
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heavy-atom-free π-twisted photosensitizers for fluorescence bioimaging and photodynamic therapy†
Darío Puchán
Sánchez‡
a,
Korentin
Morice‡
a,
Monika G.
Mutovska
b,
Lhoussain
Khrouz
c,
Pierre
Josse
a,
Magali
Allain
 a,
Frédéric
Gohier
a,
Philippe
Blanchard
a,
Cyrille
Monnereau
c,
Tangui
Le Bahers
cd,
Nasim
Sabouri
e,
Yulian
Zagranyarski
*b,
Clement
Cabanetos
*a and
Marco
Deiana
*ef
a,
Frédéric
Gohier
a,
Philippe
Blanchard
a,
Cyrille
Monnereau
c,
Tangui
Le Bahers
cd,
Nasim
Sabouri
e,
Yulian
Zagranyarski
*b,
Clement
Cabanetos
*a and
Marco
Deiana
*ef
aUniv Angers, CNRS, MOLTECH-ANJOU, SFR MATRIX, F-49000 Angers, France. E-mail: clement.cabanetos@univ-angers.fr
bFaculty of Chemistry and Pharmacy, University of Sofia, 1 James Bourchier blvd., 1164 Sofia, Bulgaria. E-mail: zagranyarskiyulian@gmail.com
cENS de Lyon, CNRS, Laboratoire de Chimie UMR 5182, F-69342 Lyon, France
dInstitut Universitaire de France, 5 rue Descartes, 75005 Paris, France
eDepartment of Medical Biochemistry and Biophysics, Umeå University, SE-901 87, Umeå, Sweden
fInstitute of Advanced Materials, Faculty of Chemistry, Wrocław University of Science and Technology, 50-370 Wrocław, Poland. E-mail: m.deiana@pwr.edu.pl
First published on 18th July 2024
Abstract
As the field of preclinical research on photosensitizers (PSs) for anticancer photodynamic therapy (PDT) continues to expand, a focused effort is underway to develop agents with innovative molecular structures that offer enhanced targeting, selectivity, activation, and imaging capabilities. In this context, we introduce two new heavy-atom-free PSs, DBXI and DBAI, characterized by a twisted π-conjugation framework. This innovative approach enhances the spin–orbit coupling (SOC) between the singlet excited state (S1) and the triplet state (T1), resulting in improved and efficient intersystem crossing (ISC). Both PSs are highly effective in producing reactive oxygen species (ROS), including singlet oxygen and/or superoxide species. Additionally, they also demonstrate remarkably strong fluorescence emission. Indeed, in addition to providing exceptional photocytotoxicity, this emissive feature, generally lacking in other reported structures, allows for the precise monitoring of the PSs’ distribution within specific cellular organelles even at nanomolar concentrations. These findings underscore the dual functionality of these PSs, serving as both fluorescent imaging probes and light-activated therapeutic agents, emphasizing their potential as versatile and multifunctional tools in the field of PDT.
1. Introduction
Phototherapeutic approaches encompass a broad spectrum of medical treatments exploiting the use of light.1–4 Among these approaches, photodynamic therapy (PDT) stands out as a well-established and extensively investigated method in the field of anticancer phototherapy.5–11 PDT harnesses the unique attributes of photosensitizers (PSs) and light to generate highly cytotoxic reactive oxygen species (ROS), specifically targeting cancer cells while preserving neighboring healthy tissues through minimally invasive to non-invasive modalities.12,13 Upon light absorption, the PS is activated, transitioning from its ground state (S0) to the lowest excited singlet state (S1). The excited S1 state subsequently evolves into an excited triplet state (T1) through a phenomenon referred to as intersystem crossing (ISC). Importantly, the extended lifetime of the generated T1 state allows it to follow two distinct evolutions. It can directly generate highly cytotoxic singlet oxygen (1O2) via a type-II mechanism or initiate a series of electron transfer reactions, through a type-I mechanism, resulting in the formation of radical intermediates such as superoxide (O2−˙) and hydroxyl radical (OH˙).14 These dual pathways collectively contribute to the induction of apoptosis and/or necrosis in cancer cells.6In the context of PSs, a conventional practice has been to introduce halogens, such as Br, I, and second or third-row transition metal ions, into a target chromophore, with the aim of boosting ISC processes, via heavy atom-induced strong spin–orbit coupling (SOC).15–18 However, this approach comes with certain limitations, such as inherent dark toxicity and potential high synthesis costs (in terms of price and number of steps).15,19 Additionally, the introduction of heavy atoms can concomitantly boost ISC from S1 → T1 and T1 → S0, resulting in a shorter lifetime of the T1 state, potentially detrimental for the generation of ROS.15,18,20
Consequently, these concerns have significantly driven researchers’ interest towards the development of heavy-atom-free PSs with optimized ISC and efficient T1 population. In recent years, three main alternative mechanisms to enhance ISC of organic PSs have been proposed which consist in:15,21
(i) Employing spin–orbit charge-transfer intersystem crossing (SOCT-ISC) by fostering, for instance, the interaction between orthogonal molecular donor–acceptor (D–A) structures.
(ii) Enhancing the SOC by involving non-bonding orbitals in the electronic transitions by replacing, for instance, the oxygen atom in the carbonyl group of conventional fluorophores with a sulfur atom.
(iii) Promoting torsional motions (twisting-induced ISC) by introducing, for instance, a nonplanar π-conjugated chromophore.
To date, a wide array of D–A dyads and thionated-based compounds that exhibit improved ISC have been reported, utilizing various structural frameworks such as anthracene,22,23 benzothioxanthene imide,24 boron dipyrromethene (BODIPY),9,20,25–28 coumarin,29 cyanine,30 imidazolium,31 naphthalimide,29,32 phenoxazine,33 pyrene,34 perylene35,36 and others.37–39 Though efficient in generating ROS, such strategies have shown adverse effects on the emissive properties of these compounds, particularly in the case of thionated derivatives.24,26,32,35 thus limiting their usefulness for imaging-guided PDT. Indeed, the inherent fluorescence properties of PS are crucial for several reasons.40 They enable real-time tumor visualization, allowing precise boundary delineation for accurate targeting and comprehensive treatment.41–44 Fluorescence aids in monitoring PS distribution and concentration within the tissue, ensuring adequate tumor accumulation before activation.41–44 This selective activation minimizes damage to healthy tissues, enhancing treatment safety and effectiveness. Additionally, fluorescent imaging is essential for dosimetry, determining the appropriate light dose for effective PDT without excessive damage.45 Post-treatment, fluorescence imaging assesses therapy response by evaluating signal changes, guiding further therapeutic decisions.46,47 This advancement facilitates precise delineation of the treatment area and enables continuous monitoring of therapeutic progress.
Moreover, PSs derived from thiones are prone to oxidation when exposed to light, leading to structural alterations. This raises safety concerns regarding their suitability for potential clinical applications.24
In contrast to these design principles, there has been a recent significant focus on the exploration of heavy-atom-free PSs that incorporate a twisted π-conjugated system.15 Indeed, considerable research efforts have been directed towards investigating the photophysical properties of extended π-conjugated helicoidal systems, which includes triply linked bay-fused diperylene bisimides,48 chromophores derived from hexa-peri-hexabenzocoronenes,49 phenanthrene-fused twisted perylenebisimide50 and BODIPY51–53 derivatives. However, despite their promise, these materials have primarily been restricted to test tubes, with only few practical applications in cellular models.52,53 Furthermore, they present several drawbacks such as their intricate synthesis, high molecular weight, limited fluorescence quantum yield (ΦF), and, for some of them, a weak absorbance in the visible spectral range (above 400 nm).15,54
Drawing on this, we reported on a rational design of a dibenzothioxanthene imide, abbreviated as DBI,55 stemming from a classic highly luminescent dye, initially reported in the late 1970's for plastic staining and textile industry, namely the benzothioxanthene imide (BTI). Characterized by a distorted π-conjugated core, resulting from the selective inclusion of an extra fused phenyl ring, that facilitates efficient ISC through a significant SOC, DBI demonstrated remarkable ROS production and phototherapeutic efficacy at nanomolar concentrations. However, although it remains fluorescent, its ΦF of ca. 8%, was found to be at the lower limit for efficient detection and localization of in vivo tumor accumulation, a key feature for therapy-guiding PSs.55
To overcome this obstacle, and provide a second generation of DBI-related molecules that attain simultaneously high fluorescence emission and effective ROS generation, we herein present derivatives, named DBXI and DBAI in which the sulfur heteroatom bridging the upper and lower part of the molecules was selectively replaced by either a less bulky chalcogen (O) or an unsubstituted amine, thus strongly affecting (i) the molecular strain, and thereby, (ii) the distortion induced SOC thus above all (iii) their relevant spectroscopic properties. By conducting a comprehensive examination that incorporates experimental, theoretical, and biological investigations, we thoroughly scrutinized the photophysical and in cellulo properties of these newly synthesized PSs, yielding highly consistent findings. With a strong visible centered emission, these compounds can be readily monitored in real-time, disclosing their accumulation within specific cellular organelles. Moreover, these PSs display minimal dark toxicity and efficiently inhibit cell proliferation in a dose-dependent manner with a half-maximum inhibitory concentration (IC50) in the nanomolar range (∼100 nM) upon light exposure associated to morphological changes in cells, leading to apoptosis. This study further highlights the key role of molecular design and promising applications of such new generation of π-distorted heavy-atom-free small PSs for imaging-guided photodynamic cancer treatment.
2. Experimental
Synthetic procedures and compounds characterization
All reagents and chemicals from commercial sources were used without further purification unless specified. Solvents were dried and purified using standard techniques. Flash chromatography was performed with analytical-grade solvents using Aldrich silica gel (technical grade, pore size 60 Å, 230–400 mesh particle size). Flexible plates Alugram Xtra SIL G UV254 from Macherey-Nagel were used for thin layer chromatography (TLC). Compounds were detected by ultraviolet (UV) irradiation (Bioblock Scientific). Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker Avance III 300 (1H, 300 MHz and 13C, 76 MHz) or a Bruker Avance DRX500 (1H, 500 MHz and 13C, 125 MHz). Chemical shifts are given in ppm relative to the residual 1H resonance of the deuterated solvent and coupling constants J in Hz. High-resolution mass spectrometry (HRMS) was performed with a JEOL JMS-700 B/E. Matrix Assisted Laser desorption/ionization was performed on MALDI-TOF MS BIFLEX III Bruker Daltonics spectrometer using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB+) as matrix (Bruker, Billerica, MA, USA). The purity of the final compounds was >95% as assessed by NMR, HRMS (mass accuracy <5 ppm) and X-ray diffraction.Crystallographic data
Crystal data were collected on a Rigaku Oxford Diffraction SuperNova diffractometer equipped with an Atlas CCD detector and micro-focus Cu-Kα radiation (λ = 1.54184 Å). The structures were solved by dual-space algorithm and refined on F2 by full matrix least-squares techniques using SHELX package (G.M. Sheldrick, ShelXT2018/2, ShelXL2018/3-2019/3). All non-hydrogen atoms were refined anisotropically and the H atoms were included in the calculation without refinement. Multiscan empirical absorption was corrected for DBI and DBAI by using CrysAlisPro program (CrysAlisPro, Rigaku Oxford Diffraction, 2019–2023). Gaussian absorption corrections were applied for DBXI. Deposition number(s) 2083069 (for DBI), 2335339 (for DBXI) and 2335340 (for DBAI) contain(s) the supplementary crystallographic data for this paper.†Photophysical characterization
Absorption spectra were obtained using a Jasco V-650 spectrophotometer for diluted solutions (∼10 or 1 μM), employing spectrophotometric grade solvents. Fluorescence spectra were acquired with a Horiba Jobin Yvon Fluorolog-3. Steady-state luminescence measurements utilized unpolarized light from a 450 W xenon continuous wave lamp as the light source, with luminescence detected at a 90° angle. These measurements were conducted in diluted solutions within a 10 mm Hellma quartz cuvette, employing a Hamamatsu R928 detector for visible light and a liquid nitrogen-cooled, solid-state Indium Gallium Arsenide detector (850–1600 nm) for near-infrared measurements of singlet oxygen phosphorescence signals. Spectra corrections accounted for variations in the excitation source light intensity and emission spectral responses.Φ F were determined in diluted chloroform (CHCl3) solutions with an absorbance below 0.1, using the following equation for approximation:
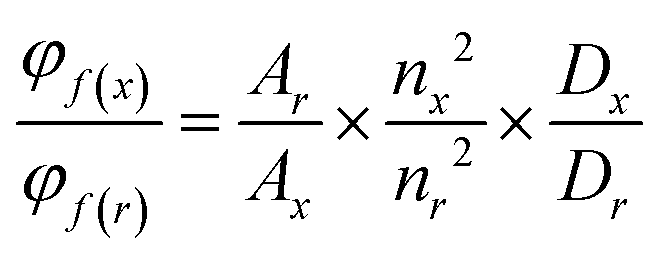 | (1) |
EPR experiments
5,5-Dimethyl-1-pyrroline N-oxide (DMPO) was purchased from TCI Chemicals. All samples were prepared under air atmosphere using capillary tubes filled with 1 × 10−4 M solutions of each chromophore with 5 × 10−3 M of the spin trap in DMSO. The irradiations were conducted using a Thorlab LED with a wavelength of 455 nm, which was directly aimed into the EPR cavity during the recording of the spectrum. All EPR assays were performed at room temperature, utilizing a Bruker E500 spectrometer that operated at the X-band frequency of 9.44 GHz, equipped with a standard cavity and a modulation frequency of 100 kHz. The settings of the instrument were adjusted as follows: microwave power at 2–2–69 mW and modulation amplitude at 1 G. The hyperfine coupling constants, including a and g values, were determined through the simulation of experimental spectra using the EasySpin toolbox in Matlab.Computational data
The Gaussian16 code56 was used to optimize the geometries at the ground and excited states along with the global hybrid functional PBE0.57 This functional was chosen because of its accuracy to reproduce spectroscopic properties of these molecules. Structural optimizations and subsequent frequency calculations for both the ground and excited states were performed using an all electron Pople triple zeta basis set with one polarization function on all atoms and one diffuse function of heavier atoms, known as 6–311+G(d,p), for H, C, N, O and S atoms. Bulk solvent effects were included using the polarizable continuum Model (PCM) of Tomasi and co-workers.58 Default radii (from the UFF, scaled by 1.1) were used. Excited state geometry was obtained by minimizing the forces of the S1 state computed at the TD-DFT level by considering the 3 first excited states. The Orca program59 was used to compute the SOC between the three first triplet states (namely T1 and T2) and the S1 state at the S1 optimized geometry using the quadric-response Tamm–Dancoff approximation of TD-DFT at the PBE0/PCM level with the Def2-TZVP basis set adapted for the Douglas–Kroll calculations. The spin–orbit coupling was computed using the Douglas–Kroll Hamiltonian along with the spin–orbit mean field approach.Live cell imaging
The day prior to treatment, a total of 10 × 104 HeLa cells were plated on a glass-bottom microwell dish (MatTek Corp.) in DMEM medium enriched with 1× penicillin–streptomycin and 10% fetal bovine serum, and they were maintained at 37 °C in a 5% CO2 environment. HeLa cells were washed with DMEM medium (twice), and then treated with DBXI (500 nM) or DBAI (500 nM) for 24 hours. After that, the cells were washed twice with 1× phosphate-buffered saline (PBS) before being incubated with the organelle-selective stains. Nuclear staining was accomplished using Hoechst 33342 (500 nM; Sigma-Aldrich, ref. no.: B2261), while ER-Tracker™ Red (500 nM; ThermoFisher Scientific, ref. no.: E34250), Lyso-Tracker™ Red (100 nM; ThermoFisher Scientific, ref. no.: L7528), or Mito-Tracker™ Red (100 nM; ThermoFisher Scientific, ref. no.: M7512) were utilized for organelle-specific staining. These dyes were dissolved in a live cell imaging solution (Molecular Probes™, ref. no.: A14291DJ) and incubated with the cells for 30 minutes at 37 °C in a 5% CO2 environment. Before imaging, the cells were washed twice with the live cell imaging solution.Imaging was conducted using a Leica SP8 FALCON confocal microscope equipped with an incubation chamber set to 37 °C and 5% CO2. Maximum intensity projections of Z-stack images were generated for data presentation. All data were subsequently processed using ImageJ software.
Intracellular detection of ROS
The day prior to treatment, 10 × 104 HeLa cells were cultured on glass-bottom microwell dishes (MatTek Corp.). Subsequently, these cells were exposed to DBXI (500 nM), DBAI (500 nM) or an equivalent volume of DMSO (0.04% v/v) and then incubated at 37 °C in 5% CO2 for 24 hours. Then, the cells were exposed to CellROX™ green reagent (5 μM; ThermoFisher Scientific, ref. no.: C10444) and Hoechst 33258 (500 nM) dissolved in the live cell imaging solution, and incubated at 37 °C for 30 minutes. Subsequently, the cells were imaged at two time points: before irradiation and 5 minutes post-irradiation, using a 480 nm supercontinuum white light laser (WLL) operating at 80% for a duration of 5 minutes.Phototoxic experiments
HeLa cells, at a density of 5 × 103 cells per well, were placed onto 96-well plates a day prior to the treatment with DBXI or DBAI. DBXI or DBAI were dissolved in complete medium at different concentrations, with DMSO reaching a maximum value of 0.5% v/v, and subsequently added to the cells for 24 hours. Where indicated, the cells underwent photo-irradiation using the EVOS® FL cell imaging system equipped with a customizable LED cube (Invitrogen, ref. no.: AMEP4651; excitation: 470/22 nm) operating at 27 mW cm−2 for 6 minutes. Following photo-irradiation, the cells were further incubated for 24 hours at 37 °C in a 5% CO2 atmosphere. After 48 hours from the initiation of DBXI or DBAI treatment, PrestoBlue™ (Invitrogen, ref. no.: A13261) was introduced to each well, and the cells were incubated at 37 °C in 5% CO2 for an additional three hours. The assessment of cell viability was performed by measuring the fluorescence signal emitted by PrestoBlue (λexc/λem: 560/590 nm) using a Synergy H4 microplate reader (Biotek).The photo-induced cell death caused by DBXI was corroborated using the LIVE/DEAD™ viability/cytotoxicity kit (Invitrogen, ref. no.: L34960). Cells were plated on glass-bottom microwell dishes (MatTek Corp.) a day prior to treatment. HeLa cells received a treatment of DBXI (500 nM) or an equivalent volume of DMSO and were then incubated at 37 °C in a 5% CO2 atmosphere for 24 hours. Where necessary, the cells underwent photo-irradiation. Following this, an additional 24-hour incubation at 37 °C in 5% CO2 was conducted. 48 hours post-DBXI treatment, live/dead fixable red stain (1 μL mL−1) was applied to the cells for 30 minutes at 37 °C before fixation with 4% paraformaldehyde (PFA). Prior to imaging, cells were washed with 1× PBS enhanced with 1% bovine serum albumin. Imaging was performed using a Leica SP8 FALCON confocal microscope, employing maximum intensity projection of Z-stack images for data representation. ImageJ software was utilized for all data processing.
Morphological changes associated with light irradiation and PSs treatment
24 hours prior to DBXI or DBAI treatment, 5 × 103 cells per well were seeded in complete medium on 96-well plates. DBXI or DBAI, at a concentration of 1 μM, or an equivalent volume of DMSO (0.08% v/v), were dissolved in complete medium and added to the cells for an additional 24 hours at 37 °C in a 5% CO2 environment. Subsequently, the cells were subjected to photo-irradiation using the EVOS® FL cell imaging system equipped with adjustable-intensity LED cubes (excitation = 470/22 nm operating at 30 mW cm−2).3. Results and discussion
Molecular design and synthesis
The synthetic route initially engineered for the synthesis DBI had to be entirely redesigned for the preparation of the new target derivatives. Starting with the oxo derivative, preparation of the latter was initiated by reacting the bromonaphtalimide 1 with the commercially available (2-methoxynaphthalen-1-yl) boronic acid 2 under Suzuki–Miyaura coupling conditions. The methoxy group of the resulting intermediate 3 was then deprotected with BBr3 prior to ring close the system under basic conditions to afford the desired DBXI derivative (Scheme 1). | ||
| Scheme 1 Synthetic routes and chemical structures of DBXI (top) and DBAI (bottom). | ||
Regarding its NH counterpart, a different approach was developed, relying on the use of the 4-bromo-5-nitro-1,8-naphthalic anhydride. Starting with its imidization in presence of the 3-aminopentane, the resulting intermediate 6 was thereafter engaged in a Suzuki–Miyaura cross coupling reaction with the commercial 1-naphthaleneboronic acid 7 to afford 8. Finally, 8 was cyclized under Cadogan conditions providing the desired DBAI compound.
The structures of both DBXI and DBAI were investigated using NMR spectroscopy and HRMS (Fig. S1–S16, ESI†). These data were confirmed through X-ray diffraction performed on crystals grown using the slow evaporation technique (Table S1, ESI† and experimental section for details).
Both molecules exhibited overall good solubility in common organic solvents, with DBAI outperforming DBXI, facilitating the investigation of their spectroscopic and photophysical properties.
Photophysical properties
The absorption and emission spectra of DBXI and DBAI were recorded in CHCl3 and compared with those of the parent DBI compound (Fig. 1). This revealed a significant impact of the substituent change on the spectral shape and the position of the maxima within the series (Fig. 1). Whereas the parent DBI exhibited structureless absorption and emission bands, with a maximum absorption (λmax) at 481 nm and maximum emission (λem) at 544 nm, the two new compounds displayed notable differences. Both DBXI and DBAI had absorption and emission profiles characterized by pronounced vibronic progressions, indicating a more localized character of the π–π* transition as typically observed in closely related naphthalene diimide molecules. Specifically, DBXI exhibited a notable blue shift, with λmax at 447 and 472 nm and λem at 494 nm, compared to DBI. Conversely, the spectral maxima of DBAI (λmax = 487 and 517 nm and λem = 543 nm) were very similar to those of the parent DBI. Additionally, DBAI demonstrated a minor transition of low intensity with a similar band structure, peaking at 420 nm. | ||
| Fig. 1 Absorption and emission spectra of 10 μM CHCl3 solutions of DBAI, DBXI, and DBI. λexc (DBAI) = 480 nm, λexc (DBXI) = 446 nm and λexc (DBI) = 480 nm. | ||
This evolution was corroborated by vertical transition energies computed at the TD-DFT level, which yielded absorption wavelengths of 461 nm for DBAI, 436 nm for DBXI, and 470 nm for DBI, along with emission wavelengths of 527 nm for DBAI, 506 nm for DBXI, and 545 nm for DBI. The more localized character of the electronic transition was further confirmed by the computation of the DCT index, which is related to the variation in electron density between the ground and excited states and quantifies the strength of a charge transfer transition. The later was found to be larger for DBI than for DBXI and DBAI, indicating a stronger charge transfer transition in DBI as illustrated in Fig. 2.60,61
 | ||
| Fig. 2 Summary of TD-DFT calculations for (A) DBAI, (B) DBXI and (C) DBI. Variation of the electron density, Δρ, presenting the electron density depletion and the electron increase in blue and green respectively (isovalue 0.004 a.u.) as long as the extract DCT index. Jablonsky diagram is presented with S0, S1 and Tn levels in black, blue and green respectively. The torsion angle of the π-systems is presented in inset. | ||
The most striking effects of changing the bridging heteroatom were observed in the photophysical properties of the molecule. While DBI exhibited very efficient intersystem crossing (ISC), with ΦΔ of approximately 0.95 and minimal residual luminescence, the ISC efficiency was significantly reduced for DBXI and DBAI to ΦΔ values of ca 0.21 and 0.33, respectively. As an antagonistic effect, the ΦF significantly increased to 0.68 for DBXI and up to 0.72 for DBAI, associated to fluorescence lifetimes of 4.95 ns for DBXI and 6.7 ns for DBAI. Quantum chemical calculations provided atomistic insights into these observations. The ISC, responsible for populating the triplet state necessary for generating 1O2, is still promoted by SOC resulting from the distortion of the π-systems, also referred to as the twisting induced triplet state population. This distortion was computed for all three molecules (Fig. 2) and observed experimentally from X-ray structures. As expected, this effect was more pronounced for DBI than for DBAI and DBXI as characterized by the largest dihedral angle (by more than 10°) measured between the upper and lower naphthyl rings (Fig. 2). This feature is directly related to the higher SOC computed for DBI, compared to the other two molecules, and ultimately the variations in 1O2 production efficiency that were experimentally evaluated.
To determine if, beyond a type-II mechanism driven by 1O2, a type-I mechanism could also play a role in generating ROS, electron paramagnetic resonance (EPR) experiments were conducted on both DBAI and DBXI. Utilizing 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a radical scavenger, capable of reacting with O2−˙, no radical adducts were detected for DBXI. In contrast, DBAI exhibited a well-resolved hyperfine splitting structure, indicative of a DMPO-superoxide adduct signal, corroborated by simulated EPR spectra (Fig. 3). These findings suggest that the photocytotoxic potential of these compounds might be exclusively mediated through a type-II mechanism for DBXI, while DBAI could involve a combined type-I and type-II mechanism.
 | ||
| Fig. 3 EPR signal of a solution of DBAI and DMPO in DMSO under 455 nm irradiation. Experimental (blue) and simulated (red) spectra of DMPO superoxide adduct. g = 2.006, aN = 12.85 G, aH = 10.37 G, aH = 1.28 G. | ||
Live cells fluorescence imaging
Organelles play pivotal roles in preserving cell structure and function, and any disruption can result in cellular dysfunction and eventual cell death.62 Achieving precise targeting of PSs to organelles offers several advantages, including the potential to lower PS dosages, mitigate side effects, reduce the risk of drug resistance, and enhance the effectiveness of PDT.11 Indeed, given that ROS typically have an exceedingly brief lifetime and a limited diffusion range in biological systems,63 PSs designed to precisely target organelles often exhibit superior PDT outcomes.64 In this context, a number of organelle-specific PSs already have been reported with localization to the nuclei,31,41,65–67 mitochondria,20,68–70 multivesicular bodies,55 lysosomes19,71–74 and endoplasmic reticulum (ER).75–79Next, we thus investigated if the high fluorescence emission demonstrated by these new PSs could be used to probe their cellular emission fingerprint by confocal laser scanning microscopy (CLSM). Live HeLa cells were treated with DBXI or DBAI at a concentration of 500 nM, followed by a 24-hour incubation. The incubation time was carefully selected based on the results of phototoxicity experiments (detailed below) to coincide precisely with the period during which light is applied to induce cell death. This synchronization ensures consistent PS accumulation and distribution within cellular compartments, thereby avoiding variability in PS concentration and localization that could affect the interpretation of the experimental outcomes. This treatment initially revealed an accumulation of the PSs in the cytoplasm, confirming that the molecules were well dissolved within the cellular environment and isolated as highly emissive species (Fig. 4–6). Thereafter, the intracellular distribution of the PSs was further explored by using four different commercially available organelle-selective live-cell trackers, namely Hoechst 33342, ER-Tracker Red, Lyso-Tracker Red, and Mito-Tracker Red to specifically label the nuclei, ER, lysosomes, and mitochondria, respectively. No relevant nuclear colocalization was observed between the PSs and Hoechst 33342, suggesting negligible uptake in the nucleus (Fig. 4–6). Also, low to moderate Pearson's correlation coefficients (PCC) were observed between the PSs and Lyso-Tracker Red, indicating their limited accumulation within the lysosomes (Fig. 4).
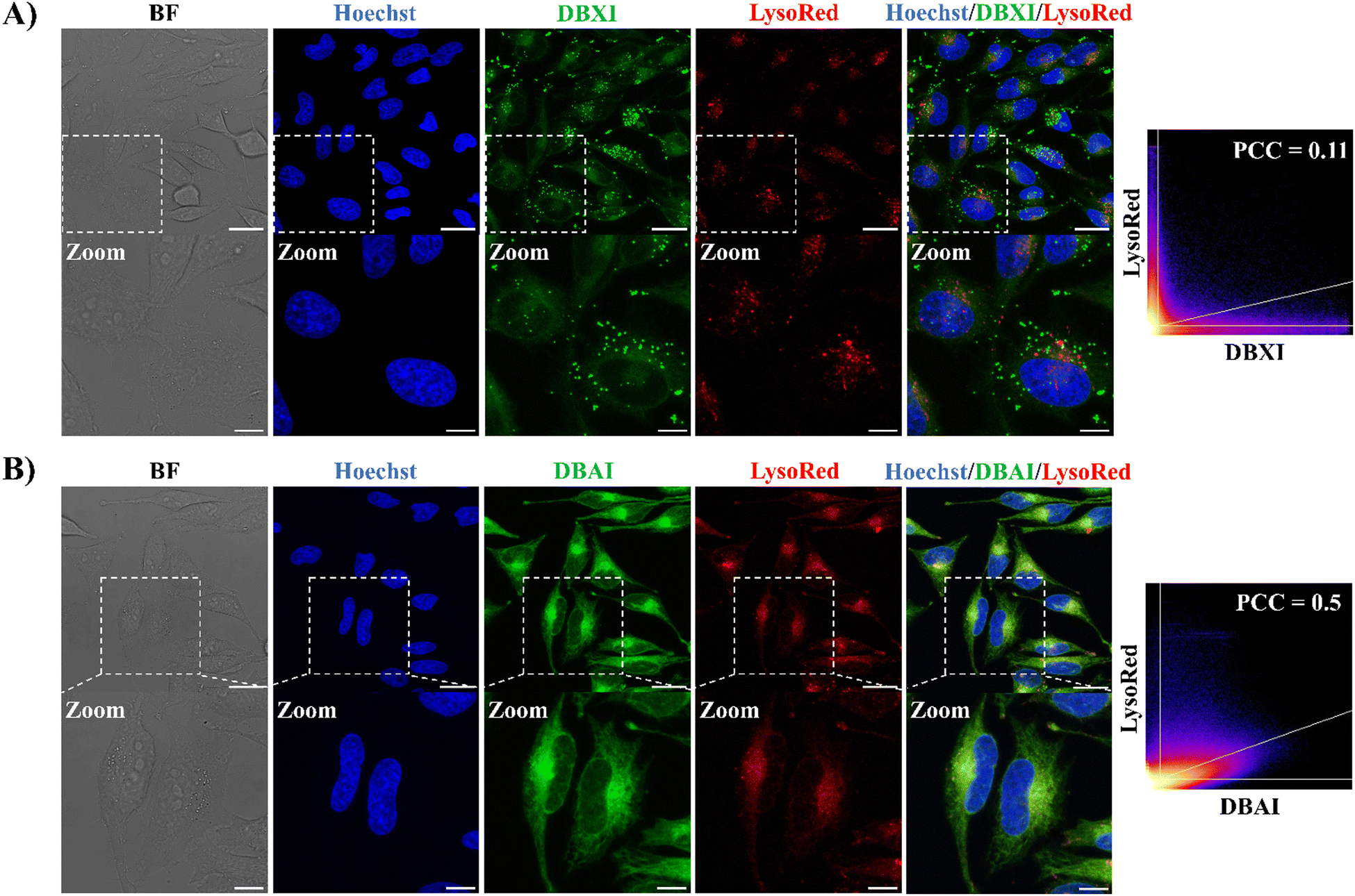 | ||
| Fig. 4 Cellular localization of DBXI and DBAI. CLSM images of live HeLa cells treated with DBXI (500 nM, green signal) (A) or DBAI (500 nM, green signal) (B) and incubated for 24 hours. After 24 hours, the cells were co-stained with the nuclear dye Hoechst 33342 (500 nM, blue signal) and Lyso-Tracker Red (100 nM, red signal). 2D scatter diagrams depicting DBXI or DBAI and Lyso-Tracker Red channels are presented, and the quantification of colocalization has been accomplished using the Pearson correlation coefficient (PCC). Image settings: λexc/λem: 405/420–470 nm for Hoechst; λexc/λem: 480/495–560 nm for DBXI or DBAI; λexc/λem: 577/600–710 nm for Lyso-Tracker Red. The scale bar is set at 25 μm for regular images (top panel) and 10 μm for the enlarged images (bottom panel; indicated zoom). | ||
 | ||
| Fig. 5 Cellular co-localization of DBXI and DBAI with ER tracker. CLSM images of live HeLa cells treated with DBXI (500 nM, green signal) (A) or DBAI (500 nM, green signal) (B) and incubated for 24 hours. After 24 hours, HeLa cells were co-stained with the nuclear dye Hoechst 33342 (500 nM, blue signal) and ER-Tracker Red (500 nM, red signal). 2D scatter diagrams depicting DBXI or DBAI and ER-Tracker Red channels are presented, and the quantification of colocalization has been accomplished using PCC. Image settings: λexc/λem: 405/420–470 nm for Hoechst; λexc/λem: 480/495–560 nm for DBXI or DBAI; λexc/λem: 587/600–710 nm for ER-Tracker Red. The scale bar is set at 25 μm for regular images and 10 μm for the enlarged images. | ||
 | ||
| Fig. 6 Cellular localization of DBXI and DBAI. CLSM images of live HeLa cells treated with DBXI (500 nM, green signal) (A) or DBAI (500 nM, green signal) (B) and incubated for 24 hours. After 24 hours, cells were co-stained with the nuclear dye Hoechst 33342 (500 nM, blue signal) and Mito-Tracker Red (100 nM, red signal). 2D scatter diagrams depicting DBXI or DBAI and Mito-Tracker Red channels are presented, and the quantification of colocalization has been accomplished using PCC. Image settings: λexc/λem: 405/420–470 nm for Hoechst; λexc/λem: 480/495–560 nm for DBXI or DBAI; λexc/λem: 580/590–715 nm for Mito-Tracker Red. The scale bar is set at 25 μm for regular images and 10 μm for the enlarged images. | ||
On the other hand, and as depicted in Fig. 5, the green fluorescence of DBXI and DBAI exhibited significant overlap with the fluorescence of ER-Tracker Red, yielding PCC values of 0.59 and 0.82 for DBXI and DBAI, respectively whereas the degree of colocalization between DBXI or DBAI and Mito-Tracker Red was lower, with PCC values of 0.3 and 0.61 for DBXI and DBAI, respectively (as seen in Fig. 6). These results highlight a good extent of PS internalization within the ER. Moreover, it is noteworthy that DBXI exhibited distinct, intense fluorescent clusters with a punctate nature in the cytoplasm, which did not overlap with the signals emitted by any of the organelle-specific trackers employed, suggesting further accumulation into vesicular bodies (Fig. 4–6). Indeed, the PCC value of 0.59 calculated for the co-localization of DBXI and ER-Tracker Red would be expected to be higher if the punctate signal of DBXI were excluded from the analysis. We believe that the slight differences in the staining patterns observed for DBXI and DBAI can be partially attributed to their differing hydrophobic properties. DBAI, which is fairly soluble, predominantly demonstrated uniform accumulation in the ER. In contrast, DBXI, due to its higher hydrophobicity, also tended to accumulate in vesicles with likely lipophilic character. These nanometer-sized spherical organelles may correspond to multivesicular bodies (MVBs) containing membrane-bound intraluminal vesicles (ILVs) or lipid droplets (LDs).
Targeting the ER holds significant importance in PDT and a number of PSs have been designed to accumulate in this organelle.11 The ER forms a complex network of membranes throughout the cytoplasm, interacting thereby closely with other organelles. It plays a vital role in several cellular processes, including lipid and protein synthesis, and the maintenance of calcium homeostasis.70–80 Hence, during the PDT treatment, the production of ROS within the ER can induce localized stress resulting in an elevation of intracellular calcium levels and an enhanced expression of specific proteins.81,82 If left unrepaired, this stress can ultimately lead to programmed cell death through apoptosis or necrosis.83–85 As a result, the ER has thus emerged as a promising therapeutic target in the context of anti-tumor strategies and addressing this organelle with our PSs is anticipated to substantially strengthen the efficacy of PDT.
In cellulo ROS generation and PDT activity
The ability of PSs to generate ROS was evaluated and tested in live HeLa cells using the CellROX™ green reagent (Fig. 7). CellROX™ functions as a DNA fluorescent probe to identify oxidative stress within the nuclei of living cells. In its reduced state, the dye exhibits minimal fluorescence. However, upon ROS-induced oxidation, it binds to DNA in both the nuclei and mitochondria, emitting intense green fluorescence.55 Our findings confirm that both DBXI or DBAI significantly increased intracellular ROS production, as evidenced by the robust green fluorescence observed in the nuclei of HeLa cells under continuous irradiation with a 480 nm supercontinuum white light laser (WLL) for 5 minutes (Fig. 7). Crucially, in control experiments involving the CellROX™ green reagent conducted without PSs or light, no nuclear fluorescent signals associated with ROS were observed (Fig. 7), confirming that the intracellular ROS generation is specifically triggered in PS-treated cells under light irradiation, thus reinforcing the potential value of these PSs for targeted applications.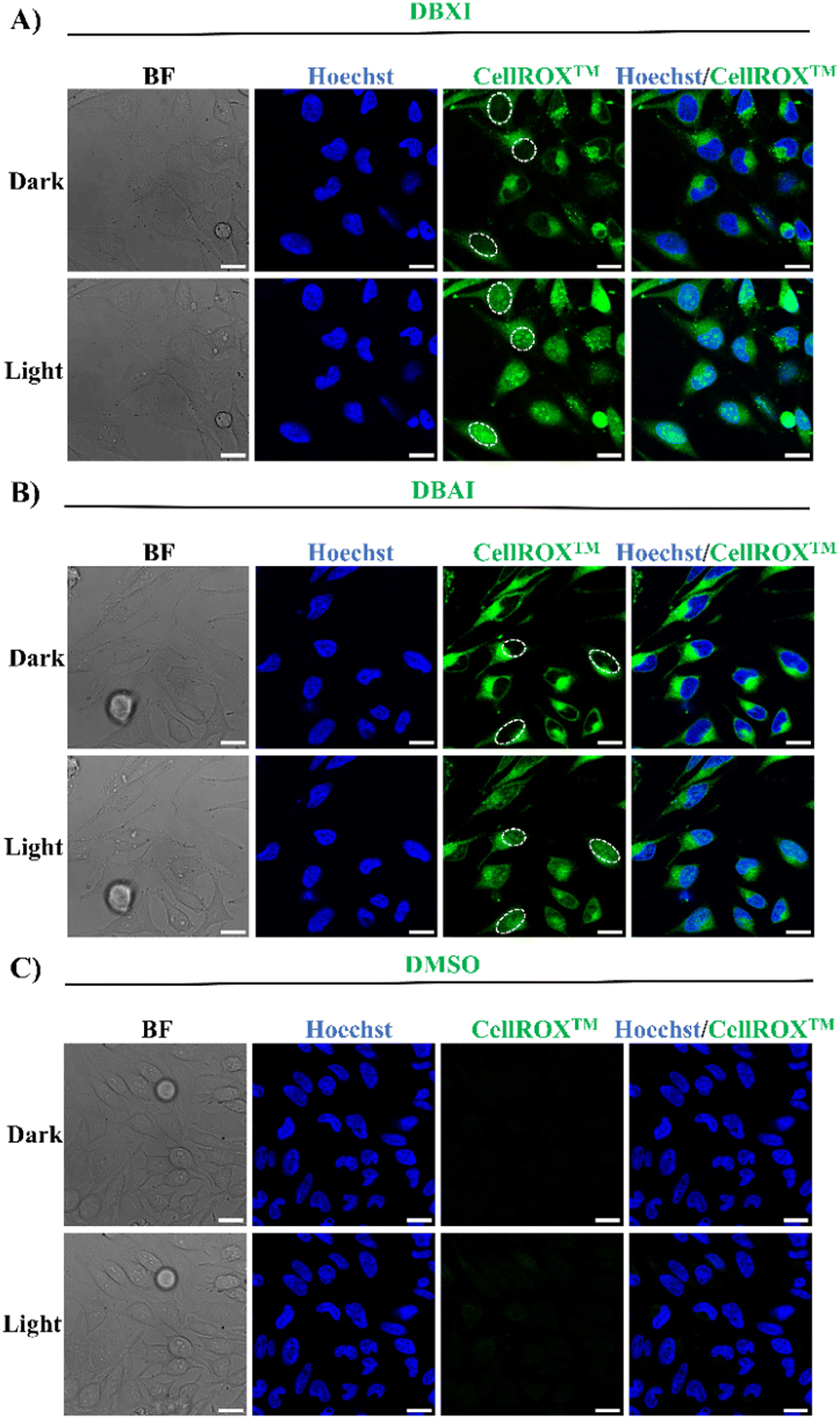 | ||
| Fig. 7 Intracellular ROS Detection. Live HeLa cells were treated with DBXI (500 nM) (A), DBAI (500 nM) (B), or DMSO (0.04% v/v) (C), and incubated for 24 hours. Subsequently, CellROX™ green reagent (5 μM) and Hoechst 33258 (500 nM) were introduced to the cells and allowed to incubate for 30 minutes at 37 °C. Finally, the cells were imaged after irradiation with a 480 nm supercontinuum white light laser (WLL) for 0 or 5 minutes. The PS-mediated production of ROS, as indicated by the activation of the nuclear fluorescence signal of CellROX™, is highlighted for some cells by the white circles. The green fluorescent signal in the cytoplasm originates from the excitation of the PSs. Control experiments with DMSO demonstrated the absence of any signal interference from CellROX™. Image settings: λexc/λem: 405/415–450 nm for Hoechst; λexc/λem: 480/495–675 nm for CellROX™. Scale bar 20 μm. | ||
Motivated by the compelling body of evidence gathered from both in vitro (test tube) and cell-based experiments, which attested to the PSs’ remarkable capacity for ROS generation, we turned our attention to a comprehensive evaluation of their potential phototoxic effects. To evaluate the potential phototoxic effects of such new PSs, PrestoBlue cell viability assay was carried out.55,86,87 This assay serves as a valuable tool for assessing if these drugs impact cell viability. Consequently, HeLa cells were first cultured with different concentrations (0–25 μM) of PSs in the absence of light for a duration of 48 hours. Notably, the cell viability remained consistently high during this period, suggesting the absence of significant dark cytotoxic effects (Fig. 8(A) and (B)). Next, to perform the photoirradiation analysis, the cells were exposed to varying PS concentrations (0–1.56 μM) for 24 hours, followed by a 6-minute irradiation under a blue LED light cube (470/22 nm) prior to be further incubated for an additional 24 hours. These experimental parameters were carefully optimized to avoid cell death due to excessive light exposure on the one hand, and to match the incubation time and overall experimental conditions employed in the dark cytotoxicity assessments on the other. As shown in Fig. 8(C) and (D), the PSs efficiently inhibited cell proliferation in a dose-dependent manner, exhibiting IC50 values of approximately 100 nM (IC50(DBXI) = 96.4 ± 26.4 nM and IC50(DBAI) = 103.2 ± 15.0 nM). It is worth noting that these IC50 values are among the most potent documented organic PSs and within the highest reported for ER-specific PSs.11,24,55 Additionally, the phototherapeutic index (PI = IC50(dark)/IC50(light)) of both compounds is at least greater than 240, establishing these compounds as very efficient PSs and far more efficient than the majority of PSs incorporating heavy atoms.
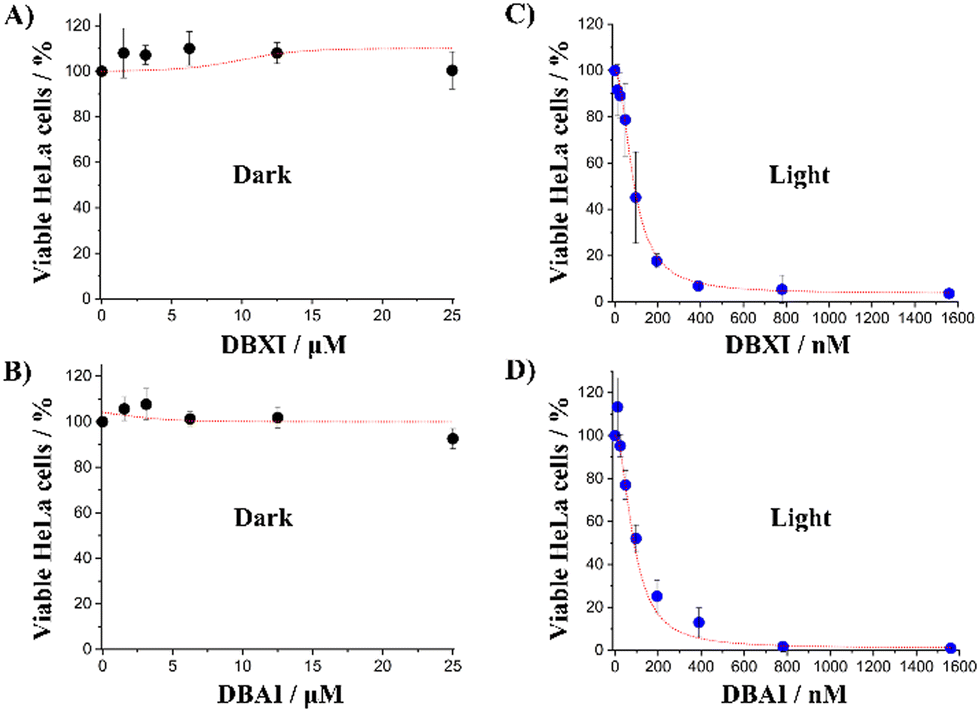 | ||
| Fig. 8 Phototoxicity studies in HeLa cells. Cell viability assay performed in the absence (A) and (B) or presence (C) and (D) of blue light (27 mW cm−2) irradiated for 6 min at different DBXI or DBAI concentrations. The error bars represent the mean ± standard deviation (n = 3). | ||
The photocytotoxic activity of DBXI and DBAI is particularly noteworthy, considering that most uncharged PSs tend to form molecular aggregates in aqueous solutions, which can adversely affect their emissive properties and the efficacy of PDT.32,88–91 Research, conducted by us and others, has shown that biomolecules such as proteins,24,89–93 nucleic acids,87,94–99 and lipids100–105 can disassemble self-assembled dyes, restoring both their ΦF and ΦΔ.106 This disassembly ensures that even if DBXI and DBAI form aggregates, they will become active and detectable at nanomolar concentrations in cellular environments. Furthermore, a direct comparison of ΦF and ΦΔ, along with IC50 values from HeLa cells for DBXI and DBAI, against structurally related analogs such as DBI,55BTI,24 and its thionated variants (T1, T2, and T3),24 highlights the significant improvements made to these compounds to achieve theranostic outcomes (Scheme 2).
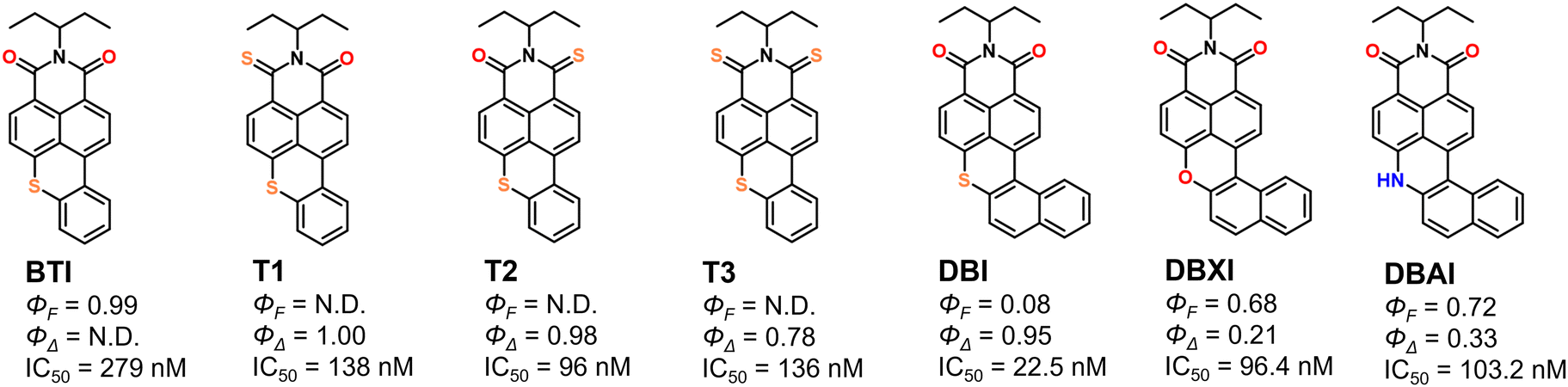 | ||
| Scheme 2 Chemical structures, ΦF, ΦΔ, and IC50 (HeLa cells) values for DBXI and DBAI from this study, along with their closely related published analogs BTI, T1, T2, T3, and DBI, tested under similar experimental conditions. BTI features a planar structure with highly emissive properties but not detectable 1O2. The phototoxicity of BTI likely occurs via a type-I mechanism involving superoxide species.24 In contrast, the ISC in T1, T2, and T3 is facilitated by a thionation strategy, which significantly influences their emissive properties.24DBI is characterized by a highly π-distorted skeleton that enhances ISC but markedly reduces its fluorescence.55 Replacing the sulfur atom in DBI with an oxygen atom (DBXI) or an unsubstituted amine (DBAI) achieves an optimal balance between optical properties and phototherapeutic activity. Not determined parameters due to the near absence of signal are marked as N.D. | ||
Finally, time-lapse microscopy experiments were conducted to observe the effects of the treatment on cell morphology. In stark contrast to control cells, HeLa cells treated with PSs exhibited noticeable alterations in morphology when subjected to continuous irradiation with blue LED light for up to 12 minutes. These alterations included characteristic signs of late-stage apoptosis, such as plasma membrane blebbing and potential nuclear enlargement, which align well with previously reported analogues (Fig. 9).20,24,55
 | ||
| Fig. 9 Time-lapse experiments were conducted to study light-induced morphological alterations in HeLa cells treated with DBXI or DBAI (1 μM). Images were taken at intervals of 0, 6 and 12 minutes after exposure to blue light (30 mW cm−2). Control experiments were carried out using HeLa cells treated with DMSO. Black arrows aim to show the appearance of apoptotic bodies. Scale bar 100 μm. | ||
These results indicate that exposing cells loaded with DBAI and DBXI to light irradiation leads to significant cytotoxicity through the generation of ROS, severely disrupting cellular functions and causing cell death. This effect, particularly with DBXI, was further validated by the live/dead viability/cytotoxicity staining assay. Fig. 10 shows that only the HeLa cells treated with DBXI and subjected to light irradiation exhibited a bright red signal, signifying cell death due to plasma membrane rupture.
 | ||
| Fig. 10 The viability of HeLa cells was assessed using the LIVE/DEAD™ assay. The cells were treated with DBXI (500 nM) or an equivalent volume of DMSO and incubated at 37 °C for 24 hours. Where noted, blue light exposure was administered at an intensity of 30 mW cm−2 for 6 minutes, succeeded by another 24-hour incubation period at 37 °C. For detecting cytotoxicity, LIVE/DEAD™ fixable red stain (1 μL mL−1) was applied to the cells for 30 minutes at 37 °C prior to fixation with PFA. The stain's excitation/emission wavelengths were set at 598/630–730 nm. Scale bar 50 μm. | ||
4. Conclusions
Highlighting the pivotal role of structural innovation, our research leverages and generalizes a twisted π-conjugated system strategy, marking significant advancements in fine-tuning the efficiency of ROS generation and fluorescence emission in PSs. This was achieved through controlled chemical modification of the dibenzothioxanthene imide scaffold, employing either a less bulky chalcogen oxygen or an unsubstituted amine. The outcome was the synthesis of two unique heavy-atom-free π-distorted PSs, each characterized by their mechanism of action and targeted ability towards the ER. Their notable capacity to activate both type-I and/or type-II photodynamic mechanisms, alongside maintaining high fluorescence emission levels, sets them apart from the majority of other PS reported in the literature. This dual functionality not only facilitates precise tracking of their distribution within live cells at nanomolar concentrations but also enables the induction of phototoxic effects at exceptionally low concentrations, with negligible dark toxicity. Through this work, we demonstrate the transformative potential of strategic structural design in surmounting the existing barriers in PDT, paving the way for future theranostic innovations with a special emphasis on shifting the optical properties to the near infra-red region.Author contributions
D. P. S., K. M. and M. G. M. synthesized and characterized the new PSs. P. J., F. G., and P. B. were involved in the supervision and discussion of the synthetic procedures. L. K. carried out EPR measurements, C. M. all photophysical characterizations and T. L. B. the computational analyses. M. A. characterized and solved the X-ray structures of the two PSs in addition to registering their CCDC numbers. Y. Z. and C. C. designed the new molecule from a synthetic point of view. N. S. was involved in the supervision and discussion of the biological data. M. D. designed and carried out biological experiments, analyzed and interpreted biological data and wrote and revised the manuscript with the contribution of N. S. C. M. and C. C.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
A patent application covering the technology presented in this manuscript has been filed.Acknowledgements
M. Deiana would like to acknowledge financial support from project no. 2022/47/P/NZ5/01156, which is co-funded by the National Science Centre and the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 945339. M. Deiana also thanks the Swedish Cancer Society for providing financial support through the postdoctoral fellowship (21 0302 PT 01 H). D. Puchán Sánchez and K. Morice acknowledge the MITI of the CNRS and the ANR (BTXI-Apogee, ANR-20-CE05-0029), respectively for their PhD grants. T. Le Bahers acknowledges the Institut Universitaire de France for funding and the SYSPROD project and AXELERA Pôle de Compétitivité for financial support (PSMN Data Center). M. Deiana and N. Sabouri acknowledge the Biochemical Imaging Center at Umeå University and the National Microscopy Infrastructure, NMI (VR-RFI 2019-00217) for providing assistance in microscopy, and the Chemical Biology Consortium Sweden (CBCS) at Umeå University for access to the Synergy H4 microplate reader. Work in N. Sabouri lab received support from the Swedish Cancer Society (22 2380 Pj 01 H), the Swedish Research Council (VR-MH 2021-02468), Knut and Alice Wallenberg foundations (KAW 2021.0173), and Biotechnology grant from the Medical faculty at Umeå University. This study received the support of the EUR LUMOMAT (programme Investissements d’Avenir ANR-18-EURE-0012) through the grant of P. Josse (Project AZA BTXI).Notes and references
- B. M. Vickerman, E. M. Zywot, T. K. Tarrant and D. S. Lawrence, Nat. Rev. Chem., 2021, 1–19 Search PubMed.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef.
- P. Kobauri, F. J. Dekker, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202300681 CAS.
- P. Cheng and K. Pu, ACS Appl. Mater. Interfaces, 2020, 12, 5286–5299 CrossRef CAS PubMed.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef PubMed.
- T. Mishchenko, I. Balalaeva, A. Gorokhova, M. Vedunova and D. V. Krysko, Cell Death Dis., 2022, 13, 455 CrossRef.
- H. Huang, S. Banerjee, K. Qiu, P. Zhang, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stavros, G. Gasser, H. Chao and P. J. Sadler, Nat. Chem., 2019, 11, 1041–1048 CrossRef CAS.
- J. An, S. Tang, G. Hong, W. Chen, M. Chen, J. Song, Z. Li, X. Peng, F. Song and W.-H. Zheng, Nat. Commun., 2022, 13, 2225 CrossRef CAS PubMed.
- K.-X. Teng, L.-Y. Niu, N. Xie and Q.-Z. Yang, Nat. Commun., 2022, 13, 6179 CrossRef CAS.
- J. Yan, T. Gao, Z. Lu, J. Yin, Y. Zhang and R. Pei, ACS Appl. Mater. Interfaces, 2021, 13, 27749–27773 CrossRef CAS PubMed.
- R. Wang, X. Li and J. Yoon, ACS Appl. Mater. Interfaces, 2021, 13, 19543–19571 CAS.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CAS.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- C. Mu, W. Wang, J. Wang, C. Gong, D. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 21515–21519 CrossRef CAS PubMed.
- V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2021, 54, 207–220 CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque, III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- M. Lan, S. Zhao, W. Liu, C.-S. Lee, W. Zhang and P. Wang, Adv. Healthcare Mater., 2019, 8, 1900132 CrossRef.
- M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10–27 RSC.
- G. He, N. Xu, H. Ge, Y. Lu, R. Wang, H. Wang, J. Du, J. Fan, W. Sun and X. Peng, ACS Appl. Mater. Interfaces, 2021, 13, 19572–19580 CrossRef CAS PubMed.
- J. Miao, Y. Huo, G. Yao, Y. Feng, J. Weng, W. Zhao and W. Guo, Angew. Chem., Int. Ed., 2022, 61, e202201815 CrossRef CAS PubMed.
- X. Zhang, Z. Wang, Y. Hou, Y. Yan, J. Zhao and B. Dick, J. Mater. Chem. C, 2021, 9, 11944–11973 RSC.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed.
- M. Lv, Y. Yu, M. E. Sandoval-Salinas, J. Xu, Z. Lei, D. Casanova, Y. Yang and J. Chen, Angew. Chem., Int. Ed., 2020, 59, 22179–22184 CAS.
- M. Deiana, P. Josse, C. Dalinot, A. Osmolovskyi, P. S. Marqués, J. M. A. Castán, L. Abad Galán, M. Allain, L. Khrouz, O. Maury, T. Le Bahers, P. Blanchard, S. Dabos-Seignon, C. Monnereau, N. Sabouri and C. Cabanetos, Commun. Chem., 2022, 5, 142 CAS.
- Y. Hou, Q. Liu and J. Zhao, Chem. Commun., 2020, 56, 1721–1724 CAS.
- Y. Xiao, X. Huang, J. Feng, Z. Ni, L. Gai, X. Xiao, X. Sui and H. Lu, Dyes Pigm., 2022, 200, 110167 CrossRef CAS.
- H. Liang, M. Lu, Z. Mahmood, Z. Li, Z. Chen, G. Chen, M.-D. Li, Y. Huo and S. Ji, Angew. Chem., Int. Ed., 2023, 62, e202312600 CrossRef CAS PubMed.
- W. Hu, M. Liu, X.-F. Zhang, M. Shi, M. Jia, X. Hu, L. Liu and T. Wang, J. Mater. Chem. C, 2020, 124, 23558–23566 CAS.
- J. Tang, L. Wang, A. Loredo, C. Cole and H. Xiao, Chem. Sci., 2020, 11, 6701–6708 RSC.
- X. Zhao, Q. Yao, S. Long, W. Chi, Y. Yang, D. Tan, X. Liu, H. Huang, W. Sun, J. Du, J. Fan and X. Peng, J. Am. Chem. Soc., 2021, 143, 12345–12354 CrossRef CAS.
- T. C. Pham, T. T. H. Hoang, D. N. Tran, G. Kim, T. V. Nguyen, T. V. Pham, S. Nandanwar, D. L. Tran, M. Park and S. Lee, ACS Appl. Mater. Interfaces, 2023, 15, 47969–47977 CrossRef CAS PubMed.
- V.-N. Nguyen, S. Qi, S. Kim, N. Kwon, G. Kim, Y. Yim, S. Park and J. Yoon, J. Am. Chem. Soc., 2019, 141, 16243–16248 CAS.
- Y. Dong, A. A. Sukhanov, J. Zhao, A. Elmali, X. Li, B. Dick, A. Karatay and V. K. Voronkova, J. Mater. Chem. C, 2019, 123, 22793–22811 CAS.
- S. Callaghan, M. A. Filatov, H. Savoie, R. W. Boyle and M. O. Senge, Photochem. Photobiol. Sci., 2019, 18, 495–504 CrossRef CAS PubMed.
- Y.-L. Lee, Y.-T. Chou, B.-K. Su, C.-C. Wu, C.-H. Wang, K.-H. Chang, J.-A. A. Ho and P.-T. Chou, J. Am. Chem. Soc., 2022, 144, 17249–17260 CrossRef CAS PubMed.
- Z. Wang, M. Ivanov, Y. Gao, L. Bussotti, P. Foggi, H. Zhang, N. Russo, B. Dick, J. Zhao, M. Di Donato, G. Mazzone, L. Luo and M. Fedin, Chem. – Eur. J., 2020, 26, 1091–1102 CrossRef CAS.
- T. C. Pham, S. Heo, V.-N. Nguyen, M. W. Lee, J. Yoon and S. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 13949–13957 CrossRef CAS.
- M. Pollum, S. Jockusch and C. E. Crespo-Hernández, J. Am. Chem. Soc., 2014, 136, 17930–17933 CrossRef CAS PubMed.
- P. Zhang, H. Kuang, Y. Xu, L. Shi, W. Cao, K. Zhu, L. Xu and J. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 42551–42557 CrossRef CAS PubMed.
- X. Yang, X. Zhang, Z. Yang, L. Cheng, X. Liu, S. Cao, H. Yue, Y. Cao, K.-N. Wang and Y. Zhang, ACS Appl. Mater. Interfaces, 2024, 16, 9816–9825 CrossRef CAS PubMed.
- K.-N. Wang, L.-Y. Liu, D. Mao, M.-X. Hou, C.-P. Tan, Z.-W. Mao and B. Liu, Angew. Chem., Int. Ed., 2022, 61, e202114600 CrossRef CAS.
- Y. Zhang, L. Pang, C. Ma, Q. Tu, R. Zhang, E. Saeed, A. E. Mahmoud and J. Wang, Anal. Chem., 2014, 86, 3092–3099 CrossRef CAS.
- Y. Tang, X. Wang, G. Zhu, Z. Liu, X.-M. Chen, H. K. Bisoyi, X. Chen, X. Chen, Y. Xu, J. Li and Q. Li, Small, 2023, 19, 2205440 CrossRef CAS.
- F. Hu, D. Mao, Kenry, X. Cai, W. Wu, D. Kong and B. Liu, Angew. Chem., Int. Ed., 2018, 57, 10182–10186 CrossRef CAS.
- M. M. Kim and A. Darafsheh, Photochem. Photobiol., 2020, 96, 280–294 CrossRef CAS.
- E. B. da Silva, M. W. M. Vasquez, B. C. de Almeida Teixeira, M. C. Neto, F. Sprenger, J. L. N. Filho, L. Almeida-Lopes and R. Ramina, Acta Neurochir., 2024, 166, 212 CrossRef PubMed.
- P. Sarbadhikary, B. P. George and H. Abrahamse, Theranostics, 2021, 11, 9054–9088 CrossRef CAS.
- Y. Wu, Y. Zhen, Y. Ma, R. Zheng, Z. Wang and H. Fu, J. Phys. Chem. Lett., 2010, 1, 2499–2502 CrossRef CAS.
- D. Reger, P. Haines, F. W. Heinemann, D. M. Guldi and N. Jux, Angew. Chem., Int. Ed., 2018, 57, 5938–5942 CrossRef CAS PubMed.
- K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776–1782 RSC.
- Y. Dong, B. Dick and J. Zhao, Org. Lett., 2020, 22, 5535–5539 CrossRef CAS.
- Z. Wang, L. Huang, Y. Yan, A. M. El-Zohry, A. Toffoletti, J. Zhao, A. Barbon, B. Dick, O. F. Mohammed and G. Han, Angew. Chem., Int. Ed., 2020, 59, 16114–16121 CrossRef CAS PubMed.
- Y. Dong, P. Kumar, P. Maity, I. Kurganskii, S. Li, A. Elmali, J. Zhao, D. Escudero, H. Wu, A. Karatay, O. F. Mohammed and M. Fedin, Phys. Chem. Chem. Phys., 2021, 23, 8641–8652 RSC.
- X. Xiao, K. Ye, M. Imran and J. Zhao, Appl. Sci., 2022, 12, 9933–9953 CrossRef CAS.
- M. Deiana, J. M. Andrés Castán, P. Josse, A. Kahsay, D. P. Sánchez, K. Morice, N. Gillet, R. Ravindranath, A. K. Patel, P. Sengupta, I. Obi, E. Rodriguez-Marquez, L. Khrouz, E. Dumont, L. Abad Galán, M. Allain, B. Walker, H. S. Ahn, O. Maury, P. Blanchard, T. Le Bahers, D. Öhlund, J. von Hofsten, C. Monnereau, C. Cabanetos and N. Sabouri, Nucleic Acids Res., 2023, 51, 6264–6285 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS.
- C. Adamo, T. Le Bahers, M. Savarese, L. Wilbraham, G. García, R. Fukuda, M. Ehara, N. Rega and I. Ciofini, Coord. Chem. Rev., 2015, 304–305, 166–178 CrossRef CAS.
- J. Yang, A. Griffin, Z. Qiang and J. Ren, Signal Transduction Targeted Ther., 2022, 7, 379 CrossRef CAS.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC.
- Z. Yu, W. Pan, N. Li and B. Tang, Chem. Sci., 2016, 7, 4237–4244 RSC.
- P. Zhang, H. Huang, S. Banerjee, G. J. Clarkson, C. Ge, C. Imberti and P. J. Sadler, Angew. Chem., Int. Ed., 2019, 58, 2350–2354 CrossRef CAS PubMed.
- Y. Yang, W. Zhu, L. Feng, Y. Chao, X. Yi, Z. Dong, K. Yang, W. Tan, Z. Liu and M. Chen, Nano Lett., 2018, 18, 6867–6875 CrossRef CAS PubMed.
- Y. Wang, S. Xu, L. Shi, C. Teh, G. Qi and B. Liu, Angew. Chem., Int. Ed., 2021, 60, 14945–14953 CrossRef CAS PubMed.
- S. Chakrabortty, B. K. Agrawalla, A. Stumper, N. M. Vegi, S. Fischer, C. Reichardt, M. Kögler, B. Dietzek, M. Feuring-Buske, C. Buske, S. Rau and T. Weil, J. Am. Chem. Soc., 2017, 139, 2512–2519 CrossRef CAS.
- W. Lv, Z. Zhang, K. Y. Zhang, H. Yang, S. Liu, A. Xu, S. Guo, Q. Zhao and W. Huang, Angew. Chem., Int. Ed., 2016, 55, 9947–9951 CrossRef CAS.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef CAS.
- Y. Tu, W. Xia, X. Wu and L. Wang, Org. Biomol. Chem., 2021, 19, 6098–6107 RSC.
- N. Niu, H. Zhou, N. Liu, H. Jiang, E. Hussain, Z. Hu and C. Yu, Chem. Commun., 2019, 55, 1036–1039 RSC.
- Z. Zhou, J. Liu, J. Huang, T. W. Rees, Y. Wang, H. Wang, X. Li, H. Chao and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20296–20302 CrossRef CAS.
- S. Li, Y. Chen, Y. Wu, S. Yao, H. Yuan, Y. Tan, F. Qi, W. He and Z. Guo, Chem. – Eur. J., 2022, 28, e202202680 CrossRef PubMed.
- B. Yuan, J. Liu, R. Guan, C. Jin, L. Ji and H. Chao, Dalton Trans., 2019, 48, 6408–6415 RSC.
- H. Ma, Y. Lu, Z. Huang, S. Long, J. Cao, Z. Zhang, X. Zhou, C. Shi, W. Sun, J. Du, J. Fan and X. Peng, J. Am. Chem. Soc., 2022, 144, 3477–3486 CrossRef CAS.
- J. S. Nam, M.-G. Kang, J. Kang, S.-Y. Park, S. J. C. Lee, H.-T. Kim, J. K. Seo, O.-H. Kwon, M. H. Lim, H.-W. Rhee and T.-H. Kwon, J. Am. Chem. Soc., 2016, 138, 10968–10977 CrossRef CAS.
- H. Deng, Z. Zhou, W. Yang, L.-S. Lin, S. Wang, G. Niu, J. Song and X. Chen, Nano Lett., 2020, 20, 1928–1933 CrossRef CAS.
- A. Spang, Curr. Opin. Cell Biol., 2018, 53, 92–96 CrossRef CAS PubMed.
- D. S. Schwarz and M. D. Blower, Cell. Mol. Life Sci., 2016, 73, 79–94 CrossRef CAS.
- A. Raffaello, C. Mammucari, G. Gherardi and R. Rizzuto, Trends Biochem. Sci., 2016, 41, 1035–1049 CrossRef CAS PubMed.
- J.-C. Simard, I. Durocher and D. Girard, Apoptosis, 2016, 21, 1279–1290 CrossRef CAS PubMed.
- M. Wang and R. J. Kaufman, Nat. Rev. Cancer, 2014, 14, 581–597 CrossRef CAS PubMed.
- T. Verfaillie, A. D. Garg and P. Agostinis, Cancer Lett., 2013, 332, 249–264 CrossRef CAS.
- J. Jamroskovic, M. Doimo, K. Chand, I. Obi, R. Kumar, K. Brännström, M. Hedenström, R. Nath Das, A. Akhunzianov, M. Deiana, K. Kasho, S. Sulis Sato, P. L. Pourbozorgi, J. E. Mason, P. Medini, D. Öhlund, S. Wanrooij, E. Chorell and N. Sabouri, J. Am. Chem. Soc., 2020, 142, 2876–2888 CrossRef CAS PubMed.
- M. Deiana, K. Chand, E. Chorell and N. Sabouri, J. Phys. Chem. Lett., 2023, 14, 1862–1869 CrossRef CAS.
- X. Li, S. Yu, Y. Lee, T. Guo, N. Kwon, D. Lee, S. C. Yeom, Y. Cho, G. Kim, J.-D. Huang, S. Choi, K. T. Nam and J. Yoon, J. Am. Chem. Soc., 2019, 141, 1366–1372 CrossRef CAS.
- X. Li, C. y Kim, S. Lee, D. Lee, H.-M. Chung, G. Kim, S.-H. Heo, C. Kim, K.-S. Hong and J. Yoon, J. Am. Chem. Soc., 2017, 139, 10880–10886 CrossRef CAS.
- D. Li, X.-Z. Wang, L.-F. Yang, S.-C. Li, Q.-Y. Hu, X. Li, B.-Y. Zheng, M.-R. Ke and J.-D. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 36435–36443 CrossRef CAS.
- H. Liu, L.-L. Lv, H. Wen, D.-M. Zhao, J. Wu, M.-R. Ke, B.-Y. Zheng, J. Li, X. Li and J.-D. Huang, ACS Appl. Mater. Interfaces, 2022, 14, 28581–28590 CrossRef CAS.
- Z. Luo, T. Lv, K. Zhu, Y. Li, L. Wang, J. J. Gooding, G. Liu and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 3131–3136 CrossRef CAS.
- L. Esteoulle, F. Daubeuf, M. Collot, S. Riché, T. Durroux, D. Brasse, P. Marchand, J. Karpenko, A. S. Klymchenko and D. Bonnet, Chem. Sci., 2020, 11, 6824–6829 RSC.
- X. Li, S. Yu, D. Lee, G. Kim, B. Lee, Y. Cho, B.-Y. Zheng, M.-R. Ke, J.-D. Huang, K. T. Nam, X. Chen and J. Yoon, ACS Nano, 2018, 12, 681–688 CrossRef CAS PubMed.
- Z. Hu, D. Wang, Q. Zhou, J. Jie and H. Su, J. Phys. Chem. B, 2024, 128, 576–584 CrossRef CAS PubMed.
- C. Okamoto, A. Momotake and Y. Yamamoto, J. Phys. Chem. B, 2023, 127, 4514–4522 CrossRef CAS PubMed.
- M. Deiana, K. Chand, J. Jamroskovic, I. Obi, E. Chorell and N. Sabouri, Angew. Chem., Int. Ed., 2020, 59, 896–902 CrossRef CAS.
- M. Deiana, K. Chand, J. Jamroskovic, R. N. Das, I. Obi, E. Chorell and N. Sabouri, Nanoscale, 2020, 12, 12950–12957 RSC.
- V. Grande, C.-A. Shen, M. Deiana, M. Dudek, J. Olesiak-Banska, K. Matczyszyn and F. Würthner, Chem. Sci., 2018, 9, 8375–8381 RSC.
- M. Collot, P. Ashokkumar, H. Anton, E. Boutant, O. Faklaris, T. Galli, Y. Mély, L. Danglot and A. S. Klymchenko, Cell Chem. Biol., 2019, 26, 600–614 CrossRef CAS.
- M. Collot, E. Boutant, M. Lehmann and A. S. Klymchenko, Bioconjugate Chem., 2019, 30, 192–199 CrossRef CAS PubMed.
- I. O. Aparin, R. Yan, R. Pelletier, A. A. Choi, D. I. Danylchuk, K. Xu and A. S. Klymchenko, J. Am. Chem. Soc., 2022, 144, 18043–18053 CrossRef CAS PubMed.
- F. Deng, L. Liu, Q. Qiao, C. Huang, L. Miao and Z. Xu, Chem. Commun., 2019, 55, 15045–15048 RSC.
- M. Collot, T. K. Fam, P. Ashokkumar, O. Faklaris, T. Galli, L. Danglot and A. S. Klymchenko, J. Am. Chem. Soc., 2018, 140, 5401–5411 CrossRef CAS PubMed.
- K. Ohira, Y. Sato and S. Nishizawa, ACS Sens., 2023, 8, 522–526 CrossRef CAS PubMed.
- K. Saczuk, M. Dudek, K. Matczyszyn and M. Deiana, Nanoscale Horiz., 2024 10.1039/D4NH00186A.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, ESI-MS, X-ray. CCDC 2083069 (for DBI), 2335339 (for DBXI) and 2335340 (for DBAI). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tb01014k |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |