
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEuropium–tannic acid nanocomplexes devised for bone regeneration under oxidative or inflammatory environments†
Daniel
Fernández-Villa
 ab,
María Rosa
Aguilar
ab and
Luis
Rojo
*ab
ab,
María Rosa
Aguilar
ab and
Luis
Rojo
*ab
aInstituto de Ciencia y Tecnología de Polímeros (ICTP) CSIC, 28006 Madrid, Spain. E-mail: rojodelolmo@ictp.csic.es; Tel: +34-915-622-900
bCentro de Investigación Biomédica en Red de Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), 28029, Madrid, Spain
First published on 2nd July 2024
Abstract
Europium ions (Eu3+) are gaining attention in the field of regenerative medicine due to increasing evidence of their osteogenic properties. However, inflammatory and oxidative environments present in many bone diseases, such as osteoporosis or rheumatoid arthritis, are known to hinder this regenerative process. Herein, we describe a straightforward synthetic procedure to prepare Eu3+–tannic acid nanocomplexes (EuTA NCs) with modulable physicochemical characteristics, as well as antioxidant, anti-inflammatory, and osteogenic properties. EuTA NCs were rationally synthesized to present different contents of Eu3+ on their structure to evaluate the effect of the cation on the biological properties of the formulations. In all the cases, EuTA NCs were stable in distilled water at physiological pH, had a highly negative surface charge (ζ ≈ −25.4 mV), and controllable size (80 < Dh < 160 nm). In vitro antioxidant tests revealed that Eu3+ complexation did not significantly alter the total radical scavenging activity (RSA) of TA but enhanced its ability to scavenge H2O2 and ferrous ions, thus improving its overall antioxidant potential. At the cellular level, EuTA NCs reduced the instantaneous toxicity of high concentrations of free TA, resulting in better antioxidant (13.3% increase of RSA vs. TA) and anti-inflammatory responses (17.6% reduction of nitric oxide production vs. TA) on cultures of H2O2- and LPS-stimulated macrophages, respectively. Furthermore, the short-term treatment of osteoblasts with EuTA NCs was found to increase their alkaline phosphatase activity and their matrix mineralization capacity. Overall, this simple and tunable platform is a potential candidate to promote bone growth in complex environments by simultaneously targeting multiple pathophysiological mechanisms of disease.
1. Introduction
Bone remodeling is a complex and dynamic process that relies on the interplay between various physiological mechanisms, cell types, and extracellular matrix (ECM) components. In healthy individuals, there is a finely regulated balance between bone formation and bone resorption, but in osteopenic diseases, dysregulations lead to a loss of bone mineral density and a higher risk of fractures.1 Given the aging of the population, it is estimated that the already high prevalence of these bone diseases (i.e., 200 million estimated cases of osteoporosis worldwide) will continue to increase in the coming decades and new therapeutic strategies are in demand.2 However, therapies envisioned to promote effective bone regeneration under complex environments need to target not only osteogenic pathways but also additional pathophysiological mechanisms underlying the diseases.In this regard, both oxidative stress (OxS) and inflammation have been described as factors contributing to bone remodeling dysregulation.3–6 On the one hand, OxS occurs when the levels of reactive species exceed the antioxidant defense system's capacity to neutralize them, damaging both the ECM and intracellular components.7,8 In the context of osteopenia, several mechanisms have been identified linking OxS and the loss of bone mass, mainly affecting osteoblasts, osteocytes, and the proteoglycans and glycoproteins composing the ECM.3–6 On the other hand, under chronic inflammation, such as that observed in rheumatoid arthritis or periodontitis, the persistent release of pro-inflammatory cytokines and chemokines leads to the differentiation and overactivation of osteoclasts, promoting bone resorption and the loss of bone mass.9,10 Furthermore, chronic inflammation and OxS are closely interconnected, interacting, and amplifying each other, leading to a harmful cycle of tissue damage and dysfunction.8,11,12
One promising alternative to simultaneously address these challenges is the use of multi-bioactive nanostructured systems that could be easily delivered in situ to specific areas of interest. In this work, tannic acid (TA), a natural-based polyphenol found in plants, was selected to prepare nano-sized vehicles due to its ability to interact with cations in a pH-dependent manner,13 and its low price and multiple properties, including antioxidant, anti-inflammatory, anti-cancer, and antibacterial ones.14–16 Moreover, some bone-regenerative effects have also been reported for TA when coating nanotubes and implants, mainly attributed to its ability to sequester bioactive metals such as strontium, zinc, or magnesium.17–19
In this regard, the use of bioactive cations for bone tissue engineering is of particular interest because of their high stability, tunability, reduced cost, and non-immunogenicity.20 However, there are fewer trivalent cations with reported regenerative properties compared to divalent ones. This phenomenon may be attributed to the adverse effects on calcification processes observed in vitro for certain trivalent cations such as Al3+, Ga3+, La3+, and Gd3+. These effects are linked to their tendency to react with the oxygen atoms of the phosphate ions forming hydroxyapatite, due to their high positive charge and small radius, which explains their effective anti-calcification behavior.21 An intriguing approach to incorporating biocompatible trivalent metals and phenolic linkers involves the preparation of metal-phenolic networks (MPNs), which can be synthesized via the coordination of phenolic ligands, mainly polyphenols, and trivalent cations such as Gd3+, Rh3+, Ru3+, V3+, or Fe2+/3+, among others.22 These MPNs offer the possibility of encapsulating specific drugs and biologically active organic molecules for drug delivery, and even for cell encapsulation.23 Furthermore, lanthanide MPNs, such as those prepared with TA and europium(III) (Eu3+), are gaining attention due to their unique properties for multiple biological applications including sensing and bioimaging.24,25 Recently, Eu3+ has gained attention due to research claiming both osteogenic and angiogenic properties, indispensable features for effective bone regeneration. However, those articles studied the effect of Eu3+ on already bioactive materials, such as bioglass or hydroxyapatite formulations,26–31 as surface coatings,32,33 or a combination of both34–36 and mainly assessing their osteogenic potential by studying their effects on the osteoblastic differentiation of mesenchymal stem cells. Thus, the exact mechanism of europium's osteogenic effects has not been elucidated yet.
For all these reasons, in this study, nano-sized complexes based on TA and Eu3+ have been devised for future intended applications in osteogenic bones with challenging environments in situ (e.g., subchondral or intraosseous administrations), whether as a mono- or as a combinational therapy using advanced injectable formulations. The hypothesis behind this approach is that the complexation of europium by tannic acid would allow for the preservation of the potent antioxidant and anti-inflammatory capabilities of the latter without compromising osteogenic processes due to its acidity, while simultaneously promoting them through the action of europium. Moreover, we have rationally designed them to present different contents of Eu3+ to further characterize the contribution of this cation to the final regenerative properties of the system. Herein, we described the synthesis and characterization of Eu3+–tannic acid nanocomplexes (EuTA NCs), as well as their biological effects in vitro. Evaluation of their anti-inflammatory and antioxidant properties has been carried out in a macrophage cell line (RAW264.7) while the test of its osteogenic potential has been performed in cultures of human osteoblasts. Altogether, the obtained results present EuTA NCs as a potent antioxidant platform where the addition of europium improves TA cytocompatibility, anti-inflammatory, and osteogenic properties, envisioning their potential applicability for bone regeneration purposes under complex conditions where regeneration is hindered.
2. Materials and methods
2.1. Reagents
EuCl3·6H2O (96.0%) was purchased from TCI; 1 M HEPES Buffer solution (pH 7.3) for molecular biology, alamarBlue® cell viability reagent, Quant-iTTM PicoGreenTM dsDNA Assay kit, T-PERTM tissue protein extraction reagent and ethylenediaminetetraacetic acid (EDTA) disodium salt dihydrate, 99+% were purchased from ThermoFisher Scientific; 33% w/v hydrogen peroxide (H2O2) and sodium hydroxide were purchased from Panreac; hydrochloric acid 37% w/v was purchased from VWR Chemicals; 4-nitrophenyl phosphate, disodium salt, and hexahydrate 98+% was purchased from Acros Organics; 2,2-diphenyl-1-picrylhydrazyl (free radical, DPPH, 95%) was purchased from Alfa-Aesar; and tannic acid (TA, Mw = 1701.2 g mol−1), iron(II) chloride tetrahydrate (FeCl2), 4-nitrophenol, Alizarin red S (AzR), cetylpyridinium chloride (CPC), 2′,7′-dichlorofluorescin diacetate (DCFH-DA), Griess reagent (modified), sodium nitrite (NaNO2) β-glycerophosphate disodium salt hydrate, (+)-sodium L-ascorbate and dexamethasone were purchased from Sigma-Aldrich. All chemicals were used as received.2.2. Synthesis of EuTA NCs
EuTA NCs were prepared via an easy, one-step method at r.t. (∼22 °C) by mixing EuCl3·6H2O and TA solutions under intense sonication. A 50 mM HEPES buffer pH 7.3 was selected to carry out the synthesis as this buffer presents a negligible binding constant for metals and the formation of tris-complexes is favored at neutral pH.13 Precursor solutions of EuCl3·6H2O and TA were prepared in distilled water and 50 mM HEPES buffer pH 7.3, respectively, and used immediately to avoid the oxidation and hydrolysis of TA. EuTA NCs formed instantaneously when adding EuCl3·6H2O dropwise to the TA solution (1 mg mL−1 final TA concentration in the mixture) under intense sonication (500 W, 38% amplitude) using a VC-505 ultrasonic processor (Sonics). EuTA NCs were then isolated from their suspensions via a centrifugation step at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm for 15 min in a Centrifuge MiniSpin® plus (Eppendorf). Next, pellets were further resuspended in distilled water or culture media and sonicated for 30 s to obtain the final EuTA NCs suspensions. When necessary, suspensions were snap-frozen with liquid N2 and freeze-dried using a LyoAlfa 6 freeze-drier (Telstar).
500 rpm for 15 min in a Centrifuge MiniSpin® plus (Eppendorf). Next, pellets were further resuspended in distilled water or culture media and sonicated for 30 s to obtain the final EuTA NCs suspensions. When necessary, suspensions were snap-frozen with liquid N2 and freeze-dried using a LyoAlfa 6 freeze-drier (Telstar).
EuTA NCs were devised to present increasing Eu:TA complexation ratios (C.R.) by producing complexes with different contents of Eu3+. As previously demonstrated, to obtain a 100% C.R. between TA and a trivalent cation, a Cat3+:TA molar ratio of 10:3 must be applied, as, at pH 7.3, TA and Eu3+ have 10 and 3 available coordination positions, respectively.37 Thus, NCs with 100, 75, 50, and 25% C.R.s were prepared by adding 10:3, 7.5:3, 5:3 and 2.5:3 Eu3+:TA molar ratios, respectively.
2.3. Characterization of EuTA NCs
Study of the chemical composition of EuTA NCs. The content of TA and Eu3+ on EuTA NCs was calculated using an indirect method. After the centrifugation step, supernatants were examined by UV-VIS (λmax = 278 nm) using a UV-VIS NanoDrop One C spectrophotometer (Thermo Scientific) and by emission spectroscopy on an inductively coupled plasma optical emission spectrometer (ICP-OES, PerkinElmer 430DV) to calculate the content of free TA and Eu3+, respectively. In this regard, the contents of TA and Eu3+ on EuTA NCs were calculated by subtracting the content of the free species from the total added during the synthesis. Calibration curves were constructed from the absorbance of a solution series prepared with TA and a europium standard solution, 1000 mg L−1 Eu in diluted nitric acid from Eu2O3 (Merck). These normalized contents were used to calculate the experimental C.R.s (eqn (1)), the reaction yields of different EuTA NCs (eqn (2)), and the concentrations to use throughout the study.
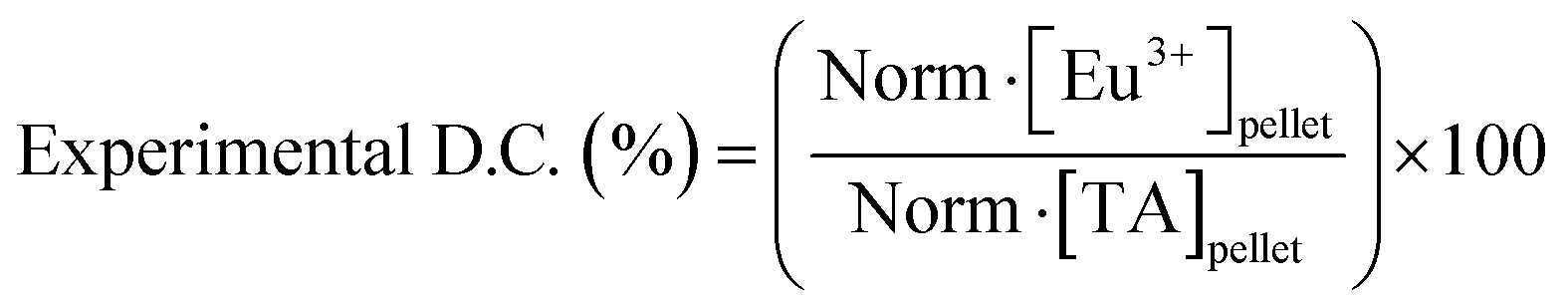 | (1) |
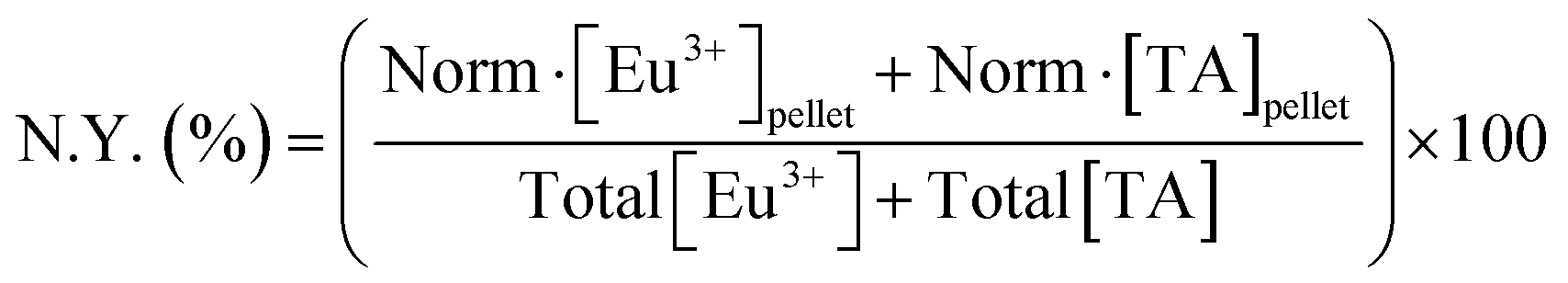 | (2) |
Furthermore, the weight attributed to europium in EuTA NCs was also validated using a direct method by analyzing the pellets instead of the supernatant. For this, different known masses of EuTA NCs were weighed and dissolved in 5 mL of concentrated HCl 37%, to ensure the rupture of tris- and bis-complexes, and then diluted to 20 mL with distilled water for the ICP-OES spectroscopic analysis.
Study of the interaction between Eu3+ and TA. Fourier-transform infrared spectroscopy (FTIR) was conducted on a PerkinElmer spectrum two spectrophotometer with an attenuated total reflectance (ATR) attachment to study the chemical compositions of freeze-dried EuTA NCs and to compare them to pristine TA and EuCl3. Analyses were performed using 32 scans and a resolution of 4 cm−1. Finally, spectra were smoothed, corrected to ATR, and normalized for drawing conclusions.
Study of the thermal degradation of EuTA NCs. TGA of freeze-dried NCs was performed using TGA Q500 apparatus (TA instruments) under dynamic N2 at a heating rate of 5 °C min−1 from 30 to 700 °C.
Study of the crystallinity of EuTA NCs. XRD was performed to analyze the crystalline composition of freeze-dried EuTA NCs. Analyses were performed on a Bruker D8 Advance instrument working with CuKα radiation (nλ = 1.542 Å) at a 0.01 step size and 0.5 s per step. D-spacing values were calculated from the Bragg law equation (nλ = 2d·sin(φ)).
Study of the UV-VIS spectroscopic properties of EuTA NCs. UV-VIS spectroscopy was performed on 2 µg mL−1 TA solution and EuTA NC suspensions to compare their maximum absorption wavelengths. Measurements were carried out with the aforementioned NanoDrop spectrophotometer and a quartz cuvette. EuTA NCs’ spectra were normalized to their highest absorbance value (i.e., A213
nm).
:chromium alloy (80:20). The particle size of EuTA NCs was estimated by measuring the diameter of multiple (n > 50) nanoparticles from FE-SEM micrographs using the measuring tool of ImageJ/FIJI software (version v1.53c).
2.4. Evaluation of the antioxidant properties
The total radical scavenging activity (RSA) of EuTA NCs was evaluated using a DPPH assay adapted from previous studies.38,39 The 1,1-diphenyl-2-picrylhydrazyl reagent was dissolved at 0.127 mM in ethanol 60%. Similarly, EuTA NCs were prepared as previously specified and resuspended in ethanol 60% after the centrifugation step. Serial dilutions of EuTA NCs were performed in the same solvent to evaluate their antioxidant properties at different concentrations. Next, 100 µL DPPH solution was added to 100 µL of EuTA NCs in the dark. The absorbance at 515 nm was monitored every 5 minutes until reaching the steady state using a multi-detection microplate reader synergy HT (BioTek Instruments). The RSA was calculated using eqn (3):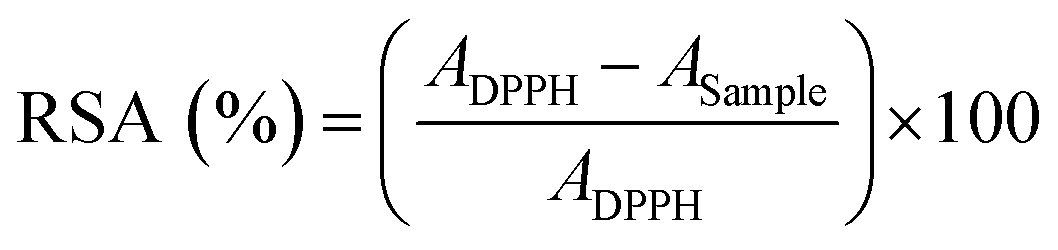 | (3) |
Hydrogen peroxide (H2O2) scavenging activity was analyzed following the protocol proposed by Pucci et al. with slight modifications.41 Briefly, 2 µL of 33% w/v H2O2 was added to 1998 µL of solutions of TA or EuTA NCs at different concentrations in distilled water. Next, the absorbance of H2O2 was immediately measured at 230 nm using the aforementioned NanoDrop spectrophotometer and a quartz cuvette. The corresponding blanks were prepared without H2O2, while control samples were prepared with distilled water. The percentage of H2O2 scavenging was calculated using eqn (4):
 | (4) |
Fe2+ chelating activity was determined using the method reported by Dinis et al. with some modifications.42 TA and EuTA NCs were prepared in distilled water and pHs were adjusted to 7.3 using 1 M NaOH when needed. Then, 37.5 µL of 2 mM FeCl2 solution was added to 300 µL of the samples at different concentrations and left to react for 15 min at r.t. in the dark. Next, 150 µL of 5 mM ferrozine was added and the final volume was adjusted to 3 mL with ethanol. The formation of ferrous chelates was spectrophotometrically calculated by measuring the absorbance at 562 nm using the aforementioned NanoDrop OneC spectrophotometer and eqn (5):
 | (5) |
2.5. Biological properties of EuTA nanocomplexes
For these studies, EuCl3 and TA precursor solutions were previously filtered using Millex-GP syringe filter units (0.22 µm), and EuTA NCs were prepared under sterile conditions working on a laminar flow cabinet. For over-time experiments, fresh EuTA NCs were prepared and added at every change of the medium.
:1 v/v with the Griess reagent kit for nitrite determination, and incubated for 15 min at r.t. in the dark. Then, absorbance at 540 nm was measured and the values were interpolated in a calibration curve constructed with known concentrations of NaNO2. In parallel, cell viability was monitored as previously described using alamarBlue® measurements and employed for normalization purposes.
:100 diluted) without EDTA. Protein extraction and subsequent assays were performed on ice to avoid protein degradation. ALP activity was determined following a previously standardized method,45 and then it was normalized according to the concentration of double-stranded DNA (dsDNA) content calculated using the PicoGreen assay dye and following the manufacturer's instructions for microtitration.
At each experimental time, wells were stained with AzR and quantification using the CPC extraction method was performed as previously described.45 Representative images were obtained under each condition before AzR extraction using an optical microscope Nikon Eclipse TE2000-S. Ca2+ concentration in the matrix was calculated using a calibration curve constructed with known concentrations of AzR in CPC buffer.
2.6. Data analysis and statistics
Results were expressed as means ± standard deviations of at least three experiments carried out with multiple replicates. Statistical analyses were performed using GraphPad Prism 8.00 software. To evaluate differences among groups, the homogeneity of variances was first confirmed and then one-way ANOVA tests, followed by a post hoc Scheffe test, were performed. In all the cases, p < 0.05 was established to be considered as statistically significant. The results were fitted when necessary to the most appropriate regression models using GraphPad Prism 8.00 software and the parameters are detailed in the ESI.†3. Results and discussion
In this work, the synthesis of EuTA NCs with different contents of Eu3+ in their structures has been reported along with the characterization of their physicochemical and biological properties, which endorse future preclinical evaluations in in vivo models in the context of osteopenic diseases under oxidative and/or inflammatory environments.3.1. Preparation of EuTA NCs with different contents of Eu3+
There are extensive reports available in the literature on the ability of TA to coordinate trivalent cations, with the ferric one (Fe3+) being the most studied, due to the 10 polyphenolic moieties present in its structure (5 catechols and 5 pyrogallols). In this sense, it has been well-documented that mono-, bis-, and tris-complexes can be formed depending on the pH of the solution.13Considering these data, the preparation of EuTA NCs was carefully devised to retrieve formulations with different contents of europium by adjusting the number of equivalents of EuCl3 added during the synthesis (Fig. 1A), which was confirmed using spectroscopical techniques as shown in Fig. 1B.
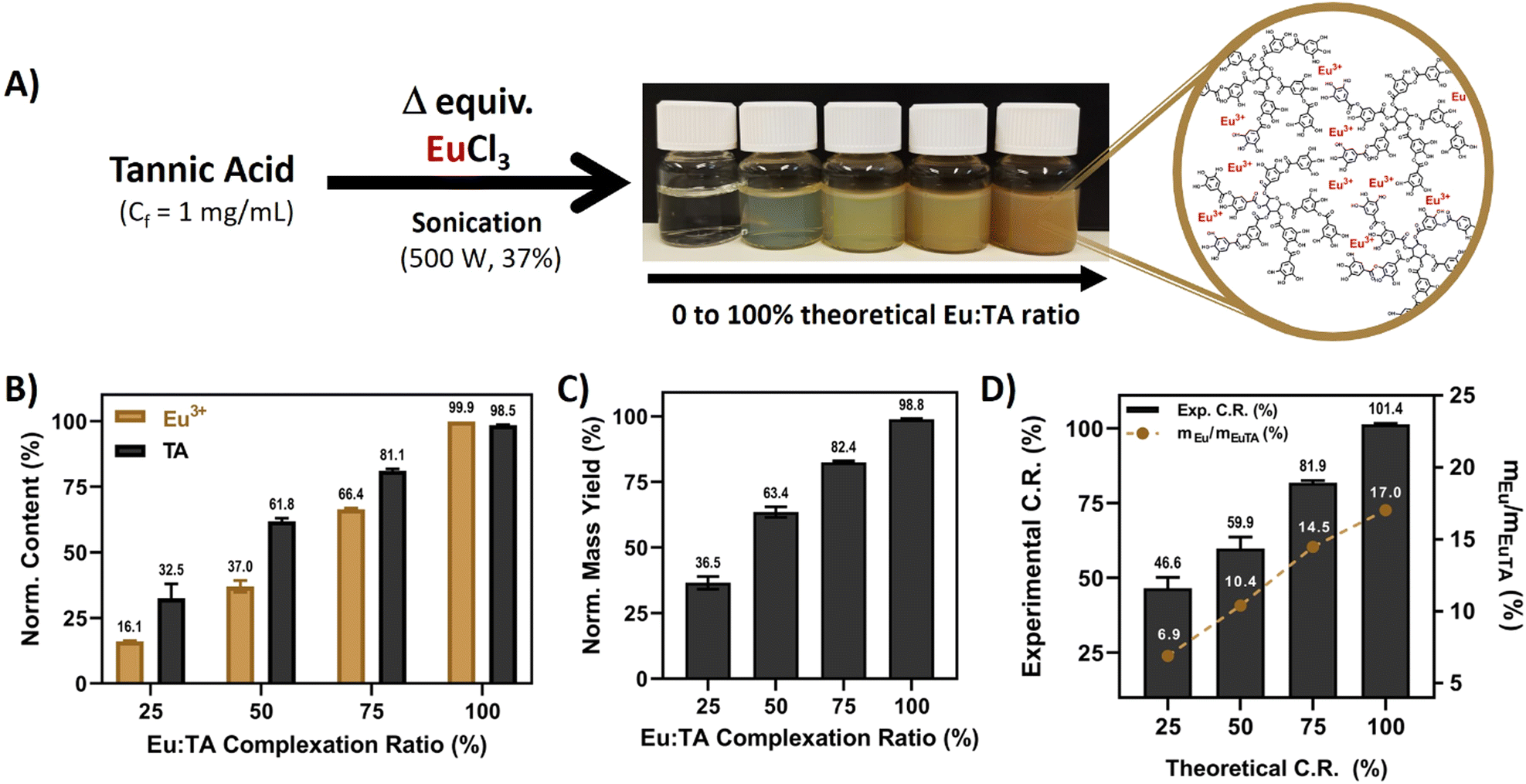 | ||
| Fig. 1 Synthesis of EuTA NCs and its compositional characterization. (A) Scheme describing the preparation of EuTA NCs and the resulting products; (B) compositional analysis of EuTA NCs as indirectly determined using ICP-OES (Eu3+ content) and UV-VIS (TA content) spectroscopies, normalized according to the maximum content of Eu3+ and TA added in the synthesis of EuTA 100%; (C) normalized yield of the different EuTA NCs normalized to the maximum theoretical yield; and (D) comparison between experimental and theoretical C.R.s and Eu contribution to weight in each EuTA NC formulation. | ||
Regarding the final products, the normalized yields for the different EuTA NCs increased while augmenting the C.R., as expected (Fig. 1C). This observation can be explained due to the lack of enough Eu3+ cations to coordinate all the TA molecules present in the mixture when added in limiting amounts and, therefore, part of the TA added is lost during the preparation. However, differences were also observed when comparing theoretical and experimental C.R. values calculated using ICP-OES. The calculated values were higher than the theoretical ones in all the cases, except for the 100% condition (Fig. 1D). This can be attributed to the high rate of the metal binding reaction with TA. In this sense, in an ideal, homogeneous state, every TA molecule would complex the same amount of Eu3+ ions. However, despite the intense sonication during the addition of Eu3+, its complexation was not homogeneous, leading to some TA molecules complexing higher amounts than the calculated average, and others, remaining free in solution, which further agrees with the lower normalized yields calculated experimentally. Thus, the process led to complexes with higher stoichiometry than expected in all the possible cases.
To date, few articles have reported Eu3+ complexation by TA, and, to the best of our knowledge, only a couple of them studied regenerative properties.33,46 Wu et al. prepared poly(L-lactic acid) nanofibers coated with different layers of TA/Eu deposits and evaluated the influence of the coating on the physicochemical properties of the material, antioxidant, and wound healing properties.33 In a later work, Wu et al. prepared pH-responsible EuTA microparticles with antioxidant and pro-angiogenic properties for myocardial infarction treatment.46 Thus, this is the first time that EuTA NCs are reported and studied in depth in the context of oxidative and inflammatory environments that hinder bone regeneration. Moreover, the mechanism of synthesis here proposed is much simpler than those in other works33,46,47 and allows for easy control of the content of Eu3+ present in the formulations by varying the volumes added, which might be of interest to preparing more complex advanced materials in the future.
3.2. Physicochemical characterization of EuTA NCs
The interactions that TA establishes with trivalent cations have been historically studied in-depth and are well-documented, occurring mainly through the hydroxylic groups of its catechol structures.13,48,49 To confirm these interactions, an ATR-FTIR spectroscopic analysis was performed. A zoomed-in region between 400 and 2000 cm−1 is shown in Fig. 2A, while the whole spectra and the corresponding frequency assignments can be found in Fig. S1 and Table S1 (ESI†), respectively. Moreover, the TA structure can be found in Fig. S2 (ESI†). Regarding TA, bands mainly corresponding to the vibrations of aromatic rings, secondary alcohols, and ester groups were identified as expected (Table S1, ESI†), agreeing with previous reports.48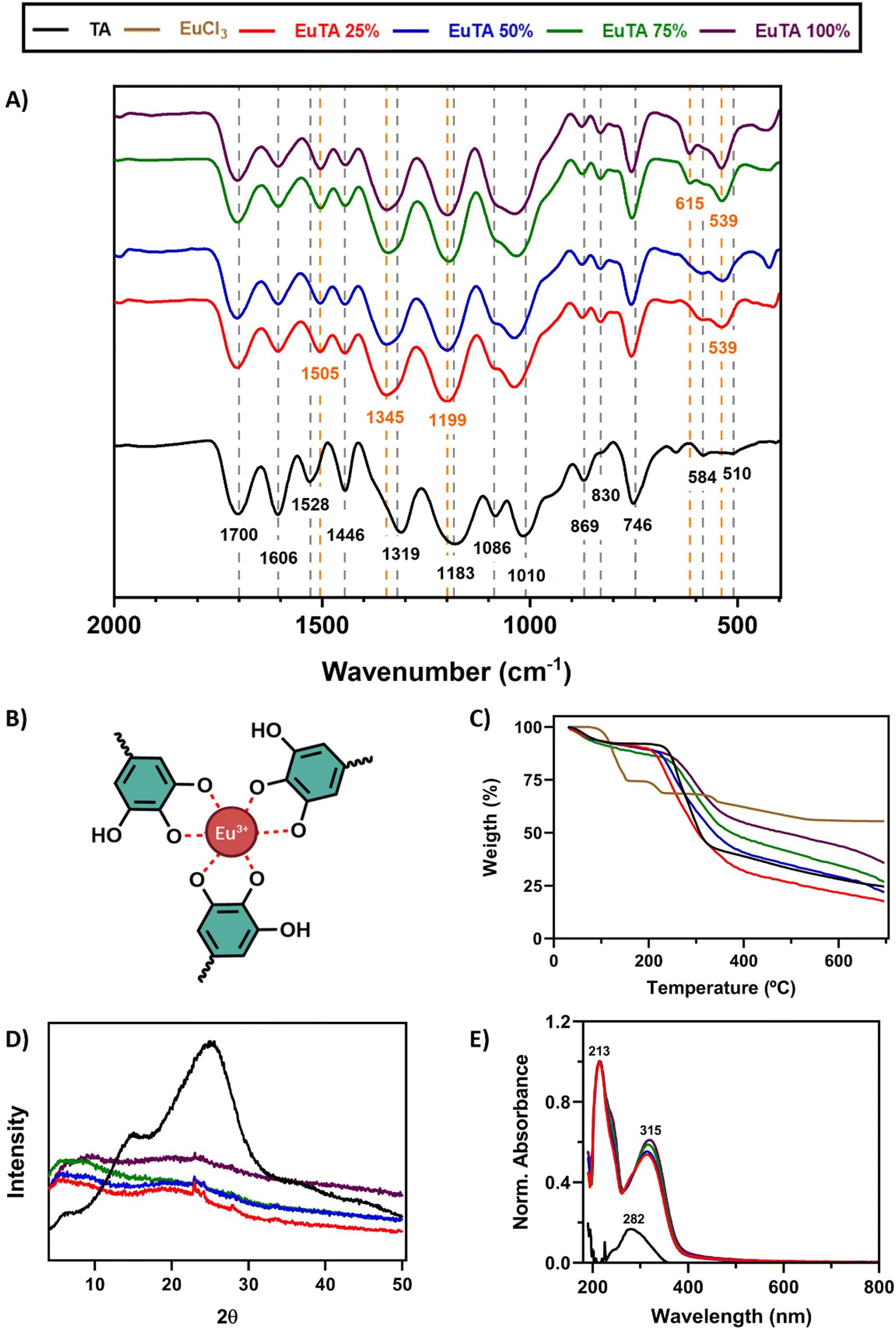 | ||
| Fig. 2 Characterization of the interaction between TA and Eu3+. (A) Zoomed-in region (2000 and 400 cm−1) of the ATR-FTIR spectra of TA and EuTA NCs; (B) scheme for the tris-complex between Eu3+ and the catechol groups (colored) of TA. Black and orange annotated wavenumbers refer to the spectrum of TA and EuTA NCs, respectively; (C) TGA analysis of TA, EuCl3, and EuTA NCs; (D) XRD analysis of TA and EuTA NCs; and (E) UV-VIS spectra of TA and EuTA NCs. | ||
A comparison of the spectra of EuTA NCs and raw TA allowed us to conclude that Eu3+ complexation exclusively took place via hydroxylic groups of the catechol structures, forming tris-complexes (Fig. 2B) as previously described for other trivalent ions.13,48,49 Briefly, the intensities of three bands (i.e., 1086, ∼583, and ∼539 cm−1) increased in a directly proportional manner to the content of europium in the NCs. These bands were also present in the FTIR spectrum of the raw EuCl3·6H2O and corresponded to the stretching and bending νEu–O, indicating the participation of oxygen atoms (Fig. S1, ESI†). However, no differences were observed for the stretching vibrations (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 1700 cm−1 meaning that the carboxyl groups that form the ester bonds between catechol groups did not participate in europium chelation.50
O) at 1700 cm−1 meaning that the carboxyl groups that form the ester bonds between catechol groups did not participate in europium chelation.50
In addition, the stretching vibration νC–O attributed to the band at 1319 cm−1 in the spectrum of TA was the one that shifted the most to higher wavenumbers (∼1345 cm−1) after europium complexation, demonstrating the involvement of hydroxyl groups in the chelation. Moreover, the stretching vibrations νCC of the phenol groups also exhibited significant variations. The band located at 1606 cm−1 decreased in intensity after europium complexation, while the band at 1528 cm−1 increased in intensity and shifted to lower wavenumbers. These two bands have been attributed to the CC double bonds of the phenolic structures in ortho/para and meta positions, respectively.51 Similarly, bands attributed to the bending vibrations (νC–H) in those aromatic structures, such as those at 1183, 869, and 830 cm−1, were also modified, shifting to higher frequencies and altering their intensities, (i.e., decreasing and increasing, respectively). Altogether, europium complexation affected the free mobility of catechol groups through the involvement of their hydroxyl groups. The complexation mechanism between Eu(III) and different tannins has been extensively studied by various authors using a wide range of techniques including potentiometry, calorimetry, XRD, and spectroscopic analysis such as XPS, NMR, and FTIR among others.46,52–54 These studies have established that during the metal chelation process, the ortho-dihydroxyl groups of the catechol ring play a crucial role in facilitating catechol-based coordination with metals. In addition, Arciszewska et al. provided an in-depth analysis of the formation mechanisms and molecular structures of Eu-tannin complexes using NMR and FTIR.54 Their research shows that in Eu(III)/catechol complexes, the hydroxyl groups of the catechol moiety are involved in metal-ion coordination, reducing the hydrogen/electron donating properties of the complex compared to the ligand. Similar conclusions have been drawn for Fe(III)/tannic acid complexes as determined by FTIR. This emphasizes the value of FTIR as a crucial technique for determining the structure of such compounds. FTIR analysis performed on the EuTA NCs corroborates these findings, supporting the notion that metal cation interaction stabilizes the aromatic system forming the tris-complex between Eu3+ and the catechol groups proposed in Fig. 2B.
The thermal properties of EuTA NCs were studied by thermogravimetric analysis (TGA, Fig. 2C). The traces of TA and EuTA NCs showed a similar tendency, with two well-defined steps of weight loss. The first decay was found at around 100 °C, which can be attributed to a dehydration step of associated water molecules. The second step of weight loss took place around 220–260 °C and can be assigned to the degradation of TA, as previously reported.41 In addition, it was found that the remanent weight at 700 °C was dependent on the content of Eu3+ in the NCs, confirming once again the different contents of Eu3+ in the samples.
The crystallinity of EuTA NCs was studied by X-ray diffraction (XRD) analysis (Fig. 2D and Fig. S3, ESI†). As shown in Fig. 2D, the diffractogram of raw TA showed two wide peaks around 2θ = 15 and 25°, thus, presenting a pronounced amorphous structure due to the great number of hydroxyl groups, which is in accordance with previous reports.46 In contrast, these characteristic peaks were markedly reduced in the diffractogram of EuTA NCs, as the exposed hydroxyl groups diminished due to the complexation of Eu3+, in accordance with the ATR-FTIR analysis and other works.46
Finally, the formation of coordination complexes between TA and Eu3+ ions was further confirmed via UV-VIS spectroscopy by the appearance of two bands with maximum absorption at 213 and 315 nm, which were not present in the spectrum of TA (Fig. 2E). UV-VIS characterization of a solution of EuCl3 did not retrieve any signal in the range analyzed (data not shown).
3.3. Characterization of the hydrodynamic properties of EuTA NCs
The morphology of EuTA NCs was characterized by different microscopical means. First, EuTA NCs dispersed in culture medium (DMEM) were evaluated by cryo-TEM to assess their morphologies under the conditions selected for the in vitro biological testing. As shown in Fig. 3A for EuTA 25% NCs, particles tend to aggregate during sample preparation, hindering the identification of individual particles and the quantification of their mean diameters. Similar results were observed for the rest of the formulations (Fig. S4, ESI†). As an alternative, the morphology of EuTA NCs dried at r.t. was explored by field emission scanning electron microscopy (FE-SEM). In all the cases, pseudospherical nanoparticles were observed, although, similarly to cryo-TEM findings, aggregates formed by the coalescence of these nanoparticles were found in most fields independently of the C.R. values (Fig. S5, ESI†). In contrast, mean diameters of 42.5 ± 9.9 nm averaging all the conditions could be measured. Histograms depicting the different sizes of the nanoparticles are shown in Fig. S6 (ESI†). Thus, as summarized in Fig. 3C and Fig. S5, S6 (ESI†), there were no significant differences in terms of mean nanoparticle size among the studied conditions.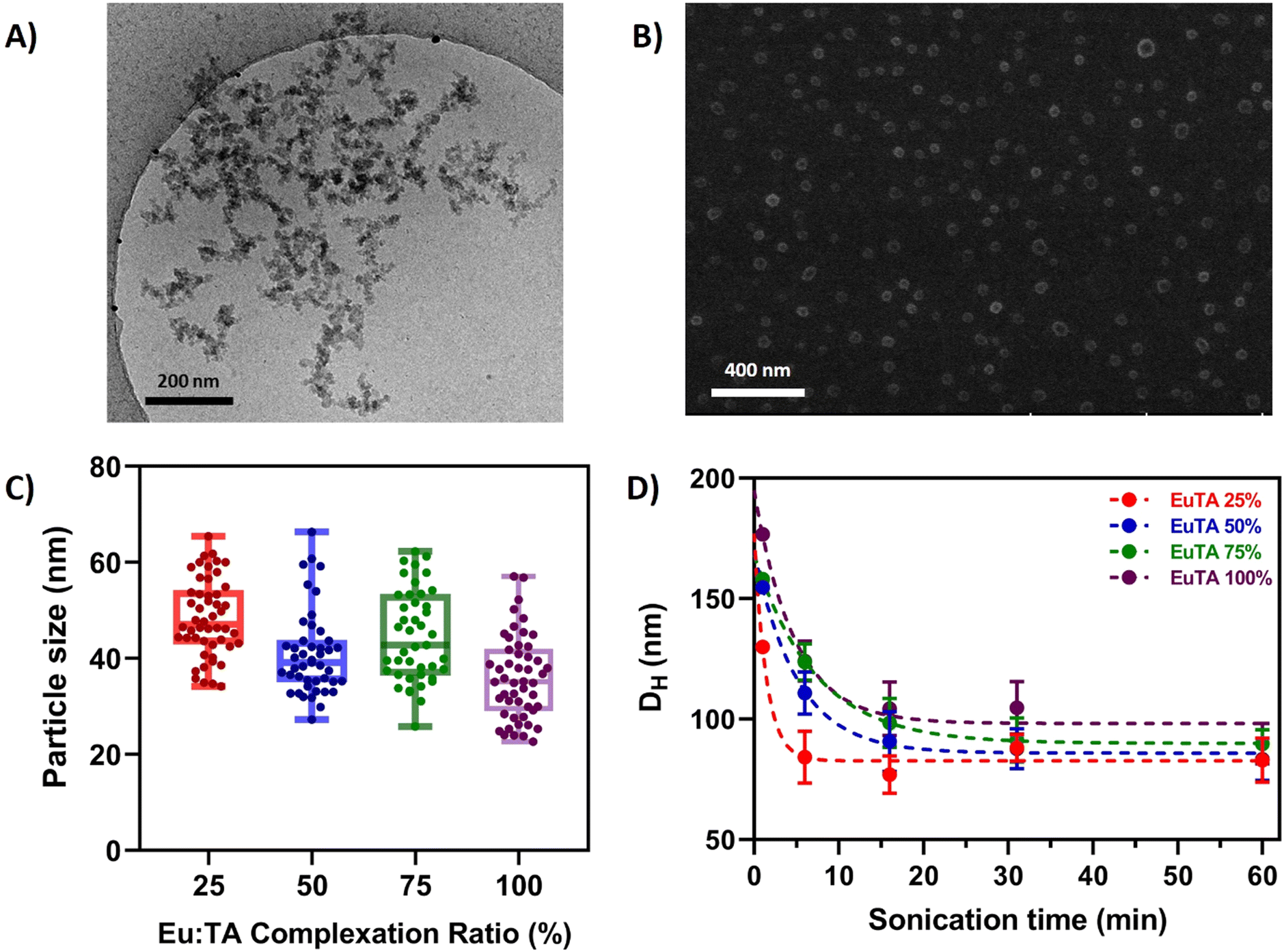 | ||
| Fig. 3 Physicochemical characterization of EuTA NCs. (A) Cryo-TEM image of EuTA 25% NCs (as a representative image of the different EuTA NCs) in DMEM; (B) FE-SEM micrograph of EuTA 25% NCs (as a representative image of the different EuTA NCs) dried from an aqueous suspension at r.t.; (C) mean particle size of different EuTA NCs calculated from FE-SEM micrographs; and (D) Dh values for EuTA NCs resuspended in distilled water and sonicated during different time periods. | ||
Next, the hydrodynamic properties of the systems were characterized by DLS (Fig. 3D and 4). In all the cases, the average hydrodynamic diameters (Dh) were at least more than twice the mean size reported for individual nanoparticles by FE-SEM. Moreover, increasing the sonication time at the resuspension step diminished the mean Dh values for all the conditions up to 100 nm after 1 hour (Fig. 3D). This finding along with the FE-SEM characterization suggests that nanoparticles tend to form stable nano-sized complexes with different numbers of nanoparticles depending on the duration of the sonication.
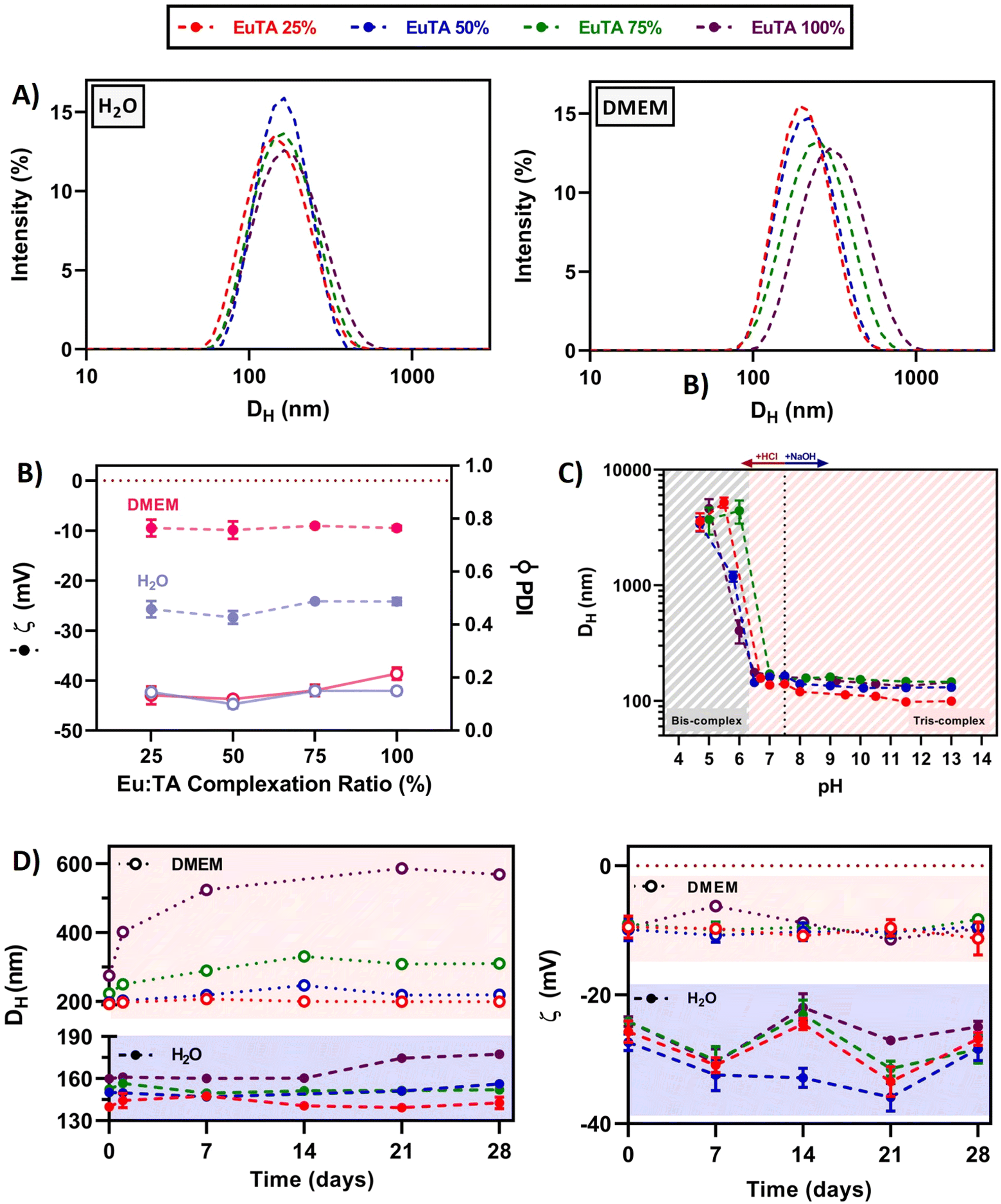 | ||
| Fig. 4 Hydrodynamic properties of EuTA NCs. (A) Particle size distribution (Dh, by intensity) of EuTA NCs resuspended in distilled water and DMEM; (B) zeta potential (ζ) values and polydispersity index (PDI) of EuTA NCs; (C) stability of EuTA NCs analyzing Dh at different pH values; and (D) stability of EuTA NCs over time including mean Dh and ζ values both in distilled water and DMEM, stored at 4 °C in the dark. | ||
In addition, EuTA NCs displayed different hydrodynamic properties depending on the solvent of resuspension. In these studies, distilled water was chosen as a traditional aqueous solvent, while culture medium (DMEM) was selected for its established use in biological cultures. On the one hand, the proposed synthetic procedure retrieved monodispersed (Dh = 150 ± 8 nm) and low-polydispersed (PDI = 0.14 ± 0.02) NCs when resuspended for 30 seconds in distilled water independently of the C.R. (Fig. 4A). In contrast, a strong dependence of Dh and PDI was found with the EuTA composition when resuspended in DMEM, increasing to average values of 222 ± 34 nm and 0.15 ± 0.04, respectively (Fig. 4A). On the other hand, the average zeta potential of EuTA NCs resuspended in distilled water (ζ = −25.4 ± 1.7 mV) rose to −9.5 ± 0.9 mV when resuspended in DMEM (Fig. 4B). These findings can be explained by the ability of serum proteins from DMEM to interact with the TA molecules present at the surface of the EuTA NCs, which are expected to be the determining factor for the negative ζ values obtained. Thus, this interaction would be responsible for the increase of the Dh and the diminishment of the surface net charge.
Regarding the stability of EuTA NCs over time, great differences were observed when comparing EuTA NCs stored in distilled water and DMEM. EuTA NCs stored in distilled water maintained their original hydrodynamic properties to a greater extent than in culture medium (Fig. 4D). In all the cases, increasing the C.R. of the formulations led to higher mean sizes, PDI, and instability over time, specially EuTA 100% NCs. A long-term stability study conducted under these conditions showed similar tendencies although physical degradation of NCs was not found in any case, indicating the suitability of distilled water for long-term storage for up to 2 months (Fig. S7, ESI†). In the case of DMEM-resuspended EuTA NCs, the long-term storage mainly affected the conditions with higher C.R. values, where colloidal aggregation was found. Regarding surface net charge, zeta potential values maintained their tendencies over the analyzed month (Fig. 4D). Smaller variations were observed for DMEM-resuspended EuTA NCs compared to the ones resuspended in distilled water, agreeing with the proposed hypothesis of serum proteins interacting with EuTA NCs.
Furthermore, it was shown that EuTA NCs were stable at neutral and basic pH values (Fig. 4C). However, their conformational structure was distorted below pH 6, leading to the presence of colloidal aggregates. This can be attributed to the type of interactions established between TA and Eu3+ ions, as previously studied.13 In this regard, when reaching values below pH 6, bis-complexes are favored over tris-complexes, and a new reorganization of interactions is arranged, leading to the aggregation of the particles and their precipitation, which is visible to the naked eye. These stability values are acceptable for the intended applications in osteopenic bone as there are some reports of pH measurements performed directly on in vivo rat models of osteoporotic bone that confirm a pH of 7.5 for this animal model and disease.55 In addition, sarcomas and other inflammatory joint diseases, where subchondral bone might be affected, present an increased acidity due to the lack of oxygen in the surroundings. In the case of solid tumors, the pH values range from 6.5–6.8, which would not distort EuTA NC integrity.56 Although in rheumatoid arthritis-affected joints, synovial cavities can reach values near 6, their possible aggregation would not imply a loss of function of the complexes.57 Indeed, the previously commented work, carried out by Wu et al. on EuTA microparticles, showed the bioactivity of their system despite their large size.46 However, its controlled delivery by means of advanced injectable formulations is currently under study to avoid any stability concerns and to optimize the bioavailability of the therapy for future intended applications.
3.4. Antioxidant properties of EuTA NCs
The antioxidant activities of TA have been previously reported, being partially attributed to the high redox potential of phenolic groups present in the molecule.49,58 In this regard, it is important to evaluate the antioxidant properties of EuTA NCs (Fig. 5) due to the partial availability of these groups after the formation of the complexes. To this end, the total radical scavenging activities and, particularly, scavenging activities against H2O2 radicals, and ferrous-chelation activities were evaluated. Studying the scavenging of H2O2 radicals has biological importance as it is formed under oxidative conditions and leads to cellular damage. Similarly, the Fe2+ removal ability is a simple approach to indirectly test the inhibition of lipid peroxidation, as Fe2+ ions catalyze Fenton reactions, which produce ˙OH radicals, resulting in lipid damage.42 Moreover, it is important to clarify that the concentrations of EuTA NCs commented on from now on (i.e., antioxidant and cell culture experiments) refer to the concentration of TA present in the samples, as calculated by UV-VIS. Thus, at each dose, the amount of TA is constant among conditions while the content of complexed Eu3+ is the variable factor.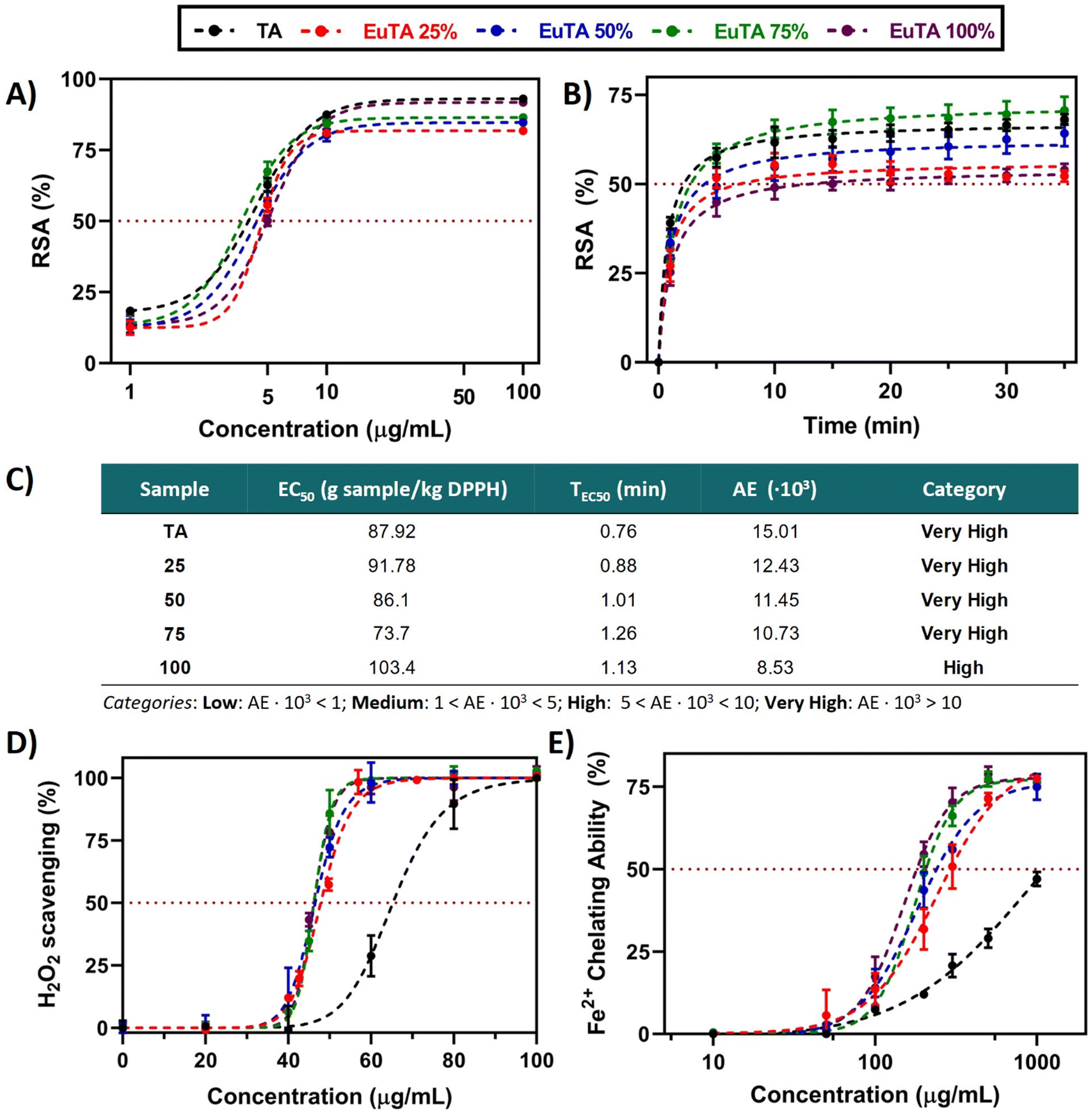 | ||
| Fig. 5 Antioxidant properties of EuTA NCs. (A) Total free radical scavenging activity (%) of TA and EuTA NCs samples at different concentrations as measured by the DPPH assay at the steady state (15 min), adjusted to a sigmoidal of the variable slope (four parameters) regression model; (B) kinetic of DPPH scavenging by TA and EuTA NCs samples prepared at their corresponding EC50; (C) main parameters for the classification of antioxidants as studied by Sánchez-Moreno et al.;40 (D) H2O2 scavenging activity (%) of TA (pH = 7.3) and EuTA NCs at different concentrations, adjusted to [agonist] vs. normalized response (variable slope); and (E) ferrous ions chelating ability (%) of TA and EuTA NCs at different concentrations, adjusted to a non-linear (sigmoidal of variable slope) regression model. The parameters for each regression model are summarized in Tables S2–S5 (ESI†). | ||
To compare the total radical scavenging activities of TA and EuTA NCs, a DDPH study was performed. Slight differences were observed in terms of potency (EC50, Fig. 5A and C) and velocity (TEC50, Fig. 5B and C). In all the cases, the kinetics reached plateaus after 10–15 min, ranging the TEC50 values between 0.76–1.26 min. Regarding potency, calculated under steady-state conditions, EC50 values for TA (4.40 ± 0.09 µg mL−1) and EuTA NCs (4.44 ± 0.62 µg mL−1) were not statistically different from each other. With these values, antiradical efficiencies (AE) were calculated showing a negative trend with the C.R., although in all the cases were found to present “high” or “very high” AE values according to the classification made by Sánchez-Moreno et al.40 A similar behavior has also been reported in nanoparticles of TA complexing Fe3+, which indicates that these changes cannot be attributed to Eu3+ itself, but to the coordination with trivalent cations in general.41 In this sense, our data agree with the hypothesis posed by Pucci et al. on the more effective exposure of the reaction sites in free TA compared to coordinated TA, as only TEC50, but not EC50, increased while augmenting the C.R.
In contrast, EuTA NCs showed higher efficacy in the chelation of hydrogen peroxide radicals compared to raw TA. As observed in Fig. 5D, EuTA NC dispersions (EC50 = 46.7 ± 0.8 µg mL−1) showed an overall better response than TA (EC50 = 65.3 µg mL−1), as this effect is independent of the content of Eu3+. Previous reports have associated this effect with a strengthening of the electron-withdrawing capacity of the antioxidant when forming nanosized metal complexes. For instance, NCs of iron–TA and iron–sapogenin showed higher scavenging activities than pure TA and sapogenin, respectively.59,60 Therefore, this effect would also be dependent on the coordination of these natural-based antioxidants with trivalent cations rather than the individual moieties by themselves. In addition, the oxidation that undergoes TA at physiological pH also influences its scavenging capacity. In this sense, europium complexation seemed to protect TA from its oxidation and hydrolysis at this pH.
In contrast, the ability of EuTA NCs to remove Fe2+ did proportionally increase with the content of Eu3+ (Fig. 5E). TA removed Fe2+via its chelation as previously reported.61 However, this process is hindered at physiological pH due to the progressive oxidation of TA. Similar to the H2O2 scavenging capacity, europium complexation improved this Fe2+-binding capacity, probably by slowing down the oxidation of TA. Nevertheless, in this case, the C.R. of EuTA NCs affected the final response of the system. These differences between assays can be explained by the incubation times of both protocols. In this sense, as the measurements for the determination of the H2O2 scavenging were instantaneous, no differences could be observed. In contrast, the ferrozine assay presents a 15-minute incubation time to ensure measurements of the steady state.
Taking these results altogether, the higher removal of H2O2 radicals and Fe2+ ions would avoid their involvement in Fenton reactions, and indirectly, inhibit lipid peroxidation, protein oxidation, and cell death to a higher extent.62 Moreover, ferroptosis, a particular type of cell death due to iron overload, has also been reported to be involved in age-related orthopedic diseases such as osteoporosis and osteoarthritis, widening the applicability of these new systems to other bone pathologies.63 These properties are crucial for reducing OxS, promoting angiogenesis, and enhancing osteogenesis, all essential processes for bone healing. Research on antioxidant biomaterials for bone regeneration showcases various innovative approaches that compare favorably against traditional methods. Polyphenolic flavonoid silymarin has demonstrated significant antioxidant and anti-inflammatory properties, effectively treating bone fractures and osteoporosis by enhancing ECM secretion and promoting osteogenic markers such as type-I collagen, osteocalcin, and Runx2.64 Similarly, biodegradable conductive polyphosphazenes have shown strong antioxidant capacity, outperforming conventional polyesters like PLGA by increasing cell viability under high OxS conditions and providing a promising alternative for developing multifunctional scaffolds for severe bone defects.65 Gelatin hydrogels, integrated with polydopamine nanoparticles and heparin, have demonstrated enhanced bone regeneration capabilities through sustained BMP-2 release, promoting cell differentiation and mineralization.66 Finally, other polyphenol-containing materials such as those prepared with TA, including TA–alendronate and Sr-based nano-constructs, have shown promise in promoting osteogenic activity and ROS scavenging. These materials activate signaling pathways such as PI3K/Akt, fostering bone regeneration and potentially treating osteoporotic defects.67
These comparative studies highlight the importance of antioxidant properties in enhancing bone regeneration, with each material offering unique benefits tailored to specific clinical needs. When compared to other antioxidant biomaterials, such as flavonoids, polyphosphazene polymers, gelatin hydrogels, bioactive cation ceramics, and tannic acid complexes, EuTA NCs show unique advantages due to intrinsic dual action where the addition of europium improves TA cytocompatibility, anti-inflammatory, and osteogenic properties, envisioning their potential applicability for bone regeneration purposes under complex conditions where regeneration is hindered.
3.5. Biological characterization of EuTA NCs
EuTA NCs have been devised to target both oxidative and inflammatory environments that hinder bone regeneration. Therefore, their in vitro biological characterization must include studies evaluating their cytocompatibility, anti-inflammatory, antioxidant, and osteogenic properties in relevant cellular models.First, cell viability studies were performed to characterize the possible cytotoxicity of the raw materials and the EuTA NCs after 24 h of incubation in this cell line. As observed in Fig. 6A, both TA and EuCl3 were cytotoxic at high concentrations (IC50 = 324.0 µg mL−1 and 388.1 µg mL−1, respectively). In contrast, EuTA NCs retrieved viability values over 70% in all the cases (IC50 > 1 mg mL−1), confirming their cytocompatibility. We hypothesize that the marked reduction in the cytotoxicity of raw TA after its complexation with Eu3+ could be due to the neutralization of their acidic properties, rather than to the presence of europium itself. In this sense, TA was deprotonated during the synthesis when dissolved in HEPES buffer pH 7.3, which allowed the complexation of europium, and after the isolation and resuspension of EuTA NCs, these protons were removed, obtaining neutral pH values in their solutions even in the absence of a pH-buffer.
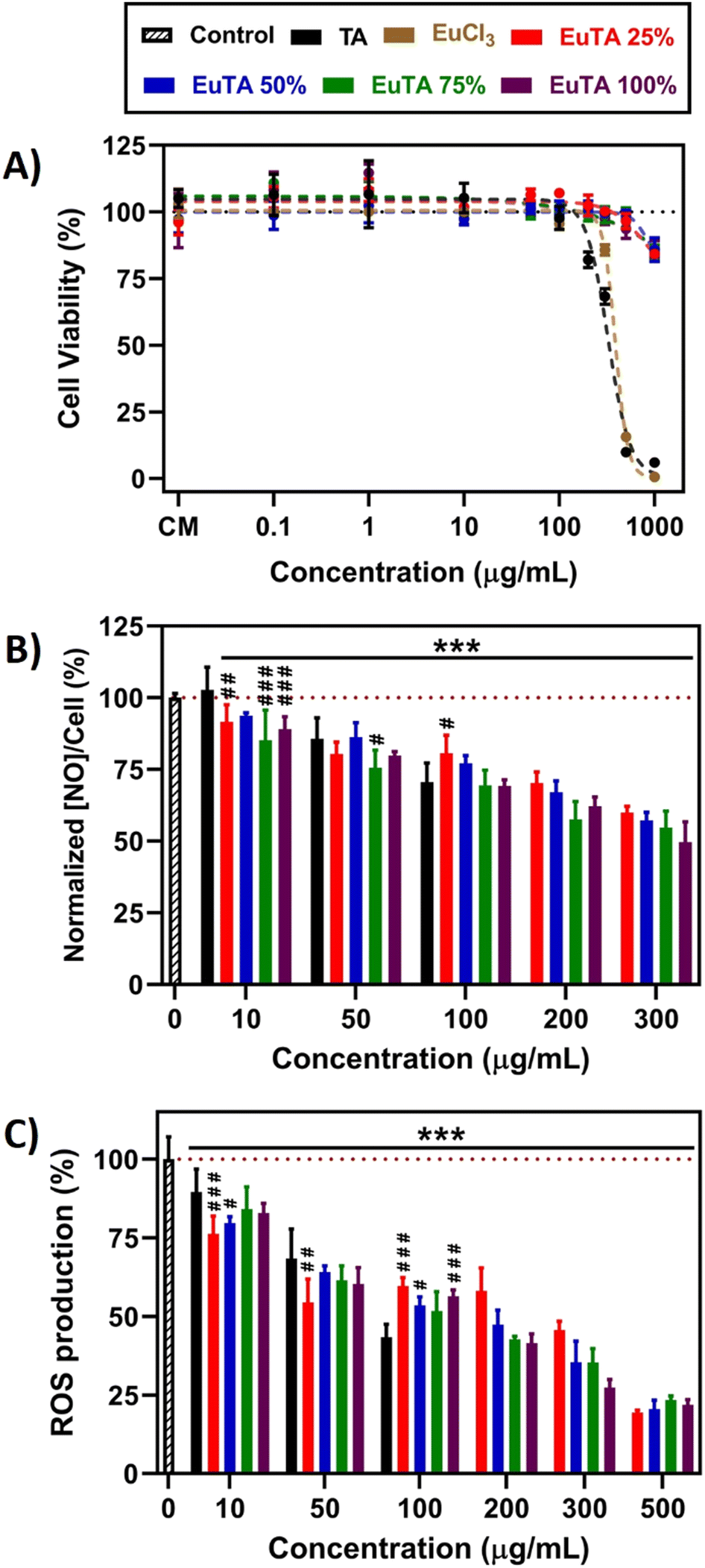 | ||
| Fig. 6 Antioxidant and anti-inflammatory properties of EuTA NCs evaluated on cultures of RAW264.7 macrophages. (A) Direct cytotoxicity assay following a 24-hour incubation with TA, EuCl3, or the different EuTA NCs and measured using the alamarBlue® method; (B) evaluation of the anti-inflammatory properties of a 24-hour incubation with TA and EuTA NCs assessed by the reduction on the production of NO by LPS-stimulated macrophages and normalized to the metabolic activity of cultures under the same conditions but without LPS-stimulation; (C) evaluation of the ROS scavenging activity of a 24-hour pre-treatment with TA and EuTA NCs before a 15-minute exposure to 100 mM H2O2 by measuring the fluorescence of DCFH-DA dye. ANOVA: * and # symbols are used to compare conditions at the same experimental times with control and TA-treated cultures, respectively. Statistical significance: *, # p < 0.05; **, ## p < 0.01; ***, ### p < 0.001. | ||
According to these results, only the effects of cytocompatible doses of each compound were further evaluated in the following tests. In addition, to characterize the contribution of the content of europium, EuTA NCs were compared in all the cases to both an untreated control and TA-treated culture at the same TA concentration.
In many inflammatory diseases, nitric oxide (NO) released by pro-inflammatory macrophages plays a crucial role in the development and progression of inflammation and, consequently, tissue destruction.69,70 We examined whether the treatment of LPS-stimulated macrophages with EuTA NCs could reduce the NO secreted, as with TA, which has been previously reported.71,72 As shown in Fig. 6B, a 24-hour incubation with TA and EuTA NCs diminished this parameter in a concentration-dependent manner. At the lowest dose (10 µg mL−1), TA did not reduce the NO secreted (102.8 ± 7.9%), while EuTA NCs did, and the better performance was from the formulations with higher C.R. values: 75% (85.2 ± 10.5%) and 100% (89.0 ± 4.3%). Analyzing the bigger picture, no statistically significant differences were found among EuTA NCs, although a trend indicating peak performance at 75% C.R. was inferred. Furthermore, NO was reduced too in cultures of resting macrophages, where no LPS was added to stimulate a pro-inflammatory environment (Fig. S8, ESI†). Thus, an improvement in the anti-inflammatory properties of TA has been shown due to Eu3+ complexation.
Targeting chronic inflammation is crucial as it can also induce the production of ROS, which can promote the activation of signaling pathways that enhance the production of pro-inflammatory mediators and exacerbate tissue damage.8,11 Hence, considering the potent antioxidant activities displayed by TA and EuTA NCs against various oxidant agents, we aimed to investigate if subjecting cells to a 24-hour exposure before an oxidant stimulus could offer further preventive effects, potentially reducing future ROS production. In this context, following the initial incubation with the antioxidants, cells were further treated for 15 min with 100 mM H2O2, and ROS production was monitored by fluorescence means (Fig. 6C and Fig. S9, ESI†). Similar to the reduction in NO levels, the prophylactic effect of TA and EuTA NCs was concentration-dependent.
Nevertheless, in this case, differences among EuTA NCs did not follow a clear trend. EuTA 75% was the only formulation that did not present significant differences with TA at the same doses. Conversely, at lower concentrations, EuTA 25% displayed a better response, showcasing a 13.3% improvement.
First, cell viability studies were performed analogously to those carried out using RAW264.7 cells. As shown in Fig. 7A, free TA was found to be the most cytotoxic of the studied treatments, presenting an IC50 of 107.7 µg mL−1. This finding does not agree with previous works performed in fetal human osteoblasts where an increase in their metabolic activity was reported at increasing concentrations of TA.76,77 However, these effects might have been overestimated due to the redox potential of TA and the fact that these assays were performed in the presence of the treatments. Therefore, the reduction of the substrate employed for the quantification was due to both the cellular metabolism and TA. In this work, all treatments were removed, and cells were washed with PBS prior to alamarBlue addition to avoid this kind of misinterpretation. Consequently, similar toxicity to that found in RAW264.7 cells was obtained, which could be attributed to the acidity at high doses. In contrast, EuCl3 and EuTA NCs only showed some toxicity at the highest dose assayed, with their IC50 values in all cases being over 1 mg mL−1.
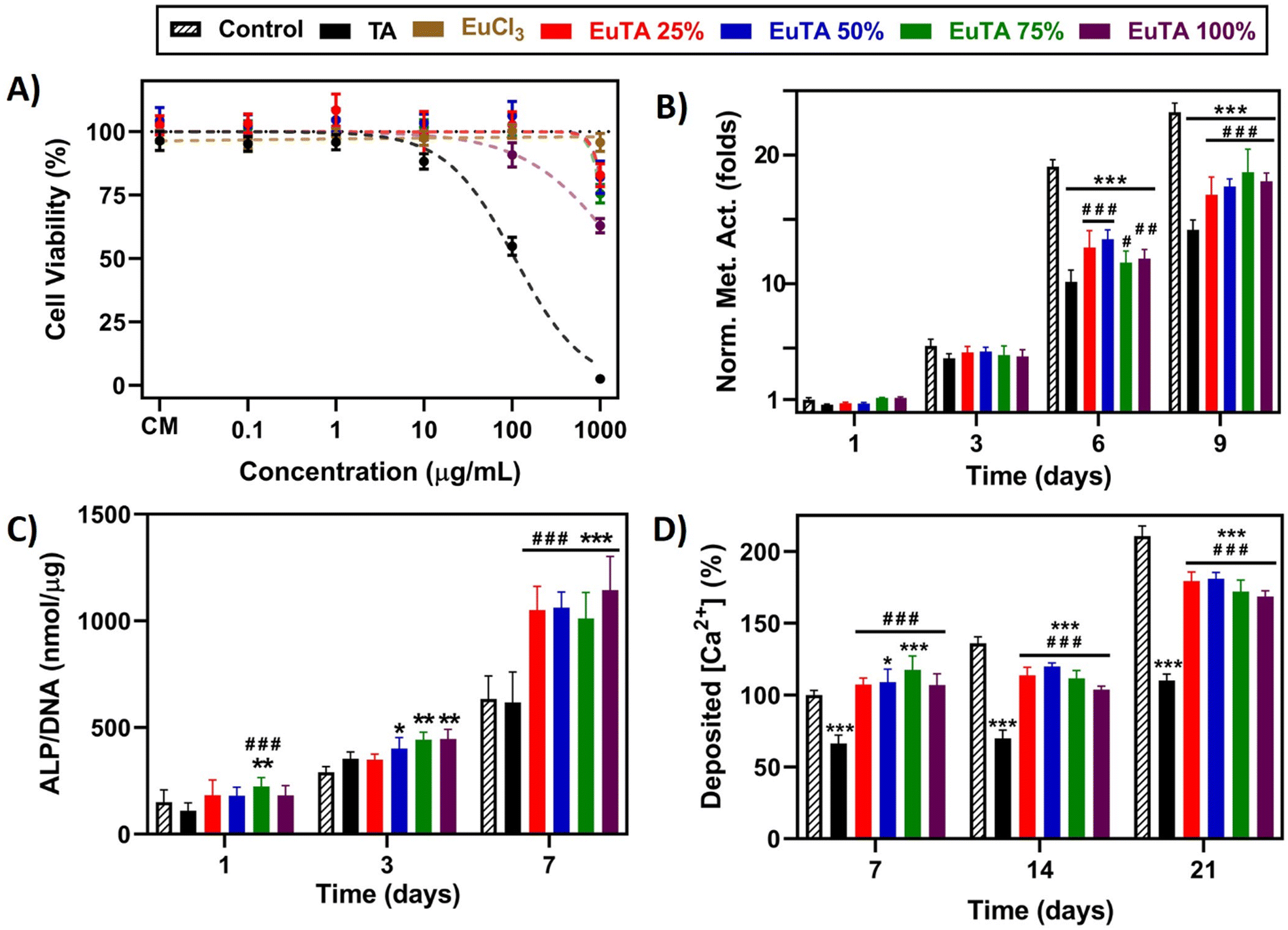 | ||
| Fig. 7 Osteogenic properties of EuTA NCs evaluated on cultures of fHOBs. (A) Direct cytotoxicity assay following a 24-hour incubation with different concentrations of TA, EuCl3, or the different EuTA NCs and measured using the alamarBlue® method; (B) cell proliferation assay of independent cultures determined by monitoring the metabolic activity via alamarBlue® measurements at different incubation times with 10 µg mL−1 of TA or EuTA NCs; (C) determination of the ALP activity of cultures treated with 10 µg mL−1 of TA or EuTA NCs at different periods of incubation; (D) quantification of AzR staining extracted from cultures treated with 10 µg mL−1 of TA or EuTA NCs at different times. ANOVAs: * and # symbols are used to compare conditions at the same experimental times with control and TA-treated cultures, respectively. Statistical significance: *, # p < 0.05; **, ## p < 0.01; ***, ### p < 0.001. | ||
Next, we evaluated the effects of cytocompatible concentrations of TA and EuTA NCs on the proliferation rate of fHOBs by monitoring their metabolism. Results obtained for treatments with 10 µg mL−1 TA or EuTA NCs are shown in Fig. 7B, while results at lower and higher doses are summarized in Fig. S10 and S11 (ESI†). On the one hand, doses over 10 µg mL−1 were shown to impair cell proliferation, although Eu3+ complexation diminished to some extent TA toxicity (Fig. S10, ESI†). Moreover, cell vacuolization was observed at higher concentrations on EuTA NCs 50, 75, and 100% after three days of treatment, which we speculate might be due to an excessive supply of Eu3+, as similar reports have been previously reported in cardiomyocytes (Fig. S11, ESI†).46 On the other hand, while there were no significant differences after short treatments (3 days), longer ones slowed fHOB proliferation depending on the dose. At 0.1 and 1 µg mL−1 there were no differences between EuTA NCs and control conditions, but there were if compared to TA (Fig. S10, ESI†). Interestingly, the administration of TA did not increase the metabolic activity of fHOB cells at any assayed dose compared to untreated cultures. In this sense, both TA and its hydrolyzable products (i.e., gallic acid and pyrogallic acid) have been reported to regulate different signaling cascades such as MAPK, PI3K/AKT, or Wnt pathways, although their precise mechanisms of action are still under research.78,79 However, as commented earlier, other hypotheses consider their metal binding capacities to be pivotal, which would increase the availability of bioactive ions involved in the formation of hydroxyapatite and osteoblast stimulation in vivo.19 Thus, further studies investigating the molecular mechanisms involved in polyphenols' bioactivity are needed to fully comprehend their biomedical potential.
Regarding these proliferation studies, the dose of 10 µg mL−1 was selected to study their osteogenic properties, as at that concentration the bioactivity of TA has been proven both throughout this article and in the available literature and it is two orders of magnitude under the IC50 values of EuTA NCs.46 The osteogenic potential of TA and EuTA NCs was assessed at early and late stages by evaluating fHOBs activity, via measurements of the alkaline phosphatase (ALP) activity (Fig. 7C), and by analyzing the matrix mineralization degree, via alizarin red (AzR) staining (Fig. 7D and Fig. S12, ESI†), respectively.
On the one hand, TA administration did not alter the ALP activity of fHOB cells compared to control cultures at any assayed time (Fig. 7C). However, it significantly diminished the concentration of Ca2+ deposited in the ECM, which agrees with its metal-sequestering properties (Fig. 7D). Therefore, the use of TA in solution, rather than in coatings, could be expected to have detrimental effects on bone regeneration applications.
In contrast, EuTA NCs showed a completely different behavior. Regarding ALP activity, small, but statistically significant, differences were observed after one and three days of treatment for the EuTA NCs with higher C.R. values (>25%) (Fig. 7C). However, after 7 days of treatment, ALP values were twice as high as the control and TA-treated cultures for all the formulations. These findings support the evidence of Eu3+ having osteogenic properties, as this one of the few reports performed on a healthy, human osteoblast cell line.
Moreover, regarding the effect of EuTA NCs on ECM mineralization, it was found that EuTAs 50 and 75% increased the amount of Ca2+ deposited by 9 and 17.5%, respectively, compared to control cultures after 7 days of treatment, thus accelerating the process of ECM maturation (Fig. 7D). In addition, although EuTA 25 and 100% did not significantly alter this parameter at 7 days compared to control cultures, all the formulations showed a better performance than free TA. However, the continuous supply of either TA or EuTA NCs for 14 and 21 days had a negative impact on matrix mineralization. Compared to the control cultures, the addition of the treatments hindered the quantity and the pattern of calcium deposition, resulting in more localized mineral nodules as opposed to a generalized deposition (Fig. S12, ESI†). Therefore, these results would indicate the use of EuTA NCs for short periods of time to alleviate OxS and inflammation while accelerating the early phases of bone growth. Moreover, evaluating the effect of combinational therapies with more potent osteogenic stimuli such as BMP-2 or BMP-7 could be of interest for maximizing their therapeutic potential.
Taken altogether, this initial study has demonstrated that short-term treatments with 10 µg mL−1 EuTA NCs are able to induce osteogenic, antioxidant, and anti-inflammatory effects, outperforming TA in all cases. According to their bioactivity and physicochemical characteristics, EuTA 75% and EuTA 50% were the ones that presented the best overall performances. The mechanisms of action of polyphenols are still under study and, given the lack of visual evidence of their internalization, different hypotheses could be raised including (i) their action occurs at the extracellular level leading to signaling cascades via different routes, or (ii) EuTA NCs are dissociated or partially hydrolyzed when entering the cells. Therefore, given their potential and these biological queries to be checked, more complex models using injectable therapies to control their spatiotemporal administration and evaluate the biochemical pathways involved in their effects are currently under study. Moreover, strategies incorporating other bioactive cations that enhance these properties to a greater extent or add novel ones might be a promising research line to exploit in future studies. In this sense, their high stability, low doses needed, and short-term application times are excellent features to provide more complex systems with these bioactive properties.
4. Conclusions
In this study, tunable EuTA NCs with varying Eu3+ contents have been developed and characterized for their physicochemical and biological properties in the context of bone regeneration under oxidative and inflammatory conditions. Complexation with Eu3+ did not compromise the bioactive properties of TA but enhanced certain antioxidant properties while reducing cellular toxicity. Additionally, EuTA NCs demonstrated superior performance to TA in promoting both early and late markers of osteoblast activity with short-term treatments. Although further research is needed to fully evaluate the regenerative potential of these systems under physiological conditions in in vivo models, these in vitro studies present EuTA NCs as suitable candidates to be applied in situ to resolve OxS and inflammation while promoting early bone regenerative processes.Author contributions
Daniel Fernández Villa: conceptualization, data curation, formal analysis, investigation, methodology, software, supervision, visualization, writing – original draft, writing – review and editing; María Rosa Aguilar: funding acquisition, investigation, project administration, resources, supervision, visualization, writing – review and editing. Luis Rojo: conceptualization, funding acquisition, investigation, project administration, resources, supervision, visualization, writing – review and editing.Data availability
Data for this article, will be available at DIGITAL.CSIC, the institutional repository of the Spanish National Research Council.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is dedicated to the memory of Blanca Vázquez-Lasa, who participated in the conceptualization and preliminary studies of this work, but lamentably passed away on 31st January 2023. This research has been funded by the Spanish MICINN (PID2020-114086RB-I00) and Comunidad Autónoma de Madrid (S2022/BMD-7406). D. F.-V. is financially supported by a predoctoral program MICINN FPU18/04683. M. R. A. and L. R. are members of PTI-SusPlast+ and Nanomedicine CSIC. The authors would like to acknowledge David Gómez, Pedro González Esperanza Benito and Pilar Posadas of the Characterization Service at the Instituto de Ciencia y Tecnología de Polímeros (CSIC) for the technical support and EM facilities of the CIB Margarita Salas (CSIC) for support in grid screening and preparation.References
- J. S. Kenkre and J. H. D. Bassett, Ann. Clin. Biochem., 2018, 55, 308–327 CrossRef CAS PubMed.
- N. Salari, H. Ghasemi, L. Mohammadi, M. hasan Behzadi, E. Rabieenia, S. Shohaimi and M. Mohammadi, J. Orthop. Surg. Res., 2021, 16, 609 CrossRef PubMed.
- C. Cervellati, G. Bonaccorsi, E. Cremonini, A. Romani, E. Fila, M. C. Castaldini, S. Ferrazzini, M. Giganti and L. Massari, Biomed Res. Int., 2014, 2014, 569563 Search PubMed.
- M. Abdollahi, B. Larijani, R. Rahimi and P. Salari, Therapy, 2005, 2, 787–796 CrossRef CAS.
- T. Iantomasi, C. Romagnoli, G. Palmini, S. Donati, I. Falsetti, F. Miglietta, C. Aurilia, F. Marini, F. Giusti and M. L. Brandi, Int. J. Mol. Sci., 2023, 24, 3772 CrossRef CAS PubMed.
- M. Chen, Y. Wang, S. Deng, Z. Lian and K. Yu, Front. Cell Dev. Biol., 2022, 10, 964130 CrossRef PubMed.
- V. Domazetovic, G. Marcucci, T. Iantomasi, M. L. Brandi and M. T. Vincenzini, Clin. Cases Miner. Bone Metab., 2017, 14, 209–216 CrossRef PubMed.
- J. Gambini and K. Stromsnes, Biomedicines, 2022, 10, 753 CrossRef PubMed.
- H. Newman, Y. V. Shih and S. Varghese, Biomaterials, 2021, 277, 121114 CrossRef CAS PubMed.
- M. A. Terkawi, G. Matsumae, T. Shimizu, D. Takahashi, K. Kadoya and N. Iwasaki, Int. J. Mol. Sci., 2022, 23, 1786 CrossRef CAS PubMed.
- S. Chatterjee, in Oxidative Stress and Biomaterials, ed. T. Dziubla and D. A. Butterfield, Elsevier Inc., 2016, pp. 35–58 Search PubMed.
- L. Kang, H. Zhang, C. Jia, R. Zhang and C. Shen, Front. Pharmacol., 2022, 13, 956355 CrossRef CAS PubMed.
- S. Çakar and M. Özacar, J. Photochem. Photobiol., A, 2019, 371, 282–291 CrossRef.
- Z. Guo, W. Xie, J. Lu, X. Guo, J. Xu, W. Xu, Y. Chi, N. Takuya, H. Wu and L. Zhao, J. Mater. Chem. B, 2021, 9, 4098–4110 RSC.
- W. Jing, C. Xiaolan, C. Yu, Q. Feng and Y. Haifeng, Biomed. Pharmacother., 2022, 154, 113561 CrossRef CAS PubMed.
- A. Bigham, V. Rahimkhoei, P. Abasian, M. Delfi, J. Naderi, M. Ghomi, F. Dabbagh Moghaddam, T. Waqar, Y. Nuri Ertas, S. Sharifi, N. Rabiee, S. Ersoy, A. Maleki, E. Nazarzadeh Zare, E. Sharifi, E. Jabbari, P. Makvandi and A. Akbari, Chem. Eng. J., 2022, 432, 134146 CrossRef CAS.
- B. Onat, S. Ozcubukcu, S. Banerjee and I. Erel-Goktepe, Eur. Polym. J., 2018, 103, 101–115 CrossRef CAS.
- C. Steffi, Z. Shi, C. H. Kong, S. W. Chong, D. Wang and W. Wang, Polymers, 2019, 11, 1256 CrossRef CAS PubMed.
- Y. Sun, Y. Qu and J. Zhao, Front. Mater., 2022, 8, 801369 CrossRef.
- M. Jiménez, C. Abradelo, J. San Román and L. Rojo, J. Mater. Chem. B, 2019, 7, 1974–1985 RSC.
- W. M. Tan, W. K. Loke, B. L. Tan, A. Wee, E. Khor and K. S. Goh, Biomaterials, 1993, 14, 1003–1007 CrossRef CAS PubMed.
- W. Xie, Z. Guo, L. Zhao and Y. Wei, Theranostics, 2021, 11, 6407–6426 CrossRef CAS PubMed.
- G. Fan, J. Cottet, M. R. Rodriguez-Otero, P. Wasuwanich and A. L. Furst, ACS Appl. Bio Mater., 2022, 5, 4687–4695 CrossRef CAS PubMed.
- W. Zhu, S. Liang, J. Wang, Z. Yang, L. Zhang, T. Yuan, Z. Xu, H. Xu and P. Li, J. Mater. Sci.: Mater. Med., 2017, 28, 1–11 CrossRef PubMed.
- H. Wang, D. Wang, J. Yu, Y. Zhang and Y. Zhou, Biomater. Sci., 2022, 10, 5786–5808 RSC.
- G. Miao, X. Chen, C. Mao, X. Li, Y. Li and C. Lin, J. Sol-Gel Sci. Technol., 2014, 69, 250–259 CrossRef CAS.
- C. Wu, L. Xia, P. Han, L. Mao, J. Wang, D. Zhai, B. Fang, J. Chang and Y. Xiao, ACS Appl. Mater. Interfaces, 2016, 8, 11342–11354 CrossRef CAS PubMed.
- M. Shi, L. Xia, Z. Chen, F. Lv, H. Zhu, F. Wei, S. Han, J. Chang, Y. Xiao and C. Wu, Biomaterials, 2017, 144, 176–187 CrossRef CAS PubMed.
- Y. Zhang, M. Hu, X. Wang, Z. Zhou and Y. Liu, Nanomaterials, 2018, 8, 961 CrossRef PubMed.
- F. Li, M. Wang, G. Pi and B. Lei, J. Biomed. Nanotechnol., 2018, 14, 756–764 CrossRef CAS PubMed.
- M. Alicka, P. Sobierajska, K. Kornicka, R. J. Wiglusz and K. Marycz, Mater. Sci. Eng., C, 2019, 99, 1257–1273 CrossRef CAS PubMed.
- R. Augustine, S. K. Nethi, N. Kalarikkal, S. Thomas and C. R. Patra, J. Mater. Chem. B, 2017, 5, 4660–4672 RSC.
- K. Wu, X. Wu, J. Guo, Y. Jiao and C. Zhou, Adv. Healthcare Mater., 2021, 10, 1–13 CAS.
- M. Sikora, K. Marcinkowska, K. Marycz, R. J. Wiglusz and A. Smieszek, Materials, 2019, 12, 3779 CrossRef CAS PubMed.
- K. Marycz, A. Smieszek, S. Targonska, S. A. Walsh, K. Szustakiewicz and R. J. Wiglusz, Mater. Sci. Eng., C, 2020, 110, 110634 CrossRef CAS PubMed.
- M. Liu, M. Shu, J. Yan, X. Liu, R. Wang, Z. Hou and J. Lin, Nanoscale, 2021, 13, 1181–1194 RSC.
- H. Fan, L. Wang, X. Feng, Y. Bu, D. Wu and Z. Jin, Macromolecules, 2017, 50, 666–676 CrossRef CAS.
- M. Huerta-Madroñal, J. Caro-León, E. Espinosa-Cano, M. R. Aguilar and B. Vázquez-Lasa, Carbohydr. Polym., 2021, 273, 118619 CrossRef PubMed.
- G. M. Pontes-Quero, L. Benito-Garzón, J. Pérez Cano, M. R. Aguilar and B. Vázquez-Lasa, Mater. Sci. Eng., C, 2021, 121, 111793 CrossRef CAS PubMed.
- C. Sánchez-Moreno, J. A. Larrauri and F. Saura-Calixto, J. Sci. Food Agric., 1998, 76, 270–276 CrossRef.
- C. Pucci, C. Martinelli, D. De Pasquale, M. Battaglini, N. Di Leo, A. Degl’Innocenti, M. Belenli Gümüş, F. Drago and G. Ciofani, ACS Appl. Mater. Interfaces, 2022, 14, 15927–15941 CrossRef CAS PubMed.
- T. C. P. Dinis, V. M. C. Madeira and L. M. Almeida, Arch. Biochem. Biophys., 1994, 315, 161–169 CrossRef CAS PubMed.
- N. Shenoy, M. Stenson, J. Lawson, J. Abeykoon, M. Patnaik, X. Wu and T. Witzig, Lab. Investig., 2017, 97, 494–497 CrossRef CAS PubMed.
- H. Kim and X. Xue, J. Vis. Exp., 2020, 2020, 1–5 Search PubMed.
- D. Fernández-Villa, G. Asensio, M. Silva, R. A. Ramírez-Jiménez, L. Saldaña, N. Vilaboa, A. Leite-Oliveira, J. San Román, B. Vázquez-Lasa and L. Rojo, Eur. J. Med. Chem., 2021, 212, 113152 CrossRef PubMed.
- K. Wu, W. Hua, X. Li and J. Lin, Chem. Eng. J., 2022, 446, 136835 CrossRef CAS.
- J. Guo, Y. Ping, H. Ejima, K. Alt, M. Meissner, J. J. Richardson, Y. Yan, K. Peter, D. Von Elverfeldt, C. E. Hagemeyer and F. Caruso, Angew. Chem., Int. Ed., 2014, 53, 5546–5551 CrossRef CAS PubMed.
- J. Iglesias, E. García de Saldaña and J. A. Jaén, Hyperfine Interact., 2001, 134, 109–114 CrossRef CAS.
- J. D. Hem, Water Supply Pap., 1960, 1459, 75–94 Search PubMed.
- G. G. Costa, C. C. S. M. Brito, A. J. Terezo, A. P. Cardoso, E. Y. Ionashiro and A. B. de Siqueira, Open Chem. J., 2018, 5, 158–171 CrossRef.
- G. Tondi and A. Petutschnigg, Ind. Crops Prod., 2015, 65, 422–428 CrossRef CAS.
- M. Taha, I. Khan and J. A. P. Coutinho, J. Inorg. Biochem., 2016, 157, 25–33 CrossRef CAS PubMed.
- P. Moreau, S. Colette-Maatouk, P. Vitorge, P. Gareil and P. E. Reiller, Inorg. Chim. Acta, 2015, 432, 81–88 CrossRef CAS.
- Ż. Arciszewska, S. Gama, M. Kalinowska, G. Świderski, R. Świsłocka, E. Gołębiewska, M. Naumowicz, M. Worobiczuk, A. Cudowski, A. Pietryczuk, C. De Stefano, D. Milea, W. Lewandowski and B. Godlewska-żyłkiewicz, Int. J. Mol. Sci., 2022, 23(2), 888 CrossRef PubMed.
- W. Liu, T. Wang, C. Yang, B. W. Darvell, J. Wu, K. Lin, J. Chang, H. Pan and W. W. Lu, Osteoporos. Int., 2016, 27, 93–104 CrossRef CAS PubMed.
- G. Di Pompo, M. Cortini, N. Baldini and S. Avnet, Cancers, 2021, 13, 1–21 CrossRef PubMed.
- F. Xiong, Z. Qin, H. Chen, Q. Lan, Z. Wang, N. Lan, Y. Yang, L. Zheng, J. Zhao and D. Kai, J. Nanobiotechnol., 2020, 18, 1–14 CrossRef PubMed.
- S. Mathew, T. E. Abraham and Z. A. Zakaria, J. Food Sci. Technol., 2015, 52, 5790–5798 CrossRef CAS PubMed.
- W. Chariyarangsitham, S. Krungchanuchat, P. Khuemjun and C. Pilapong, Arab. J. Chem., 2021, 14, 103312 CrossRef CAS.
- Q. Yang, C. Zhao, J. Zhao and Y. Ye, Pharm. Biol., 2017, 55, 428–434 CrossRef CAS PubMed.
- G. K. B. Lopes, H. M. Schulman and M. Hermes-Lima, Biochim. Biophys. Acta, Gen. Subj., 1999, 1472, 142–152 CrossRef CAS PubMed.
- H. A. Kleinveld, A. J. G. Swaak, C. E. Hack and J. F. Koster, Scand. J. Rheumatol., 1989, 18, 341–352 CrossRef CAS PubMed.
- Q. Ru, Y. Li, W. Xie, Y. Ding, L. Chen, G. Xu, Y. Wu and F. Wang, Bone Res., 2023, 11, 12 CrossRef CAS PubMed.
- A. Szwed-Georgiou, P. Płociński, B. Kupikowska-Stobba, M. M. Urbaniak, P. Rusek-Wala, K. Szustakiewicz, P. Piszko, A. Krupa, M. Biernat, M. Gazińska, M. Kasprzak, K. Nawrotek, N. P. Mira and K. Rudnicka, ACS Biomater. Sci. Eng., 2023, 9, 5222–5254 CrossRef CAS PubMed.
- Y. Huang, Z. Du, P. Wei, F. Chen, B. Guan, Z. Zhao, X. Zhang, Q. Cai, J. Mao, H. Leng and X. Yang, Chem. Eng. J., 2020, 397, 125352 CrossRef CAS.
- Y. Wu, X. Li, Y. Sun, X. Tan, C. Wang, Z. Wang and L. Ye, Bioact. Mater., 2023, 20, 111–125 CAS.
- S. Choi, H. S. Jo, H. Song, H. J. Kim, J. K. Oh, J. W. Cho, K. Park and S. E. Kim, Nanomaterials, 2021, 11(7), 1812 CrossRef CAS PubMed.
- B. Taciak, M. Białasek, A. Braniewska, Z. Sas, P. Sawicka, Ł. Kiraga, T. Rygiel and M. Król, PLoS One, 2018, 13, 1–13 CrossRef PubMed.
- J. N. Sharma, A. Al-Omran and S. S. Parvathy, Inflammopharmacology, 2007, 15, 252–259 CrossRef CAS PubMed.
- D. Fernández-Villa, R. A. Ramírez-Jiménez, I. Aranaz, N. Acosta, B. Vázquez-Lasa and L. Rojo, Int. J. Mol. Sci., 2022, 23, 10054 CrossRef PubMed.
- H. Y. Kim, H. J. Jeong, N. R. Han, J. B. Jang and H. M. Kim, J. Food Biochem., 2018, 42, 1–10 CrossRef.
- D. Song, J. Zhao, W. Deng, Y. Liao, X. Hong and J. Hou, Biochem. Biophys. Res. Commun., 2018, 503, 3078–3085 CrossRef CAS PubMed.
- V. Mouriño, J. P. Cattalini and A. R. Boccaccini, J. R. Soc., Interface, 2012, 9, 401–419 CrossRef PubMed.
- W. Qiao, D. Pan, Y. Zheng, S. Wu, X. Liu, Z. Chen, M. Wan, S. Feng, K. M. C. Cheung, K. W. K. Yeung and X. Cao, Nat. Commun., 2022, 13, 1–15 Search PubMed.
- M. Martin-Del-Campo, R. Rosales-Ibañez, K. Alvarado, J. G. Sampedro, C. A. Garcia-Sepulveda, S. Deb, J. San Román and L. Rojo, Biomater. Sci., 2016, 4, 1596–1604 RSC.
- H. Hapidin, N. A. A. Romli and H. Abdullah, Microsc. Res. Tech., 2019, 82, 1928–1940 CAS.
- H. Hapidin, N. M. Hashim, M. Z. Kasiram and H. Abdullah, Molecules, 2022, 27, 451 CrossRef CAS PubMed.
- T. H. Pham, E. Kim, N. M. Trang and G. Jeong, J. Periodontal Res., 2023, 59, 204–219 CrossRef PubMed.
- E. Torre, Phytochem. Rev., 2017, 16, 1183–1226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00697f |
| This journal is © The Royal Society of Chemistry 2024 |