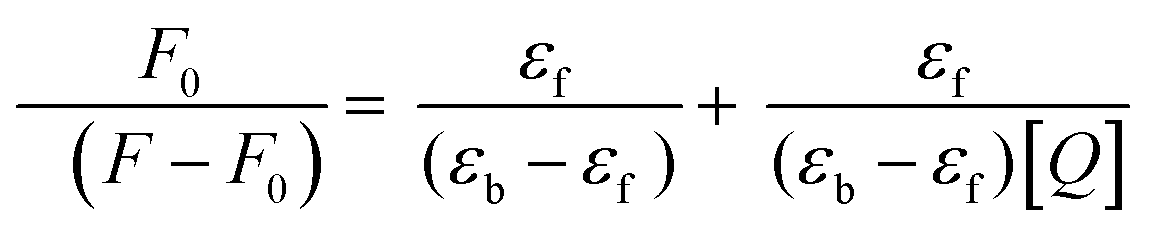DOI:
10.1039/D4TB00749B
(Paper)
J. Mater. Chem. B, 2024,
12, 9037-9049
A fluorescent “Turn-ON” probe with rapid and differential response to HSA and BSA: quantitative detection of HSA in urine†
Received
6th April 2024
, Accepted 3rd August 2024
First published on 7th August 2024
Abstract
The present study provides insight into the differential response of a benzimidazole-malononitrile fluorescent “Turn-ON” probe on interaction with two structurally similar proteins, BSA and HSA. Compound 6 shows more sensitivity towards the two SAs, which is completely lost in the case of compound 7, synthesized by substitution on 6. The aggregates of compound 6 show absorption maxima at 385 nm and weak emission maxima at 565 nm. Compound 6 forms a new emission band at 475 nm on gradual addition of BSA (200 μM) along with a slight increase in the emission band at 565 nm. However, on addition of HSA (50 μM), a new band at 475 nm is formed. In contrast to BSA, in the case of HSA, 50% quenching is observed in the emission band of compound 6 at 565 nm. The new band formed on the interaction of 6 with BSA shows four-fold more enhancement compared to HSA. Furthermore, the mechanism of interaction of 6 with serum albumin has been investigated through lifetime-fluorescence analysis, site-selective drug experiments, dynamic light scattering, FE-SEM, FT-IR, etc. Molecular docking studies and site marker drug displacement experiments reveal differential interactions of 6 towards the two structurally similar proteins. Aggregates of 6 with an average hydrodynamic size of 100–190 nm are disassembled on adding BSA and HSA, and the size of the serum albumin and 6 complex decreases to 10–20 nm, revealing the ligand's encapsulation in the serum albumin cavity. Practical applicability for the quantitative detection of HSA in human urine samples is also demonstrated. The high binding affinity, sensitivity, selectivity and differential response of probe 6 towards two serum albumins (HSA and BSA) and significant quantification of HSA in urine samples shows the potential ability of this probe in medical applications.
1. Introduction
Serum albumin (SA), including human serum albumin (HSA) and bovine serum albumin (BSA), is a water-soluble small globular protein, and the most prevalent protein in blood plasma exclusively synthesized in the liver.1 Serum albumin plays a key role in many biological functions, such as free radical scavenging, maintaining acid–base plasma, wound healing, maintenance of intravascular osmotic pressure and coagulation.2,3 Both serum albumins (HSA and BSA) show approximately 88% homology in structure and sequence.4,5 The mature secondary structure of serum albumin is a heart-shaped helical protein consisting of a single polypeptide chain with 580–585 amino acid residues and has a molecular weight of 66.5 kDa with an average life of 19–21 days.6,7 These serum albumins are composed of three α-helical domains, I, II, and III, each of which is further divided into two subdomains, A and B.8,9 SA is the most prevalent carrier protein essential for efficiently transporting a variety of endogenous and exogenous ligands to intended targets.10,11 Thus, SA serves as the primary target for any drug or medication to reach its target site and is responsible for pharmacokinetic properties.12
Serum albumin has two major drug binding sites: suldow site I, located in subdomain IIA, and suldow site II, located in subdomain IIIA, as well as subdomain IB, a recently discovered third drug binding site.13,14 Binding at site I is mainly driven by hydrophobic interaction, while at site II, binding of any drug and ligand is mediated by both electrostatic and hydrophobic interactions.15–17 All of these three drug binding sites of serum albumin have different hydrophobicity and spatial arrangements in their respective subdomains; therefore, they show a preferential affinity for specific drugs/ligands. For example, site I prefers bulky heterocyclic compounds like warfarin and indomethacin, while site II prefers aromatic carboxylic acids such as ibuprofen for binding.18,19 However, the recently discovered third site showed a preference for hemin and fusidic acid.20 The normal serum albumin range in blood plasma is 35–50 g L−1, and in urine, it is usually less than 30 mg L−1.21 Any alterations from the aforementioned range are indications of physiological malfunctioning of visceral organs, particularly the liver and kidneys. Generally, detection of the HSA level in blood plasma is employed to screen liver and kidney functioning along with other conditions including multiple myeloma, coronary heart disease, diabetes mellitus, renal disorder, neurometabolic disorder, and liver cirrhosis.22 Liver disorders like cirrhosis or hepatitis are responsible for lowering HSA levels in plasma and causing hypoproteinemia, while microalbuminuria-characterized by elevated HSA levels in urine, can be caused by diabetes mellitus, cardiovascular disease, and renal disorders.23,24 Consequently, the measurement of HSA levels in blood and urine has been used as a primary diagnostic tool for patients with severe illness. A correct diagnosis and the appropriate medication are the initial step in treating any ailment.
Most traditional diagnostic techniques used to detect HSA, including liquid chromatography, electrochemistry, mass spectrometry, proteomics, and immunoassays, suffer from several shortcomings, such as complex array-based protocols, sophisticated instrumental techniques, toxic reagents, time-consuming procedure, and high cost.25 Calorimetric methods such as using bromocresol green (BCG)26 and bromocresol purple (BCP)27 could overcome these drawbacks and have been utilized by diagnostic laboratories to measure the serum albumin level at low cost, but they often suffer from limitations like limited sensitivity and specificity, and are unable to measure trace urine albumin concentration.28 Thus, to get a definite conclusion for a diseased condition, development of molecular sensors is in high demand, because of their simple operation, rapid analysis, high sensitivity, and good selectivity. These molecular sensors can lead to a proper disclosure of a particular clinical condition at a relatively low cost. Due to structure similarity and low cost, BSA is widely used as an alternative model for HSA in many pharmacological and biochemical applications. Still, it is essential to remember that BSA showed only 75–88% similarity with HSA.29,30 Therefore, it is not appropriate to use BSA instead of HSA in many applications. Misuse of the two proteins leads to many severe injuries to patients. Despite considerable research towards the detection of serum albumin, investigation of differential recognition of the two structurally similar serum albumins is still less explored. To the best of our knowledge, no report is available where similar types of molecules showed differential behaviour and binding affinity towards two structurally similar proteins.
In 2016, Rui and co-workers reported a TPE appended malononitrile based fluorophore for selective detection of HSA, that gives a “Turn-ON” response on complexation with HSA having LOD of 2.7 nM (Table S1, ESI†).31 In 2017, the Wang group designed and synthesized a hydroxy-naphthlene bearing malononitrile fluorophore for the detection of human serum albumin in a complex biological sample. The developed probe showed 300-fold enhancement on interaction with HSA by binding at two major drug binding sites with LOD of 0.75 μM.32 In 2018, Xiaojun and co-workers reported an ICT and TICT using malononitrile-based fluorescent dyes for differential detection of serum albumin protein (LOD = 0.13 μM).33 Recently, Mahammad and co-workers reported an indole linked with dicyanoisophorone intramolecular charge transfer-based fluorophore for the detection of HSA in complex biofluids (LOD = 1.01 nM).34 In the present paper, we have developed a benzimidazole-malononitrile-based “Turn-ON” fluorescent probe 6, which shows differential behaviour and high sensitivity towards the serum albumins (HSA and BSA) without the interference of other bioanalytes, where the LODs observed were 0.2 nM for HSA and 0.86 nM for BSA. In phosphate buffer (0.1% DMSO), 6 forms aggregates with diameters of 170–190 nm. However, on addition of serum albumin, these self-assembled aggregates undergo disaggregation (size = 10 nm) and encapsulate into the hydrophobic cavity of serum albumin, causing enhancement of the fluorescence intensity. Furthermore, the mode of interaction in the secondary structure of serum albumin was studied with drug binding, lifetime fluorescence analysis, molecular docking, dynamic light scattering experiments, FE-SEM and FT-IR etc. Practical application of the probe was also demonstrated by quantitative detection of HSA in urine samples. Furthermore, for comparison, we have also developed probe 7 by blocking one of the positions of naphthalic anhydride of probe 6, where the sensitivity of probe 7 against both serum albumins is completely lost.
2. Experimental
2.1. Synthesis of probe 6
The synthesis of benzimidazole-malononitrile-based probes 6 and 7 for the detection of serum albumin is depicted in Scheme 1. The synthesis of intermediate 4 for both probes 6 and 7 is described in detail in Scheme S1 (ESI†).35 Compound 4 (500 mg, 0.83 mmol) was reacted with malononitrile (394 mg, 4.15 mmol) in ethanol in the presence of ammonium acetate (276 mg, 2.49 mmol) under reflux conditions to obtain probe 6. After completion of the reaction, water was added to the reaction mixture and orange-coloured precipitates were obtained. The precipitates were filtered, washed with water, and then oven-dried. Purification of the compound was done with column chromatography using chloroform and ethyl acetate (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). The synthesized probe was characterized by 1H NMR, 13C NMR and HRMS (Fig. S1–S3, ESI†). Orange solid; yield: 60%; 334 mg; 0.716 mmol; m.pt. 186–190 °C; 1H NMR (400 MHz, DMSO-d6): δ (ppm) 11.87 (s, 1H, ArH), 9.58 (dd, J = 8.6, 0.8 Hz, 1H, ArH), 8.57 (d, J = 7.6 Hz, 1H, ArH), 8.52–8.49 (m, 1H, ArH), 8.34 (d, J = 7.7 Hz, 1H, ArH), 7.96 (dd, J = 8.5, 7.3 Hz, 1H, ArH), 7.67–7.62 (m, 1H, ArH), 7.57–7.50 (m, 4H, ArH), 7.29 (s, 3H, ArH); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 173.2, 164.0, 163.6, 161.4, 158.7, 158.1, 149.9, 136.6, 133.0, 132.3, 132.1, 130.3, 130.2, 129.3, 129.1, 128.7, 128.0, 122.8, 116.3, 115.0, 114.8, 114.5, 81.3.
:2). The synthesized probe was characterized by 1H NMR, 13C NMR and HRMS (Fig. S1–S3, ESI†). Orange solid; yield: 60%; 334 mg; 0.716 mmol; m.pt. 186–190 °C; 1H NMR (400 MHz, DMSO-d6): δ (ppm) 11.87 (s, 1H, ArH), 9.58 (dd, J = 8.6, 0.8 Hz, 1H, ArH), 8.57 (d, J = 7.6 Hz, 1H, ArH), 8.52–8.49 (m, 1H, ArH), 8.34 (d, J = 7.7 Hz, 1H, ArH), 7.96 (dd, J = 8.5, 7.3 Hz, 1H, ArH), 7.67–7.62 (m, 1H, ArH), 7.57–7.50 (m, 4H, ArH), 7.29 (s, 3H, ArH); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 173.2, 164.0, 163.6, 161.4, 158.7, 158.1, 149.9, 136.6, 133.0, 132.3, 132.1, 130.3, 130.2, 129.3, 129.1, 128.7, 128.0, 122.8, 116.3, 115.0, 114.8, 114.5, 81.3.
 |
| | Scheme 1 Synthesis of probes 6 and 7. | |
HRMS (ESI-TOF) m/z: [M]+ Calcd for C29H14N4O3 466.1066, found 466.1296.
2.2. Synthesis of probe 7
Compound 4 (500 mg, 0.83 mmol) was refluxed with o-phenylenediamine (129 mg, 0.83 mmol) in acetic acid for 4–5 h. On completion of the reaction, the reaction mixture was poured into ice-cold water, and light-yellow precipitates of 5 were obtained. The precipitates were filtered, washed with water, and dried in an oven. Compound 5 (500 mg, 0.98 mmol) was further reacted with malononitrile (336 mg, 4.90 mmol) in ethanol in the presence of ammonium acetate (231 mg, 2.94 mmol) to form probe 7. The reaction was monitored by TLC, and after completion, water was added to the reaction mixture to obtain yellow precipitates. The compound was filtered, dried, and then purified by column chromatography using chloroform and ethyl acetate (8:2). The structure of the synthesized probe was further confirmed by 1H NMR, 13C NMR and HRMS (Fig. S4–S6, ESI†). Orange solid; yield: 58%, 318 mg; 0.589 mmol; m.pt. 210–215 °C; 1H NMR (400 MHz, CDCl3 + TFA): δ (ppm) 8.85 (d, J = 6.0 Hz, 1H, ArH), 8.73 (d, J = 7.2 Hz, 1H, ArH), 8.62 (d, J = 8.4 Hz, 1H, ArH), 8.51 (d, J = 7.8 Hz, 1H, ArH), 8.27 (d, J = 6.0 Hz, 1H, ArH), 8.15 (s, 1H, ArH), 7.73 (d, J = 7.7 Hz, 1H, ArH), 7.70–7.59 (m, 4H, ArH), 7.54 (dd, J = 13.7, 5.9 Hz, 4H, ArH), 7.47 (d, J = 7.5 Hz, 2H, ArH). 13C NMR (100 MHz, CDCl3 + TFA): δ (ppm) 173.7, 157.9, 148.7, 145.9, 135.3, 135.1, 135.0, 133.9, 133.6, 132.4, 131.5, 130.5, 129.9, 129.1, 129.0, 128.9, 128.8, 127.5, 126.5, 121.3, 118.0, 117.2, 116.7, 116.2, 115.7, 113.6, 113.3, 83.0.
HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C35H18N6O 539.1576, found 539.1624.
3. Optical properties of 6 in CH3CN–H2O binary mixtures
In 2001, Tang and his group came up with the concept of aggregation-induced emission (AIE).36 In 2002, Park's group introduced aggregation-induced emission enhancement (AIEE), which is responsible for improved emission of weak or non-emissive luminogens with the formation of their aggregates.37 Drawn-in by these fascinating phenomena, a plethora of AIEgens with diverse structures and adaptable features have been created and utilized in biological imaging, chemo- and bio-sensors, organic light-emitting devices, etc. The absorption band of 6 did not exhibit any considerable change in absorption spectrum with varying water concentrations. Therefore, the AIE properties of 6 were investigated by fluorescence measurement in ACN and H2O binary mixtures. Fig. 1 depicts an emission spectrum of 6 with varying concentrations of water. In the absence of water, i.e., in pure acetonitrile, 6 showed a weak emission band at 490 nm following excitation at 385 nm. The addition of water (up to 99%) led to an increase in fluorescence intensity with a substantial bathochromic shift of the emission maxima at around 70 nm (490 nm–560 nm). This red shift could be attributed to either formation of J-type aggregation between the monomers or due to high rigidity of the probe in an aggregated state and restricted intramolecular rotation. According to Kasha's exciton theory,38 due to the transition of a lower excitonic state, the J-type aggregation of any analyte caused the red shift of the emission maxima compared to the monomers.
 |
| | Fig. 1 Change in (A) fluorescence spectra and (B) fluorescence intensity of 6 in ACN–H2O mixture with increasing concentration of water. | |
4. Selectivity
The selectivity of ligand 6 towards HSA and BSA was investigated by a competitive fluorescence measurement experiment in the presence of various bioanalytes viz. arginine, aspartic acid, bilirubin, creatinine, cysteine, dextrose, fructose, galactose, glutamic acid, glutamine, glycine, histidine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, urea, uric acid, Na+, K+, Ca2+, Mg2+, Ni2+, and NH4+. On excitation at 385 nm, a solution of 6 (10 μM, 0.1% DMSO, pH 7.4) showed emission maxima centered at 565 nm in phosphate buffer solution. On addition of 150 μM of serum albumins and different bio-analytes to 6, the emission spectrum in phosphate buffer solution was recorded at the same wavelength (Fig. 2). Interestingly, the formation of a new band at 475 nm, which is 4-times more intense in the case of BSA than HSA, was observed with significant quenching in the emission band of 6 on addition of HSA. However, little enhancement in the emission band of 6 at 565 nm along with a new band at 475 nm was observed in the case of BSA. Other bio-analytes did not cause any significant change in the emission spectrum of compound 6. Hence, the synthesized probe 6 can be used to distinguish two structurally similar serum albumins in the presence of various bioanalytes.
 |
| | Fig. 2 Change in (A) emission spectrum and (B) emission intensity of 6 (10 μM) with different bioanalytes in phosphate buffer solution (pH = 7.4) at 298 K. | |
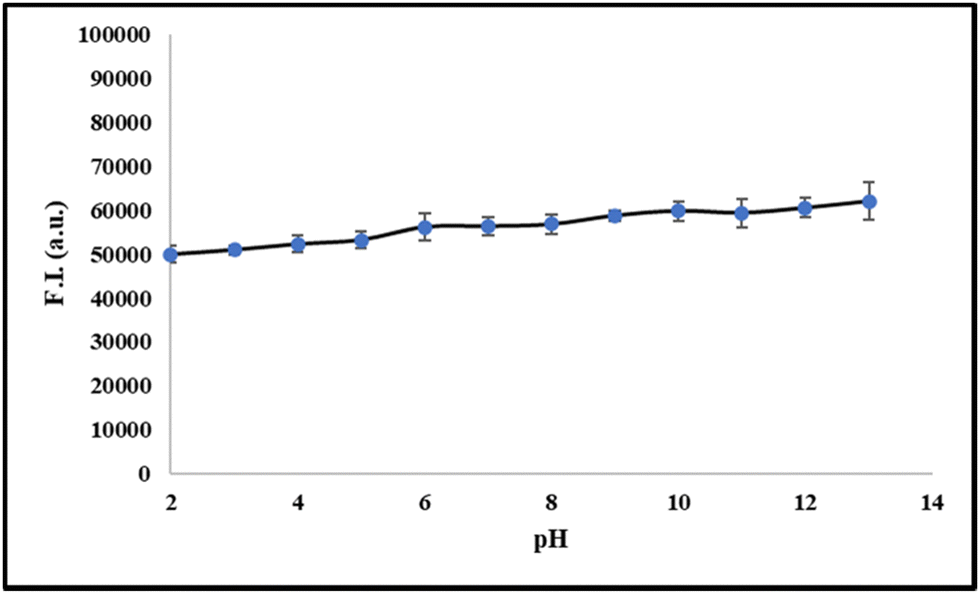
5. pH studies
To investigate the ability and stability of probe 6 for the detection of serum albumin in different pH ranges, we have conducted pH titrations of probe 6 in the pH range of 2–12. From the results, we observed that probe 6 is stable in the pH range of 6–9 and it can detect serum albumin in the physiological pH range (Fig. 3).
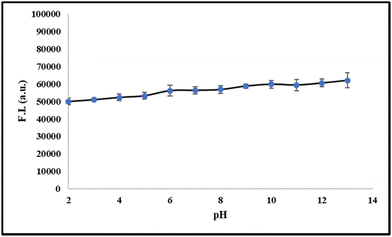 |
| | Fig. 3 Fluorescence intensity linear plot of 6 in pH range 2–12. | |
6. Optical responses of 6 and 7 towards serum albumin
The UV-vis spectra of 6 and 7 measured in phosphate buffer (pH = 7.4) exhibited low energy absorption bands at 385 and 420 nm, respectively (Fig. 4 and 5). These probes did not show any significant change on gradual addition of aliquots of serum albumins (HSA and BSA) except for enhancement of the band at 280 nm corresponding to serum albumins. The continuous addition of serum albumin to probe 6 caused a change in colour of the solution from colourless to faint-yellow, indicating a direct interaction of serum albumin with probe 6 (Fig. 4).
 |
| | Fig. 4 Change in absorption spectrum of 6 (10 μM) with increasing concentration of (A) BSA and (B) HSA in phosphate buffer solution (pH = 7.4) at 298 K. | |
 |
| | Fig. 5 Change in absorption spectrum of 7 (10 μM) with increasing concentration of (A) BSA and (B) HSA in phosphate buffer solution (pH = 7.4) at 298 K. | |
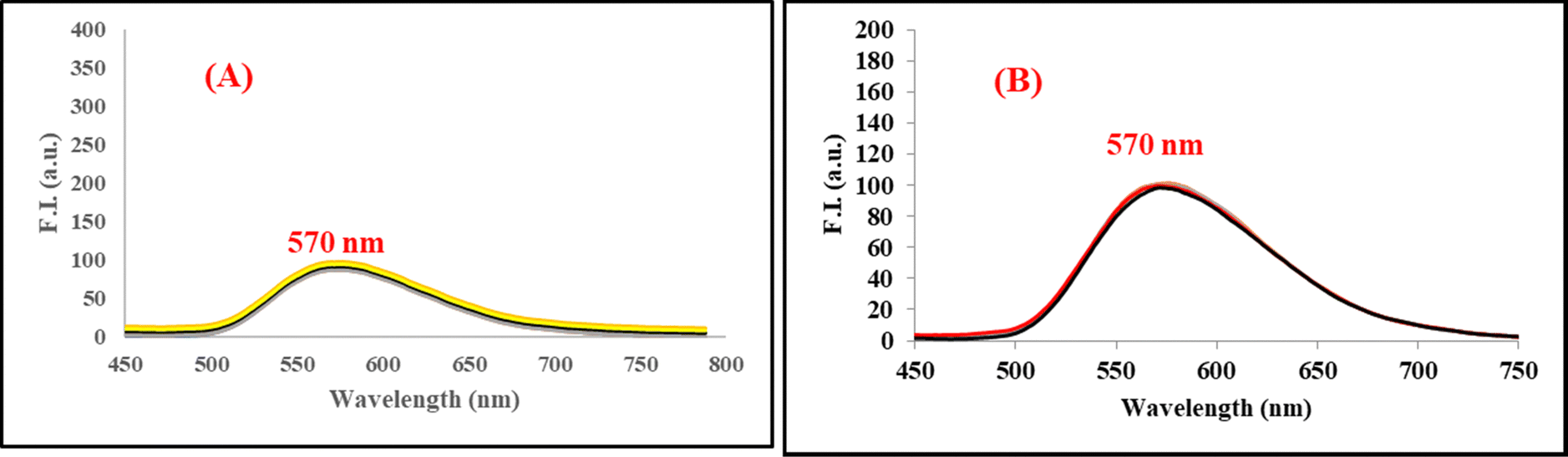
On excitation at 385 nm and 420 nm of probes 6 (lifetime = 0.24 ns) and 7 (lifetime = 0.67 ns) in phosphate buffer, a significant emission was observed at 565 and 570 nm, respectively. It was observed that probe 6 displayed significant and differential changes for both serum albumins i.e., HSA, and BSA. On addition of BSA to 6, a new band with emission maxima at 475 nm was formed. While a small increase in the emission maxima of 6 was observed at 565 nm. However, on addition of HSA, formation of a new band at 475 nm and 50% quenching of the emission band of 6 was observed in comparison to BSA. The fluorescence intensity of the new band formed at 475 nm is also different in both cases, having 4-fold more intensity for BSA than HSA (Fig. 6 and 7). The change in the emission spectrum of 6 up to addition of 200 μM of BSA was observed after that plateau was achieved. But, in the case of the emission spectrum of 6 on addition of HSA, a change was observed up to the addition of 50 μM of HSA after which negligible change was detected. The increase in fluorescence intensity can be due to the inhibition of TICT. Thus, it can be inferred that probe 6 has differential recognition and binding ability for the two structurally similar proteins. The formation of the emission band at 475 nm in the emission spectrum of 6 on addition of HSA and BSA can be attributed to disaggregation of the probe on addition of serum albumin. As also shown in the AIE experiment, red shift of the emission band from 490 nm to 560 nm was observed. The band at 490 nm corresponds to monomeric molecules while 560 nm corresponds to aggregates. However, in the case of 7 (substituting with o-phenylendiamine), the sensitivity of the probe towards serum albumins is completely lost (Fig. 7). Furthermore, the binding efficiency and ground state interaction of 6 with serum albumin were investigated from Benesi–Hildebrand eqn (1), where F0 and F are the emission values, and εf and εb are the molar extinction coefficients in the respective free and complexed forms of serum albumin proteins.39 Analyzing the linear binding plot of the Benesi–Hildebrand equation shows binding constant values of 2 × 105 M−1 for HSA and 5 × 104 M−1 for BSA (Fig. S7, ESI†).
| | 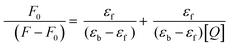 | (1) |
 |
| | Fig. 6 Change in emission spectrum of 6 (10 μM) with increasing concentration of (A) BSA and (B) HSA in phosphate buffer solution (pH = 7.4) at 298 K. | |
 |
| | Fig. 7 Change in emission spectrum of 7 (10 μM) with increasing concentration of (A) BSA and (B) HSA in phosphate buffer solution (pH = 7.4) at 298 K. | |
The limit of detection of 6 from the concentration dependent-fluorescence was investigated and found to be 8.6 × 10−10 M for BSA and 2 × 10−10 M for HSA (Fig. S8, ESI†). Furthermore, there is a significant distinguishable color change of probe 6 solution on addition of the two proteins, i.e. BSA and HSA, by the naked eye under UV light (Fig. 6, inset picture). The change in colour of the solution of probe 6 on addition of serum albumin can be credited to complex formation of the probe with serum albumin. The fluorescence enhancement is more in the case of BSA, which provides a bright yellow colour to the solution. Meanwhile, in HSA, the enhancement is observed with quenching of the band at 565 nm that could result in the light green colour fluorescence of the solution of the probe 6 and serum albumin complex.40 Thus, the synthesized probe is suitable for the differential and significant detection of two serum albumin proteins. The results obtained clearly indicate that the probe is sensitive to both the serum albumins and slightly more sensitive towards HSA.
7. Fluorescence quenching of serum albumin
The fluorescence quenching method is a significant tool to find the interaction of small molecules and proteins.41 HSA and BSA both have intrinsic fluorescence due to phenylalanine (Phe), tyrosine (Tyr), and tryptophan (Trp) residues. Change in the emission spectrum of a protein and degree of quenching on addition of a quencher suggests changes around the microenvironment of the above-mentioned residues, especially around the Trp residue.42 Thus, fluorescence measurements provide information regarding the binding mechanism of small molecular probes or drug molecules with protein macromolecules. Excitation of a protein around 280 nm is due to both Tyr and Trp residues and majorly Trp residue.43 The change in the fluorescence spectra of serum albumin with addition of probe 6 was also investigated. On excitation of serum albumin in PBS (pH = 7.4, 10 μM) at 280 nm, a significant emission band at 340 nm was observed. On gradual addition of 6, the fluorescence intensities of HSA and BSA decreased linearly up to the addition of 10 μM and 30 μM, respectively (Fig. 8). Significant quenching in the HSA and BSA emission spectrum suggested that probe 6 binds in the cavity near the tryptophan residue and showed considerable energy transfer from protein to 6. Furthermore, Stern Volmer and quenching constants were calculated by applying the Stern–Volmer equation and found to be 4.8 × 104 M−1, 4.8 × 1012 M−1 s−1 and 8.07 × 104 M−1, 8.07 × 1012 M−1 s−1 for BSA and HSA, respectively,44 which reveals that they were two times higher for HSA than BSA (Fig. S9, ESI†). Moreover, the values of quenching constant were much greater than standard values assigned for diffusion-limited quenching in water (2 × 1010 M−1 s−1).45 This suggested that there could be static quenching via. complex formation between probe 6 and serum albumin in the ground state.
 |
| | Fig. 8 Change in emission spectrum of (A) BSA (10 μM) and (B) HSA (10 μM) with increasing concentration of 6 in phosphate buffer solution (pH = 7.4) at 298 K and (C) change in emission intensity of HSA and BSA on addition of probe 6. | |
8. DFT studies
In order to understand further the “Turn-ON” fluorescence response of 6 towards HSA/BSA, and other structural and spectral properties, we performed DFT calculations to obtain the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) through structure optimization using DFT/B3LYP/6–311G (d,p) on Gaussian software. The molecule's HOMO and LUMO distribution indicate significant binding regions for receptor interactions. Other parameters, like chemical reactivity and kinetic stability, were determined from the energy band gap of the molecule. If a molecule exhibits a low energy bandgap, a higher degree of intramolecular transport of electrons from donor to acceptor is observed due to the high reactivity of the molecule. In the present case, the LUMO of 6 is predominantly concentrated over the benzimidazole ring and one nitrile group. In comparison, the HOMO is distributed over the benzimidazole ring and naphthalic anhydride, and extended to the methylene carbon and nitrogen atoms of malononitrile. Negative energy values of the HOMO (EHOMO = −7.097 eV) and LUMO (ELUMO = −6.154 eV) suggested the stability of the molecule. Probe 6 showed an energy gap of 0.942 eV ensuring significant energy transfer and thus, ultimately proved to be a significant sensing probe (Fig. 9).
 |
| | Fig. 9 The energy optimized structure of 6 (A), and computed frontier molecular orbital representations of the (B) HOMO and (C) LUMO of probe 6. | |
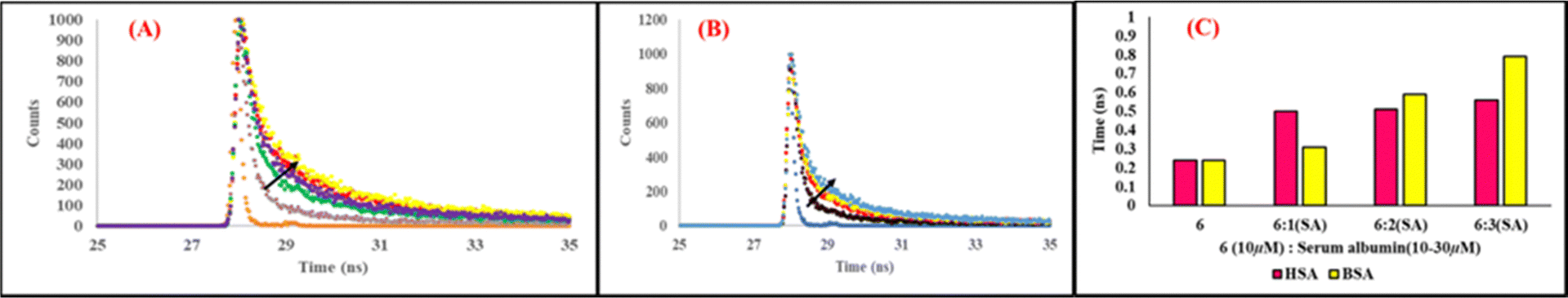
9. Time-resolved fluorescence analysis
Time-resolved fluorescence analysis is one of the most authentic experiments to explore useful information regarding excited state interaction and microenvironment around the excited probe in micro heterogeneous media. The average lifetime decay of probe 6 (10 μM) revealed triexponential decay with a lifetime of 0.58 ns (24%), 5.52 ns (33%), and 0.11 ns (41%). On addition of 10-30 μM of HSA to the solution of 6, the increase in lifetime for the first decay was from 0.58 ns (24%) to 1.13 ns (23%) and 1.25 ns (36%), for the lifetime of the second decay it was from 5.52 ns to 5.77 ns (33%) and 7.44 (38%) and the increase in lifetime of the third decay was from 0.11 ns (41%) to 0.18 ns (42%) and 0.24 ns (25%). Similarly, the fluorescence lifetime of 6 was also increased on addition of 10–30 μM BSA, for the first decay to 1.24 (31%) and 1.48 ns (38%), the increase in lifetime of the second decay was to 5.44 ns (33%) and 7.8 ns (43%) and the increase in lifetime for the third decay was to 0.18 (42%) and 0.24 ns (25%) (Table S2, ESI†). The increase in average lifetime of probe 6 from 0.24 ns to 0.56 ns and 0.96 ns on respective addition of 30 μM of HSA and BSA points to the complex formation in the excited state (dynamic quenching), binding in the hydrophobic cavity of the protein and stability in the interior of the protein microenvironment (Fig. 10), which results in inhibition of twisted internal charge transfer (TICT). Inhibition of the energy dissipated by the non-radiative pathway in the excited state is enhanced, causing an increase in lifetime of 6 and hence, favouring the fluorescence emission enhancement in the presence of serum albumin. The increase in average life of 6 on addition of 10 μM of HSA is significant and after that it becomes almost constant even at addition of 30 μM. However, in the case of the average lifetime of 6 on addition of BSA, the change remains significant throughout. This increase in average lifetime of the probe on addition of BSA is more compared to HSA. This points to the stability of the probe and serum albumin complex, which is more with BSA compared to HSA, causing the degree of TICT to be more in the case of BSA than HSA. Thus, time-resolved fluorescence analysis also suggested differential behaviour of the probe towards two serum albumins and more fluorescence enhancement in the case of BSA compared to HSA. Hence, the fluorescence “Turn-ON” response can be concluded due to the binding of the probe within the hydrophobic cavity of serum albumin proteins.46 The steric hindrance and low polarity-environment inside HSA might restrict free intramolecular rotation of the probe and ultimately inhibit TICT.
 |
| | Fig. 10 Change in lifetime spectrum of 6 (10 μM) with increasing concentration of (A) BSA and (B) HSA in phosphate buffer solution (pH = 7.4) at 298 K. (C) Change in average life of the probe on addition of serum albumin. | |
10. Site markers
To understand the binding mechanism of any probe with serum albumin, it is very crucial to know the number and position of binding sites of the probe to serum albumin. For this, we did Job's plot study of probe 6 with both the serum albumins in phosphate buffer solution by measuring the fluorescence intensity at 475 nm as a function of molar fraction of the probe. The highest fluorescence intensity was observed at 0.6, in the case of HSA suggesting 2:1 stoichiometry of the 6:HSA complex. Meanwhile, in the case of BSA the highest fluorescence intensity at 0.5, suggesting 1:1 stoichiometry the complex of 6:BSA (Fig. 11).47 To explore this further, serum albumins have two major drug binding sites, site I (warfarin binding site) and site II (ibuprofen binding site), located in the hydrophobic cavities of subdomains IIA and IIIA, respectively. Recently, a third drug binding site (bilirubin binding site) located in subdomain IB has also been recognized. To investigate the possible binding site of 6 for the two serum albumins, fluorescence titrations using complexes of serum albumins and probe 6 in the ratio of 1:1 with site-specific drugs like warfarin, ibuprofen, and bilirubin were carried out at an excitation wavelength of 280 nm.48
 |
| | Fig. 11 Job's plot based on fluorescence intensity at 475 nm with total concentration of probe 6 for (A) BSA and (B) HSA of 10 μM in phosphate buffer (pH = 7.4) at 298 K. | |
Both the HSA/BSA:6 complexes exhibited quenching in their emission spectrum on gradual addition of site markers warfarin and bilirubin (Fig. S10–S12, ESI†), and a significant blue shift of about 40 nm was also observed in the case of HSA. On the other hand, with the addition of ibuprofen to the HSA solution, a very small change is observed in the emission spectrum of HSA and 6. However, a small decrease in fluorescence intensity with a blue shift of about 20 nm was observed on addition of ibuprofen to BSA and the probe 6 complex.
Quenching in HSA:6 associated with blue shift on addition of warfarin and bilirubin is probably due to the release of the probe from the cavity of HSA by the site marker. Negligible change on addition of ibuprofen to the solution of HSA indicates that the probe has no affinity towards subdomain IIIA. However, in the case of BSA, on the addition of warfarin and bilirubin, significant quenching is observed but the emission maxima remain unaltered, which indicates the formation of the BSA:6: warfarin/bilirubin complex. On addition of ibuprofen to the BSA solution, 50% quenching associated with blue shift was observed, which reveals that the binding of 6 on BSA is close to site II in subdomain IIIA, while in HSA, binding is close to subdomain IIA and slightly towards IB. Thus, different numbers and positions of binding sites with different stoichiometry of probe 6 and serum albumin complexes can be the reason for differential behaviour of the probe towards HSA and BSA.
11. Molecular docking
Molecular docking is the most authentic technique to verify the binding domain, interaction sites, binding mode and type of interaction of any drug or ligand molecule in the protein structure. Probe 6 is docked with serum albumin to find the most probable binding mode with the lowest binding score using Autodock 1.5.7. Generally, serum albumin contains three binding sites viz. suldow: site I, located in subdomain IIA; site II, located in subdomain IIIA; and site III, located in subdomain IB. The free energy change of probe 6 was calculated in all three drug binding sites for both HSA (PDB: 1n5u) and BSA (PDB: 4f5s, A chain) after deleting water molecules, and adding polar hydrogens and Kollman charges in both the crystal structures.
Among all three sites, ligand 6 docked at subdomain IIA of HSA possessed the lowest binding energy −11.34 kcal mol−1, compared to −8.02 kcal mol−1 at subdomain IIIA and −8.82 kcal mol−1 at subdomain IB. Comparing the free binding energy of 6 at all three sites, it is concluded that the probe has preferential propensity to bind at site I in the hydrophobic cavity of subdomain IIA. Meanwhile, in BSA, docking of 6 at the three drug binding sites revealed that the ligand has more affinity towards site II (ΔG = −10.87 kcal mol−1) present in the hydrophobic cavity of subdomain IIIA, compared to −9.02 kcal mol−1 at site I and −10.35 kcal mol−1 at site III (Fig. S13, ESI†). The results obtained from molecular docking of the serum albumin crystal structures and probe 6 are in good agreement with the site marker drug displacement experiment (Table S3, ESI†).
12. FT-IR studies
FT-IR study is a sophisticated technique employed to investigate the changes in conformation of the secondary structure of the protein on interaction with probe 6 where the spectrum in the range of 1200–1700 cm−1 is monitored. In this range, a band between 1200 and 1400 cm−1 is characteristic of the amide III band, 1400 to 1500 cm−1 is for the amide II band, and 1600 to 1700 cm−1 is for the amide I band. Among all three-bands, the amide I band in the range of 1600–1700 cm−1 is further utilized to investigate the changes in the conformation of the secondary structure of the protein on interaction with the probe. The amide I band consists of β-sheets (1610–1640 cm−1), random coil (1640–1650 cm−1), α-helix (1650–1660 cm−1), β-turn (1660–1680 cm−1), and β-antiparallel (1680–1692 cm−1).49Fig. 12 shows a reduction in the absorption intensity of the protein band (500 μM) on the addition of 250 μM of probe 6. Furthermore, for quantitative analysis of the protein, the secondary structure in the absence and presence of the probe was deconvoluted in the spectral region of 1600-1700 cm−1, and the area was measured using Gaussian fitting curves. From Fig. 12, it is evident that compared to free HSA, α-helix, β-sheet, β-anti, and random coil are decreased by 1% on interaction with ligand 6. However, for BSA, the decrease in α-helix content is 3% and β-anti is 1%, while β-sheet content is increased by 1% on interaction with probe 6. These results indicated that conformational changes in the secondary structure of the protein on interaction with the probe are very minimal.
 |
| | Fig. 12 Second-derivative resolution enhancement and curve-fitted amide I region (1700–1600 cm−1) for (A) HSA (500 μM) and (B) HSA + 6 (500 + 250 μM), (C) BSA (500 μM) and (D) BSA + 6 (500 + 250 μM) in phosphate buffer solution (pH = 7.4) at 298 K. | |
13. Particle size and morphology change investigation
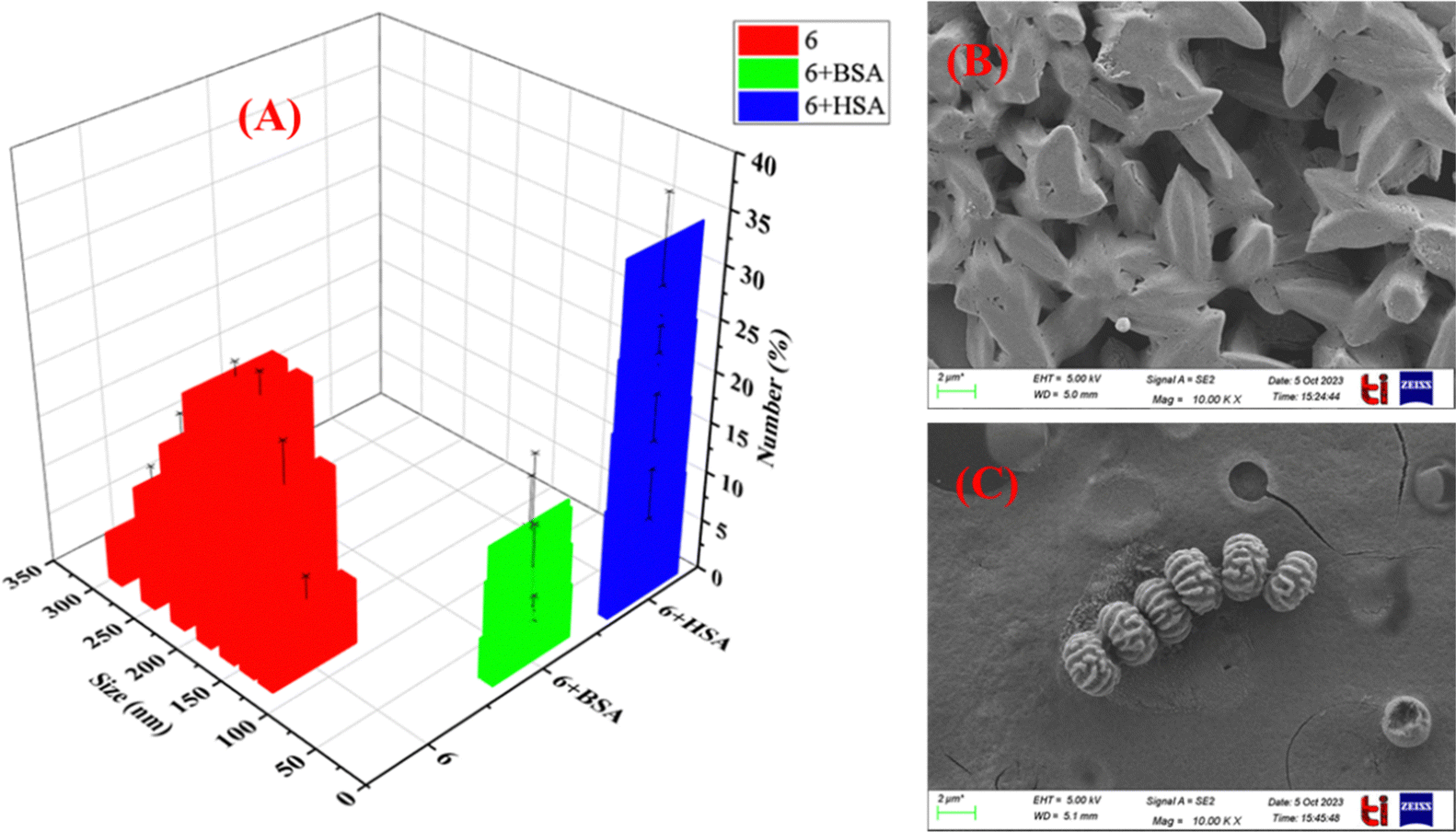
Further insight into the self-assembled particle size distribution and morphology of 6 in the absence and presence of protein was investigated by dynamic light scattering (DLS) and FE-SEM studies. The initial fluorescence studies in ACN–H2O binary mixture revealed an increase in fluorescence intensity with increased water content due to the phenomenon of aggregation-induced enhancement (AIE). DLS experiments revealed that the average hydrodynamic size for aggregates of 6 (5 μM) in 99.9% phosphate buffer solution is 190 nm. On addition of HSA (50 μM) to the solution of 6 in phosphate buffer, the particle size was extensively decreased from 190 nm to 10 nm. Similarly, on addition of BSA (50 μM) to 6 in phosphate buffer solution, the hydrodynamic size of the BSA-6 complex appeared to be 18.2 nm. The overall morphology of 6 (water-0.1% DMSO) alone and after the addition of HSA was also recorded by FE-SEM on thin films prepared by the drop casting technique on glass plates. The results revealed that the average aggregate size of 6 is around 120 nm while on addition of HSA, the aggregates of 6 disaggregate and form spherical complex structures of much smaller size of 20 nm. These results are also supported by DLS experiments. Thus, it is concluded that in 99.9% water, the size of probe 6 forms self-assembled aggregates with a size of 100–190 nm, while on addition of protein, these aggregates get disaggregated into a spherical serum albumin and probe 6 complex with a size of 10–20 nm, assigned to microencapsulation driven disassembly, responsible for the “Turn-ON” response in emission spectrum (Fig. 13).
 |
| | Fig. 13 Particle size distribution determined by (A) DLS and change in morphology and particle size distribution determined by (B) and (C) FE-SEM images in phosphate buffer solution (pH = 7.4). | |
14. Cytocompatibility
Excellent results from in vitro spectroscopic investigation studies motivated us to determine the potential biological applications of the developed probe. Therefore, we investigated the cytotoxic effects of the developed probes towards a normal cell line (HEK293) using the standard MTT assay. The cytotoxicity assay was performed following a 24 h treatment of the cell line with probes 6 and 7. From the results, it was observed that the viability of the cells treated with probes 6 and 7 is more than 78% and 67%, respectively, even at 100 μM concentration (Fig. 14). Hence, it can be concluded that this probe exhibited minimal cytotoxicity and can be further used for biological applications.32
 |
| | Fig. 14 Percentage cell viability of normal human cell line Hek293 with probes 6 and 7. | |
15. Practical application: quantification of HSA in real-urine samples
Microalbuminuria is a condition of elevated albumin level in urine and is regarded as a well-established cardiovascular risk marker, and an indication of liver and kidney diseases. Therefore, detection of human serum albumin in urine is of enormous clinical value, but methods available for direct detection of HSA in urine are less reliable due to interference of other biological metabolites.50 Significant results for the differential detection of two serum albumins in phosphate buffer solution with high sensitivity and selectivity of the developed probe motivated us to use it further to evaluate the potential for the detection of HSA in human urine. Titrations were carried out in 50-fold diluted PBS (pH = 7.4) urine samples to eliminate the autofluorescence response and pH effect. The change in emission was monitored on excitation at 385 nm. A similar change in fluorescence intensity of the emission band of probe 6 and the formation of a new band at 475 nm with the incremental addition of HSA in the concentration range of 0–50 μM was observed (Fig. 15) with a detection limit of 1.9 × 10−10 M in diluted urine. Furthermore, we conducted a quantification recovery experiment by spiking the diluted urine sample with known concentrations of HSA and monitoring the change in the emission spectrum. The analytical results of recovery of HSA (Table 1) demonstrate the potential of the probe to differentially recognize and detect serum albumin in urine at a very early stage of diagnosis.
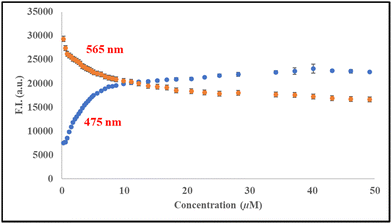 |
| | Fig. 15 Change in emission intensity of 6 (10 μM) upon addition of HSA (0–50 μM) in diluted urine (50-fold dilution) in PBS (pH = 7.4) at 298 K. | |
Table 1 Recovery study of spiked HSA in diluted urine (PBS = 7.4) using the developed fluorescent probe
| S. No. |
HSA added (μM) |
HSA found (μM) |
Recovery (%) |
| 1 |
1.0 |
1.03 |
103 |
| 2 |
2.0 |
2.1 |
105 |
| 3 |
3.0 |
2.95 |
98.3 |
| 4 |
10.0 |
10.1 |
101 |
| 5 |
25.0 |
24.8 |
99.2 |
Next, to ensure the potential of our developed probe, we evaluated the HSA content of four different urine samples diluted with PBS spiked with different concentration of HSA and compared the observed fluorescence using the same calibration plot. The calculated HSA levels in eight samples are shown in Table 2. The same samples were then clinically investigated by a diagnostic laboratory using conventional turbidimetric assay in which anti-albumin antibody is added that forms a complex with the present albumin protein. This makes the solution turbid and the absorbance intensity of the solution is measured by a spectrophotometer. The results of HSA content in the urine samples by the fluorescence and clinical methods are in close agreement (Table 2) (Fig. 16). The turbidimetric assay is suitable for concentrations above 0.07–0.15 μM (5–10 mg L−1)51 while the developed probe 6 can detect significantly up to 0.0002 μM (0.013 mg L−1). Thus, the closer agreement between the results of the two methods showed that probe 6 can detect HSA in urine samples with higher sensitivity.
Table 2 The determination of the spiked HSA level by the clinical method and the same determined by the fluorescence method with probe 6 in human urine samples
| S. No. |
Clinical method μM (mg L−1) |
Fluorescence method μM (mg L−1) (Probe 6) |
| 1 |
0.19 (12.93) |
0.11 (7.37 ± 1.5) |
| 2 |
0.23 (15.47) |
0.17 (13.34 ± 3.8) |
| 3 |
0.40 (27.83) |
0.43 (26.81 ± 4.5) |
| 4 |
0.80 (53.45) |
0.85 (56.70 ± 1.8) |
| 5 |
2.1 (140.3) |
1.99 (133.4 ± 4.3) |
| 6 |
2.3 (154.4) |
2.25 (150.4 ± 3.2) |
| 7 |
2.85 (190.3) |
3.20 (213.86 ± 1.7) |
| 8 |
2.62 (175.2) |
2.79 (186.75 ± 2.6) |
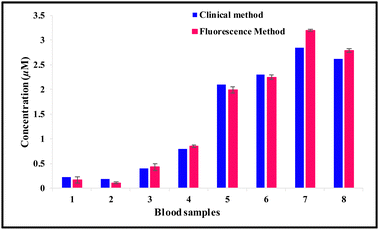 |
| | Fig. 16 Comparison of the HSA estimation data in human urine samples using the probe 6 fluorescence method with data obtained from a diagnostic laboratory using the immune turbidimetric method. | |
16. Conclusion
In conclusion, we have developed a benzimidazole-malononitrile appended fluorescent probe for differential detection of two serum albumin proteins, HSA and BSA, through spectroscopic techniques. Interestingly, the two fluorescent probes possess a similar basic structure, but the response toward serum albumin protein is entirely different. Probe 6 showed high sensitivity and differential response towards the two proteins, but substitution on 6 caused a complete loss of sensitivity (7) towards the two proteins. Probe 6 showed the formation of a new band on addition of BSA (200 μM) and HSA (50 μM). The enhancement in the new formed band is 4-fold more intense in the case of BSA than HSA. The probe also showed sensitivity towards two serum albumins without the interference of other biomolecules. It was inferred from site marker drug displacement analysis that probe 6 binds in the warfarin pocket of HSA while in BSA, the probe prefers to bind in the ibuprofen pocket. FT-IR analyses revealed the detection of serum albumin without a change in the secondary structure of the protein. An increase in average lifetime of the ligand on interaction with serum albumin proteins indicated complex formation in the excited state via dynamic quenching. Furthermore, DLS study and FE-SEM results showed disaggregation of aggregates of probe 6 into very small particles and encapsulation of the ligand into the serum albumin protein cavity. The developed probe also gave a significant response for the quantitative detection of HSA in urine samples and compared the results with the clinical method, which proves that the probe can be further used for various biomedical applications.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We gratefully acknowledge ANRF (CRG/2023/004080), CEEMS-TIET(TIET/CEEMS/Regular/2021/018) for financial support and DST-FIST(SR/FST/CS-II/2018/69) for HRMS analysis.
References
-
(a) A. Carfray, K. Patel, P. Whittaker, P. Garrick, G. Griffuths and G. Warwick, Kidney Int., 1999, 55, 2595 Search PubMed;
(b) V. Arroyo, R. Garcia-Martinezand and X. Salvatella, J. Hepatol., 2014, 61, 396–407 CrossRef CAS PubMed;
(c) F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed.
-
(a) X. Zhang, B. Yao, Q. Hu, Y. Hong, A. Wallace, K. Reynolds, C. Ramsey, A. Maeder, R. Reed and Y. Tang, Mater. Chem. Front., 2020, 4, 2548–2570 RSC;
(b) C. V. Kumar and A. Buranaprapuk, Angew. Chem., Int. Ed. Engl., 1997, 36, 2085–2087 CrossRef CAS.
- C. Y. S. Chung and V. W. W. Yam, J. Am. Chem. Soc., 2011, 133, 18775–18784 CrossRef CAS PubMed.
- P. Bolel, N. Mahapatra and M. Halder, J. Agric. Food Chem., 2012, 60, 3727–3734 CrossRef CAS PubMed.
- A. Mallick, B. Haldar and N. Chattopadhyay, J. Phys. Chem. B, 2005, 109, 14683–14690 CrossRef CAS PubMed.
-
(a) S. Huang, F. Li, C. Liao, B. Zheng, J. Du and D. Xiao, Talanta, 2017, 170, 562–568 CrossRef CAS PubMed;
(b) M. A. Razzak, J. E. Lee and S. S. Choi, Food Hydrocolloids, 2019, 91, 290–300 CrossRef CAS.
-
(a) A. Spada, J. Emami, J. A. Tuszynski and A. Lavasanifar, Mol. Pharmaceutics, 2021, 18, 1862–1894 CrossRef CAS;
(b) D. C. Carter and X. M. He, Science, 1990, 249, 302 CrossRef CAS PubMed;
(c) W. Jia, X. Jin, W. Liu, B. Zhao, M. Zhang, Y. Yang, W. Yin, Y. Zhang, Y. Liu and S. Zhou, Spectrochim. Acta, Part A, 2023, 291, 122289 CrossRef CAS.
-
(a) A. Jayaraj, H. A. Schwanz, D. J. Spencer, S. Bhasin, J. A. Hamilton, B. Jayaram, A. L. Goldman, M. Krishna, M. Krishnan and A. Shah, Endocrinology, 2021, 162, bqaa199 CrossRef PubMed;
(b) J. F. Xu, Y. S. Yang, A. Q. Jiang and H. L. Zhu, Crit. Rev. Anal. Chem., 2022, 52, 72–92 CrossRef CAS PubMed.
- S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, Protein Eng., 1999, 12, 439–446 CrossRef CAS PubMed.
-
(a) C. V. Kumar, A. Buranaprapuk, G. J. Opiteck, M. B. Moyer, S. Jockusch and N. J. Turro, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10361–10366 CrossRef CAS PubMed;
(b) J. W. Wu, P. Su, Y. Yang, J. Huang, Y. Y. Wang and Y. Yang, J. Mater. Chem. B, 2014, 2, 775–782 RSC.
-
(a) Y. J. Xu, M. M. Su, H. L. Li, Q. X. Liu, C. Xu, Y. S. Yang and H. L. Zhu, Anal. Chim. Acta, 2018, 1043, 123–131 CrossRef CAS PubMed;
(b) Z. G. Wang, X. J. Yan, H. B. Liu, D. L. Zhang, W. Liu, C. Z. Xie, Q. Z. Li and J. Y. Xu, J. Mater. Chem. B, 2020, 8(36), 8346–8355 RSC.
-
(a) Z. D. Zhivkova, Curr. Pharm. Des., 2015, 21, 1817–1830 CrossRef CAS PubMed;
(b) S. Tayyab and S. R. Feroz, Adv. Protein Chem. Struct. Biol., 2021, 123, 193–218 CAS;
(c) E. Lázaro, P. J. Lowe, X. Briand and B. Faller, J. Med. Chem., 2008, 51, 2009–2017 CrossRef PubMed;
(d) H. Yang, Q. Zeng, Z. He, D. Wu and H. Li, J. Pharm. Biomed. Anal., 2020, 178, 112962 CrossRef CAS PubMed.
-
(a) F. Zsila, Mol. Pharmaceutics, 2013, 10, 1668–1682 CrossRef CAS PubMed;
(b) X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed;
(c) M. Dockal, D. C. Carter and F. Ruker, J. Biol. Chem., 1992, 274, 29303–29310 CrossRef PubMed.
- N. Maurya, J. K. Maurya, U. K. Singh, R. Dohare, M. Zafaryab, M. Moshahid Alam Rizvi, M. Kumari and R. Patel, Mol. Pharm., 2019, 16, 952–966 CrossRef CAS PubMed.
- A. Jahanban-Esfahlan, L. Roufegarinejad, R. Jahanban-Esfahlan, M. Tabibiazar and R. Amarowicz, Talanta, 2020, 207, 120317 CrossRef CAS.
- K. Yamasaki, T. Maruyama, U. Kragh-Hansen and M. Otagiri, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1996, 1295, 147–157 CrossRef PubMed.
-
(a) D. Rout, S. Sharma, P. Agarwala, A. K. Upadhyaya, A. Sharma and D. K. Sasmal, ACS Omega, 2023, 8, 3114–3128 CrossRef CAS PubMed;
(b) E. S. Krenzel, Z. Chen and J. A. Hamilton, Biochemistry, 2013, 52, 1559–1567 CrossRef CAS PubMed;
(c) M. C. Jimenez, M. A. Miranda and I. Vaya, J. Am. Chem. Soc., 2005, 127, 10134–10135 CrossRef CAS PubMed;
(d) P. Singh, L. S. Mittal, S. Kaur, S. Kaur, G. Bhargava and S. Kumar, Sens. Actuators, B, 2018, 255, 478–489 CrossRef CAS.
- G. Lambrinidis, T. Vallianatou and A. Tsantili-Kakoulidou, Adv. Drug Delivery Rev., 2015, 86, 27–45 CrossRef CAS PubMed.
- S. Ashraf, H. Qaiser, S. Tariq, A. Khalid, H. A. Makeen, H. A. Alhazmi and Z. Ul-Haq, Curr. Res. Struct. Biol., 2023, 6, 100114 CrossRef CAS PubMed.
-
(a) F. Zsila, Z. Bikadi, D. Malik, P. Hari, I. Pechan, A. Berces and E. Hazai, Bioinformatics, 2011, 27, 1806–1813 CrossRef CAS PubMed;
(b) M. L. Hall, W. L. Jorgensen and L. Whitehead, J. Chem. Inf. Model., 2013, 53, 907–922 CrossRef CAS PubMed.
-
(a)
T. Peter, All about albumin, Academic press, San Diego, CA, 1996 Search PubMed;
(b) P. Li, Y. Wang, S. Zhang, L. Xu, G. Wang and J. Cui, Tetrahedron Lett., 2018, 59, 1390–1393 CrossRef CAS.
-
(a) D. de Zeeuw, H. H. Parving and R. H. Henning, J. Am. Soc. Nephrol., 2006, 17, 2100–2105 CrossRef CAS PubMed;
(b) S. H. Murch, D. Phillips, J. A. Walker-Smith, P. J. Winyard, N. Meadows, S. Koletzko, B. Wehner, H. A. Cheema, R. A. Risdon and J. Klein, Lancet, 1996, 347, 1299–1301 CrossRef CAS PubMed.
-
(a) J. Nompleggi and H. L. Bonkovsky, Hepatology, 1994, 19, 518–533 CrossRef PubMed;
(b) J. Wen, X. Chen, S. Wei, X. Ma and Y. Zhao, Evid. Based Complementary Altern. Med., 2022, 2245491 Search PubMed.
- P. Raja, A. P. Maxwell and D. P. Brazil, Cardiovasc. Drugs Ther., 2021, 35, 455–466 CrossRef CAS PubMed.
-
(a) M. Sasmal, A. S. Musha Islam, D. Moni, D. Maiti, A. Dutta and M. Ali, ACS Appl. Bio Mater., 2022, 5, 5854–5864 CrossRef CAS PubMed;
(b) P. F. Ruhn, J. D. Taylor and D. S. Hage, Anal. Chem., 1994, 66, 4265–4271 CrossRef CAS PubMed;
(c) A. Silver, A. Dawnay, J. Landon and W. R. Cattell, Clin. Chem., 1986, 32, 1303–1306 CrossRef CAS PubMed.
- B. T. Doumas, W. A. Watson and H. G. Biggs, Clin. Chim. Acta, 1971, 31, 87–96 CrossRef CAS PubMed.
- A. E. Pinnell and B. E. Northam, Clin. Chem., 1978, 24, 80–86 CrossRef CAS.
- B. T. Doumas and T. Peters Jr, Clin. Chem., 2009, 55, 583–584 CrossRef CAS.
- J. L. Sohl and A. G. Splittgerber, J. Chem. Educ., 1991, 68, 262–264 CrossRef CAS.
- A. Bujacz, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 1278–1289 CrossRef CAS PubMed.
- Y. Yu, Y. Huang, F. Hu, Y. Jin, G. Zhang, D. Zhang and R. Zhao, Anal. Chem., 2016, 88, 6374–6381 CrossRef CAS PubMed.
- Y. R. Wang, L. Feng, L. Xu, J. Hou, Q. Jin, N. Zhou, Y. Lin, J. N. Cui and G. B. Ge, Sens. Actuators, B, 2017, 245, 923–931 CrossRef CAS.
- J. Du, Q. Gu, J. Chen, J. Fan and X. Peng, Sens. Actuators, B, 2018, 265, 204–210 CrossRef CAS.
- M. Sasmal, A. S. Islam, D. Moni, A. Katarkar and M. Ali, J. Mater. Chem. B, 2024, 12, 4478–4488 RSC.
- Y. Feng, L. Bai, S. Wang, X. Kong, L. Cong, Q. Zhao, Q. Yang and Y. Li, Chem. Res. Chin. Univ., 2017, 33, 534–539 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
-
(a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC;
(b) S. Dey, R. Purkait, K. Pal, K. Jana and C. Sinha, ACS Omega, 2019, 4, 8451–8464 CrossRef CAS PubMed;
(c) S. B. Roy, A. Maity, T. Das and K. K. Rajak, J. Lumin., 2019, 206, 649–659 CrossRef CAS.
-
(a) E. G. McRae and M. Kasha, J. Chem. Phys., 1958, 28, 721–722 CrossRef CAS;
(b) M. Kasha, Radiat. Res., 1963, 20, 55–70 CrossRef CAS;
(c) M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
-
(a) R. L. Scott, Recl. Trav. Chim. Pays-Bas, 1956, 75, 787–789 CrossRef CAS;
(b) I. Singh, R. Rani, V. Luxami and K. Paul, Eur. J. Med. Chem., 2019, 166, 267–280 CrossRef CAS PubMed.
- P. Kavyashree, B. Chakraborty, V. Rani and A. L. Koner, J. Mater. Chem. B, 2022, 10, 5071–5085 RSC.
- Y. Wei, J. Li, C. Dong, S. Shung, D. Liu and C. W. Huie, Talanta, 2006, 70, 377–382 CrossRef CAS PubMed.
- I. Hediyeh, R. Omid, S. Roshanak and C. Jamshidkhan, J. Phys. Chem. B, 2012, 116, 1951–1964 CrossRef PubMed.
-
(a) H. M. Zhang, Y. Q. Wang and M. L. Jiang, Dyes Pigm., 2009, 82, 156–163 CrossRef CAS;
(b) A. Sulkowska, J. Mol. Struct., 2002, 614, 227–232 CrossRef CAS.
-
(a) J. R. Lakowicz and G. Webber, Biochem., 1973, 12, 4161–4170 CrossRef CAS PubMed;
(b) H. Boaz and G. K. Rollefson, J. Am. Chem. Soc., 1950, 72, 3435–3443 CrossRef CAS;
(c) A. Khan, K. Paul, I. Singh, J. P. Jasinski, V. A. Smolenski, E. P. Hotchkiss, P. T. Kelley, Z. A. Shalit, M. Kaur and S. Banerjee, Dalton Trans., 2020, 49, 17350–17367 RSC.
- Z. G. Wang, X. J. Yan, H. B. Liu, D. L. Zhang, W. Liu, C. Z. Xie, Q. Z. Li and J. Y. Xu, J. Mater. Chem. B, 2020, 8, 8346–8355 RSC.
- S. Samanta, S. Halder and G. Das, Anal. Chem., 2018, 90, 7561–7568 CrossRef CAS.
-
(a) G. Dey, A. Gupta, T. Mukherjee, P. Gaur, A. Chaudhary, S. K. Mukhopadhyay, C. K. Nandi and S. Ghosh, ACS Appl. Mater. Interfaces, 2014, 6, 10231–10237 CrossRef CAS PubMed;
(b) G. Dey, P. Gaur, R. Giri and S. Ghosh, Chem. Commun., 2016, 52, 1887–1890 RSC;
(c) Y. R. Wang, L. Feng, L. Xu, J. Hou, Q. Jin, N. Zhou, Y. Lin, J. N. Cui and G. B. Ge, Sens. Actuators, B, 2017, 245, 923–931 CrossRef CAS.
- S. Dhiman, R. Kour, G. Kumar, S. Kaur, V. Luxami, P. Singh and S. Kumar, Mater. Chem. Front., 2022, 6, 2651–2660 RSC.
-
(a) H. Dezhampanah and A. M. Moghaddam Pour, J. Biomol. Struct. Dyn., 2022, 40, 8143–8154 CrossRef CAS PubMed;
(b) H. Dezhampanah, R. Firouzi, Z. M. Shoeili and R. Binazir, J. Mol. Struct., 2020, 1205, 127557 CrossRef CAS.
-
(a) A. Bandyopadhyay, R. Hazra, D. Roy and A. Bhattacharya, Chem. – Asian J., 2024, e202301055 CrossRef CAS PubMed;
(b) W. Huang, N. Jiang, X. Lv, Y. Qu, X. Zhang and L. Wang, Dyes Pigm., 2024, 221, 111820 CrossRef CAS;
(c) X. Chao, D. Yao, Y. Qi, C. Yuan and D. Huang, Anal. Chim. Acta, 2021, 1188, 339201 CrossRef CAS PubMed;
(d) S. Huang, F. Li, C. Liao, B. Zheng, J. Du and D. Xiao, Talanta, 2017, 170, 562–568 CrossRef CAS PubMed.
- G. Viswanathan and A. Upadhyay, Adv. Chronic Kidney Dis., 2011, 18, 243–248 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*