
Rationally designed protein A surface molecularly imprinted magnetic nanoparticles for the capture and detection of Staphylococcus aureus†
Kritika
Narula
a,
Soumya
Rajpal
a,
Snehasis
Bhakta
 b,
Senthilguru
Kulanthaivel
a and
Prashant
Mishra
*a
b,
Senthilguru
Kulanthaivel
a and
Prashant
Mishra
*a
aDepartment of Biochemical Engineering and Biotechnology, Indian Institute of Technology Delhi, New Delhi, India. E-mail: pmishra@dbeb.iitd.ac.in
bDepartment of Chemistry, Cooch Behar College, West Bengal, India
First published on 4th May 2024
Abstract
Staphylococcus aureus (S. aureus), a commensal organism found on the human skin, is commonly associated with nosocomial infections and exhibits virulence mediated by toxins and resistance to antibiotics. The global threat of antibiotic resistance has necessitated antimicrobial stewardship to improve the safe and appropriate use of antimicrobials; hence, there is an urgent demand for the advanced, cost-effective, and rapid detection of specific bacteria. In this regard, we aimed to selectively detect S. aureus using surface molecularly imprinted magnetic nanoparticles templated with a well-known biomarker protein A, specific to S. aureus. Herein, a highly selective surface molecularly imprinted polymeric thin layer was created on ∼250 nm magnetic nanoparticles (MNPs) through the immobilization of protein A to aldehyde functionalized MNPs, followed by monomer polymerization and template washing. This study employs the rational selection of monomers based on their computationally predicted binding affinity to protein A at multiple surface residues. The resulting MIPs from rationally selected monomer combinations demonstrated an imprinting factor as high as ∼5. Selectivity studies revealed MIPs with four-fold higher binding capacity (BC) to protein A than other non-target proteins, such as lysozyme and serum albumin. In addition, it showed significant binding to S. aureus, whereas negligible binding to other non-specific Gram-negative, i.e. Escherichia coli (E. coli), Pseudomonas aeruginosa (P. aeruginosa), and Gram-positive, i.e. Bacillus subtilis (B. subtilis), bacteria. This MIP was employed for the capture and specific detection of fluorescently labeled S. aureus. Quantitative detection was performed using a conventional plate counting technique in a linear detection range of 101–107 bacterial cells. Remarkably, the MIPs also exhibited approximately 100% cell recovery from milk samples spiked with S. aureus (106 CFU mL−1), underscoring its potential as a robust tool for sensitive and accurate bacterial detection in dairy products. The developed MIP exhibiting high affinity and selective binding to protein A finds its potential applications in the magnetic capture and selective detection of protein A as well as S. aureus infections and contaminations.
1. Introduction
S. aureus is a commensal bacteria that colonises the human skin and is known to cause infections of the skin, pneumonia, bacteremia and infective endocarditis.1S. aureus is also a typical food-borne, zoonotic and nosocomial pathogen2 that expresses an array of extracellular toxins and enzymes that help in its virulence.3 Its isolates have shown resistance to antibiotics such as methicillin and, in some cases, carbapenems and vancomycin; hence it is a threat to both human health and animal husbandry.4Current gold standard tests for detecting S. aureus are culture based and rely on enrichment and separation on selective plates, biochemical tests and 16S rRNA sequencing.5,6 Although this approach is simple, it is typically time consuming as the process takes 4–7 days.7 In view of this, various rapid detection methods, such as ELISA,8 PCR,9 loop-mediated isothermal amplification,10 and fluorescence-based methods,11 have been developed to reduce assay time. PCR is a relatively quick method but may give false positives owing to the mispairing of primers and requires expensive equipment.12 Antibody-based tests such as ELISA are rapid but suffer from high cost, and low stability of antibodies. Hence, there is an urgent requirement for a selective, sensitive, cost-effective and stable method for the rapid detection of S. aureus from various sources. Protein A is a characteristic component of the S. aureus cell wall that binds to the Fc component of IgG antibodies and plays a critical role in defence against the host immune system. Owing to its high expression and location on the cell surface, protein A has great potential to be used as a biomarker for the specific recognition of S. aureus.
Molecularly imprinted polymers (MIPs) generally considered artificial antibodies or synthetic receptors have structure predictability, recognition specificity, and application universality.13,14 Unlike antibodies, MIPs are more stable, economical, and easy to synthesize. Earlier, protein A-imprinted polyacrylamide gel beads were synthesized using inverse suspension polymerization and bulk polymerization to detect protein A and S aureus. Bulk polymerization-based MIPs have shown an adsorption capacity of 103–104 colony-forming unit (CFU) g−1 gel beads.15,16
Recently, S. aureus whole cell imprinted polymer and turn on fluorescence probe have been synthesized for the detection of the organism.17,18 Although bulk imprinting of polymers has shown potential for small molecules, it has limited applications for larger molecules, such as proteins. Nonetheless, surface imprinting involves the restriction of a target molecule's binding site only to the surface of a nanomaterial19–23 and hence suitable for binding larger molecules, such as proteins or a whole bacterial cell. In addition, it requires fewer template molecules for imprinting and shows better selectivity, fast absorption rates, and good synthesis reproducibility.24 Further, it is prudent to enhance the translation potential of the MIP technology to the industrial scale, which is limited due to its reproducible nature and comparable high specificity of antibodies. With the intervention of in silico design, it is possible to design MIPs with optimal potential.13,25–27
Computational tools offer a more efficient and cost-effective screening of potential monomers for MIP synthesis. By simulating interactions between monomers and the target protein, we can rationally select candidates based on their binding affinity and specificity, thereby reducing the need for extensive experimental testing. Among various computational tools, molecular docking offers the estimation of monomer binding affinity with a target protein of interest with significant improvement in MIP development because it directly mimics the pre-polymerization reaction mixture.28,29 The arrangement, orientation, and interaction through ionic, hydrophobic, and hydrogen bonding of monomers at multiple (surface) residues of the protein can be predicted to essentially improve the recognition ability of MIPs.
In the present work, based on computationally guided design, protein A-binding surface imprinted magnetic nanoparticles were synthesized for the specific detection of S aureus. A set of protein-compatible monomers commonly employed for molecular imprinting were screened using molecular docking to protein A. Based on the binding energy (kcal mol−1) values and multi-point interaction analysis, high-performing monomers were selected and employed in various combinations to design target-specific imprinted layers. Unlike previous studies that used a single monomer with limited types of molecular interactions with the target, this study involves multiple types of interactions (ionic interactions, hydrogen bonding, van der Waals forces and π–π interactions), which are deployed using a multi-monomer approach, leading to optimal binding ability.30
Core–shell surface imprinting was applied for the first time for S. aureus detection. Herein, protein A was used as a template for the effective construction of an imprinted “shell” on a magnetite nanoparticle (Fe3O4) “core”. The magnetite nanoparticles synthesized were spherical and possessed a diameter of approximately 250 nm. Polymerization was performed on the surface according to the selected monomer composition. Molecular docking revealed dopamine as the best binding monomer in terms of binding energy and silane monomers in terms of improved multi-point interactions. However, the experimental analysis demonstrated silane-based MIPs with a high BC of ∼11 mg g−1 and an imprinting factor of ∼ 5, establishing the importance of interaction with multiple surface residues. In addition, MIP bonded to the S. aureus in a broad linear range of 101–107 CFU mg−1. Furthermore, at least four-fold higher binding of MIP to protein A than non-target proteins was demonstrated. Hence, synthesized MIP nanoparticles can be utilized to capture and detect S. aureus, leading to their potential applications in healthcare and the food industry.31
2. Experimental section
2.1. Chemicals
Tetraethoxysilane (TEOS), (3-aminopropyl) triethoxysilane (APTES), phenyltriethoxysilane (PTES), ureidopropyltrimethoxysilane (UPTMS), ferric chloride hexahydrate (FeCl3·6H2O), NH4OH (25% ammonium hydroxide aqueous solution), glutaraldehyde (25% aqueous solution), polyethylene glycol 2000, ammonium persulfate (APS), dopamine (DA), sodium acetate (NaAc), protein A, human serum albumin (HSA) and lysozyme were purchased from Sigma-Aldrich. Absolute ethanol was procured from Merck. A Quick Start Bradford protein assay kit was obtained from Bio-Rad.2.2. Characterization techniques
The nanoparticle size and morphology were determined using high-resolution transmission electron microscopy (HRTEM) Tecnai G2 20. The magnetic properties of the nanoparticles were investigated using PPMS from Cryogenic Limited, UK. Zeta potential measurements were performed using the Zeta Malvern Zetasizer Nano-ZS90. Surface functional groups were investigated using a Fourier-transform infrared (FTIR) spectrophotometer from Thermo Scientific Nicolet iS50 with ATR mode. UV-visible spectra were obtained using an Eppendorf BioSpectrometer. The X-ray diffraction (XRD) pattern was taken using an X-ray diffractometer from Rigaku Ultima IV, energy-dispersive X-ray (EDAX). Hitachi TM3000 was used for the elemental composition. Fluorescence studies were executed using a Zeiss Axio Observer inverted microscope. Ultrasonication was performed using Branson model 2800. Particles were freeze dried using Allied Frost Lyophilizer-FD-3.2.3. Synthesis of magnetic nanoparticles
Fe3O4 nanoparticles were synthesized using the solvothermal method.32 Briefly, FeCl3·6H2O (1.35 g), NaAc (3.6 g), and polyethylene glycol (1.0 g) were dissolved in ethylene glycol (40 mL). The mixture was stirred for 30 min. It was then sealed in a Teflon-lined stainless-steel autoclave vessel and kept for 16 h at 200 °C. The product was washed three times with ethanol and water. The particles were then freeze-dried under a vacuum.2.4. Surface modification of Fe3O4 particles
Silica coating was done to improve the stability of the magnetic core and facilitate surface functionalization.33,34 In brief, 100 mg of Fe3O4 nanoparticles were dispersed in 20 mL ethanol and 3 mL MiliQ water (resistivity of 18.2 MΩ cm−1). The above solution was treated with 1 mL of 25% ammonia aqueous solution under stirring at 40 °C. Then, 0.2 mL of TEOS was added to the mixture and stirred for 5 h, at room temperature (RT). Silica-coated magnetic nanoparticles (Fe3O4@SiO2) were captured by applying an external magnetic field, which was then washed three times and freeze-dried under a vacuum.2.5. Functionalization of Fe3O4 nanoparticles
Silica-coated magnetic nanoparticles were sequentially functionalized with APTES and glutaraldehyde to generate an aldehyde group on the surface of the nanoparticles for protein immobilization.35 To make a 3.0 mg mL−1 Fe3O4@SiO2 solution, 60 mg nanoparticles were dispersed in 20 mL of 10 mM phosphate buffer (PB), with a pH of 7.3. Then, in a round bottom flask, 22 μL of APTES was added to 20 mL of nanoparticle solution and kept under continuous stirring (400 rpm) for 2 h at RT. This was followed by washing the nanoparticles with MilliQ water. To obtain aldehyde-functionalized nanoparticles, 20 mL of amine-functionalized nanoparticles were resuspended in 1% (v/v) aqueous glutaraldehyde solution and stirred for 20 minutes at RT. These functionalized nanoparticles were washed multiple times and resuspended in PB (pH 7.4).36,372.6. Computational methods
The structural files for all monomers were obtained from the PubChem databank (Fig. S1, ESI†). The crystal structure of the monomer unit of protein A was extracted from PDB ID:1DEE using the UCSF Chimera.38 For molecular docking, Autodock 4.2.6 based on the AMBER force field was employed.39 The monomer files were converted to a pdbqt format after setting the torsional degrees of freedom based on the detected rotatable bonds. Using Autodock tools, polar hydrogens were added to the protein, and water molecules were deleted. A grid box with dimensions of 114 × 76 × 98 Å was centred on the protein. The estimated monomer-binding energy to the protein is represented as kcal mol−1. The mean binding energy of the first rank in the clustering histogram was used to compare the monomers for their high binding affinity. The remaining conformational clusters were employed to visualize all possible binding poses in the root mean square deviation (RMSD) tolerance set at 2 Å. This enables the analysis of favourable multi-point interactions on the surface of the protein consisting of hydrogen bonds, electrostatic interactions, and van der Waals interactions.2.7. Molecularly imprinted polymer (MIP) synthesis
Initially, to immobilize the protein molecules onto the nanoparticle surface, 18 μg protein A was added to 1 mL of 3 mg mL−1 aldehyde-functionalized nanoparticle solution and stirred for 3 hours. Protein A immobilized nanoparticles were resuspended in 1 mL of PB, with pH 7.3 containing different monomer amounts, as presented in Table 1. The polymerization reaction was allowed to take place for 16 h at RT on a 360° rotator, as reported.40,41 Polymerized nanoparticles were washed several times using methanol and acetic acid (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution to remove the bound template42 (Fig. 1). Non-imprinted polymer (NIP) was synthesized following the exact procedure of MIP synthesis, except for the protein A addition step.
:1) solution to remove the bound template42 (Fig. 1). Non-imprinted polymer (NIP) was synthesized following the exact procedure of MIP synthesis, except for the protein A addition step.
 | ||
| Fig. 1 Schematic illustration of the surface imprinting process to generate a ‘core–shell’ type imprinted polymer on magnetic nanoparticles for specific binding to protein A. (1) Hydrothermally synthesized iron oxide nanoparticles treated with TEOS for silica coating, (2) followed by APTES treatment to generate amine group on the surface. (3) Modification using 1% glutaraldehyde to add surface aldehyde groups. (4) Protein A immobilization by imine bond formation between free amine groups of protein and aldehyde groups on the surface of nanoparticles. (5) MIP synthesis using suitable monomer combinations. (6) Protein removal using methanol:acetic acid (9:1) to create cavities complementary to the size and shape of the template. (7) Protein A rebinding to evaluate MIP BC. | ||
2.8. Binding studies
To select the monomer combination for MIP synthesis, different combinations of monomers, as presented in Table 1, were evaluated owing to their protein binding properties. A batch experiment was performed in which 0.25 mg of MIP of each combination was suspended in 0.5 mL of 20 μg mL−1 protein A solution in PB (10 mM, pH 7.2). The mixture was then incubated for 2 h under slow rotation on a 360° rotator at RT. The supernatant was collected using magnetic separation, and the concentration of protein A in the unbound fraction was determined using the Bradford assay.43 The equilibrium adsorption amount or BC (q, mg g−1) was calculated to bind MIP, and NIP to protein A, using eqn (i):| q = (Ci − Cf)V/m, | (i) |
The imprinting factor was determined using eqn (ii):
| IF = qMIP/qNIP, | (ii) |
To investigate the selectivity, BC of MIP with non-specific proteins, such as HSA, BSA, and Lysozyme, was determined. 0.35 μM of non-specific proteins were added to 0.5 mg mL−1 of MIP and NIP each. The BC was determined as described above.
To determine the binding properties of the selected MIP, a batch experiment involving 0.25 mg of MIP and NIP each was suspended in 0.5 mL of a protein A solution in PB buffer, (10 mM, pH 7.2) at a varying concentration range of 0–40 μg mL−1. BC was determined at different protein A concentrations, and the binding isotherm was plotted.
2.9. Bacterial binding studies
To evaluate the binding characteristics of the MIP with the target bacteria, a batch experiment was conducted in which 1 mg MIP was added in 10 mL Phosphate Buffered Saline (PBS) at pH 7.4 containing 101–107 CFU mL−1S. aureus. The suspensions were kept in a shaker for 2 h at RT. The concentration of S. aureus bound to the MIP was determined by plating the unbound bacteria left in a supernatant after treatment with MIP (subtraction method). BC was determined at various bacterial concentrations, and a binding isotherm was plotted for MIP.To evaluate the selectivity of MIP against bacteria, binding studies with S. aureus, B. subtilis, and E. coli were performed and visualized using fluorescence microscopy. For this purpose, the bacteria were pre-stained with acridine orange. For staining, bacterial cells (107 CFU mL−1) were resuspended in PBS (pH 7.4) containing 100 μg mL−1 acridine orange. The suspension was incubated for 30 min at 37 °C. The excess stain was removed by washing the cells three times with PBS. Next, 200 μg of MIP was added to the 1 mL stained bacterial cells. The solution was allowed to rotate at RT for 2 h on a 360° rotator. The unbound bacteria were removed by washing with PBS three times, and the MIP was resuspended in 20 μl PBS. Finally, the sample was placed on a glass slide and visualized using a fluorescence microscope.
2.10. S. aureus detection in real samples
The validation and assessment of the MIP's applicability depend on the analysis of real samples. To conduct real sample experiments, milk samples were spiked with S. aureus. Specifically, S. aureus was added at a concentration of 106 CFU mL−1 to a milk sample diluted ten-fold with PBS at pH 7.4. Following a 2-hour incubation period with MIP and NIP, magnetic separation was done to capture MIP-bound bacteria. The unbound fraction was subsequently plated for cell counting. This approach allowed for a comprehensive evaluation of the MIP's performance using real samples.463. Results and discussion
3.1. Computational screening of functional monomers
The nature's design of the antigen–antibody complex formation, involving a unique set of interactions at multiple amino acid residues to form a specific lock-key complex, can be mimicked using molecular imprinting technology. The polymeric cavity can be tailored to project functional groups that enable complementary binding to the target analyte. Herein, S. aureus specific biomarker, protein A, with negative surface charge at pH 7.0 and predominated hydrophilicity47 is selected as a template for MIP. Protein A exists in both secretory and membrane-bound forms, thereby allowing both direct and indirect bacterial identification. The crucial step in identifying suitable functional monomers is enabled by a molecular mechanics approach where the whole protein is docked with monomers, and the intermolecular interactions are modelled. The use of monomers that can form multiple non-covalent bonds spanning the entire protein surface with comparatively higher binding energy is likely to result in an enhanced specificity of the MIP. For this, we screened a list of commonly used monomers (for protein A imprinting) to estimate their binding affinity (Table 2). Dopamine with its unique set of functional groups that enables both hydrophobic interactions (phenyl ring) and hydrophilic/ionic interactions (Table 2(I)) produced the strongest binding affinity with protein A (−4.64 kcal mol−1). Additionally, other monomers possessing phenyl rings (Table 2(III; VII)) have higher binding energy due to their ability to form strong π–π interactions with aromatic amino acids. However, the lowest binding affinity was found for the commonly used silane monomer, APTES (−2.77 kcal mol−1), and, therefore, ranked last in the group (Table 2(X)). Visualization and analysis of other distinct conformational clusters of the monomers helped to assess a network of non-covalent interactions on the entire template surface rather than just single-point binding analysis (Fig. 2). These clusters were considered only within a short range of 2 Å distance between the monomer heavy atom and amino acid to account for fairly strong interactions. The largest number of multi-point interactions was observed with each of the silane monomers, mainly UPTMS, and APTES (Table 2(IV, X)), hence validating the increased use of silanes for protein imprinting.37 The contrasting behaviour of the methacrylic acid monomer yields a higher affinity one-point interaction, but the least number of multiple interactions on the protein surface (two confirmational clusters located at the same point) precisely elaborates this analysis (Fig. 2(II)).| Rank | Monomers | Estimated binding energy (kcal mol−1) | Number of distinct conformational clusters | Number of multi point interactions |
|---|---|---|---|---|
| I | Dopamine | −4.64 | 5 | 4 |
| II | Methacrylic acid (MAA) | −3.81 | 2 | 1 |
| III | Phenyltriethoxysilane (PTES) | −3.73 | 10 | 4 |
| IV | 3-Ureidopropyltrimethoxysilane (UPTMS) | −3.30 | 9 | 8 |
| V | N-Isopropylacrylamide (NIPAM) | −3.16 | 6 | 4 |
| VI | Methacrylamide (mACR) | −3.07 | 5 | 4 |
| VII | Aminophenyl boronic acid (APBA) | −3.03 | 7 | 4 |
| VIII | Tetraethoxysilane (TEOS) | −2.85 | 7 | 5 |
| IX | Acrylamide (ACR) | −2.78 | 5 | 4 |
| X | 3-Aminopropyltriethoxysilane (APTES) | −2.77 | 9 | 6 |
 | ||
| Fig. 2 Visualization of binding poses of monomers (red) clustered on protein A (blue, helical structure). Distinct conformational clusters within RMSD-tolerance of 2 Å are represented for each monomer: I: dopamine, II: MAA, III: PTES, IV: UPTMS, V: APBA, VI: mACR, VII: NIPAM, VIII: ACR, IX: APTES, and X: TEOS. The highest number of multi-point interactions over the entire protein surface are shown in the case of PTES (III), UPTMS (IV), and APTES (IX) and least by MAA (II). | ||
Additionally, the highest ranking dopamine had lesser binding points (4) compared to the lowest-ranking APTES (6) (Table 2). PTES (Table 2(III)) was introduced as one of the monomers to cover hydrophobic regions on the protein surface. TEOS (Table 2(VIII)) is generally used as a crosslinker for silane monomers. In silico prediction shows wide range interactions (hydrogen bond, hydrophobic, and electrostatic) between these monomers (Fig. 2(III), (IV), (VIII) and (X)) and protein A, giving further validation for the use of a multi monomer approach for optimal binding efficiency (Fig. S2, ESI†). Overall, this predictive estimation motivated the selection of silane-based monomers (Fig. 2(III), (IV), (VIII) and (X)) for imprinting the target protein A. Simultaneously, we also selected three more combinations: first involves only dopamine, being the highest ranked monomer; second involves APTES and TEOS, which are the lower ranked; and third involves only TEOS as a control polymer to determine the change in BC upon the addition of other silane monomers. Simultaneously, the need for the cross-linker is addressed in each set, as dopamine and TEOS serve as cross-linkers.
Interestingly, the third set utilizing APTES as a positively charged monomer, which interacts with the negatively charged protein A, may offer valuable insights into the effective design of MIPs.
3.2. Experimental design for protein A specific MIPs
In this study, MIPs are innovatively developed as a ‘shell’ over a magnetic (Fe3O4) core offering great utility in terms of easy isolation, specific detection, and reusable nature. In addition, magnetic separation plays a major role in the enrichment of bacteria from large sample volumes, consequently increasing the sensitivity of the method.48 With the help of molecular imprinting onto magnetic nanoparticles, this can be achieved in a cost-effective and efficient manner. Protein A is a 42 kDa protein that is templated over these core–shell particles with an approximate size of 250 nm. For the magnetic core, Fe3O4 nanoparticles were synthesized using the solvothermal method. Further, nanoparticles were functionalized with a silica layer using the sol–gel method with TEOS treatment. This coating provided nanoparticles with chemical stability and prevented aggregation in the solution. Next, hydroxyl groups of SiO2 on the surface reacted with ethoxy groups of APTES to give the surface the amine group. Then, these amine groups reacted with glutaraldehyde to produce aldehyde-modified Fe3O4@SiO2 nanoparticles.36 Glutaraldehyde coating is an important step because it helps to immobilize protein A using a Schiff base reaction between an aldehyde and amino groups of the protein. Finally, for imprinting, four sets of monomer combinations as selected from the computational screening were used to synthesize MIPs under constant physical conditions (RT, pH 7.0). In the first set, dopamine was used as a monomer with various advantages, such as high water solubility, polymerization ability under RT, and several amino acid-like functional groups that interact with peptide chains. Dopamine with the highest binding energy at the lowest ΔG value (−4.64) was selected as the monomer for MIP 1. In addition, dopamine demonstrated high BC (8.14) due to the presence of several amino acid-like functionalities present in the structure. The phenyl group, hydroxyl group, free amine group and the ethyl moieties present in the dopamine molecules help to interact with the target biological molecules in a similar fashion present in natural biomolecular interactions. Phenyl groups offering π–π interactions, amines, and hydroxyl groups can help in the formation of hydrogen bonding and ionic interactions, and the ethyl and phenyl groups help to bind via hydrophobic interactions. Overall, it is found from computational studies that dopamine is a suitable monomer for protein imprinting; however, non-specific interactions could lead to poor imprinting factors (0.82) due to comparable binding with NIP as well49 (Fig. 3). | ||
| Fig. 3 Binding capacity values for different MIP combinations. Each set of MIP is made using a unique combination of monomers, i.e., MIP 1: dopamine, MIP 2: TEOS and APTES, MIP 3: TEOS, APTES, PTES, UPTMS, and control MIP i.e., MIP C: TEOS. The combination of all four silane monomers (MIP 3) resulted in better binding capacity and imprinting factor. Error bars indicate standard deviation. | ||
In the next sets, silane monomers with relatively less reactivity under various conditions (e.g., strong acids, bases, and oxidizers) were used; therefore, efficient binding sites are formed due to stable and highly crosslinked silica polymers. Various studies have reported MIPs synthesized using silane monomer combinations and achieved a high IF of 5, which was further increased to 15 via cladding using TEOS.50–53 Furthermore, organosilica hybrid materials can be easily made using molecular precursors that can participate in the hydrolysis and condensation reactions as the metal alkoxide (Scheme 1), which is a flexible way to adjust the MIP's selectivity by choosing the most appropriate functional groups to interact with the template.54 In addition, these monomers involve polymerization in the aqueous environment, which can facilitate the structural preservation of protein molecules during the process of polymerization, thereby ensuring the optimal rebinding of protein targets.
 | ||
| Scheme 1 Co-polymerization pathway of organo-silane monomers used in the molecular imprinting of protein A. (1) Hydrolysis of monomers (2) condensation reaction (3) copolymerization step leading to the formation of specific binding pocket via diverse functional groups of siloxane monomers for protein A binding. | ||
Here, for the computationally lower-ranked monomers, APTES and TEOS (Fig. 2(X), (VIII)) were selected for MIP 2; they demonstrated increased binding capacities and imprinting factors compared to dopamine imprinted MIPs (MIP 1) (Fig. 3). The improved MIP performance can be correlated with the in silico analysis, where multi-point binding of the monomers possibly leads to unique polymeric cavities that possess complementary binding sites for strongly capturing the whole protein A. These experimental results along with the computational findings inspired the use of PTES and UPTMS in a ‘third’ combination (MIP 3). Clearly, these monomers enable maximum multi-point interactions (Table 2) but also introduce new functional groups of phenyl from PTES and urea from UPTMS to generate highly specific binding sites. This effect was confirmed in the exceptional BC (10.5 mg g−1), and the highest imprinting factor of ∼5 was obtained for MIP. The relevance of diverse functional groups and multiple-monomer combinations for imprinting can be validated with the contrasting performance of MIP C, which is a control polymer formed only with TEOS serving as both a monomer and cross-linker. In the entire list of monomers, TEOS possesses the minimum functionality that distinctly affects the MIP quality, yielding the lowest BC (i.e. 4.8 mg g−1).
These results suggest that unlike the conventional drug design approach, where the ligands are ranked according to their highest binding energy, monomers should be ranked according to the multi-point interactions for effective MIP design. Protein imprinting has rightfully attracted organosilane monomers because they offer functionalities resembling amino acid side chains that bind very effectively to protein residues and directly impact the selectivity and specificity of MIPs.
3.3. Characterization of nanoparticles
To determine the actual size of the synthesized nanoparticles, visualization using HRTEM was performed. As shown in Fig. 4(a)-(i), Fe3O4 nanoparticles were spherical with a mean size of ∼ 250 nm. In Fig. 4(a)-(ii), MIP has a thin shell of silane polymer coating visible over an iron core. The XRD pattern shown in Fig. 4(b) consists of characteristic peaks (220), (311), (400), (422), (511), and (440), which are in agreement with the inverse cubic spinel phase of Fe3O4 (magnetite, JCPDS card no. 85-1436). This confirms that the crystal structure of Fe3O4 is preserved in the imprinting performed for Fe3O4 nanoparticles, silica-coated Fe3O4 nanoparticles, MIP and NIP. In Fig. 4(c)-(i), the peak at 530 cm−1 is attributed to Fe–O bond vibration, which is evident to different extents in the spectra of all the given particles. In Fig. 4(c)-(ii), the peak at 1100 cm−1 corresponds to Si–O–Si stretching, which confirms the coating of silica on the Fe3O4 nanoparticles. Next, in Fig. 4(c)-(iii), the peak at 1630 cm−1 represents the stretching vibration of the N–H, indicating the deposition of APTES from the hybrid silane polymer layer. These results suggest the successful functionalization and deposition of the imprinting layer on the magnetic nanoparticles. The presence of surface functional groups after silica coating and imprinting layer were confirmed using FTIR. EDAX analysis was performed to investigate the elemental composition present at various levels of modification. The iron and oxygen peaks in iron oxide nanoparticles are depicted in Fig. 4(d)-(i). Next, after coating with silica, a silicon peak is illustrated in Fig. 4(d)-(ii). A relative increase in the carbon atomic composition was observed in glutaraldehyde-coated nanoparticles (Table S2, ESI†). MIP and NIP nanoparticles showed a comparative increase in silicon atomic composition and a decrease in carbon atomic composition, which can be attributed to the presence of silane monomers55 (Fig. 4(d)(v) and (vi)). The magnetic properties of the nanoparticles were measured by the VSM at RT. As shown in Fig. 4(e), the saturation magnetization of Fe3O4, Fe3O4@SiO2, MIP, and NIP are 87, 84, 64, and 66 emu g−1, respectively. The decrease in the saturation magnetization is due to the presence of silica coatings over bare MNPs.56 Despite a reduction in saturation magnetization, the value is sufficient for the efficient separation of MIP in the presence of an external magnetic field. The stability and coating of nanoparticles were determined using Zeta potential values. Zeta potential values obtained for different nanoparticles are depicted in Fig. 5. The surface charge of iron oxide nanoparticles was −40.2, which further decreased to −46.4 due to the presence of added free hydroxyl groups in the silica coating. Next, after coating with APTES, it increased to −12 due to the presence of free amino groups on the surface of the nanoparticles. Then, the reaction with glutaraldehyde decreased the surface charge to −15.1. MIP showed a net negative surface charge (−30.2) due to the polymer layer of the silane monomers. A negative surface charge over the nanoparticle has been shown to facilitate the stability of these particles.57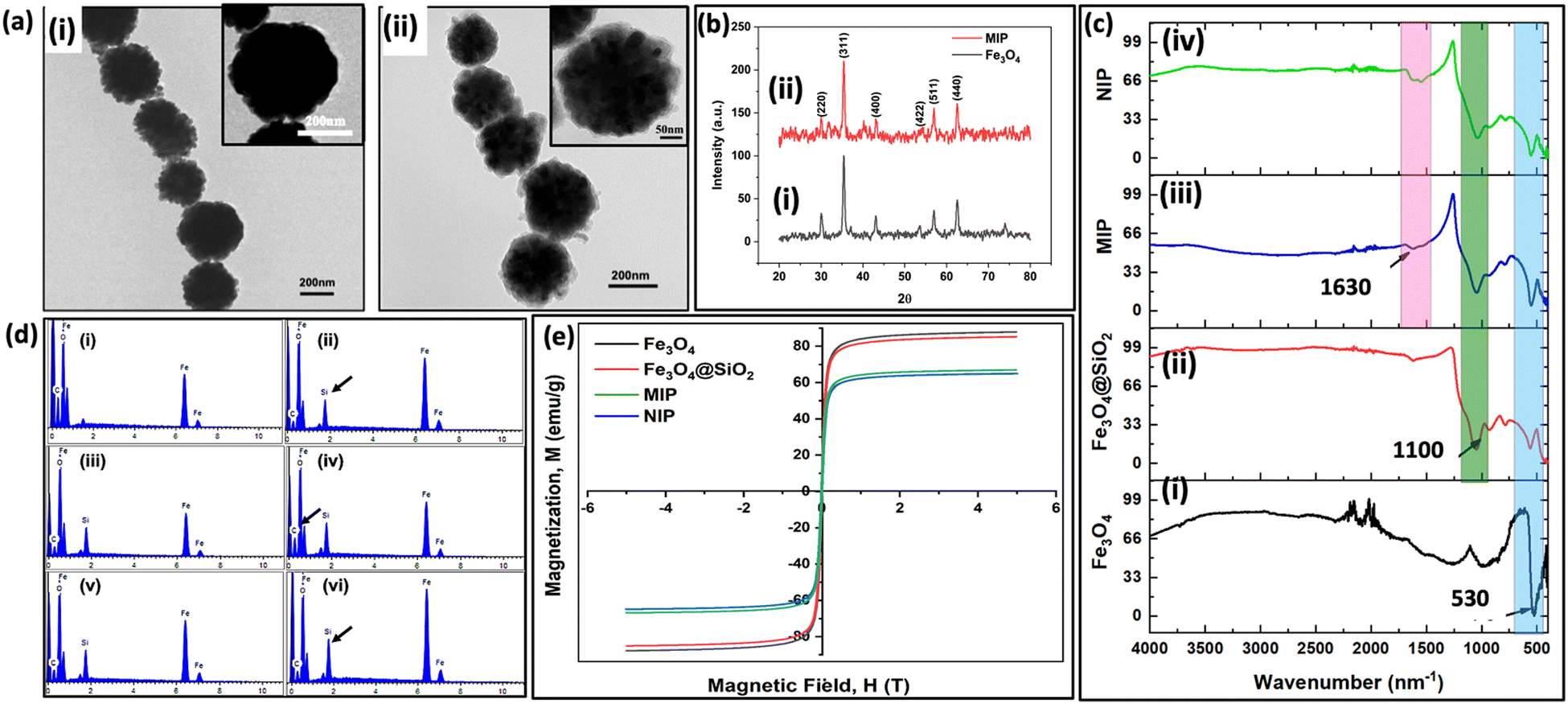 | ||
| Fig. 4 (a) TEM image of (i) Fe3O4 and (ii) MIP in a size range of 250–300 nm; (b) XRD pattern of the (i) Fe3O4 and (ii) MIP showing the magnetite nature of nanoparticles. (c) FTIR spectrum at different steps of MIP synthesis, i.e., (i) Fe3O4, (ii) Fe3O4@SiO2, (iii) MIP and (iv) NIP; (d) EDX analysis of particles after each step of chemical modification on the surface; (i) iron oxide nanoparticles (MNP), (ii) silica-coated MNP, (iii) APTES-functionalized MNP, (iv) glutaraldehyde-coated MNP, (v) MIP and (vi) NIP; (e) hysteresis loop of Fe3O4, Fe3O4@SiO2, MIP and NIP. | ||
 | ||
| Fig. 5 Zeta-potential for different particles in aqueous media. | ||
3.4. Protein binding studies
The binding isotherm of protein A binding to MIP and NIP in a concentration range of 0–40 μg mL−1 was plotted, as shown in Fig. 6. The adsorption of protein A to MIP is due to the generation of selective binding sites for protein A, indicated by the nearly five-fold lower adsorption on NIP. To further understand the adsorption properties, Langmuir (iii), Freundlich (iv) and Hill (v) equations were applied to fit the experimental results.| (Ce/qe) = (1/(qmaxKL)) + (Ce/qmax) | (iii) |
| logqe = (1/nf)logCe + logKf, | (iv) |
| qe = qmaxCno/(Cno + Knd), | (v) |
 | ||
| Fig. 6 (i) Amount of protein A (mg) bound per g of MIP and NIP particles against the initial concentration of protein A fitted using the Hill equation. (ii), (iii) Freundlich fitting of MIP and NIP binding isotherm. (iv) Langmuir fitting for MIP binding isotherm. | ||
To investigate the selectivity of the imprinted polymer, the MIP was incubated with the same concentration of different proteins—BSA, HSA, and Lysozyme. HSA and BSA are 66.5 kDa proteins with a negative net surface charge, similar to protein A. Lysozyme is a 14 kDa protein with a positive net surface charge at physiological pH.
As shown in Fig. 7, protein A showed significantly higher BC with MIP compared to HSA, BSA, and Lysozyme. Although the structures of HSA and BSA are similar, there is an amino acid sequence identity of 76%, leading to few confirmational variations. Therefore, this difference contributes to a change in the binding affinity with MIP. In addition to its high affinity and selectivity with protein A, it is expected that MIP also displays effective binding with whole S. aureus.
 | ||
| Fig. 7 BC of MIP for protein A and other non-specific proteins, i.e., BSA, HSA and lysozyme. MIP showed 10, 4 and 12-fold better BC for protein A compared to HSA, BSA and lysozyme, respectively. | ||
3.5. Bacterial binding studies
To evaluate the bacterial binding properties of MIP, bacterial concentrations ranging from 101 to 107 CFU mL−1 were used. Fig. 8 shows an increase in bacterial adsorption on MIPs in response to increased bacterial concentrations. Notably, even at the maximum bacterial concentration, surface saturation was not obtained, indicating that the produced MIP surface had sufficient binding capability for the given application.59 In addition, the magnetic core of the nanoparticles facilitated the S. aureus capture and detection at concentrations as low as 10 CFU mL−1. | ||
| Fig. 8 Adsorption isotherm of S. aureus binding to the MIP. Bound S. aureus concentration obtained by plating the unbound cells after treatment with MIP (subtraction method). | ||
To study the bacterial selectivity, MIP was incubated with S. aureus and other non-specific bacteria, i.e. B. subtilis, P. aeruginosa and E.coli. As shown in Fig. 9, S. aureus remains bound to MIP even after PBS washing. There was minimum four-fold higher fluorescence in s. aureus than other non-specific bacteria, therefore lesser binding in the case of B. subtilis, P. aeruginosa and E. coli. This study confirms that MIP formed against protein A of S. aureus shows specific binding with the target bacteria.
 | ||
| Fig. 9 Fluorescence microscopy images of different bacteria: (a) S. aureus (Gram-positive) and (b) B. subtilis (Gram positive), (c) P. aeruginosa (Gram-negative) (d) E. coli (Gram-negative) treated with MIP (0.2 mg ml−1) for 2 h, followed by washing and visualization of MIP bound bacteria. (e) Fluorescence intensity analysis of microscopy images for different bacteria using ImageJ. | ||
3.6. S. aureus detection in real samples
To assess the reliability of the method, the selective isolation of S. aureus from cows’ milk was conducted, by intentionally spiking it with a known bacterial concentration of 106 CFU mL−1. The choice of milk as the testing medium stems from the potential secretion of S. aureus into milk by dairy cows afflicted with mastitis. Table 3 illustrates a notable five-fold increase in BC of MIP compared to NIP. This enhanced binding capability facilitated the specific and efficient detection of S. aureus in milk samples. This outcome unequivocally demonstrates that the devised method exhibits robust anti-interference capabilities, enabling the accurate assessment of S. aureus concentration in complex mixtures.59| S. aureus spiked in milk sample | BC (CFU mg−1) | Recovery (%) | |
|---|---|---|---|
| MIP | 1.26 × 106 | 1.36 × 106 | 107.9 |
| NIP | 1.26 × 106 | 2.40 × 105 | 19.0 |
4. Conclusions
In this study, we synthesized protein A-imprinted magnetic nanoparticles for biomarker-based S. aureus detection. Using in silico studies, monomers were rationally selected for the imprinting process based on their interactions with protein A at multiple surface residues and fairly high binding affinity. Comparatively, dopamine showed the highest binding affinity (−4.64 kcal mol−1); however, its limited number of multi-point interactions (4) was reflected in a poor imprinting factor (0.8). In contrast to dopamine, binding at multiple points with non-covalent (reversible) bonds were displayed by silane monomers. MIP 3 displayed a high BC of 11 mg g−1 and showed selective binding and capture of protein A as well as S. aureus compared to non-specific bacteria, such as P. aeruginosa, E.coli and B. subtilis. In addition, it demonstrated a wide range (101 to 107 CFU mL−1) of S. aureus capture and detection as well as ∼100% recovery of S. aureus (106 CFU mL−1) from spiked milk samples. Instead of antibodies,60,61 cost effective, stable and selective MIPs may be employed as sensing elements capable of direct integration in different in vitro diagnostic devices.62 Similar to antigen–antibody interactions, this study clearly demonstrates the importance of multipoint interactions of monomers in the rational design of MIPs.63,64 Intervention with predictive computational modelling results in high-performing MIPs, thus reducing the need for extensive experimental design. This can accelerate the process of point-of-care device development and application, especially in resource-constrained developing nations. Biomarker imprinted bacterial detection can be applied to other ESKAPE pathogens for simultaneous detection in a multiplex platform, therefore taking a step further for the rapid detection and treatment of bacterial infections. It has further applications in the detection of food contamination, pathogen outbreaks and other infections.Author contributions
The manuscript was prepared with the involvement of all the authors. All authors have approved the manuscript's final version.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The project was financially supported by DST/NM/NNetRA/2018(G)-IITD from Department of Science and Technology (DST) and 5(1) 2017-NANO from Ministry of Electronics and Information Technology (MeitY), Govt. of India. The authors acknowledge Central Research Facilities (CRF) and Nanoscale Research Facilities (NRF) at IITD for the instrumentation facilities. SB and SK express gratitude to Ministry of Human Resource Development (MHRD), Govt. of India, for Institute postdoctoral fellowship, IIT Delhi. KN would like to thank the MHRD for the research fellowship.References
- S. Y. C. Tong, J. S. Davis, E. Eichenberger, T. L. Holland and V. G. Fowler, Clin. Microbiol. Rev., 2015, 28, 603–661 CrossRef CAS PubMed.
- F. Der Wang, Y. Y. Chen, T. L. Chen and C. Y. Liu, Am. J. Infect. Control, 2008, 36, 118–122 CrossRef PubMed.
- F. Alonzo and V. J. Torres, Microbiol. Mol. Biol. Rev., 2014, 78, 199–230 CrossRef PubMed.
- A. F. Haag, J. R. Fitzgerald and J. R. Penadés, Microbiol. Spectrum, 2019, 7, 10–1128 Search PubMed.
- N. Wellinghausen, A. J. Kochem, C. Disqué, H. Mühl, S. Gebert, J. Winter, J. Matten and S. G. Sakka, J. Clin. Microbiol., 2009, 47, 2759–2765 CrossRef CAS PubMed.
- D. J. Speers, T. R. Olma and G. L. Gilbert, J. Clin. Microbiol., 1998, 36, 1032–1034 CrossRef CAS PubMed.
- B. S. Reisner and G. L. Woods, J. Clin. Microbiol., 1999, 37, 2024–2026 CrossRef CAS PubMed.
- A. Nouri, H. Ahari and D. Shahbazzadeh, Int. J. Biol. Macromol., 2018, 107, 1732–1737 CrossRef CAS PubMed.
- O. G. Brakstad, K. Aasbakk and J. A. Maeland2, J. Clin. Microbiol., 1992, 30, 1654–1660 CrossRef CAS PubMed.
- K. T. Lim, C. S. J. Teh and K. L. Thong, Biomed. Res. Int., 2013, 895816 Search PubMed.
- W. Kong, J. Xiong, H. Yue and Z. Fu, Anal. Chem., 2015, 87, 9864–9868 CrossRef CAS PubMed.
- N. Ghanwate, P. Thakare, P. R. Bhise and S. Gawande, Sci. Rep., 2016, 6, 1–5 CrossRef PubMed.
- J. J. Belbruno, Chem. Rev., 2019, 119, 94–119 CrossRef CAS PubMed.
- P. Mishra, K. Griebenow and A. M. Klibanov, Biotechnol. Bioeng., 1996, 52, 609–614 CrossRef CAS PubMed.
- J. Pan, X. Xue, J. Wang, H. Xie and Z. Wu, Polymer, 2009, 50, 2365–2372 CrossRef CAS.
- X. Xue, J. Pan, H. Xie, J. Wang and S. Zhang, React. Funct. Polym., 2009, 69, 159–164 CrossRef CAS.
- Y. Guo, J. Li, X. Song, K. Xu, J. Wang and C. Zhao, ACS Appl. Bio Mater., 2021, 4, 420–427 CrossRef CAS PubMed.
- J. Bezdekova, K. Zemankova, J. Hutarova, S. Kociova, K. Smerkova, V. Adam and M. Vaculovicova, Food Chem., 2020, 321, 126673 CrossRef CAS PubMed.
- A. Aherne, C. Alexander, M. J. Payne, N. Perez and E. N. Vulfson, J. Am. Chem. Soc., 1996, 118, 8771–8772 CrossRef CAS.
- J. Borovička, S. D. Stoyanov and V. N. Paunov, Nanoscale, 2013, 5, 8560–8568 RSC.
- X. Shen, J. Svensson Bonde, T. Kamra, L. Bülow, J. C. Leo, D. Linke and L. Ye, Angew. Chem., Int. Ed., 2014, 53, 10687–10690 CrossRef CAS PubMed.
- G. Ertürk, D. Berillo, M. Hedström and B. Mattiasson, Biotechnol. Rep., 2014, 3, 65–72 CrossRef PubMed.
- M. Marc, Mip Synthesis, Characteristics and Analytical Application, Elsevier, 2019 Search PubMed.
- J. Liu, Q. Deng, D. Tao, K. Yang, L. Zhang, Z. Liang and Y. Zhang, Sci. Rep., 2014, 4, 1–6 Search PubMed.
- T. Cowen, K. Karim and S. Piletsky, Anal. Chim. Acta, 2016, 936, 62–74 CrossRef CAS PubMed.
- R. Boroznjak, J. Reut, A. Tretjakov, A. Lomaka, A. Öpik and V. Syritski, J. Mol. Recognit., 2017, 30, 2635 CrossRef PubMed.
- S. Suryana, Mutakin, Y. Rosandi and A. N. Hasanah, Molecules, 2021, 26, 1891 CrossRef CAS PubMed.
- M. v Sullivan, S. R. Dennison, G. Archontis, S. M. Reddy and J. M. Hayes, J. Phys. Chem. B, 2019, 123, 5432–5443 CrossRef CAS PubMed.
- D. R. Kryscio, Y. Shi, P. Ren and N. A. Peppas, Ind. Eng. Chem. Res., 2011, 50, 13877–13884 CrossRef CAS PubMed.
- S. Rajpal, S. Singh, P. Mishra and S. Bhakta, Molecularly Imprinted Polymers (MIPs): Commercialization Prospects, Meenakshi Singh, Elsevier, vol. 16, pp. 391–415 Search PubMed.
- M. K. Sandel and J. L. McKillip, Food Control, 2004, 15, 5–10 CrossRef.
- H. Deng, X. Li, Q. Peng, X. Wang, J. Chen and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 2782–2785 CrossRef CAS PubMed.
- A. Saha, K. Narula, P. Mishra, G. Biswas and S. Bhakta, Nanoscale Adv., 2023, 5, 1386–1396 RSC.
- A. H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- B. T. Thanh, N. van Sau, H. Ju, M. J. K. Bashir, H. K. Jun, T. B. Phan, Q. M. Ngo, N. Q. Tran, T. H. Hai, P. van Hung and T. T. Nguyen, J. Nanomater., 2019, 2182471 CAS.
- F. Chen, M. Mao, J. Wang, J. Liu and F. Li, Talanta, 2020, 209, 120509 CrossRef CAS PubMed.
- S. Bhakta, M. S. I. Seraji, S. L. Suib and J. F. Rusling, ACS Appl. Mater. Interfaces, 2015, 7, 28197–28206 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- A. A. Lahcen, S. G. Surya, T. Beduk, M. T. Vijjapu, A. Lamaoui, C. Durmus, S. Timur, O. Shekhah, V. Mani, A. Amine, M. Eddaoudi and K. N. Salama, ACS Appl. Mater. Interfaces, 2022, 14, 49399–49424 CrossRef CAS PubMed.
- C. Lafarge, M. Bitar, L. El Hosry, P. Cayot and E. Bou-Maroun, Mater. Today Commun., 2020, 24, 101157 CrossRef CAS.
- G. Goyal, S. Bhakta and P. Mishra, ACS Appl. Nano Mater., 2019, 2, 6747–6756 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- W. Zhang, X. She, L. Wang, H. Fan, Q. Zhou, X. Huang and J. Z. Tang, Materials, 2017, 10, 475 CrossRef CAS PubMed.
- J. Chen, S. Lei, Y. Xie, M. Wang, J. Yang and X. Ge, ACS Appl. Mater. Interfaces, 2015, 7, 28606–28615 CrossRef CAS PubMed.
- N. Idil, M. Bakhshpour, I. Perçin and B. Mattiasson, Biosensors, 2021, 11, 140 CrossRef CAS PubMed.
- Peptide calculator, https://www.bachem.com/knowledge-center/peptide-calculator/, (accessed 30 October 2022).
- J. J. Lee, K. J. Jeong, M. Hashimoto, A. H. Kwon, A. Rwei, S. A. Shankarappa, J. H. Tsui and D. S. Kohane, Nano Lett., 2014, 14, 1–5 CrossRef CAS PubMed.
- A. Lamaoui, A. A. Lahcen and A. Amine, Polymers, 2023, 15, 3712 CrossRef CAS PubMed.
- W. Li, Q. Zhang, Y. Wang, Y. Ma, Z. Guo and Z. Liu, Anal. Chem., 2019, 91, 4831–4837 CrossRef CAS PubMed.
- Z. Guo, R. Xing, M. Zhao, Y. Li, H. Lu and Z. Liu, Adv. Sci., 2021, 8, 2101713 CrossRef CAS PubMed.
- W. Li, Q. Zhang, Y. Wang, Y. Ma, Z. Guo and Z. Liu, Anal. Chem., 2019, 91, 4831–4837 CrossRef CAS PubMed.
- R. Xing, Z. Guo, H. Lu, Q. Zhang and Z. Liu, Sci. Bull., 2022, 67, 278–287 CrossRef CAS PubMed.
- R. Gao, X. Kong, X. Wang, X. He, L. Chen and Y. Zhang, J. Mater. Chem., 2011, 21, 17863–17871 RSC.
- S. Rajpal, S. Bhakta and P. Mishra, J. Mater. Chem. B, 2021, 9, 2436–2446 RSC.
- L. Liu, X. Zhu, Y. Zeng, H. Wang, Y. Lu, J. Zhang, Z. Yin, Z. Chen, Y. Yang and L. Li, Polymers, 2018, 10, 1329 CrossRef PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- X. Bi and Z. Liu, Anal. Chem., 2014, 86, 959–966 CrossRef CAS PubMed.
- J. Bezdekova, K. Zemankova, J. Hutarova, S. Kociova, K. Smerkova, V. Adam and M. Vaculovicova, Food Chem., 2020, 321, 126673 CrossRef CAS PubMed.
- S. Joseph, S. Rajpal, D. Kar, S. Devinder, S. Pandey, P. Mishra and J. Joseph, Biosens. Bioelectron., 2023, 241, 115695 CrossRef CAS PubMed.
- S. Sharma, D. Kar, A. Moudgil, S. Das and P. Mishra, Sens. Actuators, B, 2024, 407, 135486 CrossRef CAS.
- S. Rajpal and P. Mishra, Biosens. Bioelectron.: X, 2022, 11, 100201 CAS.
- S. Rajpal, B. Mizaikoff and P. Mishra, Int. J. Biol. Macromol., 2024, 266, 131101 CrossRef CAS PubMed.
- S. Rajpal, P. Mishra and B. Mizaikoff, Int. J. Mol. Sci., 2023, 24, 6785 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00392f |
| This journal is © The Royal Society of Chemistry 2024 |