
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEncapsulation of 5-fluorouracil in cholesteryl-modified cyclodextrin: thermal, spectral, and computational assessment of drug inclusion efficiency†
Paweł
Misiak
 *a,
Bartosz
Maliszewski
ab,
Zuzanna
Pawłowska
a,
Anna
Ignaczak
*c and
Agnieszka Z.
Wilczewska
*a
*a,
Bartosz
Maliszewski
ab,
Zuzanna
Pawłowska
a,
Anna
Ignaczak
*c and
Agnieszka Z.
Wilczewska
*a
aFaculty of Chemistry, University of Bialystok, Ciolkowskiego 1k, 15-245 Bialystok, Poland. E-mail: p.misiak@uwb.edu.pl; agawilcz@uwb.edu.pl
bDoctoral School of Exact and Natural Sciences, University of Bialystok, Ciolkowskiego 1k, 15-245 Bialystok, Poland
cTheoretical and Structural Chemistry Group, Department of Physical Chemistry, Faculty of Chemistry, University of Lodz, Pomorska 163/165, 90-236, Lodz, Poland. E-mail: anna.ignaczak@chemia.uni.lodz.pl
First published on 10th July 2024
Abstract
This research investigates the encapsulation of 5-fluorouracil (5-FU) within cholesteryl-modified β-cyclodextrin (CD21chol) and aims to elucidate the drug inclusion efficiency through a comprehensive analysis employing both experimental and computational techniques. The study employs thermogravimetric characterization to assess the thermal stability of the encapsulated complex and infrared measurements to explore the vibrational characteristics, providing valuable insights into the physicochemical properties. Additionally, molecular simulations are employed to evaluate the interactions between 5-FU and CD21chol on the molecular-level dynamics of drug encapsulation. This integrated approach facilitates a comprehensive understanding of encapsulation, offering valuable data for developing drug delivery systems.
Introduction
5-Fluorouracil (5-FU) is a small molecule with a 130 g mol−1 mass. It is a fluorinated derivative of uracil with unique pharmacological properties. It is a commonly used anticancer drug, mainly for colorectal cancer.1 It is an antimetabolite, and its action is to impersonate a purine or pyrimidine, which prevents it from being incorporated into DNA. Disruption of the synthesis and stability of nucleic acids leads to the cancer cell's death.2,3 Unfortunately, there are problems in therapy that are related to (I) dose limitation due to high toxicity, (II) short half-life in the body, and (III) emerging drug resistance of neoplastic cells.2,4 Due to the limitations mentioned above, encapsulation becomes an attractive strategy to improve circulation time in the bloodstream, increase biodistribution, and reduce the dose of 5-FU, which will reduce the side effects of therapy.There are many strategies for synthesizing 5-fluorouracil carriers.5,6 However, a commonly preferred option when designing drug carriers (DDS) is natural products (NPs), which are inherently better tolerated by the body than their synthetic counterparts.7 In the last 2 decades, intensive research has been conducted on 5-FU inclusion complexes with cyclodextrins. (CD).8,9 Modified cyclodextrins, including alginate complexes,10 intercalated in zinc–aluminum layered double hydroxide galleries,11 or polymer derivatives where β-CD occurs in the side chains12 are also used. Polymer nanoparticles, especially those using folic acid as a homing molecule, are among the popular strategies for transporting 5-FU to cancer cells.2,13,14 Cholesterol is a widely used NP in the synthesis of 5-fluorouracil DDS, e.g., lipid nanoemulsions,15 liposomes,16 or is directly linked to 5-FU to form a prodrug.17 It plays a pivotal role as a fundamental organic molecule in mammalian cell membranes, governing their fluidity, integration, and permeability.18,19 Due to its multifaceted significance, cholesterol is a prevalent selection for guiding molecule incorporation into cell membranes.20 Studies have shown that even a single cholesterol group at the end of a polymer chain is sufficient for effective interaction with the cell membrane.21 Despite strenuous attempts, the discomfort associated with therapy cannot be minimized, and so far, none of the formulations has undergone clinical trials.
Encapsulation of 5-FU in cholesteryl-modified cyclodextrin may provide a novel way to improve drug bioavailability by modifying its solubility and stability in the biological environment. The main goal of this work is to understand the encapsulation process of 5-FU in cholesteryl-modified cyclodextrin. Thermal, spectral, and computational assessment of drug inclusion efficiency enables understanding the behavior of the drug–cyclodextrin complex, which is a crucial step in assessing the effectiveness of this process. The proposed research may contribute to developing more effective and safer cancer therapies. Understanding the mechanisms of drug-carrier complex formation will reduce the use of drugs and limit their release into the environment, which are additional economical and ecological benefits of the study.
Experimental
Materials and methods
Nuclear magnetic resonance (NMR). The 1H and 13C NMR spectra were recorded on Bruker Avance II 400 spectrometer using CDCl3 as a solvent.
Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR). All ATR-FTIR spectra were recorded using the Thermo Scientific Nicolet 6700 FTIR spectrophotometer equipped with an ATR accessory. Spectra were ratioed against the background spectra and collected in the wavenumber range of 4000 to 500 cm−1 by co-adding 32 scans with a resolution of 4 cm−1.
Thermal analysis
Thermogravimetric analysis (TGA) was performed on the Mettler Toledo TGA/DSC1 system. The samples (2–3 mg) were placed in the aluminum oxide crucible, and an empty pan was used as a reference. The measurements took place in the inert atmosphere of argon (flow rate 40 mL min−1) at the heating rate of 10 °C min−1, in the temperature range of 50–900 °C. Differential scanning calorimetry (DSC) was performed on the Mettler Toledo Star DSC unit. The 2–3 mg samples were placed in the aluminum crucible and sealed. The empty pan was used as a reference. The measurements were carried out under inert gas conditions (flow rate 200 mL min−1), at the heating rate of 10 °C min−1, in the temperature range 25–350 °C. The melting points (Tm) of 5-Fu were determined using the Mettler Toledo STARe software based on the first heating cycle.Computational details
The theoretical part of the study included a conformational analysis for the β-CD 21-cholesteryl derivative (CD21chol) and tests on the stability of its complexes with the drug.In order to find the low energy conformers of CD21chol, in the Hyperchem program,22 the initial model of the molecule was created from β-CD by replacing a hydrogen atom in each hydroxyl group with the cholesteryl-containing moiety (Fig. S1 in the ESI†). The torsion angles defining the spatial orientation of the substituents were set so that the substituent chains had a similar orientation with respect to the CD ring and did not overlap (Fig. S2, ESI†). Next, a two-step procedure was applied, which included first the creation of many structures in the Hyperchem program and their complete optimization using the BIO+ molecular mechanics method, and next their re-optimization with the semi-empirical PM7 method in vacuo using the MOZYME procedure in the MOPAC program.23 A detailed description of the method used in this initial conformational search is given in the section Procedure S1 in the ESI.† The conformer with the lowest PM7 heat of formation was used to create the initial configurations of the complexes of CD21chol with 5-fluorouracil.
Initial studies of complexes of the CD21chol molecule (host) with 5-FU (guest) included analysis of changes in the heat of formation (Hf) of the system when the drug was introduced into the host from two opposite sides, as shown in Fig. 1. Thus, the partially relaxed ΔHf scans were performed along the Z-axis with the step 0.2 Å in two directions, denoted as upward and downward. In the article, configurations corresponding to the downward scans are marked with the symbol prime ′. First, such scans were performed for the complex 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for seven different orientations of the 5-FU monomer (Fig. 1(a)). In each step, to preserve the relative z-position of the drug with respect to the host, all coordinates of the drug and z-coordinates of three oxygen atoms belonging to the CD ring of the host were frozen. In contrast, the remaining coordinates were optimized with the PM7 method in vacuo using the MOZYME procedure. Because the complex formation converted CD21chol to a lower energy conformer, these scans were repeated five times until no more stable host structure was found, and the results were consistent. Next, similar scans were performed for the dimer (5-FU)2 (Fig. 1(b); the complex 1:2), using its lowest energy geometry reported in the literature24,25 and applying two different methods. In the first method (DIM1), at each step, only one 5-FU molecule in the dimer was moved and frozen, while the second 5-FU and CD21chol were optimized. In the second method, the entire dimer was moved and frozen during optimization. The structures corresponding to the most prominent local minima found from all these scans were fully optimized (without any constraints) with the PM7 method, and the most stable forms of complexes, 1:1 and 1:2, were selected. An additional test for the complex 1:2 was performed, which was assumed to contain one 5-FU embedded inside CD21chol and another 5-FU attached to the cholesteryl moieties on the opposite side of the host. The initial model was created from the most stable inclusion complex, 1:1, to which the second 5-FU molecule was attached at its most stable non-inclusion configuration. This type of the 1:2 complex is denoted as I/NI (inclusion/non-inclusion). It should be mentioned that another slightly more stable conformer of CD21chol was discovered from the scans performed with the dimer. Its structure was very similar to the initial one, and the heat of formation was lower only by about 2.5 kcal mol−1. Therefore, these scans were not repeated. However, after the complete optimization, this conformer was used as a reference host structure in the final calculations of the complexation enthalpies. In the scans for the complexes in the stoichiometry 1:3, the 5-FU monomer was assumed to approach the inclusion complex 1:2 (Fig. 1(c)) from two opposite sides. In this case, the host was the complex of CD21chol with the (5-FU)2 dimer having the lowest heat of formation selected in the previous step (denoted as DIM1-I2). Two different orientations of the monomer were considered, marked as x (shown in Fig. 1(c)) and y (rotated around the Z-axis by 180°). The methods used in all scans and the orientations x and y are described in detail in Procedure S2 in the ESI.† As in previous cases, the structures corresponding to minima were fully optimized after the scans. Finally, as in the case of the 1:2 complex I/NI, the possibility of forming the 1:4 complex I/NI with two dimers was examined. From the results, the complexation enthalpies Hcompl for were calculated according to the formulas (1)–(7) listed in Procedure S2d in the ESI.†
:1 for seven different orientations of the 5-FU monomer (Fig. 1(a)). In each step, to preserve the relative z-position of the drug with respect to the host, all coordinates of the drug and z-coordinates of three oxygen atoms belonging to the CD ring of the host were frozen. In contrast, the remaining coordinates were optimized with the PM7 method in vacuo using the MOZYME procedure. Because the complex formation converted CD21chol to a lower energy conformer, these scans were repeated five times until no more stable host structure was found, and the results were consistent. Next, similar scans were performed for the dimer (5-FU)2 (Fig. 1(b); the complex 1:2), using its lowest energy geometry reported in the literature24,25 and applying two different methods. In the first method (DIM1), at each step, only one 5-FU molecule in the dimer was moved and frozen, while the second 5-FU and CD21chol were optimized. In the second method, the entire dimer was moved and frozen during optimization. The structures corresponding to the most prominent local minima found from all these scans were fully optimized (without any constraints) with the PM7 method, and the most stable forms of complexes, 1:1 and 1:2, were selected. An additional test for the complex 1:2 was performed, which was assumed to contain one 5-FU embedded inside CD21chol and another 5-FU attached to the cholesteryl moieties on the opposite side of the host. The initial model was created from the most stable inclusion complex, 1:1, to which the second 5-FU molecule was attached at its most stable non-inclusion configuration. This type of the 1:2 complex is denoted as I/NI (inclusion/non-inclusion). It should be mentioned that another slightly more stable conformer of CD21chol was discovered from the scans performed with the dimer. Its structure was very similar to the initial one, and the heat of formation was lower only by about 2.5 kcal mol−1. Therefore, these scans were not repeated. However, after the complete optimization, this conformer was used as a reference host structure in the final calculations of the complexation enthalpies. In the scans for the complexes in the stoichiometry 1:3, the 5-FU monomer was assumed to approach the inclusion complex 1:2 (Fig. 1(c)) from two opposite sides. In this case, the host was the complex of CD21chol with the (5-FU)2 dimer having the lowest heat of formation selected in the previous step (denoted as DIM1-I2). Two different orientations of the monomer were considered, marked as x (shown in Fig. 1(c)) and y (rotated around the Z-axis by 180°). The methods used in all scans and the orientations x and y are described in detail in Procedure S2 in the ESI.† As in previous cases, the structures corresponding to minima were fully optimized after the scans. Finally, as in the case of the 1:2 complex I/NI, the possibility of forming the 1:4 complex I/NI with two dimers was examined. From the results, the complexation enthalpies Hcompl for were calculated according to the formulas (1)–(7) listed in Procedure S2d in the ESI.†
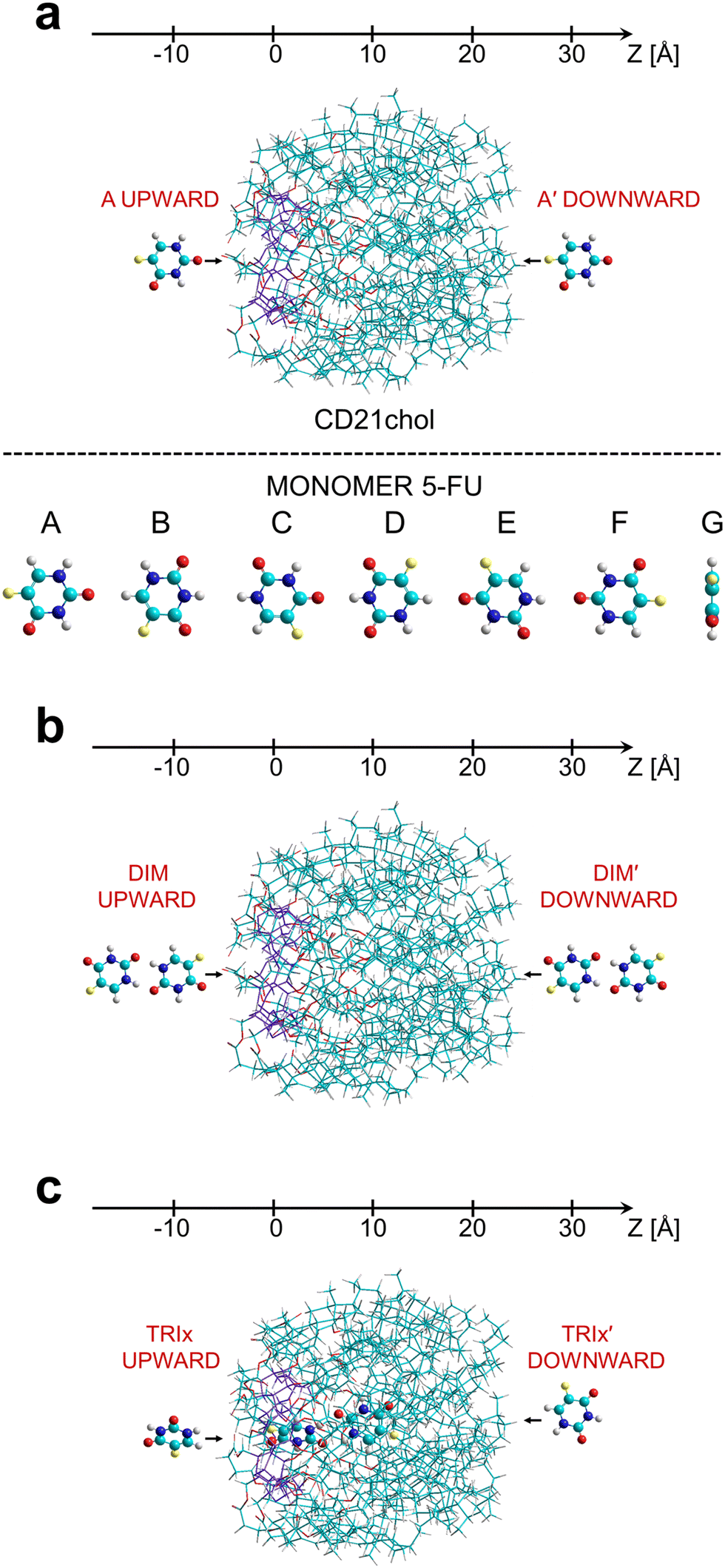 | ||
| Fig. 1 (a) The upward and downward insertion paths and seven orientations A-G of the 5-FU molecule concerning the CD21chol considered in the PM7 calculations, (b) the insertion paths of the dimer (5-FU)2, (c) the insertion paths of the monomer 5-FU in the x orientation towards the lowest energy complex of CD21chol with dimer. The CD ring is marked in violet, while the remaining atoms’ colors are carbon – cyan, nitrogen – dark blue, fluorine – yellow, oxygen – red, and hydrogen – grey. | ||
Single-point calculations were performed at the density functional theory (DFT) level in vacuo using the M062X-GD3 method and the 6-31G(d) basis set for the most stable configurations obtained with the semi-empirical method. M062X-GD3 is the hybrid metafunctional M06-2X26 with the Grimme empirical pairwise long-range (dispersion) corrections GD3.27 This method was shown in the past to yield a small root mean square error concerning the CCSD(T)/CBS reference values28 and was used earlier in the study of complexes of dimethyl-β-cyclodextrin complexes with mianserin.29 The DFT calculations were performed using the Gaussian 16 program.30 From the resulting energies, the DFT complexation energies Ecompl were obtained using formulas similar to these for Hcompl (Procedure S2d, ESI†).
For the most stable structures selected, the solvent effect on Hcompl was estimated by re-optimizing these structures in three different solvents, namely THF, methanol, and water, using the PM7 method and a conductor-like screening model (COSMO).31 The effect of the same solvents on the DFT Ecompl was evaluated by performing the SP calculations for the re-optimized structures with the polarizable continuum model (PCM).32 The DFT complexation energies obtained in vacuo for the selected structures were additionally corrected by the basis set superposition error (BSSE) calculated using the counterpoise correction.33 In the text, the BSSE-corrected complexation energies are denoted as EBSSEcompl.
Synthetic procedures
Synthesis of cholesteryl hemisuccinate (CHEMS)
Succinic anhydride (4.36 g, 43.62 mmol, 3 eq.) and DMAP (1.77 g, 14.54 mmol, 1 eq.) were added to a solution of cholesterol (5.62 g, 14.54 mmol, 1 eq.) in dry pyridine (100 mL). The reaction mixture was stirred for 6 h at 60 °C, then for 16 h at room temperature. The reaction was monitored by TLC. Upon consumption of cholesterol, the reaction mixture was washed with 10% HCl solution (250 mL) and then extracted with ethyl acetate (3 × 100 mL). The organic layer was washed with a saturated aqueous NaCl solution, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by recrystallization from methanol at 4 °C. The product was obtained as a white powder with a yield of 90%, 6.387 g.Synthesis of cholesteryl hemisuccinate chloride (CHEMS-Cl)
CHEMS (2 g, 4.1 mmol, 1 eq.) was dissolved in 20 mL of dry dichloromethane. The flask was placed in an ice bath on a magnetic stirrer. After cooling the mixture, thionyl chloride (0.46 mL, 4.9 mmol, 1.2 eq.) was added dropwise over 30 minutes. The reaction was continued for another 20 minutes at room temperature. After the reaction was completed, the solvent and the remaining thionyl chloride were evaporated on a rotary evaporator under reduced pressure.Synthesis of cholesteryl-modified cyclodextrin (CD21chol)
The β-CD (180 mg, 0.16 mmol, 1 eq.) was suspended in 17 mL of a mixture of dry pyridine and dichloromethane in a volume ratio of 7:10. The mixture was flushed with argon for 20 minutes, and the flask was placed on a magnetic stirrer. In the second flask, CHEMS-Cl was dissolved in 20 mL of dry DCM and was added dropwise to the β-CD suspension using a syringe pump at 0.22 mL min−1. The reaction was continued for 24 hours at room temperature. The product was isolated by DCM extraction, washed successively with 5% HCl (3 × 10 mL), saturated aqueous NaHCO3 solution (3 × 10 mL), saturated aqueous NaCl solution (3 × 10 mL), and water (3 × 10 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated using a rotary evaporator. The compound was dissolved in warm (65 °C) ethyl acetate (70 mL), crystallized, filtered, and dried. The product was obtained as a white powder with a yield of 50%, 0.9 g.
5-Fluorouracil encapsulation
A CD21chol solution with a concentration of 50 mg mL−1 was prepared. For this purpose, 375 mg of CD21chol was dissolved in 7.5 mL of THF. 1 mL of the prepared solution was measured into seven bottles. A solution of 5-fluorouracil with a concentration of 5.7 mg mL−1 was prepared in a flask. For this purpose, 19.95 mg of 5-FU was dissolved in 3.5 mL of methanol. The drug solution was added in different molar ratios CD21chol:5-FU: 1:1 (0.1 mL of 5-FU solution), 1:2 (0.2 mL of 5-FU solution), 1:3 (0.3 mL of 5-FU solution), 1:4 (0.4 mL 5-FU solution) 1:5 (0.5 mL 5-FU solution), 1:8 (0.8 mL of 5-FU solution), 1:10 (1 mL of 5-FU solution). Then, to equalize the concentrations in the vessels, an appropriate amount of THF was added to each solution so that the final volume was 3 mL. Mixtures were stirred on a magnetic stirrer at room temperature for 24 h. Then, 0.5 mL was taken from each solution, and the solvent was removed. The residue was analyzed using the following methods: DSC, TG, and ATR-FTIR. The remaining solution (2.5 mL) was added dropwise using a syringe pump (flow rate 0.1 mL min−1) to 25 mL of water to separate the complex from free drug molecules. The resulting suspensions were left on a magnetic stirrer for 24 h to evaporate the solvents. Then, they were centrifuged, and the supernatant was separated from the sediment. The obtained complexes (precipitates) were dried in a stove at 60 °C for 24 h and analyzed using the following methods: DSC, TG, and FTIR.
Results and discussion
Synthesis and characterization
The cholesteryl derivative of β-CD (CD21chol) was synthesized by esterification using acid chloride generated from cholesteryl hemisuccinate. In the first step, cholesterol was reacted with succinic anhydride in the presence of DMAP. In the next step, CHEMS was reacted with thionyl chloride to obtain a reactive acid chloride, which was then added dropwise to the β-CD solution. A cholesteryl derivative with all 21 arms modified was obtained. (Scheme 1) | ||
| Scheme 1 Synthesis of cholesteryl derivative of β-CD (CD21chol). | ||
Cholesteryl hemisuccinate and cholesteryl derivative of β-CD were analyzed by NMR and ATR-FTIR; full spectra are available in ESI†: 1H NMR (Fig. S3 and S4, ESI†), 13C NMR (Fig. S5 and S6 ESI†) and DEPT (Fig. S7 and S8, ESI†), and ATR-FTIR (Fig. S9b and d, ESI†). Two main changes are compared to cholesterol spectra in the 1H NMR spectrum of CHEMS (Fig. S3, ESI†). First, a new signal is observed at a chemical shift of 2.65 ppm, which comes from the H-2′,3′ protons from the succinate group. Second, the signal of the H-3 proton is shifted from 3.25 ppm to 4.64 ppm. The presence of two signals at 178.3 and 171.5 ppm of quaternary carbons in the CHEMS carbon spectrum (Fig. S5, ESI†) indicates that the reaction proceeded to obtain the succinic acid ester (monosubstituted product). In the infrared spectrum of CHEMS (Fig. S9b, ESI†), compared to cholesterol spectrum (Fig. S9a, ESI†), the signal coming from vibrations of the hydroxyl band at a region 3600–3000 cm−1 was not observed, and ester group bonds vibrations bands occurred: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1731 and 1724 cm−1, C(O)–O at 1437 and 1375 cm−1, and C–C–O at 1315 and 1254 cm−1. In the 1H NMR spectrum of CD21chol (Fig. S4, ESI†), new signals in the range of 0.60–2.70 ppm from protons, characteristic of CHEMS, appeared, which confirmed obtaining a cholesteryl derivative of β-CD. The integration ratio of signals from the protons of the β-CD core (3.40–5.70 ppm) to the protons in the cholesteryl derivative arms (4.58 ppm) indicates the substitution of all hydroxyl groups of β-CD. In the ATR-FTIR spectrum of CD21chol (Fig. S9d, ESI†), there is no band from hydroxyl bond vibrations, which is observed in the β-CD spectrum (Fig. S9c, ESI†). In addition, characteristic for CHEMS spectrum bands appeared in the region motioned above. The disappearance of the peak of COacid at 1724 cm−1 proves esterification.
O at 1731 and 1724 cm−1, C(O)–O at 1437 and 1375 cm−1, and C–C–O at 1315 and 1254 cm−1. In the 1H NMR spectrum of CD21chol (Fig. S4, ESI†), new signals in the range of 0.60–2.70 ppm from protons, characteristic of CHEMS, appeared, which confirmed obtaining a cholesteryl derivative of β-CD. The integration ratio of signals from the protons of the β-CD core (3.40–5.70 ppm) to the protons in the cholesteryl derivative arms (4.58 ppm) indicates the substitution of all hydroxyl groups of β-CD. In the ATR-FTIR spectrum of CD21chol (Fig. S9d, ESI†), there is no band from hydroxyl bond vibrations, which is observed in the β-CD spectrum (Fig. S9c, ESI†). In addition, characteristic for CHEMS spectrum bands appeared in the region motioned above. The disappearance of the peak of COacid at 1724 cm−1 proves esterification.
Determination of 5-FU inclusion efficiency
The obtained cholesteryl derivative of β-cyclodextrin was used to encapsulate 5-fluorouracil. For this purpose, solutions with different molar ratios (1:1, 1:2, 1:3, 1:4, 1:5, 1:8, and 1:10) of CD21chol and 5-FU were prepared as described in the section “5-fluorouracil encapsulation”. ATR-FTIR, thermal analysis (TGA and DSC), and quantum chemical methods were used to analyze the composition of the formed complexes.
Physicochemical studies
:1, 1:2, and 1:3 have similar shapes. In the case of systems with a higher drug content, an additional decomposition rate maximum is observed on the DTG curves. The first occurs at lower temperatures and comes from the degradation of 5-FU. The second one, with a maximum in the range of 330–340 °C, was assigned to the decomposition of CD21chol. Based on the measurements taken, it can be assumed that the sensitivity of the thermogravimetric method for examining CD21chol:5-FU complexes is limited to 4 molecules of 5-fluorouracil per one molecule of CD21chol. The curves for solutions taken after complexation look similar (Fig. 2(b) and (e)). The decomposition of 5-FU is visible only when the system is taken from a solution with a molar ratio of 1:4 and bigger. In the case of analyses of samples obtained after precipitation of carriers (Fig. 2(c) and (f)), no peaks of 5-FU decomposition were observed. This indicates the effectiveness of the proposed methodology for purifying complexes from free/non-encapsulated drug molecules by precipitation.
 | ||
| Fig. 2 TG (a)–(c) and DTG (d)–(f) curves of CD21chol, (a) and (d) physical mixtures, (b) and (e) dried solutions after complexation, (c) and (f) complexes of CD21chol:5-FU after precipitation, and 5-fluorouracil. | ||
The thermogravimetric method indicates the formation of complexes containing not more than four molecules of 5-fluorouracil, but the method's sensitivity limits this result. For this reason, research was conducted using differential scanning calorimetry. Full curves are shown in Fig. S10–S12 in the ESI.† Similarly to the above, three sets of DSC curve sections are presented (Fig. 3). In this case, the most essential area of the curves for analytical reasons is the melting peak of 5-FU, which has a maximum of 285 °C. In the case of physical mixtures, the melting peak of 5-FU could be seen starting from a molar ratio of 1:3 (Fig. 3(a)). This indicates that the sensitivity of DSC is slightly better than that of TG. Fig. 3(b) shows the curves of samples from the solutions after complexation. The melting peak of 5-FU was detected for systems 1:4 and richer in the molar ratio of the drug. No 5-FU melting peaks were observed in the curves obtained from the samples of the precipitated complexes (Fig. 3(c)), confirming the absence of free, not incorporated into the carriers 5-fluorouracil molecules.
 | ||
| Fig. 3 Sections of DSC curves show the melting peak of 5-fluorouracil measured for CD21chol, (a) physical mixtures, (b) dried solutions after complexation, (c) complexes of CD21chol:5-FU after precipitation and 5-fluorouracil. | ||
The DSC method turned out to be more sensitive than the thermogravimetric method. The analysis of the curves indicates the formation of complexes in a molar ratio of 1:1, 1:2, and 1:3 (CD21chol:5-FU). However, this result is close to the sensitivity limit of the thermal methods. Therefore, there was a need to compare the results of another method, so infrared analyses of all samples were carried out.
Infrared spectroscopy investigation
ATR-FTIR analysis of complexes of the cholesteryl derivative of β-CD and 5-fluorouracil was performed. Full spectra of 5-FU, CD21chol, physical mixtures, solutions, and complexes are shown in Fig. S13–S15 in the ESI.† First, the limit of quantification of 5-Fu was determined using the ATR-FTIR method. For this purpose, infrared spectra of CD21chol, physical mixtures of CD21chol:5-FU in various molar ratios (1:1, 1:2, 1:3, 1:4, 1:5, 1:8, and 1:10), and 5-fluorouracil were recorded (Fig. 4(a)).
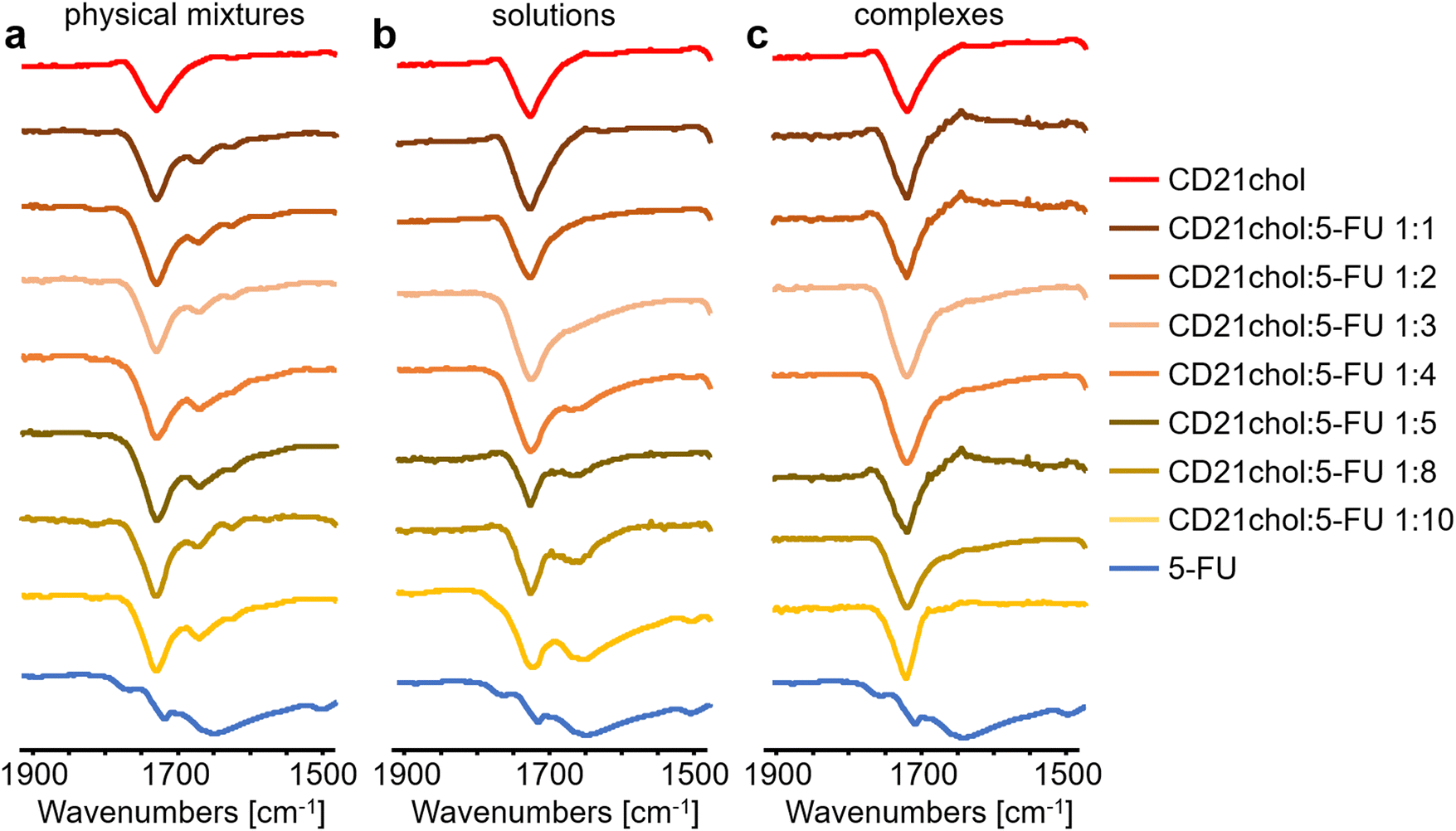 | ||
| Fig. 4 Stack of ATR-FTIR spectra of CD21chol, (a) physical mixtures, (b) dried solutions after complexation, (c) complexes of CD21chol:5-FU after precipitation, and 5-fluorouracil. | ||
In all spectra of physical mixtures, bands originating from the vibrations of CD21chol and 5-FU bonds were observed, which means that this technique can help analyze the formation of complexes in the proposed molar ratios. The reflectance intensity ratios of the band at 1731 cm−1 originating from the vibrations of the CO from the ester groups present in CD21chol and the band at 1670 cm−1 arising from the vibrations of the CO from the amide group of 5-FU were compared. Analysis of the results in Table S1 in the ESI† indicates that as the amount of drug in the mixtures increases, the vibration intensity of the COamide relative to the COester increases. In samples from solutions of molar ratios 1:1, 1:2, and 1:3, no bands from COamide vibrations were found, meaning the entire amount of initial 5-FU was encapsulated. In samples 1:4 to 1:10, the intensity of the vibration band of the carbonyl bond from the amide group relative to the other signals increases with the initial amount of drug in the solution. The results of comparing the vibration bands intensity of COamide and COester are summarized in Table S1 in the ESI.† The analysis of these results indicates a specific, limited quantity of 5-FU molecules that the CD21chol system can complex. Analysis of the spectra in Fig. 4(b) points that the limit is reached for three molecules of 5-fluorouracil. In Fig. 4(c), the vibration band originating from the carbonyl bond of the amide group of 5-FU is not observed in all spectra. This also exhibits that the proposed method of purifying from non-complexed drug molecules is effective.
ATR-FTIR turned out to be the most sensitive of the proposed methods and allowed for the analysis of all proposed systems. The examination of the infrared spectra confirmed the thesis made after the analysis of the DSC curves, which was CD21chol:5-FU complexes are formed in molar ratios of 1:1, 1:2, and 1:3. The above methods lead to the conclusion that one CD21chol molecule can complex maximum three molecules of 5-fluorouracil. Appropriate quantum chemistry methods such as PM7 and DFT were used to understand this issue better.
Computational studies
 | ||
| Fig. 5 The final most stable conformer of CD21chol found in the present work from the semi-empirical PM7 calculations in vacuo. Three different views are shown: the top view (a) and two side views (b) and (c). Atom colors are the same as in Fig. 1. | ||
The most stable structures obtained from the full PM7 optimization in vacuo for all complexes in various stoichiometries are presented in Fig. 6–8, while the corresponding complexation enthalpies and energies are shown in Fig. 9 and 10 and Table S2 in the ESI.† Sample plots of ΔHf profiles obtained from the scans usually indicate several local minima (Fig. S16–S18, ESI†). The calculations for CD21chol:5-FU in the stoichiometry 1:1 indicate that CD21chol can form stable inclusion (I) complexes with 5-FU embedded on the CD ringside as well as non-inclusion (NI) complexes with 5-FU attached to the cholesteryl groups. The complexation enthalpies for the most stable structures of CD21chol:5-FU obtained from the PM7 optimizations for all drug orientations are compared to the DFT complexation energies (calculated for the same geometries, without BSSE corrections) in Fig. S19 and Table S2 in the ESI.† For the complexes 1:1, both methods predict that the energy profit from the formation of the inclusion complexes is approximately twice as large as that for the non-inclusion complexes. The PM7 and DFT results trends are similar, but the predictions for the lowest energy structure differ. According to the PM7 values, the configurations A (Fig. 6(a)) and F′ (Fig. S20a, ESI†) are the most stable 1:1 complexes within the I and NI sets, with Hcompl of −41 and −18.3 kcal mol−1, respectively. The DFT results indicate different configurations in these sets, namely D (Fig. 6(b)) and C′ (Fig. S20b, ESI†), which have Ecompl of −31.3 and −16.7 kcal mol−1, respectively. In both A and D complexes, the monomer 5-FU is located close to the CD ring, indicating that the interaction of the drug with CD is stronger than with cholesteryl groups. In this area are oxygen atoms from the CD ring and cholesteryl groups, which can form hydrogen bonds with hydrogens in 5-FU. Such bonds and other interactions possibly contributing to the complex stabilization can be seen in the magnified views shown additionally for each molecule in Fig. 6.
 | ||
| Fig. 6 The most stable structures of complexes of CD21chol with the monomer 5-FU, indicated by the results of PM7 (a) and DFT (b) calculations in vacuo. For each complex, a top view and a side view are shown. The framed images show a magnified view of the area where 5-FU is in the complex. The CD ring and cholesteryl group fragments that may interact with the drug are rendered with thicker lines. Interactions that may contribute to stabilizing the complex (atom distances less than 2.7 Å) are indicated by dashed black lines. 5-FU is rendered with spheres and cylinders. Atom colors are the same as in Fig. 1. | ||
 | ||
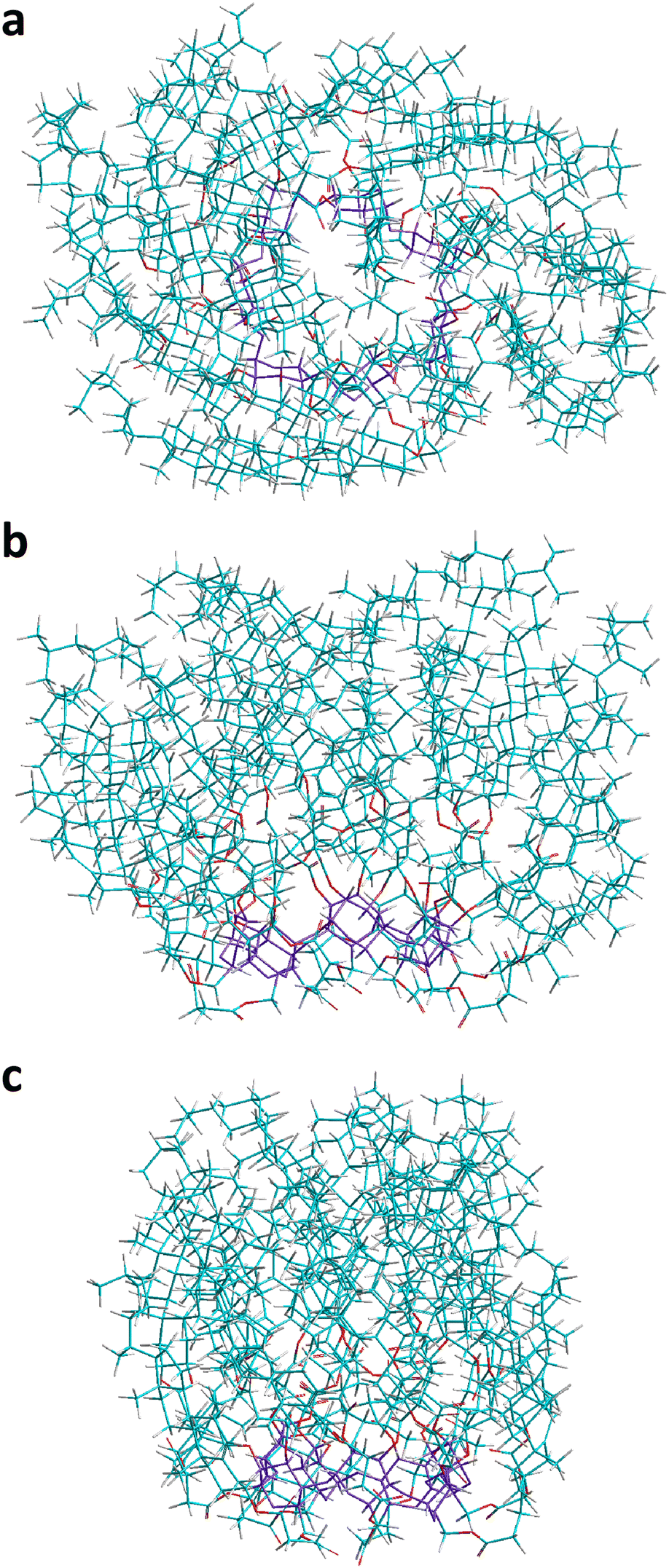
| Fig. 7 The most stable structures of complexes of CD21chol with 5-FU in stoichiometry 1:2, indicated by the results of PM7 (a, c) and DFT (b, c) calculations in vacuo. For each complex, top and side views, as well as magnified views of the area where the 5-FU molecules are located (in frames), are shown. The colors and marking method are the same as in Fig. 6. | ||
 | ||
| Fig. 8 The most stable structures of complexes of CD21chol with 5-FU in stoichiometry 1:3 (a, c) and 1:4 (c), indicated by the results of PM7 (a, c) and DFT (b, c) calculations in vacuo. For each complex, top and side views, as well as magnified views of the area where the 5-FU molecules are located (in frames), are shown. The colors and marking method are the same as in Fig. 6. | ||
 | ||
| Fig. 9 Comparison of the total complexation enthalpies (Hcompl) for the most stable complexes in different stoichiometry, obtained from the PM7 optimization in vacuo and the solvents THF, methanol, and water described with the COSMO model. Hcompl were calculated using the formulas (1)–(4) and (6) shown in Procedure S2d in the ESI.† | ||
 | ||
| Fig. 10 Comparison of the total complexation energies (Ecompl) for the most stable complexes in different stoichiometry, obtained from the DFT SP calculations in vacuo (without and with the BSSE corrections) and in the solvents THF, methanol, and water described with the PCM model (values without the BSSE corrections). | ||
Additionally, the structures corresponding to the local minima observed in the upward scans around the 8–9 Å (denoted M1 because they are approximately midway between I and NI configurations) were optimized with the PM7 method. According to the PM7 results, the most stable M1 structures correspond to the orientations A and D (Fig. S20c and d, ESI†) with Hcompl equal to −25.9 and −24 kcal mol−1, respectively, while the corresponding DFT Ecompl values are −21 and −28.8 kcal mol−1. Other local minima (M2) are observed around 17 Å in some energy profiles. In the optimized M2 structure, the 5-FU monomer is only partially embedded in the region of terminal fragments of cholesteryl groups. The lowest PM7 complexation enthalpy (after complete optimization) of −15.3 kcal mol−1 corresponds to the C orientation (Fig. S20e, ESI†), for which the DFT results predict a much larger complexation energy of −27.6 kcal mol−1. It should be mentioned that the barriers between minima observed in the energy profiles are overestimated because only partial optimization was performed for each drug position.
The pairs of favored inclusion and non-inclusion complexes 1:1 of CD21chol:5-FU, as predicted by each method, were used to test the stability of the complex in 1:2 stoichiometry in two I/NI configurations: AF′ and DC′ (Fig. 7(a) and (b)). The resulting Hcompl (I/NI) and Ecompl (I/NI) (Fig. 9 and 10) are close to the sum of their values for the two corresponding I and NI complexes 1:1, especially in the case of PM7 results. This is not surprising because, in the case of such a large host molecule, the two drug molecules in the I/NI configuration interact with CD21chol independently of each other. A comparison of interactions marked in Fig. 7(a) and (c) for the 5-FU molecule in the (I) and (NI) positions explains why the complexation enthalpy in the non-inclusion complexes is much smaller than in the inclusion ones. In the (NI) position, there are no oxygen atoms in the cholesteryl groups, and only a few of their hydrogen atoms are close enough to participate in the binding of 5-FU. It should be stressed that, in the (NI) position, the drug is exposed to the solvent. Specific interactions of 5-FU with molecules of a particular solvent may either promote its binding or, on the contrary, prevent its attachment. The role of the solvent in the formation of such complexes should be investigated at the molecular level in the future.
The experimental results suggest that, in solutions, CD21chol forms stable inclusion complexes in the stoichiometries 1:1, 1:2, and 1:3. It is also known that 5-FU can form stable dimers both in vacuo and in solvents.24,25,37 Therefore, in the following stages of our research, the dimer (5-FU)2 insertion into CD21chol and the further attachment of the third 5-FU molecule to such a complex were examined. The most stable complexes of CD21chol with the (5-FU)2 dimer (1:2) and additional 5-FU monomer (1:3) are presented in Fig. 7(c) and 8(a), (b). The results of PM7 optimization and subsequent DFT calculations performed for the complex 1:4 I/NI with two dimers (Fig. 8(c)) are also included. Some additional structures and numerical data are provided in Fig. S21, S22 and Table S3 in the ESI.† The energy gain resulting from the inclusion of the entire dimer (DIM1-I2) configuration (Fig. 7(c)) is more significant by approximately 15 kcal mol-1 than that for the inclusion complex of monomer or the partial docking of the dimer (DIM1-I1, via the CD entry; Fig. S21a, ESI†). The results of the PM7 calculations suggest that the complex with two monomers in the I/NI AF′ configuration (Fig. 7(a)) is slightly more stable than DIM1-I2, but the DFT values indicate that the formation of the complex with dimer is much more favorable (Fig. 10).
Attaching another 5-FU molecule or dimer to the DIM1-I2 complex provides an additional energy profit. The most favorable is the 1:3 complexes, in which the three drug molecules form a chain inside CD21chol (Fig. 8(a) and (b)). According to the PM7 results, this is the TRIy-I1 configuration with the 5-FU monomer docked through the CD entrance (Hcompl(MONtoDIM1) = −33.5 kcal mol−1), while the DFT values suggest that it is TRIy′-I1 with the 5-FU monomer docked through the cholesteryl groups (Ecompl(MONtoDIM1) = −43.9 kcal mol−1). Of course, the total complexation enthalpies and energies (the total energy gain from complexation of both the dimer and the monomer by CD21chol) are much larger: Hcompl(TRIMER) for TRIy-I1 is equal to −92.3 kcal mol−1, while Ecompl(TRIMER) for TRIy′-I1 is equal to −98.2 kcal mol−1. In both cases, the initially flat structure of dimer is distorted due to interaction with the additional 5-FU molecule (this is the lowest 5-FU in Fig. 8(a) and the highest one in Fig. 8(b)). Despite the initial monomer orientations in the TRIy and TRIy′ scans, in both the final TRIy-I1 and TRIy′-I1 structures, the 5-FU monomer is rotated and forms an N–H⋯OC hydrogen bond with the dimer. A similar hydrogen bond is also formed in the TRIx′-I1 complex, but this configuration is slightly less energetically favored, probably due to the interactions of the monomer with CD21chol. The three 5-FU docked inside CD21chol can also adopt a different, non-linear configuration TRIx-I2 (Fig. S22d, ESI†), which is also very stable but less than the linear arrangement (Table S3 in the ESI†). The results for the 1:4 complex of CD21chol with two dimers in the I/NI configuration (2DIM in Fig. 8(c)) indicate that such a structure can also be formed. Since only one dimer is placed inside the cavity in this complex, while the second one is attached externally and its interaction with CD21chol is weaker, the absolute values of Hcompl and Ecompl for 2DIM are smaller than for the preferred TRIy-type inclusion complexes. However, 2DIM also provides a significant energy gain, so it will likely co-exist with them.
For the structures presented in Fig. 6–8, the solvent effect on the complexation enthalpies and energies was additionally evaluated, and the results are included in Fig. 9 and 10. The numerical data is provided in Table S4 in the ESI.† As shown in Fig. 9, absolute values of Hcompl obtained in the presence of solvents are significantly smaller than in vacuo and decrease in the following order: vacuum > THF > methanol > water. The presence of solvent does not change the general trend indicated by Hcompl obtained in vacuo, so also, in a liquid phase, formation of the 1:3 complex TRIy-I1 is favored, followed by TRIy′-I1 and 2DIM. The effect of solvents on the DFT results is similar (Fig. 10), so in this case, the TRIy’-I1 complex is indicated as the most stable, both in vacuum and in solutions. It should be noted that the implicit solvent models used in these calculations do not take into account the specific interactions of solvent molecules with 5-FU, CD21chol, and their complexes, so they only allow an approximate assessment of the effect of the solvent on the complexation enthalpies (energies).
Since the DFT Ecompl were obtained from the SP calculations and thus are only approximate estimates, the above values did not include the BSSE corrections. It is not apparent whether incorporating the BSSE correction into the SP complexation energy will improve or worsen the results. This depends on whether the Ecompl obtained after optimizing all compounds would be similar or much more negative than those obtained from SP calculations. In the latter case, the SP Ecompl values can be closer to the actual values than the SP EBSSEcompl. Nevertheless, it is worth seeing how much the BSSE corrections can affect the complexation energies calculated for the tested systems. As shown in Fig. 10, the absolute EBSSEcompl values are much smaller, so the BSSE is very large and accounts for about 36 to 47% of Ecomplin vacuo. Although this reduces the differences between complexation energies for different configurations, the overall trend is maintained.
Conclusion
Thermal analysis revealed that the encapsulated 5-FU within cyclodextrins decorated with cholesteryl moiety carrier (CD21chol) maintains thermal stability, essential for potential applications in drug delivery systems. ATR-FTIR measurements further enriched our understanding by elucidating specific molecular interactions and vibrational characteristics within the encapsulated system. The observed bands confirm the successful encapsulation of 5-FU and allow for determining the maximum number of drug molecules contained in the complex. The results of quantum calculations indicate that CD21chol can form with 5-FU very stable complexes in all stoichiometries tested. Among the analyzed configurations, the inclusion complex with three 5-FU molecules arranged in a chain inside CD21chol is most energetically preferred. The total energy profit from its formation from isolated components (CD21chol, (5-FU)2 dimer, and 5-FU monomer) is vast, above 90 kcal mol−1in vacuo and 70 kcal mol−1 in solutions. Other forms, such as I/NI complexes with a dimer, a monomer, and two dimers, also show high stability and are therefore expected to be formed. It should be emphasized, however, that due to the size of the tested system, the solvent was described by an implicit model in the calculations. Since the stability of non-inclusion complexes strongly depends on the balance of drug-carrier, drug-solvent, and carrier-solvent interactions, further studies with a solvent described at the molecular level are necessary. The principal value of using computational chemistry in the present research lies in elucidating the arrangement of cholesteryl units relative to the cyclodextrin core. Notably, the binding affinity of the CD21chol towards 5-fluorouracil molecules is skewed towards the narrower end of the cone, contrary to a bare β-CD. In the classical encapsulation, guest molecules ingress from the broader end of the cone and are entrapped within its interior. However, with our cholesterol modification, conformational changes occur, leading to an arrangement of cholesteryl branches that essentially obstruct the approach of 5-FU from the narrower side. While one 5-FU molecule is entrapped within the cyclodextrin cage, the remaining reside between the cholesteryl section. The synergy between experimental and computational approaches enhances the reliability and depth of our findings, paving the way for the rational design and optimization of drug delivery systems.Author contributions
P. M.: conceptualization, visualization, investigation, writing – original draft, project administration, funding acquisition, B. M.: investigation, writing – original draft, Z. R.: investigation, A. I.: investigation, visualization, writing – original draft, A. Z. W.: conceptualization, supervision, writing – review & editing.Data availability
The data supporting this article have been included in the ESI.† Additional datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request. For inquiries related to synthetic data and experiments, please contact Paweł Misiak (p.misiak@uwb.edu.pl). Please contact Anna Ignaczak (anna.ignaczak@chemia.uni.lodz.pl) for computational data inquiries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Centre, Poland, grant no. 2020/37/N/ST5/02140. Analyses were performed in the Centre of Synthesis and Analysis BioNanoTechno of the University of Bialystok. The equipment in the Centre was funded by the EU as a part of the Operational Program Development of Eastern Poland 2007-2013, projects: POPW.01.03.00-20-034/09-00 and POPW.01.03.00-20-004/11. DFT calculations have been carried out using resources provided by the Wroclaw Centre for Networking and Supercomputing (https://wcss.pl), grant no. 443.Notes and references
- S. Vodenkova, T. Buchler, K. Cervena, V. Veskrnova, P. Vodicka and V. Vymetalkova, Pharmacol. Ther., 2020, 206, 107447 CrossRef CAS.
- G. Siemiaszko, K. Niemirowicz-Laskowska, K. H. Markiewicz, I. Misztalewska-Turkowicz, E. Dudź, S. Milewska, P. Misiak, I. Kurowska, A. Sadowska, H. Car and A. Z. Wilczewska, Cancer Nanotechnol., 2021, 12, 31 CrossRef CAS.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- X. Wang, A. Wang, W. Feng, D. Wang, X. Guo, X. Wang, Q. Miao, M. Liu and G. Xia, Mol. Pharmaceutics, 2022, 19, 2061–2076 CrossRef CAS.
- S. Cheralayikkal, K. Manoj and K. P. Safna Hussan, Heliyon, 2022, 8, e09926 CrossRef CAS.
- M. D. Blanco, O. Garcia, C. Gomez, R. L. Sastre and J. M. Teijon, J. Pharm. Pharmacol., 2000, 52, 1319–1325 CrossRef CAS PubMed.
- R. A. Khan, Saudi Pharm. J., 2018, 26, 739–753 CrossRef.
- C. Di Donato, M. Lavorgna, R. Fattorusso, C. Isernia, M. Isidori, G. Malgieri, C. Piscitelli, C. Russo, L. Russo and R. Iacovino, Molecules, 2016, 21, 1644 CrossRef CAS.
- D. L. Melnikova, Z. F. Badrieva, M. A. Kostin, C. Maller, M. Stas, A. Buczek, M. A. Broda, T. Kupka, A.-M. Kelterer, P. M. Tolstoy and V. D. Skirda, Molecules, 2020, 25, 5706 CrossRef CAS.
- C.-H. Nguyen, K.-S. Banh, C.-H. Dang, C.-H. Nguyen and T.-D. Nguyen, Arabian J. Chem., 2022, 15, 103814 CrossRef CAS.
- L. Jin, Q. Liu, Z. Sun, X. Ni and M. Wei, Ind. Eng. Chem. Res., 2010, 49, 11176–11181 CrossRef CAS.
- H. Li, S. Zhu, L. Xu, Y. Chen, X. Li and W. Wu, Transl. Cancer Res., 2020, 9, 4596–4606 CrossRef CAS PubMed.
- M. Prabaharan, J. J. Grailer, D. A. Steeber and S. Gong, Macromol. Biosci., 2009, 9, 744–753 CrossRef CAS.
- A. Kasprzak, K. Gunka, M. Fronczak, M. Bystrzejewski and M. Poplawska, ChemistrySelect, 2018, 3, 10821–10830 CrossRef CAS.
- F. K. Alanazi, N. Haq, A. A. Radwan, I. A. Alsarra and F. Shakeel, J. Drug Delivery Sci. Technol., 2014, 24, 459–463 CrossRef CAS.
- S. A. Alanazi, G. I. Harisa, M. M. Badran, N. Haq, A. A. Radwan, A. Kumar, F. Shakeel and F. K. Alanazi, Curr. Drug Delivery, 2020, 17, 898–910 CrossRef CAS.
- A. A. Radwan and F. K. Alanazi, Molecules, 2014, 19, 13177–13187 CrossRef PubMed.
- W. Zhang, Z. Mou, Y. Wang, Y. Chen, E. Yang, F. Guo, D. Sun and W. Wang, Mater. Sci. Eng., C, 2019, 97, 486–497 CrossRef CAS.
- M. P. Stewart, R. Langer and K. F. Jensen, Chem. Rev., 2018, 118, 7409–7531 CrossRef CAS PubMed.
- P. Misiak, K. H. Markiewicz, D. Szymczuk and A. Z. Wilczewska, Polymers, 2020, 12, 2620 CrossRef CAS PubMed.
- P. Misiak, K. Niemirowicz-Laskowska, K. H. Markiewicz, I. Misztalewska-Turkowicz, P. Wielgat, I. Kurowska, G. Siemiaszko, M. Destarac, H. Car and A. Z. Wilczewska, Int. J. Nanomed., 2020, 15, 7263–7278 CrossRef CAS.
- HyperChem(TM) Professional (version 8.0.10.) Hypercube, Inc, 1115 NW 4th Street, Gainesville, Florida 32601, USA, 2011 Search PubMed.
- J. J. P. Stewart, MOPAC2016 Stewart Computational Chemistry, 2016 Search PubMed.
- G. Alagona, C. Ghio and S. Monti, Int. J. Quantum Chem., 2001, 83, 128–142 CrossRef CAS.
- S. S. Ostakhov, M. Yu Ovchinnikov, G. A. Masyagutova and S. L. Khursan, J. Phys. Chem. A, 2019, 123, 7956–7964 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- A. D. Boese, ChemPhysChem, 2015, 16, 978–985 CrossRef CAS.
- A. Ignaczak, Ł. Orszański, M. Adamiak and A. B. Olejniczak, J. Mol. Liq., 2020, 315, 113767 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, 2016 Search PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- F. Ercole, M. R. Whittaker, J. F. Quinn and T. P. Davis, Biomacromolecules, 2015, 16, 1886–1914 CrossRef CAS PubMed.
- X. Liu, Z. Guo, Y. Xie, Z. Chen, J. Hu and L. Yang, J. Mol. Liq., 2018, 259, 350–358 CrossRef CAS.
- M. Shepelenko, A. Hirsch, N. Varsano, F. Beghi, L. Addadi, L. Kronik and L. Leiserowitz, J. Am. Chem. Soc., 2022, 144, 5304–5314 CrossRef CAS PubMed.
- G. Alagona, C. Ghio and S. Monti, Int. J. Quantum Chem., 2002, 88, 133–146 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00496e |
| This journal is © The Royal Society of Chemistry 2024 |