electronic structure changes, optical output, and NTQ of Li-doped Zn2GeO4. Some other prominent works explored the bright green PersL in Mn2+-doped Zn2GeO4 phosphors for applications in fluorescence latent fingerprinting, biosensing, and rewritable information storage.5,27–29 In spite of significant investigations on Zn2GeO4 phosphors for various applications, the discussion about the origin of photoluminescence, trap distribution, afterglow mechanisms, and trapping–detrapping processes is limited to few studies. Unfortunately, reports on complete mechanistic studies of defect-mediated PL, negative thermal quenching of PL in Zn2GeO4 phosphors, role of structure rigidity, and different trapping and recombination processes at different excitation wavelengths and temperatures, are missing in the literature. Short-term afterglow and low PLQY of green emission in Zn2GeO4 phosphors are still the major concerns for real-time applications.
In this regard, the substitution of Zn2+ sites by a hyper-valent rare earth ion (RE3+) can be a promising approach for introducing additional intermediate defect states and thus modulating the trap depth and probability of DAP transitions.4 Moreover, the charge mismatch and large ionic radii of RE3+ ions will induce energetically favoured formation of  and
and  vacancies to retain the charge balance. We judiciously chose Pr3+ ion for aliovalent doping in Zn2GeO4 because RE3+ incorporation at Zn2+ sites will generate additional electron and hole trap states (
vacancies to retain the charge balance. We judiciously chose Pr3+ ion for aliovalent doping in Zn2GeO4 because RE3+ incorporation at Zn2+ sites will generate additional electron and hole trap states ( and
and 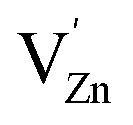 ) in Zn2GeO4. The change in trap distribution modulates the trap depths resulting in different trapping–detrapping processes which have profound effect on defect-driven photoluminescence. Gao et al.5 reported the tuning of band gap and trap depths by Pr3+ doping. Pr3+ doped inorganic materials are a great and highly favorable platform for the realization of longer and more intense persistent luminescence for multifunctional applications. Mao and his group have recently published a review article on various fascinating properties of Pr3+ doped phosphors for persistent luminescence and it covers various facets of Pr3+ ions.30 Recently, Pr3+ co-doping in Zn2GeO4:Mn2+ phosphor has been reported for enhanced storage photostimulable luminescence (PSL) due to defect engineering.5 Interestingly, no typical sharp bands of Pr3+ ions were observed in the PL and excitation spectrum, suggesting negligible radiative recombination between energy levels of Pr3+ in Zn2GeO4. Wan et al. reported similar enhancement in afterglow in Pr3+ co-doped Zn2GeO4:Mn2+ and no Pr3+ peaks were reported in the emission spectra.31 Hence, aliovalent doping of Pr3+ ions in Zn2GeO4 can be a promising approach for trap tuning and investigating trapping processes without the interference of RE3+ emissions with the host emissions.
) in Zn2GeO4. The change in trap distribution modulates the trap depths resulting in different trapping–detrapping processes which have profound effect on defect-driven photoluminescence. Gao et al.5 reported the tuning of band gap and trap depths by Pr3+ doping. Pr3+ doped inorganic materials are a great and highly favorable platform for the realization of longer and more intense persistent luminescence for multifunctional applications. Mao and his group have recently published a review article on various fascinating properties of Pr3+ doped phosphors for persistent luminescence and it covers various facets of Pr3+ ions.30 Recently, Pr3+ co-doping in Zn2GeO4:Mn2+ phosphor has been reported for enhanced storage photostimulable luminescence (PSL) due to defect engineering.5 Interestingly, no typical sharp bands of Pr3+ ions were observed in the PL and excitation spectrum, suggesting negligible radiative recombination between energy levels of Pr3+ in Zn2GeO4. Wan et al. reported similar enhancement in afterglow in Pr3+ co-doped Zn2GeO4:Mn2+ and no Pr3+ peaks were reported in the emission spectra.31 Hence, aliovalent doping of Pr3+ ions in Zn2GeO4 can be a promising approach for trap tuning and investigating trapping processes without the interference of RE3+ emissions with the host emissions.
Realizing the great potential of self-activated Zn2GeO4, this work is an effort to investigate the photo-dynamics of defect-related visible emissions of Zn2GeO4 and understand the nature of DAP recombination resulting in a complex PL band. Temperature-dependent small-angle X-ray diffraction (SAXD) patterns of Zn2GeO4 were acquired below room temperature and X-ray diffraction (XRD) patterns above RT to investigate the NTE of the Zn2GeO4 lattice. A doping strategy of aliovalent Pr3+ ions is used to trigger defect formation in Zn2GeO4 and introduce intermediate energy levels in the bandgap. The effect of Pr3+ ion doping in Zn2GeO4 on the PL properties, PLQY, defect concentration and trap depth, thermal stability and afterglow has been investigated extensively. The possible defect states in undoped and Pr3+-doped Zn2GeO4 systems, along with their defect formation energies, were probed by density functional theory (DFT) calculations. The influence of Pr3+ doping on the local structure was revealed by Raman spectroscopy. Synchrotron-based X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements were employed to probe the oxidation state of Pr and the local structure of Zn and Ge on Pr doping. DFT calculations were performed to obtain defect formation energies and to probe the formation of intermediate defect states on Pr3+ doping. Importantly, we demonstrated the excitation wavelength-dependent tunable emissions in undoped and Pr3+ doped Zn2GeO4. Steady-state and time-resolved emission spectroscopy (TRES) studies were performed for undoped and Pr3+ doped Zn2GeO4 phosphors to understand the decay kinetics of emissions from different excited species. Temperature-dependent PL studies were carried out to explore the NTQ of luminescence in Zn2GeO4 observed below RT. In-depth investigations on the temperature-dependent PL revealed the activation energies for NTQ and positive thermal quenching (PTQ) processes and different trapping–detrapping mechanisms. Further, the afterglow studies were performed to explore the mechanism involved and the role of Pr3+ in the long afterglow decay. X-ray-activated PersL studies were performed, and a long afterglow for Pr3+ doped Zn2GeO4 was captured after ceasing the X-ray excitation. The afterglow property can be a promising approach for reducing the dimming time of phosphor-converted light emitting diodes (pc-LEDs) and hence, we have demonstrated the potential of undoped Zn2GeO4 and Pr3+ doped Zn2GeO4 phosphors for pc-LEDs. The above insights into trap engineering through aliovalent Pr3+ doping in Zn2GeO4 for tuning the emission, PLQY, and afterglow will help in utilizing Zn2GeO4 phosphor for potential applications. The material synthesis, instrumentation, details about EXAFS measurements/discussions and computational methodology for DFT calculations are mentioned in the ESI† file as S1, S2, S3, and S4, respectively.
2. Results and discussion
2.1. Structural, phase, NTE, and local structure analysis
The XRD patterns of undoped Zn2GeO4 and Zn2GeO4:xPr3+ (x = 0.005, 0.01, 0.02, and 0.05) synthesized by a two-step solid-state route are presented in Fig. 1a. The XRD patterns match completely with the literature (PDF no. 011-0687) that confirmed the formation of rhombohedral structure of Zn2GeO4 lattice in all the samples. No impurity peaks were observed in the XRD pattern of Zn2GeO4. However, the introduction of Pr3+ ions at higher doping concentrations (x > 0.01) resulted in the appearance of extra peaks with weak intensities due to the presence of unreacted GeO2. The crystal structure of Zn2GeO4 (Fig. 1b) consists of GeO4 and ZnO4 tetrahedra forming four-membered and two different six-membered rings with non-equivalent Zn sites (Zn1 and Zn2). | ||
| Fig. 1 (a) XRD patterns of Zn2GeO4:xPr3+ (x = 0, 0.005, 0.01, 0.02, and 0.05) samples. (b) Crystal structure of Zn2GeO4 with ZnO4 and GeO4 tetrahedral units and two non-equivalent Zn sites in six-membered rings. (c) Raman spectrum of the 0.01Pr sample, and (d) cell volume (Å3) of Zn2GeO4 as a function of temperature in Kelvin. | ||
Raman spectroscopy was performed to study the phase formation and explore the doping effects of Pr3+ on the local structures. The peaks in the Raman spectra of Pr3+ doped Zn2GeO4 (Fig. S1a, ESI†) are similar to the Raman spectra of undoped Zn2GeO4.23 The peaks at 744 and 801 cm−1 are associated with the stretching vibrational modes of GeO4. The Zn–O–Ge stretching and Zn–O–Ge bending modes appeared at 752 and 776 cm−1 in the Zn2GeO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.01Pr3+ (0.01Pr) sample, respectively.15,23 The wave numbers of the Raman bands were obtained by Lorentzian fitting of Raman spectra (Fig. 1c), and shifts of these Raman bands as a function of Pr3+ doping concentrations are displayed in Fig. S1b (ESI†). A blue shift of Raman bands was observed on Pr3+ doping at low concentrations (x ≤ 0.01) followed by a red shift at higher doping concentrations. The blue shifts indicated increased stiffness of Zn2GeO4 crystal lattice on doping of Pr3+ at low concentrations.15 Fourier transform infrared spectroscopy (FTIR) spectra showed similar patterns in undoped and Pr3+ doped Zn2GeO4 and the characteristic peaks were observed below 800 cm−1 (Fig. S2, ESI†). The vibrational modes of the ZnO4 and GeO4 tetrahedra appeared in the range of 400–650 cm−1 and 660–850 cm−1. The EDS spectra of Pr3+ doped phosphors confirmed the presence of Zn, Ge, O, and Pr elements, and elemental mapping indicated homogenous distribution (Fig. S3a–d, ESI†). Further, energy-dispersive X-ray fluorescence (ED-XRF) confirmed the presence of Zn, Ge, and Pr elements in doped samples (Fig. S3e, ESI†).
:0.01Pr3+ (0.01Pr) sample, respectively.15,23 The wave numbers of the Raman bands were obtained by Lorentzian fitting of Raman spectra (Fig. 1c), and shifts of these Raman bands as a function of Pr3+ doping concentrations are displayed in Fig. S1b (ESI†). A blue shift of Raman bands was observed on Pr3+ doping at low concentrations (x ≤ 0.01) followed by a red shift at higher doping concentrations. The blue shifts indicated increased stiffness of Zn2GeO4 crystal lattice on doping of Pr3+ at low concentrations.15 Fourier transform infrared spectroscopy (FTIR) spectra showed similar patterns in undoped and Pr3+ doped Zn2GeO4 and the characteristic peaks were observed below 800 cm−1 (Fig. S2, ESI†). The vibrational modes of the ZnO4 and GeO4 tetrahedra appeared in the range of 400–650 cm−1 and 660–850 cm−1. The EDS spectra of Pr3+ doped phosphors confirmed the presence of Zn, Ge, O, and Pr elements, and elemental mapping indicated homogenous distribution (Fig. S3a–d, ESI†). Further, energy-dispersive X-ray fluorescence (ED-XRF) confirmed the presence of Zn, Ge, and Pr elements in doped samples (Fig. S3e, ESI†).
Temperature-dependent XRD patterns acquired below RT were used to investigate the thermal expansion (TE) behavior of Zn2GeO4 on heating. The low-temperature SAXD patterns were acquired from 123 to 300 K (Fig. S4, ESI†), and Fig. S5a (ESI†) shows the structure refinement of XRD data taken at 123 K. The lattice parameters of Zn2GeO4 determined from Rietveld refinement and lattice constants are found to be a = b = 14.099 Å, and c = 9.439 Å (α = β = 90°, and γ = 120°) at RT. The lattice volume contraction or NTE of the Zn2GeO4 lattice was revealed from the decreased cell volume and cell constants on heating the sample from 123 to 300 K (Fig. 1d). All the three a, b, and c axes shrink on heating below RT and hence, Zn2GeO4 exhibit a three-dimensional NTE (Fig. S5b, ESI†). Yuan et al.15 ascribed NTE to the “external modes” with low-frequency vibrations and the transverse vibrations of O atoms in ZnO4 and GeO4 tetrahedra forming six and four-membered rings with large spaces. However, Zn2GeO4 exhibits a very low NTE, resulting in a gradual decrease in the lattice constants and lattice volume below RT. XRD patterns were recorded above RT to study the behaviour of thermal expansion of the Zn2GeO4 lattice above the RT. XRD peaks shift towards the lower 2θ values on heating the Zn2GeO4 sample above RT (Fig. S6, ESI†). This confirmed the lattice expansion of Zn2GeO4 above RT, and thus, the NTE effect is prominent below RT. This suggested that the structural rigidity of the Zn2GeO4 lattice can efficiently suppress the thermally activated non-radiative transitions up to RT. Further, the low thermal expansion coefficient of the Zn2GeO4 lattice has attracted attention to investigating the optoelectronic properties of Zn2GeO4 above RT.
The surface composition of Zn2GeO4 and 0.01Pr samples and the influence of Pr3+ doping on the chemical and structural properties of Zn2GeO4 were investigated by XPS measurements. The survey scan and XPS spectra of Zn3d (10.7 eV), Zn2p (1022 and 1045 eV), and Ge3d (32.3) confirmed the presence of Zn and Ge in +2 and +4 oxidation states, respectively (Fig. S7, ESI†). The Zn3d, Zn2p, and Ge3d peaks in the Pr-doped Zn2GeO4 shifted to lower binding energies, which indicated a different chemical environment around these ions in both samples. Ge3d spectra with only a single component can be attributed to the Ge–O bonds in GeO4 tetrahedra. The Gaussian deconvoluted XPS spectra of the O1s peak in undoped and doped samples display two deconvoluted peaks (Fig. S8, ESI†). The former can be ascribed to lattice oxygen, while the latter may have contributions from oxygen vacancies or surface adsorbed oxygen. Moreover, due to a very low concentration of Pr3+, XPS does not reveal reliable information on its oxidation state.
The substitution site of large Pr3+ ions (0.99Å) in Zn2GeO4 was unclear as it may substitute Zn2+ ions (0.74 Å) or enter the interstitial sites of the six-membered rings. To explore the local structure and oxidation state of Pr ions in Zn2GeO4, XANES (Fig. S9, ESI†), and EXAFS measurements were carried out for Zn K-edge, Ge K-edge, Pr-L3 edge for undoped and Pr doped samples (Fig. S10, ESI†). The details about the synchrotron beamline, as well as EXAFS and XANES fitting, are discussed in S3 (ESI†). The average bond distances (R), coordination numbers (N), and disorder (Debye–Waller) factors (σ2), which give the mean square fluctuations in the distances, have been used as fitting parameters and listed for Zn K-edge, Ge K-edge, and Pr L3 edge in ESI† (Tables S1–S3), respectively. EXAFS studies revealed that the absorption edges (Fig. S10, ESI†) and the XANES features of the undoped and Pr-doped Zn2GeO4 samples (Fig S9b, ESI†) are exactly the same at the Ge-K edge, which suggested that Pr doping does not change the effective charge and local environment around Ge cations in Zn2GeO4. From XANES measurement, it is apparent that Pr cations probably occupy the Zn sites in the Zn2GeO4 structure. The σ2 values increased for Ge–O coordination shells, which suggested increased distortion in the local structure of Ge–O bonds on Pr doping due to a large size mismatch. Fig. S9c (ESI†) shows the Pr L3 edge XANES spectra of the Pr doped Zn2GeO4 samples along with the Pr6O11 standard where Pr cations exist in a mixed oxidation state of +3 and +4. The peaks at 5971 eV and 5982 eV are characteristic of Pr3+ and Pr4+ cations.32,33 The Pr L3 edge XANES spectra of Pr3+ doped Zn2GeO4 samples show a single peak at 5971 eV, suggesting that Pr cations exist in +3 oxidation states in Pr doped samples.
2.2. DFT calculations
The computational methodology for the DFT calculations is discussed in ESI† (Section S4.1). The calculated lattice parameters for Zn2GeO4 (a = b = 14.51 Å, and c = 9.69 Å) are found to be in good agreement with the experimentally and theoretical calculated values (a = b = 14.27 Å, c = 9.53 Å). Fig. 2a shows the density of states (DOS) plots for undoped Zn2GeO4, which indicates that the valence band maximum (VBM) is mainly composed of Zn(d) and O(p) orbitals, while the conduction band minimum (CBM) is hybridized states of Zn(s), Ge(s), and O(p) orbitals. The calculated band gap using PBE0 hybrid functional (4.39 eV) is found to be close to the experimental value (∼4.6 eV) and the reported value in the literature (4.50 eV).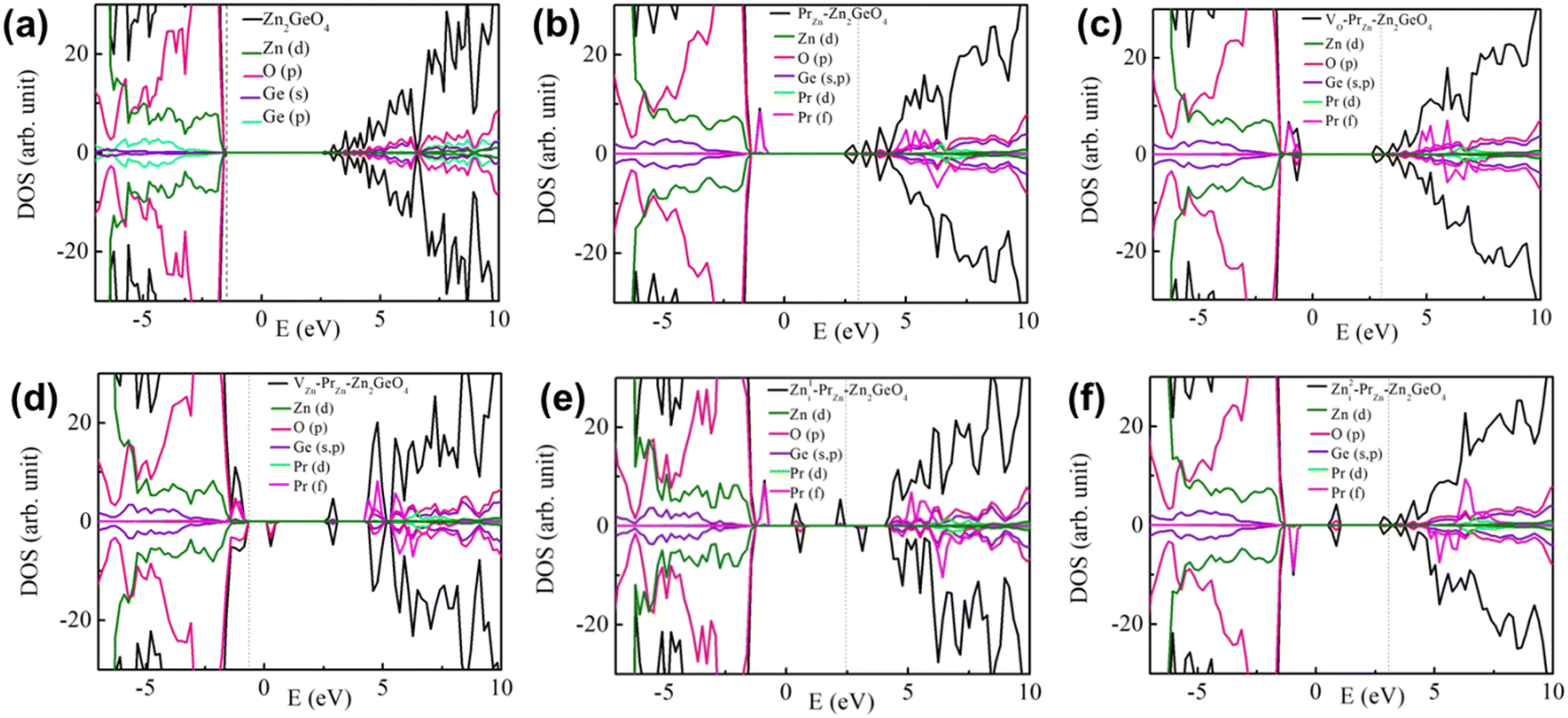 | ||
| Fig. 2 Density of states for (a) Zn2GeO4 and (b) PrZn–Zn2GeO4 in the presence of (c) one neutral oxygen vacancy, (d) neutral zinc vacancy, (e) zinc interstitial in the Zn–Zn ring position ((Zn1i)–PrZn–Zn2GeO4), and (f) zinc interstitial in the Zn–Ge ring position ((Zn2i)–PrZn–Zn2GeO4). | ||
The defect formation energies (Ef) for the Pr-doped Zn2GeO4 in the presence of different defects have been computed using the methodology discussed in ESI† (Section S4.2) and are provided in Table 1. To probe the role of different defects on the electronic structure of Zn2GeO4, we have analyzed the DOS in the presence of different defects. Notably, the Pr3+ doping in Zn2GeO4 may introduce PrZn defects when Pr substitutes Zn lattice site and Pri defects if Pr3+ ions occupy the interstitial sites in six-membered rings. The Ef is the lowest for Pr3+ substituting for Zn2+, which indicates that the substitutional doping of Pr into the Zn lattice site (PrZn) is energetically more favourable over interstitial doping (Pri), which is in line with synchrotron-based X-ray absorption spectroscopy. Herein, we have simulated the PrZn–VO, PrZn–VZn and PrZn–Zni defect complexes to evaluate the Ef and DOS. DFT results revealed that the PrZn defects are more energetically favoured than the Pri defects.
| System | Defect formation energy (eV) |
|---|---|
| PrZn–Zn2GeO4 | −3.33 |
| VO–PrZn–Zn2GeO4 | 0.25 |
| VZn–PrZn–Zn2GeO4 | 1.02 |
| Zn interstitial (Zn1i)–PrZn–Zn2GeO4 | −1.77 |
| Zn interstitial (Zn2i)–PrZn–Zn2GeO4 | −1.85 |
| Pr interstitial1 (Pr1i–Zn2GeO4) | −1.73 |
| Pr interstitial2 (Pr2i–Zn2GeO4) | −1.61 |
In our previous work,23 we calculated the Ef values in undoped Zn2GeO4 and the formation of Zni was found to be the energetically most favourable. Substitution of Zn2+ sites by a hyper-valent Pr3+ will induce energetically favoured formation of PrZn and VZn vacancies to retain the charge balance. Importantly, the defect complex formation due to the interplay between PrZn, Vo, Zni, and VZn defects can effectively alter the defect levels and formation energies. The significant reduction of Ef values on Pr doping in Zn2GeO4 for all the native defects compared to the Zn2GeO4 can be attributed to the formation of defect complexes. Impressively, the Ef for Vo decreased from 3.29 eV in undoped Zn2GeO4 to 0.25 eV for PrZn–VO complexes and the Ef for VZn reduced from 6.35 eV to 1.02 eV on PrZn–VZn complex formation. The Ef for the Zni defect at both the Zn–Zn and Zn–Ge ring interstitial positions are lowered significantly in the presence of Pr. This indicated that the presence of Pr facilitated the formation of Zni defects together with both oxygen and zinc vacancies due to formation of stable defect complexes. Hence, DFT results proposed the increase in defect formation on Pr doping, which will assist in achieving a higher probability of DAP transitions in Pr-doped Zn2GeO4.
Now, we proceed to investigate the effect of different defects on the electronic structure of PrZn–Zn2GeO4. Fig. 2b–f show the DOS for the PrZn–Zn2GeO4 unit cell with PrZn defects, and PrZn–VO, PrZn–VZn, and PrZn–Zni (Zn1i and Zn2i) defect complexes, respectively. We have also investigated the DOS for Pr doping at the interstitial sites and considered two different structures (Pr1i–Zn2GeO4 and Pr2i–Zn2GeO4) similar to the case of Zn interstitial (Fig. S11, ESI†). Various Pr-dopant-induced energy states were introduced in-between the bandgap due to the presence of PrZn defects and formation of different defect complexes. The impurity states induced after Pr doping in Zn2GeO4 have been discussed in detail in ESI† (Section S4.3) and are displayed in the band edge diagram (Fig. S12, ESI†). Subsequently, the introduction of intermediate levels in the forbidden gap on Pr3+ doping in Zn2GeO4 may result in enhanced optical response of Zn2GeO4 and the interplay between these defect levels may modulate the afterglow mechanisms.
2.3. Photoluminescence studies
The photoluminescence excitation (PLE) and steady-state PL spectra of Zn2GeO4 under different emission (500 and 535 nm) and excitation wavelengths (265 nm and 310 nm), respectively, are presented in Fig S13a–d (ESI†). Zn2GeO4 shows a broad bluish-white emission on excitation at 265 nm. The emission of undoped Zn2GeO4 under ultraviolet (UV) and near-UV (NUV) light could be ascribed to the presence of native defect centers, such as ,
,  , VGe and VZn. The
, VGe and VZn. The  and
and  defects act as the donor centers to trap electrons while VGe and VZn act as acceptor centers trapping the holes, and the DAP recombination results in visible emissions. The
defects act as the donor centers to trap electrons while VGe and VZn act as acceptor centers trapping the holes, and the DAP recombination results in visible emissions. The 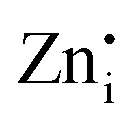 defects in Zn2GeO4 are of two types (Zn1i and Zn2i) due to the presence of two different
defects in Zn2GeO4 are of two types (Zn1i and Zn2i) due to the presence of two different  sites in Zn-Zn and Zn-Ge six-member rings, respectively. The bluish-white emission of undoped Zn2GeO4 under UV excitation has been attributed to DAP recombination.21,22,24,34,35 In our previous work,23 the blue-white and green emissions were ascribed to DAP transitions from
sites in Zn-Zn and Zn-Ge six-member rings, respectively. The bluish-white emission of undoped Zn2GeO4 under UV excitation has been attributed to DAP recombination.21,22,24,34,35 In our previous work,23 the blue-white and green emissions were ascribed to DAP transitions from  and
and 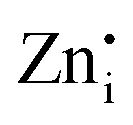 defects introduced in Zn–Zn and Zn–Ge rings, which was supported by DFT calculations. The broad PL in undoped Zn2GeO4 indicated the complex nature of radiative transitions from different native defects.36–38 In literature, the assignment of emissions to particular transitions has been controversial due to the competition between conduction bands (CB) to acceptor level transitions and DAP recombination from different donor levels.
defects introduced in Zn–Zn and Zn–Ge rings, which was supported by DFT calculations. The broad PL in undoped Zn2GeO4 indicated the complex nature of radiative transitions from different native defects.36–38 In literature, the assignment of emissions to particular transitions has been controversial due to the competition between conduction bands (CB) to acceptor level transitions and DAP recombination from different donor levels.
Notably, the PLE spectra of undoped Zn2GeO4 for the emission at 500 nm show an excitation band at 265 nm, while the PLE spectra for 535 nm emission show two excitation bands in the range of 250–290 and 300–345 nm, respectively. The former excitation band can be assigned to the host-related transition (bandgap ∼4.67 eV) from the valence band (VB) to CB. The existence of the 310 nm band revealed the direct population of defect energy levels (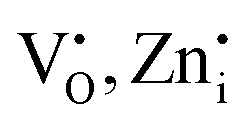 ) between the bandgap via excitation with NUV photons.34,35 The investigation of PL properties of Zn2GeO4 at band-to-band and below bandgap excitation energies could be helpful to gain insights about the excitation-dependent trap filling and recombination processes. In the past, few studies have investigated the influence of weak excitations on trap distribution and recombinations in materials and revealed the filling of only deep traps under below-band excitations.39–41
) between the bandgap via excitation with NUV photons.34,35 The investigation of PL properties of Zn2GeO4 at band-to-band and below bandgap excitation energies could be helpful to gain insights about the excitation-dependent trap filling and recombination processes. In the past, few studies have investigated the influence of weak excitations on trap distribution and recombinations in materials and revealed the filling of only deep traps under below-band excitations.39–41
The NUV excitation band of Zn2GeO4 observed in our case is rarely reported (Fig. S13b, ESI†). This suggested that a high defect concentration in Zn2GeO4 phosphors can be achieved via a two-step solid state synthetic route used in this work. In our case, a broad visible emission peak at 505 nm was observed on 265 nm excitation comprised of significant contribution from both the  donor levels to acceptor level DAP recombination along with some probability of CB to deep acceptor level charge-carrier recombination. The appearance of broad visible emissions and less intense UV or NUV emissions supported our hypothesis of a high density of native defects on high-temperature annealing (Fig. S13c, ESI†). This can be explained by fast electron trapping processes by trap states near CB in defect-rich Zn2GeO4. In earlier work, the prominent presence of strong UV emissions was seen by Dolado and group.22,34 In contrast, the below bandgap excitation will populate the defect levels selectively, and the radiative transitions from CB do not appear in the PL emission spectra excited at 310 nm as no free electrons will be present in CB. In Zn2GeO4, the
donor levels to acceptor level DAP recombination along with some probability of CB to deep acceptor level charge-carrier recombination. The appearance of broad visible emissions and less intense UV or NUV emissions supported our hypothesis of a high density of native defects on high-temperature annealing (Fig. S13c, ESI†). This can be explained by fast electron trapping processes by trap states near CB in defect-rich Zn2GeO4. In earlier work, the prominent presence of strong UV emissions was seen by Dolado and group.22,34 In contrast, the below bandgap excitation will populate the defect levels selectively, and the radiative transitions from CB do not appear in the PL emission spectra excited at 310 nm as no free electrons will be present in CB. In Zn2GeO4, the  defects are shallow donors, which can be populated on excitation above bandgap. The PL band became narrow and the percentage of blue and red emissions were decreased under weak excitation. Dolado et al.34 proposed that the radiative charge-carrier recombinations from the
defects are shallow donors, which can be populated on excitation above bandgap. The PL band became narrow and the percentage of blue and red emissions were decreased under weak excitation. Dolado et al.34 proposed that the radiative charge-carrier recombinations from the  related levels are preferred on below bandgap excitation compared to the DAP from the
related levels are preferred on below bandgap excitation compared to the DAP from the  levels and CB. However, only the UV and NUV bands were considered for the detailed investigation. Our results are consistent with the literature, and hence, we proposed that the visible emissions observed on below bandgap NUV excitations originate mainly from
levels and CB. However, only the UV and NUV bands were considered for the detailed investigation. Our results are consistent with the literature, and hence, we proposed that the visible emissions observed on below bandgap NUV excitations originate mainly from  related donor levels in undoped Zn2GeO4 with insignificant contribution from the CB and
related donor levels in undoped Zn2GeO4 with insignificant contribution from the CB and 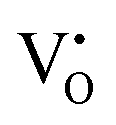 levels. Interestingly, the broad bluish-white emission band observed at 265 nm due to the existence of complex DAP recombinations was transformed into a narrow green emission band under 310 nm excitation. The changes in PL properties due to different trap filling processes led to color tunable emission on different excitations.23 The conception of the nature of different transitions emitting different visible emissions can be derived from the above observations. The blue emissions are majorly comprised of emissions from CB to acceptor level transitions and DAP transitions from the
levels. Interestingly, the broad bluish-white emission band observed at 265 nm due to the existence of complex DAP recombinations was transformed into a narrow green emission band under 310 nm excitation. The changes in PL properties due to different trap filling processes led to color tunable emission on different excitations.23 The conception of the nature of different transitions emitting different visible emissions can be derived from the above observations. The blue emissions are majorly comprised of emissions from CB to acceptor level transitions and DAP transitions from the 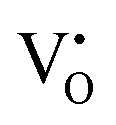 donor, the green emissions are majorly from the DAP transitions from two different
donor, the green emissions are majorly from the DAP transitions from two different  donors, and the green-yellow emissions are a result of the DAP recombination involving deep donor or acceptor levels. The interactions between
donors, and the green-yellow emissions are a result of the DAP recombination involving deep donor or acceptor levels. The interactions between  and Zni levels may form
and Zni levels may form  defect complexes and result in deep defect levels in bandgap along with the anti-site defects. The bluish-white emission of Zn2GeO4 under UV excitation with a high PLQY (∼42%) can be promising for solid-state lighting applications.23 However, the low PLQY of green emission (∼9%) obtained in undoped Zn2GeO4 on below band NUV excitation was a major drawback. The green Zn2GeO4 phosphors excitable with NUV photons and high PLQY would have more potential for LED applications.
defect complexes and result in deep defect levels in bandgap along with the anti-site defects. The bluish-white emission of Zn2GeO4 under UV excitation with a high PLQY (∼42%) can be promising for solid-state lighting applications.23 However, the low PLQY of green emission (∼9%) obtained in undoped Zn2GeO4 on below band NUV excitation was a major drawback. The green Zn2GeO4 phosphors excitable with NUV photons and high PLQY would have more potential for LED applications.
Aiming at tailoring the trap distribution, we carried out Pr3+ doping in Zn2GeO4. The defect formation on the substitution of two Zn2+ ions by two Pr3+ ions and three Zn2+ ions by two Pr3+ ions in the case of low and high doping concentrations can be explained with the help of Kröger–Vink notations by the following processes:35
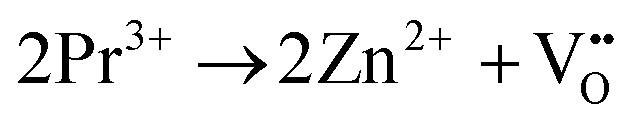 | (1) |
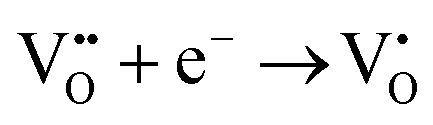 | (2) |
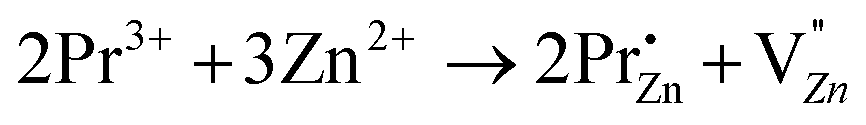 | (3) |
 | (4) |
Additionally, the distortion in the [GeO4] and [ZnO4] local structure endowed by both size and charge mismatch on Pr3+ aliovalent doping further boosted the formation of  and VGe. The increased defect formation may improve the defect emissions as DAP recombinations would be enhanced on Pr3+ doping.
and VGe. The increased defect formation may improve the defect emissions as DAP recombinations would be enhanced on Pr3+ doping.
The PLE and PL emission spectra of all the Pr3+ doped samples were recorded under the 265 nm excitation and below bandgap (310 nm) excitation energies (Fig. S14 and S15, ESI†). The Pr3+ doped samples show a broad emission band peak at 535 nm when excited at 265 nm. A small doping concentration of Pr3+ (x = 0.005) resulted in decreased PL intensity, but a subsequent increase was observed when the Pr3+ doping concentration was increased up to 0.01. The intensity decreased on further increase in the doping concentration (>0.01) as a consequence of concentration quenching. In addition to DAP recombinations, the probability of a weak energy transfer (ET) from the host to Pr3+ ions could not be ruled out. In our case, no sharp emission peaks for f → f transitions of Pr3+ ions were observed in any of the Pr3+ doped samples. This revealed the absence of efficient radiative transitions between 4f levels of Pr3+ ions in the Zn2GeO4 host. Similar observations have been reported in Pr3+ doped Zn2GeO4:Mn2+.5,31 He et al.35 have reported a weak ET from Zn2GeO4 host to Eu3+ ions resulting in defect-dominated emission along with less intense Eu3+ emissions. Based on the above observations, the 0.01Pr sample with an optimum concentration of Pr3+ (x = 0.01) was considered for the comparative investigation of the doping effect on PL, defect distribution, and optoelectronic properties with respect to Zn2GeO4.
It can be observed from the excitation spectra of undoped and 0.01Pr phosphors (Fig. 3a) that the 0.01Pr exhibited an intense shoulder absorption band when emission was monitored at both the 500 and 535 nm. This can be explained by the DFT results, which revealed that Pr3+ doping introduced various additional defect states between the VB and CB. The 0.01Pr phosphor is rich in midgap defect states and impurity levels, and broad PLE spectra can be attributed to the direct activation of these levels by UV light. Subsequently, the probability of electron excitation in CB is inappreciable in 0.01Pr phosphor, and probably, only the defect states would be populated even on bandgap excitation (265 nm). The normalized emission spectra of undoped and 0.01Pr samples excited at 265 nm (Fig. 3b) demonstrated a red shift in the emission peak on Pr3+ doping from 500 to 535 nm, and a slightly resolved emission spectra could be observed in the Pr3+ doped samples. This suggested that Pr3+ doping prompted the DAP transitions, resulting in green emissions more selectively. Consequently, the blue emissions became less intense as no free electrons were available in CB for CB-acceptor level transitions. Clearly, below bandgap NUV excitations (310 nm) also contributed to the final green emission in Pr3+ doped samples. In Pr3+ doped Zn2GeO4, weak below bandgap excitations can populate the donor levels via excitations from VB or acceptor levels, and the PL emission will be solely due to DAP transitions. In our interest, a remarkable four-fold enhancement in the intensity of the green emission band peaking at 535 nm on Pr3+ doping under excitation at 310 nm is seen (Fig. 3c). We kept the samples in air for a month and tested the stability of our material by monitoring the optical properties at regular intervals. We recorded the emission spectra of the samples on the 1st, 5th, 15th, and 30th day. No significant decrease in photoluminescence output was observed in the samples. The same has been provided in the ESI† file as Fig. S16.
 | ||
| Fig. 3 (a) PLE spectra of undoped and 0.01Pr samples at different emission wavelengths, (b) and (c) PL spectra of undoped and 0.01Pr sample at 265 nm and 310 nm excitation, respectively. (d) Emission photographs captured by a Nikon camera under 254 and 365 nm UV lamp. (e) Emission spectra of 0.01Pr at different excitation wavelengths. (f) Green emission intensity changes as a function of Pr3+ doping concentrations, and (g) EPR spectra of Zn2GeO4 and 0.01Pr samples at 100 K. | ||
To visualize the color tunability of emissions on Pr3+ doping in Zn2GeO4, the emission images of samples under a UV lamp at 254 and 365 nm are shown in Fig. 3d and CIE color coordinate diagrams are provided as Fig. S17a and b (ESI†). The excitation-dependent PL emission spectra of 0.01Pr display color tunability due to different trapping mechanisms at different excitation energies (Fig. 3e and Fig. S18, ESI†). The increase in the excitation wavelength from 250 to 350 nm decreased the full width at half maximum (FWHM) of emission spectra. At higher excitation energies, the broad visible emission was observed, which is supposed to have a significant contribution from both the  donor levels to acceptor level DAP recombinations and charge carrier recombinations from CB to deep acceptor levels. In contrast, the below bandgap excitations may populate the defect levels selectively, and the probability of radiative transitions from CB is negligible as no free electrons will be present in CB due to higher concentrations of intermediate defect states. Hence, the PL emission band became narrow as it comprised only DAP recombinations from the
donor levels to acceptor level DAP recombinations and charge carrier recombinations from CB to deep acceptor levels. In contrast, the below bandgap excitations may populate the defect levels selectively, and the probability of radiative transitions from CB is negligible as no free electrons will be present in CB due to higher concentrations of intermediate defect states. Hence, the PL emission band became narrow as it comprised only DAP recombinations from the 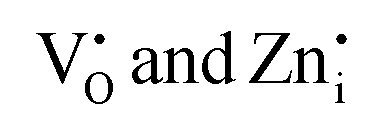 donor levels.
donor levels.
To illustrate the effect of Pr3+ doping in the green emissions, the intensity changes as a function of Pr3+ doping concentrations are shown in Fig. 3f. A two-fold increase in the PLQY (∼19%) of green emission under NUV excitation was attained on Pr3+ doping in Zn2GeO4 (Fig. S19, ESI†). The increased green emission can be explained by DFT results, indicating that Pr3+ doping will facilitate the formation of  and Zni defect-related levels, which can act as electron traps and VZn as hole traps. The high Ef of VZn reported for undoped Zn2GeO4 suggested that the formation of VO and Zni defects is more favourable than VZn.23 On Pr3+ doping, an appreciable reduction in the Ef of VZn was revealed by theoretical calculations due to the formation of VZn–PrZn defect complexes. The increased DAP transitions from
and Zni defect-related levels, which can act as electron traps and VZn as hole traps. The high Ef of VZn reported for undoped Zn2GeO4 suggested that the formation of VO and Zni defects is more favourable than VZn.23 On Pr3+ doping, an appreciable reduction in the Ef of VZn was revealed by theoretical calculations due to the formation of VZn–PrZn defect complexes. The increased DAP transitions from  related donor levels to
related donor levels to  acceptor levels became more prominent on Pr3+ doping, which explained the enhanced green emission in Pr3+ doped Zn2GeO4.
acceptor levels became more prominent on Pr3+ doping, which explained the enhanced green emission in Pr3+ doped Zn2GeO4.
EPR measurements of undoped and Pr3+ doped Zn2GeO4 samples were carried out at 100 K to understand the types of native defects (Fig. 3g). The EPR signal at g ∼1.955 is composed of paramagnetic 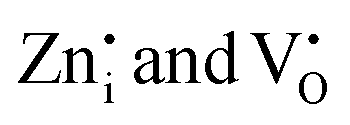 defects. The signal at g ∼2.003 is usually reported to
defects. The signal at g ∼2.003 is usually reported to  in most of the host matrices.42 However, the presence of this signal in Zn2GeO4 can be proposed due to
in most of the host matrices.42 However, the presence of this signal in Zn2GeO4 can be proposed due to 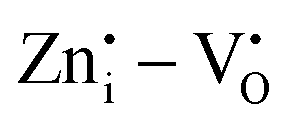 defect complex formation or isolated cation vacancies (
defect complex formation or isolated cation vacancies ( ).43–46 The intensity of the EPR signal due to
).43–46 The intensity of the EPR signal due to 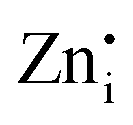 and
and 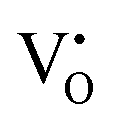 defects (g ∼1.955) enhanced significantly in 0.01Pr samples that indicated large defect formation on Pr3+ doping. This observation is in line with DFT calculations and also supports the PL results.
defects (g ∼1.955) enhanced significantly in 0.01Pr samples that indicated large defect formation on Pr3+ doping. This observation is in line with DFT calculations and also supports the PL results.
Based on the previous literature and our experimental and theoretical results, we proposed an energy band model (Fig. 4a) and respective transitions involving the defect states, which resulted in the broad visible PL of Zn2GeO4. The luminescence decay profiles of Zn2GeO4 and 0.01Pr phosphors excited at 265 and 310 nm and emissions monitored at 500 and 535 nm are shown in Fig. S20a and b (ESI†). The decay curves were best fitted with a three-exponential decay function:
| I(t) = I0 + A1e−t/τ1 + A2e−t/τ2 + A3e−t/τ3 | (5) |
 | ||
| Fig. 4 (a) Energy band diagram demonstrating radiative transitions and DAP recombinations for visible emissions in Zn2GeO4. Normalized TRES at different delay times of 100 μs to 180 ms for (b) Zn2GeO4 and (c) 0.01Pr samples. (d) Schematic for the trap-to-trap hopping mechanism. | ||
To further understand the decay kinetics, TRES of Zn2GeO4 and 0.01Pr phosphors were acquired with the integration time of 200 ms at different delay times in the range of 100 μs to 180 ms (Fig. S21a and c, ESI†). TRES studies can be helpful in resolving overlapping emissions originating from different electron–hole recombinations involving CB, defect states, and VB. At small delay times, all the emission components due to different excited species are supposed to be present, as the lifetime values for visible emissions of undoped and doped Zn2GeO4 are >1 ms (Table S4, ESI†). The normalized TRES spectra at different delay times for Zn2GeO4 and 0.01Pr phosphors under 265 nm excitation are provided in Fig. 4b and c. In the beginning, the TRES spectra of Zn2GeO4 excited at 265 nm consisted of a significant contribution from weak NUV emissions along with visible emissions up to a delay time of 100 μs. The NUV and blue emissions decayed faster, and the intensity decreased significantly on longer delay times (>100 μs). The same can be visualized by a change in color coordinates of emission from blue towards the blue-green region on increasing delay times (Fig. 4b and Fig. S21b, ESI†). In the case of Pr3+ doped Zn2GeO4, a broad emission consisting of all emissions was observed at a 100 μs delay time and emission components became more resolved on an increase in delay times (Fig. 4c). The TRES of the 0.01Pr sample suggested that the green emissions have longer decay times compared to blue emissions and only green emissions could be observed at longer delay times (Fig. S21c and d, ESI†). The TRES studies of the 0.01Pr sample at below bandgap excitation (310 nm) displayed similar behaviour and showed individual green emissions on long delay times > 300 μs (Fig. S22, ESI†).
Such long decay times (in ms scale) can be explained by charge carrier redistribution in defect states due to the involvement of trap-to-trap tunnelling (or hopping) processes.41,47,49 The excited electrons in CB and defect states can directly undergo emissive electron–hole recombinations with short decay times (∼μs) or photogenerated carriers may be trapped in stable non-radiative defects. With time, the captured carriers relax via tunnelling towards the luminescent sites, which is followed by the visible emissions due to DAP recombinations with a longer time component (∼ms) (schematic shown in Fig. 4d). The TRES supported the proposed hypothesis that the blue emissions have contributions from CB and defect states while the green emission bands originated solely from DAP transitions of charge carriers in defect levels. The TRES provided strong evidence for spatial correlation between  -induced traps and emissive donor defect centres. Therefore, the 0.01Pr sample exhibits a time-dependent trap distribution due to high trap density in the vicinity of emissive levels that facilitated hopping mechanisms.
-induced traps and emissive donor defect centres. Therefore, the 0.01Pr sample exhibits a time-dependent trap distribution due to high trap density in the vicinity of emissive levels that facilitated hopping mechanisms.
2.4. Persistent luminescence and Thermoluminescence (TL) studies
To better understand the trapping and de-trapping processes, the PersL decay curves were measured on irradiation of Zn2GeO4 and 0.01Pr phosphors with UV lamps at 265 and 310 nm, respectively (Fig. S23, ESI†). The afterglow decay curves measured at 265 nm charging for blue-white emissions are shown in Fig. 5a. The afterglow decay curves monitored at 535 nm for the green emission band on charging the phosphors at 310 nm for 100 s are shown in Fig. 5b. For both the excitations, the afterglow intensity was improved on Pr3+ doping in Zn2GeO4. To further explore the mechanisms of decay processes, the experimental afterglow curves were fitted into the power-law exponent function (∝t−α) (Fig. 5a and b). By fitting, the power-law exponent (α) was found to be approximately in the range of 0.56–1.06. This behavior is consistent with the TRES findings and lifetime decay observations which suggested that the visible emissions are controlled by the tunnelling of charge carriers between trap states.41,47,48 On excitation, electrons and holes are captured by traps with some probability of filling charge carriers in stable non-luminous sites. The tunneling of charge carriers in non-luminescent traps populate luminescent defect sites, and are responsible for long afterglow. The schematic illustration for the PersL mechanism is presented in Fig. S24 (ESI†). To calculate the afterglow decay times (τ), the decay curves were fitted with eqn (5) and the τ values are tabulated in Table S5 (ESI†). It was observed that the afterglow decay times increased on Pr3+ doping when the samples were charged at bandgap excitation (265 nm). However, for the 0.01Pr phosphor charged at a below bandgap excitation (310 nm), the τ values were lower compared to the undoped sample. This can be explained by excitation wavelength-dependent charge distribution in traps.40 Hence, it can be inferred that the tunnelling processes are dominant at higher excitation energies, resulting in photogenerated carriers with more energy. The migration tendency is higher, which increases the probability of trapping at non-radiative states.41 The effective filling of charge carriers in all trap states can be achieved by increasing the excitation energies. The feasibility of the same was explored with higher energy X-ray excitations to attain long PersL. | ||
| Fig. 5 PersL decay curves of Zn2GeO4 and 0.01Pr phosphors irradiated at (a) 265 nm and (b) 310 nm for 100 s (the red lines are the fitting of decay with power-law exponent function). (c) TL glow curves for Zn2GeO4 and 0.01Pr phosphors recorded in the range of 273–600 K on charging at 265 nm for 100 s. (d) X-ray activated PersL images recorded by camera at different time intervals, and (e) X-ray activated PersL decay curve for the 0.01Pr sample. | ||
TSL studies were performed to reveal the features of trap states, trap depth, and distribution.9Fig. 5c depicts the TL glow curves of Zn2GeO4 and 0.01Pr phosphors after charging by UV irradiation at 265 nm for 100 s. The broad TL emissions on heating of phosphors confirmed the large defect formation in the samples. Two and three components in the TL curves were obtained for Zn2GeO4 and 0.01Pr samples by Gaussian deconvolution shown in Fig. S25 (ESI†). The trap depth (Et) can be estimated with the help of the following equation:9
| Et = Tm/500 | (6) |
In order to understand the effect of different excitations on the trap depth and distribution, the comparison of TL glow curves upon charging the samples with 265 and 310 nm has been provided in Fig. S26a (ESI†). Upon charging with 310 nm, TL glow curves are similar for undoped and 0.01Pr samples, but the TL intensity is significantly higher for 0.01Pr. This indicated a much more effective trap filling in the Pr3+ doped sample that resulted in enhanced PersL on charging with 310 nm. The samples irradiated with higher excitation energy (265 nm) resulted in intense TL, which indicated more efficient trap filling as compared to 310 nm. It can be inferred that higher excitation energy can fill a higher concentration of traps, leading to long PersL. However, the filling of relatively shallow traps upon 310 nm charging resulted in intense PersL at room temperature with a shorter afterglow time. The TL glow curves on X-ray irradiation of undoped and 0.01Pr samples, shown in Fig. S26b (ESI†), revealed a much higher concentration of filled traps in the Pr3+ doped sample with a rich trap distribution. The broad TL of 0.01Pr explained the long PersL obtained on Pr3+ doping due to the slow release of charge carriers from multiple trap states on gaining thermal energy during the afterglow phenomenon.
To explore the PersL on charging the phosphors with higher excitation energy, the Zn2GeO4 and 0.01Pr phosphors were irradiated with X-rays for 1 min and the PersL intensities were captured by a camera at different decay instants after ceasing X-ray excitation (Fig. 5d). The radioluminescence (RL) spectra of undoped and Zn2GeO4:0.01Pr samples are provided in the ESI† file as Fig. S27. The 0.01Pr sample showed intense RL compared to the undoped sample, and the spectral features on X-ray excitation were similar to UV excitation. The 0.01Pr sample displayed an intense X-ray-activated PersL, which could be observed for 18 hours in the dark, while PersL in undoped Zn2GeO4 completely ceased after 38 min. The X-ray-activated PersL decay curve for the 0.01Pr sample is presented in Fig. 5e. The long PersL decay times of 0.01Pr can be ascribed to prominent tunnelling mechanisms as the PersL decay curve can be well fitted into a power-law exponent function with α = 1.02 (Fig. S28, ESI†). The RL afterglow curve for the 0.01Pr sample after ceasing off the X-ray light is provided in Fig. S27b (ESI†), and the decrease in FWHM of RL can be attributed to different de-trapping processes as a function of time. Moreover, the additional defect states introduced on Pr3+ doping increased the defect density significantly, as revealed by TL, EPR and DFT studies. The high energy X-ray excitations can result in the distribution of photogenerated charge carriers in all the trap states.50 Thus, the long PersL in Zn2GeO4 could be achieved by tailoring the defect states in between the bandgap by doping.
Table S6 (ESI†) lists some of the recent X-ray persistent phosphors and compares their performance with ZnGe2O4 and 0.01 Pr. Based on this table (Table S6, ESI†), we could find that for some of the phosphors, X-ray persistent luminescence duration was more than 18 h, but they are reported for ultraviolet-B (UVB)/UVC emission and that too in fluoride-based hosts. In both ways, their usability is quite curtailed for commercial lighting owing to non-visible light production and the use of toxic and corrosive fluoride-based hosts. There are two very recent review articles on X-ray persistent phosphors, which can be referred and may be quite useful for new researchers.51,52 In terms of UV persistent phosphors, Zn2GeO4 and 0.01Pr phosphors were inferior to some of the recent long afterglow lead halide perovskite materials.53,54 But the 0.01Pr phosphor excels because it demonstrated remarkable enhancement in X-ray persistent luminescence for more than 18 h with very high radioluminescence intensity.
2.5. AC impedance analysis
Decay kinetics suggested the role of tunnelling of charge carriers between trap states in longer afterglow times. To further support the same, the electrochemical impedance (EIS) of Zn2GeO4 was analysed before and after Pr3+ doping. From the impedance curves shown in Fig. S29 (ESI†), it can be observed that the impedance of the Zn2GeO4 sample reduced on Pr3+ doping. The radius of curvature of the 0.01Pr curve decreased compared to the undoped Zn2GeO4 curve. The impedance drop in Zn2GeO4 with the incorporation of Pr3+ ions revealed the widening of electron channels, which, in turn,55 reduced the resistance to electron movement in the 0.01Pr phosphor. Hence, the increased electron migration rate in the 0.01Pr sample resulted in enhanced afterglow performance.2.6. Temperature-dependent PL studies
To explore the variation in the defect-related emission of Zn2GeO4 as a function of temperature, the temperature-dependent PL spectra of Zn2GeO4 and 0.01Pr phosphors were acquired from 80 to 300 K under excitation at 265 nm (Fig. 6a). It was observed that PL intensity increased on increasing the temperature that revealed the abnormal NTQ of PL in Zn2GeO4 below RT. The 0.01Pr phosphor (Fig. 6b) exhibited the thermally enhanced PL as the emission intensity increased on increasing the temperature, demonstrating the NTQ effect in doped Zn2GeO4 as well. Among the various competitive processes, multi-phonon assisted crossover and non-radiative relaxation are one of the mechanisms for thermal quenching.56 Hence, the NTE effect of Zn2GeO4 can be one of the reasons for the observed NTQ behavior below RT due to the enhanced structural rigidity of the Zn2GeO4 lattice up to RT. Liu et al. demonstrated the improved thermal stability of luminescence by controlling the structural rigidity.25 The insignificant non-radiative relaxations due to low-frequency phonon modes in Zn2GeO4 can efficiently reduce the thermal quenching. At 80 K, well-resolved PL spectra could be observed, while on heating, the emission band became broader with a gradual blue shift. This shift can be explained by the existence of charge trapping in defects, and the population of these defect states varies with temperature.22 In Zn2GeO4, the intensity at 300 K is enhanced by 362% of the intensity observed at 80 K (Fig. S30, ESI†). | ||
| Fig. 6 (a) and (b) Temperature-dependent PL spectra in the range of 80–300 K under 265 nm excitation for Zn2GeO4 and 0.01Pr samples, respectively. (c) and (d) Temperature-dependent PL spectra in the range of 80–300 K under 310 nm excitation for undoped and Pr doped samples, respectively. (e) and (f) Contour plots demonstrating the intensity variation with temperature for undoped and Pr doped samples at 265 and 310 nm excitations, respectively. | ||
As mentioned earlier, the emissions under below band gap excitations originate from defect-related DAP transitions. In order to have a clear picture of trap distribution and changes in PL features under low energy excitations, temperature-dependent PL measurements of Zn2GeO4 and 0.01Pr phosphors were performed from 80 to 300 K under NUV excitation (310 nm). Surprisingly, the PL emissions in Zn2GeO4 exhibited a completely opposite behavior under below bandgap NUV excitation (Fig. 6c). A normal PTQ of green emission band was observed on weak excitation that resulted in a continuous decrease of PL intensity from 80 to 300 K. The temperature-dependent PL in Zn2GeO4 displayed divergent NTQ and PTQ processes below RT, which indicated the existence of different activation barriers, trapping and detrapping processes under high (265 nm) and weak excitation energies (310 nm). The temperature-dependent PL of the Pr3+ doped Zn2GeO4 phosphor on below band NUV excitations showed exciting results (Fig. 6d). Pr3+ doping resulted in the thermal stability of the green emission of Zn2GeO4 up to 180 K, followed by a gradual NTQ of PL up to 220 K. A normal PTQ trend was observed on further increase in temperature (>220 K). The thermal stability of the green PL band in 0.01Pr phosphors can be explained as a result of a balance between the thermal activation of DAP transitions and thermal quenching. The intensity variation of PL as a function of temperature in undoped Zn2GeO4 and 0.01Pr phosphors can be visualized in the contour plots in Fig. 6e and f under 265 and 310 nm excitations, respectively. The thermal stability of the green band in Pr3+ doped phosphor can be clearly seen in Fig. 6f as the luminous intensity remains almost constant. The PL intensity at 300 K in 0.01Pr was 85% of the intensity observed at 80 K (Fig. S30, ESI†). In contrast, the green band intensity of Zn2GeO4 in the absence of Pr3+ doping diminished to 64% of the initial intensity at 80 K.
The main transitions for the relaxations of photo-generated carriers (e− and h+) between the bandgap and trap states are schematically shown in Fig. 7a. The recombinations from CB have been excluded to understand the dynamics of only defect-mediated PL due to DAP recombinations. The different transitions are:9,41,57,58 (1) non-radiative capture of electrons by traps, (2) thermal excitation of electrons from traps, (3) thermal excitation of holes from VB, (4) non-radiative capture of holes by VB, (5) tunnel transitions of carriers and relaxations from non-radiative traps (Trap I and Trap II) to luminescent defect sites, (6) radiative DAP transitions, (7) non-radiative recombination process assisted by phonons, and (8) non-radiative relaxations to killer sites. In addition, the thermal hopping of electrons from acceptor levels to spatially separated trap states induces the generation of holes in the acceptor levels. Hence, the NTQ processes in native defect phosphors could be induced via thermally activated hopping mechanisms due to the increased probability of DAP transitions (Fig. 7b).
 | ||
| Fig. 7 Schematic illustration of (a) trapping–detrapping processes of photo-generated carriers, and (b) thermal hopping mechanism. | ||
The temperature-dependence studies of luminescence from defect-rich Zn2GeO4 can reveal the different activation barriers and thermal quenching mechanisms for emissions originating from DAP transitions. The above trapping and detrapping mechanisms are competitive, and the dominance of one over the other in particular temperature zones led to NTQ and PTQ of visible emissions. The temperature dependence of PL in semiconductors with multiple NTQ and PTQ processes has been explained in the literature by Shibata et al. by fitting the experimental data in a multi-level model:59
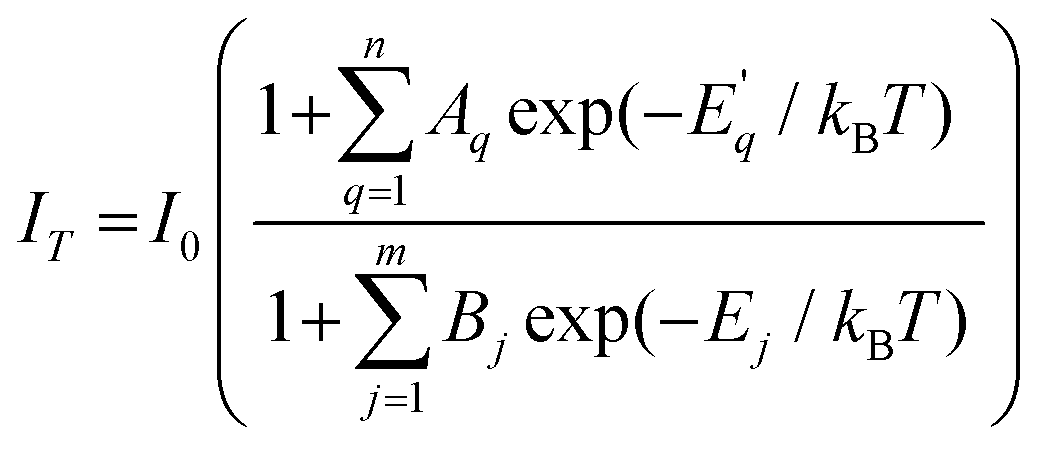 | (7) |
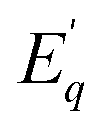 is the activation energy for the q-th radiative transition mechanism, and Ej is the activation of j-th non-radiative transition, Aq and Bj are constants, n and m are the total number of intermediate radiative and non-radiative processes, respectively. The exponential terms in the numerator and denominator represent a multi-level model for NTQ and PTQ processes, respectively. Fig. 8a, b shows the ratios of integrated PL intensity (IT/I0) as a function of 1/kBT in the temperature range from 80 to 300 K for Zn2GeO4 and 0.01Pr samples excited at the bandgap energy (265 nm), respectively. The NTQ of PL emissions under bandgap transitions was best fitted with a single-barrier model with n = 1 for Zn2GeO4 and 0.01Pr samples (eqn (8)). The calculated activation energies are summarized in Table 2.
is the activation energy for the q-th radiative transition mechanism, and Ej is the activation of j-th non-radiative transition, Aq and Bj are constants, n and m are the total number of intermediate radiative and non-radiative processes, respectively. The exponential terms in the numerator and denominator represent a multi-level model for NTQ and PTQ processes, respectively. Fig. 8a, b shows the ratios of integrated PL intensity (IT/I0) as a function of 1/kBT in the temperature range from 80 to 300 K for Zn2GeO4 and 0.01Pr samples excited at the bandgap energy (265 nm), respectively. The NTQ of PL emissions under bandgap transitions was best fitted with a single-barrier model with n = 1 for Zn2GeO4 and 0.01Pr samples (eqn (8)). The calculated activation energies are summarized in Table 2. | (8) |
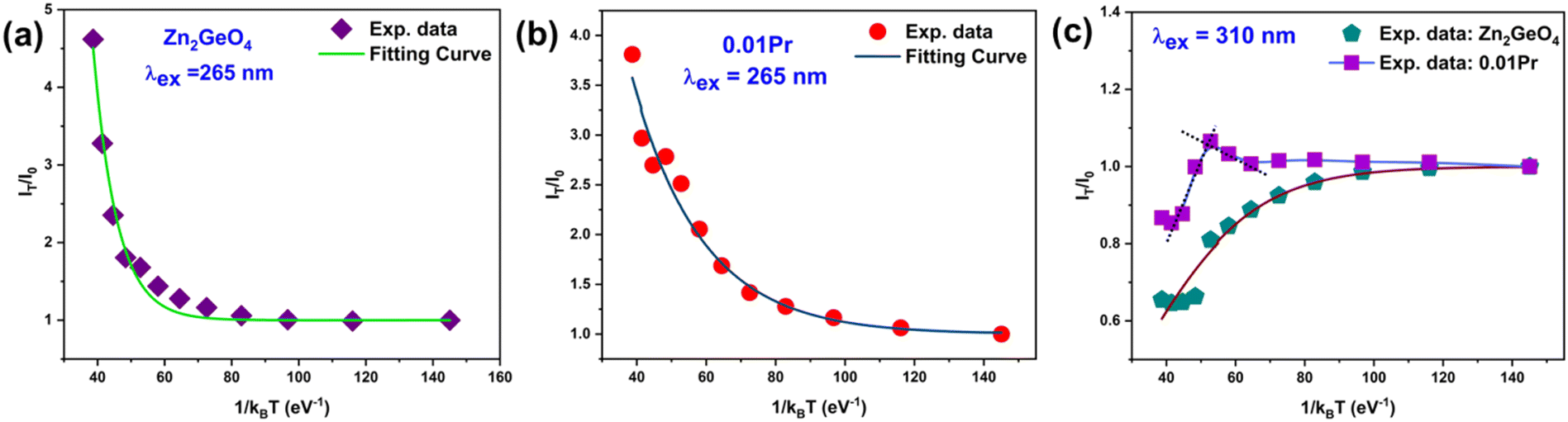 | ||
| Fig. 8 Ratio of integrated PL intensity (IT/I0) as a function of 1/kBT for (a) Zn2GeO4 and (b) Zn2GeO4:0.01Pr3+ excited at 265 nm, and (c) undoped and Pr doped Zn2GeO4 samples excited at 310 nm, respectively. | ||
| Sample (λex) | NTQ | PTQ |
|---|---|---|
| E′1 (meV) | E 1 (meV) | |
| Zn2GeO4 (265 nm) | 141 | — |
| Zn2GeO4 (310 nm) | — | 68 |
| 0.01Pr (265 nm) | 48 | — |
| 0.01Pr (310 nm) | 4 | 17 |
For the NTQ profile of undoped Zn2GeO4, the calculated activation energy ( ) is 141 meV. For bandgap excitation, the electrons excited to the CB and holes generated in VB can be captured by shallow and deep traps (Fig. 7a). The calculated barrier height of 141 meV can be explained by the thermal excitation of electrons from trap centres to CB. This process is thermally activated, and hence, the NTQ effect of the PL band can be attributed to enhanced transitions from CB to acceptor levels. This explained the gradual blue shift of PL emission peak in Zn2GeO4 on increasing the temperature (Fig. 6a). In addition, the increment of holes in acceptor levels by thermally activated excitation of holes from VB to Zn and Ge vacancies will initiate more DAP recombinations. Simultaneously, an increase in temperature may trigger the release of charge carriers from non-radiative defects to luminescent donor sites (
) is 141 meV. For bandgap excitation, the electrons excited to the CB and holes generated in VB can be captured by shallow and deep traps (Fig. 7a). The calculated barrier height of 141 meV can be explained by the thermal excitation of electrons from trap centres to CB. This process is thermally activated, and hence, the NTQ effect of the PL band can be attributed to enhanced transitions from CB to acceptor levels. This explained the gradual blue shift of PL emission peak in Zn2GeO4 on increasing the temperature (Fig. 6a). In addition, the increment of holes in acceptor levels by thermally activated excitation of holes from VB to Zn and Ge vacancies will initiate more DAP recombinations. Simultaneously, an increase in temperature may trigger the release of charge carriers from non-radiative defects to luminescent donor sites ( ). The above processes dominate over the non-radiative recombinations of charge carriers below RT that may be ascribed to the increased stiffness due to NTE of the Zn2GeO4 lattice up to 300 K.25,26 In 0.01Pr, the small barrier (48 meV) for NTQ that resulted in the enhanced PL intensity with temperature indicated the contribution from trap-to-trap tunnelling of charge carriers.41,47,49,60 Notably, the activation barrier for NTE in 0.01Pr is lower compared to the undoped sample, which can be attributed to improved structural rigidity on Pr3+ doping indicated by Raman spectroscopy. The decay kinetic studies by TRES measurements also suggested the prominent role of indirect tunneling pathways for populating the radiative trap states in Pr3+ doped samples.
). The above processes dominate over the non-radiative recombinations of charge carriers below RT that may be ascribed to the increased stiffness due to NTE of the Zn2GeO4 lattice up to 300 K.25,26 In 0.01Pr, the small barrier (48 meV) for NTQ that resulted in the enhanced PL intensity with temperature indicated the contribution from trap-to-trap tunnelling of charge carriers.41,47,49,60 Notably, the activation barrier for NTE in 0.01Pr is lower compared to the undoped sample, which can be attributed to improved structural rigidity on Pr3+ doping indicated by Raman spectroscopy. The decay kinetic studies by TRES measurements also suggested the prominent role of indirect tunneling pathways for populating the radiative trap states in Pr3+ doped samples.
Fig. 8c displays the IT/I0 as a function of 1/kBT in the temperature range from 80 to 300 K for Zn2GeO4 and 0.01Pr samples excited at below bandgap energy (310 nm). The PL intensity remained almost constant for T < 120 K and above 120 K, and a strong PTQ of the Zn2GeO4 emission band was observed under below-bandgap NUV excitations. The PTQ process can be modeled by using the expression with a single non-radiative process as below:61,62
IT = I0/(1 + A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) exp(−E1/kBT)) exp(−E1/kBT)) | (9) |
The activation energy of the TQ process of Zn2GeO4 emissions is 68 meV and this can be attributed to non-radiative relaxations when charge carriers have enough energy for crossover due to thermal activation.56,61 In contrast, for the 0.01Pr sample, the PL intensity remained constant up to 180 K and then increased gradually, showing a weak NTQ effect. After T > 220 K, the PL intensity decreased abruptly due to PTQ processes. The activation energies of each component were obtained by fitting the data into eqn (7) with n and m = 1, and calculating the slope values after valid approximations proposed by Shibata et al.59 The lower activation energies for NTQ (4 meV) suggested the prominent role of thermal hopping pathways (Fig. 7b) for generating sufficient holes for DAP transitions in Pr3+ doped samples. The activation energy for the PTQ (17 meV) process is the barrier for non-radiative relaxations of charge carriers to killer sites. A higher barrier for TQ may be observed at higher temperatures due to the involvement of thermal ionization processes from deep traps.
To study the behavior of the bluish-white emission band of undoped Zn2GeO4 and green emission band of 0.01Pr samples on heating the samples above RT, the temperature-dependent PL spectra for Zn2GeO4 and 0.01Pr samples were recorded in the range of 298–408 K (Fig. 9a and c). Above RT, the PL intensity decreased on heating, which revealed positive thermal quenching (PTQ) of bluish-white emission. The TQ is observed due to the increased non-radiative charge carrier recombinations, which can be assisted by positive thermal expansion (PTE) of the Zn2GeO4 lattice above RT which was confirmed by high-temperature XRD studies.
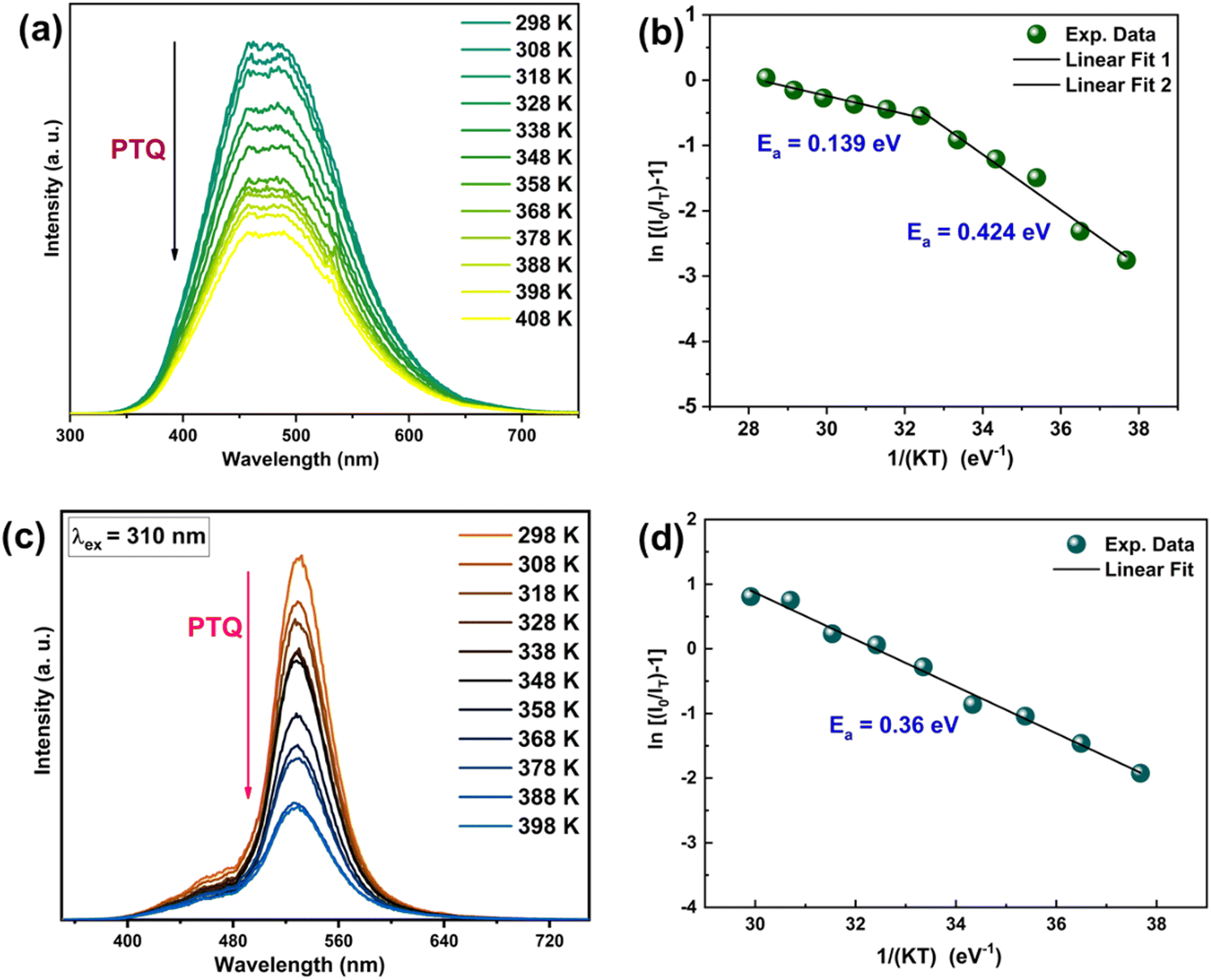 | ||
| Fig. 9 Temperature-dependent PL spectra for the Zn2GeO4 sample in the range of 298–408 K under excitation at (a) 265 nm. (b) ln(I0/IT − 1) versus 1/kT for the Zn2GeO4 phosphor. (c) Temperature-dependent PL spectra for the 0.01Pr sample in the range of 298–408 K under excitation at 310 nm, and (d) ln(I0/IT − 1) versus 1/kT for the 0.01Pr phosphor. | ||
The resistance of Zn2GeO4 to TQ can be evaluated by using the Arrhenius expression:
| IT = I0/(1 + Aexp(−Ea/kT)) | (10) |
2.7. LED application
The LED fabrication was performed to demonstrate the potential of our phosphor material for solid state lighting applications. Fig. 10(a and b) show the electroluminescence (EL) spectra of undoped and Pr3+ doped Zn2GeO4 samples using 280 nm LED chips. The performance of pc-LED was evaluated, and the parameters for pc-LED are presented in Table 3. A crucial point to be noted is that the CRI value is slightly reduced in the case of the 0.01Pr3+-doped phosphor-based LED compared to that of the undoped one. This is reasonable since the emission peak of the 0.01Pr3+-doped sample slightly shifted towards the higher wavelength side and has narrower emission width compared to that of the undoped sample, as shown in the normalized emission spectral comparison (Fig. 3b), and in the EL spectral comparison (Fig. S31, ESI†). The reason for this has been discussed in the photoluminescence section of this manuscript. Because of such red shifting and narrowing of the emission width, the emission curve of the Pr3+-doped sample covered a lower visible region compared to the emission curve of the undoped sample. That is the main reason why the Pr3+ doped sample showed a slightly lower CRI compared to that of the undoped sample. The respective color coordinates of EL obtained in both the phosphors are marked in the color coordinate diagram (Fig. 10c). Moreover, the phosphors exhibiting long afterglow decay times (in ms) can be promising for pc- LED to compensate for the dimming time and flicker effects.63 The potential application of Zn2GeO4 and 0.01Pr phosphors in solid-state lighting can be supported by high color rendering index (CRI) and correlated color temperature (CCT) values. The optimum CCT for daylight and CRI values were obtained in the 0.01Pr sample. Hence, trap tuning in Zn2GeO4 can be a promising approach for developing bluish-white and green-emitting phosphors for warm pc-LED applications. | ||
| Fig. 10 EL spectra of (a) undoped and (b) Pr3+ doped Zn2GeO4 on 280 nm LED excitation, and (c) the CIE color coordinate diagram. | ||
| Samples | CIE | CRI | CCT |
|---|---|---|---|
| Zn2GeO4 | (0.2940, 0.3765) | 76 | 7004 |
| Zn2GeO4:0.01Pr | (0.3315, 0.4393) | 72 | 5546 |
3. Conclusion
Here, detailed studies of NTE in the Zn2GeO4 phosphor and luminescent properties have been carried out to understand the optical response of self-activated Zn2GeO4. A doping strategy of hypervalent Pr3+ was employed to realize controllable trap tuning, defect-related trap depths, and improved PLQY with long afterglow. The decay kinetics of visible emissions was studied by time-resolved fluorescence spectroscopy (TRES) to understand the photophysics of complex PL emissions of Zn2GeO4. This knowledge is crucial for optimum trap tuning and improving the PLQY and PersL performance by solving the afterglow mechanisms. Interestingly, a spectral shift from blue to green was observed in the emission spectra within different time windows. A two-fold enhancement in PLQY of green emission was achieved on Pr3+ doping. Importantly, the Pr3+ doping increased shallow traps that facilitated efficient PersL at RT. The existence of trap-to-trap tunnelling mechanisms imparts longer afterglow decay lifetimes, as confirmed by fitting into the power-law decay function. The Pr3+ doped Zn2GeO4 phosphor demonstrated a long X-ray-activated PersL at room temperature for up to 18 hours. The thermal behavior of the emission band at bandgap and below bandgap energies was investigated extensively to reveal the different trapping–detrapping processes. The LED fabrication was performed to display the electroluminescent spectrum of phosphors, and the trap tuning in Zn2GeO4 can be a promising strategy for developing warm pc-LEDs.Data availability
All data required to reproduce the results in the paper are available in main text and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. Balhara and S.K. Gupta would like to acknowledge Apurav Guleria, RPCD for Raman measurements, N. Rawat for TL measurements, Kathi Sudarshan, RCD for help in Rietveld analysis, Dr Ruma Gupta for EDAX and elemental mapping, Dr Manoj Mohapatra for EPR measurements, Dr Dibyendu Bhattacharyya and Dr Biplab Ghosh for help in getting EXAFS beamline, Dr Rohan Phatak for high temperature XRD, and Dr Aviru Kumar Basu, INST for XPS measurements. We acknowledge Dr P S Sarkar, TPD, for providing an X-ray tube and Dr Meghnath Sen, RSSD, for TL measurements on X-ray irradiated samples. This work was funded by Bhabha Atomic Research Centre (BARC) which is a government of India funded research institute.References
- J. Zhang, Z. Song, P. Cai and X. Wang, Structures, photoluminescence, and principles of self-activated phosphors, Phys. Chem. Chem. Phys., 2023, 25(3), 1565–1587 RSC.
- N. Pathak, P. S. Ghosh, S. K. Gupta, S. Mukherjee, R. M. Kadam and A. Arya, An Insight into the Various Defects-Induced Emission in MgAl2O4 and Their Tunability with Phase Behavior: Combined Experimental and Theoretical Approach, J. Phys. Chem. C, 2016, 120(7), 4016–4031 CrossRef CAS.
- Y. Shi, X. Zhang, X. Chen and Y. Zhang, Trap-tuning in afterglow perovskite crystals through alkali metal ion doping, Chem. Commun., 2022, 58(72), 10048–10051 RSC.
- J. Zhou, G. Zheng, X. Liu, G. Dong and J. Qiu, Defect engineering in lanthanide doped luminescent materials, Coord. Chem. Rev., 2021, 448, 214178 CrossRef CAS.
- F. Gao, Q. Pang, D. Gao, C. Jia, H. Xin, Y. Pan, Y. Wang and S. Yun, Mn2+-Activated Photostimulable Persistent Nanophosphors by Pr3+ Codoping for Rewritable Information Storage, ACS Appl. Nano Mater., 2023, 6(4), 3054–3064 CrossRef CAS.
- C. Guo, K. Tian, L. Wang, F. Liang, F. Wang, D. Chen, J. Ning, Y. Zhong and Y. Hu, Approach of Fermi level and electron-trap level in cadmium sulfide nanorods via molybdenum doping with enhanced carrier separation for boosted photocatalytic hydrogen production, J. Colloid Interface Sci., 2021, 583, 661–671 CrossRef CAS.
- Y. Kuang, Y. Li, B. Chen, S. Zhao, M. Chen, S. Lian and J. Zhang, Regulating anti-thermal quenching to zero thermal quenching for highly efficient blue-emitting Eu2+-doped K-beta-alumina phosphors, J. Mater. Chem. C, 2023, 11(17), 5874–5881 RSC.
- Y. Xu, Z. Zhou, P. Yu and Y. Wang, Al-doped CuS/Znln2S4 forming Schottky Junction Induced Electron Trap Centers for Photocatalytic Overall Water Splitting, Chem. Eng. J., 2023, 144275 CrossRef CAS.
- Y. Chen, B. Yu, J. Gou and S. F. Liu, Zero-thermal-quenching and photoluminescence tuning with the assistance of carriers from defect cluster traps, J. Mater. Chem. C, 2018, 6(40), 10687–10692 RSC.
- K. Huang, N. Le, J. S. Wang, L. Huang, L. Zeng, W. C. Xu, Z. Li, Y. Li and G. Han, Designing next generation of persistent luminescence: recent advances in uniform persistent luminescence nanoparticles, Adv. Mater., 2022, 34(14), 2107962 CrossRef CAS.
- Y. Li, M. Gecevicius and J. Qiu, Long persistent phosphors—from fundamentals to applications, Chem. Soc. Rev., 2016, 45(8), 2090–2136 RSC.
- X. Wang, X. Zhang, S. Yan, H. Liu and Y. Zhang, Nearly-Unity Quantum Yield and 12-Hour Afterglow from a Transparent Perovskite of Cs2NaScCl6:Tb, Angew. Chem., Int. Ed., 2022, 61(40), e202210853 CrossRef CAS PubMed.
- S. Wang, Z. Song and Q. Liu, Recent progress in Ce3+/Eu2+-activated LEDs and persistent phosphors: focusing on the local structure and the electronic structure, J. Mater. Chem. C, 2023, 11, 48–96 RSC.
- H. Miao, Y. Zhou, P. Wang, Z. Huang, W. Zhaxi, L. Liu, F. Duan, J. Wang, X. Ma and S. Jiang, High-temperature negative thermal quenching phosphors from molecular-based materials, Chem. Commun., 2023, 59(9), 1229–1232 RSC.
- H. Yuan, Q. Gao, P. Xu, J. Guo, L. He, A. Sanson, M. Chao and E. Liang, Understanding negative thermal expansion of Zn2GeO4 through local structure and vibrational dynamics, Inorg. Chem., 2021, 60(3), 1499–1505 CrossRef CAS PubMed.
- J. Dolado, F. Malato, J. Segura-Ruiz, R. Martínez-Casado, M. Taeño, I. Snigireva, P. Hidalgo, B. Méndez and G. Martínez-Criado, Interplay between Crystal Structure and Optical Response in Plateau–Rayleigh Zn2GeO4/SnO2 Heterostructures, Adv. Photonics Res., 2023, 4(8), 2300063 CrossRef CAS.
- X. Li, Y. Feng, M. Li, W. Li, H. Wei and D. Song, Smart hybrids of Zn2GeO4 nanoparticles and ultrathin g-C3N4 layers: synergistic lithium storage and excellent electrochemical performance, Adv. Funct. Mater., 2015, 25(44), 6858–6866 CrossRef CAS.
- Z. Ma, X. Liu, X. Wang, Z. Luo, W. Li, Y. Nie, L. Pei, Q. Mao, X. Wen and J. Zhong, Manipulating the d-band center enhances photoreduction of CO2 to CO in Zn2GeO4 nanorods, Chem. Eng. J., 2023, 468, 143569 CrossRef CAS.
- S. Yan, L. Wan, Z. Li and Z. Zou, Facile temperature-controlled synthesis of hexagonal Zn2GeO4 nanorods with different aspect ratios toward improved photocatalytic activity for overall water splitting and photoreduction of CO2, Chem. Commun., 2011, 47(19), 5632–5634 RSC.
- H. Zhao, X. Wang, J. Feng, Y. Chen, X. Yang, S. Gao and R. Cao, Synthesis and characterization of Zn2GeO4/Mg-MOF-74 composites with enhanced photocatalytic activity for CO2 reduction, Catal. Sci. Technol., 2018, 8(5), 1288–1295 RSC.
- J. Dolado, J. García-Fernández, P. Hidalgo, J. González-Calbet, J. Ramírez-Castellanos and B. Méndez, Intense cold-white emission due to native defects in Zn2GeO4 nanocrystals, J. Alloys Compd., 2022, 898, 162993 CrossRef CAS.
- J. Dolado, B. Rodríguez, R. Martínez-Casado, I. Píš, E. Magnano, P. Hidalgo and B. Méndez, Li-doping effects on the native defects and luminescence of Zn2GeO4 microstructures: Negative thermal quenching, Acta Mater., 2023, 245, 118606 CrossRef CAS.
- S. K. Gupta, K. Sudarshan, B. Modak and R. Gupta, Interstitial Zinc Boosted Light Tunability, Afterglow, and Ultrabright White Emission in Zinc Germanate (Zn2GeO4), ACS Appl. Electron. Mater., 2023, 5(2), 1286–1294 CrossRef CAS.
- G. Gao, M. Peng and L. Wondraczek, Spectral shifting and NIR down-conversion in Bi3+/Yb3+ co-doped Zn2GeO4, J. Mater. Chem. C, 2014, 2(38), 8083–8088 RSC.
- G. Liu, M. S. Molokeev and Z. Xia, Structural rigidity control toward Cr3+-based broadband near-infrared luminescence with enhanced thermal stability, Chem. Mater., 2022, 34(3), 1376–1384 CrossRef CAS.
- Y. Wei, H. Yang, Z. Gao, X. Yun, G. Xing, C. Zhou and G. Li, Anti-Thermal-Quenching Bi3+ Luminescence in a Cyan-Emitting Ba2ZnGe2O7:Bi Phosphor Based on Zinc Vacancy, Laser Photonics Rev., 2021, 15(1), 2000048 CrossRef CAS.
- R. M. Calderón-Olvera, E. Arroyo, A. M. Jankelow, R. Bashir, E. Valera, M. Ocaña and A. I. Becerro, Persistent Luminescence Zn2GeO4:Mn2+ Nanoparticles Functionalized with Polyacrylic Acid: One-Pot Synthesis and Biosensing Applications, ACS Appl. Mater. Interfaces, 2023, 15(17), 20613–20624 CrossRef.
- D. Gao, F. Gao, Q. Kuang, X. Zhang, Z. Zhang, Y. Pan, R. Chai and H. Jiao, Zinc Germanate nanophosphors with persistent luminescence for multi-mode imaging of latent fingerprints, ACS Appl. Nano Mater., 2022, 5(7), 9929–9939 CrossRef CAS.
- B. B. Srivastava, S. K. Gupta, Y. Li and Y. Mao, Bright persistent green emitting water-dispersible Zn2GeO4:Mn nanorods, Dalton Trans., 2020, 49(22), 7328–7340 RSC.
- X. Wang and Y. Mao, Recent advances in Pr3+-activated persistent phosphors, J. Mater. Chem. C, 2022, 10(10), 3626–3646 RSC.
- M. Wan, Y. Wang, X. Wang, H. Zhao and Z. Hu, The properties of a novel green long afterglow phosphor Zn2GeO4:Mn2+,Pr3+, Opt. Mater., 2014, 36(3), 650–654 CrossRef CAS.
- M. Anenburg, A. D. Burnham and J. L. Hamilton, Quadrivalent praseodymium in planetary materials, Am. Mineral., 2020, 105(12), 1802–1811 CrossRef.
- V. Sadykov, N. Eremeev, Z. Vinokurov, A. Shmakov, V. Kriventsov, A. Lukashevich, A. Krasnov and A. Ishchenko, Structural studies of pr nickelate-cobaltite-Y-doped ceria nanocomposite, J. Ceram. Sci. Technol., 2017, 8(1), 129–140 Search PubMed.
- J. Dolado, R. Martínez-Casado, P. Hidalgo, R. Gutierrez, A. Dianat, G. Cuniberti, F. Domínguez-Adame, E. Díaz and B. Méndez, Understanding the UV luminescence of zinc germanate: The role of native defects, Acta Mater., 2020, 196, 626–634 CrossRef CAS.
- H. He, Y. Zhang, Q. Pan, G. Wu, G. Dong and J. Qiu, Controllable synthesis of Zn2GeO4:Eu nanocrystals with multi-color emission for white light-emitting diodes, J. Mater. Chem. C, 2015, 3(21), 5419–5429 RSC.
- J. Dolado, P. Hidalgo and B. Méndez, Correlative study of vibrational and luminescence properties of Zn2GeO4 microrods, Phys. Status Solidi A, 2018, 215(19), 1800270 CrossRef.
- P. Hidalgo, A. López, B. Méndez and J. Piqueras, Synthesis and optical properties of Zn2GeO4 microrods, Acta Mater., 2016, 104, 84–90 CrossRef CAS.
- Z. Liu, X. Jing and L. Wang, Luminescence of native defects in Zn2GeO4, J. Electrochem. Soc., 2007, 154(6), H500 CrossRef CAS.
- X. Chen, X. Wu, L. Yue, L. Zhu, W. Pan, Z. Qi, S. Wang and J. Shao, Negative thermal quenching of below-bandgap photoluminescence in InPBi, Appl. Phys. Lett., 2017, 110, 5 Search PubMed.
- D. Kulesza, A. J. Bos and E. Zych, The effect of temperature and excitation energy of the high-and low-spin 4f→ 5d transitions on charging of traps in Lu2O3:Tb,M (M = Ti,Hf), Acta Mater., 2022, 231, 117852 CrossRef CAS.
- X. Wang, Z. Feng, J. Shi, G. Jia, S. Shen, J. Zhou and C. Li, Trap states and carrier dynamics of TiO2 studied by photoluminescence spectroscopy under weak excitation condition, Phys. Chem. Chem. Phys., 2010, 12(26), 7083–7090 RSC.
- J. Ren, X. Xu, H. Zeng, G. Chen, D. Kong, C. Gu, C. Chen, Z. Liu and L. Kong, Novel Self-Activated Zinc Gallogermanate Phosphor: The Origin of its Photoluminescence, J. Am. Ceram. Soc., 2014, 97(10), 3197–3201 CrossRef CAS.
- M. D'Arienzo, M. Redaelli, B. Di Credico, S. Polizzi, R. Scotti and F. Morazzoni, New insights into the sensing mechanism of shape controlled ZnO particles, RSC Adv., 2016, 6(58), 52987–52997 RSC.
- V. Ischenko, S. Polarz, D. Grote, V. Stavarache, K. Fink and M. Driess, Zinc oxide nanoparticles with defects, Adv. Funct. Mater., 2005, 15(12), 1945–1954 CrossRef CAS.
- S. Pramanik, S. Mukherjee, S. Dey, S. Mukherjee, S. Das, T. Ghosh, P. Ghosh, R. Nath and P. K. Kuiri, Cooperative effects of zinc interstitials and oxygen vacancies on violet-blue photoluminescence of ZnO nanoparticles: UV radiation induced enhanced latent fingerprint detection, J. Lumin., 2022, 251, 119156 CrossRef CAS.
- D. Savchenko, A. Vasin, O. Kuz, I. Verovsky, A. Prokhorov, A. Nazarov, J. Lančok and E. Kalabukhova, Role of the paramagnetic donor-like defects in the high n-type conductivity of the hydrogenated ZnO microparticles, Sci. Rep., 2020, 10(1), 17347 CrossRef CAS PubMed.
- J. Kong, W. Zheng, Y. Liu, R. Li, E. Ma, H. Zhu and X. Chen, Persistent luminescence from Eu3+ in SnO2 nanoparticles, Nanoscale, 2015, 7(25), 11048–11054 RSC.
- W. Sun, R. Pang, H. Li, D. Li, L. Jiang, S. Zhang, J. Fu and C. Li, Investigation of a novel color tunable long afterglow phosphor KGaGeO4:Bi3+: luminescence properties and mechanism, J. Mater. Chem. C, 2017, 5(6), 1346–1355 RSC.
- A. Barzykin and M. Tachiya, Mechanism of charge recombination in dye-sensitized nanocrystalline semiconductors: Random flight model, J. Phys. Chem. B, 2002, 106(17), 4356–4363 CrossRef CAS.
- Y. Zhang, Y. Liang, X. Shan, D. Chen, S. Miao, R. Shi, F. Xie and W. Wang, X-ray-Excited Long-Lasting Narrowband Ultraviolet-B Persistent Luminescence from Gd3+-Doped Sr2P2O7 Phosphor, Inorg. Chem., 2022, 61(50), 20647–20656 CrossRef CAS PubMed.
- H. Suo, X. Zhang and F. Wang, Controlling X-ray-activated persistent luminescence for emerging applications, Trends Chem., 2022, 4(8), 726–738 CrossRef CAS.
- C. Richard and B. Viana, Persistent X-ray-activated phosphors: mechanisms and applications, Light: Sci. Appl., 2022, 11(1), 123 CrossRef CAS PubMed.
- S. Ge, H. Peng, Q. Wei, X. Shen, W. Huang, W. Liang, J. Zhao and B. Zou, Realizing Color-Tunable and Time-Dependent Ultralong Afterglow Emission in Antimony-Doped CsCdCl3 Metal Halide for Advanced Anti-Counterfeiting and Information Encryption, Adv. Opt. Mater., 2023, 2300323 CrossRef CAS.
- W. Zheng, X. Li, N. Liu, S. Yan, X. Wang, X. Zhang, Y. Liu, Y. Liang, Y. Zhang and H. Liu, Solution-grown chloride perovskite crystal of red afterglow, Angew. Chem., 2021, 133(46), 24655–24660 CrossRef.
- F. Peng, T. Seto and Y. Wang, First Evidence of Electron Trapped Ln2+ Promoting Afterglow on Eu2+, Ln3+ Activated Persistent Phosphor-Example of BaZrSi3O9: Eu2 +, Sm3 +, Adv. Funct. Mater., 2023, 2300721 CrossRef CAS.
- Y.-C. Lin, M. Bettinelli and M. Karlsson, Unraveling the mechanisms of thermal quenching of luminescence in Ce3+-doped garnet phosphors, Chem. Mater., 2019, 31(11), 3851–3862 CrossRef CAS.
- M. A. Reshchikov, Two-step thermal quenching of photoluminescence in Zn-doped GaN, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85(24), 245203 CrossRef.
- A. J. van Bunningen, A. D. Sontakke, R. van der Vliet, V. G. Spit and A. Meijerink, Luminescence Temperature Quenching in Mn2+ Phosphors, Adv. Opt. Mater., 2023, 11(6), 2202794 CrossRef CAS.
- H. Shibata, Negative thermal quenching curves in photoluminescence of solids, Jpn. J. Appl. Phys., 1998, 37(2R), 550 CrossRef CAS.
- D. Huntley, An explanation of the power-law decay of luminescence, J. Phys.: Condens. Matter, 2006, 18(4), 1359 CrossRef CAS.
- J. Dolado, P. Hidalgo and B. Méndez, Kinetic Study of the Thermal Quenching of the Ultraviolet Emission in Zn2GeO4 Microrods, Phys. Status Solidi RRL, 2022, 16(6), 2100613 CrossRef CAS.
- J. J. Joos, D. Van der Heggen, L. I. Martin, L. Amidani, P. F. Smet, Z. Barandiarán and L. Seijo, Broadband infrared LEDs based on europium-to-terbium charge transfer luminescence, Nat. Commun., 2020, 11(1), 3647 CrossRef CAS.
- Y.-F. Liu, P. Liu, L. Wang, C.-E. Cui, H.-C. Jiang and J. Jiang, A two-step solid-state reaction to synthesize the yellow persistent Gd3Al2Ga3O12:Ce3+ phosphor with an enhanced optical performance for AC-LEDs, Chem. Commun., 2017, 53(77), 10636–10639 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc03731b |
| This journal is © The Royal Society of Chemistry 2024 |