
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarrier-free chemo-phototherapeutic nanomedicines with endo/lysosomal escape function enhance the therapeutic effect of drug molecules in tumors†
Xue
Feng
a,
Calum M.
Brown
b,
Hongdi
Wang
a,
Saima
Kashif
a,
Sam
Roberts
a,
Li
Yan
 c,
Tasnim
Munshi
d,
Philip J. W.
Hands
b,
Wenjun
Zhang
e and
Xianfeng
Chen
*a
c,
Tasnim
Munshi
d,
Philip J. W.
Hands
b,
Wenjun
Zhang
e and
Xianfeng
Chen
*a
aSchool of Engineering, Institute for Bioengineering, University of Edinburgh, The King's Buildings, EH9 3JL Edinburgh, UK. E-mail: Michael.Chen@ed.ac.uk
bSchool of Engineering, Institute for Integrated Micro and Nano Systems, University of Edinburgh, The King's Buildings, EH9 3FF Edinburgh, UK
cCollege of Health Science and Environmental Engineering, Shenzhen Technology University, Shenzhen 518118, China
dSchool of Chemistry, University of Lincoln, Brayford Pool, Lincoln, Lincolnshire LN6 7TS, UK
eDepartment of Materials Science and Engineering, City University of Hong Kong, 83 Tat Chee Ave, Kowloon Tong, Hong Kong SAR
First published on 12th June 2024
Abstract
Carrier-free nanomedicines offer advantages of extremely high drug loading capacity (>80%), minimal non-drug constituent burden, and facile preparation processes. Numerous studies have proved that multimodal cancer therapy can enhance chemotherapy efficiency and mitigate multi-drug resistance (MDR) through synergistic therapeutic effects. Upon penetration into the tumor matrix, nanoparticles (NPs) are anticipated to be uptaken by cancer cells, primarily through clathrin-meditated endocytosis pathways, leading to their accumulation in endosomes/lysosomes within cells. However, endo/lysosomes exhibit a highly degradative environment for organic NPs and drug molecules, often resulting in treatment failure. Hence, this study designed a lysosomal escape mechanism with carrier-free nanomedicine, combining the chemotherapeutic drug, curcumin (Cur), and the photothermal/photodynamic therapeutic drug, indocyanine green (ICG), for synergistic cancer treatment (ICG-Cur NPs) via a facile preparation process. To facilitate endo/lysosomal escape, ICG-Cur NPs were modified with metal-phenolic networks (MPNs) of different thickness. The results indicate that a thick MPN coating promotes rapid endo/lysosomal escape of ICG-Cur NPs within 4 h and enhances the photothermal conversion efficiency of ICG-Cur NPs by 55.8%, significantly improving anticancer efficacy in both chemo- and photo-therapies within 3D solid tumor models. This finding underscores the critical role of endo/lysosomal escape capacity in carrier-free drug NPs for therapeutic outcomes and offers a facile solution to achieve it.
1. Introduction
Cancer remains one of the deadliest diseases in the world today, and chemotherapy stands as a cornerstone in cancer treatment.1 Despite remarkable advancements in cancer therapeutics, multi-drug resistance (MDR) poses a persistent challenge and can lead to chemotherapy failure. When cancer cells are exposed to a drug repeatedly, they develop resistance to it, often extending to multiple drugs with different formulations.2 To overcome MDR, several strategies have been reported.One effective strategy is combining various therapeutic modalities (e.g., radiotherapy, immunotherapy, photothermal and photodynamic therapies) with chemotherapy, known as synergistic therapy. Numerous therapeutic combinations have been achieved in nanomedicines, where nanoparticles effectively encapsulate multiple components and deliver them concurrently to cancer cells.3–9 In addition, nanocarriers aid in enhancing the pharmacokinetics and tumor accumulation of encapsulated drugs.10 Clathrin-mediated endocytosis serves as the primary cellular uptake pathway for NPs (generally above 50 nm). In the process, NPs are initially internalized into endocytic vesicles (early endosomes), which mature into late endosomes, and eventually lysosomes.11,12 Lysosomes maintain a highly acidic environment (∼pH 5) and are rich in enzymes, which degrade drugs and organic NPs before they can diffuse into the cytoplasm.13 As many cancer therapies rely on therapeutic agents to act on cytoplasmic and nuclear targets, a drug delivery strategy involves maximizing the exposure of anticancer drugs within cells via endosomal disruption.13,14 To avert therapeutic nanomedicine failure, imparting NPs with the endo/lysosomal escape capabilities is crucial. Metal-phenolic networks (MPNs), the coordination complexes of phenolic ligands and metal ions, were first discovered in 2013 by Caruso et al. as versatile and safe coatings for in vivo applications.15 In 2019, they reported that MPN coating facilitates the endo/lysosomal escape of nanomedicines due to their pH-buffering capacity. MPNs induce a proton sponge effect in endo/lysosomes, disrupting endo/lysosomal membranes.16 Compared with conventional nanomedicines using organic and inorganic nanomaterials as drug carriers to deliver drugs, carrier-free nanomedicines offer advantages of extremely high drug loading capacity (>80%), minimal non-drug constituent burden, and facile preparation processes.17–22 Therefore, herein, we aim to demonstrate using MPN coating to facilitate carrier-free nanomedicine's endo/lysosomal escape and further improve their therapeutic efficacy. We designed to combine Cur and ICG as carrier-free nanomedicines (ICG-Cur NPs) for potential synergistic chemotherapy and photothermal/photodynamic therapy.23–27 The chemical structure of ICG, containing benzene rings, facilitates its interaction with Cur via π–π stacking, leading to nanoparticle self-assembly via hydrophobic interactions. Besides, the amphiphilic property of ICG improves the solubility of curcumin. Unlike many previously reported carrier-free nanomedicines requiring surface modification, ICG-Cur NPs do not necessitate the use of surfactants to be stable in solution.28,29 Subsequently, we employed MPNs to functionalize the ICG-Cur NPs by assembling Fe3+ and tannic acid (TA) complexes on the NP surface (termed as ICG-Cur/MPN) (Scheme 1). Our study demonstrated that MPN networks readily form on the surface of pure drug NPs with adjustable thicknesses and morphologies (thin/smooth or thick/rough). Notably, this study represents the first exploration of the impact of MPN thickness on the endo/lysosomal escape efficiency of carrier-free drug NPs. Finally, we compared the chemotherapeutic and phototherapeutic effects of ICG-Cur NPs and MPN-coated ICG-Cur NPs using a 3D tumor model, aiming to explore whether endo/lysosomal escape and MPN networks could improve the anticancer efficacy of drug NPs.
 | ||
| Scheme 1 Illustration of the preparation of the ICG-Cur/MPN NP. | ||
2. Methods
2.1 Materials
Indocyanine green (ICG) was bought from Cambridge Bioscience. Curcumin (≥65%, HPLC), tannic acid, iron(III) chloride hexahydrate (FeCl3·6H2O), and cell counting kit-8 (CCK-8) were ordered from Sigma-Aldrich. LysoTracker™ Red DND-99, Hoechst 33342 (10 mg mL−1), and propidium iodide (PI) were purchased from ThermoFisher.2.2 Synthesis of ICG-Cur NPs
Carrier-free ICG-Cur NPs were produced by the co-assembly method using small molecular drugs. Briefly, 1 mL of 1 mg mL−1 ICG water solution was added in a glass vial, and then 100 μL of 10 mg mL−1 curcumin was dropwisely added to the ICG solution under sonication. The drug mixture was kept under sonication for 20 min and transferred to an orbital shaker (200 rpm) at room temperature for aging 14 h. The assembled NPs were collected by centrifugation at 21500 rpm for 30 min. The collected ICG-Cur NPs were re-dispersed into ultra-pure water and then purified by ultracentrifugation with a membrane of 10 KDa molecular cut-off weight (Amicon® Ultra; Merck Millipore Ltd) to remove free molecules. The yield of ICG-Cur NPs was determined to 34.7%.2.3 Functionalization with MPN (Fe3+/TA) coating
The assembly of Fe3+/TA networks on ICG-Cur NPs was based on a previous method with adaption.30 In this study, the thickness of MPN coating (thin or thick) was adjusted by tuning feeding ratios of ingredients. First, TA and FeCl3·6H2O aqueous solutions were prepared separately at concentrations of 40.8 mg mL−1 and 10 mg mL−1, respectively. To obtain a thin MPN coating, 3 μL of TA solution and 3 μL of FeCl3·6H2O solution were sequentially added to 900 μL of 0.5 mg mL−1 ICG-Cur NPs at room temperature. After each addition, the mixture was vortexed for 1 min, followed by 1 min ultrasonication. Next, the mixed solution was continually ultrasonicated for 10 min to allow sufficient film formation. The ICG-Cur/MPN NPs were collected and purified by two centrifugation/redispersion cycles with ultra-pure water to remove excess TA and Fe3+. A thick MPN coating was produced using the same protocol, except for increasing both the feeding of TA and Fe3+ from 3 to 10 μL.2.4 Characterization of synthesized NPs
To determine the Cur and ICG encapsulation efficiencies (EE) and loading efficiencies (LE) of synthesized NPs, the NPs were first immersed into DMSO to extract Cur and ICG, and the UV-Vis absorbance intensities of Cur (at 425 nm) and ICG (at 800 nm) were analyzed by a Nanodrop spectrophotometer (Nanodrop 2000c, ThermoFisher™ Scientific). The standard curve equations of Cur and ICG were obtained by measuring the absorbance of their solutions with different drug concentrations in DMSO (Cur/DMSO: y = 0.0158x + 0.0047, R2 = 0.999; ICG/DMSO: y = 0.02x + 0.0737, R2 = 0.9996). The Cur and ICG concentrations in NPs were calculated from calibration curves. The EE and LE of drugs (Cur or ICG) were calculated according to the following formulas: | (1) |
 | (2) |
2.5 In vitro drug release study
The drug release profile of IGC-Cur, ICG-Cur/thin MPN, and ICG-Cur/thick MPN NPs were examined using the dialysis method in 1 × PBS buffer with two different pH values (7.4 and 5). First, 1 mL of NP solution was sealed in a dialysis bag with a molecular cut-off of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da and immersed into 20 mL of the release buffer with 0.5% (w/v) tween 80 to increase the solubility of the released hydrophobic Cur. The drug release process was performed at 37 °C with constant shaking (180 rpm) in the dark. Then, at desired time intervals, 1 mL of the release medium were collected for UV-Vis measurements to determine the amount of the released drugs, and 1 ml of fresh medium were replenished to the release medium. The standard curves of Cur/PBS and ICG/PBS solution with 0.5% tween 80 at neutral and acid pH conditions were plotted by measuring the absorbance of different concentrations of Cur and ICG at the wavelength of 425 nm and 800 nm, respectively (Fig. S1, ESI†).
000 Da and immersed into 20 mL of the release buffer with 0.5% (w/v) tween 80 to increase the solubility of the released hydrophobic Cur. The drug release process was performed at 37 °C with constant shaking (180 rpm) in the dark. Then, at desired time intervals, 1 mL of the release medium were collected for UV-Vis measurements to determine the amount of the released drugs, and 1 ml of fresh medium were replenished to the release medium. The standard curves of Cur/PBS and ICG/PBS solution with 0.5% tween 80 at neutral and acid pH conditions were plotted by measuring the absorbance of different concentrations of Cur and ICG at the wavelength of 425 nm and 800 nm, respectively (Fig. S1, ESI†).
2.6 Cell culture
MCF-7 and MDA-MB-231 (human breast adenocarcinoma cancer) cell lines were bought from ATCC®. Cells were maintained in DMEM medium containing 10% FBS, 1% penicillin G and 1% streptomycin and cultured at 37 °C in a humidified 5% CO2 atmosphere.2.7 3D cell culture
30 μL of 3 × 104 MCF-7 cells in DMEM containing 20% FBS were seeded on the inverted lid of the 65 mm Petri dish to form a drop, and 20 drops were produced per dish. The Petri dish was filled with PBS to prevent the evaporation of drops. The lid was then inverted onto the dish and incubated at 37 °C/5% CO2 for 4–6 days until spheroids had formed (as observed by optical microscopy).2.8 Cell uptake and lysosome escape of NPs
MCF-7 or MDA-MB-231 cells were seeded in the glass-bottom 24-well confocal plate at a density of 30000 cells per well in 500 μL of growth medium for 30 h to allow adhesion. After adhesion, the culture medium was replaced with 300 μL of fresh medium (without FBS) containing ICG-Cur NPs, or ICG-Cur/MPN (thin or thick coating) NPs at a Cur concentration of 0.2 μg mL−1. After 4 h of the co-incubation, the medium containing NPs were replaced with fresh medium to stop the further uptake of NPs by cells. Then, the cells were ready to stain with LysoTracker Red for observing (4 h) or continue the co-incubation to the desired time points (8 h, 12 h and 24 h co-incubations at 37 °C). For the staining of endo/lysosomes, the old culture medium was replaced by 300 μL of fresh medium containing 50 nM LysoTracker Red and incubated for 1 h at 37 °C. Cells were then gently washed with PBS three times and fixed with 4% paraformaldehyde for 20 min, then washed again. Cell nuclei were stained with 1 μg mL−1 Hoechst 33342 for 10 min and washed twice with PBS. Finally, the cell uptake and endo/lysosomal escape of NPs were observed using confocal laser scanning microscopy (CLSM, ZEISS, 880, Germany). Channel information of CLSM: Hoechst 33342 (blue, excitation = 405 nm, detection = 415–470 nm); curcumin (green, excitation = 488 nm, detection = 510–570 nm); LysoTracker Red (red, excitation = 561 nm, detection = 605–690 nm). The colocalization of NPs and endo/lysosomes was quantitatively evaluated by Pearson's correlation coefficient (PCC) values of Cur vs. endo/lysosomes, which were obtained from Imaris software (>50 randomly picked cells were analyzed). Data are presented as mean ± standard deviation.
2.9 In vitro cytotoxicity assessment of NPs
The cytotoxicity of NPs was first assessed by the CCK-8 cell viability assay according to manufacturer's suggested procedures. Briefly, 1 × 104 cells per well of MCF-7 cells were seeded in 96-well plates in 200 μL of growth medium and incubated overnight at 37 °C/5% CO2. Then, the old medium in each well was replaced with 150 μL of fresh DMEM medium without FBS containing free Cur, ICG-Cur NPs, or ICG-Cur/thick MPN NPs at curcumin concentrations of 5, 10 and 30 μg mL−1. After incubating for 24 h or 48 h, cells were carefully washed with pre-warmed culture medium twice, and 100 μL of fresh medium were added to each well. Next, 10 μL of CCK-8 solution was added to each well and incubated with cells for 4 h at 37 °C. The plate was read at 490 nm (O.D.) using a plate reader (CLARIOstar® plus), and the cell viability was calculated. The data are expressed at the mean values ± standard deviation of five measurements.Then, the apoptosis and necrosis of cells were investigated by Hoechst 33342/PI staining. In brief, MCF-7 cells were seeded in glass-bottom 24-well confocal plate at a density of 50000 cells per well in 500 μL of growth medium for 30 h to allow adhesion and then replaced with fresh medium without FBS. Free Cur, ICG-Cur NPs, or ICG-Cur/thick MPN NPs were added to the well at a dose of 30 μg mL−1 Cur concentration and co-incubated with cells for 24 h at 37 °C. The staining solution was prepared by mixing Hoechst and PI in serum-free DMEM culture medium with a final concentration is 5 μg mL−1 for each dye. After that, the cells were stained with the staining solution at 37 °C, and the staining time was 15 min. After gently washing twice with the culture medium, the colorless DMEM medium (no phenol red) was added to the well, and the cells were imaged using the CLSM (ZEISS, 880).
2.10 Measurement of the efficacy of laser-induced hyperthermia of NP suspensions
1 mL of free ICG + Cur, ICG-Cur NP, or ICG-Cur/thick MPN NP PBS suspensions were added to 1.5 mL centrifuge tubes (containing 20 or 40 μg mL−1 ICG). The tubes were irradiated by an 808 nm laser (LRD-0808 collimated diode laser system, Laserglow Technologies; accessorized with an optical beam expander – BE02-05-B, Thorlabs) at 1.5 W cm−2. The temperature changes of the NP suspension areas along with the irradiation time were measured (area maximum temperatures) and recorded by a thermography camera (FLIR C3).The photothermal conversion efficiency of NPs (at ICG concentration of 40 μg mL−1) was calculated according to the previous study,31 by using the eqn (3):
 | (3) |
Then, the value of hA can be determined by the eqn (4):
 | (4) |
| t = τsln(θ) | (5) |
 | (6) |
2.11 Evaluation of the reactive oxygen species (ROS) generation of NPs in cells with laser irradiation
The ROS generation abilities of free ICG, ICG-Cur NPs, or ICG-Cur/thick MPN NPs were assessed using a ROS probe dye (DCFH-DA). Specifically, MCF-7 or MDA-MB-231 cells were seeded in glass-bottom 24-well confocal plate at a density of 50000 cells per well in 400 μL of growth medium for 30 h to allow adhesion. Subsequently, the culture medium was replaced with fresh DMEM medium without FBS for control groups; free ICG and Cur, ICG-Cur NPs, or ICG-Cur/thick MPN NPs for therapeutic groups (at the ICG concentration of 1 μg mL−1; at the Cur concentration of 1.12 μg mL−1). After an 8 h incubation, the cells were washed with PBS and then replaced with a fresh culture medium containing 30 μM DCFH-DA and further incubated for another 30 min at 37 °C. Following loading with the DCFH-DA, the cells were washed twice with PBS, cultured with a serum-free DMEM medium (no phenol red), and irradiated with an 808 nm laser at a power intensity of 1 W cm−1 for 1 min. Finally, the ROS in cells were visualized using CLSM (ZEISS, 880). The integrated fluorescent density in cells from the ROS signals was quantified by Image J.
2.12 Hoechst/PI staining of 3D cells to evaluate the apoptosis of tumor spheroids with laser irradiation
The apoptosis and necrosis of cells in 3D cell cultures were assessed using Hoechst 33342/PI staining. For the 3D cell staining, after cell spheroids were formed in hanging drop culture, the spheroids were carefully transformed to glass-bottom 24-well confocal plates. Free ICG + Cur, ICG-Cur NPs, or ICG-Cur/thick MPN NPs were added to the well at Cur concentrations of 5 μg mL−1. Then, cell spheroids and NPs were co-incubated for 4 days at 37 °C. After incubation, cell spheroids were irradiated with the 808 nm laser at a power intensity of 1.5 W cm−1 for 3 min and continued to incubation for 24 h (n = 3) (the control groups were directly incubated for 24 h without laser treatment). The cell spheroids were then stained with the Hoechst 33342/PI staining solution at 37 °C (5 μg mL−1 for each dye), and the staining time was 30 min for cell spheroids. After gently washing twice with the culture medium, the colorless serum-free DMEM medium (without phenol red) was added to the well, and the cells were imaged using the CLSM (ZEISS, 880).2.13 Statistical analysis
Statistical analyses were conducted using one-way analysis of variance (ANOVA) for multiple comparisons in Excel software (version 2016). P-values less than 0.05 were considered statistically significant. Statistical differences are defined as ***P < 0.001, **P < 0.01, *P < 0.05, and ns.3. Results and discussion
3.1 Preparation and characterization of ICG-Cur/MPN (Fe3/TA) NPs
Carrier-free ICG-Cur NPs were fabricated by simply mixing the Cur/DMSO solution and ICG/water solution under ultrasonic stirring and following aging process on a shaker. The ICG molecules and Cur molecules co-assemble into NPs via π–π stacking, hydrophobic interaction and hydrogen-bonding interaction.32 After a purification process to remove the organic solvent, the prepared ICG-Cur NPs could be directly redispersed in water without any coating. Although lacking a surface coating, these NPs exhibited high stability, as evidenced by a slight increase in hydrodynamic particle size increase from 87.7 ± 5.3 nm (PDI = 0.263) to 93.0 nm (PDI = 0.274) at 10 days after preparation. Additionally, the NP aqueous solution remained transparent with no observable precipitates at 4 °C during the 5 months observation period. Subsequently, the surface of NPs was functionalized with the MPN by assembling TA and ferric irons (Fe3+) (Fig. 1A). The encapsulation efficiencies (EE) of Cur and ICG in ICG-Cur NPs were 36.3% and 33.1%, respectively, as determined by the UV-Vis analysis. The loading efficiencies (LE) of Cur and ICG were 54.5 vs. 45.5 ± 2.6% in ICG-Cur NPs and 56.5 vs. 43.5 ± 5.5% in ICG-Cur/MPN NPs, respectively. It indicates that the loading efficiencies of Cur and ICG in NPs remained relatively consistent before and after MPN coating. | ||
| Fig. 1 Characterization of ICG-Cur/MPN NPs. (A) Schematic illustration of the fabrication of ICG-Cur/MPN NPs. (B) TEM images of ICG-Cur NPs (a) and (b), ICG-Cur NPs with the thin MPN coating (c) and (d), and with the thick MPN coating (e) and (f). The inset images show the corresponding NP aqueous suspensions. Scale bars = 100 nm. Data represent mean values ± standard deviation, n = 30. (C) Number size distributions and (D) zeta potentials of ICG-Cur NPs and ICG-Cur/MPN (thin and thick coatings) NPs measured by DLS. Data represent mean values ± standard deviation, n = 3. (E) UV-Vis spectra of ICG-Cur NPs and ICG-Cur/thick MPN NPs. (F) Cumulative Cur release curves of ICG-Cur/thin MPN and ICG-Cur/thick MPN NPs at pH = 7.4 and 5. | ||
The size and morphology of NPs before and after MPN coating were characterized by TEM (Fig. 1B). The ICG-Cur NPs showed a uniform spherical morphology with an average size of 71.2 ± 11.1 nm (Fig. 1B(a) and (b)). After coating with MPNs, TEM images revealed that the thickness of MPN could be controlled. ICG-Cur/thin MPN NPs showed a thin and smooth shell around the drug NP core and increased its average size to 97.6 ± 6.9 nm (Fig. 1B(c) and (d)). ICG-Cur/thick MPN NPs displayed a more visible and thick shell structure with a rough surface morphology and an average size of 98.7 ± 14.4 nm (Fig. 1B(e) and (f)). Additionally, it was observed that the color of NP solutions became darker with increasing thickness of the MPN (inset in Fig. 1B and Fig. S2, ESI†). The MPN modification resulted in the increase of the hydrodynamic size of drug NPs as well. ICG-Cur, ICG-Cur/thin MPN and ICG-Cur/thick MPN possessed Z-average diameters of 87.7 ± 5.3 nm (PDI = 0.341 ± 0.07), 123.9 ± 5.4 nm (PDI = 0.15 ± 0.013) and 139.8 ± 1.65 nm (PDI = 0.139 ± 0.02), respectively. Besides, the number-average hydrodynamic sizes of ICG-Cur, ICG-Cur/thin MPN and ICG-Cur/thick MPN were measured to be 83.2 ± 2.7 nm, 92.6 ± 2.7 nm and 93.1 ± 4.6 nm (Fig. 1C), which are closer to their TEM measured sizes (Fig. S3, ESI†).33,34 The ICG-Cur NPs displayed a negative surface zeta potential of −32.8 ± 0.9 mV. The MPN coating induced NPs to shift to more negative charges of −39.9 ± 1.5 mV for thin coating and −45.7 ± 1.0 mV for thick coating (Fig. 1D). The UV-Vis absorption spectra of both ICG-Cur and ICG-Cur/thick MPN NPs showed strong curcumin absorbance bands at 425 nm and ICG absorbance bands at 814 nm (Fig. 1E). While MPN-coated NPs had increased absorption at around 565 nm, the characteristic band of the forming of TA-Fe3+ networks (Fig. S4, ESI†).
To investigate the release profiles of Cur and ICG from NPs, the uncoated, thin MPN coated and thick MPN coated ICG-Cur NPs were placed in dialysis containers with buffers at different pH values, in which 7.4 is the pH of the blood circulation environment, and 5.0 is the pH of the intracellular endo/lysosomal environment. It can be observed in Fig. 1F and Fig. S5A (ESI†), at pH 7.4, Cur exhibited a gradual release profile, stabilizing after 45 h. However, at pH 5.0, the release of Cur dramatically increased after 45 h for both types of MPN-coated NPs. The enhanced release at pH 5.0 is likely due to the disassembly of the Fe3+/TA complex (MPN coating) at an acidic condition.15 At pH 7.4, Fe3+ and TA molecules can form a stable tris-complex. While at pH < 6, most of the phenolic hydroxyl groups on TA are protonated, thereby accelerating the destabilization and disassembly of networks. Attractively, we found that the release rate of Cur from thick MPN coated NPs was faster than from both the ICG-Cur NPs and thin MPN coated NPs (Fig. 1F, Fig. S5A and B, ESI†). At the time of 150 h, 91.6% of Cur was released from ICG-Cur/thick MPN NPs at pH 5.0, compared to only 73.3% of and 47.2% from ICG-Cur/thin MPN and ICG-Cur NPs. One hypothesis for this observation is that the initially disassembled Fe3+ from the MPN coating may attract Cur molecules from the drug NP structure because of their affinity with Cur, thereby accelerating the structure disintegration of carrier-free NPs and increasing the drug release.35–37 However, the release profiles of ICG from NPs were low at both pH 7.4 and pH 5.0 (Fig. S5C and D, ESI†). These findings may suggest amphiphilic ICG molecules might maintain the assembled structure during the release of Cur. Consequently, ICG-Cur/MPN NPs are promising self-delivery carriers for intracellular drug delivery as the MPN coating can delay the drug release at physiological pH and readily degrade at intracellular acidic pH for exposing the inner therapeutic drugs.
3.2 Intracellular uptake and endo/lysosomal escape of ICG-Cur/MPN NPs
Cur has intrinsic fluorescent emission at around 520 nm. Thus, the intracellular uptake process of ICG-Cur/MPN NPs could be tracked by visualizing the green fluorescence of Cur. To compare the endo/lysosomal escape abilities, ICG-Cur NPs, ICG-Cur/thin MPN and ICG-Cur/thick MPN NPs were incubated with breast cancer cell lines for 4 h, 8 h, 12 h and 24 h, and observed using confocal microscopy. The endo/lysosomes were stained with LysoTracker red to exhibit red fluorescence.The total endocytic uptake of nanoparticles typically lasts for 2–6 h, and the risk of the degradation of drug NPs becomes higher with the longer time they are trapped in lysosomes.38 As shown in the images (Fig. 2A), all three types of NPs show fluorescence at sites of endo/lysosomes in colocalization inspections (yellow points represent overlapped green and red signals in the merged images) at 4 h after incubation with MCF-7 cells, indicating that they are internalized by the clathrin-mediated endocytosis pathway. However, most ICG-Cur/thick MPN NPs had already escaped from lysosomes after only 4 h of incubation and spread into the cytoplasm. ICG-Cur/thin MPN NPs were observed to escape from lysosomes after 8 h of incubation, and ICG-Cur NPs began to escape from lysosomes after 12 h of incubation. The faster endo/lysosome escape capability of the thick MPN-coated NPs compared to that of the original ICG-Cur NPs and thin MPN-coated NPs can be more clearly seen in the zoom-in images (Fig. 2B).
 | ||
| Fig. 2 Endo/lysosomal escape of NPs in MCF-7 cell line. (A) CLSM images of MCF-7 cells incubated with ICG-Cur NPs, ICG-Cur/thin MPN and ICG-Cur/thick MPN NPs at 4 h, 8 h, and 12 h. Nuclei (blue fluorescence) were stained by Hoechst 33342, and lysosomes (red fluorescence) were stained by LysoTracker red. Green fluorescence is from Cur in NPs. Scale bars = 30 μm. (B) Representative zoom-in images of merged CLSM images in figure (A), including ICG-Cur NPs, ICG-Cur/thin MPN NPs and ICG-Cur/thick MPN NPs treated MCF-7 cells at various times. Scale bar = 30 μm. (C) Calculated Pearson's correlation coefficient (PCC) values of corresponding NPs vs. endo/lysosomes. Data represent mean values ± standard deviation (n > 50). (*p < 0.05; **p < 0.01; ns: no statistical significance). | ||
To quantify the colocalization of NPs and endo/lysosomes, PCC values between green and red fluorescence signals in images were calculated (Fig. 2C). For all three NPs, the PCC values were decreased over time from 4 h to 12 h, suggesting NPs gradually exited from endo/lysosomes. At 4 h and 8 h, ICG-Cur and CG-Cur/thin MPN NPs showed high PCC values above 0.5, indicating that NPs have high degrees of colocalization with endo/lysosomes, although thin MPN coating slightly improved the endo/lysosome escape ability of ICG-Cur NPs. In contrast, even at 4 h, the PCC value of ICG-Cur/thick MPN NPs was already lower than 0.5, indicating their low degree of colocalization with lysosomes. However, as the incubation time increased to 12 h, the thin MPN coating significantly enhanced the capacity of NPs to transport out of lysosomes, with a PCC value of only 0.19, which is even lower than that of the thick MPN coating (PCC = 0.29). This indicates that the thin MPN facilitates the escape of most drug NPs from lysosomes after 12 h co-incubation. However, it does not necessarily confirm that a higher amount of thin MPN NPs escaped than thick MPN NPs at 12 h. This discrepancy can be attributed to the nature of LysoTracker Red, a pH-dependent dye characterized with weak base property, which specially accumulates in acidic late endosomes and lysosomes. The low pH in these organelles allows LysoTracker becomes protonated and generates fluorescence signal.39 But when the membrane of lysosomes was damaged, as is the case with the mechanism of MPN, the lysosomal proton gradient is gradually lost as the acidic compartments were released out, resulting in the missing label by LysoTracker red at a certain point.40–42 Therefore, in the thick MPN groups, only intact lysosomes that still contain NPs are tracked, leading to higher PCC value. This interpretation is supported by the observation at 24 h (Fig. S6, ESI†), where an increased spread of Cur molecules (green signals) into the cytoplasm was evident for all three types of NPs. Despite this, the PCC values of all groups rose above 0.5, and obviously lower number of lysosomes were tracked in fluorescent images, particularly in the thick MPN NPs group. Therefore, we emphasized the initial escape time point, where PCC values are below 0.5, to compare the endo/lysosomal escape efficacy of the NPs.
Remarkably, the accelerated endo/lysosomal escape of thick MPN-coated NPs was also observed in the MDA-MB-231 cell line (Fig. S7, ESI†). ICG-Cur/thick MPN NPs showed more distinct green and red fluorescence signals than ICG-Cur NPs in merge images after 4 h and 8 h of incubations. In conclusion, the MPN coating facilitates the fast endo/lysosome escape of carrier-free ICG-Cur drug NPs in different cell lines, and this facilitation effect is magnified with increased thickness of MPN coating.
3.3 In vitro chemotherapy of ICG-Cur/MPN NPs
To assess the anticancer ability of carrier-free ICG-Cur NPs with or without the MPN coating, the cytotoxicity of NPs was initially measured using a cell survival assay. The preceding section has demonstrated that the ICG-Cur/thick MPN NPs showed faster endo/lysosomal escape than ICG-Cur/thin MPN NPs. Subsequently, the cell survival assay confirmed that ICG-Cur/thick MPN NPs also induced more cell death than ICG-Cur/thin MPN NPs (Fig. S8, ESI†). To ascertain whether the higher cytotoxicity is attributable to the increased ferric iron content in the thick MPN coating, we evaluated the cytotoxicity of MPN coating itself (Fig. S9, ESI,† 48 h no laser). The MPN (Fe3+ + TA) showed no obviously toxicity to the cancer cells, even at the highest concentration tested (cell viability above 90%). This concentration is equivalent to the MPN content in ICG-Cur/thick MPN NPs at the Cur concentration of 30 μg m−1 (Table S1, ESI†). Thus, the enhanced chemotherapeutic effect of drug NPs can be primarily own to the fast endo/lysosomal escape facilitated by the thick MPN coating. Consequently, the thick MPN coated NP group was selected for the following therapeutic experiments.To compare the cytotoxicity of ICG-Cur NPs and ICG-Cur/MPN NPs, cells were incubated with NPs and free Cur at different Cur concentrations ranging from 5 to 30 μg mL−1 for 24 h and 48 h. As presented in Fig. 3A and B, the cell viability of the MCF-7 cells treated with two NP formulations and free Cur followed does-dependent and time-dependent pattern. Free Cur began to exhibit toxicity against MCF-7 cells at the concentration of 10 μg mL−1. Additionally, it was noted that free Cur possessed higher cytotoxicity than NP formulations in this 2D cell test because the free drugs directly interact with cells through passive diffusion, whereas NP formulations require a drug release process inside the cells.28 When comparing the two NP formulations, ICG-Cur NPs showed negligible toxicity to cancer cells at low concentrations (5 and 10 μg mL−1) but started to show toxicity when concentrations increased to 30 μg mL−1. In contrast, after coating with MPN, the cytotoxicity of NPs was significantly enhanced since the MPN coating had been proven to facilitate the endo/lysosomal escape of NPs in the previous section.
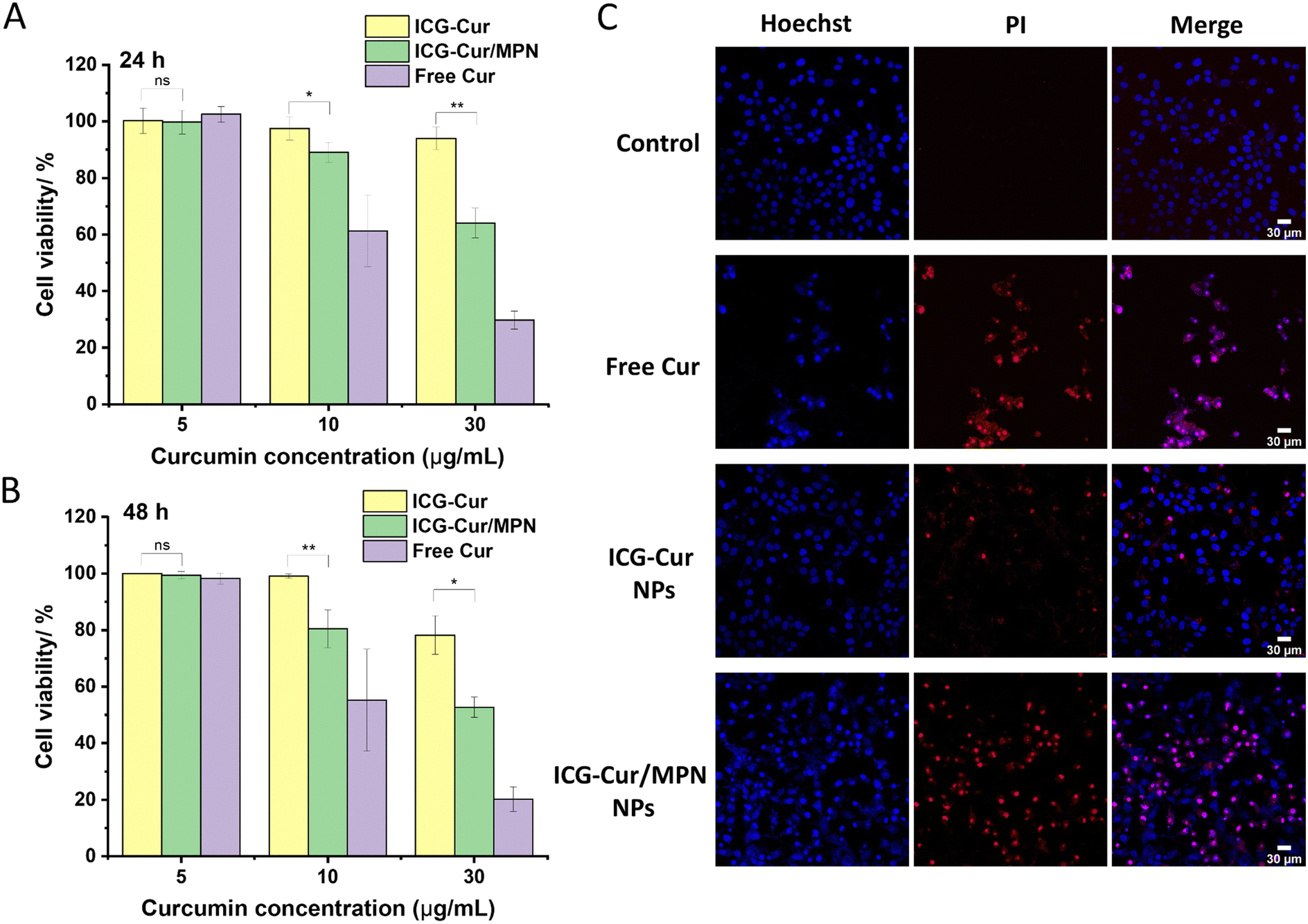 | ||
| Fig. 3 Cell viabilities of MCF-7 cells after being incubated with ICG-Cur NPs, ICG-Cur/thick MPN NPs and free Cur molecules for (A) 24 h and (B) 48 h. Data represent the mean value of measurements ± standard deviation. (*p < 0.05; **p < 0.01; ns: no statistical significance.) (C) MCF-7 cell death induced by free Cur, ICG-Cur NPs, and ICG-Cur/thick MPN NPs. Hoechst 33342 (blue fluorescence) and PI (red fluorescence) were used for labelling live and apoptosis/necrosis cells. Scale bars = 30 μm. | ||
To further confirm the effect of MPN coating on promoting the anticancer ability of ICG-Cur NPs, cell apoptosis/necrosis after co-incubation with NPs was assessed by co-staining with Hoechst 33342 and PI. The Hoechst 33342 stains all living and dead cells, but early apoptotic cells will exhibit brighter blue and condensed chromatin after staining. PI staining indicates late apoptotic or necrotic cells.43 In the experiment, MCF-7 cells were treated with drug NPs and free Cur at a Cur concentration of 30 μg mL−1. As displayed in Fig. 3C, ICG-Cur/MPN NPs dramatically increased the cytotoxicity of ICG-Cur NPs to MCF-7 cells and induced more cells to undergo apoptosis (red PI signals). Moreover, it can be observed that only in free Cur and ICG-Cur/MPN NPs treated cells, the blue fluorescent signals leaked out from the cell nuclei, suggesting the breakage of cell nuclei caused by these two treatments. Overall, both the cell survival assay and Hoechst/PI staining method proved that the thick MPN coating could highly enhance the anticancer effect of carrier-free ICG-Cur NPs in the in vitro 2D cell culture condition.
3.4 In vitro phototherapy of ICG-Cur/MPN NPs
ICG can be stimulated by an 808 nm NIR laser to simultaneously conduct photothermal therapy (PTT) and photodynamic therapy (PDT) for killing cancer cells.26,27 Here we first assessed the photothermal conversion efficiency of ICG-Cur/MPN NPs in vitro. As shown in Fig. 4A, the NP structure notably enhanced the photothermal conversion of ICG compared to its free molecular form. Subsequently, we observed that the temperature elevation efficiencies of ICG-based NPs were proportional to both the duration of the laser irradiation and the concentration of ICG. At an ICG concentration of 20 μg mL−1, the highest temperatures reached by ICG-Cur NPs and ICG-Cur/MPN NPs were 44.4 °C and 48.0 °C, respectively, after 6 min NIR irradiation (Fig. 4B). With an increase in ICG concentration to 40 μg mL−1, the maximum temperatures achieved by ICG-Cur NPs and ICG-Cur/MPN NPs were 60.5 °C and 66.3 °C, respectively, after 5 min NIR irradiation (Fig. 4C). Although ICG-Cur NPs and ICG-Cur/MPN NPs exhibited similar temperature rise trends before 4.5 min for the 20 μg mL−1 groups and before 1.5 min for the 40 μg mL−1 groups, the MPN coating enhanced the maximum temperature of ICG drug NPs under laser irradiation. This enhancement is attributed to the tannic acid-based MPN coating itself possessing superior light-to-heat conversion ability under 808 nm laser irradiation.44,45 Based on the photothermal conversion efficiency (η) calculation method and the data obtained from Fig. S10 (ESI†), the 808 nm laser η of ICG-Cur/thick MPN NPs was determined to be 37.7% which is approximately 55.8% higher than that of ICG-Cur NPs (24.2%). These results suggest that the synergistic photothermal effects between ICG and MPN significantly boost the PTT efficiency of ICG-Cur/MPN NPs. | ||
| Fig. 4 (A) IR thermal images of free ICG + Cur molecules, ICG-Cur NPs, and ICG-Cur/MPN NPs under 808 nm laser at 1.5 W cm−2. The temperature increasing curves of different samples exposed to 808 nm laser at the ICG concentration of (B) 20 μg mL−1, and (C) 40 μg mL−1. (D) Fluorescent images of ROS generation (green fluorescence) of different samples in MCF-7 and MDA-MB-231 tumor cells, in the absence and presence of 1.0 W cm−2 808 nm laser irradiation. Scale bars = 100 μm. | ||
To evaluate the phototherapy potential of MPN coating itself, a cell survival assay was conducted using only the MPN (Fe3+ + TA). The tested concentrations of MPN correspond to the concentrations of Fe3+ and TA in ICG-Cur/MPN NPs when their drug (Cur) concentrations vary from 2 to 30 μg mL−1 (Table S1, ESI†). As shown in Fig. S9 (ESI†), when the cells were totally cultured with MPN for 24 h after irradiating with 808 nm laser for 3 min at the 8 h post-coculture, MPN began to show slight toxicity at the Fe3+ concentration at 0.6 μg mL−1 (89% viability), and the cytotoxicity gradually enhanced with the increase of the of Fe3+ concentrations. At the concentration of Fe3+ of 11.7 μg mL−1 (= the MPN mass in ICG-Cur/thick MPN NPs at Cur concentration of 30 μg mL−1), cancer cell viability decreased to 57.6%. However, since MPN is not toxicity without the laser irradiation, the remaining viable cells continued to proliferate, resulting in the recovery of cell viability after an additional 24 h co-culture (48 h plus laser). Thus, even without ICG, MPN alone can serve as an effective phototherapy agent for killing cancer cells. ROS generation is a fundamental parameter for estimating PDT efficiency. Next, we evaluated the intracellular ROS generation efficiency of various NPs following laser treatment using DCFH-DA as a probe, which is oxidized into green fluorescent dichlorofluorescein (DCF) by ROS. As illustrated in fluorescent images (Fig. 4D and Fig. S11, ESI†) and quantitative analysis results (Fig. S12, ESI†), laser treatment activated the ROS generation of ICG in all conditions (free ICG, ICG-Cur NPs and ICG-Cur/MPN NPs). However, both MCF-7 and MDA-MB-231 cells treated with ICG-Cur/MPN NPs plus a laser displayed significantly stronger green fluorescence compared to free ICG and ICG-Cur NPs plus laser groups. These results signify that the MPN coating has the potential to enhance the PDT efficiency of original drug NPs. Moreover, it is noteworthy that even in the absence of laser irradiation, the MPN-coated NPs presented higher ROS generation in both cells than non-MPN-coated NPs. This phenomenon may be attributed to two reasons. Firstly, the anticancer mechanism of Cur, which is known to stimulate redox reactions in cancer cells via mitochondria.46 Thus, it appears MPN coating could enhance this mechanism by promoting the Cur release from NPs, as discussed in the previous section. Secondly, a portion of Fe3+ released from MPN may be reduced to Fe2+ within cells, then Fe2+ can contribution to the generation of ROS through Fenton reaction.47
3.5 Chemo-phototherapeutic evaluation in the 3D tumor model
The multicellular spheroid is a type of 3D cell culture model that closely resembled the structure and properties of a solid tumor. In this study, media containing free Cur + ICG molecules, ICG-Cur NPs, and ICG-Cur/MPN NPs were prepared to incubate with MCF-7 cell spheroids, aiming to investigate their anticancer abilities for solid tumors both with or without laser treatment. The anticancer effect was also assessed by Hoechst 33342/PI co-staining. As shown in Fig. S13 (ESI†), different from the results observed in 2D cell cultures, the chemotherapeutic effectiveness of ICG-Cur/MPN NPs (at both 5 μg mL−1 and 10 μg mL−1 of Cur) and ICG-Cur NPs (at 10 μg mL−1 of Cur) was certainly better than that of free Cur molecules after 6 days of co-incubation. The diminished chemotherapeutic efficacy observed in the free Cur groups within the 3D tumor model is likely attributable to the constrained penetration of free drug molecules within solid tumors. Quantitative analysis of results without laser treatment (Fig. 5A and B) revealed that MPN-coated NPs induced more cell deaths in tumor spheroids at relatively deeper intervals of 90 μm and 120 μm compared to free Cur and original ICG-Cur NPs. These findings suggest MPN-coated NPs possess enhanced permeation within tumor tissue, effectively targeting and eliminating cancer cells in the deeper regions. | ||
| Fig. 5 Cytotoxicity of free ICG + Cur, ICG-Cur NPs, and ICG-Cur/MPN NPs (the concentration of Cur is 5 μg mL−1) for 3D MCF-7 breast cancer spheroids, in the absence and presence of 808 nm laser irradiation at the power density at of 1.5 W cm−1 for 3 min. (A) Cell spheroids were co-stained with Hoechst 33342 (blue fluorescence) and PI (red fluorescence) to indicate live and apoptosis/necrosis cells, respectively. Fluorescence images of different transverse sections (depth increases with 15 μm) and reconstructed 3D stacks of PI channel of cell spheroids (0–150 μm). Control groups were spheroids cultured in DMEM medium without NPs application. Scale bar, 200 μm. (B) The mean fluorescence intensity of PI in tumor spheroids in (A) and Fig. S14 (ESI†) at 60 μm, 90 μm, and 120 μm depths of tumor spheriods. | ||
However, as observed from Fig. 5A and B, although MPN coating improved the therapeutic effect of free Cur, the chemotherapy strategy only induced cell apoptosis in the surface area of tumor spheroids. At the deeper area (120 μm), the PI intensities (indicating dead cells) of the three treatment groups were close to the control group. The significant enhancements in therapeutic efficacy of all free Cur + ICG, ICG-Cur NP, and ICG-Cur/MPN NP groups were only observed upon laser treatment, evidenced by substantially stronger red PI fluorescence in both surface and deeper areas of 3D tumor models. Hence, the combination of phototherapy with chemotherapy emerges as crucial for achieving highly efficient tumor inhibition. Under laser irradiation, ICG-Cur/MPN NPs still showed the best anticancer performance compared to the other groups across all depths of spheroids (60 μm, 90 μm, and 120 μm; Fig. 5B). Conversely, ICG-Cur NPs plus laser showed markedly weaker efficiency in killing cancer cells. In conclusion, these findings underscore the pivotal role of MPN coating in improving the chemo- and photo-anticancer effects of carrier-free drug NPs.
4. Conclusions
In this study, a type of carrier-free NP was designed to integrate a chemotherapeutic agent (Cur) and a phototherapeutic agent (ICG) for the synergistic treatment of tumors. Leveraging the complementary physical and chemical properties of Cur and ICG, these two therapeutic molecules were co-assembled into stable, water-soluble NPs with uniform size and morphology. This preparation strategy holds particular promise for developing carrier-free nanomedicines from highly hydrophobic drugs such as Cur. Subsequently, we successfully decorated ICG-Cur NPs with the MPN network of different thicknesses to maximize the delivery of drugs into the cytosol from the clathrin-mediated endocytotic internalization pathway. Our results revealed that following cellular uptake, the ICG-Cur NPs and ICG-Cur/thin MPN NPs remained sequestered within endo/lysosomes for up to 12 hours and 8 hours, respectively. In contrast, thick MPN-coated NPs exhibited rapid escape from endo/lysosomal within 4 h, and the acidic lysosomal environment led to the disassembly of MPN for the controlled release of drug Cur. Therefore, achieving endo/lysosomal escape ability may necessitate a certain thickness of MPN. Additionally, we observed that the thick MPN accelerated the Cur release which might be due to the Fe3+-Cur chelation effect. Some studies have reported that the iron chelation would interfere the anticancer activity of Cur molecules,48,49 but this influence appears to be mitigated by the high loading efficiency of Cur in carrier-free NPs. Consequently, the thick MPN coating remarkably promoted the chemotherapy efficiency of ICG-Cur NPs in monolayer cell experiments.However, when assessing the therapeutic effects of NPs in 3D tumor spheroid models, chemotherapy alone displayed limited toxicity towards cancer cells, especially those residing in deeper tumor regions. The substantial anticancer effect was only observed with ICG-Cur/thick MPN NPs combined with laser treatment. These findings indicate the importance of multimodal therapy for tumors and affirm that the MPN coating can significantly amplify the chemotherapeutic effect of Cur and the phototherapeutic effect of ICG.
In conclusion, our study demonstrated that that MPN exerts multiply effects on the therapeutic performance of carrier-free ICG-Cur NPs, including influencing the drug release profile, facilitating endo/lysosomal escape, promoting ROS generation, and enhancing photothermal conversion efficiency.
Author contributions
XF: conceptualization, data curation, formal analysis, investigation, methodology, and writing – original draft; CMB, HW, SK, SR: formal analysis, methodology, and writing – review and editing; LY, TM, PJWH, WZ: formal analysis, supervision, and writing – review and editing; and XC: conceptualization, supervision, funding acquisition, resources, writing – review and editing, and project administration.Data availability
The data underlying this study are available in the published article and its Supporting Information.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Stephen Mitchell and David Kelly at the School of Biological Sciences Electron Microscopy unit for assistance with TEM. The authors acknowledge the support of the Wellcome Multi User Equipment Grant (WT104915MA).References
- Z. Jing, Q. Du, X. Zhang and Y. Zhang, Chem. Eng. J., 2022, 446, 137147 CrossRef CAS.
- Q. Cui, J. Q. Wang, Y. G. Assaraf, L. Ren, P. Gupta, L. Wei, C. R. Ashby, D. H. Yang and Z. S. Chen, Drug Resist. Updates, 2018, 41, 1–25 CrossRef.
- J. Zhang and P. X. Ma, Angew. Chem., Int. Ed., 2009, 48, 964–968 Search PubMed.
- X. Nan, X. Zhang, Y. Liu, M. Zhou, X. Chen and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 9986–9995 CrossRef CAS.
- J. Zhang, Y. C. Liang, X. Lin, X. Zhu, L. Yan, S. Li, X. Yang, G. Zhu, A. L. Rogach, P. K. N. Yu, P. Shi, L. C. Tu, C. C. Chang, X. Zhang, X. Chen, W. Zhang and C. S. Lee, ACS Nano, 2015, 9, 9741–9756 CrossRef CAS.
- N. Wang, Z. Deng, Q. Zhu, J. Zhao, K. Xie, P. Shi, Z. Wang, X. Chen, F. Wang, J. Shi and G. Zhu, Chem. Sci., 2021, 12, 14353–14362 RSC.
- M. He, Y. Song, W. Xu, X. Zhang and C. M. Dong, Adv. Funct. Mater., 2023, 33, 2304216 CrossRef CAS.
- Y. Ding, C. Du, J. Qian and C. M. Dong, Polym. Chem., 2019, 10, 4825–4836 RSC.
- H. Lv, Y. Zhu, J. Xue, X. Jia and J. Chen, Langmuir, 2022, 38, 15766–15775 CrossRef CAS.
- D. Yu, P. Peng, S. S. Dharap, Y. Wang, M. Mehlig, P. Chandna, H. Zhao, D. Filpula, K. Yang, V. Borowski, G. Borchard, Z. Zhang and T. Minko, J. Controlled Release, 2005, 110, 90–102 CrossRef CAS.
- V. T. Cong, J. L. Houng, M. Kavallaris, X. Chen, R. D. Tilley and J. J. Gooding, Chem. Soc. Rev., 2022, 51, 7531–7559 RSC.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, Bioconjugate Chem., 2019, 30, 263–272 CrossRef CAS.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS.
- E. S. Lee, K. Na and Y. H. Bae, J. Controlled Release, 2005, 103, 405–418 CrossRef CAS.
- H. Ejima, J. J. Richardson, K. Liang, J. P. Best, M. P. Van Koeverden, G. K. Such, J. Cui and F. Caruso, Science, 2013, 341, 154–157 CrossRef CAS.
- J. Chen, J. Li, J. Zhou, Z. Lin, F. Cavalieri, E. Czuba-Wojnilowicz, Y. Hu, A. Glab, Y. Ju, J. J. Richardson and F. Caruso, ACS Nano, 2019, 13, 11653–11664 CrossRef CAS.
- Y. Liu, X. Zhang, M. Zhou, X. Nan, X. Chen and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 43498–43507 CrossRef CAS.
- S. Karaosmanoglu, Y. Zhang, W. Zhou, D. Ouyang and X. Chen, Bioengineering, 2022, 9, 815 CrossRef CAS.
- S. Karaosmanoglu, M. Zhou, B. Shi, X. Zhang, G. R. Williams and X. Chen, J. Controlled Release, 2021, 329, 805–832 CrossRef CAS.
- M. Zhou, W. Wei, X. Chen, X. Xu, X. Zhang and X. Zhang, Nanomedicine, 2019, 20, 102008 CrossRef CAS.
- M. Zhou, X. Zhang, X. Xu, X. Chen and X. Zhang, ACS Appl. Bio Mater., 2018, 1, 289–297 CrossRef CAS.
- C. Yu, M. Zhou, X. Zhang, W. Wei, X. Chen and X. Zhang, Nanoscale, 2015, 7, 5683–5690 RSC.
- V. Zoi, V. Galani, G. D. Lianos, S. Voulgaris, A. P. Kyritsis and G. A. Alexiou, Biomedicines, 2021, 9, 1086 CrossRef CAS.
- Y. M. Attia, D. M. El-Kersh, R. A. Ammar, A. Adel, A. Khalil, H. Walid, K. Eskander, M. Hamdy, N. Reda, N. E. Mohsen, G. M. Al-Toukhy, M. T. Mansour and M. M. Elmazar, Chem. – Biol. Interact., 2020, 315, 108865 CrossRef CAS.
- L. Yang, D. Li, P. Tang and Y. Zuo, Oncol. Lett., 2020, 19, 83–92 CAS.
- H. Wang, X. Li, B. W. C. Tse, H. Yang, C. A. Thorling, Y. Liu, M. Touraud, J. B. Chouane, X. Liu, M. S. Roberts and X. Liang, Theranostics, 2018, 8, 1227 CrossRef CAS.
- W. Li, H. Zhang, X. Guo, Z. Wang, F. Kong, L. Luo, Q. Li, C. Zhu, J. Yang, Y. Lou, Y. Du and J. You, ACS Appl. Mater. Interfaces, 2017, 9, 3354–3367 CrossRef CAS.
- W. Wei, X. Zhang, X. Chen, M. Zhou, R. Xu and X. Zhang, Nanoscale, 2016, 8, 8118–8125 RSC.
- H. Y. Zhang, C. Yong Sun, M. Adu-Frimpong, J. Nan Yu and X. Ming Xu, Int. J. Pharm., 2019, 555, 270–279 CrossRef CAS.
- J. Chen, J. Li, J. Zhou, Z. Lin, F. Cavalieri, E. Czuba-Wojnilowicz, Y. Hu, A. Glab, Y. Ju, J. J. Richardson and F. Caruso, ACS Nano, 2019, 13, 11653–11664 CrossRef CAS.
- S. A. Chechetka, Y. Yu, X. Zhen, M. Pramanik, K. Pu and E. Miyako, Nat. Commun., 2017, 8, 1–19 CrossRef.
- H. A. Fargher, T. J. Sherbow, M. M. Haley, D. W. Johnson and M. D. Pluth, Chem. Soc. Rev., 2022, 51, 1454–1469 RSC.
- S. Starikova, C. Jones, W. R. Forman, M. James Jee, J. Anthony Tyson, M. D. Schneider, T. G. F. Souza, V. S. T. Ciminelli and N. D. S. Mohallem, J. Phys.: Conf. Ser., 2016, 733, 012039 CrossRef.
- P. Eaton, P. Quaresma, C. Soares, C. Neves, M. P. de Almeida, E. Pereira and P. West, Ultramicroscopy, 2017, 182, 179–190 CrossRef CAS.
- F. Mohammed, F. Rashid-Doubell, S. Cassidy and F. Henari, Spectrochim. Acta, Part A, 2017, 183, 439–450 CrossRef CAS.
- A. Beneduci, G. A. Corrente, T. Marino, D. Aiello, L. Bartella, L. Di Donna, A. Napoli, N. Russo, I. Romeo and E. Furia, J. Mol. Liq., 2019, 296, 111805 CrossRef CAS.
- Y. Jiao, J. Wilkinson IV, E. Christine Pietsch, J. L. Buss, W. Wang, R. Planalp, F. M. Torti and S. V. Torti, Free Radical Biol. Med., 2006, 40, 1152–1160 CrossRef CAS.
- J. Gilleron, W. Querbes, A. Zeigerer, A. Borodovsky, G. Marsico, U. Schubert, K. Manygoats, S. Seifert, C. Andree, M. Stöter, H. Epstein-Barash, L. Zhang, V. Koteliansky, K. Fitzgerald, E. Fava, M. Bickle, Y. Kalaidzidis, A. Akinc, M. Maier and M. Zerial, Nat. Biotechnol., 2013, 31, 638–646 CrossRef CAS.
- B. Zhitomirsky, H. Farber and Y. G. Assaraf, J. Cell. Mol. Med., 2018, 22, 2131–2141 CrossRef CAS.
- I. Eriksson, L. Vainikka, H. L. Persson and K. Öllinger, Methods Protoc., 2023, 6, 72 CrossRef CAS.
- I. Maejima, A. Takahashi, H. Omori, T. Kimura, Y. Takabatake, T. Saitoh, A. Yamamoto, M. Hamasaki, T. Noda, Y. Isaka and T. Yoshimori, EMBO J., 2013, 32, 2336–2347 CrossRef CAS.
- F. Wang, M. G. Bexiga, S. Anguissola, P. Boya, J. C. Simpson, A. Salvati and K. A. Dawson, Nanoscale, 2013, 5, 10868–10876 RSC.
- K. J. Kelly, R. M. Sandoval, K. W. Dunn, B. A. Molitoris and P. C. Dagher, Am. J. Physiol.: Cell Physiol., 2003, 284, C1309–C1318 CrossRef CAS.
- C. Zhang, L. Huang, D. W. Sun and H. Pu, J. Hazard. Mater., 2022, 426, 127824 CrossRef CAS.
- Q. Shi, K. Wu, X. Huang, R. Xu, W. Zhang, J. Bai, S. Du and N. Han, Colloids Surf., A, 2021, 618, 126475 CrossRef CAS.
- C. Yu, B. Yang and M. Najafi, Basic Clin. Pharmacol. Toxicol., 2021, 129, 397–415 CrossRef CAS.
- T. Nakamura, I. Naguro and H. Ichijo, Biochim. Biophys. Acta, Gen. Subj., 2019, 1863, 1398–1409 CrossRef CAS.
- K. Nakano, T. Nakayachi, E. Yasumoto, S. R. Morshed, K. Hashimoto, H. Kikuchi, H. Nishikawa, K. Sugiyama, O. Amano, M. Kawase and H. Sakagami, Anticancer Res., 2004, 24, 711–718 CAS.
- N. E. Rainey, A. Moustapha, A. Saric, G. Nicolas, F. Sureau and P. X. Petit, Cell Death Discovery, 2019, 5, 1–15 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00465e |
| This journal is © The Royal Society of Chemistry 2024 |