
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSteric protection of near-infrared fluorescent dyes for enhanced bioimaging
Sai Shradha Reddy
Kommidi
 ,
Kirk M.
Atkinson
and
Bradley D.
Smith
*
,
Kirk M.
Atkinson
and
Bradley D.
Smith
*
Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: smith.115@nd.edu
First published on 31st July 2024
Abstract
Near-fluorescent (NIR) dyes that absorb and emit light in the wavelength range of 650–1700 nm are well-suited for bioimaging due to the improved image contrast and increased penetration of the long-wavelength light through biological tissue. However, the imaging performance of NIR fluorescent dyes is limited by several inherent photophysical and physicochemical properties including, low fluorescence quantum yield, high chemical and photochemical reactivity, propensity to self-aggregate in water, non-specific association with off-target biological sites, and non-optimal pharmacokinetic profiles in living subjects. In principle, all these drawbacks can be alleviated by steric protection which is a structural process that surrounds the fluorophore with bulky groups that block undesired intermolecular interactions. The literature methods to sterically protect a long-wavelength dye can be separated into two general strategies, non-covalent dye encapsulation and covalent steric appendage. Illustrative examples of each method show how steric protection improves bioimaging performance by providing: (a) increased fluorescence brightness, (b) higher fluorophore ground state stability, (c) decreased photobleaching, and (d) superior pharmacokinetic profile. Some sterically protected dyes are commercially available and further success with future systems will require experts in chemistry, microscopy, cell biology, medical imaging, and clinical medicine to work closely as interdisciplinary teams.
 From left to right: Sai Shradha Reddy Kommidi, Kirk M. Atkinson and Bradley D. Smith | Sai Shradha Reddy Kommidi received her MS from the National Institute of Technology Rourkela and PhD from the University of Notre Dame in 2023 for research on supramolecular complexation of azo dyes by cucurbit[7]uril and hypoxia-responsive fluorescent cyanine probes. Kirk M. Atkinson received his BS from University of Richmond and PhD from the University of Notre Dame in 2024 for work on functionalized near-infrared cyanine dyes for fluorescence imaging and diagnostics. Bradley D. Smith is the Emil T. Hofman Professor of Science at the University of Notre Dame, and he conducts research in the fields of supramolecular chemistry and molecular imaging. His research group has created fluorescent molecular probes for imaging cancer, cell death, and microbial infections in living animals. Many of these fluorescent probes are commercially available for preclinical research applications. |
1. Introduction
Fluorescence bioimaging is often greatly enhanced by using biocompatible dyes and nanoparticles that absorb and emit light in the near-infrared (NIR) range of 650–1700 nm.1,2 This wavelength range is sometimes called the NIR transparency window due to the low background absorption and autofluorescence from endogenous molecules and the decreased scattering of NIR light by biological tissue. These favorable characteristics facilitate high-contrast and high-resolution imaging with relatively deep penetration through biological tissue. The specific focus of this Perspective article is on low molecular weight, NIR organic dyes and the scope does not include fluorescent proteins, polymers, pigments, or inorganic materials, such as quantum dots, gold nanorods or lanthanide metal complexes. Also not covered is the large body of work on supramolecular capture of relatively short wavelength, visible fluorescent dyes for applications in sensing and diagnostics.3Long-wavelength organic dyes are well-suited for biological and clinical applications for several reasons.4 They are typically biocompatible and exhibit narrower peaks with higher brightness and photostability than fluorescent proteins. Moreover, the relatively small size of organic dyes usually allows the pharmacokinetic properties to be modulated by rational structural modification. The exact combination of dye molecular properties needed for optimal bioimaging performance depends on the requirements of the technique. For example, modern cell microscopy often employs intense, focused light and requires fluorescent probes with narrow absorption/emission bands, high brightness, high photostability, and low phototoxicity. Probe photobleaching is less of a concern for in vivo imaging which uses low intensity illumination light, and the deep-tissue penetration of NIR light is a major asset. But a molecular probe that is labeled with a NIR dye must possess acceptable pharmacokinetic properties as judged by assays that measure absorption, distribution, metabolism, excretion, and toxicity (ADMET).5 Shown in Scheme 1 is an abridged summary of fluorophores that can absorb/emit in the NIR wavelength range.6–11 Readers interested in a more complete list of NIR dyes are directed to recent review articles.1,2,12,13 The spectral properties of these NIR dyes, such as the excitation and emission maxima, can be systematically altered by synthetic methods that locate substituents (auxochromes) at specific sites on the dye fluorochrome. Additional fluorochrome structure modification strategies are known to improve dye photostability.14–16
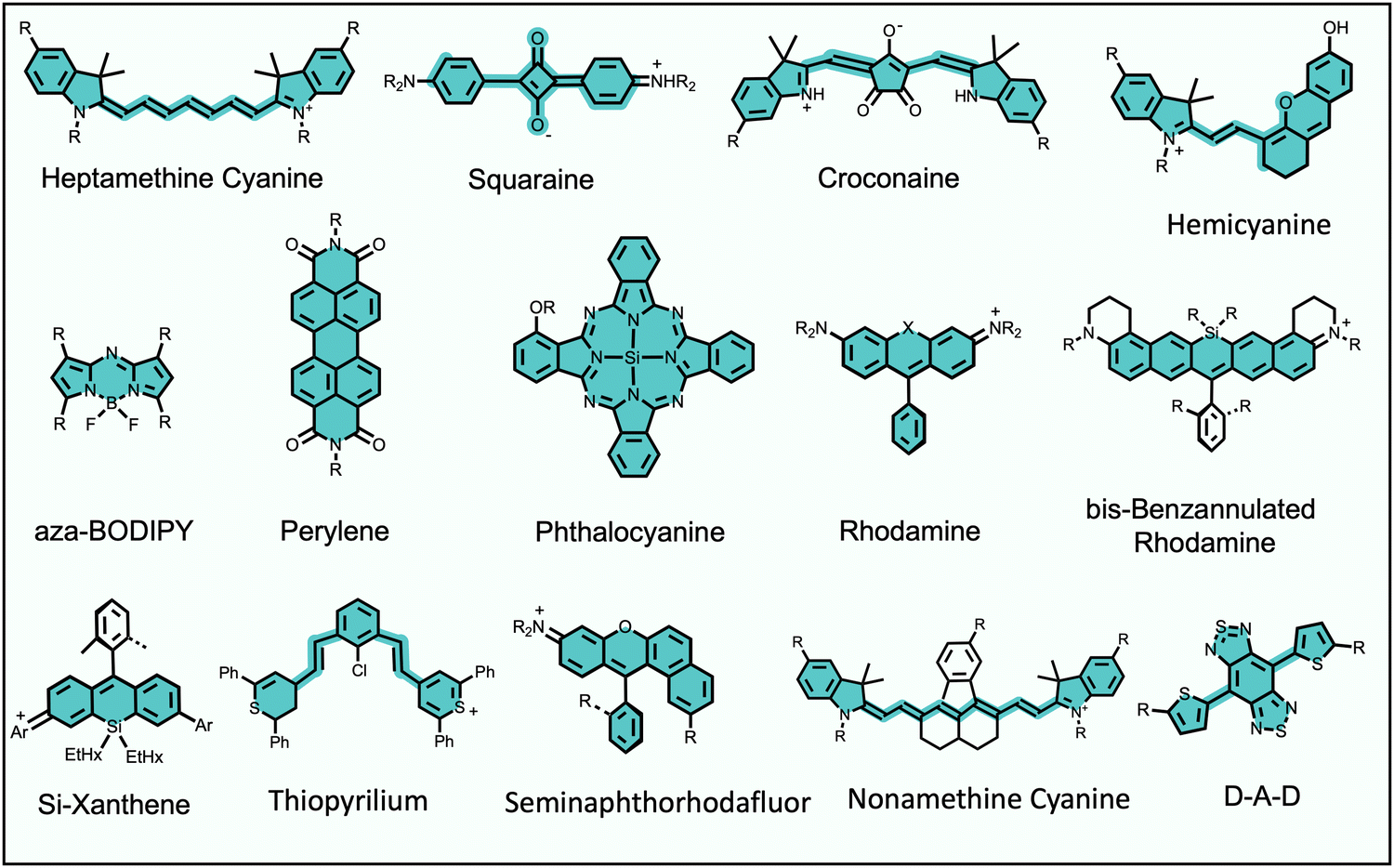 | ||
| Scheme 1 Representative fluorophores that can absorb/emit in the NIR wavelength range. | ||
2. Factors that limit bioimaging performance of long-wavelength fluorophores
Fluorescence brightness is a key performance property that is often defined as the product of dye molar absorptivity and fluorescence quantum yield. As a rule, fluorescence quantum yield decreases with increased dye absorption wavelength and is due to an increased rate constant for non-radiative relaxation of the dye electronic singlet excited state, knr, caused by various processes such as internal conversion, intersystem crossing, or nuclear reorganization.17 Intuitively, it is logical to link these processes to the relatively high structural flexibility of a NIR dye and an increased number of bond rotations or vibrations. However, a recent study reported that structural rigidification of a NIR cyanine dye did not increase the fluorescence quantum yield.18 Moreover, non-radiative relaxation for excited-sate NIR dyes strongly depends on the solvent and is enhanced in protic solvents, especially water.19 In other words, the rate constant for dye fluorescence quenching by protic solvent, ks, is unusually high and a dominant excited state relaxation pathway (Scheme 2(a)). Researchers have attributed this correlation to a dye fluorescence quenching mechanism that involves fluorescence resonance energy transfer (FRET) from the electronic excited state dye to high energy –OH vibrational transitions in the region 700–900 nm.19 Although FRET quenching by an individual water molecule is inefficient due to the very small spectral overlap, the energy transfer effect is additive and the number of energy-accepting water molecules that solvate a dye surface is high (i.e., the solvation shell of a long-wavelength dye contains 100 to 200 water molecules).20 This FRET quenching pathway explains why the fluorescence quantum yield declines with increased dye emission wavelength (i.e., there is increased spectral overlap with energy accepting –OH vibration band). It also suggests that the fluorescence intensity of a long-wavelength dye will likely be enhanced by molecular designs that separate the fluorophore surface from the surrounding water molecules.20 This idea is consistent with the common observation that dye fluorescence intensity is increased by a supramolecular process that dehydrates the dye surface, such as dye insertion into a hydrophobic membrane core or dye binding to a complementary protein pocket.21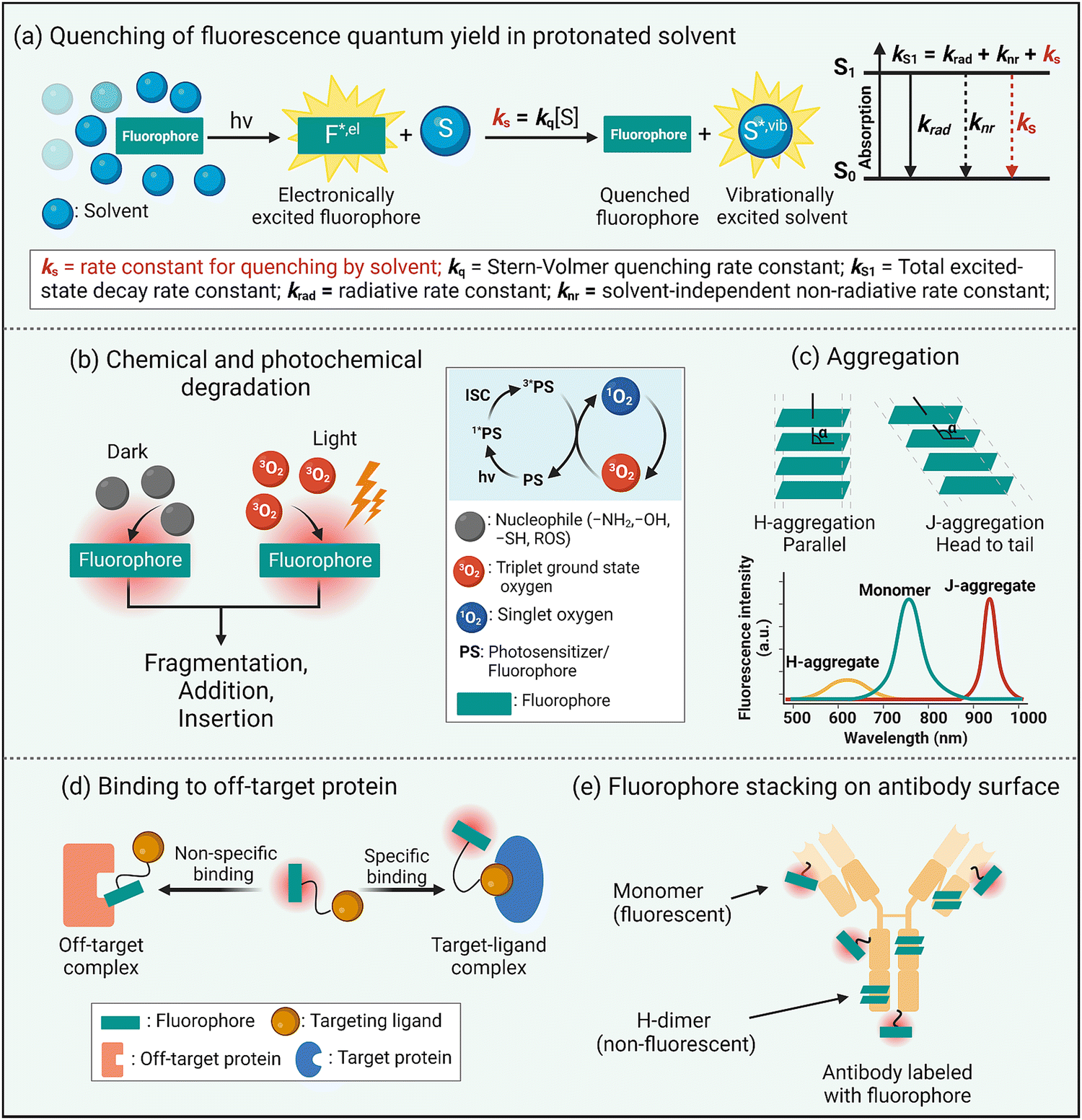 | ||
| Scheme 2 Limitations of long-wavelength fluorophores. | ||
Another inherent property of NIR dyes is relatively high chemical reactivity. This is because long wavelength dyes have a relatively high energy HOMO (highest occupied molecular orbital) and low energy LUMO (lowest unoccupied molecular orbital) that favor a range of ground-state or photoexcited chemical reactions that involve heterolytic or homolytic mechanisms leading to dye degradation. In biological media, NIR dyes can be attacked by nucleophiles or electrophiles or they can undergo photobleaching reactions that can either increase molecular weight (dimerization, insertion, or addition photoreactions)22,23 or reduce the molecular weight (fluorochrome fragmentation or truncation) (Scheme 2(b)).24,25 In general terms, photobleaching reduces a dye's photon budget, a term that refers to the overall number of detected photons and determines the maximum amount of extractable imaging information. A high photon budget is crucial for effective single molecule imaging, since the achievable spatial resolution scales inversely with the square root of the number of photons emitted per single fluorophore.26,27
A common chemical outcome for photoexcited dyes is reaction with molecular oxygen (photo-oxidation) and this can be initiated by two distinct processes, (a) single electron transfer from the excited state dye to molecular oxygen, or (b) energy transfer from the excited state dye to generate highly reactive excited state singlet oxygen.28 While dye photo-oxidation is undesired for biological imaging, photogeneration of reactive oxygen species (ROS) is central to the clinical phototherapeutic method of photodynamic therapy (PDT) where the dye is employed as an oxygen photosensitizer (Scheme 2(b)). In this regard, effective PDT requires an efficient oxygen photosensitizer with long-wavelength spectral properties and enough stability to produce the phototherapeutic effect combined with the pharmacokinetic capacity to be completely cleared from the patient so there are no long-term side-effects such as ongoing light photosensitivity.
The extended π-electron fluorochromes that are inherent to the structures of NIR dyes often (but not always29) produce a large region of hydrophobic surface area that is also polarizable. These structural properties favor dye self-aggregation in water leading to greatly altered spectral profiles. There are two main types of dye self-aggregation processes: H-aggregation and J-aggregation.30 As shown in Scheme 2(c), H-aggregates align the transition dipoles in a parallel, co-facial orientation usually producing an absorption band that is broadened and blue-shifted relative to the monomeric state. Most importantly, H-aggregated fluorophores exhibit reduced fluorescence due to static quenching, which is undesired for fluorescence imaging but can improve other modalities such as photoacoustic imaging.31 In contrast, the dyes in J-aggregates have the dye transition dipoles aligned “head-to-tail” resulting in red-shifted absorption emission bands that are usually emissive. There is growing interest in developing rational methods to self-assemble long-wavelength fluorophores as stable J-aggregates with desirable, red-shifted peaks and potentially high fluorescence quantum yields.32 But typically dye self-aggregation is viewed as an undesired, complicating process in bioimaging that needs to be attenuated or eliminated by molecular design or environmental control. A related bioimaging problem caused by dye hydrophobicity is non-specific association with various biological sites that have a lipophilic surface or pocket (Scheme 2(d)). In cells, this leads to bioconjugate accumulation at off-target intracellular sites, and in living subjects there are various pharmacokinetic problems including enhanced extravasation from the bloodstream into non-target tissue, slowed excretion from the living subject, and enhanced accumulation in the liver via lipid clearance pathways.33
A manifestation of the propensity of long wavelength dyes to self-aggregate in water becomes apparent during the process of dye conjugation to the surface of a protein such as an antibody. Consider a conjugation reaction that attaches reactive dye molecules to the amino groups on lysine side-chains. Once a dye molecule is bonded to a lysine, it becomes a docking site that directs a second reactive dye to a proximal lysine, producing a protein surface that is labeled with dyes that have H-dimer properties (Scheme 2(e)).34 Not only is the protein-dye conjugate weakly fluorescent, but the appended H-dimer dyes create a localized patch of hydrophobicity that promotes off-site targeting and enhanced in vivo accumulation in the liver.
3. Two general methods to sterically protect long-wavelength fluorophores
Steric protection of a long-wavelength dye is a structural process that surrounds the fluorochrome with bulky appendages or non-covalently associated molecules that do not degrade the fluorescence properties but prevent undesired intermolecular interactions. Steric protection is much more than a method to solubilize hydrophobic dyes in water. Effective steric protection: (a) increases fluorescence brightness by inhibiting various fluorescence quenching processes including collisional quenching and FRET to the surrounding hydration sphere; (b) improves fluorophore ground state stability by blocking chemical attack; (c) improves photochemical stability by inhibiting oxygen photosensitization or subsequent photo-oxidation reactions; (d) increases fluorescence brightness and improves pharmacokinetic profiles by preventing dye self-aggregation in water or dye stacking on a protein surface. It also needs to be stated that a potential detraction of the steric protection strategy is an increase in the size and molecular weight of a fluorescent dye. In some bioimaging applications, this might introduce potential problems such as a slower rate of molecular diffusion, less-specific targeting by a bioconjugate, decreased fluorophore density at a targeted surface, or decreased rate of cell permeation. Therefore, the most useful steric protection methods must significantly enhance dye performance properties without producing a large increase in dye molecular size.Scheme 3 summarizes two general methods that have been employed to sterically protect a NIR dye, covalent steric appendage, and non-covalent encapsulation. The following sections describe illustrative examples of these two approaches and summarize the bioimaging performance gains. Since this a tutorial-style review, our goal is to illustrate the basic concepts rather than present an exhaustive summary of all research, and we apologize to researchers whose relevant published work is not discussed.
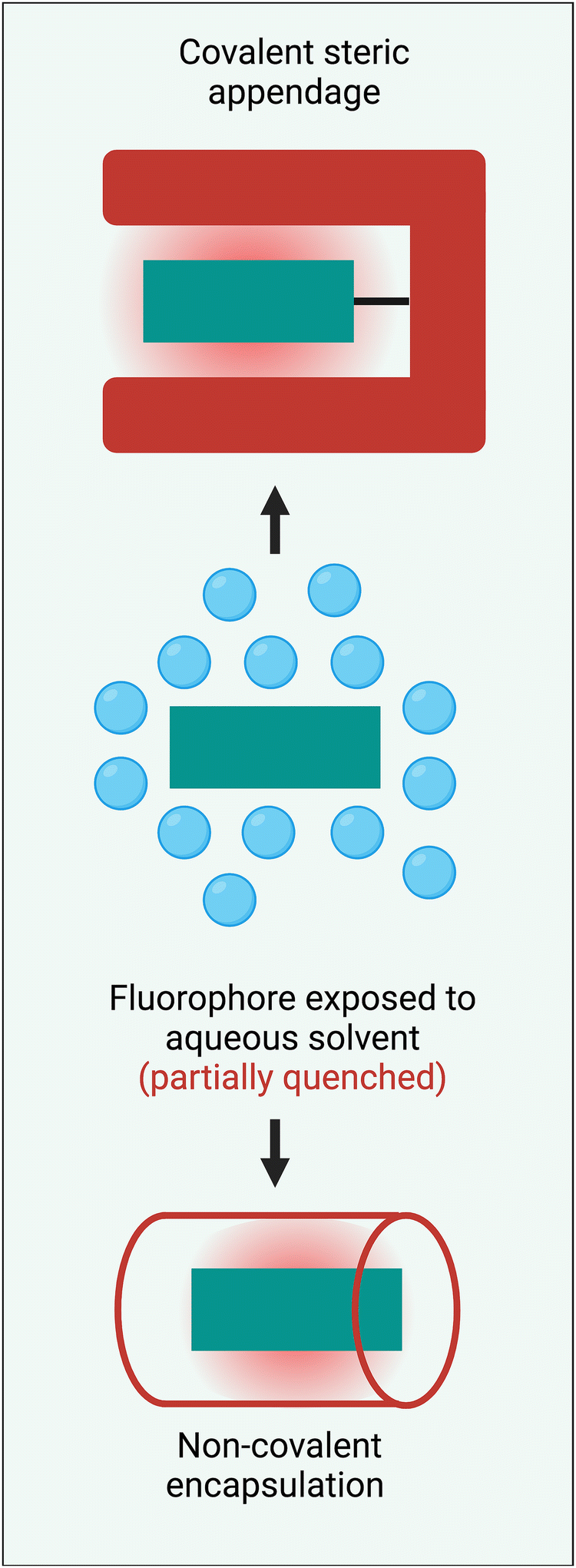 | ||
| Scheme 3 Two general methods to sterically protect a NIR fluorophore are: (top) covalent steric appendage, and (bottom) non-covalent encapsulation. | ||
4. Fluorophore protection within nanoparticles
One of the simplest ways to solubilize a hydrophobic NIR dye in water is to assemble it as a nanoparticle or incorporate it within the non-polar core of a micelle or liposome bilayer membrane.30,35 An additional feature with liposomes is the option of trapping hydrophilic dyes in the aqueous liposome interior (Scheme 4(a)). The instability of these self-assembled nanoparticles in biological media is a drawback that can be obviated by using a hyperbranched covalent polymer as a unimolecular micelle with internal pores that can accommodate and protect dye molecules as cargo. The surface polarity of the hyperbranched covalent polymer can be tuned to solubilize dyes in organic or aqueous solvents (Scheme 4(b)).36 Dyes have also been loaded within nanoparticles that are comprised of one or more proteins, such as albumin, which are inherently biocompatible (Scheme 4(c)).37 Alternatively, there are technical straightforward methods for trapping and isolating fluorescent dyes within the core of silica nanoparticles (Scheme 4(d)).38 Finally there has been elegant work to create ordered self-assemblies of dye-labeled conjugates.39 A notable recent example is the nanotube assembly shown in Scheme 4(e), where a cyclic peptide with appended dye is incorporated into a multicomponent hydrogen-bonded nanotube that is surrounded by hydrophilic polyethylene glycol chains that protect the surface-located fluorophores.40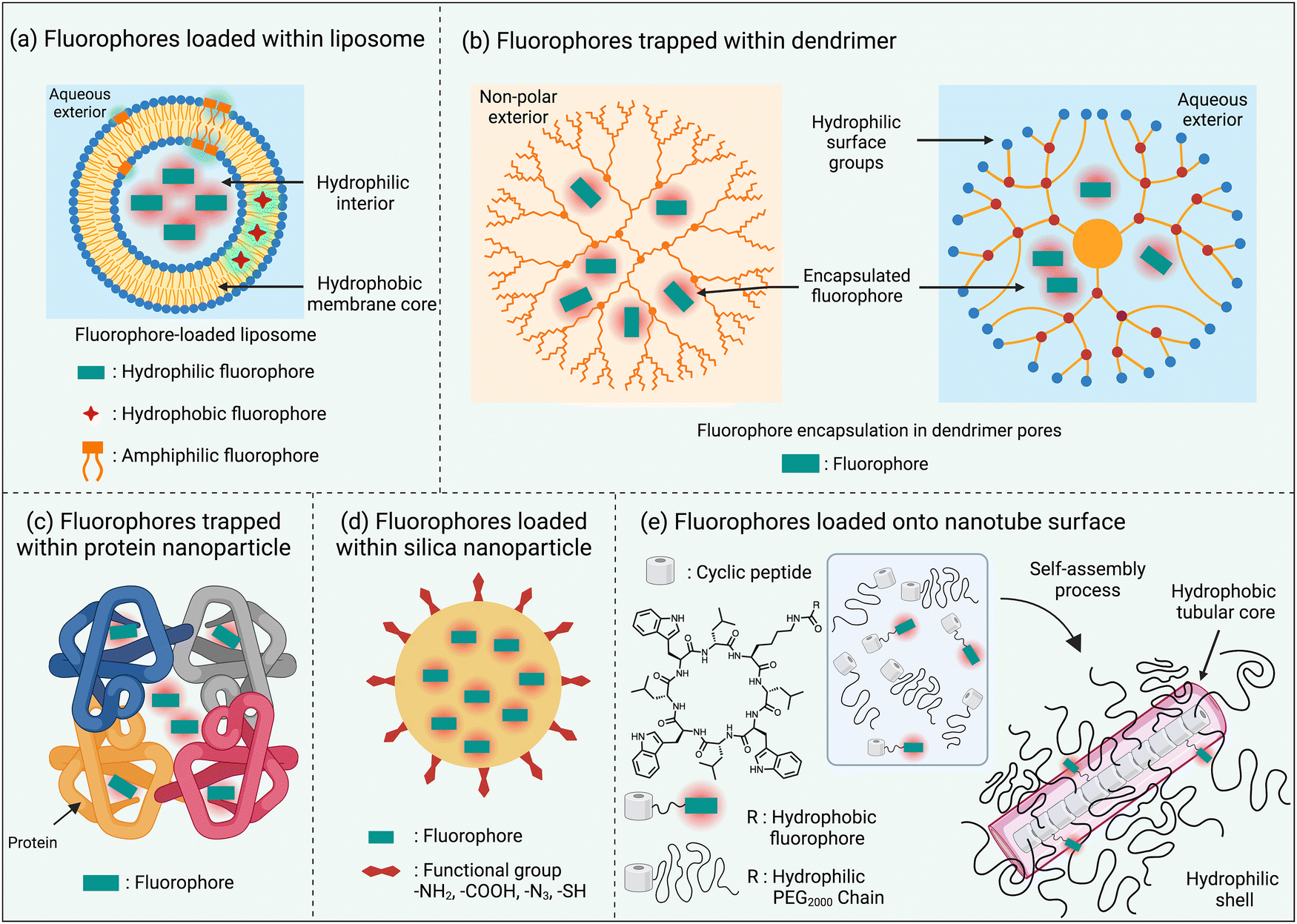 | ||
| Scheme 4 Fluorophore encapsulation and protection by nanoparticles. | ||
5. Fluorophore protection as host/guest complex or rotaxane
Steric protection of fluorophores can also be achieved by non-covalent encapsulation of a dye to form a highly stable host/guest complex or a mechanically interlocked rotaxane (Scheme 3 bottom).41 The surrounding host molecule protects the dye from the aqueous solvent which enhances dye photophysical properties and inhibits chemical attack of the dye. Shown in Scheme 5 are four representative examples of this non-covalent encapsulation strategy.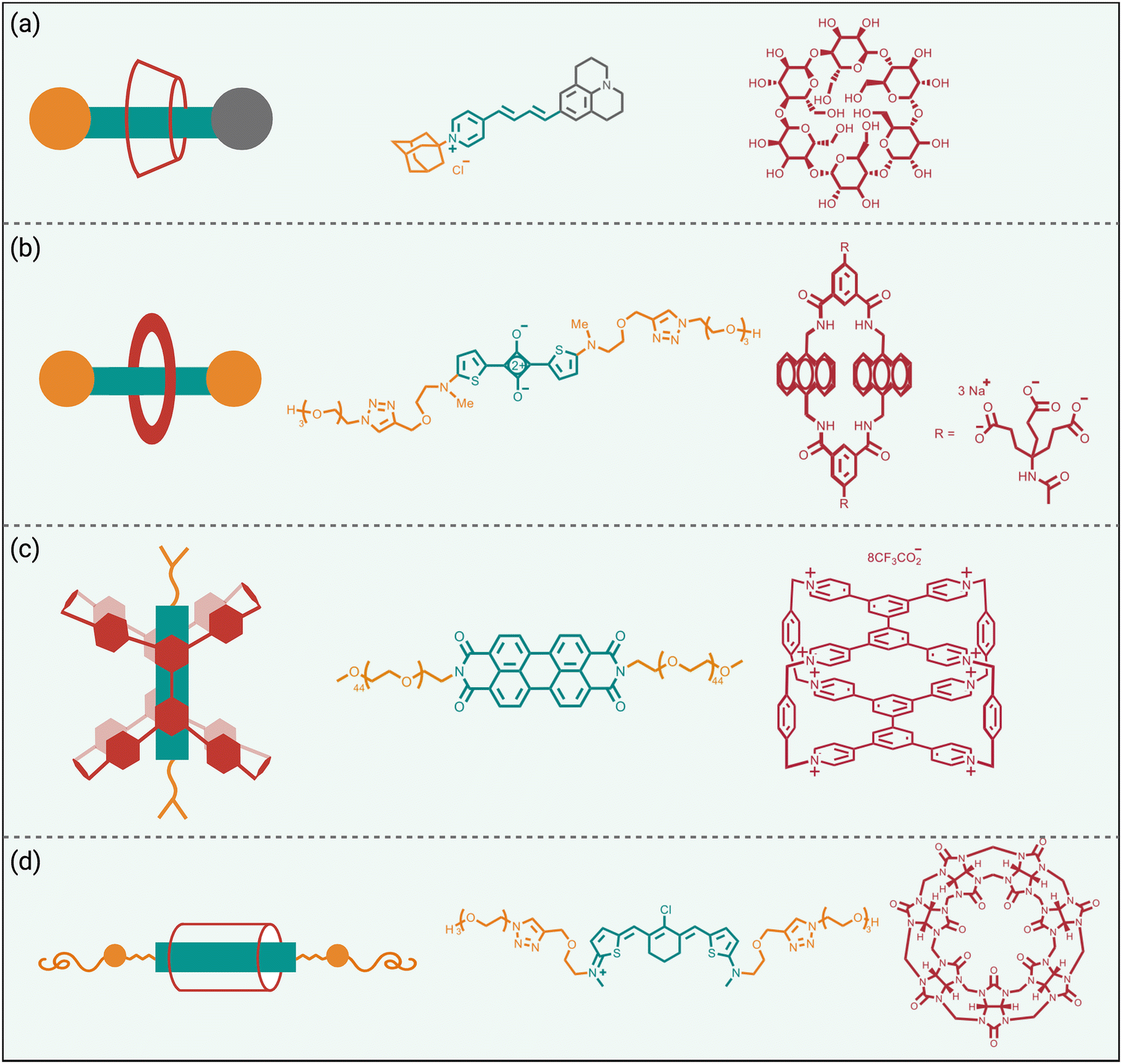 | ||
| Scheme 5 Fluorophore protection by non-covalent encapsulation. The chemical structures within each panel are, (left) fluorophore/host complex, (middle) encapsulated fluorophore component, (right) surrounding macrocyclic host component. | ||
In 2000, Anderson and coworkers reported the first mechanically interlocked cyanine rotaxane (Scheme 5(a)), that was comprised of a dumbbell-shaped fluorophore surrounded by a cyclodextrin wheel.42 Cyanine encapsulation produced a red-shift in the dye absorption and emission maxima bands, enhanced dye fluorescence quantum yield in some but not all solvents, and increased photostability. Starting in 2004, the Smith group developed permanently interlocked squaraine rotaxane dyes for enhanced biological imaging,43 and in recent years a commercial version, SeTau-647™, has been employed routinely by the microscopy community as a deep-red fluorescent label for single-molecule tracking experiments.44 In 2015, the Smith group showed that tetralactam macrocycles could be rapidly threaded by squaraine dyes to produced highly stable complexes (pseudo-rotaxanes) with red-shifted absorption/emission bands and enhanced fluorescence quantum yield.45 Moreover, the surrounding macrocycle protected the encapsulated dye from attack by thiol nucleophiles. The kinetics for macrocycle threading were very sensitive to the steric size of the N-alkyl substituents on the ends of the squaraine,46 and structural optimization led to a spontaneous self-assembly system that could generate multivalent cancer targeted fluorescent probes with sufficient kinetic stability for effective cancer imaging in living subjects.47 The most recent version of this “synthavidin technology” uses a macrocycle with expanded sidewalls that can bind squaraines with very high affinities (Ka ∼ 1010 M) and produce highly photostable squaraine complexes.48
Perylene diimides are another well-known class of long-wavelength dyes with very high brightness but large hydrophobic surface areas that favor dye self-aggregation. Building on their experience developing cationic cyclophanes as host molecules, the Stoddart group prepared the tetracationic cyclophane, known as ExBox+4, and found that it captured a perylene diimide dye with picomolar affinity in water.49 The free dye exhibited extensive self-aggregation in water with a low fluorescence quantum yield of 4%. However, upon addition of ExBox+4 the solution color transitioned from dark red to bright orange, and the fluorescence quantum yield increased to 63% while the fluorescence lifetime was extended from 4.7 to 7.3 nanoseconds.
The synthetic accessibility and favorable physicochemical properties of cyanine dyes makes them attractive choices for biological imaging. But a problem with NIR cyanine dyes that have highly extended fluorochromes is a propensity to cross over the “cyanine limit” and favor a ground state with an unsymmetric distribution of π-electron density that produces low fluorescence brightness. The Smith group demonstrated that supramolecular encapsulation of suitable NIR cyanine dyes by cucurbit[7]uril (CB7) can alter the π-electron distribution within the cyanine fluorochrome and produce greatly enhanced fluorescence brightness.50 The cyanine dye, depicted in Scheme 5(d), consists of a pentamethine structure with terminal 2-aminothiophene heterocycles and a chlorocyclohexenyl ring at its center. Without CB7, the dye in water assumes a polar ground state with an unsymmetrical distribution of π-electron density and localized positive charge, resulting in a broad absorption profile and low fluorescence quantum yield. However, upon encapsulation within CB7, the fluorophore adopts a more symmetrical π-electron state, resulting in a sharper absorption band and a 12-fold increase in fluorescence brightness in the deep NIR region. This study showed how supramolecular encapsulation of a cyanine dye by CB7 can control the surrounding solvation sphere and mitigate the practically important “cyanine limit” problem.
6. Fluorophore protection by covalent appendages
While fluorophore encapsulation by nanoparticles or host/guest complexation has the attraction of technical simplicity, the practical use of these nanoscale constructs in certain bioimaging applications is limited for two major reasons, (a) the pharmacokinetic and targeting properties are often defined by the nanoscale size and cannot be altered much by structural modification, and (b) it can be a synthetic challenge to synthesize a bioconjugate with a precise number of targeting units. One conceptual way to obviate these drawbacks is to revise the molecular design concept and focus on a single dye molecule that is synthetically endowed with covalent appendages that provide steric protection and a separate reactive functional group for bioconjugation (Scheme 3 top). Early work on visible and organic-soluble dyes has shown that the size and location of the covalent appendages are key design elements.51–54 If the project goal is to generate a hydrophilic NIR dye to act as a tracer with minimal affinity for any biological surface, then the covalent appendages can simply be multiple hydrophilic chains.55,56 However, if the sterically protected dye is intended to become a fluorescent label for bioconjugation, then the dye appendages must have sufficient size and location on the dye structure to protect the dye, but they must not prevent bioconjugation or inhibit the bioconjugate from associating with its intended biological target.57Shown in Scheme 6 are three illustrative examples of fluorophore protection by covalent appendages. The first example (Scheme 6(a)), reported by the team of Ha and Zimmerman, is a three-component structure composed of a NIR furan-fused BODIPY fluorescent dye with an appended biotin group as a targeting unit and a hydrophilic dendritic polyglycerol as a protecting appendage.58 Single molecule fluorescence studies showed long-lasting fluorescence emission with a very low level of blinking. In comparison, a homologous dye molecule that lacked the protecting polyglycerol exhibited a significantly higher level of blinking under the same imaging conditions. This finding suggests that steric protection of dyes could be an effective way to systematically alter blinking dynamics which could be exploited for the newly proposed technique of blink-based multiplexing, where individual emitters are classified using a single excitation laser based on blinking dynamics, rather than color.59,60
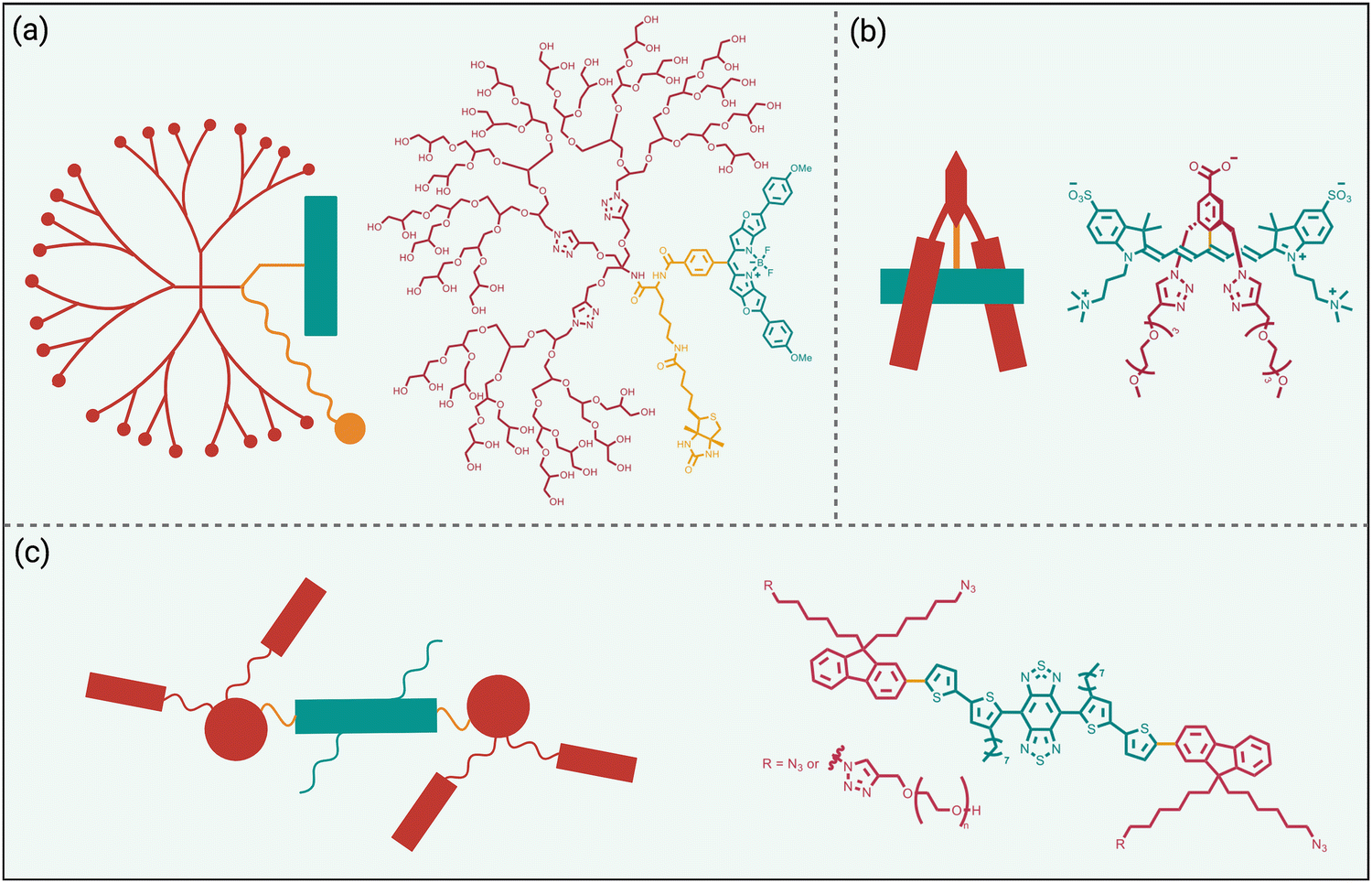 | ||
| Scheme 6 Fluorophore protected by covalently appended, (a) hydrophilic dendritic polymer, (b) two projecting hydrophilic arms, (c) four projecting hydrophilic arms. | ||
The example in Scheme 6(b) is also a three-component molecular construct with two projecting hydrophilic arms extended over both faces of a conjugatable cyanine heptamethine dye.61 Moreover, the dye structure contains a balanced mix of cationic and anionic groups, a design feature that is known to suppress non-specific interactions. Compared to an unshielded counterpart, the sterically protected dye exhibited higher water solubility, higher stability, and negligible dye self-aggregation. A cancer targeting peptide-dye bioconjugate permitted NIR fluorescence imaging of a tumor in a living mouse with a very high tumor-to-background ratio and ultralow retention in background tissue. A notable advantage with this sterically shielded dye emerged during comparison studies that attached the dye to lysine residues on the surface of IgG antibodies. As summarized above in Scheme 2(e), the classic problem that arises during IgG labeling with an unshielded NIR dye is attachment at proximal lysine positions which leads to stacked appended fluorochromes (H-dimer) that are quenched. In contrast, the shielding arms on the dye in Scheme 6(b) ensured that each copy of the dye was appended to a spatially separated region on the antibody surface and thus there was no fluorochrome stacking. The outcome was an ultrabright NIR fluorescent dye-antibody conjugate for use at very low doses for diagnostics or imaging applications.34
Over the last decade, the commercial availability of InGaAs cameras that can detect fluorescence in the NIR-II region of 900–1700 nm (also known as shortwave infrared or SWIR) has spurred a major research effort to develop fluorescent NIR-II dyes for next-generation bioimaging. Shown in Fig. 1 is an illustration of the substantial improvement in mouse image contrast that is gained by moving from conventional NIR imaging using a dye with 800 nm fluorescence to NIR-II imaging using a dye with 1100–1700 nm fluorescence.10 NIR-II dyes typically have highly extended π-conjugated fluorochromes and therefore possess the accompanying physicochemical properties that are described above.62 The Dai group developed the NIR-II dye in Scheme 6(c) which has a structural sequence of shielding unit, donor, acceptor, donor, shielding unit (S–D–A–D–S).63 The shielding unit at each end of the S–D–A–D–S dye contains a pair of hydrophilic arms that provide water solubility and prevent dye association with off-target sites. The dye structure also has two non-polar alkyl chains that surround the central fluorochrome to create a non-polar microenvironment that inhibits interaction of the fluorochrome with quenching water solvent molecules (see Scheme 2(a)). In aqueous solution, the shielded NIR-II dye produced an emission peak at 1048 nm with a remarkably high fluorescent quantum yield of 5.3%, whereas the fluorescent quantum yield for a homologous version of the dye that lacked the two central alkyl chains was 0.1%. This “double layer” approach to dye steric protection is a promising strategy for producing bright fluorescence in water. However, a potential caveat with the central non-polar alkyl layer is the possibility of undesired association with hydrophobic biomolecules, and thus in vivo imaging experiments using bioconjugates that are labeled with this NIR-II dye might incur off-target accumulation and a non-optimal pharmacokinetic profile.
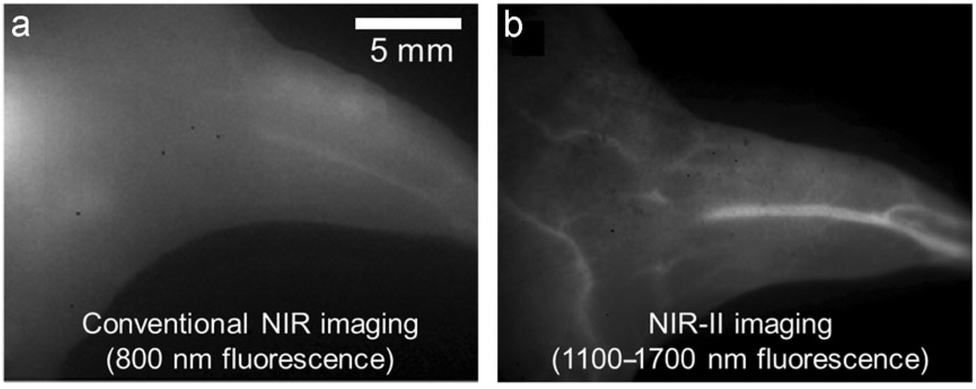 | ||
| Fig. 1 Mouse hind leg vasculature images acquired using fluorescent molecular probes that enable, (a) conventional NIR imaging (800 nm fluorescence), or (b) NIR-II imaging (1100–1700 nm fluorescence).10 Reprinted with permission from Wiley. Copyright©2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
7. Fluorophore protection by surrounding covalent straps or loops
The previous section highlighted the steric protection gained by appending multiple copies of a linear or branched hydrophilic polymer chain to a NIR dye. In recent years, emerging work has shown that the dye protection effect can be improved if both ends of a protecting polymer chain are anchored to the dye to create a protecting strap or loop whose reduced flexibility generates a more formidable steric barrier (Scheme 7).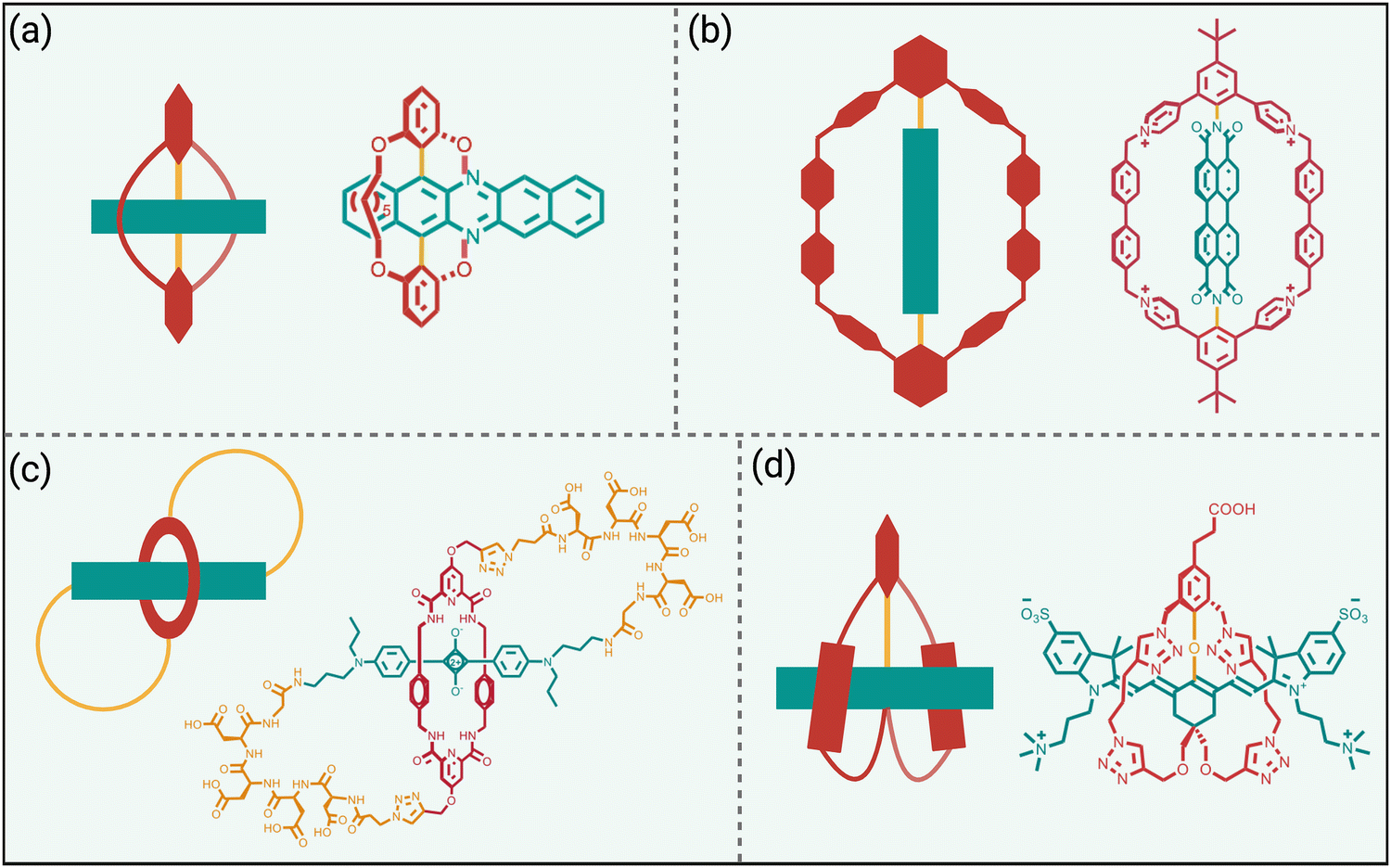 | ||
| Scheme 7 Fluorophore protected by covalent double straps or loops. | ||
The first example (Scheme 7(a)), reported by the team of Freudenberg and Bunz, is a doubly strapped azapentacene that exhibits greatly enhanced photostability compared to a counterpart that only has linear covalent appendages.64 While this azapentacene molecule was not designed for bioimaging applications, it nicely highlights the capacity of the two protective straps to inhibit bimolecular reactions with an extended π-electron system that is inherently reactive.
As noted above, perylene diimide dyes have a strong tendency to self-aggregate in water which restricts their phototherapeutic applications. The team of Wei, Xu, and Zhao mitigated this problem by developing a “Gemini box” design that surrounds the perylene diimide with two molecular straps that contain cationic aromatic subunits (Scheme 7(b)).65 The two straps conferred high water solubility and eliminated dye self-aggregation in water even in a concentrated aqueous solution up to 2 mM. The doubly strapped dye exhibited high fluorescence and was employed for lysosome-targeted live-cell imaging. In addition, the protected perylene diimide was highly stabilized as a radical anion compared to an analogue that lacked the two straps. The dye radical anion produced an intense NIR absorption that permitted efficient photothermal heating with 808 nm irradiation and no deterioration in photothermal performance was observed after twenty heating–cooling cycles. Phototherapeutic utility was demonstrated by achieving photothermal inaction of reductive E. coli bacteria.
The molecular design in Scheme 7(c), reported by Smith and coworkers, is a self-threaded molecular figure-eight architecture that contains a sterically protected squaraine fluorophore.66 The molecule was synthesized by a general method that grafted a peptide sequence at each end of a squaraine rotaxane intermediate. The covalently appended peptidyl loops served two useful purposes, they bolstered the buried squaraine within the core of the threaded structure which enhanced dye protection from attack by nucleophiles, and they acted as rigidified recognition motifs for selective imaging of biological targets within complex biomedical samples such as cell culture, tissue histology slices, or living subjects.67,68 Moreover, the peptidyl loops were unusually resistant to proteolytic degradation and thus these figure-eight molecules acted as fluorescent mimics of physically entangled peptides such as lasso peptides or knottins.
As previously mentioned, steric protection of NIR-II dyes is likely to rectify the inherent performance problems of high reactivity, low fluorescence quantum yield, and a strong propensity to undergo self-aggregation in water. The doubly strapped heptamethine cyanine dye in Scheme 7(d) is not only sterically protected but it is equipped with a carboxyl group for bionjugation.69 In aqueous buffer, the fluorescence emission peaked at 1015 nm with a quantum that was low (0.014%) but still more than double the value measured for an unstrapped homologue of the dye. In addition, the unstrapped homologue showed absorption band that indicated self-aggregation in water whereas the doubly strapped dye only exhibited an emissive monomer band. The double strapped homologue was also much more resistance to degradative, nucleophilic attack by glutathione. Bioconjugation of the dye to a cancer targeting antibody produced a fluorescent probe that enabled fluorescence NIR-II imaging of a tumor in a living mouse. The modular synthetic method provides a future opportunity to produce bioresponsive versions of this doubly strapped cyanine dyes for deployment as next-generation fluorogenic probes for more effective NIR-II imaging of living subjects.
8. Concluding remarks
Steric protection of fluorescent NIR dyes for high performance bioimaging has progressed over the last twenty-five years from bench top proof-of-concept experiments to commercially available dyes for general use in cells and living subjects (e.g., squaraine rotaxane SeTau-647™, shielded heptamethine cyanine s775z). Steric protection can be achieved by two distinct molecular design strategies, non-covalent dye encapsulation or covalent steric appendage, and each method has different strengths and weaknesses. An attractive feature of non-covalent dye encapsulation is the technical simplicity of a “mix and use” procedure. In principle, a suitable fluorophore/host complex can be generated by simply combining the fluorophore with a complementary host molecule or nanoparticle. But there are two potential drawbacks with dye encapsulation. One is the relatively large molecular size of the complex which might produce undesired pharmacokinetic or targeting properties that weaken bioimaging performance. The other is the synthetic challenge to equip the complex with a precise number of targeting units. Compared to non-covalent encapsulation, the strategy of steric protection by covalent appendages requires increased expertise in dye synthesis. Furthermore, the covalent appendages must have sufficient size and location to protect the dye, but they must not so large that they prevent subsequent bioconjugation reactions or inhibit biological targeting.Looking to the future, there is little doubt that transformative new fluorescence imaging techniques will continue to emerge and many of these new techniques will require NIR dyes with optimized photophysical, physicochemical, and pharmacokinetic properties. A notable example is the current burst of research publications on next-generation fluorescent NIR-II dyes,1,2,35 which includes some recent reports describing innovative, low molecular weight fluorophores that are delocalized carbocations with very small HOMO–LUMO energy gaps.11,29,70–73 These new NIR-II dyes have very promising photophysical and physicochemical properties, but they are susceptible to nucleophilic attack and they self-aggregate in water – two problems that can be solved by steric protection. The field of super-resolution imaging using single-molecule localization microscopy needs new classes of spontaneously blinking NIR dyes with very high photon budgets,74 and the example in Scheme 6(a) suggests that steric protection could be an effective way to systematically modulate the rate of photoblinking. Technical success in each of these future NIR bioimaging endeavors will require experts in chemistry, microscopy, cell biology, and clinical medicine to work closely as synergistic interdisciplinary teams, and the practical impact of their efforts will be amplified by effective coordination with commercial vendors to ensure that the new dyes are distributed to the broader bioimaging community.4
Author contributions
Sai Shradha Reddy Kommidi was involved in conceptualization, data curation, original draft writing, and preparation of graphics. Kirk M. Atkinson was involved in manuscript editing and preparation of graphics. Bradley Smith supervised all aspects of the work. All authors have read and approved the final manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH grant R35GM136212.References
- X. Zhao, F. Zhang and Z. Lei, Chem. Sci., 2022, 13, 11280–11293 RSC.
- J. Wu, Z. Shi, L. Zhu, J. Li, X. Han, M. Xu, S. Hao, Y. Fan, T. Shao, H. Bai, B. Peng, W. Hu, X. Liu, C. Yao, L. Li and W. Huang, Adv. Opt. Mater., 2022, 10, 2102514 CrossRef CAS.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed.
- C. Ran, J. R. Mansfield, M. Bai, N. T. Viola, A. Mahajan and E. J. Delikatny, Mol. Imaging Biol., 2023, 25, 240–264 CrossRef PubMed.
- M. Beckers, N. Sturm, F. Sirockin, N. Fechner and N. Stiefl, J. Med. Chem., 2023, 66, 14047–14060 CrossRef CAS PubMed.
- L. Yuan, W. Lin, S. Zhao, W. Gao, B. Chen, L. He and S. Zhu, J. Am. Chem. Soc., 2012, 134, 13510–13523 CrossRef CAS PubMed.
- S. Khopkar and G. Shankarling, Dyes Pigm., 2019, 170, 107645 CrossRef CAS.
- V. G. Bandi, M. P. Luciano, M. Saccomano, N. L. Patel, T. S. Bischof, J. G. P. Lingg, P. T. Tsrunchev, M. N. Nix, B. Ruehle, C. Sanders, L. Riffle, C. M. Robinson, S. Difilippantonio, J. D. Kalen, U. Resch-Genger, J. Ivanic, O. T. Bruns and M. J. Schnermann, Nat. Methods, 2022, 19, 353–358 CrossRef CAS PubMed.
- L. G. Wang, I. Munhenzva, M. Sibrian-Vazquez, J. O. Escobedo, C. H. Kitts, F. R. Fronczek and R. M. Strongin, J. Org. Chem., 2019, 84, 2585–2595 CrossRef CAS PubMed.
- Z. Tao, G. Hong, C. Shinji, C. Chen, S. Diao, A. L. Antaris, B. Zhang, Y. Zou and H. Dai, Angew. Chem., Int. Ed., 2013, 52, 13002–13006 CrossRef CAS PubMed.
- J. Li, Y. Dong, R. Wei, G. Jiang, C. Yao, M. Lv, Y. Wu, S. H. Gardner, F. Zhang, M. Y. Lucero, J. Huang, H. Chen, G. Ge, J. Chan, J. Chen, H. Sun, X. Luo, X. Qian and Y. Yang, J. Am. Chem. Soc., 2022, 144, 14351–14362 CrossRef CAS PubMed.
- J. Mu, M. Xiao, Y. Shi, X. Geng, H. Li, Y. Yin and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202114722 CrossRef CAS PubMed.
- H. Dai, Q. Shen, J. Shao, W. Wang, F. Gao and X. Dong, Innovation, 2021, 2 Search PubMed.
- G. Jiang, H. Liu, H. Liu, G. Ke, T. B. Ren, B. Xiong, X. B. Zhang and L. Yuan, Angew. Chem., Int. Ed., 2024, 63, e202315217 CrossRef CAS PubMed.
- Y. Zhang, J. Ling, T. Liu and Z. Chen, Curr. Opin. Chem. Biol., 2024, 79, 102439 CrossRef CAS PubMed.
- Q. Zheng and L. D. Lavis, Curr. Opin. Chem. Biol., 2017, 39, 32–38 CrossRef CAS PubMed.
- H. Friedman, E. Cosco, T. Atallah, S. Jia, E. Sletten and J. Caram, Chem, 2021, 7, 3359–3376 CAS.
- S. S. Matikonda, G. Hammersley, N. Kumari, L. Grabenhorst, V. Glembockyte, P. Tinnefeld, J. Ivanic, M. Levitus and M. J. Schnermann, J. Org. Chem., 2020, 85, 5907–5915 CrossRef CAS PubMed.
- J. Maillard, K. Klehs, C. Rumble, E. Vauthey, M. Heilemann and A. Fürstenberg, Chem. Sci., 2021, 12, 1352–1362 RSC.
- R. S. Gamage and B. D. Smith, Chem. Biomed. Imaging, 2024, 2, 384–397 CrossRef CAS PubMed.
- R. S. Gamage and B. D. Smith, Langmuir, 2022, 38, 11950–11961 CrossRef CAS PubMed.
- D. H. Li and B. D. Smith, Chem. – Eur. J., 2021, 27, 14535–14542 CrossRef CAS PubMed.
- H. Li, X. Li, W. Shi, Y. Xu and H. Ma, Angew. Chem., Int. Ed., 2018, 57, 12830–12834 CrossRef CAS PubMed.
- A. P. Demchenko, Methods Appl. Fluoresc., 2020, 8, 22001 CrossRef CAS PubMed.
- A. P. Gorka, R. R. Nani and M. J. Schnermann, Org. Biomol. Chem., 2015, 13, 7584–7598 RSC.
- L. Möckl and W. E. Moerner, J. Am. Chem. Soc., 2020, 142, 17828–17844 CrossRef PubMed.
- R. E. Thompson, D. R. Larson and W. W. Webb, Biophys. J., 2002, 82, 2775–2783 CrossRef CAS PubMed.
- Q. Zheng, S. Jockusch, Z. Zhou and S. C. Blanchard, Photochem. Photobiol., 2014, 90, 448–454 CrossRef CAS PubMed.
- K. Yan, Z. Hu, P. Yu, Z. He, Y. Chen, J. Chen, H. Sun, S. Wang and F. Zhang, Nat. Commun., 2024, 15, 2593 CrossRef CAS PubMed.
- H. Piwoński, S. Nozue and S. Habuchi, ACS Nanosci. Au, 2022, 2, 253–283 CrossRef PubMed.
- K. Wei, Y. Wu, P. Li, X. Zheng, C. Ji and M. Yin, Nano Res., 2023, 16, 970–979 CrossRef CAS.
- X. Hu, C. Zhu, F. Sun, Z. Chen, J. Zou, X. Chen and Z. Yang, Adv. Mater., 2024, 36, 2304848 CrossRef CAS PubMed.
- S. M. Usama, E. R. Thapaliya, M. P. Luciano and M. J. Schnermann, Curr. Opin. Chem. Biol., 2021, 63, 38–45 CrossRef CAS PubMed.
- C. L. Schreiber, D. H. Li and B. D. Smith, Anal. Chem., 2021, 93, 3643–3651 CrossRef CAS PubMed.
- R. Wei, Y. Dong, X. Wang, J. Li, Z. Lei, Z. Hu, J. Chen, H. Sun, H. Chen, X. Luo, X. Qian and Y. Yang, J. Am. Chem. Soc., 2023, 145, 12013–12022 CrossRef CAS PubMed.
- S. Padilla-Coley, H. Xu, J. Morsby, H. Gao and B. D. Smith, Biomacromolecules, 2020, 21, 2165–2175 CrossRef CAS PubMed.
- H. H. Han, A. C. Sedgwick, Y. Shang, N. Li, T. Liu, B. H. Li, K. Yu, Y. Zang, J. T. Brewster, M. L. Odyniec, M. Weber, S. D. Bull, J. Li, J. L. Sessler, T. D. James, X. P. He and H. Tian, Chem. Sci., 2020, 11, 1107–1113 RSC.
- X. Wu and W. Zhu, Chem. Soc. Rev., 2015, 44, 4179–4184 RSC.
- Z. Li, P.-Z. Liang, T.-B. Ren, L. Yuan and X.-B. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202305742 CrossRef CAS PubMed.
- H. Lu, Y. Wang, S. K. Hill, H. Jiang, Y. Ke, S. Huang, D. Zheng, S. Perrier and Q. Song, Angew. Chem., Int. Ed., 2023, 62, e202311224 CrossRef CAS PubMed.
- E. Arunkumar, C. C. Forbes and B. D. Smith, Eur. J. Org. Chem., 2005, 4051–4059 CrossRef CAS.
- J. E. H. Buston, J. R. Young and H. L. Anderson, Chem. Commun., 2000, 905–906 RSC.
- J. J. Gassensmith, J. M. Baumes and B. D. Smith, Chem. Commun., 2009, 6329–6338 RSC.
- T. A. Tsunoyama, Y. Watanabe, J. Goto, K. Naito, R. S. Kasai, K. G. N. Suzuki, T. K. Fujiwara and A. Kusumi, Nat. Chem. Biol., 2018, 14, 497–506 CrossRef CAS PubMed.
- E. M. Peck, W. Liu, G. T. Spence, S. K. Shaw, A. P. Davis, H. Destecroix and B. D. Smith, J. Am. Chem. Soc., 2015, 137, 8668–8671 CrossRef CAS PubMed.
- W. Liu, E. M. Peck, K. D. Hendzel and B. D. Smith, Org. Lett., 2015, 17, 5268–5271 CrossRef CAS PubMed.
- C. L. Schreiber, C. Zhai, J. M. Dempsey, H. H. McGarraugh, B. P. Matthews, C. R. Christmann and B. D. Smith, Bioconjugate Chem., 2020, 31, 214–223 CrossRef CAS PubMed.
- F. Balduzzi, P. Stewart, S. K. Samanta, T. J. Mooibroek, T. Hoeg-Jensen, K. Shi, B. D. Smith and A. P. Davis, Angew. Chem., Int. Ed., 2023, 62, e202314373 CrossRef CAS PubMed.
- W. Liu, S. Bobbala, C. L. Stern, J. E. Hornick, Y. Liu, A. E. Enciso, E. A. Scott and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 3165–3173 CrossRef CAS PubMed.
- D. H. Li and B. D. Smith, J. Org. Chem., 2022, 87, 5893–5903 CrossRef CAS PubMed.
- Z. Lei, X. Li, Y. Li, X. Luo, M. Zhou and Y. Yang, J. Org. Chem., 2015, 80, 11538–11543 CrossRef CAS PubMed.
- N. Chen, W. Zhang, S. Chen, Q. Wu, C. Yu, Y. Wei, Y. Xu, E. Hao and L. Jiao, Org. Lett., 2017, 19, 2026–2029 CrossRef CAS PubMed.
- J. Wang, X. Fang, X. Guo, Q. Wu, Q. Gong, C. Yu, E. Hao and L. Jiao, Org. Lett., 2021, 23, 4796–4801 CrossRef CAS PubMed.
- A. Otsuka, K. Funabiki, M. Matsui, H. Minoura, T. Yoshida, H. Mase and N. Sugiyama, Chem. Lett., 2008, 37, 176–177 CrossRef CAS.
- B. Li, M. Zhao, L. Feng, C. Dou, S. Ding, G. Zhou, L. Lu, H. Zhang, F. Chen, X. Li, G. Li, S. Zhao, C. Jiang, Y. Wang, D. Zhao, Y. Cheng and F. Zhang, Nat. Commun., 2020, 11, 3102 CrossRef CAS PubMed.
- D. H. Li, R. S. Gamage and B. D. Smith, J. Org. Chem., 2022, 87, 11593–11601 CrossRef CAS PubMed.
- K. Huth, T. Heek, K. Achazi, C. Kühne, L. H. Urner, K. Pagel, J. Dernedde and R. Haag, Chem. – Eur. J., 2017, 23, 4849–4862 CrossRef CAS PubMed.
- S. K. Yang, X. Shi, S. Park, T. Ha and S. C. Zimmerman, Nat. Chem., 2013, 5, 692–697 CrossRef CAS PubMed.
- A. G. Seabury, A. J. Khodabocus, I. M. Kogan, G. R. Hoy, G. A. DeSalvo and K. L. Wustholz, Commun. Chem., 2024, 7, 18 CrossRef CAS PubMed.
- E. Sandberg, B. Demirbay, A. Kulkarni, H. Liu, J. Piguet and J. Widengren, J. Phys. Chem. B, 2024, 128, 125–136 CrossRef CAS PubMed.
- D. H. Li, C. L. Schreiber and B. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 12154–12161 CrossRef CAS PubMed.
- M. M. M. Swamy, Y. Murai, K. Monde, S. Tsuboi and T. Jin, Bioconjugate Chem., 2021, 32, 1541–1547 CrossRef CAS PubMed.
- Q. Yang, H. Ma, Y. Liang and H. Dai, Acc. Mater. Res., 2021, 2, 170–183 CrossRef CAS.
- L. Ahrens, O. Tverskoy, S. Weigold, M. Ganschow, F. Rominger, J. Freudenberg and U. H. F. Bunz, Angew. Chem., Int. Ed., 2021, 60, 9270–9273 CrossRef CAS PubMed.
- F. Yang, R. Li, W. Wei, X. Ding, Z. Xu, P. Wang, G. Wang, Y. Xu, H. Fu and Y. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202202491 CrossRef CAS PubMed.
- D. Li, C. L. Schreiber and B. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 12154–12161 CrossRef CAS PubMed.
- C. Zhai, C. L. Schreiber, S. Padilla-Coley, A. G. Oliver and B. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 23740–23747 CrossRef CAS PubMed.
- C. L. Schreiber, C. Zhai and B. D. Smith, Photochem. Photobiol., 2022, 98, 354–361 CrossRef CAS PubMed.
- D.-H. Li, R. S. Gamage, A. G. Oliver, N. L. Patel, S. Muhammad Usama, J. D. Kalen, M. J. Schnermann and B. D. Smith, Angew. Chem., Int. Ed., 2023, 62, e202305062 CrossRef CAS PubMed.
- M. Grzybowski, O. Morawski, K. Nowak and P. Garbacz, Chem. Commun., 2022, 58, 5455–5458 RSC.
- W. E. Meador, T. A. Lewis, A. K. Shaik, K. H. Wijesinghe, B. Yang, A. Dass, N. I. Hammer and J. H. Delcamp, J. Org. Chem., 2024, 89, 2825–2839 CrossRef CAS PubMed.
- X. Lu, X. Zhuang, Y. Dong, C. Chen, R. Wei, W. Chen, H. Li, X. Luo, X. Qian and Y. Yang, Chem. Mater., 2024, 36, 949–958 CrossRef CAS.
- X. Lu, L. Yin, X. Zhuang, R. Wei, C. Yao, Y. Zhang, Y. Wang, X. Luo, X. Qian and Y. Yang, Ind. Eng. Chem. Res., 2024, 63, 1239–1247 CrossRef CAS.
- B. Zhao, D. Guan, J. Liu, X. Zhang, S. Xiao, Y. Zhang, B. D. Smith and Q. Liu, Nano Lett., 2024, 24, 4936–4944 CAS.
| This journal is © The Royal Society of Chemistry 2024 |