
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boosting stability: a hierarchical approach for self-assembling peptide structures†
Denys
Balandin‡
ab,
Natalia
Szulc‡
c,
Dominika
Bystranowska
 d,
Marlena
Gąsior-Głogowska
e,
Roksana
Kruszakin
f and
Monika
Szefczyk
*a
d,
Marlena
Gąsior-Głogowska
e,
Roksana
Kruszakin
f and
Monika
Szefczyk
*a
aDepartment of Bioorganic Chemistry, Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, Wrocław 50-370, Poland. E-mail: monika.szefczyk@pwr.edu.pl
bDepartment of Medicinal Chemistry, Otto Loewi Research Center, Medical University of Graz, Neue Stiftingtalstrasse 6, 8010, Graz, Austria
cDepartment of Physics and Biophysics, Wrocław University of Environmental and Life Sciences, Norwida 25, Wrocław 50-375, Poland
dDepartment of Biochemistry, Molecular Biology and Biotechnology, Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, Wrocław 50-370, Poland
eDepartment of Biomedical Engineering, Faculty of Fundamental Problems of Technology, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, Wrocław 50-370, Poland
fLaboratory of Instrumental Analysis and Preparation, Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Rudolfa Weigla 12, Wrocław 53-114, Poland
First published on 18th September 2024
Abstract
The primary objective of this study was to implement a hierarchical approach to enhance the conformational stability of a selected group of peptides by incorporating trans-(1S,2S)-2-aminocyclopentanecarboxylic acid (trans-ACPC). The influence of residue mutation on the peptide structures was investigated using circular dichroism, analytical ultracentrifugation, and vibrational spectroscopy. The resulting nanostructures were examined via transmission electron microscopy. The incorporation of trans-ACPC led to increased conformational stability and self-assembling propensity in peptides containing constrained β-amino acid residues.
Introduction
Soft materials science encounters several significant challenges. On one hand, the complexity of structure and behavior is sought after due to the potential for achieving sophisticated applications and functions.1 However, soft materials often exhibit complex hierarchical structures and behaviors that are difficult to predict and control owing to their dynamic nature.2 This dynamism is evident in their continual changes in structure and properties under various stimuli, complicating efforts to comprehend and manage their behavior over time.3 Furthermore, the development of soft materials that are biocompatible, biodegradable, and environmentally friendly for diverse applications, including biomedical and environmental fields, poses ongoing challenges.4 Moreover, soft materials frequently possess complex structures across multiple length scales, necessitating advanced experimental and computational techniques for precise characterization.5,6 Additionally, the integration of experimental data and theoretical models across various length and time scales to accurately predict the behavior of soft materials remains a significant difficulty.7,8 Ultimately, overcoming the hurdle of establishing reliable, straightforward, and cost-effective methods for processing and fabricating soft materials with desired properties remains an obstacle.9–11 Tackling these challenges requires an interdisciplinary approach that integrates expertise from fields such as chemistry, physics, materials science, biology, and engineering, as well as advancements in experimental techniques, computational methods, and theoretical modeling.To address these challenges, our focus has been on employing a hierarchical approach for constructing self-organizing peptide-based nanomaterials. Peptides possess a unique combination of biocompatibility, tunable properties, dynamic behavior, versatility, and biodegradability that make them promising candidates for a wide range of soft material applications.12–14 Moreover, the self-assembly capability of peptides allows for the creation of nanostructures with high efficiency, versatility, dynamic adaptability, and potential for complexity making it an appealing method for the cost-effective and scalable fabrication of nanomaterials across various fields.15,16 Furthermore, the hierarchical approach to constructing self-assembling peptide-based nanostructures facilitates the rational design and control over higher-ordered structure formation. To address the structure stability issue, our efforts have focused on incorporating cyclic-beta amino acid residues into the peptide sequence, resulting in well-defined structures in solution known as foldamers. Ultimately, several experimental methods have been employed to comprehensively characterize the obtained structures at various organizational levels.
Precisely controlling a peptide's secondary structure by incorporating constrained β-residues proved to be instrumental in designing novel materials with specific properties and functions. Trans-(1S,2S)-2-aminocyclopentanecarboxylic acid (trans-ACPC) serves as a versatile building block in peptide design, significantly influencing not only the peptide's structure but also its self-organization and self-assembly properties.17,18 When employed as a building block in β-peptide monomers, trans-ACPC homo-oligomers have demonstrated spontaneous self-assembly into various microstructures.19,20 Additionally, mixed α/β-peptides have exhibited the capability to form well-defined nanostructures in solutions.21 The ability of trans-ACPC-containing peptides to maintain their self-assembly characteristics despite the presence of bulky side chains or hydrophobic sequences makes them particularly attractive for the development of advanced materials. Integrating trans-ACPC into peptide materials has also been found to confer stimulus-responsive properties. For instance, foldectures containing trans-ACPC can respond to external dynamic magnetic fields, exhibiting real-time mechanical motions.22 This magneto-responsive behavior has been further explored by embedding foldectures into hydrogel containers, demonstrating magnetosome-inspired magnetotactic behavior and the translation of dynamic magnetic fields into instantaneous motions at both microscopic and macroscopic scales. Moreover, the ribosomal synthesis of bioactive foldameric peptides containing cyclic β-amino acids, such as trans-ACPC, has been reported, indicating the potential for de novo discovery of bioactive materials with desired properties.23 Thus, incorporating constrained β-residues into peptides emerges as a promising strategy in material science for creating novel materials with controlled morphologies and properties.
In this study, we present our efforts to implement a hierarchical approach aimed at improving the conformational stability and enhancing the self-organization propensity of a selected group of peptides. Both objectives were pursued through the incorporation of the helix-stabilizing and promoting trans-ACPC residue into the peptide sequence. The impact of residue mutation on the peptide structures was assessed using circular dichroism (CD) and attenuated total reflectance Fourier-transform infrared spectroscopy (ATR-FTIR). The self-organization propensity of the peptides was evaluated through sedimentation velocity analytical ultracentrifugation (SV-AUC). The resulting nanostructures were examined via transmission electron microscopy (TEM). These analyses were conducted immediately after dissolution and following sample incubation.
Results and discussion
The primary aim of this study was to investigate the structural and conformational diversity of designed peptides and their propensity for self-organization. Specifically, we sought to examine the impact of residue mutation, particularly the introduction of cyclic β-amino acids, on the conformational stability and self-assembly propensity of coiled-coil-based peptides. The peptides (Table S1, ESI†) were obtained using solid-phase peptide synthesis on a fully automated synthesizer, purified using a preparative reverse phase high-performance liquid chromatography, and then subjected to qualitative analysis (Fig. S1 and Table S2, ESI†).Model peptides CAM 1 and CAM 2 (Table 1 and Fig. 1), originally designed by Pandya et al., were reported to undergo heteroaggregation following a “sticky-end” assembly process to form fibrils.24 However, these peptides exhibited structured behavior in aqueous solution at pH 7 only at 5 °C. Our goal was to enhance the conformational stability of these peptides. Initially, we modified model peptides CAM 1 and CAM 2 with trans-ACPC, known for its helix-stabilizing and helix-promoting properties.25 Following the hierarchical approach of α/β peptide-based nanofibril formation previously introduced,17,18 and the general principles of dimeric coiled-coil design,26trans-ACPC was incorporated at the outer positions of each repeating heptad to enhance the self-assembly properties of the studied sequences (Fig. 1). This positioning of trans-ACPC in the designed peptides is expected to facilitate additional interactions between coiled-coils and enhance the stability of the formed nanostructures, thanks to the conformational rigidity of the cyclic β-residue.
| Peptide | Sequence |
|---|---|
| gabcdef | |
| CAM 1 |

|
| CAM 1X |

|
| Di 1 |

|
| Di 1X |

|
| CAM 2 |

|
| CAM 2X |

|
| Di 2 |
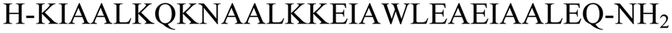
|
| Di 2X |

|
 | ||
| Fig. 1 Helical wheels of the studied peptides with mutations highlighted. | ||
Circular dichroism (CD) was utilized to determine the secondary structure and conformational stability of the synthesized peptides. CD signatures indicative of α-helical structures are characterized by evident negative minima at 208 and 222 nm. The CD spectra of freshly dissolved peptides, as reported by Pandya et al., exhibited a negative peak solely at 222 nm. As the samples mature, the intensity of this peak doubled, and a weak peak emerged at 208 nm.24 CD spectra of model peptides CAM 1 and CAM 2 recorded at 25 °C revealed two minima at λ = 204 and 222 nm, with a ratio of R(θ222/θ204) < 1, indicating a disrupted α-helix structure (Fig. 2). CD spectra of peptides containing trans-ACPC, CAM 1X and CAM 2X, displayed only one minimum at λ = 204 and 205 nm, respectively (Fig. 2A), suggesting random coil formation. Conformational stability was assessed based on CD measurements in water at pH 7, and thermal unfolding curves followed at 208 nm were used to determine the melting temperatures. Peptides CAM 1 and CAM 2 exhibited low stability, with a cooperative melting transition occurring at temperatures 33.6 ± 0.9 °C and 23.8 ± 0.9 °C, respectively (Fig. 2B). The data for peptides CAM 1X and CAM 2X were not obtained as the CD spectra did not indicate α-helical fold.
 | ||
| Fig. 2 (A) CD spectra and (B) thermal unfolding curves, followed by CD signal at 208 nm with calculated melting temperatures indicated in the legend for the peptides CAM 1, CAM 2, CAM 1X, and CAM 2X dissolved in water at pH 7 (Cpep = 0.2 mM). | ||
Since the introduction of trans-ACPC into the structure of peptides CAM 1 and CAM 2 did not yield the expected outcome (peptides CAM 1X and CAM 2X did not show helix formation), we endeavored to improve stability of the model peptides through amino acid residue mutations. The following modifications were made to obtain peptides Di 1 and Di 2: in CAM 1, negatively charged aspartic acid residues in position b were replaced by small, hydrophobic alanine. In position f, glutamine residue was replaced with lysine to enhance solubility. Tyrosine in position f was replaced by alanine, and UV chromophore tryptophan was introduced at position c. Polar serine was replaced by hydrophobic alanine at position c. CAM 2 was modified as depicted in Fig. 1, so that the resulting Di 2 differs from Di 1 only in the positioning of one asparagine residue. CD spectra of the obtained peptides exhibited α-helix structure with two more evident minima compared to CAM 1 and CAM 2, specifically at λ = 209 and 222 nm for Di 1 and at 207 and 222 nm for Di 2 (Fig. 3A). The improvement in stability can also be observed in the case of the modified peptides. The melting temperature of peptide Di 1 was 57.2 ± 0.2 °C (compared to 33.6 for CAM 1) and for peptide Di 2 was 41.2 ± 0.8 °C (compared to 23.8 for CAM 2) (Fig. 3B).
 | ||
| Fig. 3 (A) CD spectra and (B) thermal unfolding curves, followed by CD signal at 208 nm with calculated melting temperatures indicated in the legend for the peptides CAM 1, CAM 2, Di 1 and Di 2 dissolved in water at pH 7 (Cpep = 0.2 mM). | ||
To enhance both stability and self-aggregation properties, Di 1 and Di 2 underwent further modification with trans-ACPC residues. These residues were introduced in the f positions of peptides Di 1 and Di 2 to produce Di 1X and Di 2X, respectively (Fig. 1). This resulted in foldameric helix formation characterized by low minima at 208 nm (Di 1X) and 207 nm (Di 2X), with a shallow negative peak at 222 nm (Fig. 4A). Calculated melting temperatures indicated improved stability of the obtained peptides compared to peptides Di 1 and Di 2 (Fig. 4B). The melting temperature for Di 1X was 72.0 ± 0.3 °C (compared to 57.2 for Di 1), and for Di 2X was 52.7 ± 1.2 °C (compared to 41.2 for Di 2).
 | ||
| Fig. 4 (A) CD spectra and (B) thermal unfolding curves, followed by CD signal at 208 nm with calculated melting temperatures indicated in the legend for the peptides Di 1, Di 2, Di 1X and Di 2X dissolved in water at pH 7 (Cpep = 0.2 mM). | ||
More detailed information about the secondary structure of the studied peptides was obtained through analysis of attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra. Spectroscopic data were registered for the peptides in powder form (Fig. S2, ESI†), as well as in solution immediately after dissolving (Fig. S3, ESI†), and after 7 (Fig. S4, ESI†) and 30 days (Fig. S5, ESI†) of incubation at 37 °C (98.6 F). Initially, all studied peptides exhibited maxima of the amide I bands in the range of 1655–1651 cm−1, typically indicative of α-helical peptides.27 The maximum of the amide II band, located around 1548–1544 cm−1, also supports the presence of a helical structure.28,29 Upon dissolution in water, no significant changes in the positions of amide bands were observed, suggesting structural stability in aqueous solution. However, α,β-peptides exhibited lower frequencies for their amide I band maximum compared to model peptides, potentially indicating the presence of trans-ACPC.17 This preservation of amide band positions was similarly observed for peptides incubated at 37 °C for 7 and 30 days. Comprehensive insights into the secondary structure were gained through second-derivative and deconvolution procedures, enabling separation of overlapping components within the respective region (Fig. S6–S8, ESI†).30,31
We observed four subcomponents within the amide I band: helical structures (at 1655 cm−1), turns and β-antiparallel (at 1678 cm−1), β-sheets (ranging from 1635 to 1625 cm−1), and intermolecular aggregates (ranging from 1624 to 1610 cm−1), alongside other residues such as aspartic acid and glutamic acid.32–34 Immediately after dissolution, most peptides exhibited a mixture of helical structures and turns/β-antiparallel subcomponents (Fig. 5 and Fig. S3, ESI†). The presence of intermolecular aggregates was generally low, suggesting limited aggregation/self-assembly processes. After 7 days of incubation at 37 °C, there was a noticeable trend towards increased formation of β-sheet/intermolecular aggregate formation for some peptides, indicating initial stages of aggregation (Fig. 5 and Fig. S9, ESI†). Following 30 days of incubation, the content of helical structures tended to decrease for most peptides, suggesting a potential transition to more ordered structures. Concurrently, the content of β-sheets/intermolecular aggregates often increased, signifying further aggregation. Specific peptides, such as CAM 1X, Di 1X, Di 2X, demonstrated a significant rise in β-sheets/intermolecular aggregates content, indicating potential stabilization of certain conformations. Peptides with trans-ACPC generally showed a higher propensity for β-sheet/intermolecular aggregate formation compared to their non-modified counterparts, particularly after 30 days (Fig. 5 and Fig. S10, ESI†). This suggests that trans-ACPC not only stabilizes helical structures but may also promote the self-assembly of peptides into more ordered aggregates, which could be advantageous for the formation of stable nanostructures.
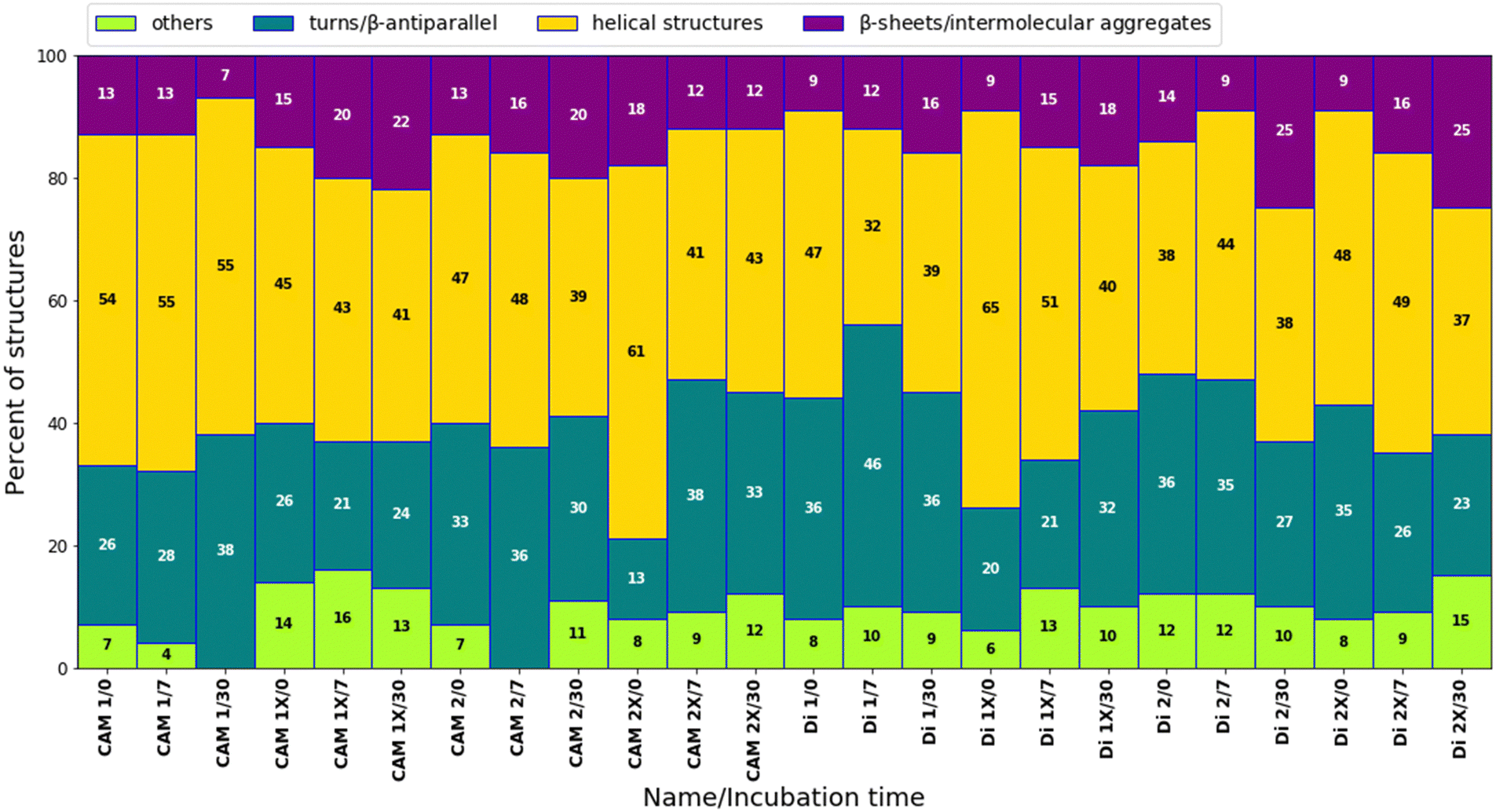 | ||
| Fig. 5 Percentages of subcomponents in the amide I band relative to the total area (1750–1590 cm−1). The data were obtained by deconvoluting the ATR-FTIR spectra of the studied peptides directly after dissolution, as well as after 7 and 30 days of incubation at 37 °C (Cpep = [0.317–0.327] mM). | ||
At the outset, the trans-ACPC containing peptides exhibited a significantly lower percentage of β-turns, indicating the presence of the elongated structures through the β-sheet mechanism as previously suggested.30,31 Interestingly, despite the random coil structure evidenced by CD studies, CAM 1 and CAM 2 peptides exhibited significant helicity as revealed by deconvolution studies. Furthermore, we observed a decrease in helicity for the model CAM 1 peptide over time, along with an increase in the β-turn sub-band area. This trend was not observed for the CAM 2 peptide, as the overall contribution of subcomponents to the amide I band remained consistent over time. The tendency for stable secondary structure is evident in the modified Di 1 and Di 2 peptides. In Di 1X and Di 2X, an overall increase in the helical component suggests the effect of cyclic β-amino acid incorporation as a helix-promoting residue.
Both CD and FTIR data analyses suggest that the studied peptides predominantly adopt a helical structure. However, for CAM 1X and CAM 2X, the peptides may exist in solution in an equilibrium between very low-stability helices (as indicated by FTIR) and random coils (as suggested by CD). Additionally, the thermal denaturation CD data for Di 1X (Fig. 4) indicates a potential two-step melting process, suggesting a more complex conformation, which is consistent with FTIR findings. FTIR analysis also revealed that the incorporation of trans-ACPC into Di 1X stabilizes specific secondary structures, potentially leading to distinct structural transitions during thermal unfolding. This two-step melting behavior further supports the presence of multiple stable conformations within the peptide, reinforcing the hypothesis of a more intricate conformational landscape in Di 1X compared to its unmodified counterparts. The high percentage of helices observed by FTIR (Fig. 5) may suggest the presence of multiple types of helices in solution, each with varying stabilities.
Overall, the trend suggests that the introduction of trans-ACPC promotes the formation and stabilization of ordered secondary structures over time. This finding aligns with the hierarchical approach of α/β peptide-based nanofibrils formation and the goal of enhancing the stability and self-assembly properties of these peptides.
SV-AUC with UV detection at 280 nm was applied as an additional method to gather further information on the structural properties of Di peptides. The experiments were conducted at three peptide concentrations of 50 μM, 80 μM, and 100 μM. Fig. S9 (ESI†) displays the sedimentation coefficient distributions obtained from the c(s) analysis method. All analyzed peptides exhibited a single peak with a similarly narrow and spike-like continuous sedimentation coefficient distribution c(s). Both, in the absence (Fig. S9A and B, ESI†) and presence (Fig. S9C and D, ESI†) of trans-ACPC, the value of the sedimentation coefficients of the main peaks corresponded to a mass of the dimer (Table 2). Therefore, this is likely to indicate that the incorporation of trans-ACPC did not significantly affect the oligomerization state of Di peptides compared to its unmodified version (Table 2). According to the collected data, no significant changes in the shape of the peptides were also observed. The f/f0 coefficient values oscillated in a range of 1.34–1.42 with no relevant variations resulting from either the changes of concentrations of the peptides or the incorporation of the trans-ACPC moieties. In summary, based on the hydrodynamic properties determined by SV-AUC we can conclude that Di peptides exist in solution as moderately extended molecules with a propensity for dimerization, at least up to a peptide concentration of 100 μM, regardless of the presence of trans-ACPC.
| Peptide | c (μM) | rmsd | s 20,w | f/f0 | MWapp (kDa) | Oligomerization state |
|---|---|---|---|---|---|---|
| Numbers in brackets indicate the percentage of each fraction and are given considering 100% for the sum of the main indicated types of sedimenting species. rmsd – root-mean-square deviation; s20,w – sedimentation coefficient in the standard conditions (i.e. water, 20 °C); f/f0 – frictional ratio (the ratio of the actual frictional coefficient to that for an anhydrous sphere with equal volume); MWapp – apparent molecular weight derived from SV-AUC experiments. | ||||||
| Di 1 | 50 | 0.004855 | 0.755 | 1.39 | 6548 | Dimer (100%) |
| 80 | 0.005119 | 0.782 | 1.35 | 6658 | Dimer (100%) | |
| 100 | 0.005646 | 0.788 | 1.35 | 6735 | Dimer (100%) | |
| Di 2 | 50 | 0.005530 | 0.632 | 1.40 | 5079 | Dimer (100%) |
| 80 | 0.005890 | 0.690 | 1.38 | 5688 | Dimer (100%) | |
| 100 | 0.006112 | 0.703 | 1.41 | 6051 | Dimer (100%) | |
| Di 1X | 50 | 0.006235 | 0.754 | 1.35 | 6524 | Dimer (100%) |
| 80 | 0.006333 | 0.782 | 1.35 | 6919 | Dimer (100%) | |
| 100 | 0.006337 | 0.792 | 1.34 | 6950 | Dimer (100%) | |
| Di 2X | 50 | 0.006991 | 0.659 | 1.42 | 5750 | Dimer (100%) |
| 80 | 0.007149 | 0.688 | 1.37 | 5794 | Dimer (100%) | |
| 100 | 0.008047 | 0.705 | 1.36 | 5970 | Dimer (100%) | |
Overall, structural studies using CD and ATR-FTIR have revealed that the tested peptides exhibit varying tendencies to form helices in solution. The introduction of subsequent rational mutations allowed for the generation of peptides with increased conformational stability. SV-AUC data indicate that the studied Di peptides predominantly exist as dimers in solution. Following incubation, most of the studied peptides exhibited changes in band composition, suggesting the formation of aggregates, therefore transmission electron microscopy (TEM) imaging was used to visualize the formed nanostructures.
The TEM micrographs revealed distinct morphological changes in peptide structures over 30 minutes post-dissolution (Fig. 6 and Fig. S10 and S11, ESI†) and after 48 hours of incubation at 37 °C (Fig. 6 and Fig. S12, S13, ESI†). The diameter size distribution for the studied peptides is shown in Fig. S14 and S15 (ESI†). Initially, at 30 minutes, the peptides exhibited primarily amorphous aggregates and early fibrillar formations, indicating the onset of self-assembly processes. Notably, peptide CAM 1X showed fibrillar structures immediately after dissolution. The median diameter of CAM 1X increased slightly from 6.94 nm to 9.82 nm, indicating the growth and densification of these fibrous structures, likely due to the maturation of fibrillar assemblies. CAM 2X featured small aggregates that grew larger and more defined over time, with the median diameter slightly decreasing from 22.24 nm to 16.90 nm, suggesting assembly into more distinct and potentially functional structures. Conversely, Di 1X transitioned from spherical nanostructures to more elongated and interconnected forms. The significant reduction in median diameter, from 36.47 nm to 7.67 nm, highlights a major restructuring into smaller and highly uniform structures. Di 2X displayed dense clustered nanostructures that dispersed into more fibrous forms over time.
 | ||
| Fig. 6 Temporal evolution of nanostructure morphologies: TEM micrographs were taken 30 minutes post-dissolution and after 48 hours of incubation at 37 °C during the self-assembly process. | ||
Initially, the CAM group showed a quicker progression towards elongated fibril structures, whereas the Di group presented a higher proportion of amorphous aggregates. This suggests that CAM peptides inherently possess a greater propensity for rapid fibril formation. In both groups, peptides with the trans-ACPC (CAM 1X, CAM 2X, Di 1X, Di 2X) demonstrated improved self-assembly characteristics, exhibiting more defined, regular, and finer fibrils by the 48-hour mark. This indicates that the trans-ACPC likely enhances the stability and regularity of fibril formation. By 48 hours, both groups showed significant structural maturation, but the CAM group maintained a more uniform and denser fibrillar network compared to the Di group, which still showed some heterogeneity in fibril distribution and morphology. The CAM and Di groups exhibited diverse transformations in their peptide structures over time, each displaying unique patterns of aggregation, network formation, and structural evolution. These transformations were reflected in changes in median diameters and distributions (Fig. S14 and S15, ESI†). Increase in median size and variability suggest aggregation and growth, while decreases typically indicate a breakdown or structural transformation leading to more uniformity.
Overall, the introduction of trans-ACPC enhances the self-assembly properties of the peptides, leading to more structured and regular fibrillar networks. CAM peptides show a slightly more uniform and efficient path to fibril maturation. However, over time, the same effect can be observed for the Di group, but the fibrils are created from amorphous oligomers, which exhibit higher elasticity, especially in Di 2X peptides. The structures of the Di group are significantly shorter compared to CAM peptides and are bonded together.
The findings were further validated using a thioflavin T (ThT) assay to observe the kinetics of peptide aggregation. CAM 1X and CAM 2X showed substantial increases in fluorescence, particularly between 28.8 and 33.0 hours, with the sigmoidal shape of the kinetics curves (see Fig. S16 and Table S3, ESI†) strongly suggesting fibrils formation. In contrast, the Di group exhibited negligible ThT fluorescence, characterized by a plateau. This observation is further supported by TEM results (see Fig. 6 and Fig. S11, S13, ESI†), which show the presence of more amorphous aggregates.
The turbidity measurements (Table S4, ESI†) also corroborate the ThT assay results, confirming that the CAM group samples effectively formed fibrils, as evidenced by increased turbidity and ThT fluorescence. Conversely, the Di group samples formed globular aggregates that do not bind ThT effectively, resulting in lower turbidity increase. These findings are crucial for understanding the different aggregation pathways and the structural nature of the aggregates formed in these samples.
In summary, we aimed to test the hierarchical approach for the structure stabilization and nanostructures formation of two peptides that were reported to form heterodimers using a “sticky-end” approach at low temperatures but possess low conformational stability at room temperature, as indicated by their melting temperatures of 34 and 24 °C, respectively.24 In previous articles, we demonstrated that replacing alpha residues with cyclic beta residues in the outer positions of coiled-coils (b, c, f) can stabilize peptide structures.17,18 However, for peptides CAM 1 and CAM 2, additional residue mutations were necessary. The straightforward introduction of trans-ACPC in the f position of the model coiled-coils led to poorly structured peptides with some helical features (CAM 1X and CAM 2X). This indicates that, apart from the core interactions at the a and d positions, the outer positions also significantly impact the fold, particularly in terms of structure stability. In our case, replacing selected positions with alanine significantly improved the melting temperatures, to 57 and 41 °C for Di 1 and Di 2, respectively. The subsequent introduction of trans-ACPC residues at the f positions resulted in the formation of Di 1X and Di 2X peptides with high melting temperatures of 72 and 53 °C, respectively, indicating high conformational stability. Importantly, Di 2 differs from Di 1 only in the positioning of one asparagine residue (N9 vs. N23, respectively). The difference in their stability may be attributed to the preferential pairing of N23 in the coiled-coil structure, rather than N9.
In line with the design assumptions, the tested peptides aggregated, but they differed in the kinetics of this process and the morphology of the resulting structures. As expected, Di 1X and Di 2X with their high conformational stability, formed fibrils due to the hydrophobic interactions of trans-ACPC in neighboring helices, creating a “cyclopentyl zipper”.17,18 Interestingly, CAM 1X and CAM 2X, which do not possess well-defined helical structures in solution, formed well-defined fibrils. This suggests that trans-ACPC promotes stable and predictable interactions among neighboring peptides, regardless of their secondary structure stability. Furthermore, peptides within the CAM group exhibit better-defined fibers compared to those within the Di group. This distinction is more pronounced than the difference observed between peptides with and without trans-ACPC. It suggests that the development of well-defined nanostructures may be influenced by interactions at the b, c, and f positions, indicating that a different mechanism of fiber formation may be at play.
In summary, the introduction of trans-ACPC residues into the sequence effectively stabilized and promoted helix formation. However, the influence of other positions within the coiled-coils is also crucial for both structural stabilization and nanostructure formation. The modifications applied to CAM and Di peptides demonstrate the effectiveness of strategic residue mutations, underscoring the significance of the overall network of interactions.
Conclusions
The results indicate that, beyond the core interactions at the a and d positions, the outer positions also significantly affect the folding and stability of the structure. The presence of trans-ACPC promotes stable and predictable interactions among neighboring peptides, independent of their secondary structure stability, forming what can be described as a “cyclopentyl zipper.” Additionally, interactions at the b, c, and f positions seem to contribute to the formation of well-defined nanostructures and a distinct mechanism for fiber formation. Overall, the incorporation of trans-ACPC residues effectively stabilizes and encourages helix formation. However, the roles of other positions within the coiled-coils are equally important for both structural stabilization and nanostructure assembly. The modifications made to CAM and Di peptides demonstrate the effectiveness of strategic residue mutations and emphasize the significance of the comprehensive network of interactions that can be utilized to design self-organizing peptide-based nanomaterials with specific functions and applications.Experimental
Peptide synthesis
All commercially available reagents and solvents were purchased from Merck or Sigma-Aldrich, and used without further purification. Fmoc-(1S,2S)-2-aminocyclopentanecarboxylic acid (trans-ACPC) was purchased from Synnovator, Inc. The peptides were obtained with an automated solid-phase peptide synthesizer (Liberty Blue, CEM) using H-Rink amide ChemMatrix resin (loading: 0.59 mmol g−1). Fmoc deprotection was achieved using 20% piperidine in DMF for 1 min at 90 °C. A double-coupling procedure was performed with a 0.5 M solution of DIC and a 0.25 M solution of OXYMA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in DMF for 4 min at 90 °C. Cleavage of the peptides from the resin was accomplished with the mixture of TFA/TIS/H2O (95:2.5:2.5) after 3 h of shaking. The crude peptide was precipitated with ice-cold Et2O and centrifuged (9000 rpm, 15 min, 2 °C). Peptides were purified using preparative HPLC (Knauer AZURA ASM 2.1 L system with a C18 Thermo Scientific, Hypersil Gold 12 μm, 250 mm × 20 mm column) in a water/acetonitrile (0.05% TFA) eluent system. The purified peptide fractions were lyophilized, analyzed by MS and analytical HPLC, and stored at −70 °C prior to further studies.
:1) in DMF for 4 min at 90 °C. Cleavage of the peptides from the resin was accomplished with the mixture of TFA/TIS/H2O (95:2.5:2.5) after 3 h of shaking. The crude peptide was precipitated with ice-cold Et2O and centrifuged (9000 rpm, 15 min, 2 °C). Peptides were purified using preparative HPLC (Knauer AZURA ASM 2.1 L system with a C18 Thermo Scientific, Hypersil Gold 12 μm, 250 mm × 20 mm column) in a water/acetonitrile (0.05% TFA) eluent system. The purified peptide fractions were lyophilized, analyzed by MS and analytical HPLC, and stored at −70 °C prior to further studies.
Analytical high-performance liquid chromatography (HPLC)
Analytical HPLC was performed using UltiMate 3000 LC System Dionex with Waters C18 1.7 μm 50 × 2.1 mm column and following program: eluent A: 0.05% TFA in H2O, eluent B: 0.05% TFA in acetonitrile, flow 0.5 mL min−1: A: t = 0 min, 90% A; t = 30 min, 10% A or using Shimadzu System with Kinetex 5 μm EVO C18 100 Å 150 × 4.6 mm column and following program: eluent A: 0.05% TFA in H2O, eluent B: 0.05% TFA in acetonitrile, flow 0.5 mL min−1: A: t = 0 min, 90% A; t = 25 min, 10% A.Mass spectrometry (MS)
Peptides were studied by the WATERS LCT Premier XE System, consisting of a high-resolution mass spectrometer with a time of flight (TOF) using electrospray ionization (ESI).pH adjustment
The peptides were dissolved in water and the pH was adjusted to 7 by adding 0.1 M NaOH solution.Circular dichroism (CD)
CD spectra were recorded using JASCO J-1500 at 20 °C between 240 and 190 nm in water pH = 7 with following parameters: 0.2 nm resolution, 1.0 nm bandwidth, 20 mdeg sensitivity, 0.25 s response, 50 nm min−1 scanning speed, 5 scans, 0.02 cm cuvette path length. The CD spectra of the solvent alone was recorded and subtracted from the raw data. The peptides were dissolved in water and pH was adjusted to 7 by adding 0.1 M NaOH solution. Typically, the samples were prepared by dilution of peptides stock solution to obtain peptide concentration of 0.2 mM. The CD intensity is given as mean residue ellipticity (θMRE [deg × cm2 × dmol−1]) calculated using the following equation (eqn (1)):18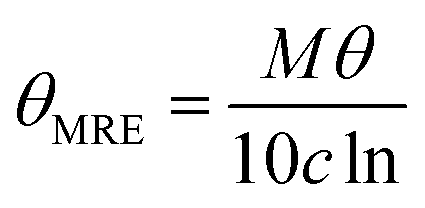 | (1) |
Temperature denaturation measurements using CD
To examine the thermal unfolding of the peptides, stock solutions were diluted to 0.2 mM and measurement at 208 nm in water pH = 7 was performed. The temperature was increased from 4 to 96 °C in increments of 2 °C. Ellipticity measurements were recorded with 1 mm path length cuvette, others parameters remained unchanged. For each thermal denaturation experiment, the data were fit to a two-state folding model adapted and described by Kreitler et al.35 using OriginPro 9.0 and equation (eqn (2)):35 | (2) |
Sedimentation-velocity analytical ultracentrifugation (SV-AUC)
SV-AUC experiments were performed on the Beckman Coulter Proteome Lab XL-I ultracentrifuge (software version 6.0, Beckman Coulter Inc.) equipped with an An-60Ti rotor. The concentrations of the peptides were 50 μM, 80 μM and 100 μM. The samples were suspended in the PB buffer. Analyses were done at 20 °C and 50000 rpm using a step size of 0.003 cm, a delay time of 0 s. Parameters obtained with SEDNTRP36 were as follows: peptide partial specific volumes (0.77204, 0.77204, 0.77751, 0.77751 mL g−1 for Di 1, Di 2, Di 1X, Di 2X, respectively), buffer density (1.0056 g mL−1) and buffer viscosity (1.0199 mPa s). Partial specific volumes of the peptides were estimated and corrected for the presence of trans-ACPC, N-terminal acetyl and C-terminal amine moieties using published molar increment values of chemical groups.37 Time-corrected data38 were analyzed with SEDFIT software (version 16.1c) using the built-in continuous sedimentation coefficient distribution model, c(s). Maximum-entropy regularization of the c(s) models was set to a confidence level of 0.68.39,40
Vibrational spectroscopy
Attenuated total reflectance—Fourier-transform infrared (ATR-FTIR) studies were conducted using a Thermo Scientific FT-IR spectrometer (USA) equipped with a Golden Gate Mk II ATR accessory with a heated diamond top-plate (PIKE Technologies), continuously purged with dry air. ATR-FTIR spectra were acquired in the range of 3600–400 cm−1. Each spectrum was the result of coadding 512 interferograms with a resolution of 4 cm−1. Immediately before sampling, the background spectrum of the diamond/air was recorded as a reference (512 scans, 4 cm−1). Peptide concentrations of [0.317–0.327] mM were used to achieve a good signal-to-noise ratio. All measurements were conducted at a constant temperature of 25 °C (77 °F). Additionally, air-dried films were prepared by dropping 10 μL of peptide aqueous solution directly onto the diamond surface and allowing it to dry. All spectra were analyzed using OriginPro 2021 (OriginLab Corporation, Northampton, Massachusetts, United States). The analysis included baseline correction, smoothing using the Savitzky–Golay filter (polynomial order 2, window size 19), and normalization relative to the amide I band. In the amide region (1750–1490 cm−1), the spectra were deconvoluted into subcomponents using the Lorentz function based on their second derivative spectra. The deconvolution was performed using the Peak Analyzer tool in OriginPro 2021, achieving an R-square value of 0.999.Transmission electron microscopy (TEM)
The samples were prepared using the negative staining technique. 7 μL drop of [0.159–0.164] mM solution was applied on glow-discharge the formvar-carbon film coated 600 mesh copper grids. After 2 minutes of incubation, an excess of the material was blotted. 2% Uranyl acetate was applied for 1 minute. The samples were allowed to dry. Imaging was performed using a transmission electron microscope (TEM) Jeol JEM F-200 was acquired 30 minutes after dissolving and after 48 hours of incubation at 37 °C (98.6 °F). Statistical analysis of the TEM images was used to determine the average width of the nanostructures. All samples exhibit non-normal distributions across several statistical tests (Shapiro-Wilk test, Kolmogorov-Smirnov test and D’Agostino's K-squared test). Central tendency and dispersion for the diameter size distribution were described with median and standard deviation values.ThT fluorescence kinetic assay
Kinetic experiments were performed at 22 °C using a CLARIOstar Plus microplate reader (BMG LABTECH) with a 96-well plate (BRANDplates®). ThT excitation and emission were monitored at 440 nm and 480 nm, respectively. The plate was agitated for 30 seconds every 5 minutes throughout the 46-hour measurement period. The final concentrations used were 50 μM for ThT and 100 μM for the peptides. The experiments were conducted in duplicate, and the resulting fluorescence values were normalized to a 0–1 scale (Fig. S16 and Table S3, ESI†).Turbidity assay
Turbidity of the peptide solutions was assessed by measuring absorbance at 340 nm immediately after dissolution and again after 46 hours. Throughout this 46-hour period, the plate was continuously mixed. The experiments were conducted at 22 °C using a CLARIOstar Plus microplate reader (BMG LABTECH) with a 96-well plate (BRANDplates®). The analyzed absorbance is the average value from four wells, and the standard deviation in all samples was zero (Table S4, ESI†).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
Authors declare no potential conflict of interest.Acknowledgements
The work was financially supported by the National Science Centre, Poland, Grant No. 2017/26/D/ST5/00341 (to M. S.)References
- A. M. Kushner and Z. Guan, Angew. Chem., Int. Ed., 2011, 50, 9026–9057 CrossRef CAS.
- J. N. B. D. Pelin, C. J. C. Edwards-Gayle, A. M. Aguilar, A. Kaur, I. W. Hamley and W. A. Alves, Soft Matter, 2020, 16, 4615–4624 RSC.
- G. L. Eakins, J. K. Gallaher, R. A. Keyzers, A. Falber, J. E. A. Webb, A. Laos, Y. Tidhar, H. Weissman, B. Rybtchinski, P. Thordarson and J. M. Hodgkiss, J. Phys. Chem. B, 2014, 118, 8642–8651 CrossRef CAS PubMed.
- X. Zhang, C. Gong, O. U. Akakuru, Z. Su, A. Wu and G. Wei, Chem. Soc. Rev., 2019, 48, 5564–5595 RSC.
- S. H. Yoo and H.-S. Lee, Acc. Chem. Res., 2017, 50, 832–841 CrossRef CAS.
- C. J. C. Edwards-Gayle and J. K. Wychowaniec, Characterization of Peptide-Based Nanomaterials in Peptide Bionanomaterials, ed. M. A. Elsawy, Springer, Cham, 2023, pp. 255–308 Search PubMed.
- E. Gatto, C. Toniolo and M. Venanzi, Nanomaterials, 2022, 12, 466 CrossRef CAS PubMed.
- A. Khedr, M. A. N. Soliman and M. A. Elsawy, Design Rules for Self-Assembling Peptide Nanostructures in Peptide Bionanomaterials, ed. M. A. Elsawy, Springer, Cham, 2023, pp. 1–52 Search PubMed.
- X.-y Yang, H.-m Cao and X. Li, Mater. Today Commun., 2024, 38, 108563 CrossRef CAS.
- A. Mujeeb and A. F. Miller, Peptide Bionanomaterials Global Market: The Future of Emerging Industry in Peptide Bionanomaterials, ed. M. A. Elsawy, Springer, Cham, 2023, pp. 539–555 Search PubMed.
- E. Mathew, E. Weaver, R. Cazoria-Luna, E. Utomo, E. Larrañeta and D. A. Lamprou, Advanced Manufacturing of Peptide Nanomaterials in Peptide Bionanomaterials, ed. M. A. Elsawy, Springer, Cham, 2023, pp. 335–366 Search PubMed.
- S. Guo, J. Wang, Q. Wang, J. Wang, S. Qin and W. Li, Heliyon, 2024, 10, e26009 CrossRef.
- B. J. Pepe-Mooney and R. Fairman, Curr. Opin. Struct. Biol., 2009, 19, 483–494 Search PubMed.
- Y. Wang, W. Zhang, C. Gong, B. Liu, Y. Li, L. Wang, Z. Su and G. Wei, Soft Matter, 2020, 16, 10029–10045 Search PubMed.
- Y. Lin and C. Mao, Front. Mater. Sci., 2011, 5, 247–265 Search PubMed.
- A. K. Das and P. K. Gavel, Soft Matter, 2020, 16, 10065–10095 Search PubMed.
- M. Szefczyk, N. Szulc, M. Gąsior-Głogowska, A. Modrak-Wójcik, A. Bzowska, W. Majstrzyk, M. Taube, M. Kozak, T. Gotszalk, E. Rudzińska-Szostak and Ł. Berlicki, Nanoscale, 2021, 13, 4000–4015 Search PubMed.
- M. Szefczyk, N. Szulc, M. Gąsior-Głogowska, D. Bystranowska, A. Żak, A. Sikora, O. Polańska, A. Ożyhar and Ł. Berlicki, Soft Matter, 2023, 19, 3828–3840 RSC.
- K. Kulkarni, N. Habila, M. P. Del Borgo and M.-I. Aguilar, Front. Chem., 2019, 7, 70 CrossRef PubMed.
- S. Rinaldi, Molecules, 2020, 25, 3276 CrossRef PubMed.
- M. Szefczyk, Nanoscale, 2021, 13, 11325–11333 RSC.
- S. Kwon, B. Kim, H. K. Lim, K. Kang, S. H. Yoo, J. Gong, E. Yoon, J. Lee, I. S. Choi, H. Kim and H.-S. Lee, Nat. Commun., 2015, 6, 8747 CrossRef CAS.
- T. Katoh, T. Sengoku, K. Hirata, K. Ogata and H. Suga, Nat. Chem., 2020, 12, 1081–1088 CrossRef CAS PubMed.
- M. J. Pandya, G. M. Spooner, M. Sunde, J. R. Thorpe, A. Rodger and D. N. Woolfson, Biochem., 2000, 39, 8728–8734 CrossRef CAS.
- W. S. Horne, J. L. Price and S. H. Gellman, Proc. Natl. Acad. Sci., 2008, 105, 9151–9156 CrossRef CAS.
- P. B. Harbury, T. Zhang, P. S. Kim and T. Alber, Science, 1993, 262, 1401–1407 CrossRef CAS.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef PubMed.
- B. M. Murphy, J. D'Antonio, M. C. Manning and W. Al-Azzam, Curr. Pharm. Biotechnol., 2014, 15, 880–889 Search PubMed.
- J. Zhao and J. Wang, J. Phys. Chem. B, 2015, 119, 14831–14839 CrossRef PubMed.
- V. A. Lorenz-Fonfria and E. Padros, Spectrochim. Acta, Part A, 2004, 60, 2703–2710 CrossRef.
- J. Seo, W. Hoffmann, S. Warnke, X. Huang, S. Gewinner, W. Schollkopf, M. T. Bowers, G. Von Helden and K. Pagel, Nat. Chem., 2017, 9, 39–44 CrossRef.
- J. De Meutter and E. Goormaghtigh, Eur. Biophys. J., 2021, 50, 641–651 CrossRef PubMed.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141–173 CrossRef CAS PubMed.
- D. F. Kreitler, D. E. Mortenson, K. T. Forest and S. H. Gellman, J. Am. Chem. Soc., 2016, 138, 6498–6505 CrossRef CAS.
- T. M. Laue, B. Shah, T. M. Ridgeway and S. L. Pelletier, Computer-aided Interpretation of Analytical Sedimentation Data for Proteins, in Analytical Ultracentrifugation in Biochemistry and Polymer Science, ed. S. E. Harding, J. C. Hortong and A. J. Rowe, Royal Society of Chemistry, Cambridge, U.K., 1992, pp. 90–125 Search PubMed.
- H. Durchschlag and P. Zipper, J. Appl. Crystallogr., 1997, 30, 803–807 CrossRef CAS.
- H. Zhao, R. Ghirlando, G. Piszczek, U. Curth, C. A. Brautigam and P. Schuck, Anal. Biochem., 2013, 437, 104–108 CrossRef CAS PubMed.
- P. Schuck, Biophys. J., 1998, 75, 1503–1512 CrossRef.
- P. Schuck, Biophys. J., 2000, 78, 1606–1619 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01545b |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |