
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine-substituted hydroxyapatite nanoparticles and activation of derived ceramics for range verification in proton therapy
R. Magro
Hernández
 abc,
A.
Muñoz-Noval
cd,
J. A.
Briz
e,
J. R.
Murias
e,
A.
Espinosa-Rodríguez
e,
L. M.
Fraile
e,
F.
Agulló-Rueda
f,
M. D.
Ynsa
ab,
C.
Tavares de Sousa
ab,
B.
Cortés-Llanos
a,
G. García
López
a,
E.
Nácher
g,
V.
García-Tavora
h,
N.
Mont i Geli
i,
A.
Nerio
h,
V. V.
Onecha
e,
M.
Pallàs
i,
A.
Tarifeño
g,
O.
Tengblad
h,
M. Manso
Silván
*ab and
S.
Viñals
be
abc,
A.
Muñoz-Noval
cd,
J. A.
Briz
e,
J. R.
Murias
e,
A.
Espinosa-Rodríguez
e,
L. M.
Fraile
e,
F.
Agulló-Rueda
f,
M. D.
Ynsa
ab,
C.
Tavares de Sousa
ab,
B.
Cortés-Llanos
a,
G. García
López
a,
E.
Nácher
g,
V.
García-Tavora
h,
N.
Mont i Geli
i,
A.
Nerio
h,
V. V.
Onecha
e,
M.
Pallàs
i,
A.
Tarifeño
g,
O.
Tengblad
h,
M. Manso
Silván
*ab and
S.
Viñals
be
aCentro de Microanálisis de Materiales, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: miguel.manso@uam.es
bDepartamento de Física Aplicada e Instituto de Ciencia de Materiales Nicolás Cabrera, Universidad Autónoma de Madrid, 28049 Madrid, Spain
cDepartamento de Física de Materiales, Universidad Complutense de Madrid, 28040 Madrid, Spain
dIMDEA Nanociencia, C/Faraday, 9, 28049 Madrid, Spain
eGrupo de Física Nuclear, Universidad Complutense de Madrid, 28040 Madrid, Spain
fInstituto de Ciencia de Materiales de Madrid, Consejo Superior de Investigaciones Científicas, 28049 Madrid, Spain
gInstitut de Física Corpuscular (IFIC), 46980 Valencia, Spain
hInstituto de Estructura de la Materia, Consejo Superior de Investigaciones Científicas, 28019 Madrid, Spain
iUniversitat Politècnica de Catalunya, 08034 Barcelona, Spain
First published on 15th October 2024
Abstract
Osteosarcoma is a radioresistant cancer, and proton therapy is a promising radiation alternative for treating cancer with the advantage of a high dose concentration in the tumor area. In this work, we propose the use of iodine-substituted hydroxyapatite (IHAP) nanomaterials to use iodine (127I) as a proton radiation tracer, providing access to range verification studies in mineralized tissues. For this purpose, the nanomaterials were synthesized at four iodine concentrations via hydrothermal synthesis. The materials were characterized via different microstructural techniques to identify an optimal high iodine concentration and pure apatite phase nanomaterial. Finally, such pure IHAP powders were shaped and irradiated with proton beams of 6 and 10 MeV, and their activation was demonstrated through subsequent decay analysis. The materials could be integrated into phantom structures for the verification of doses and ranges of protons prior to animal testing and clinical proton therapy treatments of tumors located deep under combined soft and calcified tissues.
1. Introduction
Osteosarcoma (OSA) is one of the most aggressive tumors and has the highest incidence in children and adolescents under 20 years of age.1 This type of tumor presents a high risk of metastasis, and consequently, the standard treatment usually begins with chemotherapy, followed by complete surgical resection, and finally, the tumor area is controlled with radiotherapy (RT).2–4 Although this is the usual treatment, OSA is a neoplasm that responds poorly to radiation because of its high radioresistance.5 In fact, studies using photon beams have reported local control rates of 60% when irradiated with 60 Gy, a dose much higher than that commonly used in breast or prostate cancer (approximately 8 Gy).6Proton therapy (pT) consists of a treatment where the tumor is irradiated with a proton beam, allowing enhancement of the dose received in the region while protecting the surrounding tissues.7 This aspect is extremely beneficial in the treatment of OSA considering the high doses needed. The difference in the deposition of energy against photons is due to the way in which protons interact with matter and how they deposit a dominant fraction of their energy within a restricted range around the Bragg peak.8 The difference in the deposition of energy against photons is due to the way in which protons interact with matter and how the energy deposited per unit of depth is clearly dominant at the Bragg peak. A big advantage with respect to photon irradiation is that the proton beam energy is fully extinct at the end of the Bragg peak, while photons travel beyond tumors irradiating healthy tissues. In the last decade, great efforts have been devoted to studying the effectiveness of pT in OSA treatment, with local control rates of 82% and 72% at three and five years, respectively.9
One of the main uncertainties associated with pT is the deposited dose derived from the range of protons in the tissue.10 This aspect is crucial in bones because of their greater density than soft tissues in the body.11 One of the most attractive approaches for proton range verification is the use of biocompatible contrast agents that, once irradiated, emit different particles in response to the nuclear activation they have undergone. An example of this type of contrast agent is 127I.12 This isotope of iodine has a low energy threshold to open the activation channel, a large effective cross section (200 mb at 10 MeV) and, in addition, is routinely used as a contrast agent for various medical procedures.13 For online verification of the proton range, the induced nuclear reaction is 127I(p,n)127mXe. The half-life of 127mXe is 69.2(9) s, and it decays by gamma deexcitation with two characteristic rays of 124.7 keV and 172.4 keV.14 These gamma rays are emitted with a short enough half-life to assess activation during irradiation.
While most pT preclinical studies are based on cellular irradiation, this research proposes the use of synthetic bone as a mannequin to determine the dose deposited in this type of tissue and obtain information about the range of protons in complex tissues comprising hard and soft materials.15 Hydroxyapatite (HAP) is a calcium phosphate present in human hard tissues with highly specific morphology and composition. It has a hexagonal structure and a stoichiometric Ca/P ratio of 1.67, resembling that of bone apatite.16 This compound is biocompatible and has various applications, such as for bone tissue engineering, orthopedic and dental implant coatings, and drug delivery.17–19 Mimicking what happens with F− substitution in HAP for tooth hardening or natural carbonate integration in biosynthesized HAP (carbonate integrates substituting both OH− and PO4−), several forms of iodine/iodate apatites have been described, in addition to the predicted formable and stable stoichiometric forms.20–22 Despite the tendency of iodine atoms to transform into volatile I2 under heat treatment, apatite structures can effectively trap iodine ions. HAP has been used to confine iodine atoms in its structure, either by replacing OH− with I− or PO4− with IO3− ions,23 as performed for the retention of radioactive 129I byproducts.24 However, because of the high retention rate and reduced volatility of this isotope, more complex matrices containing Pb cations and VO4 molecular anions have been proposed.25
The objective of this work is to synthesize and characterize samples of iodine-substituted HAP (IHAP) containing different concentrations of iodine via a hydrothermal process that inhibits the gas-phase losses induced at high temperatures, as observed via precipitation and sol–gel methods, and with increased I stoichiometry with respect to implanted samples.26–28 These often require post-nucleation thermal annealing, which is less prone to be required in hydrothermal processes, which take place at considerably higher temperatures allowed by high-pressure autoclave confinement. Furthermore, the objective of this study was to test the ability of 127I ceramic constructs fabricated via isostatic pressing as contrast agents for pT in OSA treatments via proton beam-induced gamma rays as a target experiment.
2. Experimental methods
2.1 Iodine-substituted hydroxyapatite synthesis
For the synthesis of the IHAP nanocrystals, the main reagents used were calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), potassium iodate 99% (KIO3), ortho-phosphoric acid 85% (H3PO4), ammonia 25% (NH3) and deionized water as the reaction media. All reactants were acquired from Panreac (ITW Reagents, Spain).The synthesis process was carried out via a hydrothermal method previously designed for substituted apatite and adapted to our hydrothermal processing unit.29,30 Four batches were performed to obtain different concentrations of iodine in each sample, where the only modified variable was the iodine concentration. To produce the samples, 3 main solutions were made: a solution of 1.7853(1) g of Ca(NO3)2·4H2O dissolved in 35.0(1) ml of deionized water; a second solution of 0.0824(1) g of KIO3 in 17.5(1) mL of deionized water; and a third solution of 127.83(1) μL of H3PO4 in 17.5(1) mL of deionized water. After these three solutions were prepared, they were mixed in a hydrothermal reactor, and the pH of the medium was controlled and homogenized via NH3 dilution (754(1) μL of 25% NH3 in 20(1) mL of deionized water) to keep the pH of the reaction medium equal to 10. The pH of the reaction medium was controlled using pH measuring test strips. Table 1 specifies the dosing of the KIO3 reagent used in each of the batches. The autoclave was sealed so that it was completely watertight and placed in a furnace at 150 °C for a minimum of 12 h. After 12 h, the autoclave was removed from the furnace, and the precipitate was recovered by performing a vacuum wash with deionized water via a water tube, a Kitasato and a Büchner funnel. The washing process was carried out 3 times. Finally, the material was placed in a Petri dish and allowed drying under atmospheric conditions.
| KIO3 mass (g) | |
|---|---|
| HAP | 0 |
| HAP-I008 | 0.0824(1) |
| HAP-I050 | 0.5014(1) |
| HAP-I325 | 3.2500(1) |
For irradiation experiments, the synthesized ceramic powders were shaped via a hydraulic press (2000 psi), resulting in a circular tablet with a diameter of 1 cm. This treatment was enough to produce a densified form for manipulation and irradiation, not requiring complex post-processing (such as spark plasma annealing or zeolite compounding), as previously reported to avoid 129I ion release from nuclear waste ceramic forms.31,32
2.2 Characterization of the samples
The different characterization techniques and conditions used to carry out the characterization were as follows:To analyse the morphology of the samples, scanning electron microscopy (SEM) was performed with a Hitachi S-3000 N instrument (20 kV voltage). For the local microstructure, high-resolution transmission electron microscopy (HRTEM) studies were carried out with a JEM-3000F instrument from the national electron microscopy center (CNME-UCM) (a column voltage of 300 kV).33 The powder samples were highly diluted in acetone and dispersed on carbon-coated cellulose membranes assembled on Cu grids. The selected area diffraction pattern was also obtained during the HRTEM measurements to check the local crystallinity of the samples.
The samples were studied via X-ray diffraction (XRD) to determine the global crystalline phases present in the synthesized samples. This study was conducted at SIDI-UAM with a Bruker D8 diffractometer (θ/2θ geometry, Cu anode and no monochromatic (Kα) radiation). The data were analysed with the software Match!
Further characterization via spectroscopic techniques was carried out to determine the presence of iodine in the samples. Vibrational analysis was performed via Raman spectroscopy to identify the relative presence of the molecular ions integrated in the synthesized materials. Raman spectra were obtained via irradiation with an infrared laser (λ = 780 nm) to avoid fluorescence of the sample using a Renishaw Ramascope 2000 spectrometer. The laser power was maintained at a low value to avoid sample damage.
Finally, the samples were analysed via particle-induced X-ray emission (PIXE) using a 2 MeV proton external microbeam to determine the composition of each of the samples quantitatively.34 The data were analysed with GUPIX-NEW software, and the concentration of elements from silicon onwards (i.e., Z ≥ 14) was determined quantitatively, assuming a matrix containing light elements (hydrogen and oxygen) with stoichiometric equivalences.35 The concentrations of phosphorus, calcium and iodine were determined in this way for the samples.
2.3 Irradiation of the samples
The experiment is based on the detection and characterization of the gamma rays emitted by the 127mXe nucleus produced in the proton-induced reaction. For the detection of these gamma rays, a set of LaBr3(Ce) scintillation detectors was used. The activation experiment consists of irradiating the samples with protons of 6 or 10 MeV from an external microbeam. The beam spot was considered punctual since its size was less than 100 μm in diameter. The irradiation lasted 5 minutes, reaching accumulated charges of 1000 nC at 10 MeV and 1500 nC at 6 MeV, which led to an ion density of 1016 cm−2. This regime is, in view of the huge in-depth distribution of the protons (several mm), well below the monolayer regime, which allows predicting a non-significant modification of the bombarded HAP ceramics. The decay curve was acquired for 10 additional minutes. However, owing to the collimation of the beam, strong activation of the beamline occurred. Surrounding the HAP sample, a set of two LaBr3(Ce) scintillator crystals, placed at ∼65 degrees with respect to the beam, were used to detect the gamma rays emitted from the deexcitation of 127mXe. The two LaBr3(Ce) detector crystals were coupled to R9779 Hamamatsu photomultiplier tubes.12 Both crystals have truncated cone shapes, with a height of 38.1 mm and top and bottom diameters of 25.4 mm and 38.1 mm, respectively. The energy resolution of these detectors was better than 4% at 662 keV (energy of calibration with an Eu source). The setup is shown in the picture in Fig. 1. | ||
| Fig. 1 Irradiation and the detector setup at the CMAM. The two LaBr3(Ce) detectors look at the HAP sample at 65° with respect to the proton beam. The two extra HPGe and LaCl3 detectors were not used for the described analyses. | ||
3. Results and discussion
3.1 Microstructure of hydrothermally synthesized nanomaterials
XRD analyses confirmed the presence of the HAP phase in all synthesized samples. However, several differences can be found in the diffractograms of the samples depending on the iodine fraction. The chart in Fig. 2 shows the different phases detected in the diffractograms of each sample. In the pure HAP sample (Fig. 2, bottom), there are small traces of calcium carbonate (CaCO3), characterized by particularly fine spectral peaks.18,36 The presence of this precipitate in the sample can be explained by the presence of carbonate in the water in the hydrothermal reactor, with no concurrence for integration in the structure with the KIO3 solution in this sample. This provoked a reaction with the CO3−2 present in the autoclave, precipitating this phase. The main peaks of the calcium carbonate phase are marked with a square. The peaks of the HAP phase, common to all diffractograms, are identified with circles.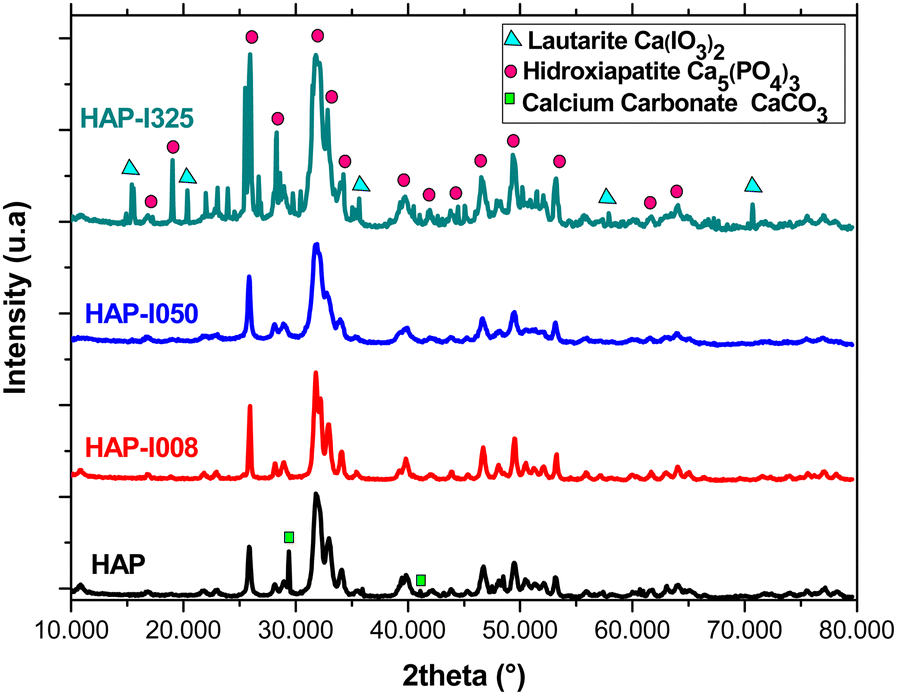 | ||
| Fig. 2 XRD analysis. From bottom to top, diffractograms corresponding to HAP, HAP-I008, HAP-I050 and HAP-I325. | ||
In samples HAP-I008 (Fig. 2, the second diagram from the bottom) and HAP-I050 (Fig. 2, the second diagram from the top), only the HAP phase was observed. Sample HAP-I325 (Fig. 2, top) contains a lautarite (Ca(IO3)2) phase.37 The main peaks of this phase are marked with triangles. Lautarite is an intermediate phase in the production of HAP in the presence of carbonate. Lautarite first precipitates during synthesis and later reacts with the PO4− group.23 Since, in sample HAP-I350, the reactive KIO3 was introduced in saturation, the limiting reagent was H3PO4, limiting the transformation of lautarite and causing this transient phase to remain in the precipitate.
HRTEM analysis revealed that pure HAP crystals are not acicular crystals, as would be expected for these types of compounds, but rather have cylindrical morphology (Fig. 3A and B) with a mean aspect ratio of 3.3 ± 0.5. This change in morphology contrasts with that of typical HAP products, which are usually obtained via precipitation methods.38 However, the hydrothermal synthesis of the samples has led to a material with a less preferred orientation and marked morphology. Although none of the processed samples present the typical acicular crystals of HAP obtained by precipitation, sample HAP-I008 was observed to present morphology closer to what is expected for precipitated HAP (Fig. 3C). An aspect ratio of 11 ± 2 was estimated for this sample.
 | ||
| Fig. 3 HRTEM images of the pure HAP (top pictures A and C) and HAP-I008 (bottom pictures B and D) samples. The arrows indicate (110) planes. | ||
Even so, it is clearly observed that the apatite nanocrystals tend to agglomerate in all the samples, and a preferential growth direction can be identified. The formation of agglomerated crystallites is a common phenomenon in the synthesis of apatites via hydrothermal processes that can be improved by additional mechanical stimuli, alternative solvents, or organic acid addition to modify the zeta potential of the particles during their growth.39–41 When we analyse the images at higher magnifications, we can observe the crystallographic planes of the crystals and determine their interplanar distances. In this case, an interplanar distance of 3.12 Å corresponds to the (110) planes, as shown in Fig. 3B and D with an arrow, which clearly indicates that the preferential growth direction is along the c axis of the HAP crystals.
Fig. 4 shows the SAD pattern of sample HAP-I008 and an overlapping XRD pattern of sample HAP. This graphical composition allows verification that the pattern obtained is consistent with the apatite crystal structure. Both patterns are made by nonlocal diffraction, but the presence of Airy rings is clearly reinforced by local diffraction from selected nanocrystals within the focused aggregate. This confirms the overall good quality of the synthesized nanocrystals.
 | ||
| Fig. 4 Electron diffraction pattern of sample HAP-I008 overlapped with an X-ray diffractogram. | ||
Fig. 5 shows the Raman spectra obtained from the synthesized samples. The key HAP bands can be detected at 580–590 cm−1, assigned to PO4 vibrations; at 530–545 cm−1, attributed to OH ions; and at 960 cm−1, related again to PO4 vibrations.42,43 The global spectral results show that an increase in the amount of iodine used for synthesis reduces the intensity and definition of the PO4 and OH bands. In fact, the higher the concentration of iodine in the sample preparation, the more intense the signal at approximately 780 cm−1 was assigned to the presence of I–O bonds in the structure.44 In sample HAP-I325, owing to the presence of the lautarite phase, the peaks of the I–O bond appear perfectly defined since the presence of iodine is related, in this case, to a pure iodine-containing structure, as already identified by X-ray diffraction (XRD).45 This agrees with the lower intensity of the P–O bond in this sample, at approximately 960 cm−1. For the other samples, the gradual increase in the intensity of the I–O bond can be observed, from the total absence for the HAP sample to a clear wide band for the HAP-I050 sample. The width of the peak is related to the diversity of conformations of the I–O bonds, suggesting multiple substitutions at the PO4 and OH sites.
 | ||
| Fig. 5 Raman spectra of HAP (top), HAP-I008 (second from the top), HAP-I050 (second from the bottom) and HAP-I325 (bottom). The most relevant IO3 and PO4 bands are indicated with dashed lines. | ||
PIXE was selected as a previous reference technique for the detection of engineered or collateral doping in HAP, the study of resorption/ossification kinetics and the classification of biomineralized HAP forms and allows the quantitative identification of the elements present in the samples.46–51Fig. 6 shows the PIXE spectra of different samples in the energy range concerning the peaks of the principal elements (P, Ca and I). The overall composition of each sample can be derived from the data in Table 2. The results show that, in all the samples, the amount of calcium and phosphorus evolves in coherence with the iodine content. The pure HAP sample presents a Ca/P stoichiometric ratio of 1.57, which is slightly below the theoretical value of 1.67. The iodine concentration increases with sample HAP-I008 (in red) and HAP-I050 (in yellow) until it reaches a maximum in sample HAP-I325 (in green). A histogram detailing the concentration of the main elements in different samples clearly illustrates the decrease in the P content in correlation with the increase in the I content, which suggests partial PO4 substitution by IO3 ions.23 The degree of loss of P and increase in I in the HAP-I325 sample are consistent with the detection of the lautarite phase in the XRD analysis. Overall, the iodine concentration increased from 0% (HAP sample) to 6(1)% (HAP-I325) with a decreasing P content, which confirms PO4 ion displacement by IO3. It is also worth noting that the integration of iodate substituting phosphate creates a deficiency of a negative charge in the structure, which points to a decrease of the Ca/(P + I) ratio as the concentration of iodate increases, as confirmed by the results (Table 2, last line). Further analyses by ICP-MS/ICP-OES could contribute to a much more precise determination of the elemental composition and a refinement of the optimal stoichiometry of the preparation around the HAP-I050 sample.
 | ||
| Fig. 6 PIXE spectra of pure HAP, HAP-I008, HAP-I050 and HAP-I325. The characteristic peaks identified are indicated in the spectra, and the derived concentrations are detailed in the table inset. | ||
| (at%) | HAP | HAP-I008 | HAP-I050 | HAP-I325 |
|---|---|---|---|---|
| Ca | 22.5(0.4) | 16.24(0.05) | 17.54(0.08) | 14.88(0.07) |
| P | 13.7(0.4) | 15.1(0.2) | 13.5(0.2) | 12.3(0.2) |
| I | — | 0.8(0.3) | 3.6(0.3) | 5.9(0.3) |
| O | 59 | 65 | 65 | 67 |
| H | 5 | 2 | 0 | 0 |
| Ca/P + I | 1.6 | 1.0 | 1.0 | 0.8 |
3.2 Activation of the samples
The activation of the samples was performed on isostatically pressed tablets of the most relevant sample. Sample selection was made looking for a compromise between the highest I content and the purity of the apatite phase. While the high I content may facilitate the activation, the decrease in the concentration of the original ions, especially Ca and PO4, makes the structure deviate from the real stopping power of HAP. Thus, as a first estimate, the HAP-I050 sample containing the highest I content within a pure apatite phase was selected for the activation experiments. The activation of the HAP-I050 sample was carried out by irradiation with 6- and 10-MeV proton beams for 5 min, with an average current intensity of 7.8(1) nA. Fig. 7 shows the peaks from the two-characteristic gamma rays emitted from the deexcitation of 127mXe, measured with the two LaBr3(Ce) scintillator detectors after excitation with 6 and 10 MeV proton beams (at the bottom and on the top of Fig. 7, respectively). | ||
| Fig. 7 Activation spectra of the HAP-I050 sample with 6- and 10-MeV proton beams (bottom and top frames, respectively). In blue, the decay spectrum acquired the next 10 s after the activation beam was cut. In red, the decay spectrum acquired 10 s after 4 half-lives of decay. The dominant peak at 511 keV arises from the annihilation of the positrons emitted by the activated materials of the beamline, most likely the copper nose at the exit of the beam. | ||
The two peaks at 134.7 and 172.4 keV, obtained by acquiring the spectrum during 10 s after extinction of the proton excitation, demonstrate the effective activation of the iodine integrated in the apatite structure.12 The identification of radiation could also be achieved by the characteristic decay time, acquiring a spectrum after 4 half-lives had occurred (10 s of acquisition after 4 min and 12 s of beam cut-off). One can, in fact, see the bottom curve in each graph with a noticeably decreased intensity for both characteristic peaks in Fig. 7. The different intensities of the decay peaks for the two incident beam energies result from the different activation cross sections, which are greater at 10 MeV than at 6 MeV.
These results indicate that pure apatite preparation with an iodine concentration of 4% can be efficiently activated, suggesting the use of these nanomaterials for the fabrication of bone-like contrast agents, which can be used for proton range verification with phantom structures to reinforce theoretical predictions and provide alternative methods for animal testing.
4. Conclusions
The characterization and activation of an apatite nanomaterial containing 127I have been successfully carried out. The selected hydrothermal process has been studied with different iodine concentrations, reaching an optimal process for the synthesis of pure apatite phase nanocrystals with two levels of integration of iodine. A drastic increase in the iodine concentration results in the presence of a secondary lautarite phase. The nanocrystals maintain the preferential z-axis growth direction characteristic of apatite crystals, as supported by HRTEM, but at a reduced rate with respect to other apatite precipitation preparations. However, the incorporation of iodine promoted the characteristic acicular shape with respect to the undoped samples. The nanocrystals also present a natural tendency to aggregate, which is the characteristic of this type of nanomaterial.The incorporation of iodine by dissolved iodate ions results in competition with both phosphate and carbonate ions, as illustrated by XRD, Raman spectroscopy and PIXE measurements. The former (phosphate) clearly decreases its degree of integration in the apatite structure, and the second (carbonate) is critically reduced, with the disappearance of a secondary calcium carbonate phase identified in the pure hydroxyapatite sample. The iodate ion also integrates the structure substituting OH ions, reaching a maximal atomic percent concentration of 4%.
To satisfy the main objective of the present work, that is, to produce iodine-containing structures that can be activated by proton irradiation, apatite nanocrystals with higher iodine contents have been used to produce compact ceramic tablets by isostatic pressing. These ceramics were irradiated with 6- and 10-MeV proton beams. The activation of the sample was confirmed by the detection of radiation after the beam excitation cut-off. The identification of the 127I(p,n)127mXe nuclear reaction was identified through the characteristic decay energy of 127mXe and the follow-up of the decay at different times after beam extinction. Further improvement of the measurements could be achieved by additionally detecting the neutrons produced in the reaction. The results point to the possibility of designing IHAP ceramic constructs that reproduce real bone affected by OSA and performing real cancer treatments on proton-therapy phantoms including both the synthetic soft tissue and hard tissue parts.52 The insertion of iodine-doped apatite ceramics is especially attractive for verifying the range of protons in the tissue by monitoring, using an array of detectors, the decay radiation induced by the primary proton beam.
Data availability
Data are available on request from the authors.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The current research was funded through grants PID2020-112770RB-C21, PDC2022-133382-I00, PID2020-112770RB-C22 and PID2023-151371OB-C22 by MCIN/AEI/10.13039/501100011033 and FEDER, EU, grant FLASHOnChip (PLEC2022-009256) by MCIN, ASAP-CM (P2022/BMD-7434) from DGI-Comunidad de Madrid and CIPROM/2021/064 from Generalitat Valenciana. Beamtime at the CMAM through experiment EUB004/22 is acknowledged.References
- L. Mirabello, R. J. Troisi and S. A. Savage, Int. J. Cancer, 2009, 125, 229–234 CrossRef CAS PubMed.
- A. Misaghi, A. Goldin, M. Awad and A. A. Kulidjian, SICOT-J, 2018, 4, 12 CrossRef PubMed.
- Z. G. Nie and H. Peng, Oncol. Lett., 2018, 16, 6502–6514 Search PubMed.
- S. Cole, D. M. Gianferante, B. Zhu and L. Mirabello, Cancer, 2022, 128, 2107–2118 CrossRef PubMed.
- R. Schwarz, O. Bruland, A. Cassoni, P. Schomberg and S. Bielack, Cancer Treat. Res., 2009, 152, 147–164 Search PubMed.
- G. N. Machak, S. I. Tkachev, Y. N. Solovyev, P. A. Sinyukov, S. M. Ivanov, N. V. Kochergina, A. D. Ryjkov, V. V. Tepliakov, B. Y. Bokhian and V. V. Glebovskaya, Mayo Clin. Proc., 2003, 78, 147–155 CrossRef PubMed.
- P. Fossati, U. Ricardi and R. Orecchia, Cancer Treat. Rev., 2009, 35, 79–96 CrossRef CAS PubMed.
- A. R. Smith, Phys. Med. Biol., 2006, 51, R491–504 CrossRef CAS PubMed.
- I. F. Ciernik, A. Niemierko, D. C. Harmon, W. Kobayashi, Y. L. Chen, T. I. Yock, D. H. Ebb, E. Choy, K. A. Raskin, N. Liebsch, F. J. Hornicek and T. F. Delaney, Cancer, 2011, 117, 4522–4530 CrossRef PubMed.
- S. España, D. Sánchez-Parcerisa, P. Ibáñez, V. Sánchez-Tembleque, J. M. Udías, V. V. Onecha, A. Gutierrez-Uzquiza, C. M. Bäcker, C. Bäumer, K. Herrmann, P. F. Costa, B. Timmermann and L. M. Fraile, Radiat. Phys. Chem., 2021, 182, 109385 CrossRef.
- T. F. DeLaney, A. V. Trofimov, M. Engelsman and H. D. Suit, Cancer Control, 2005, 12, 27–35 CrossRef PubMed.
- A. Espinosa Rodriguez, V. V. Onecha, V. Sánchez-Tembleque, C. Gutiérrez-Neira, M. García-Díez, P. Ibáñez, S. España, D. Sánchez-Parcerisa, J. M. Udías and L. M. Fraile, Radiat. Phys. Chem., 2021, 185, 109485 CrossRef CAS.
- A. J. Koning, S. Hilaire and M. C. Duijvestijn, AIP Conf. Proc., 2005, 769, 1154–1159 CrossRef.
- W. G. Myers, J. R. Dahl and M. C. Graham, J. Nucl. Med., 1990, 31, 489–492 CAS.
- N. Peters, V. T. Taasti, B. Ackermann, A. Bolsi, C. V. Dahlgren, M. Ellerbrock, F. Fracchiolla, C. Gomà, J. Góra, P. C. Lopes, I. Rinaldi, K. Salvo, I. S. Tarp, A. Vai, T. Bortfeld, A. Lomax, C. Richter and P. Wohlfahrt, Radiother. Oncol., 2023, 184, 109675 CrossRef PubMed.
- S. Teixeira, M. A. Rodriguez, P. Pena, A. H. De Aza, S. De Aza, M. P. Ferraz and F. J. Monteiro, Mater. Sci. Eng. C, 2009, 29, 1510–1514 CrossRef CAS.
- N. Kantharia, S. Naik, S. Apte, M. Kheur, S. Kheur and B. Kale, Bone, 2014, 34, 171 Search PubMed.
- M. Manso-Silvan, M. Langlet, C. Jimenez, M. Fernandez and J. M. Martinez-Duart, J. Eur. Ceram. Soc., 2003, 23, 243–246 CrossRef CAS.
- J. Hernandez-Montelongo, D. Gallach, N. Naveas, V. Torres-Costa, A. Climent-Font, J. P. Garcia-Ruiz and M. Manso-Silvan, Mater. Sci. Eng., C, 2014, 34, 245–251 CrossRef CAS PubMed.
- A. Kuczumow, J. Nowak and R. Chalas, Radiat. Phys. Chem., 2011, 80, 1129–1134 CrossRef CAS.
- W. T. Butler, H. H. Ritchie and A. Bronckers, Dent. Enamel, 1997, 205, 107–117 CAS.
- T. Q. Hartnett, M. V. Ayyasamy and P. V. Balachandran, MRS Commun., 2019, 9, 882–890 CrossRef CAS.
- L. Campayo, A. Grandjean, A. Coulon, R. Delorme, D. Vantelon and D. Laurencin, J. Mater. Chem., 2011, 21, 17609–17611 RSC.
- M. U. Hassan and H. J. Ryu, J. Nucl. Mater., 2019, 514, 84–89 CrossRef CAS.
- F. Audubert, J. Carpena, J. L. Lacout and F. Tetard, Solid State Ionics, 1997, 95, 113–119 CrossRef CAS.
- M. Manso, M. Langlet, C. Jimenez and J. M. Martinez-Duart, Biomol. Eng., 2002, 19, 63–66 CrossRef CAS PubMed.
- M. Manso, M. Langlet, M. Fernandez, L. Vazquez and J. M. Martinez-Duart, Mater. Sci. Eng., C: Biomimetic Mater., Sens. Syst., 2003, 23, 451–454 CrossRef.
- S. Miro, D. Grebille, D. Chateigner, D. Pelloquin, J. P. Stoquert, J. J. Grob, J. M. Costantini and F. Studer, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 227, 306–318 CrossRef CAS.
- S. Kannan, J. H. G. Rocha, S. Agathopoulos and J. M. F. Ferreira, Acta Biomater., 2007, 3, 243–249 CrossRef CAS PubMed.
- R. Pulido, N. Naveas, T. Graber, R. J. Martin-Palma, F. Agullo-Rueda, I. Brito, C. Morales, L. Soriano, L. Pascual, C. Marini, J. Hernandez-Montelongo and M. M. Silvan, Dalton Trans., 2021, 50, 10765–10778 RSC.
- A. Coulon, D. Laurencin, A. Grandjean, S. Le Gallet, L. Minier, S. Rossignol and L. Campayo, J. Eur. Ceram. Soc., 2016, 36, 2009–2016 CrossRef CAS.
- Y. Watanabe, T. Ikoma, H. Yamada, Y. Suetsugu, Y. Komatsu, G. W. Stevens, Y. Moriyoshi and J. Tanaka, ACS Appl. Mater. Interfaces, 2009, 1, 1579–1584 CrossRef CAS PubMed.
- M. Kars, A. Gómez-Herrero, A. Rebbah and L. C. Otero-Diáz, Mater. Res. Bull., 2009, 44, 1601–1607 CrossRef CAS.
- A. Redondo-Cubero, M. Borge, N. Gordillo, P. Gutiérrez, J. Olivares, R. Casero and M. Ynsa, Eur. Phys. J. Plus, 2021, 136, 175 CrossRef.
- J. Campbell, N. Boyd, N. Grassi, P. Bonnick and J. Maxwell, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 3356–3363 CrossRef CAS.
- J. Kawano, N. Shimobayashi, A. Miyake and M. Kitamura, J. Phys.: Condens. Matter, 2009, 21, 425102 CrossRef PubMed.
- S. Ghose, C. Wan and O. Wittke, Acta Crystallogr., Sect. B: Struct. Sci., 1978, 34, 84–88 CrossRef.
- I. Mobasherpour, M. S. Heshajin, A. Kazemzadeh and M. Zakeri, J. Alloys Compd., 2007, 430, 330–333 CrossRef CAS.
- R. E. Riman, W. L. Suchanek, K. Byrappa, C. W. Chen, P. Shuk and C. S. Oakes, Solid State Ionics, 2002, 151, 393–402 CrossRef CAS.
- X. Y. Guo and P. Xiao, J. Eur. Ceram. Soc., 2006, 26, 3383–3391 CrossRef CAS.
- G. Karunakaran, G. S. Kumar, E. B. Cho, Y. Sunwoo, E. Kolesnikov and D. Kuznetsov, Ceram. Int., 2019, 45, 970–977 CrossRef CAS.
- W. P. Griffith, J. Chem. Soc. A, 1970, 286–291 RSC.
- D. G. A. Nelson and J. D. B. Featherstone, Calcif. Tissue Int., 1982, 34, S69–S81 Search PubMed.
- M. Zhang, E. R. Maddrell, P. K. Abraitis and E. K. H. Salje, Mater. Sci. Eng. B, 2007, 137, 149–155 CrossRef CAS.
- E. Alici, T. Schmidt and H. D. Lutz, Z. Anorg. Allg. Chem., 1992, 608, 135–144 CrossRef CAS.
- E. L. Solla, P. GonzaleZ, J. Serra, S. Chiussi, B. Leon and J. García-López, Appl. Surf. Sci., 2007, 254, 1189–1193 CrossRef CAS.
- E. Jallot, J. L. Irigaray, H. Oudadesse, V. Brun, G. Weber and P. Frayssinet, Eur. Phys. J.: Appl. Phys., 1999, 6, 205–215 CrossRef CAS.
- E. Jallot, J. L. Irigaray, G. Weber and P. Frayssinet, Surf. Interface Anal., 1999, 27, 648–652 CrossRef CAS.
- G. Weber, G. Robaye, F. Braye, H. Oudadesse and J. L. Irigaray, Nucl. Instrum. Methods Phys. Res., Sect. B, 1994, 89, 200–203 CrossRef CAS.
- R. K. Dutta, R. B. Roijers, P. H. A. Mutsaers, P. Pauwels, J. J. M. de Goeij and G. J. van der Vusse, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 231, 257–262 CrossRef CAS.
- E. Jallot, H. Benhayoune, G. Weber, G. Balossier and P. Bonhomme, J. Phys. D: Appl. Phys., 2000, 33, 321–326 CrossRef CAS.
- S. Vinals, G. Garcia, R. Magro and M. Manso, J. Instrum., 2023, 18, C03025 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |