
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme-responsive nanoparticles: enhancing the ability of endolysins to eradicate Staphylococcus aureus biofilm†
Mariana
Blanco Massani
 a,
Dennis
To
a,
Susanne
Meile‡
b,
Mathias
Schmelcher
b,
David
Gintsburg
a,
Débora C.
Coraça-Huber
c,
Anna
Seybold
d,
Martin
Loessner
b and
Andreas
Bernkop-Schnürch
*a
a,
Dennis
To
a,
Susanne
Meile‡
b,
Mathias
Schmelcher
b,
David
Gintsburg
a,
Débora C.
Coraça-Huber
c,
Anna
Seybold
d,
Martin
Loessner
b and
Andreas
Bernkop-Schnürch
*a
aCentre for Chemistry and Biomedicine (CCB), Department of Pharmaceutical Technology, Institute of Pharmacy, University of Innsbruck, Innrain 80/82, 6020 Innsbruck, Austria. E-mail: Andreas.Bernkop@uibk.ac.at
bInstitute of Food, Nutrition and Health, ETH Zürich, Schmelzbergstrasse 7, 8092 Zürich, Switzerland
cBiofilm Lab, Experimental Orthopedics, University Hospital for Orthopaedics and Traumatology, Medical University Innsbruck, Müllerstrasse 44, 1. Floor, 6020 Innsbruck, Austria
dDepartment of Zoology, University of Innsbruck, 6020, Innsbruck, Austria
First published on 12th September 2024
Abstract
Stimuli-responsive nanomaterials show promise in eradicating Staphylococcus aureus biofilm from implants. Peptidoglycan hydrolases (PGHs) are cationic antimicrobials that can be bioengineered to improve the targeting of persisters and drug-resistant bacteria. However, these molecules can be degraded before reaching the target and/or present limited efficacy against biofilm. Therefore, there is an urgent need to improve their potency. Herein, PGH–polyphosphate nanoparticles (PGH–PP NPs) are formed by ionotropic gelation between cationic PGHs and anionic polyphosphate, with the aim of protecting PHGs and delivering them at the target site triggered by alkaline phosphatase (AP) from S. aureus biofilm. Optimized conditions for obtaining M23–PP NPs and GH15–PP NPs are presented. Size, zeta potential, and transmission electron microscopy imaging confirm the nanoscale size. The system demonstrates outstanding performance, as evidenced by a dramatic reduction in PGHs’ minimum inhibitory concentration and minimum bactericidal concentration, together with protection against proteolytic effects, storage stability, and cytotoxicity towards the Caco-2 and HeLa cell lines. Time-kill experiments show the great potential of these negatively charged delivery systems in overcoming the staphylococcal biofilm barrier. Efficacy under conditions inhibiting AP proves the enzyme-triggered delivery of PGHs. The enzyme-responsive PGH–PP NPs significantly enhance the effectiveness of PGHs against bacteria residing in biofilm, offering a promising strategy for eradicating S. aureus biofilm.
1. Introduction
Opportunistic Staphylococcus aureus, causative of extensive morbidity and mortality globally, can be acquired during implant surgery, and compromised immunity may lead to persistent infections at the implant–tissue interface.1 Pathogenesis of S. aureus infections in native tissues is linked to biofilm formation on implant devices, which have a propensity to chronicity through increased antimicrobial resistance and immune evasion.1,2 The S. aureus biofilm matrix is mainly composed of exopolysaccharides, extracellular DNA (eDNA) and several proteins showing pI lower than 7.4.3 This dense matrix hinders the penetration of antibacterial drugs.4In the current antibiotic crisis, the use of enzybiotics like phage endolysins have emerged as a potential alternative to treat S. aureus infections.2,5–7 These peptidoglycan hydrolases (PGHs) are part of the fascinating development of biotechnology inspired by phage nature.8 They degrade the peptidoglycan from within the cell at the end of the lytic cycle to disrupt the cell wall and release phage progeny.9,10 This can effectively kill bacteria allowing for host-specific antimicrobial activity, together with the improved targeting of drug-resistant bacteria and metabolically inactive persisters.5,6
PGHs can be engineered to enhance their antimicrobial efficacy.9 This was proven for M23LST(L)_SH3b2638A (M23) and CHAPGH15_SH3bALE1 (GH15) against multiple staphylococcal strains in different infection models.2,5,6,11
Even when the activity of endolysins against biofilm has been demonstrated,7,12 their full potential has not been reached yet. In particular, instability and susceptibility towards peptidases and proteases cause their rapid degradation.13
Furthermore, because of the cationic charge of antimicrobial proteins, they show poor penetration in microbial biofilms exhibiting a pronounced anionic net charge.14 Therefore, bioavailability at the target site is too low to eradicate the pathogen residing in biofilm.
Drug delivery through stimuli-responsive materials represents an effective antibacterial strategy to overcome bioavailability limitations, while fighting the current antibiotic crisis. Viewing Nature as a source of inspiration, stimuli-responsive materials are designed to mimic the features of the natural environment of a cell.15 Stimuli-responsive antibacterial materials (SRAMs) eradicate bacteria at the target site in a self-defensive manner upon stimuli caused by bacteria.
SRAMs comprising liposomes16 and poly(N-isopropylacrylamide) nanoparticles17 have been used for the delivery of endolysins. However, none of these materials showed improvement of efficacy dramatically reducing the concentrations required for biofilm eradication.
Polyphosphate nanoparticles (PP NPs) are a type of SRAMs undergoing changes in their chemical and/or physical properties upon degradation by alkaline phosphatase (AP) enzymatic activity.18 This delivery strategy proved effective for delivering different cationic molecules and allowing them to bypass anionic obstacles such as the mucus barrier, or microbial biofilm to reach their action target.14,19–22 Since AP is expressed in planktonic and biofilm cells from staphylococci,23,24 we reasoned that PGH polyphosphate nanoparticles (PGH–PP NPs) constitute a ‘raise one's game’ approach for protecting PGHs and delivering them on site upon trigger by the enzyme. Furthermore, to the best of our knowledge, exploitation of AP-response for the design of a S. aureus biofilm-targeting delivery system has not been performed yet.
Accordingly, we screened different conditions for optimizing the production of M23–PP NPs and GH15–PP NPs. Size, zeta potential, TEM imaging, minimum inhibitory concentration (MIC), minimum bactericidal concentration (MBC), stability under storage, and cytotoxicity of the NPs toward Caco-2 and HeLa cell lines were studied. As a proof of concept, time-kill experiments and protection against proteolytic effects were assessed to show the great technological potential of these delivery systems. To prove the target specific activity through AP mediated release, changes in antimicrobial efficacy under conditions inhibiting this enzyme were studied.
2. Results and discussion
2.1 Formation of PGH–PP NPs
Based on our previous experience and the literature, key factors influencing polyelectrolyte complexation with antimicrobial proteins or peptides include charge density, ionic strength, concentration, and the ratio between the protein or peptide and the polyelectrolyte.14,25 Table S1 (ESI†) shows the systematic screening performed here to obtain the PGH–PP NPs. Microparticles were obtained at concentrations of 2.5 and 5 μM of M23 and GH15 for all the endolysin-to-PP ratios studied (Table S1, ESI†). In contrast, a PGH concentration of 1.25 μM was suitable for the formation of NPs (Fig. 1(A) and (B)). As described in Table S1 (ESI†), at pH 1.92, the M23 endolysin formed NPs with a charge of 20.85 ± 7.73 mV and a size of 189 ± 112 nm. After dropping the endolysin solution (1.25 μM) into different concentrations of PP solution, a shift in zeta potential towards more negative values was observed for increasing polyphosphate concentrations (Fig. 1(A)). This confirmed the formation of NPs with PP on its surface owing to its dominating concentration over protein.14 | ||
| Fig. 1 Mean size (nm) and SD (bars), PDI (dots) and zeta potential (mV) of (A) M23–PP NPs and (B) GH15–PP NPs as a function of the protein-to-polyphosphate ratio. Encapsulation of each PGH is schematically represented by a negatively charged sphere. Schematics are not drawn to scale. | ||
A molar ratio of protein to polyphosphate of 1 to 7 was chosen for the formation of M23–PP NPs, resulting in a polydispersity index (PDI) lower than 0.3 (Fig. 1(A)), which indicates uniformity in size distribution and that they are moderately polydisperse.13,26
M23 is a protein with 14 negatively charged (Asp + Glu) and 16 positively charged residues (Arg + Lys). NPs of this PGH were formulated at pH 1.92. On one hand, this pH increases the number of charges per molecule of M23 to around +26 as predicted in silico; it is on the other hand in the buffering region of PP.14 Therefore, an increased repulsive potential between the positive charges of M23 and protonated phosphate may lead to a less compact structure with a mean particle size distribution of 229 ± 119 nm (Fig. 1(A)). Additionally, as discussed in the ESI† (Fig. S1, S3 and S4), M23–PP NPs seem to have a mesh arrangement of coiled M23 intercalated with PP. As will be further discussed, this may have a positive impact on antimicrobial activity.
For GH15, a ratio of protein to polyphosphate of 1 to 5 led to a PDI of 0.36 ± 0.04 and a zeta potential of −31.15 ± 5.05 mV (Fig. 1(B)). It is worth noting that the zeta potential of GH15–PP NPs was not significantly changed upon the increase of PP from a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to 1:5. As discussed in the ESI† (Fig. S1, S3 and S4), this could suggest polyphosphate being saturated at the surface of the particles already at a 1:3 ratio of GH15 to PP, and, therefore, more accessible for cleavage by AP. Moreover, GH15 contains 17 negatively charged residues and 21 positively charged residues per molecule, and GH15–PP NPs are formulated at a neutral pH, in which polyphosphate is negatively charged. This enhances its crosslinking ability leading to stable colloidal particles in the nano-size range (148 ± 94 nm), as confirmed by TEM (Fig. S1, ESI†). As will be further discussed, this seems to endow the particles with more stability over storage time.
:3 to 1:5. As discussed in the ESI† (Fig. S1, S3 and S4), this could suggest polyphosphate being saturated at the surface of the particles already at a 1:3 ratio of GH15 to PP, and, therefore, more accessible for cleavage by AP. Moreover, GH15 contains 17 negatively charged residues and 21 positively charged residues per molecule, and GH15–PP NPs are formulated at a neutral pH, in which polyphosphate is negatively charged. This enhances its crosslinking ability leading to stable colloidal particles in the nano-size range (148 ± 94 nm), as confirmed by TEM (Fig. S1, ESI†). As will be further discussed, this seems to endow the particles with more stability over storage time.
The nano-size ranges, PDI and zeta potential obtained after encapsulation of PGHs with polyphosphate are in the ranges of those obtained for other antimicrobial cation–PP NPs.14 Importantly, at least 200-fold lower concentrations of PGHs were used here compared to ionotropic gelation using other polymers.13
2.2 Technological properties of PGH–PP NPs
The technological properties of PGH–PP NPs are presented in Fig. 2. After encapsulating the enzymes in NPs, the minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) of free M23 and GH15 were 8- and 4-fold lower, respectively (Fig. 2(A)). These results show an improvement in the efficacy of PGHs after encapsulation with polyphosphate. Similar results were also found after encapsulating synergistically acting CHAPK endolysin and lysostaphin in SRAMs comprising poly(N-isopropylacrylamide) nanoparticles,17 or using antimicrobial polymers like alginate and chitosan for endolysin encapsulation.13 Here, consistent with the literature, PP alone presented an inhibitory effect against S. aureus ATCC 25923, probably owing to its chelating properties.27 However, at the concentrations used in our study (4.375 μM and 3.125 μM, respectively, for M23–PP NPs and GH15–PP NPs), a bacteriostatic mode of action against this bacterium was found (Table S2, ESI†). A synergistic bactericidal mode of action of antimicrobials was previously reported when combining bacteriostatic and bactericidal agents.28 Here, the synergistic bactericidal mode of action of PGH–PP NPs may be attributed to the large surface area-to-volume ratio of NPs that may provide better interactions of PP with the cell wall, together with the high macromolecular concentration in the M23–PP complexes,29 plus the protective effects of NPs as will be further discussed (Section 2.3). | ||
| Fig. 2 (A) MIC, MBC and size and zeta potential as a function of storage time at 4 °C for (B) M23 and M23–PP NPs and (C) GH15 and GH15–PP NPs. Values are means of at least 3 replicates ± SD. | ||
The size and zeta potential of PGH–PP NPs were determined after storage at 4 °C. M23–PP NPs showed an increase of size with a mean particle size of 1702 nm after 168 h (7 days) and no significant changes in zeta potential (Fig. 2(B)). This indicates the agglomeration of NPs over time that may occur due to the presence of protonated phosphate at the pH of this formulation. For GH15–PP NPs, agglomeration was not evident (Fig. 2(C)). Compared to alginate–chitosan NPs encapsulating PGHs,13 PP–M23 seems to be less stable for avoiding self-aggregation in colloidal suspension for prolonged periods of time. This may limit the shelf life of M23–PP NPs. However, the MIC measured at the end of the storage time was 39.1 nM for M23–PP NPs (7 days at 4 °C) and 156.3 nM for GH15–PP NPs (4 weeks at 4 °C). Hence, agglomeration had no or very limited impact on the antimicrobial stability of the PGH–PP NPs.
Cytotoxicity towards Caco2 and HeLa cells showed that, for all the samples, the viability was above 78%, indicating a nontoxic effect at the assayed concentrations (Fig. S2, ESI†). This is in line with previous results on the application of M23 and GH15 in vivo and in vitro.2,5,6
2.3 Protective effect of NPs towards degradation by proteases
Since S. aureus produces serine proteases as a virulence factor,1 PGHs have to be protected from degradation by these enzymes to avoid efficacy loss. Upon treatment of free M23 with TrypLE, a strong decrease in the antimicrobial efficacy of the endolysin, which is depicted by an increase in MIC, was observed (Fig. 3; the M23/Tr sample compared to the M23/Cont sample). In contrast, after M23–PP NPs were treated with TrypLE, changes in MIC were not significant compared to the control (Fig. 3; M23–PP NPs/Tr and PP NPs/Cont, respectively). As depicted in the figure, this suggests the protection of M23 towards proteolytic degradation and consequently inactivation by serine protease when the endolysin is encapsulated in NPs. | ||
| Fig. 3 MIC changes upon treatment of M23 and M23–PP NPs with serine protease. The indicated values are means (n = 6) ± SD. ***p < 0.001 compared to the respective control. Cont: control, Tr: TrypLE treatment. | ||
2.4 Time-kill assays of PGH–PP NPs
Characterization of S. aureus biofilm regarding composition (Fig. S5) is presented in the ESI.† To prove the efficacy of PGH–PP NPs against staphylococcal biofilm, a concentration lower than the effective MBC (Fig. 2(A)) for free enzyme was chosen. The value is 156.3 nM of endolysin for free M23 and M23–PP NPs (Fig. 4(A)–(D)), and 625 nM for GH15 and GH15–PP NPs (Fig. 5(A)–(D)). In contrast to the self-sterilizing effect observed for other acidic polymers,30 polyphosphate alone presented no antimicrobial effect on bacteria residing in biofilm (Fig. S6, ESI†). | ||
| Fig. 4 (A) Time-kill assay, (B) cell damage, (C) biofilm dispersal, and (D) AP activity in biofilm after different treatments. The indicated values are means of at least 2 replicates ± SD. ***p < 0.001 compared to the control (water treated) at the same time. Red arrows in panel (B) show distortion of cell wall morphology. | ||
 | ||
| Fig. 5 (A) Time-kill assay, (B) cell damage, (C) biofilm dispersal, and (D) AP activity in biofilm after different treatments. The indicated values are means of at least 2 replicates ± SD. **p < 0.05 compared to the control (water treated) at the same time. Red arrows in panel (B) show distortion of cell wall morphology. | ||
Pathogens can adapt to antimicrobial materials and antibiotics.30,31 Therefore, it is imperative to generate strategies dispersing biofilm that immediately kill dispersed bacteria.32,33 In contrast to stimuli-responsive delivery of phage endolysins from liposomes,16 the strategy developed here for the delivery of PGHs did not cause any deleterious effect on killing efficacy. Furthermore, in agreement with our findings on the effect against planktonic bacteria (Fig. 2(A)), PGH–PP NPs outperformed the efficacy of free endolysins. This was observed in terms of the decrease in S. aureus counts in biofilm (Fig. 4(A) and 5(A), respectively, for M23 and GH15) and cell damage (Fig. 4(B) and 5(B)). Free PGHs caused limited (Fig. 4(A)) or no (Fig. 5(A)) killing effect at any time point, respectively, for free M23 or GH15. In contrast, total eradication of S. aureus from biofilm was obtained already at 1 h of treatment with M23–PP NPs (Fig. 4(A)). For GH15–PP NPs, an increase in killing efficacy was noted after 4 h of treatment (Fig. 5(A), (p < 0.05)) compared to GH15. The surface charge of materials plays a key role in governing initial electrostatic interactions at the nano-bio interface.33 These interactions are in turn of paramount importance to reach antimicrobial efficacy.34 Since S. aureus biofilm is composed of proteins with an isoelectric point lower than physiological pH,3 a negative charge of biofilm impairing the action of positively charged PGHs could explain the obtained results, a problem that is visibly circumvented by a shift to a negative charge (Fig. 1) after encapsulation in polyphosphate NPs.14
It should be noted from the particle sizes that the probably high macromolecular concentration in the M23–PP complexes (229 ± 119 nm) than GH15–PP (148 ± 94 nm) seems to be correlated with the stronger efficacy of M23–PP NPs (Fig. 4(A), compared to Fig. 4(B)).
Additionally, as could be observed from the analysis of AP triggered changes in PGH–PP NPs (Fig. S3, ESI†), together with the enzyme triggered phosphate release (Fig. S4, ESI†), a shift in net charge driven by the effective cleavage by AP is crucial for achieving the full potential of these NPs. This, combined with the increased efficacy of M23 compared to GH15 against planktonic and bacteria residing in biofilm (Fig. 2),2,5 makes M23–PP NPs a promising formulation to effectively eradicate S. aureus biofilm already after 1 h of incubation (Fig. 4(A)).
SEM analyses showed the spherical morphology of S. aureus with a size around 1 μm, being embedded in an extracellular matrix (Fig. 4(B) and 5(B), time 0 control). Exposure of bacteria to water did not affect the bacterial structure, but partially eliminated the extracellular matrix from biofilm (Fig. 4(B) – 2 h control and Fig. 5(B) – 4 h control). This is consistent with the literature, which shows that S. aureus and other bacterial species can survive without changes in morphology and/or viability in the biofilm state on the surface of osteosynthesis devices.35,36
Engineered M23 is composed of an M23 endopeptidase domain from lysostaphin and an SH3b cell wall binding domain (CBD) from phage 2638A and showed improved efficacy at reducing multiple staphylococcal strains to undetectable levels in human serum.5,11 Similarly, GH15, consisting of a CHAP endopeptidase domain from LysGH15 and an SH3b CBD from ALE1, showed efficacy under conditions found in different intracellular compartments, and intracellular activity against S. aureus strains in multiple eukaryotic infection models.6,11 Consistent with their lytic mode of action, free M23 and GH15 damaged the cells by causing distortion of cell wall morphology (marked with red arrows in Fig. 4(B) and 5(B)), leading to the presence of extracellular debris. This effect was more evident in the case of M23 and PGH–PP NPs compared to GH15 treatment. Biofilm dispersal was observed for all free PGHs and PGH–PP NPs (Fig. 4(C) and 5(C)). The results obtained here are similar to those previously reported for the combination of M23 with the chimeric endolysin GH15 at a 10-fold higher concentration than that used here.2 This confirms the great potential of these NPs for the delivery of endolysins.
Harnessing the production of enzymes by microorganisms has been a strategy used to modulate antimicrobial release. The main biological function of AP is to supply inorganic phosphate (Pi).37 This enzyme is constitutively expressed on the surface of S. aureus and has been reported to participate in dephosphorylation of different proteins.24,38 In turn, for S. aureus biofilm, AP regulated biofilm formation was suggested, and a correlation between the phosphodiesterase activity of the enzyme and increased biofilm formation has been established.23 We wanted to assess the effect of PGH–PP NPs on AP activity (Fig. 4(D) and 5(D)). No increase of activity upon treatment with NPs was observed for either M23–PP NPs (Fig. 4(D)) or GH15–PP NPs (Fig. 5(D)). This observation is in line with a constitutive mechanism of AP expression.24,38 As the presence of AP is essential for the proper function of PP NPs, altogether these results validate the efficacy of polyphosphate NPs against the biofilms of microorganisms in which the expression of AP is constitutive.
2.5 Enzyme-triggered antistaphylococcal activity
The triggered release of a therapeutic agent relying on external stimuli can be demonstrated by a gating strategy, i.e. assaying the effect when the trigger is present or absent.16,17 The antibiofilm activity of M23 and M23–PP NPs in the presence (Fig. 6(A)) and absence of the AP inhibitor (Fig. 6(B)) is schematically presented in Fig. 6(C) and (D), respectively. For all the samples, a decrease in AP activity between 0.0010 and 0.0015 DEA ml−1 was observed (Fig. S7, ESI†) in the presence of inorganic phosphate (Pi). This is consistent with the role of Pi as an AP activity inhibitor.18,37 When M23–PP NPs were applied in the presence of Pi, no significant differences in lytic activity were observed compared to the control + Pi (Fig. 6(A)). In contrast, significant staphylococcal lytic activity (p < 0.001) was observed for M23–PP NPs without the AP inhibitor (Fig. 6(B)). These results show that a sufficiently high AP activity is necessary for M23–PP NPs to achieve the killing of S. aureus in biofilm. As shown in Fig. 6(C) and (D), this suggests that only in the presence of AP, M23 would be released from M23–PP NPs to effectively kill the bacteria, confirming the enzymatically triggered release. An increase of the efficacy of M23 in the presence of Pi was observed (Fig. 6(A)). A transient protection of the positive charges of M23 by Pi could explain this result. However, the effect was not as remarkable as that observed for M23–PP NPs, highlighting the suitability of polyphosphate for encapsulating endolysins.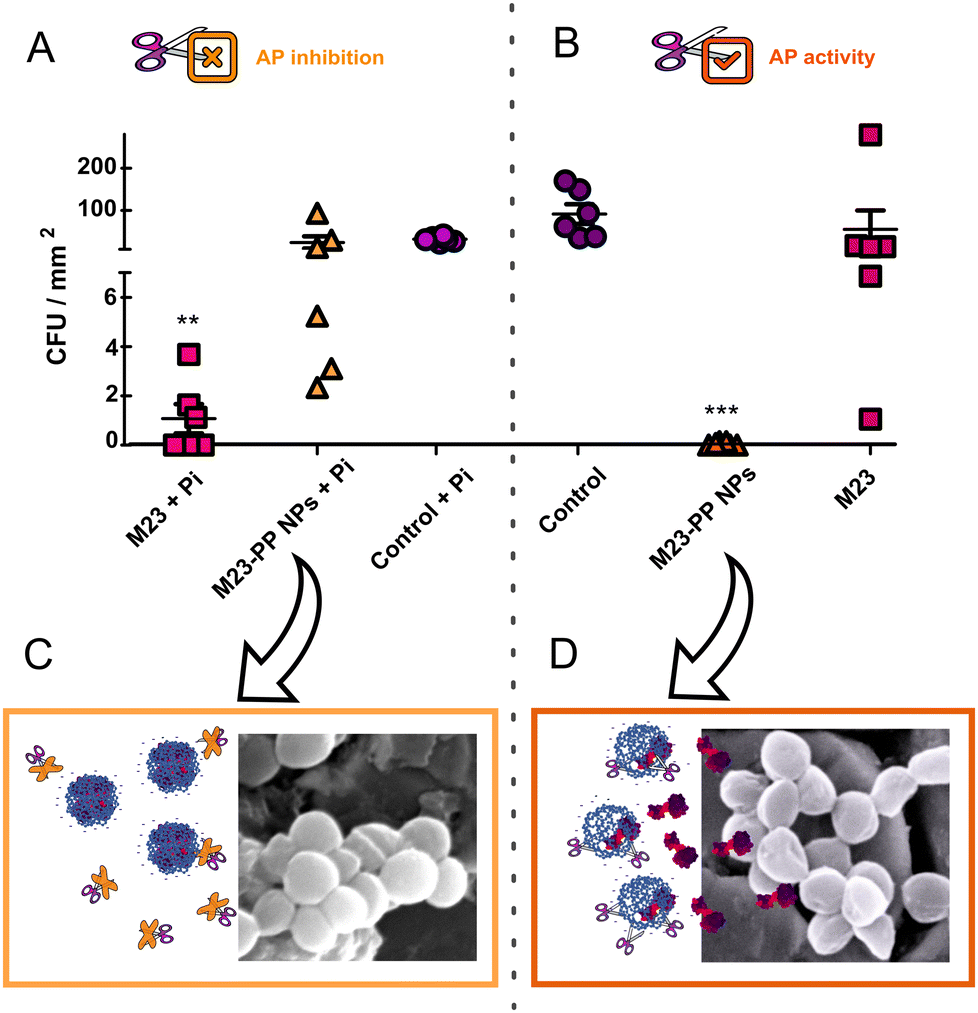 | ||
| Fig. 6 CFU counts after treatment of S. aureus biofilm with M23, M23–PP NPs and control in the (A) presence and (B) absence of Pi. (C), (D) Schematic representation of the efficacy of M23–PP NPs under AP inhibition and in the presence of the enzyme, respectively. The indicated values are means (n = 6) ± SD. **p < 0.05, ***p < 0.001 compared to the control without Pi. | ||
3. Conclusions
The difficulty of eradicating S. aureus residing in biofilm poses a major threat to patients having implant related infections. In line with the current hub for fighting the antibiotic crisis, in this study we designed SRAMs by combining interdisciplinary approaches to avoid drug resistance development. On the one hand, molecular biology and biotechnology converge for the design of engineered PGHs decreasing the chance for resistance development.39 Moreover, unlike most antibiotics, PGHs do not rely on active bacterial metabolism for antimicrobial activity; hence, they can fight S. aureus drug-resistant bacteria and metabolically inactive persisters, together with those residing in biofilm.2,5,6,12 On the other hand, microbiology and pharmaceutical technology converge with nanotechnology to formulate enzyme-responsive NPs that improve the efficacy of PGHs. We successfully developed PGH–PP NPs by a one-step ionotropic gelation method. PP NPs protected PGHs from the environmental proteases that could be present as a virulent factor of S. aureus. Moreover, target specific activity through AP mediated release proved to efficiently eradicate S. aureus biofilm. Aided by nanotechnology, we were able to deliver and dramatically improve the efficacy of PGHs against bacteria residing in biofilm through an enzyme-responsive delivery system.The present work intends to contribute to the growing field of stimuli-responsive materials to fight the current antibiotic crisis. In this fight, cost-effective approaches that provide high efficiency and attractive biocompatibilities for antibacterial materials are required. We anticipate that the approach developed here does not have the bottleneck in scaling up the technology that is faced by other stimuli-responsive delivery systems.40 Moreover, given the specificity of GH15 and M23 towards S. aureus and S. epidermidis,5,6 PGH–PP NPs seem to be a promising delivery system for avoiding the formation of staphylococcal biofilm on implant surfaces without targeting surrounding non-target microbial flora and host tissues.
Author contributions
Mariana Blanco Massani: conceptualization, experimental design, overall data analyses and result interpretation, writing original draft, correction, proofreading, funding acquisition; Dennis To: methodology, cytotoxicity experiments, SEM characterization, visualization, writing – review & editing; Susanne Meile: chimeric phage endolysin production, manuscript correction, proofreading; Mathias Schmelcher: chimeric phage endolysin production, conceptualization and supervision, funding acquisition, manuscript correction, proofreading; David Gintsburg: technical support in phosphate release experiments, proofreading; Anna Seybold: EFTEM analyses; Débora C. Coraça-Huber: SEM analyses, proofreading; Martin Loessner: chimeric phage endolysin production and conceptualization, funding acquisition, manuscript correction, proofreading; Andreas Bernkop-Schnürch: writing – review & editing, funding acquisition, supervision.Data availability
The data related to this study are available in the published article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Mariana Blanco Massani acknowledges the support of the European Union's Horizon 2020 Research and Innovation Programme under Marie Skłodowska-Curie IF [NanoBioRS-101025065]. The authors would like to acknowledge Pharm Annika Postina and Pharm Ilaria Polidori for the provision of mammalian cells, and Pharm Luca Richter for his insights in protein complexation.References
- B. P. Howden, S. G. Giulieri, T. Wong Fok Lung, S. L. Baines, L. K. Sharkey, J. Y. H. Lee, A. Hachani, I. R. Monk and T. P. Stinear, Nat. Rev. Microbiol., 2023, 21, 380–395 CrossRef CAS PubMed.
- E. T. Sumrall, M. I. Hofstee, D. Arens, C. Röhrig, S. Baertl, D. Gehweiler, M. Schmelcher, M. J. Loessner, S. Zeiter, R. G. Richards and T. F. Moriarty, Antibiotics, 2021, 10, 1186 CrossRef CAS PubMed.
- L. Foulston, A. K. W. Elsholz, A. S. Defrancesco and R. Losick, mBio., 2014, 5, 1–9 CrossRef PubMed.
- J.-S. Baek, C. H. Tan, N. K. J. Ng, Y. P. Yeo, S. A. Rice and S. C. J. Loo, Nanoscale Horiz., 2018, 3, 305–311 RSC.
- A. M. Sobieraj, M. Huemer, L. V. Zinsli, S. Meile, A. P. Keller, C. Röhrig, F. Eichenseher, Y. Shen, A. S. Zinkernagel, M. J. Loessner and M. Schmelcher, mBio, 2020, 11, 1–16 CrossRef PubMed.
- C. Röhrig, M. Huemer, D. Lorgé, S. Luterbacher, P. Phothaworn, C. Schefer, A. M. Sobieraj, L. V. Zinsli, S. Mairpady Shambat, N. Leimer, A. P. Keller, F. Eichenseher, Y. Shen, S. Korbsrisate, A. S. Zinkernagel, M. J. Loessner and M. Schmelcher, mBio, 2020, 11, 1–19 CrossRef PubMed.
- M. Behera, G. Singh, A. Vats, Parmanand, M. Roshan, D. Gautam, C. Rana, R. K. Kesharwani, S. De and S. M. Ghorai, Int. J. Biol. Macromol., 2024, 254, 127969 CrossRef CAS PubMed.
- A. Harms and M. J. Loessner, Curr. Opin. Microbiol., 2023, 72, 102278 CrossRef PubMed.
- H. Haddad Kashani, M. Schmelcher, H. Sabzalipoor, E. Seyed Hosseini and R. Moniri, Clin. Microbiol. Rev., 2017, 31, e00071 Search PubMed.
- M. Tolba, M. U. Ahmed, C. Tlili, F. Eichenseher, M. J. Loessner and M. Zourob, Analyst, 2012, 137, 5749–5756 RSC.
- M. Schulz, S. Calabrese, F. Hausladen, H. Wurm, D. Drossart, K. Stock, A. M. Sobieraj, F. Eichenseher, M. J. Loessner, M. Schmelcher, A. Gerhardts, U. Goetz, M. Handel, A. Serr, G. Haecker, J. Li, M. Specht, P. Koch, M. Meyer, P. Tepper, R. Rother, M. Jehle, S. Wadle, R. Zengerle, F. Von Stetten, N. Paust and N. Borst, Lab Chip, 2020, 20, 2549–2561 RSC.
- N. Olsen, E. Thiran, T. Hasler, T. Vanzieleghem, G. Belibasakis, J. Mahillon, M. Loessner and M. Schmelcher, Viruses, 2018, 10, 438 CrossRef PubMed.
- J. Kaur, A. Kour, J. J. Panda, K. Harjai and S. Chhibber, AAPS PharmSciTech, 2020, 21, 1–15 CrossRef PubMed.
- Z. B. Akkuş-Dağdeviren, A. Saleh, C. Schöpf, M. Truszkowska, D. Bratschun-Khan, A. Fürst, A. Seybold, M. Offterdinger, F. Marx and A. Bernkop-Schnürch, J. Colloid Interface Sci., 2023, 646, 290–300 CrossRef PubMed.
- H. W. Ooi, S. Hafeez, C. A. Van Blitterswijk, L. Moroni and M. B. Baker, Mater. Horiz., 2017, 4, 1020–1040 RSC.
- S. Portilla, L. Fernández, D. Gutiérrez, A. Rodríguez and P. García, Antibiotics, 2020, 9, 1–8 CrossRef PubMed.
- H. Hathaway, J. Ajuebor, L. Stephens, A. Coffey, U. Potter, J. M. Sutton and A. T. A. Jenkins, J. Controlled Release, 2017, 245, 108–115 CrossRef CAS PubMed.
- B. Le-Vinh, Z. B. Akkuş-Dağdeviren, N. N. Le, I. Nazir and A. Bernkop-Schnürch, Adv. Ther., 2022, 5, 2100219 CrossRef CAS.
- A. Bernkop-Schnürch, Adv. Drug Delivery Rev., 2018, 136–137, 62–72 CrossRef PubMed.
- A. Saleh, Z. B. Akkuş-Daǧdeviren, S. Haddadzadegan, R. Wibel and A. Bernkop-Schnürch, Biomacromolecules, 2023, 24, 2587–2595 CrossRef CAS PubMed.
- C. Federer, H. V. Spleis, S. Summonte, J. D. Friedl, R. Wibel and A. Bernkop-Schnürch, J. Pharm. Sci., 2022, 111, 2270–2279 CrossRef CAS PubMed.
- A. Saleh, Z. B. Akkuş-Dağdeviren, J. D. Friedl, P. Knoll and A. Bernkop-Schnürch, Heliyon, 2022, 8, e10577 CrossRef CAS PubMed.
- K. M. Danikowski and T. Cheng, Curr. Microbiol., 2018, 75, 1226–1230 CrossRef CAS PubMed.
- G. Satta, L. D’Andrea, G. Grazi, O. Soro and P. E. Varaldo, Int. J. Syst. Bacteriol., 1993, 43, 813–818 CrossRef CAS PubMed.
- B. C. Borro and M. Malmsten, Adv. Colloid Interface Sci., 2019, 270, 251–260 CrossRef CAS PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- E. Lorencová, P. Vltavská, P. Budinský and M. Koutný, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2012, 47, 2241–2245 CrossRef PubMed.
- M. C. Verdi, C. Melian, P. Castellano, G. Vignolo and M. Blanco Massani, Int. J. Food Sci. Technol., 2020, 55, 267–275 CrossRef CAS.
- S. Gao, A. Holkar and S. Srivastava, Polymers, 2019, 11, 1097 CrossRef CAS PubMed.
- B. S. T. Peddinti, F. Scholle, M. G. Vargas, S. D. Smith, R. A. Ghiladi and R. J. Spontak, Mater. Horiz., 2019, 6, 2056–2062 RSC.
- M. Blanco Massani, J. Klumpp, M. Widmer, C. Speck, M. Nisple, R. Lehmann and M. Schuppler, Biometals, 2018, 31, 1101–1114 CrossRef CAS PubMed.
- H.-J. Jian, A. Anand, J.-Y. Lai, B. Unnikrishnan, H.-T. Chang, S. G. Harroun and C.-C. Huang, ACS Appl. Mater. Interfaces, 2023, 15, 26457–26471 CrossRef CAS PubMed.
- H.-J. Jian, J. Yu, Y.-J. Li, B. Unnikrishnan, Y.-F. Huang, L.-J. Luo, D. Hui-Kang Ma, S. G. Harroun, H.-T. Chang, H.-J. Lin, J.-Y. Lai and C.-C. Huang, Chem. Eng. J., 2020, 386, 123913 CrossRef CAS.
- Y. Fu, L. Yang, J. Zhang, J. Hu, G. Duan, X. Liu, Y. Li and Z. Gu, Mater. Horiz., 2021, 8, 1618–1633 RSC.
- H.-C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575 CrossRef CAS PubMed.
- L. Knabl, B. Kuppelwieser, A. Mayr, W. Posch, M. Lackner, D. Coraca-Huber, A. Danita, M. Blauth, C. Lass-Flörl and D. Orth-Höller, MicrobiologyOpen, 2019, 8, e00658 CrossRef PubMed.
- E. R. Kantrowitz, Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2011, pp. 1–13 Search PubMed.
- K. Okabayashi, M. Futai and D. Mizuno, Jpn. J. Microbiol., 1974, 18, 287–294 CrossRef CAS PubMed.
- V. S. Gondil, K. Harjai and S. Chhibber, Endolysins as emerging alternative therapeutic agents to counter drug-resistant infections, Elsevier B.V., 2020, vol. 55 Search PubMed.
- T. Rajasooriya, H. Ogasawara, Y. Dong, J. N. Mancuso and K. Salaita, Adv. Mater., 2023, 1–34 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01122h |
| ‡ Current address: Department of Immunology and Microbiology, Anschutz Medical Campus, University of Colorado, USA. |
| This journal is © The Royal Society of Chemistry 2024 |