
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous photoactivation of a fluoroquinolone antibiotic and nitric oxide with fluorescence reporting†
Tassia J.
Martins‡
a,
Cristina
Parisi‡
a,
Juliana Guerra
Pinto
b,
Isabelle de Paula Ribeiro
Brambilla
b,
Barbara
Melilli
c,
Danilo
Aleo
c,
Juliana
Ferreira-Strixino
b and
Salvatore
Sortino
 *a
*a
aPhotoChemLab, Department of Drug and Health Sciences, University of Catania, I-95125, Italy. E-mail: ssortino@unict.it
bLaboratory of Photobiology Applied to Health, Research and Development Institute, University of Vale do Paraíba, Urbanova I-2911, Brazil
cMEDIVIS S.r.l., Tremestieri Etneo, 95030 Catania, Italy
First published on 9th July 2024
Abstract
The achievement of smart pharmaceuticals whose bioactivity can be spatiotemporally controlled by light stimuli is known as photopharmacology, an emerging area aimed at improving the therapeutic outcome and minimizing side effects. This is especially attractive for antibiotics, for which the inevitable development of multidrug resistance and the dwindling of new clinically approved drugs represent the main drawbacks. Here, we show that nitrosation of the fluoroquinolone norfloxacin (NF), a broad-spectrum antibiotic, leads to the nitrosated bioconjugate NF–NO, which is inactive at the typical minimum inhibitory concentration of NF. Irradiation of NF–NO with visible blue light triggers the simultaneous release of NF and nitric oxide (NO). The photouncaging process is accompanied by the revival of the typical fluorescence emission of NF, quenched in NF–NO, which acts as an optical reporter. This permits the real-time monitoring of the photouncaging process, even within bacteria cells where antibacterial activity is switched on exclusively upon light irradiation. The mechanism of photorelease seems to occur through a two-step hopping electron transfer mediated by the lowest triplet state of NF–NO and the phosphate buffer ions or aminoacids such as tyrosine. Considering the well-known role of NO as an “unconventional” antibacterial, the NF–NO conjugate may represent a potential bimodal antibacterial weapon activatable on demand with high spatio-temporal control.
Introduction
Multidrug resistance (MDR) in bacteria is the prime example of bacterial adaptation and the pinnacle of evolution and remains one of the major public health threats of the 21st century.1–3 This concern and the dwindling of new clinically approved antibacterial drugs (only two new classes of antibiotics have been introduced into the clinic over the last two decades), represent the main obstacles to successfully treating bacterial diseases.4 Another critical issue that makes the above scenario even worse is the occurrence of severe adverse off-target effects.5 They arise especially for long-term antibacterial treatments and eventually can lead to autoimmune disorders, obesity and allergy.6–8 Therefore, developing novel therapeutic approaches to fight MDR and reduce the exposure to antibiotic drugs in the body is desperately needed and represents one of the most challenging goals in human health care.9 In this regard, the emerging field of photopharmacology holds high potential to improve the therapeutic outcome while minimizing the off-target side effects.10–13 Antibiotic photopharmacology is based on the temporary inactivation of conventional antibiotics by their covalent linking with a photoresponsive moiety. This component can be designed to operate either irreversibly or reversibly. In the former case it is a photocleavable group, namely a photocage, which liberates the antibiotic under its active form upon light excitation. In the latter case it is a photoisomerizable component, namely an optical photoswitch, which remains always linked to the antibiotic, permitting the activity of the whole molecular construct to be reversibly switched on and off using light sources of different wavelengths.10–13 In both cases, using light as a remote activation mechanism permits the biological activity of the antibiotic to be spatio-temporally controlled with high precision. Light is a non-invasive stimulus, easy to manipulate and ensures the confinement of the site of action of the photoactivatable drug at the illuminated area by the appropriate positioning of the light beam and the accurate control of the therapeutic doses by tuning the light intensity and the irradiation time.14 Compared with the better-known antibacterial photodynamic therapy (PDT),15 based on the generation of the highly cytotoxic singlet oxygen,16 that has already found applications in clinics,17 the above-mentioned photopharmacological approaches are not dependent on the presence of oxygen, representing a significant advantage to also treating anaerobic bacteria, where PDT fails.Fluoroquinolones (FQs) are a successful class of broad-spectrum, synthetic antibiotics.18,19 Although widely used for treating general bacterial infections, FQs have seen a significant rise in antibiotic-resistant bacterial strains.20 Besides, several safety concerns regarding their therapeutic use are still being raised by the FDA.21 FQs have also been demonstrated to induce undesired photosensitivity side effects, including phototoxicity and photocarcinogenesis, upon undesired and uncontrolled environmental light absorption.22–25 These processes are primarily associated with photodefluorination reactions,26–29 a very uncommon event in the arena of fluoroaromatics due to the strength of the C–F bond (dissociation energy ca. 523 kJ mol−1).30
Photopharmacological approaches have successfully faced the above limitations by controlling the antibacterial activity of FQs, exploiting both photoswitches and photocages activatable by biocompatible visible light.31–40
In the frame of novel antibacterial modalities, nitric oxide (NO) represents a promising alternative to conventional antibiotic drugs. Besides playing a multifaceted role in the bioregulation of vital functions in living bodies,41,42 this inorganic free radical has been demonstrated to be an excellent antibacterial with a broad spectrum of action.43–45 Interestingly, NO exhibits important advantages over conventional antibiotics, such as (i) absence of MDR,46 (ii) multi-target reactivity,47 (iii) confinement of its cytotoxic action over distance <200 μm, as a result of its half-life of ca. 1 s.41
Due to its transient nature, the antibacterial effects of NO are strictly dependent on its generation site and, analogously to other therapeutics, also from its doses.48 This dependence has made the NO precursors activatable by light stimuli, usually named NO photodonors (NOPDs), appealing given the high spatiotemporal control light-triggering offers.49–52
Based on these considerations, in this contribution, we devised a smart bioconjugate in which a FQ antibacterial is temporarily inactivated by integrating a nitroso group within its molecular skeleton. This work aims to exploit the FQ chromophore as a photocage capable of generating NO and simultaneously restoring the FQ in its active form upon light excitation. Besides being motivated by the critical need to achieve novel photopharmaceuticals, the present work has been further encouraged by merging our ongoing interest in developing NOPDs49,53,54 and our past interest in studying the photoreactivity of FQs.29,55–59 As a proof of concept, we focused herein on norfloxacin (NF), a FQ used in the treatment of several bacterial diseases, including urinary tract infections in humans,60 enteritis in dogs,61 and chronic respiratory disease in chickens.62 This choice is not random but based on the following: (i) the nitroso-derivative (NF–NO) formed by biotransformation has been reported to have minimum inhibitory concentrations (MICs) higher than that of NF for several different bacteria,63 (ii) the photochemical behaviour of NF has been well elucidated.58,64,65
Here, we show that the nitroso-derivative NF–NO, in which both NF and NO are reciprocally caged, is inactive towards ATCC S. aureus under dark conditions at a concentration in which NF is active (Scheme 1). Irradiation of NF–NO with visible blue light completely suppresses the photodefluorination, the main photodegradation pathway of NF and common to many FQs, and, in contrast, triggers the release of NO liberating NF in its active form. It is also shown that the characteristic fluorescence emission of NF is quenched in the NF–NO and restored after the release event (Scheme 1). This makes NF a proper optical reporter, permitting to follow its release and that of the equivalent amount of NO, in real-time, even within bacteria cells. Insights into the photorelease mechanisms are also reported.
 | ||
| Scheme 1 Molecular structure of the most abundant prototropic species of NF–NO and NF present at physiological pH of 7.4. Irradiation of the inactive and low fluorescent NF–NO leads to the simultaneous generation of NO and the highly fluorescent NF. | ||
Results and discussion
Spectroscopic properties and photolysis
NF–NO was easily obtained by nitrosation of NF in one step (see ESI†). Fig. 1 shows the spectroscopic features of NF–NO and, for the sake of comparison, those of NF in phosphate buffer at neutral pH. Like other structurally related FQs,65 due to the ionization of the carboxylic group and the 4′-amino of the piperazinyl ring, NF may exist in three different prototropic forms in aqueous solution depending on pH and possessing distinct absorption and emission properties.57 Given that the isoionic pH is ca. 7.4,28 the zwitterionic form dominates at physiological pH (see Scheme 1). Nitrosation of the piperazinyl ring makes the related nitrogen less basic and thus, it is non-protonated at physiological pH. Therefore, NF–NO presents as an anion under these experimental conditions (Scheme 1).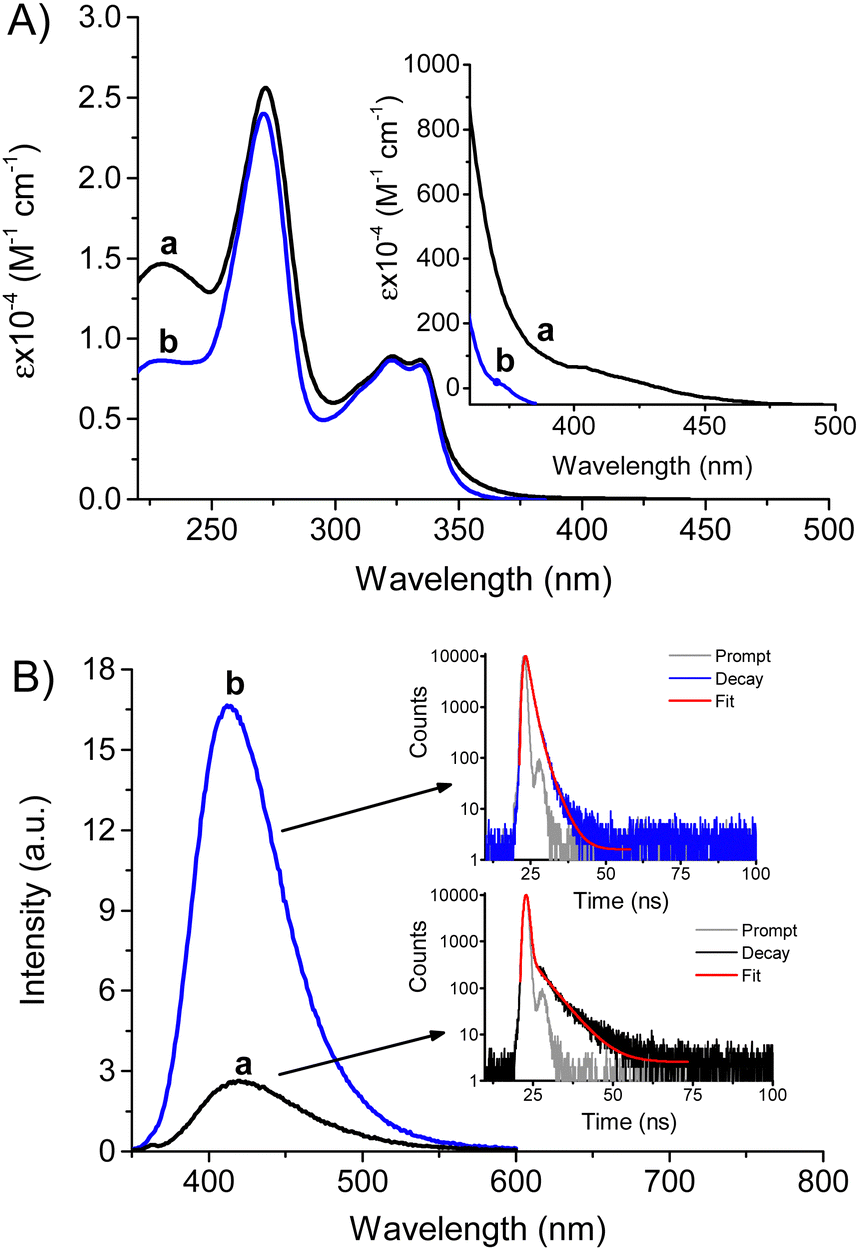 | ||
| Fig. 1 (A) Absorption spectra of NF–NO (a) and NF (b) in PBS solution (10 mM, pH 7.4). The inset shows a magnified view of the absorption region beyond 360 nm. (B) Fluorescence emission spectra (λexc = 324 nm) of NF–NO (a) and NF (b) in PBS solution (10 mM, pH 7.4), T = 25 °C. The insets show the fluorescence decay (λexc = 370 nm; λem = 415 nm) and the related bi-exponential fitting. | ||
The absorption spectra shown in Fig. 1A show that despite the spectral profiles of NF–NO changes only slightly if compared to NF, it exhibits a new absorption band in the UV region at 230 nm and, although not very intense, an extension of its absorption in the visible region well beyond 400 nm (see inset Fig. 1A). This latter feature permits NF–NO excitation with visible blue light (vide infra).
Similar to other FQs,65NF shows an intense blue emission at physiological pH characterized by fluorescence quantum yield ΦF = 0.11 and a bi-exponential decay with a dominant component (ca. 90%) having a lifetime τ = 1.5 ns (spectrum b and related inset in Fig. 1B). This emission is significantly quenched in NF–NO (ΦF = 0.015) showing a red-shift of the emission maximum of ca. 15 nm and a shortening of the lifetime to τ = 0.5 ns (spectrum a and related inset in Fig. 1B). NF–NO remains stable for at least one month in the dark under these experimental conditions, as proven by the unaltered absorption and emission spectra.
As anticipated, the absorption of NF–NO extending in the visible region beyond 400 nm permits excitation of this compound with blue light. Fig. 2A reports the absorption spectral changes observed upon blue light irradiation of an air-equilibrated solution of NF–NO. They show the bleaching of all the main absorption bands, which is more evident in the band at 230 nm. Besides, the band at 275 nm slightly shifted to the blue. This spectral evolution is in line with the formation of NF as a stable photoproduct, which is characterized by a less intense absorption of the 230 nm band and absorption maximum of the 275 nm band blue-shifted if compared to NF–NO (see spectra a and b in Fig. 1A). Accordingly, the evolution of the fluorescence emission spectra upon irradiation shows a significant revival of the blue emission accompanied by a blue-shift of the emission maximum from 425 nm to 411 nm, all features typical of the NF fluorophore (Fig. 2B). The high fluorescence contrast in the samples before and after irradiation is also visible even at the naked eye (see images in Fig. 2B).
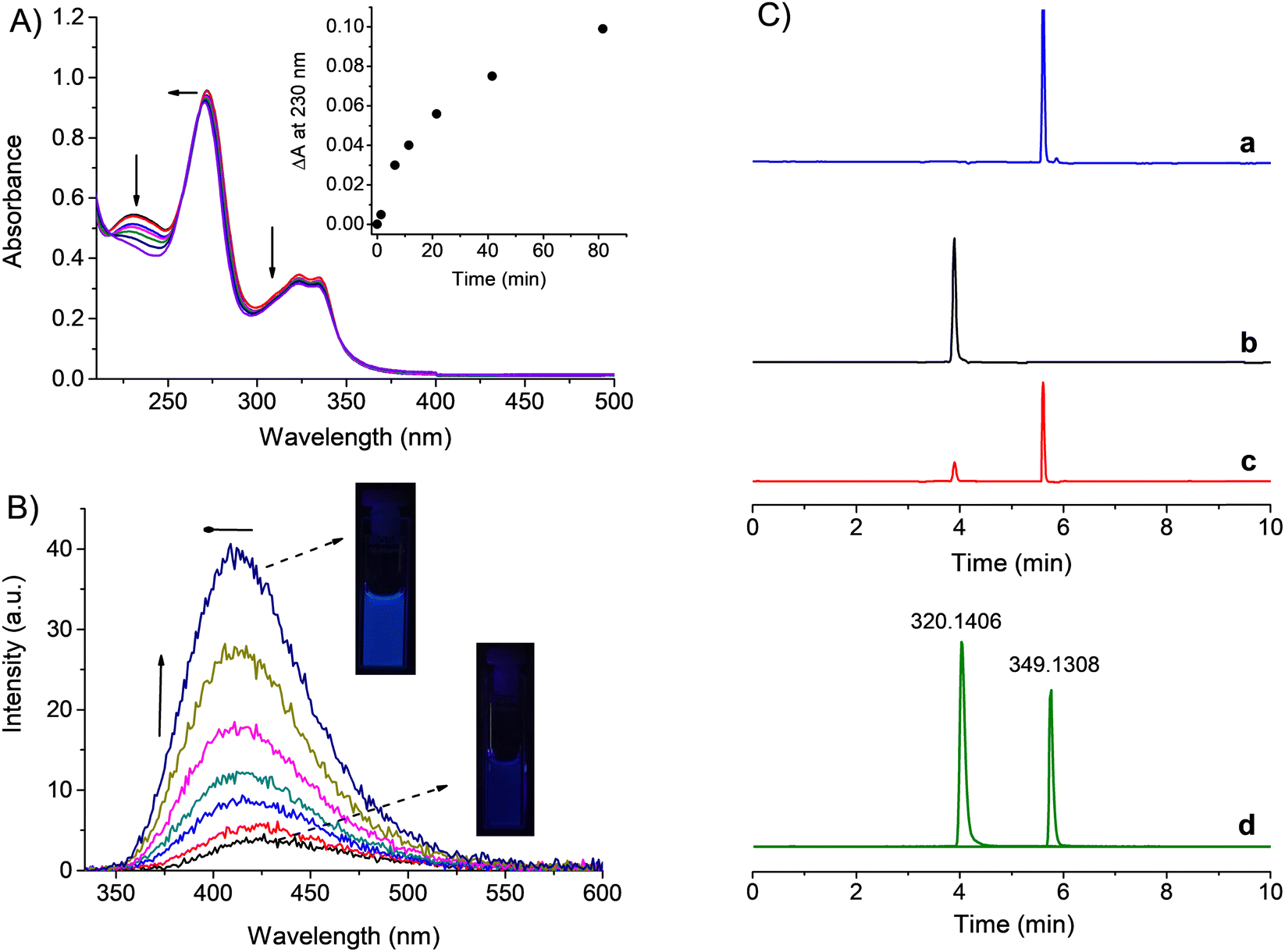 | ||
| Fig. 2 (A) Absorption spectral changes observed upon exposure of a PBS solution (10 mM, pH 7.4) of NF–NO (40 μM) at λexc = 420 nm (ca. 20 mW cm−2) for time intervals from 0 to 81 min. The arrows indicate the course of the spectral profile with the illumination time. The inset shows the difference absorbance changes at λ = 230 nm. (B) Evolution of the fluorescence emission spectra corresponding to the sample of (A) and recorded at λexc = 324 nm. The inset shows the actual images of the solution acquired before and after the photolysis. (C) UHPLC traces related to solutions of NF–NO (a), NF (b) and NF–NO after 15 min irradiation at λexc = 420 nm (ca. 20 mW cm−2) (c) recorded at λ = 278 nm; (d) shows UHPLC traces recorded at m/z values of NF and NF–NO. T = 25 °C. | ||
UHPLC-MS analysis was then carried out with the authentical NF–NO and NF samples. The chromatograms reported in Fig. 2C unambiguously confirmed this latter as the main stable photoproduct.
NF photouncaging implies, of course, the photogeneration of stoichiometric amounts of NO. Direct and indirect methods demonstrated photogeneration of this radical species through an ultrasensitive NO electrode and the typical diaminonaphthalene (DAN) assay (see ESI†), respectively. Amperometric monitoring allows NO to be directly detected with nM concentration sensitivity. As shown in Fig. 3A, NF–NO is stable in the dark but releases NO upon blue light excitation. According to an exclusively photoregulated process, NO generation stops as the light is turned off and restarts again as the illumination is switched on. Besides, the DAN assay, one of the most sensitive and selective fluorescence-based methods for indirect NO detection as nitrite (the main oxidation product of NO), showed the formation of fluorescence emission and excitation spectra typical for the fluorescent probe after its reaction with nitrite, only in the case of the irradiated samples (Fig. 3B).
 | ||
| Fig. 3 (A) NO release profile observed for an air-equilibrated PBS solution (10 mM, pH 7.4) of NF–NO (40 μM) upon alternate cycles of light irradiation at λexc = 405 nm. T = 25 °C. (B) Fluorescence emission (solid lines) and excitation (dotted lines) spectra obtained after fluorimetric assay of PBS solutions (10 mM, pH 7.4) of NF–NO (40 μM) before (a) and (b) and after (c) and (d) 30 min irradiation with blue light at 420 nm. The emission wavelength for the excitation spectra was 410 nm whereas the excitation wavelength for the emission spectra was 360 nm. T = 25 °C. | ||
An important aspect to be highlighted is that the high fluorescence contrast ratio between NF and NF–NO (see Fig. 2B) makes the former a suitable optical self-reporter,53 giving readable and real-time information about the amount of NF and NO photogenerated. The quantum yield for the uncaging process, Φu, was estimated to be ca. 0.1. Although affected by a quite large error due to the low absorbance value at the excitation wavelength, the order of magnitude of Φu is worthy of note. In fact, despite the low absorption of NF–NO in the blue region, the high efficiency of the photouncaging process permits the release of sufficient amounts of NF and NO (μM range) within moderate time irradiation. Note that the absorption and emission photolysis profiles reported in Fig. 2 were very similar in the N2-saturated solution (data not shown), suggesting that the efficiency and nature of the photochemical reaction are oxygen-independent.
Photouncaging mechanism
To elucidate the photouncaging mechanism, we carried out time-resolved absorption measurements using nanosecond laser flash photolysis, a powerful tool to gain insights into the spectroscopic and kinetic properties of transient intermediates produced by light excitation. However, before discussing these results, for the sake of clarity, we consider it helpful to briefly recall the critical aspects of the photodegradation mechanism of NF that was previously reported.63–65 This FQ undergoes photodefluorination mediated by the lowest excited triplet. In the presence of a phosphate buffer, the loss of fluoride is triggered by the formation of the NF radical anion generated by an unexpected photoinduced electron transfer between the NF triplet state and the hydrogen phosphate dianion of the buffer, which acts as a reducing agent. The NF radical anion is well detectable with laser flash photolysis because it is characterized by a longer lifetime and absorption maximum in a different spectral region than the triplet precursor (Scheme 2A).63–65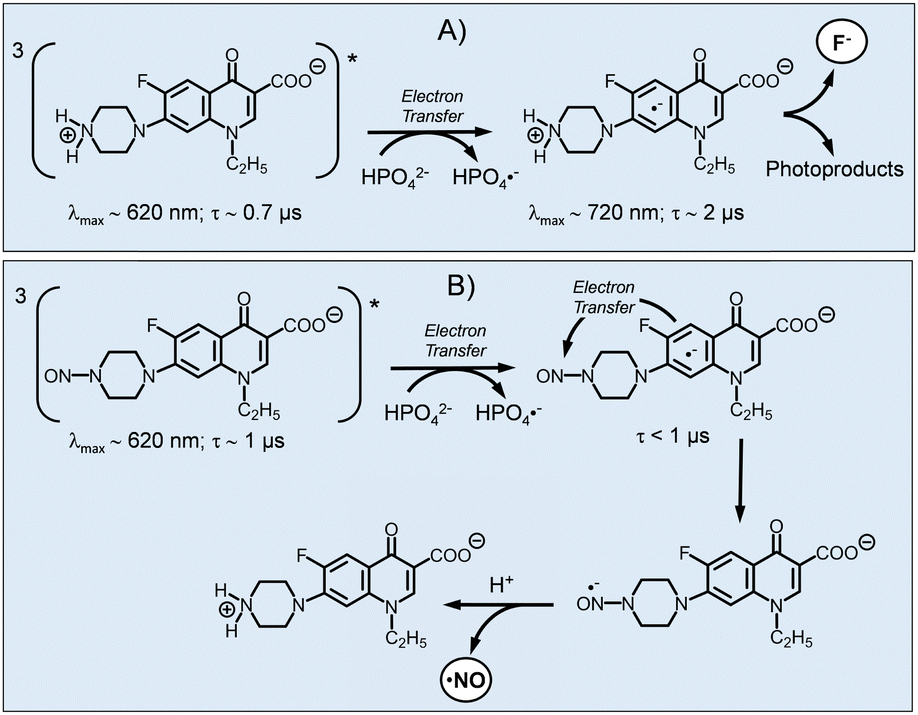 | ||
| Scheme 2 (A) Photodefluorination mechanism of NF in PBS solution at pH 7.4. (B) Proposed mechamism for NF and NO photouncaging from NF–NO in PBS solution at pH 7.4. | ||
In the case of NF–NO, a different scenario was observed. Fig. 4 shows the transient spectra observed with elapsing time after the initial excitation laser pulse of an NF–NO solution in PBS. The spectrum taken at the shortest delay time shows a maximum at ca. 620 nm, which agrees with the features of the lowest triplet state of NF. The spectral evolution reveals that no new transient species are formed concurrently with the triplet decay. Therefore, there is no evidence for the radical anion with a maximum of 720 nm. The triplet state decays mono-exponentially with a lifetime τ ∼ 1 μs (inset Fig. 4) in reasonably good agreement with triplet decay reported for NF. Analogously to NF, this decay was dependent on the PBS with bimolecular rate constants kq(PBS) ∼ 8 × 107 M−1 s−1. This value is similar to that reported for NF63 and confirms the occurrence of the bimolecular photoinduced electron transfer between the NF–NO triplet and the phosphate ions.
 | ||
| Fig. 4 Transient absorption spectra observed 0.1 μs (■), 0.5 μs (○) and 8 μs (▲) after 355 nm laser excitation (E355 ∼ 10 mJ pulse−1) of N2-saturated PBS solution (10 mM, pH 7.4) of NF–NO (40 μM). The inset shows the decay trace monitored at 610 nm and the related first-order fitting. | ||
Note that, the photogeneration of NO cannot be the result of a direct excitation of the nitrosated piperazinyl moiety since it does not absorb the blue excitation light. Therefore the photouncaging mechanism illustrated in Scheme 2B and discussed in the following is proposed.
Nitrosation of the piperazinyl ring is reasonably expected not significantly to change neither the oxidation potential nor the energy of the triplet state with respect to the values reported for NF. Therefore, also in this case, the triplet quenching by PBS must necessarily involve the formation of NF–NO radical anion. We believe that the lack of observation of this species might be attributable to a second, intramolecular electron transfer between this radical anion and the nitroso group leading to a radical anion centred on the nitroso group and occurring on a time scale faster than the triplet decay. This process is expected to be exergonic or almost thermoneutral (the reduction potentials of aliphatic nitrosamine are in the range −0.5 V to −1.5 V vs Ag/AgCl, depending on the conditions66,67 whereas that of the aromatic core of NF is reported to be ca. −1.4 V vs. Ag/AgCl63) and can be also favoured by the close spatial proximity between the FQ core and the nitroso-piperazinyl moiety. The formation of NO-centred radical anion can be reasonably responsible for the release of NO and, after protonation, the restoring of NF. This hypothesis is supported by the fact that radical anions of nitroso-derivatives possess a lower dissociation energy of the N–NO bond than their neutral form, encouraging fast NO detachment.68 This proposal accords well with the negligible photogeneration of NO observed when photolysis was carried out in neat water in the absence of PBS. This finding confirms the key role of the buffer as external reducing agent in the photouncaging process and rules out a photouncaging mechanism involving the formation of a NO-centered radical anion by an intramolecular electron transfer between the photoexcited FQ core and the nitroso-piperazinyl moiety.
The mechanism of Scheme 2B implies a photodecaging strictly dependent on the presence of phosphate buffer as reducing agent as initiator of the stepwise process. This condition may, in principle, represent a not negligible limitation. Therefore, we wondered if the photouncaging process can be triggered by biological reducing agents analogously to what observed for phosphate ions. To this end, we devised laser flash photolysis experiments with NF–NO in neat water in the absence of PBS but in the presence of a tyrosine (TyrOH), used as representative example of an amino acid commonly present in biological samples. The rationale behind this experiment is that TyrOH not only is a better reducing agent than phosphate ion but, if an electron transfer with the NF–NO triplet occurs, it is expected to generate a tyrosyl radical (TyrO˙). Analogously to others phenoxyl radicals, this species is well detectable by transient spectroscopy due to its absorption around 400 nm (where the triplet absorption of NF–NO is negligible) and the decay in the tens of microseconds time regime,69 and would elegantly confirm the proposed mechanism.
Fig. 5A shows the transient spectra taken at 2 different delay time with respect to the initial laser pulse. They clearly show that the decay of the NF–NO triplet at 620 nm is accompanied by a build-up of a new absorption showing the characteristic features of the TyrO˙ radical70 which decays via second-order kinetic on longer temporal scale. As shown in the Fig. 5B, although affected by the fluorescence emission on this short time scale, the decay of the triplet monitored at 620 nm matches very well the build-up of the tyrosyl radical at 390 nm with an observed rate constant kobs ∼ 1.6 × 106 s−1. This confirms that TyrO˙ radical is generated by intermolecular electron transfer between NF–NO triplet and TyrOH and, according with literature, after the concomitant fast deprotonation of the very acid TyrOH˙+ radical cation (pKa = −2) (see inset Fig. 5A).71
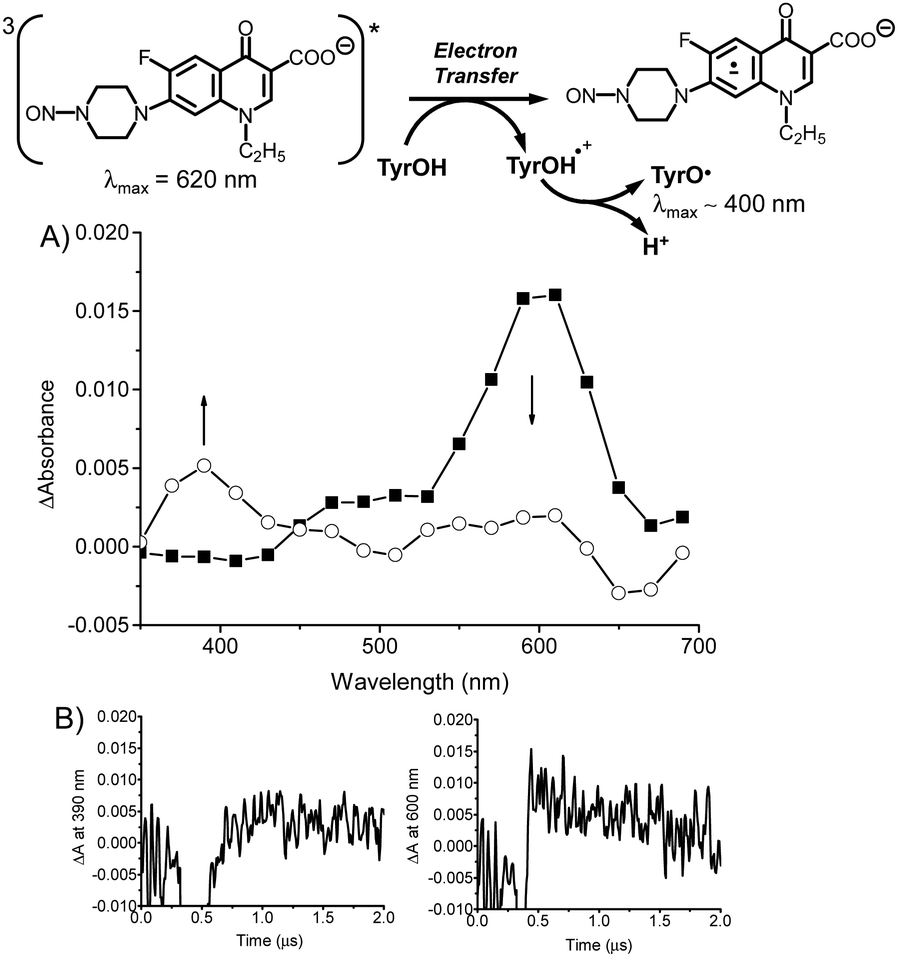 | ||
| Fig. 5 (A) Transient absorption spectra observed 0.2 μs (■) and 1.5 μs (○) after 355 nm laser excitation (E355 ∼ 10 mJ pulse−1) of N2-saturated aqueous solution of NF–NO (40 μM) in the presence of TyrOH (800 μM). T = 25 °C. The inset illustrates the bimolecular electron transfer involved. (B) Build-up (left) and decay (right) traces monitored at 390 and 600 nm, respectively. | ||
Based on this value and the concentration of the TyrOH used in the experiments, a bimolecular quenching constant for this electron transfer process of kq(TyrOH) ∼ 1.9 × 109 M−1 s−1 can be estimated. Note that, this value is more than one order of magnitude higher than that observed for the phosphate buffer (kq(PBS) ∼ 8 × 107 M−1 s−1) and it is in excellent agreement with the highly favoured thermodynamic associated to this process if compared with the almost thermoneutral quenching by phosphate (the potential of the couple TyrOH˙+/TyrOH70,71 is at least 0.5 V lower than that of the couple HPO4˙−/HPO42−).
After this initial electron transfer involving TyrOH, the uncaging process is expected to proceed in the same way as reported in Scheme 2. This was confirmed by the steady-state photolysis experiments carried out in the presence of TyrOH. The intense absorption of TyrOH below 270 nm did not allow to follow the bleaching of the 230 nm band typical of NF–NO (see Fig. 2A). However, Fig. S7 (ESI†) shows the fluorescence intensity increase accompanied by the shift of the emission maximum upon photolysis with blue light. This photolysis profile is the same as observed in the presence of PBS (see Fig. 2B), and it provided unambiguous evidence for the uncaging of NF under these conditions. In parallel, the trapping experiment performed with the DAN confirmed the simultaneous generation of NO (Fig. S8, ESI†).
The last quantitative aspect that deserves to be highlighted is the reliability of the value of ca. 0.1 estimated for Φu (vide supra) for a triplet-mediated reaction. If this is the case, the quantum yield for the triplet formation, ΦT, should be >Φu. To this end, we determined the ΦT value by performing a laser power effect on the top ΔA of the NF–NO triplet and, for comparison, of NF, for which a value of ΦT > 0.5 has been reported. The slopes of the data set reported in Fig. S9 (ESI†) are proportional to the product ΦT × εT–T, where εT–T is the triplet molar absorption coefficient. By taking into account that the solutions are iso-absorbing at the excitation wavelength and that large changes in the εT–T are reasonably unlikely, being substantially unchanged the band profiles, a ΦT > 0.17 for NF–NO can be directly estimated by the different slopes of the straight lines obtained from the linear portion of the plots.
Biological studies
Previous studies have reported that the MIC of NF–NO is 2 to 38-fold higher than that of NF for several different bacteria.63 This was also confirmed by preliminary biological studies performed in this work in which NF–NO and, for comparison, NF were tested against ATCC S. aureus in the dark and under blue light irradiation. As shown in Table 1, the MIC value of NF–NO is ca. 4-fold higher than that found for NF. Interestingly, while the MIC value of NF did not change upon irradiation, that of NF–NO reduced by about 50% after only 10 min irradiation, according to the release of the more active NF and NO.| Sample | MIC (μg mL−1) dark | MIC (μg mL−1) light |
|---|---|---|
| NF | 0.39 | 0.39 |
| NF–NO | 1.56 | 0.78 |
As emphasized above, another remarkable point of this work is the possibility of real-time monitoring of the release process thanks to the highly fluorescent NF, photoliberated by the low fluorescent NF–NO, which acts as self-reporting. To demonstrate that the reduction of the MIC of NF–NO is due to the photouncaging of NF, we carried out fluorescence microscopy imaging experiments with a 24-hour biofilm of ATCC strain of S. aureus assembled on a glass coverslip and incubated with NF–NO. Fig. 6 shows no detectable fluorescence in the non-irradiated sample (panel a). In contrast, a significant blue emission, typical for NF, appears after irradiation of the sample (panel b), analogously to what was observed for the PBS solution in the absence of bacteria (see Fig. 2B and related pictures). Moreover, since the emission of NF presents a tail that extends up to the green region (see Fig. 2B), the fluorescence in bacteria can also be monitored in the green channel (panel c).
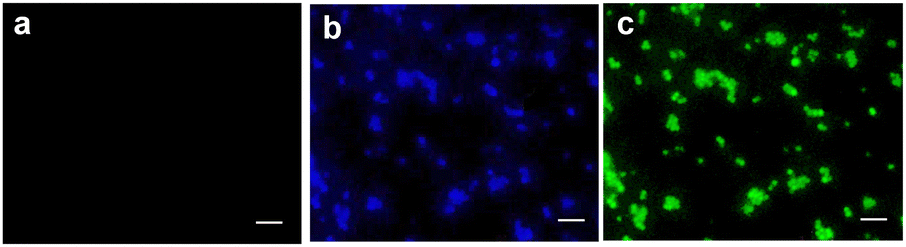 | ||
| Fig. 6 Confocal fluorescence microscopy images of ATCC strain of S. aureus biofilm incubated with NF–NO in the dark (a) and after 15 min irradiation with blue light (b) and (c), and monitored in the blue (a) and (b) and the green channel (c). λexc = 405 nm. Scale bar: 5 μm. | ||
These findings unequivocally demonstrate the formation of the active NF and, consequently, the release of NO under these experimental conditions, suggesting an excellent preservation of the photobehaviour of NF–NO even in the presence of bacterial cells. It should be stressed that the retention of the photochemical properties after incubation of photoactivatable components with biological systems is not trivial. Several factors, such as the micropolarity of the medium, steric constraints, and specific interactions, may often lead the specific photoresponsive compound forming supramolecular assemblies exhibiting significant changes in the primary photochemical process.
Conclusions
We successfully demonstrated that the temporary inactivation of the FQ antibacterial NF through its conversion into the nitrosated derivative NF–NO might represent an intriguing photopharmacological approach to simultaneously liberate the active NF and the unconventional antibacterial NO with high spatiotemporal precision under the exclusive control of visible blue light. The mechanism of photorelease probably occurs through a two-step inter-and intramolecular electron transfer initiated by the lowest triplet state of NF–NO and the phosphate buffer ions as reducing agents. This mechanism not only permits the simultaneous generation of a conventional and an “unconventional” antibacterial species under light control but significantly suppresses the photodefluorination process typical for NF and other FQs, which is the primary process responsible for the phototoxic and photocarcinogenic side effects. Another remarkable value of this approach is that the typical fluorescence emission of NF is quenched in NF–NO, and it is wholly restored upon NF photouncaging, making NF an effective optical self-reporter to monitor the photorelease in real-time with the aid of fluorescence techniques. Notably, the photouncage mechanism also operates without phosphate buffer but in the presence of biologically relevant reducing components such tyrosine, and is well preserved in bacteria cells where a significant reduction of the MIC of NF–NO is observed upon irradiation due to the release of NF and NO. To our knowledge, this represents the first example of FQ acting as a photocage to uncage itself and generate NO.The presence of piperazinyl appendages possessing secondary amine is quite common in the structure of many FQs. Besides, the photochemical reactivity of this class of compounds has been, in most cases, well-elucidated. Taking this into account, the present work might, in principle, open up unexplored opportunities in antibiotic photopharmacology, exploiting the nitrosation as a facile route to cage NO, switching OFF the pharmacological action of the FQs, and using light as a remote activation mechanism to uncage the active antibiotic drug and NO with high spatiotemporal control. Studies in this direction will be the object of further investigations.
Data availability
Data for this article, including [Fig. 1–6] are available at [Zenodo at https://Zenodo.org].Conflicts of interest
No conflicts to declare.Acknowledgements
We thank HORIZON-MSCA-2021-PF-01, 101057562 – SUPREME for financial support to MC fellow T. J. M. and research funding. JGP – FAPESP – 2022/10251-0, IPRB – Coordination for the Improvement of Higher Education Personnel – Brazil (CAPES) – Finance Code 001.Notes and references
- World Health Organization. Antimicrobial resistance. A manual for developing national action plans. chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://iris.who.int/bitstream/handle/10665/204470/9789241549530_eng.pdf? sequence = 1(accessed May 17, 2024).
- Antimicrobial Resistance: Tackling a Crisis for the Future Health and Wealth of Nations, https://amr-review.org/(accessed May 17, 2024).
- World Health Organization, Antimicrobial Resistance: Global Report on Surveillance; World Health Organization, https://www.who.int/publications/i/item/9789241564748 (accessed May 17, 2024).
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool and S. C. Johnson, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- Q. A. Duong, L. F. Pittet, N. Curtis and P. Zimmermann, J. Infect., 2022, 85, 213–300 CrossRef CAS.
- G. Dubourg, J. C. Lagier, C. Robert, F. Armougom, P. Hugon, S. Metidji, N. Dione, N. P. M. Dangui, A. Pfleiderer, J. Abrahao, D. Musso, L. Papazian, P. Brouqui, F. Bibi, M. Yasir, B. Vialettes and D. Raoult, Int. J. Antimicrob. Agents, 2014, 44, 117–124 CrossRef CAS.
- D. V. Patangia, C. A. Ryan, E. Dempsey, R. P. Ross and C. Stanton, MicrobiologyOpen, 2022, 11, 1–23 CrossRef.
- F. S. Del Fiol, V. M. Balcão, S. Barberato-Fillho, L. C. Lopes and C. C. Bergamaschi, Front. Pharmacol., 2018, 9, 1–10 CrossRef.
- D. C. Nwobodo, M. C. Ugwu, C. O. Anie, M. T. S. Al-Ouqaili, J. C. Ikem, U. V. Chigozie and M. Saki, J. Clin. Lab. Anal., 2022, 36, 1–10 Search PubMed.
- M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 10978–10999 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- I. M. Welleman, M. W. H. Hoorens, B. L. Feringa, H. H. Boersma and W. Szymanski, Chem. Sci., 2020, 11, 11672–11691 RSC.
- M. J. Fuchter, J. Med. Chem., 2020, 63, 11436–11447 CrossRef CAS PubMed.
- S. Sortino, J. Mater. Chem., 2012, 22, 301–318 RSC.
- M. R. Hamblin and T. Hasan, Photochem. Photobiol. Sci., 2004, 3, 436–450 CrossRef CAS.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, CA - Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- A. M. Emmerson and A. M. Jones, J. Antimicrob. Chemother., 2003, 51, 13–20 CrossRef CAS.
- P. C. Appelbaum and P. A. Hunter, Int. J. Antimicrob. Agents, 2000, 16, 5–15 CrossRef CAS.
- E. R. Vlieghe, J. A. Jacobs, M. Van Esbroeck, O. Koole and A. VanGompel, J. Travel Med., 2008, 15, 419–425 CrossRef PubMed.
- U.S. Food and Drug Administration, FDA updates warnings forfluoroquinolone antibiotics, https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm513183.htm (accessed May 17, 2024).
- J. Ferguson, Photochem. Photobiol., 1995, 62, 954–958 CrossRef CAS.
- G. Klecak, F. Urbach and H. Urwyler, J. Photochem. Photobiol., B, 1997, 37, 174–181 CrossRef CAS PubMed.
- A. A. Chatelat, S. Albertini and E. Gocke, Mutagenesis, 1996, 11, 497–504 CrossRef PubMed.
- M. Makinen, P. D. Forbes and F. Stenbaeck, J. Photochem. Photobiol., B, 1997, 37, 182–187 CrossRef CAS PubMed.
- L. J. Martinez and C. F. Chignell, J. Photochem. Photobiol., B, 1998, 45, 51–59 CrossRef CAS PubMed.
- L. J. Martinez, G. Li and C. F. Chignell, Photochem. Photobiol., 1997, 65, 599–602 CrossRef CAS PubMed.
- L. J. Martinez, R. H. Sik and C. F. Chignell, Photochem. Photobiol., 1998, 67, 399–403 CrossRef CAS PubMed.
- S. Sortino, G. De Guidi, S. Giuffrida, S. Monti and A. Velardita, Photochem. Photobiol., 1998, 67, 167–173 CAS.
- G. Zhang and P. Wan, J. Chem. Soc., Chem. Commun., 1994, 1, 19–20 RSC.
- W. A. Velema, J. P. van der Berg, W. Szymanski, A. J. Driessen and B. L. Feringa, ACS Chem. Biol., 2014, 9, 1969–1974 CrossRef CAS PubMed.
- W. A. Velema, J. P. van der Berg, M. J. Hansen, W. Szymanski, A. J. M. Driessen and B. L. Feringa, Nat. Chem., 2013, 5, 924–928 CrossRef CAS PubMed.
- W. A. Velema, M. J. Hansen, M. M. Lerch, A. J. M. Driessen, W. Szymanski and B. L. Feringa, Bioconjugate Chem., 2015, 26, 2592–2597 CrossRef CAS PubMed.
- M. Wegener, M. J. Hansen, A. J. Driessen, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2017, 139, 17979–17986 CrossRef CAS.
- S. Bhunia, A. Das, S. K. Jana, S. Mandal and S. Samanta, Bioconjugate Chem., 2024, 35, 92–98 CrossRef CAS PubMed.
- S. Bhunia, S. K. Jana, S. Sarkar, A. Das, S. Mandal and S. Samanta, Chem. Eur. J, 2024, 30, e2023036 CrossRef PubMed.
- G. Du, J. Fu, Y. Zheng, F. Hu, X. Shen, B. Li, X. Zhao and Z. Yu, Org. Biomol. Chem., 2023, 21, 1021–1026 RSC.
- P. Kumari, A. Kulkarni, A. K. Sharma and H. Chakrapani, ACS Omega, 2018, 3, 2155–2160 CrossRef CAS PubMed.
- Y. Shi, V. X. Truong, K. Kulkarni, Y. Qu, G. P. Simon, R. L. Boyd, P. Perlmutter, T. Lithgow and L. S. Forsythe, J. Mater. Chem. B, 2015, 3, 8771–8774 RSC.
- E. Contreras-García, C. Lozano, C. García-Iriepa, M. Marazzi, A. H. Winter, C. Torres and D. Sampedro, Pharmaceutics, 2022, 14, 2–13 CrossRef PubMed.
- L. J. Ignarro, Nitric Oxide: Biology and Pathobiology, Academic Press, 2010 Search PubMed.
- L. J. Ignarro, Arch Pharm Res., 2009, 32, 1099–10100 CrossRef CAS PubMed.
- G. G. Fang, Nitric Oxide and Infections, Kluwer Academic/Plenum Publishers, 1999 Search PubMed.
- F. C. Fang, J. Clin. Invest., 1997, 99, 2818–2825 CrossRef CAS.
- D. O. Schairer, J. S. Chouake, J. D. Nosanchuk and A. J. Friedman, Virulence, 2012, 3, 271–279 CrossRef.
- P. R. Gardner, A. M. Gardner, L. A. Martin and A. L. Salzman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10378–10383 CrossRef CAS PubMed.
- F. C. Fang, Nat. Rev. Microbiol., 2004, 2, 820–832 CrossRef CAS.
- Q. Jia, A. J. Janczuk, T. Cai, M. Xian, Z. Wen and P. G. Wang, Expert Opin. Ther. Patents., 2002, 12, 819–826 CrossRef CAS.
- S. Sortino, Chem. Soc. Rev., 2010, 39, 2903–2913 RSC.
- P. C. Ford, Coord. Chem. Rev., 2018, 376, 548–564 CrossRef CAS.
- N. L. Fry and P. K. Mascharak, Acc. Chem. Res., 2011, 44, 289–298 CrossRef CAS.
- N. Ieda, Y. Oka, T. Yoshihara, S. Tobita, T. Sasamori, M. Kawiguchiand and H. Nakagawa, Sci. Rep., 2019, 9, 1–8 CrossRef CAS.
- A. Fraix, C. Parisi, M. Seggio and S. Sortino, Chem. – Eur. J., 2021, 27, 12714–12725 CrossRef CAS.
- A. Fraix, N. Marino and S. Sortino, Topics in Curr. Chem., 2016, 370, 225–257 CAS.
- S. Sortino and G. Condorelli, New J. Chem., 2002, 26, 250–258 RSC.
- S. Sortino, G. De Guidi and S. Giuffrida, New J. Chem., 2001, 25, 197–199 RSC.
- S. Sortino, Photochem. Photobiol., 2006, 82, 64–70 CrossRef CAS PubMed.
- S. Monti, S. Sortino, E. Fasani and A. Albini, Chem. – Eur. J., 2001, 7, 2185–2196 CrossRef CAS.
- G. Condorelli, G. De Guidi, S. Giuffrida, S. Sortino, R. Chillemi and S. Sciuto, Photochem. Photobiol., 1999, 70, 280–286 CAS.
- R. A. Lawrenson and J. W. Logie, J. Antimicrob. Chemother., 2001, 48, 895–901 CrossRef CAS.
- A. Bhaumik, Ind. Vet. J., 1997, 74, 246–247 CAS.
- L. H. Sumano, C. L. Ocampo, G. W. Brumbaugh and R. E. Lizarraga, Br. Poult. Sci., 1998, 39, 42–46 CrossRef CAS.
- M. D. Adjei, T. M. Heinze, J. Deck, J. P. Freeman, A. J. Williams and J. B. Sutherland, Appl. Environ. Microbiol., 2006, 72, 5790–5793 CrossRef CAS.
- E. Fasani, M. Mella, S. Monti and A. Albini, Eur. J. Org. Chem., 2001, 391–397 CrossRef CAS.
- A. Albini and S. Monti, Chem. Soc. Rev., 2003, 32, 238–250 RSC.
- X. Su, L. Bromberg, K.-J. Tan, T. F. Jamison, L. P. Padhye and T. A. Hatton, Environ. Sci. Technol. Lett., 2017, 4, 161–167 CrossRef CAS.
- S. Toma, A. Omosebi, X. Gao, K. Abad, S. Bhatnagar, D. Qian, K. Liu and J. G. Thompson, Chemosphere, 2023, 333, 1–11 CrossRef.
- X. Q. Zhu, J.-Q. He, Q. Li, M. Xian, J. Lu and J.-P. Cheng, J. Org. Chem., 2000, 65, 6729–6735 CrossRef CAS.
- P. K. Das, M. V. Encinas, S. Steenken and J. C. Scaiano, J. Am. Chem. Soc., 1981, 103, 4162–4166 CrossRef CAS.
- C.-Y. Lu and Y.-Y. Liu, Biochem. Biophys. Acta, 2002, 1571, 71–76 CAS.
- J. J. Warren, J. R. Winkler and H. B. Gray, FEBS Lett., 2012, 586, 596–602 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic and experimental procedures. See DOI: https://doi.org/10.1039/d4tb01291g |
| ‡ Equally contributed. |
| This journal is © The Royal Society of Chemistry 2024 |