
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding selective sensing of human serum albumin using a D–π–A probe: a photophysical and computational approach†
Anamika
Bandyopadhyay
and
Anupam
Bhattacharya
 *
*
Department of Chemistry, Birla Institute of Technology and Science-Pilani (Hyderabad Campus), Hyderabad-500078, India. E-mail: anupam@hyderabad.bits-pilani.ac.in; Tel: +91-40-66303522
First published on 25th September 2024
Abstract
The human serum albumin (HSA) level is a valuable indicator of an individual's health status. Therefore, its detection/estimation can be used to diagnose several diseases. In this work, we have developed a series of donor–π–acceptor probes, which were found to selectively detect HSA over BSA (bovine serum albumin). Among these probes, A4, which bears the trifluoroacetyl group, showed the highest selectivity for HSA, with limits of detection and quantification being 1.36 nM and 2.59 nM, respectively. CD spectroscopy of the HSA–A4 ensemble indicated an increase in the α-helicity of the protein, while the displacement assays revealed the localization of the probe in the hemin site of HSA. The probe works on the principle of excited state intramolecular charge transfer (ICT). Its selectivity was also validated computationally. Docking experiments confirmed the preference of the probe for the hemin binding IB site of HSA, as observed from the fluorescence displacement assay results, and a comparison of docking scores demonstrated the greater preference of A4 for HSA compared to BSA. Computational experiments also showed a change in preference for HSA amino acid residues exhibited by the excited state of probe A4 (Tyr161, Met123, Pro118, and Leu115) when compared to its ground state (Arg186 and His146). Hydrophobic interactions dominated the excited state protein–probe ensemble, whereas there was significant involvement of the water bridges along with the hydrophobic interactions in the ground state ensemble. Probe A4 was also assessed for its practical utility and found to successfully sense HSA in urine at extremely low concentrations. Moreover, the A4–HSA ensemble was employed for hemin sensing with a detection limit of 0.23 μM.
Introduction
Human serum albumin (HSA), the most abundant plasma protein in human blood (∼7.5 × 10−4 M), has multiple functions in the human body.1 These functions range from modulating oncotic pressure to transporting endogenous and exogenous compounds, exhibiting antioxidant properties, improving the solubility and lifetime of hydrophobic compounds, and incapacitating certain toxic substances to orchestrate fluid distribution in different body compartments.1–6 Microalbuminuria is a condition with mildly elevated albumin levels in urine, whereas massive proteinuria is responsible for nephrotic syndrome, leading to edema, hyperlipidemia, neoplasia, and diabetes.7,8 To avoid detrimental conditions like these, timely detection of HSA becomes necessary.Structurally speaking, this protein consists of three homologous domains: I, II, and III. Each domain is further fractionated into two subdomains, A and B, consisting of 4 and 6 α-helices, respectively.1 In clinical practice, HSA is commonly measured using the bromocresol green (BCG) and bromocresol purple (BCP) assays because of the ease of availability.9 Nevertheless, the drawback of these techniques is their indiscriminate nature. Several reports suggest an overestimation of plasma albumin by the BCG method; likewise, the BCP method is less conclusive in hemodialysis patients. Van de Logt et al., in a recent research study, showed that the BCG method overestimated the plasma and serum albumin concentrations in patients suffering from hypoalbuminemia due to liver cirrhosis, sepsis, or membranous nephropathy.10 This research highlighted that based on the outcomes of the BCG assay, close to two-thirds of the patients with membranous nephropathy would receive prophylactic anticoagulant therapy, which was unnecessary. This is why efforts are being invested in developing sensors that can determine the albumin concentration in the serum and urine with greater accuracy.
The other existing methods of serum albumin quantification, such as immunoassays, colorimetric assays, etc., have several disadvantages associated with them, such as the expensive and time-intensive nature of immunoassays, subjectivity or predilection in color interpretation, and limited specificity and selectivity of colorimetric assays.11,12
Fluorescence methods are being explored as an alternative due to their advantages of low cost and real-time detection.13–23 Most recent reports show a turn-on fluorescence response upon interaction of the probes with one or more pockets of HSA.14–19 However, many of these probes suffer from problems like short absorption and emission wavelengths, poor sensitivity and selectivity, and partial solubility in water.
We have previously reported a D–A (donor–acceptor) TICT-based probe, NPNF [2-(4-nitrophenyl)naphtho[2,1-b]furan], which is HSA selective but with moderate sensitivity and micromolar range detection values.15 Besides, the emission maximum of NPNF is in the yellow region, which limits its practical application in biological systems. In this work, we wanted to understand the relationship between ICT exhibited by D–π–A systems and their corresponding ability to act as serum albumin probes. Several donor–π–acceptor systems were designed (A1–A4) by varying the acceptor moieties while keeping the donor moiety constant [Fig. 1]. A π-spacer in the probes, A1–A4, was introduced to decrease the HOMO–LUMO band gap, thereby facilitating charge transfer. Further lowering of the band gap was ensured by the electron-withdrawing acceptor group. The most electron-withdrawing group ensures the formation of the lowest band gap and aids in charge transfer.24–30 An ester-containing acceptor was considered due to its well-known cell permeability.31 On the other hand, the CF3 group was chosen for its documented hydrogen-bond-accepting and tetrel bond-forming abilities.32 A better interaction with the hydrophobic pocket of the protein was also anticipated due to the hydrophobic nature and relatively small size of CF3. Likewise, the CN group is a known bioisostere of the carbonyl group and has been used in numerous pharmaceuticals.33 A transition was made from donor–acceptor systems to donor–π–acceptor systems to improve their selectivity/sensitivity toward serum albumins.
 | ||
| Fig. 1 D–π–A systems developed for this work. | ||
Materials and methods
Analytical grade reagents and solvents were procured from commercial sources and were used without further purification. 100–200 mesh silica gel was used for column chromatography. A Bruker AV NEO-400 MHz spectrometer was used for 1H NMR (400 MHz), 13C (100 MHz), and 19F NMR measurements. Chemical shifts were reported in ppm, and TMS was used as the internal standard. Spectra of A1, A2, A3 and A4 were recorded in CDCl3. PBS buffer was used as the solvent in all the experiments. Phosphate buffer was used exclusively for CD measurements. A JASCO V-650 spectrophotometer and a Horiba Fluorolog-3 were used for recording absorption and emission spectra. The Horiba DeltaFlex modular fluorescence lifetime system was used to carry out fluorescence lifetime measurements. An Agilent LC-MS/MS Q-TOF 6540 was used to record the mass of the synthesized probes. The pH measurements were performed on an ELICO L1 120 pH meter. Natural amino acids, proteins, and metals used in the study were obtained from various commercial sources.Synthesis of A1, A2 and A3 [Fig. S1, ESI†]
To a solution of (E)-3-(4-(dimethylamino)phenyl)acrylaldehyde (2.02 mmol) in EtOH (8 mL) at 25 °C was added the appropriate active methylene compound (2.02 mmol) and piperidine (200 μL). The reaction mixture was allowed to stir at room temperature and checked periodically by TLC. Upon completion of the reaction, the solvent was removed under reduced pressure. The resulting residue was chromatographed on silica gel using various combinations of ethyl acetate/hexanes to obtain the desired compounds.1H NMR (400 MHz, CDCl3): 1.34 (3H, t, J = 8 Hz) 1.41 (3H, t, J = 8 Hz), 3.05 (6H, s), 4.28 (2H, q, J = 8 Hz), 4.40 (2H, q, J = 8 Hz), 6.68 (2H, d, J = 8 Hz), 7.01 (1H, d, J = 16 Hz), 7.15 (1H, m), 7.42 (2H, d, J = 8), 7.58 (1H, d, J = 12 Hz).
13C NMR (101 MHz, CDCl3): 165.9, 165.4, 151.5, 147.4, 146.1, 131.4, 129.6, 123.7, 121.1, 118.8, 111.9, 61.0, 40.2, 14.3.
HRMS-ESI(+) m/z: calculated for C18H24NO4+ [M + H+]: 318.1705; found: 318.1712.
1H NMR (400 MHz, CDCl3): 1.28 (3H, t, J = 8 Hz), 2.98 (6H, s), 4.23 (2H, q, J = 8 Hz), 6.58 (2H, d, J = 12 Hz), 6.97 (1H, dd, J = 14.4 Hz, 12 Hz), 7.08 (1H, d, J = 14.8 Hz), 7.39 (2H, J = 8 Hz), 7.87 (1H, J = 12 Hz).
13C NMR (101 MHz, CDCl3): 163.4, 156.6, 152.5, 150.3, 130.9, 122.6, 118.1, 115.8, 111.9, 99.2, 61.8, 40.1, 14.3.
HRMS-ESI(+) m/z: calculated for C15H19N2O2+ [M + H+]: 271.1447; found: 271.1448.
1H NMR (400 MHz, CDCl3): 3.10 (6H, s), 6.68 (2H, d, J = 8 Hz), 7.02 (1H, dd, J = 14.2 Hz, 12 Hz), 7.18 (1H, d, J = 16 Hz), 7.48 (2H, d, J = 12 Hz), 7.51 (1H, d, J = 8 Hz).
13C NMR (101 MHz, CDCl3): 160.6, 153.0, 151.6, 131.6, 121.8, 117.3, 115.0, 113.1, 111.9, 40.1.
HRMS-ESI(+) m/z: calculated for C14H14N3+ [M + H+]: 224.1188; found: 224.1185.
Synthesis of A4 [Fig. S1, ESI†]
Piperidine and EtOH were not used. Instead, acetic anhydride was used as the solvent under reflux conditions. The rest of the procedure and stoichiometry remained the same.1H NMR (400 MHz, CDCl3): 3.05 (6H, s), 6.61 (2H, d, J = 12 Hz), 7.14 (1H, dd, J = 14.4 Hz, 12 Hz), 7.31 (1H, d, J = 14.8 Hz), 7.45 (2H, d, J = 8 Hz), 7.68 (1H, d, J = 12 Hz).
13C NMR (101 MHz, CDCl3): 157.2, 156.0, 153.6, 132.5, 122.7, 120.9, 117.9, 117.3, 115.1, 112.0, 40.2.
19F NMR (376 MHz, CDCl3): 70.87 (s), 74.98 (s).
HRMS-ESI(+) m/z: calculated for C16H14NO2F6+ [M + H+]: 366.0929, found mass: 366.0929.
Spectral studies
The stock solution of probes A1 to A4 (10−2 M) was prepared in DMSO. PBS (pH = 7.4, 20 mM) was the solvent for all absorption and emission studies. Stock solutions of metal ions (10−2 M), amino acids (10−2 M), and proteins (10−3 M) were prepared in Milli Q water. The absorption spectra of all four probes were recorded within the range of 250–700 nm. The emission spectra of each probe were recorded in different solvents, amino acids, proteins, and metal ions in a cuvette with a path length of 1 cm and at 25 °C. The excitation wavelength of A1 was set at 423 nm, and the emission maximum was obtained at 590 nm. Likewise, the excitation wavelengths of A2, A3, and A4 were set at 476, 492, and 540 nm, respectively. Their emission maxima were at 592, 587, and 619 nm.Theoretical calculations
The ground state S0 geometry of the probes (A1–A4) was optimized using density functional theory (DFT), the PBE functional, and the 6-31+g(d) basis set. The conductor-like polarizable continuum model (CPCM) was employed to simulate the water environment around these molecules.The excited state S1 calculation was conducted only for A4 using the TD-DFT (time-dependent) method. All these calculations were performed using the Gaussian 09 package.
Fluorescence quantum yield measurements
The quantum yield for A4 was measured in toluene, PBS, and in the presence of HSA with PBS as solvent. Rhodamine 6G was used as the standard. The quantum yields were calculated using the formula φx/φs = [As/Ax][nx2/ns2][Dx/Ds], where x: sample, s: reference standard, A: optical density value at the wavelength of excitation, n: refractive index of the solvent, and D: area under the fluorescence spectrum recorded.Calibration curves and detection limit determination
Calibration curves were plotted to estimate HSA using each of the probes. The linear detection of probes: HSA was monitored in the 0 to 5 μM range for A1 and A2. In comparison, linearity was monitored at an even lower concentration of HSA (0 to 0.5 μM) in the cases of A3 and A4. The limit of detection and the limit of quantification were calculated using 3σ/S and 10σ/S rules (where σ is the standard deviation of three and ten blank measurements and S stands for the slope of the curve obtained by plotting fluorescence intensity against analyte concentration).The measured absorbance of A4 had an optical density value of >0.05 at both the excitation maxima 540 nm (O.D. = 0.18) and emission maxima 619 nm (O.D. = 0.07). So, it was necessary to introduce corrections for the resultant inner filter effects. The corrected fluorescence intensity was obtained using
Fcorr = Fobs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) antilog(ODex + ODem)/2 antilog(ODex + ODem)/2 |
Anisotropy measurements
Single-point anisotropy of A4 and the A4–HSA conjugate was measured at an excitation wavelength of 540 nm and an emission wavelength of 619 nm. Each sample was measured ten times, and the results were averaged.TCSPC data
Fluorescence lifetimes of A4 and HSA-bound A4 were determined using TCSPC. The analysis of the decay plots was done using the Horiba EZ time software. χ2 values were used to determine the goodness of the fit. The average singlet excited state lifetime values for the biexponential decays were calculated using the formula 〈τf〉 = α1τ1 + α2τ2, where α1 + α2 = 1 and α1 and α2 are the relative amplitudes and τ1 and τ2 are the lifetimes of the two components.Establishment of the binding site(s) of A4 in HSA
A4 was dissolved in DMSO to obtain a 10 mM stock solution. Aspirin (subdomain II A), ibuprofen (subdomain III A), and hemin (subdomain IB), which are long-established drugs known to bind to HSA, were selected for the experiment. 10 mM stock solutions of these drugs were made in DMSO. Displacement of A4 from the A4–HSA ensemble was monitored by adding these drugs at increasing concentrations of up to 100 μM.Circular dichroism
Circular dichroism spectra were recorded in the far-UV region on a JASCO J1500 CD spectrometer equipped with a thermoelectric cell holder at 25 °C and using a cuvette of path length 1 mm. A HSA concentration of 1 μM and ligand concentrations of 2.5 μM, 5 μM, and 7.5 μM were used during the experiments. The scan rate was maintained at 50 nm min−1. The spectral range was 200 to 250 nm, and the ellipticity range was −120 to 200 mdeg. The spectra shown are an average of three consecutive scans. The analysis of the spectra was performed using the BESTSEL application.Molecular docking
Schrodinger Suite 2022-1 was employed to prepare the crystal structure of the protein obtained from the RCSB PDB, generate a grid, prepare the ligand, and perform molecular docking.Protein preparation was carried out using the ‘protein preparation workflow’. For the protein HSA, the resolutions of the structures obtained from PDB were as follows: 3.2 Å for 1O9X and 2.70 Å for 2I2Z and 2BXG. The following changes were introduced before running the protein preparation. The simulation pH was maintained at 7.4. The protein pre-processing step involved filling in the missing side chains and introducing disulfide bonds wherever appropriate. The option ‘optimize H-bond assignments’ took care of allotting appropriate orientation of the hydrogen atoms that are part of the protein's amino acids and the water molecules (solvent). It also assigned the correct protonation state to each amino acid in the protein by calculating the pKa value. The ‘clean up’ section ensured that the protein was brought to a local energy minimum and also involved removing water molecules that are farther away (beyond 5 Å), as these will not be involved in the protein–ligand interaction.
Grid generation was done to identify the binding pocket in the protein. The application-based tool that was used to generate the grid was Glide. The prepared protein (receptor) had a ligand (e.g., hemin/aspirin/ibuprofen, etc.) bound to it. Hence, identifying and removing the existing ligand from the grid was necessary. So, in the ‘grid generation panel,’ the options ‘pick to identify the ligand’ and ‘show markers’ were selected. Ligand selection was done by selecting any one atom of the ligand. Following this, the job was run.
Ligand preparation was the next step, where the .mol file of the ligand A4 was introduced into the workspace. The ‘LigPrep’ application was used to prepare the ligand. This application generates the ionization and stereoisomers wherever applicable, and the pH is set to 7.4 ± 0.0 before running the job.
Molecular docking was the final step, where the ‘Ligand Docking’ application of ‘Glide’ was used.
For the protein BSA, the resolution of the structure 4F5S obtained from the PDB was 2.47 Å. Protein preparation, grid generation, ligand preparation, and molecular docking were carried out using the same procedure adopted for HSA.
The site score was calculated using the formula: site score = 0.0733√n + 0.6688e − 0.20p, where n = the number of site points (capped at 100), e = enclosure space, and p = hydrophilic score.
A score greater than 1 suggested a promisingly good site, and a score of 0.8 was used to distinguish a binding site from a non-binding one.
Molecular dynamics simulation and analysis
Molecular dynamics simulation was performed using Desmond to study the A4–HSA interaction. The ‘System Builder’ application in Maestro created the simulation system. The protein 1O9X–ligand A4 complex was centrally placed in a cubic box 10 Å × 10 Å × 10 Å to create a hydration model. The solvent model chosen to compute and mimic the bulk water properties was the SPC (single point charge) model. Accordingly, the force field applied in the experiment was OPLS4 (optimized potential for liquid simulation) due to its compatibility with the SPC water model. To cater to the negative charge of −13 on the protein (developed due to the different protonation states of the amino acid residues after the protein minimization step), the counter ion +13 Na was added to neutralize the system. A similar procedure was followed for ligand A4* (excited state structure obtained using the TD-DFT method). Simulation studies were also performed for the apoprotein 1O9X.Finally, the molecular dynamics application in Maestro was used to perform simulations. The following parameters were set up before running the simulation: the simulation time was set to 100 ns; the trajectory was recorded at intervals of 10 ps; and the approximate number of frames generated was 10000. The ensemble class chosen for the experiment was NPT; the temperature was maintained at 300 K, and pressure was set at 1.01325 bar. Maestro's ‘System Interaction Diagram’ application was used to analyze the MD simulations data.
Calculation of root mean square deviation (RMSD) and root mean square fluctuation (RMSF)
where N = the number of atoms in the atom selection, tref = reference time (the first frame is used as the reference frame, at time t = 0), and r′ = position of selected atoms in frame x after superimposing it onto the reference frame, where frame x is recorded at time tx.

RMSD shows to what extent one protein structure differs from another. The extent is calculated by measuring the average distance between the protein atoms. In our experiment, the approximate number of frames monitored during the simulation was 10000. Each frame was compared to the reference structure (structure at time t = 0).
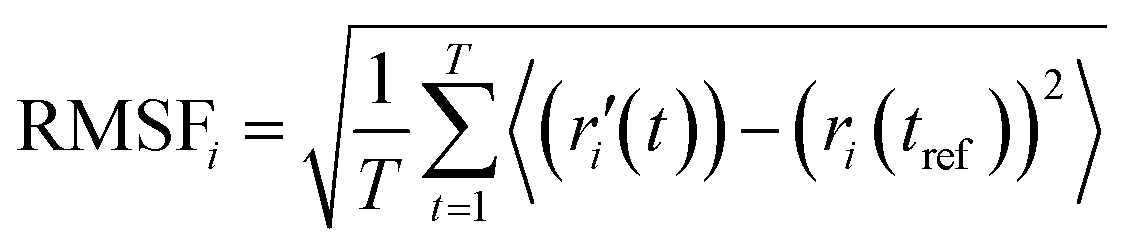 〉 = the average square distance taken over the selection of atoms in the residue.
〉 = the average square distance taken over the selection of atoms in the residue.
RMSF shows the local changes that occur in the protein structure. It measures the average atomic displacement of atoms in the protein structure. It provides information about the flexibility of the different regions within the protein.
Results and discussion
All synthesized probes were thoroughly characterized, and DFT calculations were performed. Molecular electrostatic potential (MEP) plots were obtained to understand the charge transfer interaction of probe molecules [Fig. 2]. Frontier molecular orbital analysis showed the localization of the HOMO mainly over the N,N-dimethylamino phenyl moiety and the π-linker. On the other hand, the LUMO was primarily localized on the electron-withdrawing portion of the compounds, with a marginal extension over the π-bridge [Fig. S15, ESI†]. There was a gradual reduction in the HOMO–LUMO energy gap on moving from probe A1 to A4 [Table S2, ESI†]. | ||
| Fig. 2 Electrostatic potential maps of compounds A1, A2, A3, and A4 obtained using the PBEPBE/6-31+g(d) level of theory: red colour – high electron density region and blue colour – electron deficient region. | ||
Photophysical studies of the molecules
Each molecule's absorption and emission spectra were monitored in solvents of different polarities [Table 1 and Tables S3–S5, ESI†]. The absorption spectra showed a double maxima absorption with a higher energy π → π* transition and a lower energy n → π* transition. Upon increasing acceptor strength from A1 to A4, the lower energy n → π* absorption range gradually shifted from a shorter to a longer wavelength range [Fig. S17, ESI†], as articulated computationally [Table S2, ESI†] and experimentally using the Tauc plot [Fig. S16, ESI†].34,35| Solvent | Dielectric constant | λ nm (absorbance) for A4 | ε (M−1 cm−1) | λ nm (emission) for A4 | Stokes shift (nm) |
|---|---|---|---|---|---|
| Toluene | 2.38 | 525 | 47090 |
599 | 74 |
| Dioxane | 2.25 | 519 | 46830 |
603 | 84 |
| THF | 7.58 | 539 | 44960 |
615 | 76 |
| EtOAc | 6.02 | 528 | 46500 |
611 | 83 |
| CH2Cl2 | 8.93 | 544 | 36890 |
614 | 70 |
| DMF | 36.7 | 567 | 44650 |
629 | 62 |
| DMSO | 46.7 | 574 | 57660 |
634 | 60 |
| Water | 80.1 | 515 | 18200 |
623 | 108 |
The emission spectrum of the diketo-containing probe A1 ranged between 450 and 650 nm, whereas that of A4 ranged from 550 to 700 nm. It was observed that A1 exhibited positive solvatochromism. The emission maxima shifted bathochromically as we moved from toluene to water. Probe A4 showed positive solvatochromism when moving from toluene to DMSO. However, the emission maximum in the case of water was slightly hypsochromically shifted [Fig. 3]. The effect of substituents in this series of molecules is perceptible from the shifts obtained in the absorption and emission spectra. In all the cases, the donor and the length of the π bridge remain constant, so the observed redshift in the spectra indicates a stronger intramolecular charge transfer in A4 compared to A1.
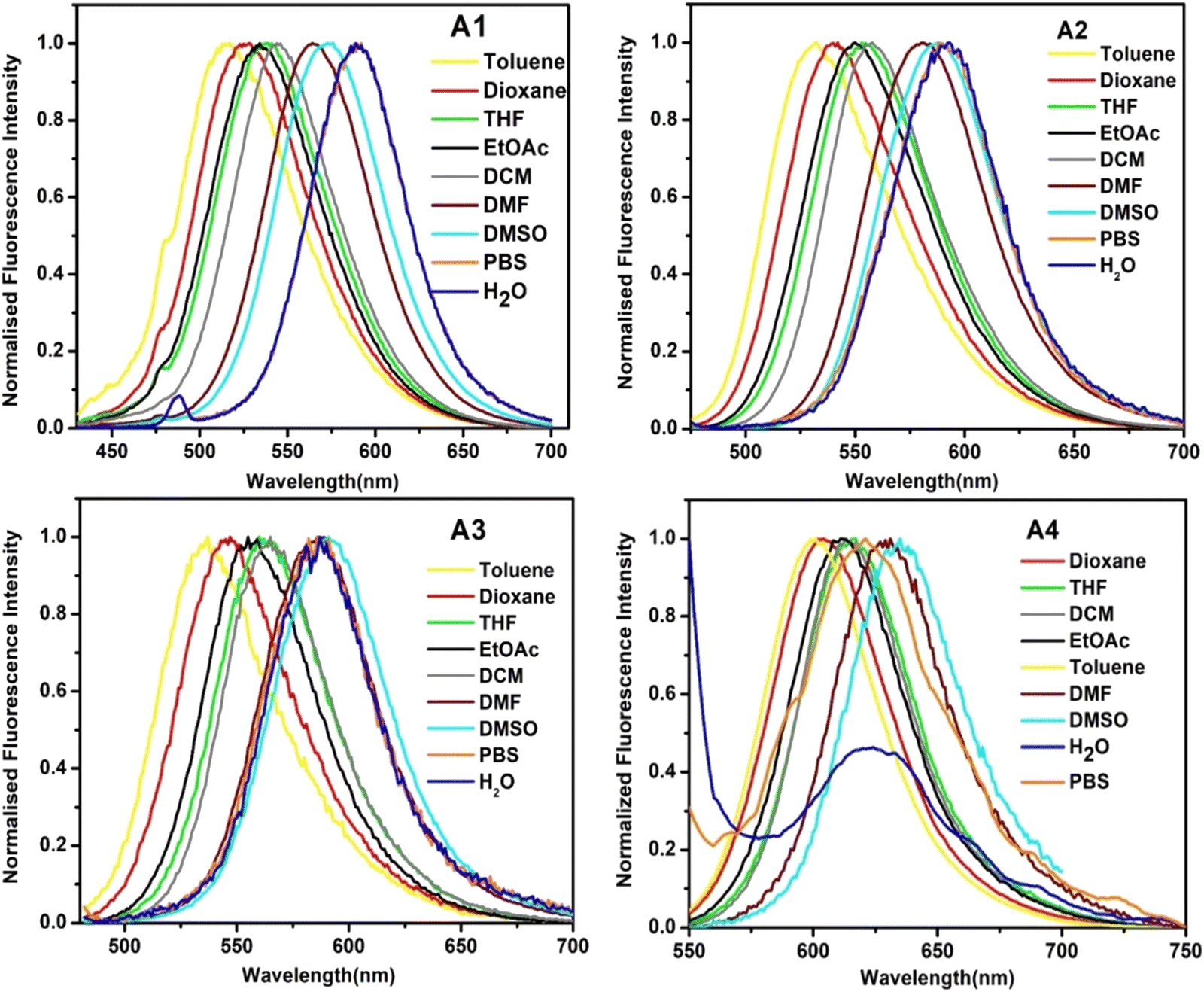 | ||
| Fig. 3 The emission spectra of A1, A2, A3, and A4 in solvents of different polarities. | ||
Based on the outcome of the preliminary investigations, we decided to monitor the fluorescence behavior of the compounds in solvents of variable polarities. For this experiment, each probe was dissolved in different fractions of THF and PBS. Upon transitioning from THF to PBS, probes A1 and A2 exhibited an initial rise in fluorescence intensity followed by a gradual quenching, with emission maxima at PBS:THF (7:3) and PBS:THF (4:6), respectively. Meanwhile, A3 was highly emissive in THF and showed a steady drop in fluorescence with an increasing percentage of water. A4 displayed a similar trend to those of A1 and A2 [Fig. S18, ESI†]. Most probes that exhibit AIEE (aggregation-induced emission enhancement) show a drastic fluorescence quenching even in the presence of very little percentage of water, followed by a sudden/gradual emission intensity enhancement, which either continues to rise or fall again.23,36–38 Unlike these results, all the probes (except A3) showed an enhancement in emission upon the introduction of the first aliquot of PBS (PBS:THF ratio of 1:9).
A subsequent study focused on the effect of the viscosity changes on the emission intensities of probes [Fig. 4]. An enhancement was observed as the solvent medium was gradually changed from PBS (η = 1.002 mPa s) to glycerol (η = 521.03 mPa s) due to the restriction of intramolecular rotation. These changes were most pronounced in the case of A4, where a fluorescence enhancement accompanied by a blue shift of 52 nm was observed upon marginal change in the viscosity (PBS to 10% glycerol–PBS). The possible reason could be stabilizing the locally excited state in the viscous medium.39,40 In a low-viscosity medium such as water, the intramolecular rotation results in a quick relaxation of the excited state via a non-radiative path, which causes a significant lowering of emission. But, in a higher viscosity medium, free rotation is arrested, and the molecules’ mode of energy loss is emissive. A low polarity and high viscosity environment is also observed in the hydrophobic pockets of HSA.39 Given the high sensitivity of A4 to minute changes in polarity and viscosity, its suitability for HSA sensing was tested upon completion of the preliminary studies.
 | ||
| Fig. 4 Fluorescence spectra of A1, A2, A3, and A4 (10 μM) at different ratios of the glycerol/PBS mixture. | ||
Studying the behavior of A1–A4 against serum albumins and various analytes of biological importance
Upon completion of the preliminary studies, molecules A1–A4 were explored for sensing serum albumins, BSA, and HSA. While all the molecules showed fluorescence enhancement when incubated with BSA/HSA, the response towards HSA was better than that towards BSA in all cases [Fig. S19, ESI†]. Compound A4 gave the best result with ∼182-fold enhancement against HSA, compared to ∼56-fold enhancement against BSA [Fig. 5]. All the other molecules, i.e., A1, A2, and A3, failed to show a clear selectivity for HSA over BSA [Fig. S19, ESI†]. Subsequently, studies were conducted to evaluate the selectivity of these probes against various plausible interferents, such as amino acids, essential cations present in the human body, and proteins of different molecular weights and functions. The studies revealed the retention of selectivity towards serum albumins for all the probes, with A4 retaining its selectivity despite excess concentration (100 μM) of the competing analytes. While the initial response time of each probe was very low (<30 seconds), it took ∼90 seconds for all the probes to produce a complete emission enhancement [Fig. S24, ESI†]. | ||
| Fig. 5 Fluorescence emission spectra and the corresponding bar plot showing the results of selectivity experiments of A4 (5 μM) against amino acids/cations (100 μM) and proteins (10 μM). The results were obtained after applying corrections for the inner filter effects. | ||
As previously discussed, the enhancement in the fluorescence intensity of A4 upon interaction with HSA was due to the polarity and viscosity-sensitive nature of A4. The initial non-fluorescence of A4 in a polar medium like PBS is due to the stabilization of the intramolecular charge transfer state of A4via the dipole–dipole interaction between the probe and the surrounding media. In contrast, the stability of the ICT state in a non-polar medium, or the hydrophobic pocket of a protein, is significantly lowered. Subsequent stabilization of the locally excited state results in the blue-shifted fluorescence enhancement. Additionally, the restricted motion of the probe inside the protein pocket reduces the non-radiative relaxation significantly, thereby contributing to the turn-on emission.
Given the preference shown by the probes towards HSA, we decided to monitor the changes in the absorption spectra upon interaction with HSA. These measurements showed broad structure-less absorption bands with peak maxima at 416 nm, 475 nm, 490 nm, and 523 nm for A1, A2, A3, and A4, respectively [Fig. S20, ESI†]. Upon increasing HSA concentrations, all four probes displayed a bathochromic shift. In the case of A1 and A3, the bathochromic shift was also associated with a mild enhancement in the absorbance value. A2 and A4, on the other hand, exhibited larger bathochromic shifts of 23 nm and 29 nm, along with a lowering of the absorbance values. The accompanying bathochromic shift indicates that the interaction of A4 with the protein favors a much lower energy absorption. It is important to note that a similar redshift in absorption spectra is reported by amyloid-detecting probes, which showed planarization of the probe molecule with greatly restricted motion.41
Finally, fluorescence titration experiments were carried out to quantify the sensitivity of each of these probes toward HSA using limit of detection (LOD) and limit of quantification (LOQ) values [Fig. 6 and Fig. S21–S23, ESI†]. Probes A1 and A2 showed a good linear correlation between fluorescence intensity and concentration of HSA (0–5 μM). In the case of A3 and A4, linear detection was possible in the 0 to 0.5 μM concentration range [Fig. S23 and Fig. 6, ESI†]. The LOD and LOQ values were calculated for each probe. A1, A2, A3, and A4 showed LOD values at 0.128 μM, 0.594 μM, 14 nM, and 1.36 nM, respectively. The LOQ values were 0.55 μM, 7.49 μM, 58 nM, and 2.59 nM for A1, A2, A3, and A4, respectively [Table 2]. Lower LOD/LOQ values and an emission wavelength >600 nm prompted us to select A4 as the preferred probe for further studies. The linearity range in which A4 detects HSA is 0.0001 g dL−1 to 0.02 g dL−1. It is essential to monitor extremely low concentrations of HSA (<0.00365 g dL−1) as such concentrations are associated with in-hospital mortality in acute coronary syndromes (ACS).42–44
 | ||
| Fig. 6 Fluorescence titration experiment showing the interaction of probe A4 (5 μM) in the presence of HSA (0 to 0.5 μM) in PBS buffer (pH = 7.4). Calibration curve at λem = 587 nm for calculating the detection limit for HSA using A4. | ||
| Probes | LOD | LOQ |
|---|---|---|
| A1 | 0.128 μM | 0.55 μM |
| A2 | 0.594 μM | 7.49 μM |
| A3 | 14 nM | 58 nM |
| A4 | 1.36 nM | 2.59 nM |
Before starting further studies, the fluorescence response of A4 and A4–HSA conjugates was also studied at various pH values [Fig. S25, ESI†], revealing an optimal performance at neutral and basic pH (pH 8–9) towards HSA sensing.
Time-resolved fluorescence studies
Time-resolved fluorescence studies were deemed essential to develop an idea about the effect of the immediate environment on the fluorescence lifetime of the probe.45–48 The free probe A4 gave a single exponential fitting with a lifetime of 1.01 ns [Table 3 and Fig. S26, ESI†], whereas biexponential fitting was required in the case of A4–serum albumin conjugates. In the case of HSA, τ2 was 1.61 ns with an amplitude (α2) of ∼20%, and τ1 was 610 ps with an α1 of ∼80%. A similar trend was also observed with BSA, with τ2 ∼1.54 ns and α2 ∼16% and τ1 ∼370 ps and α1 ∼84%. The enhancement of the lifetime of A4 in the presence of serum albumins can be attributed to the motional restrictions [Table 3], with the associated alpha values indicative of the fraction of the bound probe. In addition, the data also revealed that the lifetime components in HSA are higher when compared to BSA, plausibly due to the localization of the probe in a more hydrophobic region in HSA. Also, a longer lifetime of A4 in HSA indicated a higher degree of rigidity experienced by A4, which causes a slower decay rate to the ground state through a radiative pathway.49 Upon adding the proteins to the probe, the newly developed component plausibly corresponded to a solvation component as it is ultrafast (on the picosecond scale).| Sample | λ max (nm) | τ 1 (ns) | τ 2 (ns) | τ av (ns) | α 1 | α 2 | χ 2 |
|---|---|---|---|---|---|---|---|
| A4, 5 μM | 619 | 0.0006 | 1.01 | 0.00069 | 5.41 × 10−20 | 1 | 1.16 |
| A4 + 10 μM HSA | 619 | 0.61 | 1.61 | 0.813 | 0.804 | 0.195 | 1.05 |
| A4 + 10 μM BSA | 619 | 0.37 | 1.53 | 0.56 | 0.835 | 0.164 | 0.99 |
Steady-state anisotropy experiments
When the excited fluorophore emits light, it retains some memory of the polarization of the light it absorbed. This feature is exploited while measuring fluorescence anisotropy. Small molecules tumble randomly in solution and get depolarized easily. However, the ease of tumbling depends significantly on the probe's environment. In a viscous medium or a restricted environment, the probe is not free to rotate or tumble; hence, the emission retains the polarization better, and the resultant anisotropy is high. A4 exhibited near zero anisotropy in the PBS buffer owing to its free rotational movements. Upon addition of the serum albumins, the anisotropy value increased, which indicated the incorporation of the probe in a much-restricted environment of the protein binding pocket(s) [Table 4].| Ligand/ligand–HSA | Anisotropy | Polarization | Trails |
|---|---|---|---|
| A4 (1 μM) | 0.0607 | 0.08841 | 10 |
| A4 + HSA (5 μM) | 0.1678 | 0.23222 | 10 |
| A4 + HSA (10 μM) | 0.16779 | 0.2322 | 10 |
| A4 (1 μM) | 0.10472 | 0.14926 | 10 |
| A4 + BSA (5 μM) | 0.17546 | 0.24196 | 10 |
| A4 + BSA (10 μM) | 0.16946 | 0.23433 | 10 |
Studies on the changes in the HSA structure upon interaction with A4
It was important to understand if the tertiary structure of the protein had a key role in the enhancement of the emission intensity of A4. For this, endopeptidase trypsin was utilized to hydrolyze HSA. It was observed that upon increasing the concentration of trypsin, the fluorescence emission gradually dropped [Fig. S27(a), ESI†]. However, the fluorescence intensity was not entirely quenched. This could possibly be due to A4 tethering to the hydrophobic environment of the cleaved protein.In addition, the hydrolysis of the protein was also confirmed by monitoring its emission spectrum. It showed quenching of the tryptophan emission accompanied by a blue shift, which indicated a change in the environment surrounding the lone tryptophan of HSA [Fig. S27(b), ESI†].
The change in intrinsic fluorescence of tryptophan residues within serum albumins was also monitored upon the interaction with A4. Quenching of the fluorescence signal was observed in the case of both serum albumins plausibly due to the binding-induced conformational changes or due to changes in the vicinity of the interaction site. The inner filter effect (hence quenching without interaction) was ruled out as A4 absorbed in the 400 to 600 nm region. In the case of HSA, there are many tyrosines and a lone tryptophan Trp-214. A mild redshift indicated that the tryptophan was exposed to a more polar setting upon such an interaction [Fig. S28(a), ESI†]. A more significant quenching in the case of BSA plausibly originated due to the interaction of the probe with both the surface tryptophan (Trp-134) and the deep-seated tryptophan (Trp-213). No significant change in the polarity was observed upon the interaction of A4 with BSA [Fig. S28(b), ESI†].
CD spectroscopy was performed to determine the changes in the secondary structure of HSA upon its interaction with A4. A pure α-helix conformation generally displays negative bands at 208 nm and 222 nm due to π to π* and n to π* transitions of the non-bonding electrons of the oxygen of the peptide carbonyl in the CD spectra.50 The spectrum showed an initial decrease in α-helicity, followed by an increase. The overall change in percentage alpha helicity, calculated using BestSel, was from 68% to 70.5% upon treating one equiv. of HSA with 7.5 equiv. of the probe [Fig. 7].
 | ||
| Fig. 7 Circular dichroism spectra of HSA with different molar ratios of A4 at 298 K. The concentration of HSA was 1 μM in phosphate buffer at pH 7. | ||
Displacement assays and the recognition mechanism
The displacement assay used three classical drugs: aspirin, ibuprofen, and hemin for sites I, II, and IB, respectively. A4 was incubated with HSA and then increasing concentrations of site-specific drugs were introduced into this ensemble. When introduced, aspirin and ibuprofen caused minor enhancement in the fluorophore emission [Fig. 8 and Fig. S29, ESI†]. This enhancement was primarily due to the secondary binding of these drugs to their respective binding sites that brought about conformational changes and resulted in the tighter binding of A4 to HSA.51 Introduction of hemin, however, led to the complete quenching of the probe's fluorescence at 100 μM, which indicated the binding of the probe to the IB site of the protein.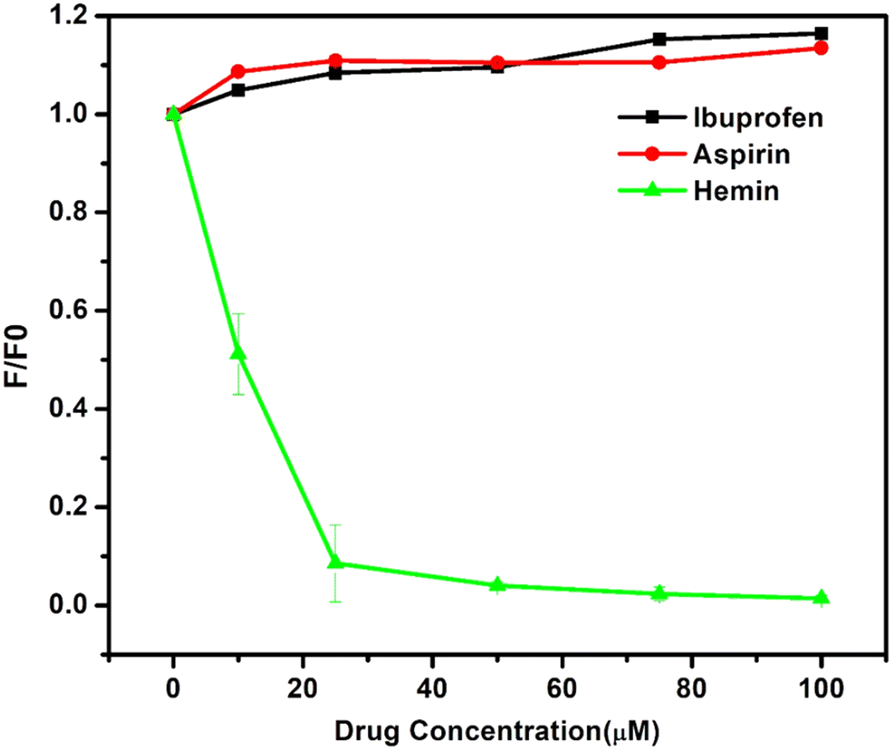 | ||
| Fig. 8 Displacement assay of A4 where site-specific drugs were tested against the complex of HSA–A4. The expanded version of aspirin and ibuprofen is included in the ESI.† | ||
Molecular docking experiments
After determining the binding site of A4 using displacement assay, we performed molecular docking to understand the binding interactions between A4 and HSA at the atomic level. Similar studies were also performed on A4 and BSA to understand the plausible reason for the difference in the behavior of both serum albumins towards A4.HSA–A4 docking
For appropriate site selection, the following proteins with PDB IDs were chosen: PDB ID: 1O9X for site A (hemin binding site), located in domain IB; PDB ID: 2I2Z for site B (aspirin binding site), located in domain IIA; and PDB ID: 2BXG for sites C and D (ibuprofen binding sites), located in domains III A and II B respectively.The detailed tabulated report showed the binding affinities of the best-docked poses to be −7.491, −4.90, −6.184, and −5.01 kcal mol−1 at sites A, B, C, and D, respectively [Table 5, Fig. 9 and Fig. S30–S32, ESI†]. This indicated that site A, the hemin binding site (IB), was the most preferred binding site, which aligned with the fluorescence displacement assay results.
| PDB ID | Interacting amino acids | Docking score (kcal mol−1) | Types of interactions |
|---|---|---|---|
| 1O9X (for site A) | Phe 134, Leu 135, Tyr 138, Met 123, Leu 182, Tyr 1616, Ser 193, Lys 190, Gly 189, Arg 186, Leu 115, Phe 149, Ile 142 | −7.491 | π–π stacking interactions with Tyr 161, charged interactions (positive) with Lys 190 and Arg 186, polar interactions with Ser 193, and hydrophobic interactions with Tyr 138, Leu 135, Phe 134, Met 123 and Tyr 161 |
| 2I2Z (for site B) | Lys 195, Gln 196, Glu 153, Tyr 150, Val 241, Leu 238, Ser 287, Ile 290, Ala 291 | −4.90 | Polar interactions with Ser 287 and Gln 196, charged (positive) interactions with Arg 257 and Lys 195, charged (negative) interactions with Glu 153, and hydrophobic interactions with Val 241, Leu 238, Tyr 150, Ala 291, Ile 290, Phe 223, and Leu 260 |
| 2BXG (for site C) | Leu 430, Leu 453, Val 433, Gly 434, Cys 437, Cys 438, Cys 392, Asn 391, Ile 388, Leu 387, Lys 414, Tyr 411, Arg 410, Leu 407 | −6.184 | H-bonding interactions with Lys 414 and Arg 410, polar interactions with Ser 489 and Asn 391, hydrophobic interactions with Cys 392, Ile 388, Leu 387, Cys 438, Cys 437, Val 433, Leu 453, Leu 430, Leu 457, Phe 488, Tyr 411 and Leu 407 |
| 2BXG (for site D) | Glu 354, Lys 351, Ala 350, Leu 347, Asp 324, Leu 327, Gly 328, Phe 330, Leu 331, Ala 213, Arg 209, Phe 206 | −5.01 | Charged (negative) interactions with Glu479, Asp 324, and Glu 354, H-bonding interactions with Lys 351, cation–π interactions with Arg 209, polar interactions with Ser 480, and hydrophobic interactions with Leu 481, Leu 347, Ala 350, Phe 206, Ala 210, Ala 213, Leu 331, Phe 330, and Leu 327 |
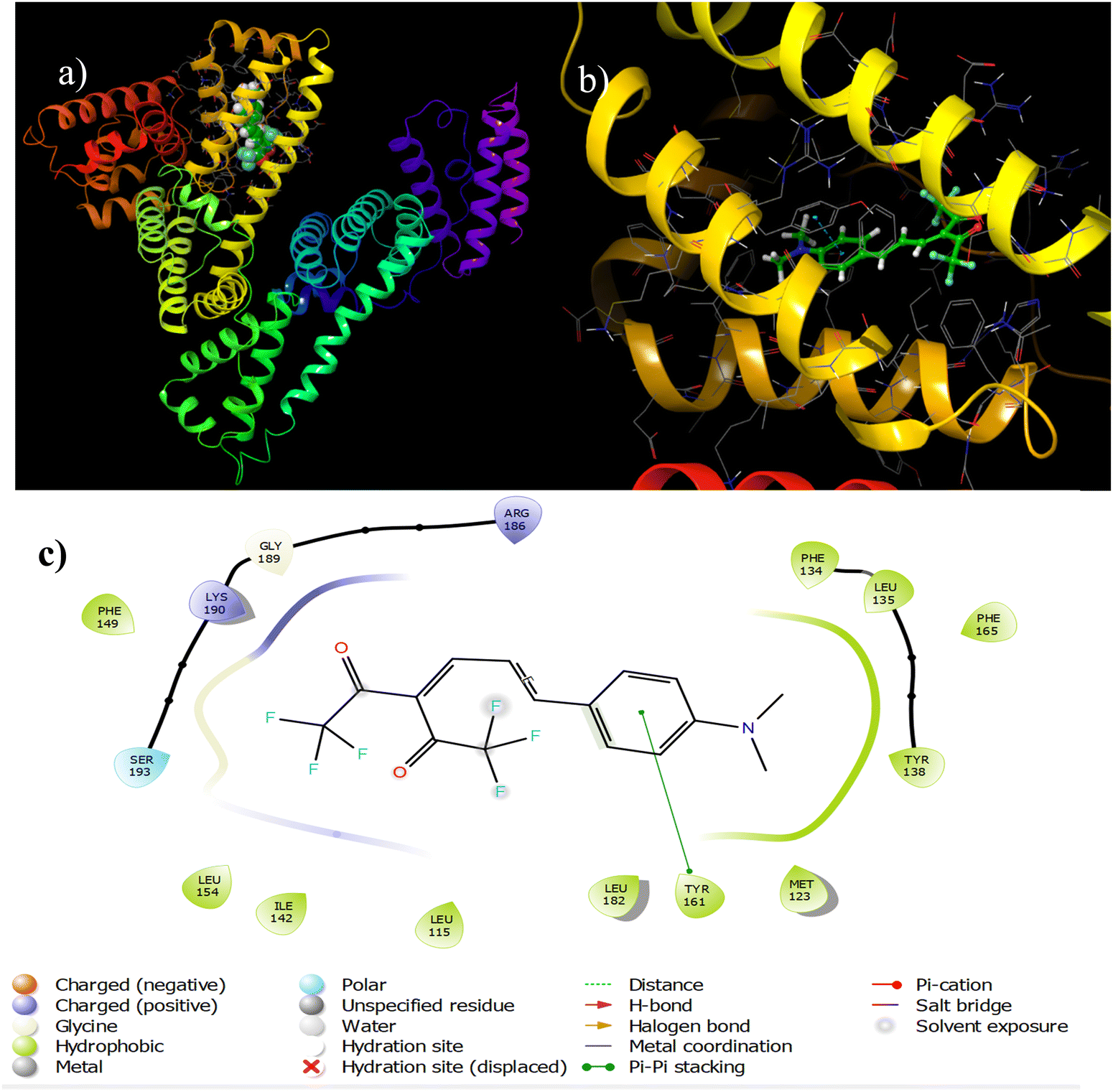 | ||
| Fig. 9 Binding interactions between A4 and 1O9X (site A). | ||
Determination of most probable binding sites in BSA using ‘SiteMap’
The most favorable binding sites in BSA were identified using Schrodinger's ‘SiteMap’ application [Fig. S33, ESI†]. The Site Score was used to distinguish the potential sites from the other regions of the protein. Using the ‘SiteMap’, five different sites were identified with scores of 1.039, 1.063, 1.108, 0.983, and 1.038 for sites 1, 2, 3, 4 and 5, respectively. For BSA–A4 docking, the scores obtained were −5.399, −4.487, −4.013, −3.211, and −2.333 kcal mol−1 for sites 1, 2, 3, 4, and 5, respectively [Table S10 and Fig. S34–38, ESI†]. This demonstrated that site 1 is the most favorable interaction site and also substantiated the greater binding affinity of A4 for HSA than BSA.Molecular dynamics simulations
Although docking provides substantial information about the protein–ligand interaction, it doesn’t offer a holistic view of the binding interaction as it assumes a rigid protein structure. On the other hand, molecular dynamics simulates the motion of both the probe and the protein over time, thus revealing a more representative view of the interactions.Simulation experiments were performed for the apoprotein (only HSA) and the holoprotein [HSA–A4 (ground state)/HSA–A4* (excited state)]. Since the absorption and emission interactions of A4 with HSA were studied in this work, computationally monitoring the interactions of A4/A4* with HSA was deemed worthwhile.
Parameters thoroughly studied to understand the A4/A4* binding to HSA were root mean square deviation (RMSD), root mean square fluctuation (RMSF), protein secondary structure elements (PSSE), and the ligand torsion profile.
Root mean square deviation (RMSD)
RMSD helps quantify the structural similarity between two protein structures, with lower RMSD values indicating greater similarity and higher values implying greater divergence from the original/reference structure. Similar calculations are also performed for the ligand in the protein pocket.Initially, the RMSD fluctuation of about 5.6 Å units was observed for the apoprotein [Fig. S39, ESI†]. It displayed an increasing trend as the simulation progressed from 0 to 100 ns. However, in the case of the HSA–A4 conjugate, the protein RMSD (the left y-axis in Fig. S40, ESI†) increased in the time frame of 0–20 ns, which subsequently converged beyond 20 ns and led to the RMSD fluctuation in the range of 4.8 Å (Fig. S40, ESI†). Compared to the apoprotein, the decrease in protein RMSD in the case of the HSA–A4 conjugate indicated stabilization of the protein structure upon interaction with A4. Ligand RMSD of the HSA–A4 interaction initially rose in the 0–20 ns time frame, following which it stabilized and fluctuated around 7 Å. This observation (the right y-axis in Fig. S40, ESI†) helps us conceptualize the stability of the ligand in the protein pocket. It is important to note that A4, a fairly flexible ligand, has a higher RMSD value compared to the protein by virtue of its immanent conformational freedom. The fluctuations in the ligand structure are possibly due to binding pose variations owing to the conformational changes occurring in the ligand while executing multiple interactions with the amino acid residues during the course of the simulation.
In the case of the HSA–A4* ensemble, the protein RMSD rose in the timeframe of 0–10 ns and then fluctuated around 6 Å [Fig. 10]. This indicates a significant difference between the reference structure and the other structures that the protein adopts upon interacting with A4* during the simulation. The ligand RMSD shows that A4* fluctuates around 6 Å; this closely matches the protein RMSD. Note that the RMSD is lower in the case of the excited state structure of the probe due to its greater rigidity.
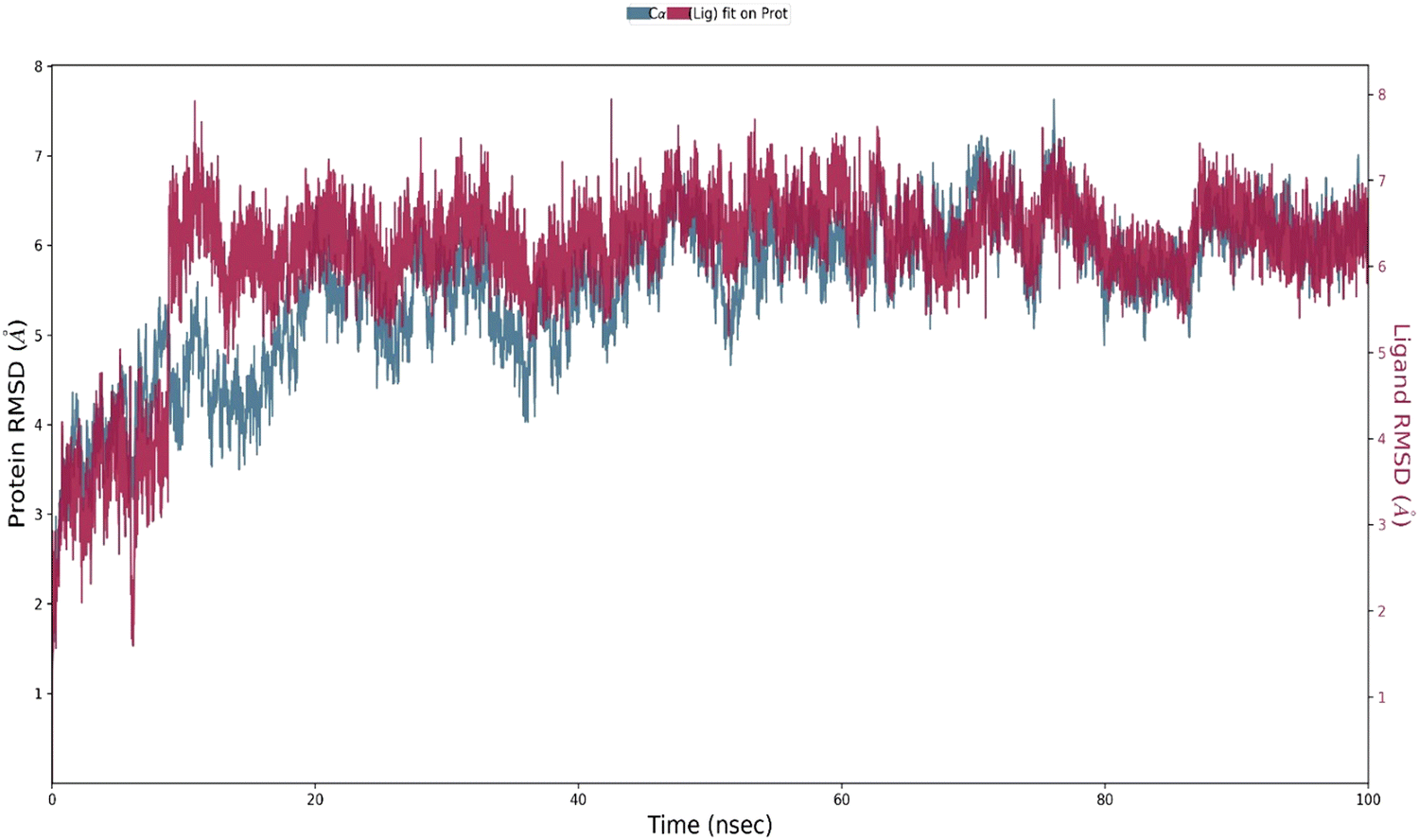 | ||
| Fig. 10 The RMSD plot obtained for the HSA–A4* ensemble. The protein Cα and probe A4 are shown in blue and red colors, respectively. | ||
Root mean square fluctuation (RMSF)
The protein RMSF plot sheds light on the flexibility/mobility of atoms in the protein structure and helps elucidate the effectiveness of HSA–A4 and HSA–A4* binding and stabilization. The protein residues that interacted with probe A4 are marked using vertical green lines in Fig. S42 (ESI†). The RMSF value of HSA residues is lowered when A4/A4* interacts with the protein [Fig. S41–S43, ESI†]. This reduction in flexibility may be attributed to stabilizing interactions between the ligand and the protein, such as hydrogen bond formation, hydrophobic interactions, or electrostatic interactions.52The ligand RMSF sheds light on the behavior of the ligand within the protein pocket to which it is bound. Both ligand structures A4 and A4* [Fig. S44 and S45, ESI†] showed fluctuations around 2.5 Å; however, it was observed that A4* has a lower RMSF than A4, which implied an increased rigidity of A4*.
PSSE analysis
PSSE plots of apoprotein, A4, and A4* bound protein were monitored during simulation [Fig. S46, ESI†]. The orange-colored bars represent the α-helix. A marginal yet steady rise in the α-helicity (65.26% to 65.52% to 65.90%) was observed as we moved from apoprotein to A4–HSA to A4*–HSA. Noticeably, a similar observation was registered in the CD spectrum, i.e., a marginal rise in α-helix content. This observation may corroborate the lowering of RMSF of the protein upon binding to A4 and A4*.Ligand torsion plot analysis
The ligand torsion profile depicts the conformational evolution of each rotatable bond in the ligand as it moves during simulation. A 2-D structure showing the ligand with different color codes for the single bonds is shown in Fig. 11. The radial plot depicts the conformation of the rotatable bond throughout the simulation. The center of the dial represents the start of the simulation. With the progression of time, the torsion evolution spreads outwards. The bar plot summarises the data of the radial plot by binning the probability density of the torsions from the simulation, thereby providing us with a quantitative picture. The bar plot and the radial plots follow the same color code. The x-axis in the bar plot represents the dihedral angles of the ligand, and the y-axis represents the ligand population assuming/adopting that particular angle during the simulation. Multiple uniform peaks in the bar plot and the corresponding completely colored radial plot show the ease of free rotation and the subsequent conformations these moieties adopt while interacting with the protein. On the other hand, bonds that exhibit a rather restricted rotation exhibit radial plots colored only in a specific region.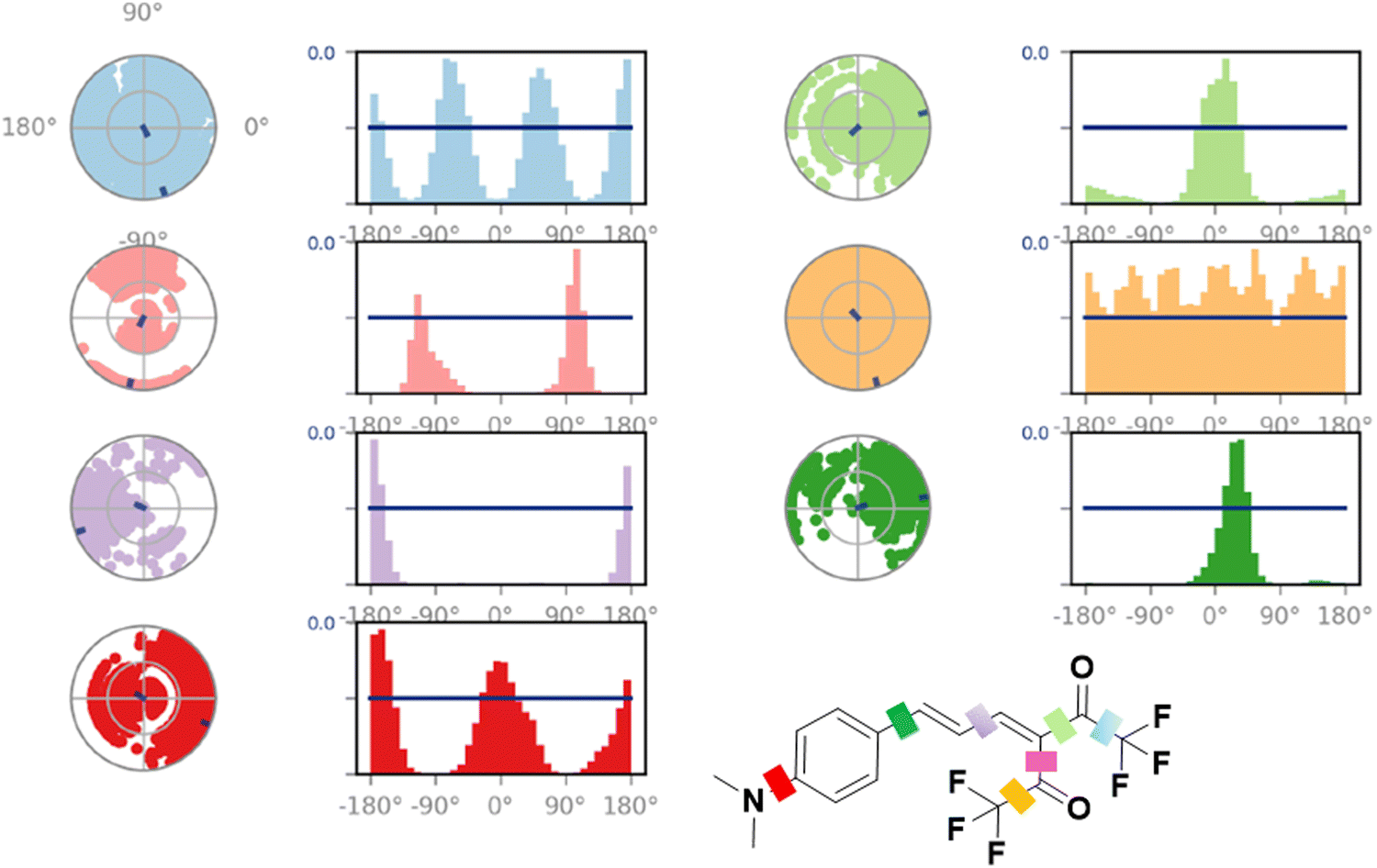 | ||
| Fig. 11 Torsion plots of the A4* ligand inside the protein pocket. | ||
Upon comparing A4 and A4* plots, we see a significant conformational difference in the bond between the phenyl ring and the π linker, depicted in a dark green color. In the case of A4, this bond shows a 180° rotation clockwise and counterclockwise [Fig. S47, ESI†], but in the case of A4*, the rotation/twisting of this bond is restricted severely [Fig. 11]. The torsional potential of A4* is lowest in the range of 0 to 60°, as evident from the bar plot. A holistic view of the torsion plots of A4 and A4* shows that the latter adopts and maintains a more rigid structure throughout the simulation. This observation reveals an overall increase in the restriction of motion in A4* during its interaction with HSA compared to that observed in the case of A4. Owing to this, the intramolecular charge transfer process may be inhibited, which results in the subsequent stabilization and emission from the locally excited state, as observed in the fluorescence experiments.
The simulation data shed light on the stabilizing interactions between the ligand A4/A4* and HSA, which helped lower the RMSD and RMSF of the protein. Ligand RMSF and ligand torsion plot analyses show a comparatively higher rigidity of A4* than that of A4 in the protein pocket during the course of the simulation. A comparison was made between A4 and A4* interactions with the amino acid residues inside the hemin binding pocket. This showed that the ground state structure A4 had one strong cation–π interaction with arginine 186 that was persistent throughout the simulation (90% of the time) [Fig. S50, ESI†]. On the other hand, A4* showed multiple prominent hydrophobic interactions for a comparatively shorter time span [Fig. 12].
 | ||
| Fig. 12 Histogram representing protein residues interacting with ligand A4* in each trajectory frame during the simulation. | ||
The protein–ligand contact histogram
The HSA–A4 contacts can be understood from the protein–ligand contact histogram. For the interaction between A4 and HSA [Fig. S50, ESI†], it was observed that Arg186 and His146 bind to A4 90% of the time and 30% of the time, respectively, with minor contributions from Arg145, Ile142, Leu115, and Pro110 [Fig. S50 and S51, ESI†]. The water bridges reflect the interaction between ligand A4 and HSA mediated by water. These are shown using dark blue labels in the protein–ligand histogram plot. The geometric criteria currently accepted for a ligand–water or a protein–water hydrogen bond are a distance of 2.8 Å between the donor and acceptor atoms (D–H⋯A), a donor angle ≥110° between the donor–hydrogen–acceptor atoms (D–H⋯A), and an acceptor angle ≥90° between the hydrogen–acceptor–bonded atom (H–A⋯B).53 Water bridges were mainly formed by His146 and Arg186, Leu182 and Ser193. The protein–ligand contact plot showed that Arg186 interacts with the ligand A4 more or less throughout the simulation time, followed by His146 and Arg145 [Fig. S52, ESI†]. Cation–π interactions were observed between positively charged guanidine nitrogens of Arg186 and Arg145 residues and the phenyl ring of A4 [Fig. S51a and b, ESI†]. It is well known that cation–π interactions are fairly strong electrostatic interactions, with approximately 1.5–2.5 kcal mol−1 energy released during the interaction. The known order of the strength of the interaction between the protein and the ligand is salt bridges > H-bonds/halogen bonds > cation–π > π–π.54 This study also showed that the C-2 in the imidazole ring of His146 formed a tetrel bond with one of the fluorines in the CF3 moiety of A4 [Fig. S51(a), ESI†]. Interestingly, the likelihood of this interaction was hypothesized when the probe structure was initially proposed. The Lig plot also shows the various hydrophobic interactions with the amino acid residues in the vicinity, i.e., Tyr161, Leu182, Leu185, Ile142, Phe157, Phe149, Pro110, and Leu112 [Fig. S51(b), ESI†].The binding mode of ligand A4* differed significantly from that observed for A4. Here, the major hydrophobic interactions are shown by Tyr161, Met123, Pro118, and Leu115 [Fig. S53, ESI†]. The water bridges and H-bonding are shown by Arg186 and His146 [Fig. 12 and 13]. In the case of A4*, it is also important to note the lowered interaction fraction compared to that observed for A4. For example, Tyr161, which shows the highest interaction fraction among all the other participating residues, interacts with A4* for only close to 50% of the simulation time [Fig. 12].
 | ||
| Fig. 13 Snapshot of a trajectory frame showing HSA–A4* interaction and its corresponding ligand plot. | ||
Quantification of HSA in urine
As discussed initially, the extent of kidney damage can be evaluated from the serum albumin concentration in urine. Most reported probes use extremely diluted urine as the emission wavelength of the probe overlaps with the region where many other biological entities present in the urine tend to auto-fluoresce. This is mainly observed in the lower wavelength regions. Considering the probe's ability to sense HSA in the nanomolar range and its near-red emitting ability, we used it to quantify serum albumin in real urine samples. Titration experiments were conducted using real human urine spiked with very low concentrations of HSA ranging between 0.05 and 0.3 μM. The subsequent introduction of A4 led to a linear rise in the fluorescence intensity of the HSA–A4 composite [Fig. 14 and Fig. S55, ESI†]. This observation shows the suitability of A4 to detect and estimate HSA in urine.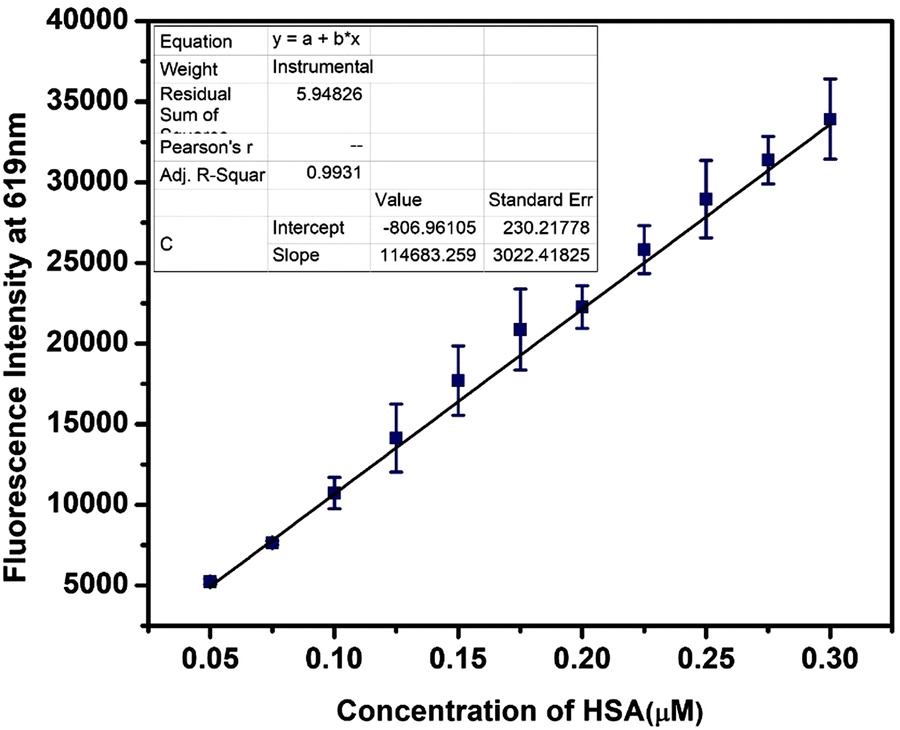 | ||
| Fig. 14 The change in the emission intensity of A4 (5 μM) upon the addition of HSA (0 to 0.3 μM) in a 10-fold diluted urine sample in PBS (pH = 7.4). | ||
Detection of hemin
Hemin, an iron-containing porphyrin compound derived from red blood cells, serves multiple functions, such as regulating ion channels, controlling gene expression, controlling hemoglobin synthesis, and preventing cardiac dysfunction.55 It also assists as an electron transfer medium due to the reversible redox interconversion of Fe(III)/Fe(II) and is used to treat iron deficiency. The bacteriostatic effects of hemin are also well known.56–58 Excess of hemin can also have harmful effects, as it oxidizes proteins, nucleic acids, cell membrane components, and lipids and causes cell lysis and death.59 There are reports on hemin-mediated oxidation of low-density lipids that increase the risk of atherosclerosis.55Several methods for hemin detection have been proposed, including spectroscopic, electrochemical, voltammetric, fluorimetric, and chemiluminescence techniques.60 Here, we propose hemin detection using the A4–HSA ensemble. This proposition is based on the previous displacement assay results where hemin could displace A4 from its complex with HSA, resulting in a turn-off fluorescence. Before using the A4–HSA ensemble, it was necessary to determine the stoichiometry of binding between A4 and HSA. The fluorescence titration experiment of the probe with HSA showed that the probe binds to the protein in a 1:1 ratio [Fig. S56, ESI†]. These results were further corroborated by a linear Stern–Volmer plot and the slope value of the modified Stern–Volmer plot [Fig. S57 and S58, ESI†]. Once the binding stoichiometry was obtained, the 1:1 HSA–A4 complex was used for hemin detection.
Fluorescence quenching of HSA–A4 by gradual addition of hemin
Herein, equal concentrations (5 μM) of HSA and A4 were mixed in PBS buffer and incubated for 20 minutes, and then emission was recorded. Upon stabilization of the fluorescence signal, hemin was added gradually to this solution up to 10 μM concentration, which resulted in the quenching of the signal [Fig. 15(a) and Fig. S59, ESI†]. | ||
| Fig. 15 (a) The fluorescence quenching of the HSA–A4 ensemble upon incremental addition of hemin. (b) Change in the emission intensity of the A4–HSA ensemble upon addition of various cations (100 μM) in PBS (pH 7.4). | ||
Hemin, being the more potent ligand, displaces the weaker ligand A4 from the binding pocket of HSA. The initial weak ligand (A4)–protein (HSA) ensemble is emissive; when the ensemble breaks, the fluorescence gets quenched as A4 is displaced by hemin. The hemin–HSA complex, on the other hand, is non-emissive in the region where the A4–HSA ensemble emits.
A4, when present alone, does not show any quenching or enhancement in its emission signal intensity (A4 inherently is non-fluorescent in PBS). The fluorescence intensity (A4–HSA–hemin ensemble) was then plotted against the hemin concentration, which exhibited a linear decrease [Fig. S59, ESI†]. It also allowed the calculation of the LOD and LOQ values of 0.23 μM and 0.66 μM, respectively.
The HSA–A4 ensemble was tested against well-known entities such as amino acids, ions, glucose, urea, and hemoglobin [Fig. 15b]. While a few showed an emission enhancement, none showed quenching of the signal except hemin. Similar experiments were also performed where hemin was introduced into the media containing A4–HSA along with the above-mentioned entities. In all the cases, the fluorescence remained quenched except when Fe2+ was present [Fig. S60, ESI†]. This result demonstrates the selectivity of HSA–A4 towards hemin and its suitability for hemin detection in biological systems.
Conclusions
In this work, we have demonstrated the efficacy of an ICT exhibiting D–π–A system, A4, for nanomolar detection (1.36 nM) of HSA. The selective site of binding of the probe within HSA was shown via displacement assay and was further substantiated by docking studies. Molecular docking experiments also highlighted A4's preference for HSA over BSA. The simulation experiments clearly show the tetrel bond-forming ability of the CF3 group available in the molecule in accordance with the initial design hypothesis of the probe. The simulation experiments were also conducted using the excited state structure of the ligand, A4*. These experiments showed that compared to A4, A4* maintained a more rigid structure throughout the simulation and exhibited greater hydrophobic interactions with the amino acid residues. The probe was applied to detect serum albumin levels in spiked urine samples, and the A4–HSA ensemble was used to detect hemin. The limit of detection for hemin was observed to be 0.23 μM. Given the importance of ascertaining serum albumin levels in clinical studies, work is currently undertaken in our lab to structurally modify the developed scaffold to further enhance its sensitivity and selectivity towards HSA.Data availability
All the data obtained during this study are included in the main manuscript and the ESI† portion of this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DST-SERB is acknowledged for the research grant CRG/2022/002175. The authors would like to thank the Central Analytical Laboratory and the high-performance computing facility of the BITS-Pilani Hyderabad campus for helping to obtain the spectroscopic and computational data and Dr Abhijit Kayal, a Senior Scientist from Schrodinger Inc., for constant support with our queries related to the software.References
- G. J. Quinlan, G. S. Martin and T. W. Evans, Albumin: biochemical properties and therapeutic potential, Hepatology, 2005, 41(6), 1211 CrossRef CAS PubMed.
- T. R. Guizado, Analysis of the structure and dynamics of human serum albumin, J. Mol. Model., 2014, 20(10), 2450 CrossRef PubMed.
- G. Fanali, A. di Masi, V. Trezza, M. Marino, M. Fasano and P. Ascenzi, Human serum albumin: from bench to bedside, Mol. Aspects Med., 2012, 33(3), 209 CrossRef CAS PubMed.
- C. E. Ha and N. V. Bhagavan, Novel insights into the pleiotropic effects of human serum albumin in health and disease, Biochim. Biophys. Acta, 2013, 1830(12), 5486 CrossRef CAS PubMed.
- S. P. Clavant, T. M. Osicka and W. D. Comper, Albuminuria: Its Importance in Disease Detection, Lab. Med., 2007, 38(1), 35–38 CrossRef.
- B. K. Paul, N. Ghosh and S. Mukherjee, Interplay of Multiple Interaction Forces: Binding of Norfloxacin to Human Serum Albumin, J. Phys. Chem. B, 2015, 119(41), 13093 CrossRef CAS PubMed.
- M. Bernardi, P. Angeli, J. Claria, R. Moreau, P. Gines, R. Jalan, P. Caraceni, J. Fernandez, A. L. Gerbes, A. J. O'Brien, J. Trebicka, T. Thevenot and V. Arroyo, Albumin in decompensated cirrhosis: new concepts and perspectives, Gut, 2020, 69(6), 1127–1138 CrossRef CAS PubMed.
- J. J. Ho, A. S. Adnan, Y. C. Kueh, N. J. Ambak, H. Van Rostenberghe and F. Jummaat, Human albumin infusion for treating edema in people with nephrotic syndrome, Cochrane Database Syst. Rev., 2019, 7(7), CD009692 Search PubMed.
- S. Choi, E. Y. Choi, D. J. Kim, J. H. Kim, T. S. Kim and S. W. Oh, A rapid, simple measurement of human albumin in whole blood using a fluorescence immunoassay (I), Clin. Chim. Acta, 2004, 339(1–2), 147 CrossRef CAS PubMed.
- A. E. van de Logt, S. R. Rijpma, C. H. Vink, E. Prudon-Rosmulder, J. F. Wetzels and M. van Berkel, The bias between different albumin assays may affect clinical decision-making, Kidney Int., 2019, 95(6), 1514–1517 CrossRef CAS PubMed.
- A. Regeniter and W. H. Siede, Peaks and tails: Evaluation of irregularities in capillary serum protein electrophoresis, Clin. Biochem., 2018, 51, 48–55 CrossRef CAS PubMed.
- M. Lettieri, P. Palladino, S. Scarano and M. Minunni, Protein-templated copper nanoclusters for fluorimetric determination of human serum albumin, Mikrochim. Acta, 2021, 188(4), 116 CrossRef CAS PubMed.
- A. Bandyopadhyay, A. Kumar Shukla, P. Singh, A. Mahale, S. Sharma Irukuvajjula, R. Vadrevu, O. P. Kulkarni, M. Dixit and A. Bhattacharya, Nickel-assisted selective detection of histidine and histidine-rich proteins via an ON-OFF-ON fluorescent probe and its imaging in live cells, J. Photochem. Photobiol., A, 2023, 443 Search PubMed.
- X. Liu, S. Qi, J. Mao, Y. Zhang, B. Dong, B. Song and L. Ji, A supramolecular ratiometric fluorescent probe for the detection of human serum albumin, Dyes Pigm., 2024, 224 CAS.
- A. Bandyopadhyay, R. Hazra, D. Roy and A. Bhattacharya, HSA over BSA: Selective detection of Human Serum Albumin via a naphtho [2,1-b] furan-based system, Chem. – Asian J., 2024, 19(5), e202301055 CrossRef CAS PubMed.
- K. P, B. Chakraborty, V. Rani and A. L. Koner, Rationally designed far-red emitting styryl chromones and a magnetic nanoconjugate for strip-based 'on-site' detection of metabolic markers, J. Mater. Chem. B, 2022, 10(26), 5071–5085 RSC.
- Y.-R. Wang, L. Feng, L. Xu, J. Hou, Q. Jin, N. Zhou, Y. Lin, J.-N. Cui and G.-B. Ge, An ultrasensitive and conformation sensitive fluorescent probe for sensing human albumin in complex biological samples, Sens. Actuators, B, 2017, 245, 923–931 CrossRef CAS.
- H. Xu, G. Shen, C. Peng, X. Han, L. Duan and T. Cheng, BODIPY-based fluorescent probe for selective detection of HSA in urine, Dyes Pigm., 2022, 197 Search PubMed.
- Y. J. Xu, M. M. Su, H. L. Li, Q. X. Liu, C. Xu, Y. S. Yang and H. L. Zhu, A fluorescent sensor for discrimination of HSA from BSA through selectivity evolution, Anal. Chim. Acta, 2018, 1043, 123–131 CrossRef CAS PubMed.
- Z. Huang, H. Wang and W. Yang, Gold Nanoparticle-Based Facile Detection of Human Serum Albumin and Its Application as an INHIBIT Logic Gate, ACS Appl. Mater. Interfaces, 2015, 7(17), 8990 CrossRef CAS PubMed.
- J. Du, Q. Gu, J. Chen, J. Fan and X. Peng, A novel fluorescent probe for the ratiometric recognition of protein based on intramolecular charge transfer, Sens. Actuators, B, 2018, 265, 204–210 CrossRef CAS.
- H. Li, Q. Yao, J. Fan, J. Du, J. Wang and X. Peng, An NIR fluorescent probe of uric HSA for renal diseases warning, Dyes Pigm., 2016, 133, 79–85 CrossRef CAS.
- Y. Tu, Y. Yu, Z. Zhou, S. Xie, B. Yao, S. Guan, B. Situ, Y. Liu, R. T. K. Kwok, J. W. Y. Lam, S. Chen, X. Huang, Z. Zeng and B. Z. Tang, Specific and Quantitative Detection of Albumin in Biological Fluids by Tetrazolate-Functionalized Water-Soluble AIEgens, ACS Appl. Mater. Interfaces, 2019, 11(33), 29619–29629 CrossRef CAS PubMed.
- H. Yanai, Y. Terajima, F. Kleemiss, S. Grabowsky and T. Matsumoto, Reversing the Bond Length Alternation Order in Conjugated Polyenes by Substituent Effects, Chemistry, 2023, 29(15), e202203538 CrossRef CAS PubMed.
- S. Mayarambakam, M. R. Busireddy, K. Sekar and V. J. Rao, Facile synthesis of A-π-D-π-A architecture organic small molecules. Experimental and theoretical investigation of the effect of a π-conjugated spacer, Asian J. Org. Chem., 2023, 12, 5 CrossRef.
- S. Namuangruk, K. Sirithip, R. Rattanatwan, T. Keawin, N. Kungwan, T. Sudyodsuk, V. Promarak, Y. Surakhot and S. Jungsuttiwong, Theoretical investigation of the charge-transfer properties in different meso-linked zinc porphyrins for highly efficient dye-sensitized solar cells, Dalton Trans., 2014, 43(24), 9166 RSC.
- O. Britel, A. Fitri, A. T. Benjelloun, A. Slimi, M. Benzakour and M. McHarfi, Theoretical investigation of the influence of π-spacer on photovoltaic performances in carbazole-based dyes for dye-sensitized solar cells applications, J. Photochem. Photobiol., A, 2022, 428 Search PubMed.
- Y. Sekkat, A. Fitri, O. Britel, A. T. Benjelloun, M. Benzakour and M. McHarfi, A theoretical study on the role of the pi-spacer in the thoughtful design of good light-absorbing dyes with phenothiazine for efficient dye-sensitized solar cells (DSSCs), J. Mol. Model., 2023, 30(1), 5 CrossRef PubMed.
- M. Bourass, A. El Alamy and M. Bouachrine, Structural and photophysical studies of triphenylamine-based nonlinear optical dyes: effects of π-linker moieties on the D-π-A structure, C. R. Chim, 2019, 22(5), 373–385 CrossRef CAS.
- Y. Chen, X. Wan and G. Long, High Performance Photovoltaic Applications Using Solution-Processed Small Molecules, Acc. Chem. Res., 2013, 46(11), 2645–2655 CrossRef CAS PubMed.
- V. G. Klein, A. G. Bond, C. Craigon, R. S. Lokey and A. Ciulli, Amide-to-Ester Substitution as a Strategy for Optimizing PROTAC Permeability and Cellular Activity, J. Med. Chem., 2021, 64(24), 18082–18101 CrossRef CAS PubMed.
- S. Scheiner, Comparison of CH⋯O, SH⋯O, Chalcogen, and Tetrel Bonds Formed by Neutral and Cationic Sulfur-Containing Compounds, J. Phys. Chem. A, 2015, 119(34), 9189 CrossRef CAS PubMed.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore, J. Med. Chem., 2010, 53(22), 7902 CrossRef CAS PubMed.
- K. S. M. Salih, ChemistryOpen, 2022, 11, e202100237 CrossRef CAS PubMed.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef PubMed.
- A. Ekbote, S. H. Han, T. Jadhav, S. M. Mobin, J. Y. Lee and R. Misra, Stimuli responsive AIE active positional isomers of phenanthroimidazole as non-doped emitters in OLEDs, J. Mater. Chem. C, 2018, 6(8), 2077–2087 RSC.
- L. Li, R. Wang, L. Wang and L. Huang, Development of phenothiazine-based fluorescent probe with aggregation induced emission (AIE) for detection of hydrazine and its application in imaging of living cells, J. Mol. Struct., 2022, 1249 Search PubMed.
- K. S. Jagadhane, S. R. Bhosale, D. B. Gunjal, O. S. Nille, G. B. Kolekar, S. S. Kolekar, T. D. Dongale and P. V. Anbhule, Tetraphenylethene-Based Fluorescent Chemosensor with Mechanochromic and Aggregation-Induced Emission (AIE) Properties for the Selective and Sensitive Detection of Hg(2+) and Ag(+) Ions in Aqueous Media: Application to Environmental Analysis, ACS Omega, 2022, 7(39), 34888–34900 CrossRef CAS PubMed.
- S. Pei, J. Li, N. Kang, G. Zhang, B. Zhang, C. Zhang and S. Shuang, Synthesis of a new environment-sensitive fluorescent probe based on TICT and application for detection of human serum albumin and specific lipid droplets imaging, Anal. Chim. Acta, 2022, 1190, 339267 CrossRef CAS PubMed.
- S. Pei, J. Li, C. Zhang, G. Zhang, Y. Zhou, L. Fan, W. Wang, S. Shuang and C. Dong, TICT-Based Microenvironment-Sensitive Probe with Turn-on Red Emission for Human Serum Albumin Detection and for Targeting Lipid Droplet Imaging, ACS Biomater. Sci. Eng., 2022, 8(1), 253–260 CrossRef CAS PubMed.
- G. Kuang, N. A. Murugan, Y. Zhou, A. Nordberg and H. Ågren, Computational Insight into the Binding Profile of the Second-Generation PET Tracer PI2620 with Tau Fibrils, ACS Chem. Neurosci., 2020, 11(6), 900–908 CrossRef CAS PubMed.
- E. Gremese, D. Bruno, V. Varriano, S. Perniola, L. Petricca and G. Ferraccioli, J. Clin. Med., 2023, 12 Search PubMed.
- J. Fan, W. Sun, Z. Wang, X. Peng, Y. Li and J. Cao, Chem. Commun., 2014, 50, 9573–9576 RSC.
- A. Kurtul, S. N. Murat, M. Yarlioglues, M. Duran, A. H. Ocek, C. Koseoglu, I. E. Celık, A. Kilic and O. Aksoy, Angiology, 2016, 67, 34–40 CrossRef CAS PubMed.
- B. K. Paul, N. Ghosh and S. Mukherjee, Interplay of Multiple Interaction Forces: Binding of Norfloxacin to Human Serum Albumin, J. Phys. Chem. B, 2015, 119(41), 13093–13102 CrossRef CAS PubMed.
- A. Albini and S. Monti, Photophysics and photochemistry of fluoroquinolones, Chem. Soc. Rev., 2003, 32(4), 238–250 RSC.
- M. Y. Berezin and S. Achilefu, Fluorescence Lifetime Measurements and Biological Imaging, Chem. Rev., 2010, 110(5), 2641–2684 CrossRef CAS PubMed.
- D. De and A. Datta, Unique Effects of Aerosol OT Lamellar Structures on the Dynamics of Guest Molecules, Langmuir, 2013, 29(25), 7709–7714 CrossRef CAS PubMed.
- D. Sarkar, A. Mahata, P. Das, A. Girigoswami, D. Ghosh and N. Chattopadhyay, Deciphering the perturbation of serum albumins by a ketocyanine dye: A spectroscopic approach, J. Photochem. Photobiol., B, 2009, 96(2), 136–143 CrossRef CAS PubMed.
- B. Ranjbar and P. Gill, Circular dichroism techniques: biomolecular and nanostructural analyses- a review, Chem. Biol. Drug Des., 2009, 74(2), 101 CrossRef CAS PubMed.
- R. Choudhury, A. K. Sharma, P. Paudel, P. Wilson and A. B. Pereira, In situ generation of a Zwitterionic fluorescent probe for detection of human serum albumin protein, Anal. Biochem., 2022, 646, 114630 CrossRef CAS PubMed.
- H. Maity, M. Maity, M. M. G. Krishna, L. Mayne and S. W. Englander, Protein folding: The stepwise assembly of foldon units, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(13), 4741–4746 CrossRef CAS PubMed.
- S. Beura and P. Chetti, In-silico strategies for probing chloroquine based inhibitors against SARS-CoV-2, J. Biomol. Struct. Dyn., 2021, 39(10), 3747–3759 CrossRef CAS PubMed.
- D. A. Dougherty, The cation-pi interaction, Acc. Chem. Res., 2013, 46(4), 885 CrossRef CAS PubMed.
- T. H. Fereja, S. A. Kitte, W. Gao, F. Yuan, D. Snizhko, L. Qi, A. Nsabimana, Z. Liu and G. Xu, Artesunate-luminol chemiluminescence system for the detection of hemin, Talanta, 2019, 204, 379–385 CrossRef CAS PubMed.
- Y. Lin, M. Li, L. Huang, W. Shen and Y. Ren, Involvement of heme oxygenase-1 in β-cyclodextrin–hemin complex-induced cucumber adventitious rooting process, Plant Cell Rep., 2012, 31(9), 1563–1572 CrossRef CAS PubMed.
- B. Panijpan, C. Mohan Rao and D. Balasubramanian, Interaction of antimalarial drugs with hemin, Biosci. Rep., 1983, 3(12), 1113–1117 CrossRef CAS PubMed.
- J. M. Combrinck, T. E. Mabotha, K. K. Ncokazi, M. A. Ambele, D. Taylor, P. J. Smith, H. C. Hoppe and T. J. Egan, Insights into the Role of Heme in the Mechanism of Action of Antimalarials, ACS Chem. Biol., 2013, 8(1), 133–137 CrossRef CAS PubMed.
- Z. Guo, B. Li, Y. Zhang, Q. Zhao, J. Zhao, L. Li, L. Feng, M. Wang, X. Meng and G. Zuo, Acid-treated Graphitic Carbon Nitride Nanosheets as Fluorescence Probe for Detection of Hemin, ChemistrySelect, 2019, 4(28), 8178–8182 CrossRef CAS.
- L. Zhao, F. Chen, W. Huang, H. Bao, Y. Hu, X.-A. Huang, T. Deng and F. Liu, A fluorescence turn-on assay for simple and sensitive determination of hemin and blood stains, Sens. Actuators, B, 2020, 304 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01229a |
| This journal is © The Royal Society of Chemistry 2024 |