
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocompatible metal–organic frameworks as promising platforms to eradicate HIV reservoirs ex vivo in people living with HIV†
José A.
Lebrón‡
 a,
Francisco J.
Ostos‡
bc,
Marta
Martínez-Santa
a,
Francisco
García-Moscoso
d,
Manuel
López-López
e,
María L.
Moyá
a,
Eva
Bernal
a,
Sara
Bachiller
bc,
Gabriel
González-Ulloa
bc,
David
Rodríguez-Lucena
d,
Tania
Lopes-Costa
d,
Rut
Fernández-Torres
f,
Ezequiel
Ruiz-Mateos
c,
José M.
Pedrosa
d,
Mohammed
Rafii-El-Idrissi Benhnia
*bc and
Pilar
López-Cornejo
*a
a,
Francisco J.
Ostos‡
bc,
Marta
Martínez-Santa
a,
Francisco
García-Moscoso
d,
Manuel
López-López
e,
María L.
Moyá
a,
Eva
Bernal
a,
Sara
Bachiller
bc,
Gabriel
González-Ulloa
bc,
David
Rodríguez-Lucena
d,
Tania
Lopes-Costa
d,
Rut
Fernández-Torres
f,
Ezequiel
Ruiz-Mateos
c,
José M.
Pedrosa
d,
Mohammed
Rafii-El-Idrissi Benhnia
*bc and
Pilar
López-Cornejo
*a
aDepartment of Physical Chemistry, Faculty of Chemistry, University of Seville, C/Prof. García González 1, 41012 Seville, Spain. E-mail: pcornejo@us.es
bDepartment of Medical Biochemistry, Molecular Biology, and Immunology, School of Medicine, University of Seville, 41009 Seville, Spain
cInstitute of Biomedicine of Seville, IBiS/Virgen del Rocío University Hospital/CSIC/University of Seville, Clinical Unit of Infectious Diseases, Microbiology and Parasitology, 41013 Seville, Spain
dDepartment of Physical, Chemical and Natural Systems, University Pablo de Olavide, Ctra. Utrera Km. 1, 41013, Seville, Spain
eDepartment of Chemical Engineering, Physical Chemistry and Materials Science, Campus ‘El Carmen’, Faculty of Experimental Sciences, University of Huelva, 21071, Huelva, Spain
fDepartment of Analytical Chemistry, Faculty of Chemistry, University of Seville, c/Prof. García González, 1, 41012, Seville, Spain
First published on 17th April 2024
Abstract
The HIV attacks the immune system provoking an infection that is considered a global health challenge. Despite antiretroviral treatments being effective in reducing the plasma viral load in the blood to undetectable levels in people living with HIV (PLWH), the disease is not cured and has become chronic. This happens because of the existence of anatomical and cellular viral reservoirs, mainly located in the lymph nodes and gastrointestinal tract, which are composed of infected CD4+ T cells with a resting memory phenotype and inaccessible to antiretroviral therapy. Herein, a new therapeutic strategy based on nanotechnology is presented. Different combinations of antiretroviral drugs (bictegravir/tenofovir/emtricitabine and nevirapine/tenofovir/emtricitabine) and toll-like receptor agonists were encapsulated into metal–organic frameworks (MOFs) PCN-224 and ZIF-8. The encapsulation efficiencies of all the drugs, as well as their release rate from the carriers, were measured. In vitro studies about the cell viability, the hemocompatibility, and the platelet aggregation of the MOFs were carried out. Epifluorescence microscopy assays confirmed the ability of ZIF-8 to target a carboxyfluorescein probe inside HeLa cell lines and PBMCs. These results pave the way for the use of these structures to eliminate latent HIV reservoirs from anatomical compartments through the activation of innate immune cells, and a higher efficacy of the triplet combinations of antiretroviral drugs.
1. Introduction
The human immunodeficiency virus (HIV) is a lentivirus whose infection triggers profound immune deficits. The virus preferentially infects CD4+ T-cells, rapidly destroying massive numbers of these cells in the gastrointestinal tract and causing a state of immunodeficiency.1,2 This leads to the development of acquired immune deficiency syndrome (AIDS) and ultimately death. The introduction of combined antiretroviral therapy (cART) has been one of the most significant advances in medicine to date. In most people living with HIV (PLWH), its use has been observed to effectively reduce the plasma viral load in the blood by suppressing HIV replication, increasing circulating CD4+ T-cell counts and reducing hypergammaglobulinaemia.3 This has led to a significant decrease in AIDS-related morbidity and mortality, transforming a terminal illness into a chronic disease and leading to increased survival and life quality of PLWH.3–6 Recently, cART has demonstrated effective virological control and a reduction in the risk of transmission. It is generally tolerated well, and its short-term side effects are acceptable.Although HIV infection has become a chronic disease in treated patients,5–7 and despite long-term persistent viral suppression, prolonged highly active cART is not curative and does not fully restore the immune function.3,4,8–10 The HIV persists in lymph nodes and other tissues of infected patients,11 as well as other remote sites such as the brain, reproductive tract and bone marrow.12 The major barrier to sterilizing and curing HIV is the early establishment of latency after infection, defined as the persistence integration of the replication competent intact viral DNA into the host genome in the absence of virion production.13–15
The use of drug delivery nanosystems for HIV treatment seems to be a promising strategy that improves the therapy efficacy of these patients. It is well-known that nanocarriers can usually increase the solubility of the drugs in water, protect the encapsulated compounds against degradation, and provide a controlled drug release. With regard to HIV treatment, the use of nanocarriers can enhance the pharmacological properties of the antiretroviral agents, decrease their toxic effects, increase their biodistribution, reduce treatment fatigue, and others.16 Polymeric, solid lipid, inorganic and magnetic nanoparticles and liposomes, niosomes or ethosomes have been developed as platforms for enhancing the biodistribution of HIV/AIDS drugs in the organism.17–21 In this regard, lipid nanoparticles containing 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and N-(carbonylmethoxypolyethyleneglycol-2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine were prepared to incorporate lopinavir and ritonavir, virus protease inhibitors with more than 90% of encapsulation efficiency, simultaneously into the hydrophilic reverse transcriptase inhibitor tenofovir. This formulation demonstrated an enhancement of intracellular drug concentrations in the blood and lymph nodes in primates.22,23 Vavia et al. have prepared lipid nanoparticles containing antiretroviral drugs and nanoselenium to act more efficiently against HIV infection.24–27 These nanoparticles showed a wide range of sizes and polydisperse nature, which allows them to simultaneously target multiple HIV reservoirs. The formulation was found to exhibit high cellular uptake and efficacy against HIV, further enhancing its potential in multi-reservoir targeting.
Liposomes are long-circulating nanosystems. A decrease in the size of the liposomes and the inclusion of cholesterol into these nanostructures are factors that can provoke an increase in the survival time in the systemic circulation long enough to reach and bind to their targets.28 Several liposomal formulations, varying in size and cholesterol content, carrying the synthetic polymeric peptide SPC3, were prepared. The antiviral efficacy of the SPC3-liposomes measured in vitro improved that produced by free SPC3.29 Similar results were observed for other drugs such as the viral protease inhibitor L-689 502.30
Metal–organic frameworks (MOFs) are tridimensional materials formed by the coordination of metal ions with different organic ligands.31 This interaction generates crystalline solids with different internal channels that give rise to specific properties such as high porosity, diverse functionality, and tunable structure. The size and the morphology of the MOFs also depend on the synthesis method and the working conditions.32 Solvent, temperature, reagent concentrations and duration of the synthesis can be modified in the preparation process. The nature of the metal also influences the cytotoxic properties of these supramolecular architectures.33 Significant efforts are focused on the preparation of biocompatible MOFs.
These new findings may have potential applications for the early treatment of HIV infection, as well as other viral infections. In this regard, nanosized mesoporous iron carboxylate metal–organic frameworks were used as drug delivery nanosystems of azidothymidine triphosphate, an inhibitor of both HIV-1 reverse transcriptase and DNA polymerase gamma. Strong interactions between the accessible Lewis acid unsaturated iron sites of these platforms and the phosphate groups of the drug resulted in an encapsulation efficiency close to 100%.34 The anti-HIV activity was also explored in vitro in this work using human activated peripheral blood mononuclear cells (PBMCs). These loaded nanoparticles were able to penetrate inside HIV-infected cells and deliver the drug into them. More recently, nanoparticles of iron(III) polycarboxylate metal–organic frameworks were synthesized to improve anti-HIV therapies.35 The results demonstrated the capacity of these nanocarriers to store the drugs up to two months with their physicochemical properties intact after freeze-drying. Besides, they developed their antiretroviral activity in vitro in primary cultures of human monocyte-derived macrophages. However, MIL-100 shows a slow degradation for 8 days in PBS, losing their constituent trimesate linkers, transforming into inorganic buildings, and become amorphous.36,37
MOFs have also been prepared with other purposes related to HIV/AIDS. Wang et al. prepared a Ti3C2Tx modified ZIF-8 as an electrochemical luminescence biosensor to detect the HIV-1 Tat protein.38 In addition, derivative NiCO-MOFs were synthetized as platforms for the detection of cell-associated HIV–DNA.39
In the present work, the MOFs ZIF-8 (zeolitic imidazolate framework-8) and PCN-224 (porphyrin Zr metal–organic framework), in the form of nano- and micro-crystals, respectively, were prepared to be used as carriers for the prevention and treatment of HIV-infection. To understand the outstanding features of MOFs used with different sizes and, therefore, distinct administration routes to the patient, physicochemical and in vitro biological characterization was carried out. The cytotoxicity of these MOFs was evaluated in different non-primary cell lines related to the immune system as well as in PBMCs isolated from healthy donors. To gain further information about their biocompatibility, an in vitro hemolytic effect and platelet aggregation assays were performed. The triplet combinations of antiretroviral drugs (cARTs), bictegravir (BIC)/tenofovir (TFV)/emtricitabine (FTC) or nevirapine (NVP)/tenofovir (TFV)/emtricitabine (FTC), and toll-like receptor (TLR) agonists TLR-7 (GS-9620) and -9 (CpG-(ODN 2216) class A), Scheme 1, were encapsulated into these MOFs. The percentage of encapsulation efficiency (% EE) for each drug was estimated and a new method for the determination of the % EE of TLR agonists was developed. The influence of the carrier size on the encapsulation process was studied. The release of the antiretroviral drugs was also determined.
 | ||
| Scheme 1 Molecular structures of ARTs and TLR agonists. | ||
Results showed a high encapsulation efficiency of the drugs and a slow controlled release, which could increase the therapeutic effect in PLWH. A high biocompatibility of the systems in all in vitro experiments was observed.
The use of MOFs containing cARTs and TLR agonists in the same framework, or the combination of solutions containing cART-loaded and TLR-loaded MOFs, can synergistically improve the therapeutic activity of the drugs by decreasing the plasma viral load in PLWH. This can result in a new procedure to enhance the innate immune response, providing a further effective strategy to decrease HIV reservoirs in CD4+ T cells from PLWH.
2. Results and discussion
2.1. Characterization of MOFs
 | ||
| Fig. 1 HR-TEM bright-field images (a) and (b) and SEM images (c) and (d) of ZIF-8 nanoparticles with different magnifications. (e) and (f) Scanning electron microscopy images of PCN-224 powder. | ||
The nano-MOF material was isolated as a white crystalline powder and was also characterized by powder X-ray diffraction (PXRD) (Fig. 2(A)). ZIF-8 was obtained as a unique pure phase, as the experimental diffraction pattern does not differ from the simulated one. The crystallite size was obtained from such XRD patterns by Scherrer's equation, being smaller (20.9 nm) than the particle size, revealing the polycrystalline nature of the particles.
 | ||
| Fig. 2 (A) Experimental (red line) and simulated (black line) PXRD diffractograms of ZIF-8. (B) X-ray diffraction patterns of the as-synthesized PCN-224 and simulated from the PCN-224 crystal structure data. (C) FT-IR spectra of ZIF-8 (grey line) and 2-methylimidazole (black line). (D) Transmittance FT-IR spectra of the H4TCPP ligand (black line) and PCN-224 MOF (grey line). | ||
The coordination between the Zn centers and the nitrogen atoms of the ligands (2-methylimidazole) was further confirmed by FT-IR (Fig. 2(C)). As can be seen, both 2Me-Him and ZIF-8 exhibit the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching (1592 cm−1), whereas bands between 1300–1500, 1250–900 and 800–600 cm−1 can be assigned to the imidazolate ring stretching, in-plane bending and out-of-plane bending, respectively.41,42 The narrow band at 425 cm−1, attributed to the Zn–N stretching, and the absence of the typical N–H stretching (3150 cm−1) in the FT-IR spectrum of ZIF-8 indicate the correct coordination between the metal and the ligand.41,42 Additionally, the absence of some bands in the spectrum of ZIF-8 compared to that of the free ligand could be explained by the restriction of some vibration modes in the MOF due to the new coordination environment.
N stretching (1592 cm−1), whereas bands between 1300–1500, 1250–900 and 800–600 cm−1 can be assigned to the imidazolate ring stretching, in-plane bending and out-of-plane bending, respectively.41,42 The narrow band at 425 cm−1, attributed to the Zn–N stretching, and the absence of the typical N–H stretching (3150 cm−1) in the FT-IR spectrum of ZIF-8 indicate the correct coordination between the metal and the ligand.41,42 Additionally, the absence of some bands in the spectrum of ZIF-8 compared to that of the free ligand could be explained by the restriction of some vibration modes in the MOF due to the new coordination environment.
The micro-sized material was isolated as dark purple powder and was characterized by X-ray diffraction (XRD), as shown in Fig. 2(B). The pristine material exhibits virtually the same diffraction pattern as the simulated one. It is worth noting that the peak at 5.6° is absent in the experimental diffractogram. This fact has been previously reported in the literature43,44 and is possibly caused by the highly defined geometrical shape of the particles, leading them to exhibit preferred orientations,45 which can eventually obscure the corresponding plane (211). The coordination between the Zr(IV) centers and the carboxylate groups of the porphyrins was confirmed by Fourier-transform infrared spectroscopy (FT-IR), as shown in Fig. 2(D). The H2TCPP FT-IR spectrum was included for comparison purposes, and as can be seen, the ligand exhibits two intense bands at 1685 cm−1 and 1270 cm−1 that correspond to the CO stretching and C–O stretching modes, respectively, of the carboxylic acid groups. Upon the coordination of TCPP-4 with the metal nodes, these bands disappear entirely in the FT-IR spectrum of the PCN-224. Moreover, the absence of the carbonyl bands in the PCN-224 structure indicates that no solvent molecules (DMF) are occluded into the MOF cavities.
2.2. In vitro viability assays
The biocompatibility of MOFs, as well as of any drug carrier, turns out to be one of the main concerns for their clinical use. In this regard, estimation of their cytotoxicity is required as a useful initial step for biocompatibility assessment. Cytotoxicity measurements of ZIF-8 and PCN-224 were obtained in this work using a CyQUANT™ lactate dehydrogenase (LDH) assay to measure the release of an intracellular stable enzyme, LDH, from cells with damaged plasma membranes to the cell culture medium as a marker of cytotoxicity/cell death. Several non-primary human cell lines and primary human cells were selected based on the organs or cells of interest in the clinical assay, and the cytotoxicity assay must include cells that represent the exposure route and organs targeted by nanoparticles. The results showed a non-significant or low cytotoxicity effect of ZIF-8 and PCN-224 within the concentration range studied in both primary and most non-primary human cells at 6 h and 36 h of incubation, respectively, compared to the negative control, as can be seen in Fig. 3 and Fig. S1 (ESI†). The cell viability decreases significantly at higher concentrations of ZIF-8 as shown in Fig. 3(A), compared to the negative control, for Jurkat (0 vs. 250 or 500 μg mL−1, P = 0.002 or 0.0003, respectively) and U937 cell lines (0 vs. 125, 250 or 500 μg mL−1, P = 0.04, 0.02 or 0.03, respectively). PCN-224 shows a significant decrease in the cell viability of only U937 cell lines as shown in Fig. 3(B) compared to the negative control (0 vs. 3.9, 7.8, 15.6, 31.3, 62.5, 125, 250 or 500 μg mL−1, P = 0.01, 0.006, 0.002, 0.002, 0.0008, 0.0008, 0.0008, or 0.006, respectively). However, the observed decrease in the cell viability is minimal and the maximum decrease is 16.7% or 12.6% for ZIF-8 in Jurkat or U937 cell lines and 24.3% for PCN-224 in U937 cell lines. Our data suggest that the MOFs ZIF-8 and PCN-224 have no or minimal cytotoxicity effect and do not affect cell growth in all cell lines studied and in human PBMCs (Fig. S1, ESI†). | ||
| Fig. 3 Cell viability values by the LDH assay in the presence of different MOF concentrations for 36 h in various cell lines: (A) ZIF-8 and (B) PCN-224. Each MOF concentration was measured in triplicates and the experiments were repeated thrice independently. Error bars represent standard deviation values. The two-way ANOVA Dunnett multiple comparison test was applied for comparing different concentrations of MOFs with the negative control (cells treated with PBS, 0). *P < 0.05, **P < 0.01 and *** P < 0.001. In panel A, * represents the comparison in the U937 cell line, and ** and *** represent the comparison in the Jurkat cell line. | ||
The following cytotoxicity technique is based on the reaction of free amines in the cytoplasm of the dead cell with the dye used that moves into damaged cells resulting in very bright fluorescence. According to the results, no significant cytotoxicity was also seen either when evaluating the cell death by flow cytometry in human PBMCs treated with ZIF-8 or PCN-224 and stained with the live/dead fixable aqua dye. The results showed cell viability rates higher than 90.6% and 73.6% (0 vs. 500 μg mL−1, P = 0.07) for ZIF-8 and 96.8% and 92.5% for PCN-224 (0 vs. 500 μg mL−1, P = 0.9) at both 6 h and overnight incubation, respectively (Fig. S2B–E, ESI†). However, the treatment with DMSO at 6 h (Fig. S2A, ESI†) or overnight (data not shown) was very cytotoxic and showed 82% or 98% of dead cells, respectively (Fig. S2A, ESI†).
Several studies have been found in the bibliography in relation to the toxicity of the MOFs studied in this work. Thus, F. Hao et al.46 showed that the cytotoxicity of PCN-224 decreases when their concentration increases in J774A.1 cell lines, as a model of macrophages. Nevertheless, Y. Zhang et al.47 demonstrated that its cytotoxicity has a lower effect in in vitro measurements using the HT-29 human colon adenocarcinoma cell line. M. Hoop et al.48 studied the biocompatibility of the MOF ZIF-8 in different cell lines, spanning kidneys, skin, breast, blood, bones, and connective tissue cells. ZIF-8 showed no significant cytotoxicity up to a value of 30 μg mL−1, but the cell viability decreased to 10% for both 75 and 100 μg mL−1 due to the influence of released Zn2+ ions on the mitochondrial ROS production. M. Zheng et al.49 also reported that the cell viability in the presence of ZIF-8 was over 70% in HeLa cells for 48 h up to a value of 50 μg mL−1. L. R. de Moura Ferraz et al.50 confirmed that ZIF-8 exhibited a low cell death in PBMCs up to a concentration value of 100 μg mL−1 for 24 and 48 h.
Given the concerns that the LDH release assay only provides information about the early event in necrosis and late event in apoptosis, a more comprehensive and complementary assay was performed using flow cytometry. Thus, a fluorescent reactive dye has been used to provide information distinguishing live cells from dead cells based on cell membrane integrity.51 Using this experimental approach, information about different cellular apoptosis stages and necrosis at the moment of treatment with ZIF-8 and PCN-224 has been obtained in human PBMCs at 6 h and overnight incubation, compared to the negative control (Fig. 4). A more exhaustive study on the possible toxicity caused by ZIF-8 and PCN-224 has been carried out. Thus, apoptosis and/or necrosis induced by ZIF-8 were investigated after 6 h and overnight incubation. The results showed that around 2–3% of PBMCs treated with increasing concentrations of ZIF-8 for 6 h were in early process of apoptosis and about 2–4% were late apoptotic cells. Necrosis rates varied between 9 and 13% in the whole range of ZIF-8 concentrations used. No differences were seen when comparing cell viability between ZIF-8-treated cells and untreated cells, and the latter was used as a negative control group (Fig. 4(B) upper panel and Fig. 4(C)). Early and late apoptosis rates were slightly higher after overnight incubation with ZIF-8 nanoparticles (Fig. 4(B) lower panels and Fig. 4(D)). Around 4–5% cells were early apoptotic cells, 3–5% cells were late apoptotic cells and 9–13% cells were necrotic cells, showing a minimum apoptotic and necrotic effects of the MOF after overnight treatments. As was expected, the treatment with DMSO at 6 h or overnight showed strong apoptotic effects: the early apoptotic cells were less than 1.5%, the late apoptotic cells were 47–66%, and the necrotic cells were 3–6% (Fig. 4). Our results agree to those shown by S. Jin et al.,52 H. Yang et al.53 and X. Chen et al.54 in different types of cell lines, which showed maximum total apoptosis rates of 8.09%, 7.29% and 16.3%, respectively. These results demonstrate that ZIF-8 nanoparticles present a low apoptosis and/or necrosis in human primary immune cells and can be considered safe carriers of drugs after in vitro evaluation.
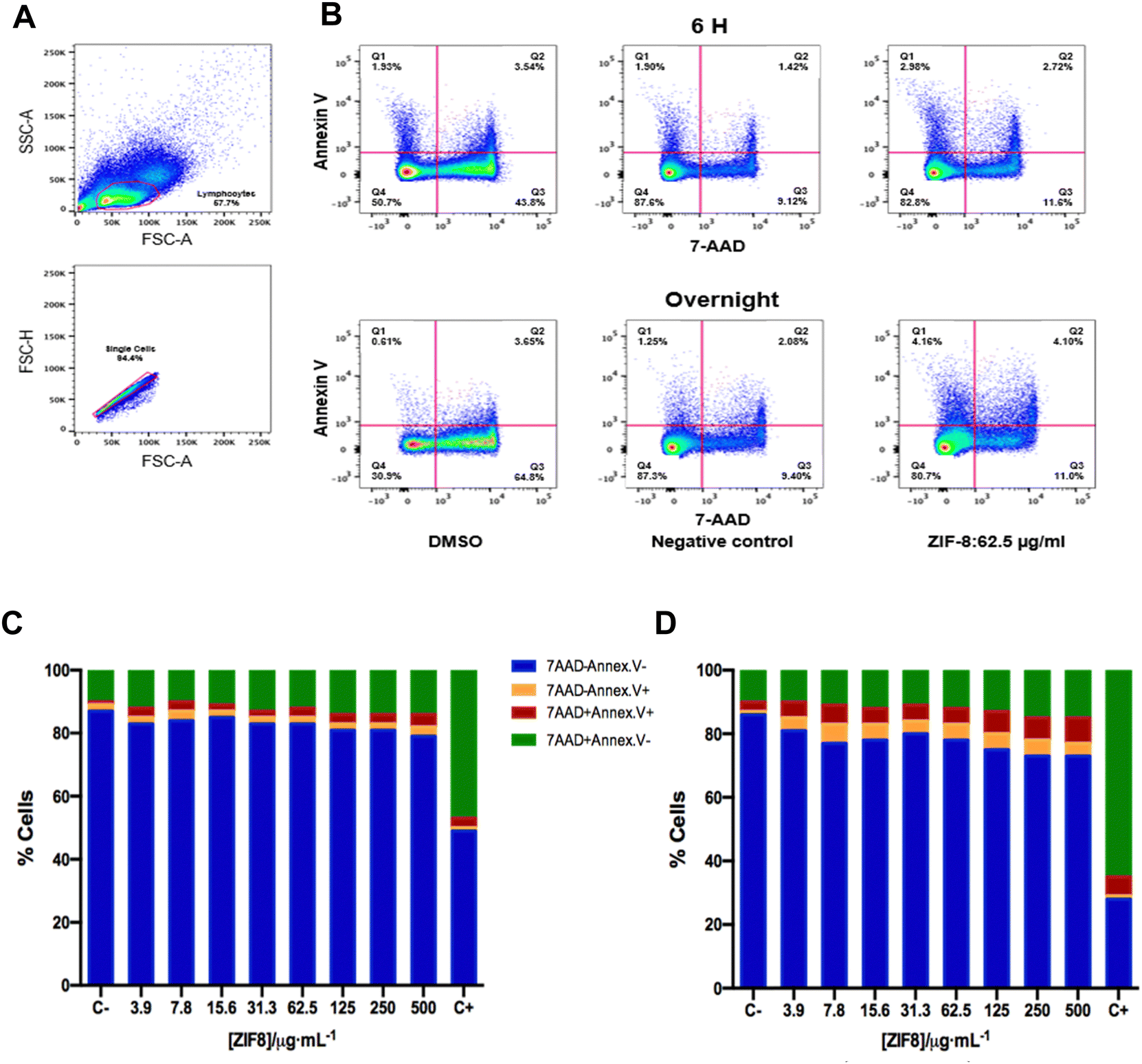 | ||
| Fig. 4 Apoptosis and necrosis induced by ZIF-8. (A) and (B) Pseudocolor dot plots showing the gating strategy used for the identification of apoptotic cells (PBMCs) in a representative sample. (A) Mononuclear cells were selected according to their size (FSC-A) and complexity (SSC-A). (B) The cells using annexin V/7-AAD staining are represented as live cells, annexin V−/7-AAD− staining (lower-left, Q4), early apoptotic cells, annexin V+/7-AAD− staining (upper-left, Q1), necrotic or dead cells, annexin V+/7-AAD+ staining (upper-right, Q2), and late apoptotic cells, annexin V−/7-AAD+ staining (lower-right, Q3) at 6 h and overnight incubation with DMSO (used as the positive control, C+), ZIF-8 (62,5 μg mL−1) and 1× PBS (used as the negative control, C−). (C) and (D) Bar graphs show the percentage of viable cells (blue), early apoptotic cells (orange), necrotic or dead cells (red) and late apoptotic cells (green) at 6 h (C), and overnight (D) incubation with different concentrations of ZIF-8. | ||
The apoptotic/necrotic effect of PCN-224 could not be analysed by flow cytometry due to its fluorescence properties, as it overlaps with the emission spectra of 7-aminoactinomycin D (7-AAD) and most fluorochromes used in a FACS Canto or LSRFortessa cytometer.
Altogether, the in vitro viability results obtained demonstrated that PCN-224 and ZIF-8 MOFs could be potential biocompatible candidates as carriers of antiretroviral drugs for preventing and treating any microbial infection and HIV infection in humans.
2.3. In vitro hemolysis assays
Some metal complexes may cause a hemolytic effect due to the interactions with the phospholipid bilayer located in the erythrocyte membrane, which induce membrane destabilization and cause a hemolytic anaemia condition.55 To gain further information about the safety of the materials investigated here, hemolysis experiments were performed to explore the in vitro hemocompatibility of MOFs in human red blood cells (RBCs). The hemolytic effect was determined as a function of intracellular hemoglobin release.56Fig. 5(A) shows the results obtained for each of the systems. ZIF-8 does not show any significant hemolytic activity within the concentration range investigated compared to the negative control (0 vs. 3.9, 7.8, 15.6, 31.3, 62.5, 125, 250 or 500 μg mL−1, P > 0.99), while PCN-224 shows no significant hemolytic activity at concentrations lower than 500 μg mL−1, where the hemolysis percentages remain below 4%.57 However, the treatment with ACK lysing buffer was very hemolytic and showed a significant difference compared to different MOF concentrations (P < 0.0001). According to the ASTM E2524-08 standard, hemolysis <5% is not considered significant. Similar results agree to those found by other authors in the literature.58–60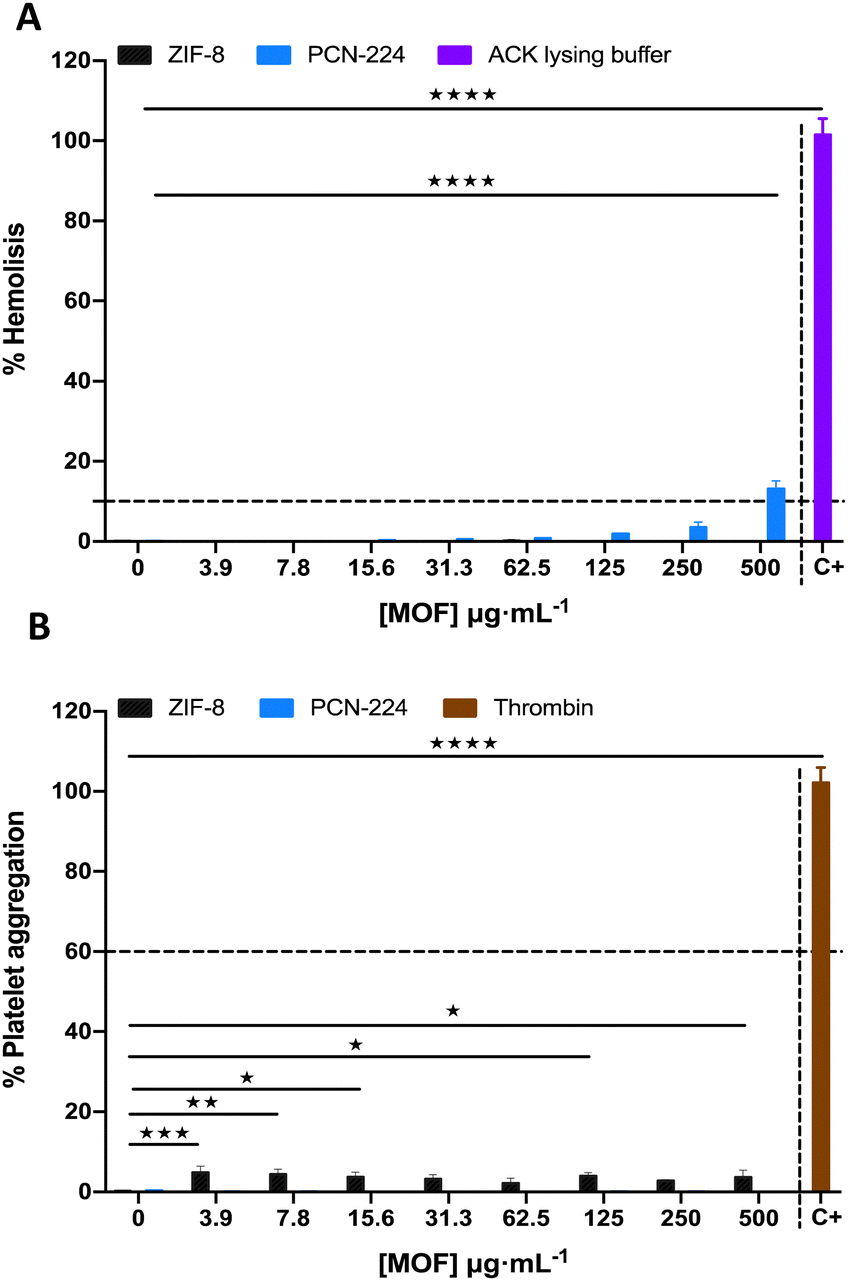 | ||
| Fig. 5 (A) % Hemolysis induced by PCN-224 and ZIF-8 for 4 h at 37 °C in 5% CO2. Each MOF concentration was measured in duplicate, and the tests were repeated by triplicate independently. ACK lysing buffer (C+) and 1× PBS (0 μg mL−1) were used as the positive and negative controls, respectively. (B) % Platelet aggregation induced by PCN-224 and ZIF-8 for 5 min at 37 °C in 5% CO2. Each MOF concentration was measured in duplicate, and all data are representative of three or more experiments. Thrombin (C+) and 1× PBS (0 μg mL−1) were used as the positive and negative controls, respectively. The dashed line indicates the limit of the hemolytic effect or platelet aggregation. Error bars represent standard deviation values. The two-way ANOVA Dunnett multiple comparisons test was applied for comparing different MOF concentrations with the negative and positive controls (ACK lysing buffer or thrombin). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. | ||
2.4. Platelet aggregation
The main function of platelets is to control the hemostasis at sites of injury or inflammation in our vascular system. Nevertheless, excessive activation of platelets, leading to platelet aggregation, is considered an important factor in thrombotic diseases such as heart attacks or strokes. Platelet aggregation is a risk factor when percentages remain above 60%.61As can be seen in Fig. 5(B), the platelet aggregation process has been evaluated in the systems investigated. ZIF-8 induces very low platelet aggregation within the concentration range studied over 5% platelet aggregation percentages and remains below the limit of platelet aggregation risk, 60%, even showing a significant difference by comparing different MOF concentrations to the negative control (0 vs. 3.9, 7.8, 15.6, 31.3, 62.5, 125, 250 or 500 μg mL−1, P = 0.0008, 0.004, 0.03, 0.08, 0.59, 0.01, 0.23, or 0.03, respectively). Meanwhile, PCN-224 did not show aggregating ability within the concentration range studied (P > 0.99). However, the thrombin has a high aggregation capacity of more than 60% and showed a significant difference compared to different MOF concentrations (P < 0.0001). The results obtained for ZIF-8 are in accordance with results in the literature.62 In contrast, no comparison could be made with the results obtained for PCN-224 because no data were found in the bibliography.
These results confirm that ZIF-8 and PCN-224 could be potential candidates for being used as therapeutic platforms in HIV/AIDS treatment.
2.5. Encapsulation of cARTs and TLR agonists into ZIF-8 and PCN-224
Different combinations of antiretroviral agents are being used to achieve virological suppression in PLWH.63,64 Integrase strand transfer inhibitors, nonnucleoside reverse transcriptase inhibitors, nucleos(t)ide reverse transcriptase inhibitors or pharmacologically boosted protease inhibitors are some of the agents combined in these treatments. Here, the two cARTs bictegravir/tenofovir/emtricitabine (BIC/TFV/FTC) and nevirapine/tenofovir/emtricitabine (NVP/TFV/FTC) have been studied.64,65 Emtricitabine and tenofovir are pyrimidinic and purine bases, respectively, of the group of nucleos(t)ide reverse transcriptase inhibitors; bictegravir is an integrase enzyme inhibitor that prevents the integration of DNA into CD4+ T cells; and nevirapine is a nonnucleoside inhibitor whose use was declined due to the availability of a more tolerable therapy, but it is still useful in cases of treatment failure and intolerance to other therapies.Their encapsulation into MOFs could reduce the adverse effects such as skin rashes, diarrhoea, headaches, nephrotoxicity, kidney stone formation, diabetes, etc.66 Administration of small concentrations of cARTs to the patients, due to a controlled release of the drugs into the organism as well as their transport to target cells in anatomical compartments, may diminish the side effects of the drugs and reduce HIV reservoir more efficiently.
On the other hand, TLRs are responsible for recognizing different pathogens and trigger responses aimed at eliminating them. The new strategy of encapsulating TLR agonists in the same nanovehicles containing cARTs, or even the use of solution mixtures containing antiretroviral-loaded MOFs and TLR agonist-loaded MOFs, may not only reduce the HIV viral load, but also reduce or eliminate the HIV reservoirs that are the real cause of the failure to clear the virus in the tissues of PLWH.
The encapsulation efficiency values, % EE, obtained from the encapsulation of the mentioned cARTs and TLR-7 and -9 agonists into ZIF-8 and PCN-224 are collected in Table 1.
| MOFs | cARTs | TLR agonists | |||||
|---|---|---|---|---|---|---|---|
| TFV | FTC | NVP | BIC | TLR-7 | TLR-9 | ||
| Values in this table are given as mean value ± standard deviation. The experiments were repeated thrice independently. cART-1 and cART-2 represent the combined antiretroviral therapy TFV + FTC + NVP and TFV + FTC + BIC, respectively. | |||||||
| ZIF-8 | cART-1 | 98 ± 3 | 89 ± 2 | 96 ± 5 | — | 100 ± 3 | 90 ± 4 |
| cART-2 | 76 ± 5 | 99 ± 2 | — | 88 ± 7 | |||
| PCN-224 | cART-1 | 68 ± 2 | 76 ± 3 | 71 ± 4 | — | 100 ± 2 | 60 ± 3 |
| cART-2 | 88 ± 2 | 76 ± 3 | — | 87 ± 6 | |||
High % EEs were obtained for all the drugs in the MOFs. There is no difference in the % EE value of the TLR-7 agonist in the MOFs, but the encapsulation of the TLR-9 agonist results to be higher in the ZIF-8 (nanometric framework) than in the PCN-224 (micrometric framework). With respect to the antiretrovirals, % EE values are slightly larger in the nano-MOF ZIF-8 than those in the micrometric PCN-224 for most drugs except for BIC were obtained. Therefore, encapsulation of the cARTs and TLR agonists investigated in this work seems to be slightly dependent on the size of the framework or the pore size of the MOF, being higher for the nano-MOF than for the PCN-224 microstructure.
Combinations of antiretroviral agents were previously encapsulated into PLGA, PLGA-PEG, lipid, or glucose-coated gold nanoparticles.67–71 The % EE values obtained in all cases agreed with those obtained here with the ZIF-8 and PCN-224 MOFs investigated. There are only a few works in the literature about the encapsulation of cARTs into MOFs. Two active triphosphorylated antiretroviral agents, azidothymidine triphosphate and lamivudine triphosphate, were encapsulated into the nano-MOF MIL-100 (Fe). The % EE value obtained was about 80%,35 which also agrees with our results. Horcajada et al.72 studied the encapsulation of the antiretrovirals azidothymidine triphosphate and cidofovir into different iron(III) carboxylate nano-MOFs with distinct porous sizes. The results demonstrated that, for the same type of MOFs, an increase of the size of the porous increases the % EE values. This behaviour is opposite to those found here. This demonstrates the importance of the different interactions between the antiretrovirals and the carriers in the encapsulation process.
The presence of the antiretroviral drugs NVP or BIC has also not been found to have much influence on the encapsulation values of TFV or FCT. This demonstrates that there is no interaction between the antiretrovirals into the MOF structure, probably because the location of each antiretroviral into the carriers is different.
2.6. In vitro drug (cART) release from ZIF-8 and PCN-224
The release of the cART encapsulated into the MOFs PCN-224 and ZIF-8 was measured at three different pHs, simulating the potential administration route that could be used in vivo for each framework (see Tables S1, S2, ESI† and Fig. 6). The release of the antiretroviral efavirenz from the zeolitic framework ZIF-8 was previously measured by Tabosa et al. at different pH values.73 The results were higher at an acid medium, increasing from 75% at pH = 6.8 to 90% at pH = 4.5. | ||
| Fig. 6 (A)–(I) Release profiles of antiretrovirals and TLR agonists from ZIF-8 and PCN-224 at different cARTs and pHs. The lines correspond to the best fit using eqn (6). (J)–(K) Rate constants corresponding to the delivery of antiretrovirals at different pHs and cARTs. The experiments were repeated thrice independently. | ||
This clear pH sensitivity has not been observed in the in vitro measurements in the present work for the antiretroviral agents investigated. The results in PCN-224 showed a low percentage of the release of all the antiretroviral agents, without any dependence on neither the cART nor the pH. Some differences were only found for the nanoparticle ZIF-8. Although the release does not depend on the pH in the nano-MOF, a higher release was found for TFV (>90%) for both cARTs. Also, the percentage of NVP released from ZIF-8 is higher (>98–99%) than that for BIC (1–2%). Therefore, the release of some antiretroviral agents (TFV and NVP) is favoured in the nano-MOF ZIF-8 in comparison with the micro-sized structure PCN-224. In contrast, the release of BIC and FTC does change upon varying neither the framework nor the medium pH.
Similar results were found for the TLRs-7 and -9 agonists. Their release was influenced neither by the MOF nature nor by the acidity of the medium, for pH = 2.2 and 5.5. The release of the TLR-9 agonist is much higher than that of TLR-7; that is, the interaction of the TLR-7 agonist with both frameworks seems to be stronger than that of the TLR-9 agonist. A different behaviour was found for the TLR agonists at pH = 7.4 in PCN-224. There is no release of the Toll agents at physiological pH.
In general, the release of the different drugs depends on their interactions (with hydrophobic and π–π interactions being the strongest) with the MOFs as well as on their location in the pores of the carriers. Although the sizes of such pores are larger in PCN-224 than those in ZIF-8 (∼2 nm and ∼1.2 nm, respectively),74,75 a higher quantity of the antiretrovirals TFV and NVP is released from ZIF-8 than that from PCN-224, for all the pH values studied. A larger size of the framework pores does not facilitate the release of the cART. The same relationship was found between the % EE and the pore size. According to these results, not only the interactions of antiretrovirals (or TLR agents) with the MOFs play a main role in the release (or encapsulation) of the drugs, but also their location in the MOFs seems to be important in the release process.
Fig. 6(A)–(I) shows the release profiles obtained for the systems studied. In both, PCN-224 and ZIF-8, the release profiles correspond to first order kinetic processes. The release rate constants were calculated at all the working conditions by using eqn (1):
| % Drugreleased = a(1 − exp(−kt)) |
| Drug = antiretroviral or TLR agonist | (1) |
Fig. 6(J) and (K) and Table S2 (ESI†) show the rate constant values obtained.
Different results were observed for the antiretrovirals and the TLR agents. With respect to the cARTs, changes in the medium pH, as well as variations in the combination of the antiretrovirals, provoke variations in the release way of the drug from both frameworks.
A different behaviour was observed for the antiretrovirals in the nanostructure ZIF-8. So, for example, a rapid release of TFV and NVP in cART-2 occurs from this nano-framework, and faster at pH = 5.5 than at pH = 7.4. This is an excellent result considering that the HIV reservoirs are mainly located in the intestines at a pH range of 5–6. No difference was observed for the other antiretroviral combination (cART-1) with the pH.
In general, a slower release rate in PCN-224 is observed for the antiretroviral agents at the different pHs in comparison with ZIF-8. This could be related to a different location of the drug in the carrier structure, as well as the different interactions between the drug and the carrier. In any case, a progressive release of the drug is an advantage in the therapeutic effect.
With respect to the TLR agonists, the rate constant of the TLR-7 agonist release is about 10 times higher in ZIF-8 than that in PCN-224. No dependence on the pH value is found for either the PCN-224 or the ZIF-8 structure. The TLR-9 agonist is released faster than the TLR-7 agonist in both MOFs for all the different pH values.
In the case of the PCN-224 microstructure, the release of BIC and FTC in cART-1 occurs at a higher rate constant at pH = 2.2 than at pHs 5.5 and 7.4, i.e. this antiretrovirals would be released mainly in the stomach. No differences are observed for the other cART and pH in PCN-224.
It is worth noting that the release rate constant of TLR-7 and -9 agonists is higher at pH = 5.5 than at pH = 2.2 in PCN-224 and higher for pH = 7.4 than for pH = 5.5 in ZIF-8. The rate constant corresponding to the release of the TLR agonists increases upon augmenting the pH value for the MOFs studied.
2.7. Internalization into cells
The main purpose of this study is to test if the encapsulated TLR ligands and antiretroviral drugs into MOFs could be potentially targeted lymph nodes to induce a powerful early immune response and bring forth an effective strategy to eradicate of the HIV reservoirs in CD4+ T cells of infected patients. For this, we need to know if the MOF assemblies can be internalized by themselves into PBMCs, which are the main source of CD4+ T cells. The intake and internalization of the loaded and unloaded ZIF-8 in PBMCs as a cellular model of human HIV reservoirs and HeLa cell lines have been analysed by using epifluorescence microscopy. The probe 5(6)-carboxyfluorescein (CF), encapsulated into ZIF-8, was used as the dye. Fluorescent dyes are used to study the internalization of loaded forms as well as the intracellular delivery and release thanks to the lower leakage rate and excellent pH sensitivity of the probes.76 Weinstein et al., reported that CF loaded into liposomes shows endocytic uptake by lysosomal pathway, and the subsequent diffusion of the protonated dye from the acidic endosomal lumen to the cytoplasm, resulting in a self-quenching phenomenon.76Our data show that the cellular internalization of carboxyfluorescein-loaded ZIF-8 was successful in HeLa cell lines (Fig. 7(A) and (B)) and human PBMCs (Fig. 8(A) and (B)) at a physiological temperature (37 °C) for 1 h and 6 h, and 2 h time point treatment, respectively. For PBMCs, the fluorescence of the probe appears to be uniformly dispersed through the cell suggesting a homogenous cytoplasmic pattern covering the cytoplasm including the nucleus at 37 °C for 2 h time point treatment (Fig. 8(A)).
 | ||
| Fig. 7 Internalization of the ZIF-8 nanostructure in cells. Quantification of the mean fluorescence intensity (MFI) of fluorescence images of fixed HeLa cell lines (A) and (B) in the absence of treatment (cells) or after treatment with ZIF-8 nanoparticle free (ZIF-8), probe alone (5-(6)-carboxyfluorescein dye, CF) and CF-loaded-ZIF-8 at 4 °C and 37 °C for 30 min, 1 h and 6 h time points of incubation. HeLa cells in panels A (1 h of incubation at 37 °C) and B were processed for epifluorescence microscopy at 30 min, 1 h and 6 h. The scale bar in all panels of HeLa cell lines is 50 μm. For each image of different treatment conditions (B), the fluorescence intensity was quantified and subtracted the background signal of the cells alone. MFI (A.U.) was calculated as the mean of three different fields in the glass slides. | ||
 | ||
| Fig. 8 Internalization of the ZIF-8 nanostructure into cells. Quantification of the mean fluorescence intensity (MFI) of fluorescence images of PBMCs (A) and (B) in the absence of treatment (cells) or after treatment with ZIF-8 nanoparticle free (ZIF-8), probe alone (5-(6)-carboxyfluorescein dye, CF) and CF-loaded-ZIF-8 at 4 °C and 37 °C for 30 min, 1 h, 2 h and 6 h time points of incubation. PBMCs in panels A (2 h of incubation at 37 °C) and B were processed for epifluorescence microscopy at 30 min, 1 h, 2 h and 6 h. The scale bar in all panels of PBMCs is 200 μm. For each image of different treatment conditions (B), the fluorescence intensity was quantified and subtracted the background signal of the cells alone. MFI (A.U.) was calculated as the mean of three different fields in the glass slides. | ||
As expected, negligible cellular fluorescence was found after incubation of PBMCs with free ZIF-8 nanoparticles or the probe alone at 37 °C for all time points. Insignificant cellular fluorescence was observed at this temperature after 2 h incubation (Fig. 8(B)). A punctate diffuse pattern of fluorescence throughout the cytoplasmic cell was observed in HeLa cell lines from approximately 1 h of the incubation and maintained until 6 h time points at 37 °C, and a negligible cellular fluorescence after incubation of HeLa cell lines with free ZIF-8 nanoparticles or the probe alone at 37 °C for all time points (Fig. 7(A) and (B)). Data showed that uptake and internalization efficiencies of the probe are considerably higher when the HeLa cell lines were treated with the nano-sized structure ZIF-8 loaded probe for 1 h incubation and maintained for approximately 6 h at 37 °C. In addition, the intake and the internalization efficiency of the loaded ZIF-8 in PBMCs and HeLa cell lines were demonstrated to be very low at 4 °C, due to a low cell activity, for different time points of incubation using epifluorescence microscopy (Fig. 8(A) and (B)). Altogether, data suggest that the cellular uptake and internalization of CF-loaded ZIF-8 were time- and temperature-dependent in both cell types and agree with previous behaviours observed in which nanostructures showed high cellular uptake and likely results in the endosomal release of the organelle-specific drugs to the cells.77 Thereby, data provide the basis for developing an efficient nanoplatform using ZIF-8 as the long acting and stable drug delivery vehicle of antiretroviral drugs in combination with latency reversing agents, such as TLR-7 and -9 agonists, to improve the treatment and prevention of HIV infection.34,35
It is worth noting that PCN-224 is a micrometre-sized MOF. Therefore, the internalization process could only be studied with ZIF-8.
3. Materials and methods
3.1. Materials
Zinc(II) nitrate hexahydrate (Zn(NO3)2·6H2O >99%), 2-methylimidazole (2-Me-Him) and zirconium(IV) chloride (ZrCl4 >99%) were from Sigma-Aldrich (St. Louis, MO, USA). Meso-tetra(4-carboxyphenyl) porphyrin (H4TCPP) was obtained from Frontier Scientific (Newark, DE, USA). The antiretrovirals tenofovir and nevirapine agents were from Sigma Aldrich (St. Louis, MO, USA), bictegravir was from CSN pharm (Arlington Heights, IL, USA) and emtricitabine was from Abcam (Cambridge, UK). TLR-7 and -9 agonists were purchased from Cayman Chemical Company (Ann Arbor, MI, USA) and InvivoGen (San Diego, CA, USA). Other reagents and solvents were purchased as reagent-grade and used without further purification.The buffer solutions used in the study were prepared with HCl/NaCl at pH = 2.2 and NaH2PO4/Na2HPO4 at pHs = 5.5 and 7.4. The pH was adjusted using a pH-meter basic 20+ from Crison (Barcelona, Spain).
All solutions were prepared with deionized water (from a Millipore Milli-Q system, Burlington, U.S.A.) with a conductivity <10−6 S m−1.
3.2. Synthesis methods for MOFs
3.3. Methods of characterization
Powder X-ray diffraction (PXRD) analysis was performed on approximately 10–15 mg of each sample at room temperature using a Bruker D8 Discover diffractometer at 50 kV and 1000 mA for Cu kα (λ = 1.5418 Å), in the range 5°–50° 2θ with a step of 0.02° per 0.5 s.The shape and size of the MOF particles were examined by scanning electron microscopy (SEM), using a FEI Teneo instrument and, specifically for ZIF-8, it was worked in transmission mode (STEM). For this purpose, a droplet of an aqueous suspension of particles was deposited onto a copper grid coated with a thin carbon layer and dried in air. The particle size distributions obtained from the SEM/STEM images were analysed with the ImageJ software.
Fourier transformation infrared spectra (FT-IR) were recorded using an IFS-66a (Bruker). The data were collected from 450 cm−1 to 3500 cm−1 in transmittance mode. The samples were prepared by depositing a MOF suspension drop on top of the Si substrate and dried in an oven at 50 °C.
3.4. In vitro cytotoxicity assays
The cytotoxicity of MOFs was in vitro estimated using the CyQUANT™ LDH cytotoxicity assay.79 Several cell lines from commercial supplier (ATCC®, Manassas, VA, USA) were used, including HeLa (human cervical carcinoma epithelial cells), U937 (human leukaemia monocytic cells), THP-1 (human leukaemia monocytic cells), and Jurkat (human T leukaemia cells) cell lines. The cytotoxicity of these systems was also evaluated in human PBMCs. PBMCs were isolated from the whole blood of three healthy donors, using the Ficoll-Hypaque density gradient centrifugation method80 from Sigma Aldrich (St. Louis, MO, USA). Each cell line was seeded at 10 × 104 cells per well (or at 20 × 104 cells per well for PBMCs) in Nunc flat-bottomed 96-well plates (ThermoFisher Scientific, Waltham, MA, USA) using complete the D-10 or R-10 medium [Dulbecco's modified Eagle medium (DMEM) or Roswell Park Memorial Institute (RPMI) supplemented with 10% of fetal bovine serum (FBS), and penicillin, streptomycin, and L-glutamine], incubated at 37 °C in 5% CO2, and used the same day for PMBCs and the following day (75 to 90% confluence) for all cell lines. FBS used in all experiments was heat inactivated (56 °C, 30 min) prior to use to eliminate complement activity. MOF solutions at different concentrations, two-fold dilutions (500 to 3.9 μg mL−1, final concentration), were added to each well and the plates were incubated for 6 h for PBMCs, or 36 all cell lines, at 37 °C in 5% CO2. The D-10 or R-10 medium alone was used as the negative control. 10 μL of 10× lysis buffer and 10 μL of sterile ultrapure water were added to each set of wells in triplicates and used as the maximum LDH activity and spontaneous LDH activity, respectively. Later, the medium from each well was collected by centrifugation of the plate and used to test the cytotoxicity of the MOF solutions using the CyQUANT™ LDH Cytotoxicity Assay Kit according to manufacturer's suggestion (Invitrogen™ from Thermo Fisher Scientific, Waltham, MA, USA). Cytotoxicity was measured by fluorescence using a CLARIOstar® analyzer (BMG LABTECH, Allmendgrün, Ortenberg, Germany). Each MOF concentration was measured in triplicate and the tests were repeated thrice independently. The cell viability was calculated by using eqn (2): | (2) |
The cell viability values were also checked by the trypan blue (Sigma Aldrich, MO, USA) method81 and no significant differences were observed.
3.5. Flow cytometry
The cell death in PBMCs treated with ZIF-8 and PCN-224 was assessed by flow cytometry using a live/dead fixable aqua dead cell stain (Life Technologies, Carlsbad, CA, USA). Dead cells with damaged membranes are permeable to the live/dead fixable aqua dead cell stain, due to a reaction with free amines in the cytoplasm, producing an intense fluorescent staining. In contrast, this dye cannot pass through cell membranes in viable cells because their membranes are intact.82 For cell death evaluation, 10 × 105 PBMCs were exposed to increasing concentrations, two-fold dilutions (500 to 3.9 μg mL−1, final concentration), of ZIF-8 and PCN-224, respectively, for 6 h and overnight at 37 °C with 5% CO2. 1× PBS (Gibco™ from Thermo Fisher Scientific, Waltham, MA, USA) and dimethyl sulfoxide, 10% DMSO (PanReac AppliChem, Barcelona, Spain) were used as the negative and positive controls, respectively. Then, cells were washed twice with 1× PBS, stained with 5 μL of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 dilution live/dead fixable aqua dead cell stain for 5 minutes in the dark and incubated with 100 μL of binding buffer for 25 more minutes. Cells were analysed using a FACS Canto flow cytometer (BD, Franklin Lakes, NJ, USA) using the FACS Diva software (BD, Franklin Lakes, NJ, USA). 20 × 104 events were acquired per sample. Data were analysed using the FlowJo software v10 (Treestar, Ashland, OR, USA). The cell viability was estimated as described below, eqn (3):
:20 dilution live/dead fixable aqua dead cell stain for 5 minutes in the dark and incubated with 100 μL of binding buffer for 25 more minutes. Cells were analysed using a FACS Canto flow cytometer (BD, Franklin Lakes, NJ, USA) using the FACS Diva software (BD, Franklin Lakes, NJ, USA). 20 × 104 events were acquired per sample. Data were analysed using the FlowJo software v10 (Treestar, Ashland, OR, USA). The cell viability was estimated as described below, eqn (3):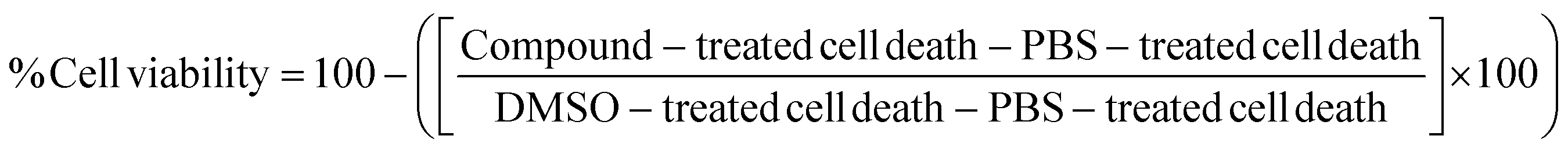 | (3) |
For the assessment of the apoptotic and/or necrotic effect on PBMCs, 10 × 105 cells were treated with increasing concentrations (3.9–500 μg mL−1) of MOFs for 6 h and overnight at 37 °C with 5% CO2. 1× PBS and DMSO were used as the negative and positive controls, respectively. PBMCs were washed twice with cold 1× PBS, resuspended in 100 μL of 1× binding buffer and incubated with 2.5 μL of PE-annexin V and 10 μL of 7-AAD (BD, Franklin Lakes, NJ, USA) for 15 min at room temperature in the dark. Annexin V/7-AAD staining allows discrimination of early apoptosis from late apoptosis and necrosis. Annexin V binds to phosphatidylserine (PS), which is expressed on the outer layer of the plasma membrane of early apoptotic cells.83 7-AAD is a vital dye that can only access the DNA cells in late apoptotic cells and necrotic cells, where the membrane integrity is lost.84 Cells were analysed using a FACS Canto cytometer and the FACS Diva Software (BD, Franklin Lakes, NJ, USA). The gating strategy is shown in Fig. 4(A) and (B). Cell death was evaluated on annexin V vs. 7-AAD dot plots using the FlowJo software v10 (Treestar, Ashland, OR, USA). Acquired data results were shown in a dot-plot format with a quadrant marker, as was recommended by the manufacturer and four different populations were taken into account: annexin V−/7-AAD− cells were considered live cells; annexin V+/7-AAD− cells were considered early apoptotic cells; annexin V+/7-AAD+ cells were considered necrotic or dead cells; and annexin V−/7-AAD+ cells were considered late apoptotic cells.85
3.6. In vitro hemolysis assays
The hemocompatibility of MOFs was determined in human red blood cells (RBCs). Blood samples were extracted from three healthy human donors in vacutainer tubes containing EDTA (BD, Franklin Lakes, NJ, USA). RBCs were isolated by centrifugation at 1800 rpm for 5 min. The supernatant was discarded, and RBCs were washed twice with PBS 1×. Subsequently, 1 mL of the washed RBCs was suspended in 9 mL of 1× PBS and carefully homogenized. The MOF solutions were evaluated at the same concentration values used for in vitro viability assays. ACK (ammonium–chloride–potassium) lysing buffer (Gibco™ from Thermo Fisher Scientific, Waltham, MA, USA) and 1× PBS (Gibco™ from Thermo Fisher Scientific, Waltham, MA, USA) were used as the positive and negative controls, respectively. To evaluate the hemolysis effect, 180 μL of the RBC stock solution was seeded with 20 μL of the different systems into Nunc flat-bottomed 96-well plates (Thermo Fisher Scientific, Waltham, MA, USA), and were incubated at 37 °C in 5% CO2 for 4 h. Then, 96-well plates were centrifuged, and the supernatants were transferred to another flat-bottomed 96-well plates. Each MOF concentration was measured in duplicate and the tests were repeated thrice independently. Finally, the absorbance was read at 540 nm, and the hemolysis percentage was calculated by following eqn (4):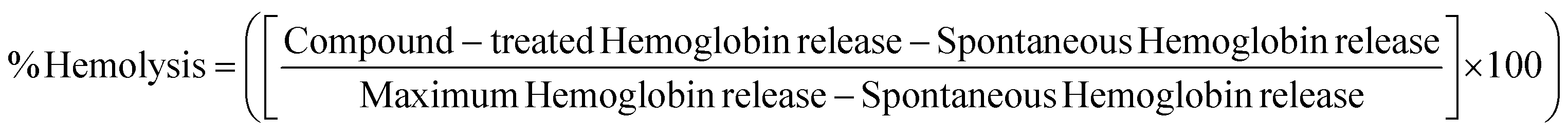 | (4) |
3.7. Platelet aggregation
The effect of PCN-224 and ZIF-8 on platelet aggregation was examined in platelet-rich plasma (PRP). Blood samples were isolated from three healthy human donors in vacutainer tubes containing sodium citrate (BD, Franklin Lakes, NJ, USA). PRP was isolated by centrifugation at 1000 rpm for 15 min. Erythrocytes were discarded, and the supernatant containing PRP was used. 0.2 U.S. of thrombin (MP Biomedicals™, Santa Ana, CA, USA) and 1× PBS (Gibco™ from Thermo Fisher Scientific, Waltham, MA, USA) were used as the positive (C+) and negative (C−) controls, respectively. To evaluate the platelet aggregation, 50 μL of PRP was exposed to 50 μL of the different systems into Nunc flat-bottomed 96-well plates (Thermo Fisher Scientific, Waltham, MA, USA), and were incubated at 37 °C in 5% CO2 for 5 min. Each MOF concentration was measured in duplicate, and the tests were repeated thrice independently. Finally, the absorbance was read at 620 nm, and the platelet aggregation percentage was calculated by using eqn (5):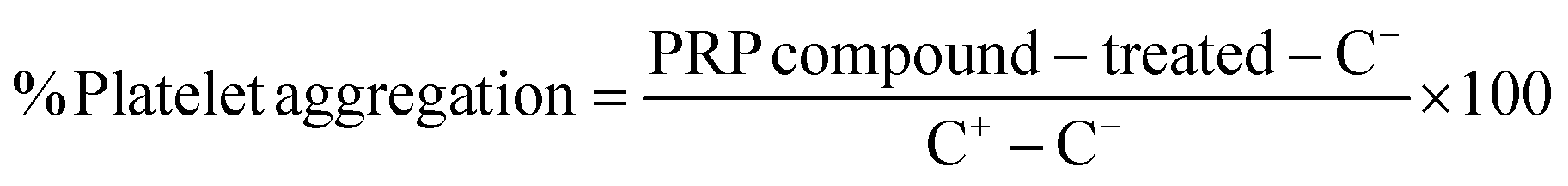 | (5) |
3.8. Encapsulation of cARTs and TLR agonists
An amount of the triplet combination of antiretroviral agents, bictegravir/tenofovir/emtricitabine (cART-1) or nevirapine/tenofovir/emtricitabine (cART-2), was dissolved in 2 mL of aqueous solution and added to 1.0 mg of the MOFs under study. The concentrations of each drug in the mixtures were 30 ppm (ARTs), 25 ppm (TLR-7 agonist) and 6.4 ppm (TLR-9 agonist). The mixture was stirred for 24 h in a vortex at 500 rpm and centrifuged for 15 min at 12500 rpm. After this time, the solid (cART/TLR agonists/MOFs) was separated by ultracentrifugation at 12500 rpm for 15 min and dried under vacuum by using an Eppendorf Concentrator plus for 40 min. The quantity of antiretroviral drugs remaining in the supernatant was quantified by liquid chromatography measurements.
A Xevo G2S-Q-TOF-MS (Waters, Micromass, Manchester, UK) coupled to an acquity H-class ultra-performance liquid chromatography (UPLC) instrument (Waters, Milford, MA, USA) with an orthogonal Z-spray lock spray and electrospray interface (ESI) was used. The chromatographic separation was achieved on an Cortecs® C18 analytical column (100 mm × 2.1 mm i.d., 1.6 μm particle size) (Waters, Milford, MA, USA). A gradient elution was carried out with a mobile phase that consisted of aqueous formic acid solution (0,1% v/v) (A) and acetonitrile (B) at 0.3 mL min−1 for 10 minutes. The gradient started at 99% A for 1 min, then the A composition was decreased linearly from 99% to 80% A in 1 min to subsequently decreasing A until 60% in 3 min more, maintaining a final isocratic step at 60% A and 40% B for 2.5 min more returning then to initial conditions in 1.5 min before a new injection. The injection volume was 10 μL. The column and autosampler tray were maintained at 30 °C and 5 °C, respectively.
The samples were eluted into the mass spectrometer through the turbo ion spray, operating in a positive mode to detect the analytes using full mass acquisition mode, according to the experimental parameters depicted in Table S3 (ESI†). The detection of the analyte ions was confirmed by monitoring the corresponding [M + H]+ ions detailed in Table S3 (ESI†).
The MassLynx V4.1 software was used as the data process platform while quantitation data were processed by the TargetLynxTM V4.1 accurate mass of [M + H]+ analytes obtained in full scan mode with a target retention time tolerance of 0.200 min and a mass tolerance of 5 ppm.
Under these conditions, analytical figures of merit were evaluated in terms of linearity, repeatability, intermediate precision and limits of detection and quantification (LOQ) (see Fig. S4, ESI†). The linearity response was studied using standard solutions of the target compounds within the range 1–1000 μg L−1, in triplicate. Calibration curves were constructed using a least-square linear regression analysis obtaining a correlation coefficient of >0.996 for all the compounds in the range 1–500 μg L−1 for TFV, FTC and NVP and in the range of 10–1000 μg L−1 for BIC. Method LODs and LOQs were calculated as the minimum concentration of analyte giving peaks whose signal to noise ratios are 3 and 10, respectively; therefore, LOQs were 1.0, 2.1, 0.9 and 9.6 μg L−1 for TFV, FTC, NVP and BIC, respectively, and LODs were 0.5, 0.63, 0.55 and 4.6 μg L−1 for TFV, FTC, NVP and BIC, respectively. Repeatability (intraday) and intermediate precision, expressed as the relative standard deviation, were evaluated at three different concentration levels of each compound 5 (15 for BIC), 50 and 250 μg L−1, in one single day for repeatability (in quintuplicate) and one day per week among two months for intermediate precision (in triplicate). Repeatability showed relative standard deviations lower than 4.3, 3.8, 5.6 and 2.1% for TNF, EMT, NVP and BIC, respectively, and intermediate precision results were lower than 6.3, 5.5, 7.1 and 4.2% for TFV, FTC, NVP and BIC, respectively.
The percentage of encapsulated TLR agonists was determined as shown in Scheme 2. A quantity of TLR-7 (or -9) agonist was added to an aqueous solution containing the MOFs. After 24 h of stirring, the solution was centrifuged at 13500 rpm for 15 min and the loaded MOF was decanted. The amounts of TLR-7 and -9 agonists remaining in the supernatant were determined from emission intensity measurements by using RedSafe as the dye. The concentration of RedSafe was 200× in all the assays and the excitation and emission wavelengths were 419 nm and 537 nm, respectively. These measurements were carried out using a Hitachi F-2500 spectrofluorimeter interfaced to a PC for the recording and handling of the spectra and connected to a flow Lauda thermostat to maintain the temperature at 298.0 ± 0.1 K. A standard fluorescence quartz cell of 10 mm path length was used.
 | ||
| Scheme 2 Preparation of samples to determine the quantity of encapsulated TLR agonists into MOFs. | ||
Calibration curves were constructed for the determination of TLR agonists in different media. The concentration of the TLR-9 agonist remaining in the supernatant was determined directly from the calibration curve, constructed at a fixed RedSafe concentration of 200× and different TLR-9 agonist concentrations. The determination of the TLR-7 agonist was performed in a different manner. The dye RedSafe shows a modification in the emission intensity at different TLR-9 agonist concentrations, but no modification is observed when the concentration of the TLR-7 agonist is changed. Therefore, the calibration curve for the determination of the TLR-7 agonist was constructed in the presence of the fixed TLR-9 agonist and RedSafe concentrations (1 μg mL−1 and 200×, respectively) and different concentrations of the TLR-7 agonist.
The encapsulation efficiency (% EE) of each of the drugs in the MOFs was determined from eqn (6).
 | (6) |
The experimental values are the average of three replicates.
3.9. Release of cARTs
The release of each antiretroviral agent was determined adding a quantity of 1.5 mg of each loaded MOF to 2 mL of a buffer solution with the desired pH value. The mixture was stirred by magnetic agitation at 300 rpm at 310 K. At determined time intervals, the mixture was centrifuged at 12500 rpm for 15 min. An aliquot of the supernatant was removed and replaced with an equal quantity of the same buffer. The concentration of the drug was measured by UPLC measurements as was previously mentioned. The experimental values were the average of three replicates, the precision being within 5%.
The release was determined at different pHs, depending on the MOF. So, this was measured at pH values of 2.0, 5.5 and 7.4 for the micrometric structure PCN-224 and 5.5 and 7.4 for the nanoparticle ZIF-8. The pH values mimic the pH of the stomach (2.0), the small intestine and the oesophagus (5.5) and the blood stream (7.4), respectively. Given the size of the two MOFs studied, PCN-224 may be orally administered, whereas the route of administration for the nano-MOF ZIF-8 will be intravenous. This explains why the release for the nanocarrier ZIF-8 was not measured at a pH = 2.2.
3.10. Epifluorescence microscopy
The ZIF-8 cellular uptake was assessed by epifluorescence microscopy86 in HeLa cell lines and human PBMCs. 1 × 105 HeLa cells were seeded in a 24-well tissue culture plate containing sterile coverslips using complete D-10 medium and used after 4-hour incubation to allow adherence of the cells. Then, the cells were treated with free ZIF-8 nanostructures (62 μg mL−1), loaded the probe (5-(6)-carboxyfluorescein, 2 mM), ZIF-8 alone, or probe alone (the latter at the same concentration as that loaded in the ZIF-8 MOF) at 4 °C and 37 °C with 5% CO2 for 30 minutes, 1 h, and 6 h incubation. Cells alone were used as a control to eliminate their background fluorescence. Cells were washed twice with 1× PBS and fixed in 4% p-formaldehyde (PFA) for 30 min. The fluorescent probe was previously encapsulated into the MOF ZIF-8 with 50% efficiency of encapsulation and dialyzed against distilled deionized water to remove the excess of the probe before use.PBMCs were seeded at 1 × 106 cells per well into 24-well plates using the complete R-10 medium and treated as described above for HeLa cell lines at 4 °C and 37 °C with 5% CO2 for 30 min, 1 h, 2 h, and 6 h incubation. Then, cells were washed twice with 1× PBS and fixed in 4% PFA for 30 min. Cell were washed twice with 1× PBS, resuspended using the complete R-10 medium and seeded on poly-L-lysine-coated glass coverslips in 24-well tissue culture plates to allow cell attachment. Poly-L-lysine (100 μg mL−1) was incubated previously for 1 h at room temperature to allow adherence to coverslips in 24-well plates. Finally, coverslips of both cell types were mounted with ProLong Diamond Antifade Mounting with DAPI (Thermo Fisher Scientific, Waltham, MA, USA) to stain the cell nucleus onto glass slides and ZIF-8 uptake was evaluated using an Olympus microscope using 40×/0.75 magnification lens. Mean fluorescence intensity (MFI) was quantified using the Fiji ImageJ software (W. Rasband, National Institutes of Health, NIH). In each treatment condition, the fluorescence intensity was quantified and subtracted the background signal of the cells alone. Mean fluorescence intensity (MFI, A. U.) was calculated as the mean of three different fields in each preparation.
4. Statistical analysis
Tests were performed using the Prism 7.0 software program (GraphPad, San Diego, CA). Statistics were determined using the two-way ANOVA Dunnett multiple comparisons test comparing different MOF concentrations with the negative and positive controls with 95% confidence bounds. Unless otherwise indicated, data represent the mean ± standard deviation (SD), with p < 0.05 considered statistically significant.5. Conclusions
Two MOFs of different sizes (PCN-224 and ZIF-8) were synthesized and characterized by X-ray diffraction, FT-IR spectroscopy, and SEM imaging. In vitro studies of the cell viability, hemocompatibility and platelet aggregation of MOFs were also performed. The results demonstrated the efficacy of these particles as carriers for different therapeutic agents. The routes of administration are different because of their sizes: ZIF-8 is a nanoparticle that can be administered orally or intravenously, while PCN-224 is a microparticle that can only be administered orally.Combinations of antiretrovirals and TLR agents were encapsulated into the MOFs, each with a different function: the former eliminate the circulating virus, while the latter activate the latent virus in the reservoirs so that the antiretrovirals can continue to work. The presence of antiretrovirals and TLR agonists in the same structure is a new way to fight against HIV by eliminating all these reservoirs that are difficult to access.
The high % EE values obtained for all the therapeutic agents were slightly more favorable for the nanostructured ZIF-8 than those for the micro-framework PCN-224. The release rate constants also depended on the working conditions (combination of antiretrovirals, nature of the MOF and pH). According to the results, a decrease in the pore size of the metal–organic frameworks favors both the encapsulation and the release of the drugs. The hydrophobic and the π–π interactions of the drugs with the MOF structures, as well as the location of the drugs into the framework of the carriers, also play an important role.
Epifluorescence microscopy assays have confirmed the ability of the nanostructured ZIF-8 to target drugs inside HeLa cell lines and PBMCs.
These findings will contribute towards improving the efficacy of antiretrovirals and TLR agonists using nanotechnology. The encapsulation of therapeutic agents diminishes their toxicity, increases their bioavailability by enhancing their efficacy, prolongs the half-life in the human organism and promotes the presence of high drug concentrations at the site of infection.
Abbreviations
| MOFs | Metal–organic frameworks |
| HIV | Human immunodeficiency virus |
| cART | Combination antiretroviral therapy |
| HR-TEM | High-resolution transmission electron microscopy |
| SEM | Scanning electron microscope |
| TLR | Toll-like receptor |
| BIC | Bictegravir |
| TFV | Tenofovir |
| FTC | Emtricitabine |
| NVP | Nevirapine |
| PBMCs | Peripheral blood mononuclear cells |
Author contributions
F. J. Ostos and J. A. Lebrón: conceptualization, data curation, formal analysis, methodology, validation, investigation, writing – review and editing, visualization, formal analysis, resources, and software. M. Martínez-Santa, M. López-López, M. L. Moyá, and J. M. Pedrosa: methodology, software, investigation, formal analysis, supervision, formal analysis, and visualization. F. García-Moscoso, D. Rodríguez-Lucena, T. Lopes-Costa, and G. González-Ulloa: methodology, validation, investigation, and software. E. Ruiz-Mateos, E. Bernal, S. Bachiller, and Rut Fernández-Torres: formal analysis, investigation, and visualization. M. Rafii-El-Idrissi Benhnia and P. López-Cornejo: conceptualization, methodology, data curation, formal analysis, investigation, validation, writing – original draft, writing – review and editing, project administration, resources, funding acquisition, and supervision.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financied by the Consejería de Economía, Conocimiento, Empresas y Universidad de la Junta de Andalucía (P20-01234, FQM-206, and FQM-274), the European Union (Feder Funds), and the University of Seville (Ayudas Servicios Generales de la Universidad, CITIUS, 2023/00000306). The authors acknowledge the postdoctoral grant of F. J. Ostos (PAIDI-DOCTOR, Junta de Andalucía with European Social Fund, DOC_00963), J. A. Lebrón (Fundación ONCE funded by the Fondo Social Europeo) and S. Bachiller (Ramón y Cajal 2021 research grant, RYC-2021-031161-I, MCIN/AEI/10.13039/501100011033, European Union NextGenerationEU/PRTR).References
- S. G. Lim, A. Condez and L. W. Poulter, Clin. Exp. Immunol., 1993, 92, 448–454 CrossRef CAS PubMed.
- R. Su, T. Zhang, H. Wang, G. Yan, R. Wu, X. Zhang, C. Gao, X. Li and C. Wang, Inmunology, 2024, 171, 313–443 Search PubMed.
- J. Popovic-Djordjevic, C. Quispe, R. Giordo, A. Kostic, J. S. K. Stankovic, P. V. T. Fokou, K. Carbone, M. Martorell, M. Kumar, G. Pintus, J. Sharifi-Rad, A. O. Docea and D. Calina, Eur. J. Med. Chem., 2022, 233, 114217 CrossRef CAS PubMed.
- P. Thoueille, E. Choong, M. Cavassini, T. Buclin and L. A. Decosterd, J. Antimicrob. Chemother., 2022, 77, 290–302 CrossRef CAS PubMed.
- C. Y. Chiu, J. J. Chang, A. I. Dantanarayana, A. Solomon, V. A. Evans, R. Pascoe, C. Gubser, L. Trautman, R. Fromentin, N. Chomont, J. H. McMahon, P. U. Cameron, T. A. Rasmussen and S. R. Lewin, J. Immunol., 2022, 208, 54–62 CrossRef CAS PubMed.
- J. X. Velasco-Hernandez, H. B. Gershengorn and S. M. Blower, Lancet, 2002, 2, 487–497 CrossRef CAS PubMed.
- S. G. Deeks, S. R. Lewin and D. V. Havlir, Lancet, 2013, 382, 1525–1533 CrossRef PubMed.
- H. Valdez, E. Connick, K. Y. Smith, M. M. Lederman, R. J. Bosch, R. S. Kim, M. Clair, D. R. Kuritzkes, H. Kessler, L. Fox, M. Blanchard-Vargas and A. Landay, AIDS, 2002, 16, 1859–1866 CrossRef CAS PubMed.
- G. Méndez-Lagares, M. C. Romero-Sánchez, E. Ruiz-Mateos, M. Genebat, S. Ferrando-Martínez, M. A. Muñoz-Fernández, Y. M. Pacheco and M. Leal, J. Infect. Dis., 2013, 207, 1221–1225 CrossRef PubMed.
- R. S. De Pablo-Bernal, R. Ramos, M. Genebat, J. Cañizares, M. Rafii-El-Idrissi Benhnia, M. A. Muñoz-Fernández, Y. M. Pacheco, M. I. Galvá, M. Leal and E. Ruiz-Mateos, J. Infect. Dis., 2016, 213, 999–1007 CrossRef CAS PubMed.
- J. Chen, T. Zhou, Y. Zhang, S. Luo, H. Chen, D. Chen, C. Li and W. Li, Front. Cell. Infect. Microbiol., 2022, 12, 945956 CrossRef CAS PubMed.
- L. F. Abadi, F. Damiri, M. Zehravi, R. Joshi, R. Pai, M. Berrada, E. E. S. Massoud, M. H. Rahman, S. Rojekar and S. Cavalu, Polymers, 2022, 14, 3090 CrossRef PubMed.
- M. K. Rothenberger, B. F. Keele, S. W. Wietgrefe, C. V. Fletcher, G. J. Beilman, J. G. Chipman, A. Khoruts, J. D. Estes, J. Anderson, S. P. Callisto, T. E. Schmidt, A. Thorkelson, C. Reilly, K. Perkey, T. G. Reimann, N. S. Utay, K. N. Makamdop, M. Stevenson, D. C. Douek, A. T. Haase and T. W. Schacker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1126–E1134 CrossRef CAS PubMed.
- A. Sigal, J. T. Kim, A. B. Balazs, E. Dekel, A. Mayo, R. Milo and D. Baltimore, Nature, 2011, 477, 95–98 CrossRef CAS PubMed.
- M. J. Buzón, M. Massanella, J. M. Llibre, A. Esteve, V. Dahl, M. C. Puertas, J. M. Gatell, P. Domingo, R. Paredes, M. Sharkey, S. Palmer, M. Stevenson, B. Clotet, J. Blanco and J. Martinez-Picado, Nat. Med., 2010, 16, 460–465 CrossRef PubMed.
- M. Sapuła, M. Suchacz, A. Załęski and A. Wiercińska-Drapało, Viruses, 2022, 14, 122 CrossRef PubMed.
- M.-S. Yadavar-Nikravesh, S. Ahmadi, A. Milani, I. Akbarzadeh, M. Khoobi, R. Vahabpour, A. Bolhassani and H. Bakhshandeh, Adv. Powder Technol., 2021, 32, 3161–3173 CrossRef CAS.
- B. Eshaghi, J. Fofana, S. B. Nodder, S. Gummuluru and B. M. Reinhard, ACS Appl. Mater. Interfaces, 2022, 14, 2488–2500 CrossRef CAS PubMed.
- S. Mandal, P. K. Prathipati, M. Belshan and C. J. Destache, Antiviral Res., 2019, 167, 83–88 CrossRef CAS PubMed.
- G. Singh and R. S. Pai, Sci. World J., 2014, 30, 583090 Search PubMed.
- M. J. Faria, C. M. Lopes, J. das Neves and M. Lúcio, Pharmaceutics, 2021, 13, 1294 CrossRef CAS PubMed.
- J. P. Freeling, J. Koehn, C. Shu, J. Sun and R. J. Ho, AIDS Res. Hum. Retroviruses, 2015, 107, 107–114 CrossRef PubMed.
- J. C. Kraft, L. A. McConnachie, J. Koehn, L. Kinman, J. Sun, A. C. Collier, C. Collins, D. D. Shen and R. J. Y. Ho, J. Controlled Release, 2018, 275, 229–241 CrossRef CAS PubMed.
- S. Rojekar, L. F. Abadi, R. Pai, K. Mahajan, S. Kulkarni and P. R. Vavia, Eur. J. Pharm. Sci., 2021, 164, 105916 CrossRef CAS PubMed.
- S. Rojekar, R. Pai, L. F. Abadi, K. Mahajan, M. K. Prajapati, S. Kulkarni and P. Vavia, Int. J. Pharm., 2021, 607, 120986 CrossRef CAS PubMed.
- S. Rojekar, L. F. Abadi, R. Pai, M. K. Prajapati, S. K. Pradeep and R. Vavia, AAPS PharmSciTech, 2022, 23, 230 CrossRef CAS PubMed.
- K. Mahajan, S. Rojekar, D. Desai, S. Kulkarni, G. Bapat, S. Zinjarde and P. Vavia, AAPS PharmSciTech, 2021, 22, 171 CrossRef CAS PubMed.
- H. Daraee, A. Etemadi, M. Kouhi, S. Alimirzalu and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 381–391 CrossRef CAS PubMed.
- N. Yahi, J. Fantini, S. Baghdiguian, K. Mabrouk, C. Tamalet, H. Rochat, J. Van Rietschoten and J. M. Sabatier, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 4867–4871 CrossRef CAS PubMed.
- E. Pretzer, D. Flasher and N. Düzgüneş, Antiviral Res., 1997, 34, 1–15 CrossRef CAS PubMed.
- C. R. Quijia, R. C. Alves, G. Hanck-Silva, R. C. G. Frem, G. Arroyos and M. Chorilli, Crit. Rev. Microbiol., 2021, 48, 161–196 CrossRef PubMed.
- C. Jeyaseelan, P. Jain, D. Soin and D. Gupta, Inorg. Nano-Metal Chem., 2021, 1, 1463–1475 Search PubMed.
- N. Ji, Y. Hong, Z. Gu, L. Cheng, Z. Li and C. Li, J. Agric. Food Chem., 2018, 66, 4244–4250 CrossRef PubMed.
- V. Agostini, T. Chalati, P. Horcajada, H. Willaime, R. Anand, N. Semiramoth, T. Baati, S. Hall, G. Maurin, H. Chacun, K. Bouchemal, C. Martineau, F. Taulelle, P. Couvreur, C. Rogez-Kreuz, P. Clayette, S. Monti, C. Serre and R. Gref, Adv. Healthcare Mater., 2013, 2, 1630–1637 CrossRef PubMed.
- M. T. Marcos-Almaraz, R. Gref, V. Agostoni, C. Kreuz, P. Clayette, C. Serre, P. Couvreur and P. Horcajada, J. Mater. Chem. B, 2017, 5, 8563–8569 RSC.
- C. R. Quijia, C. Lima, C. Silva, R. C. Alves, R. Frem and M. Chorilli, J. Drug Delivery Sci. Technol., 2021, 61, 102217 CrossRef CAS.
- A. M. Chávez, A. Rey, J. López, P. M. Álvarez and F. J. Bertrán, Sep. Purif. Technol., 2021, 255, 117660 CrossRef.
- Y. Wang, W. Sun, Y. Li, X. Zhuang, C. Tian, F. Luan and X. Fu, Microchem. J., 2021, 167, 106332 CrossRef CAS.
- Z. Jia, Y. Ma, L. Yang, C. Guo, N. Zhou, M. Wang, L. He and Z. Zhang, Biosens. Bioelectron., 2019, 133, 55–63 CrossRef CAS PubMed.
- J. Cravillon, S. Müncer, S.-J. Lohmeier, A. Feldhoff, K. Huber and M. Wiebcke, Chem. Mater., 2009, 21, 1410–1412 CrossRef CAS.
- M. Adnan, K. Li, L. Xu and Y. X. Yan, Catalysts, 2018, 8, 96 CrossRef.
- R. Zamiri, A. Rebelo, G. Zamiri, A. Adnani, A. Kuashal, M. S. Belsley and J. M. F. Ferreira, RSC Adv., 2014, 4, 20902–20908 RSC.
- N. Huang, S. Yuan, H. Drake, X. Yang, J. Pang, J. Qin, J. Li, Y. Zhang, Q. Wan, D. Jiang and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 18590–18597 CrossRef CAS PubMed.
- Y. Zong, S. Ma, J. Gao, M. Xu, J. Xue and M. Wang, ACS Omega, 2021, 6, 17228–17238 CrossRef CAS PubMed.
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed.
- F. Hao, Z.-Y. Yan and X.-P. Yan, Sci. Total Environ., 2022, 833, 155309 CrossRef CAS PubMed.
- Y. Zhang, Q. Wang, G. Chen and P. Shi, Inorg. Chem., 2019, 58, 6593–6596 CrossRef CAS PubMed.
- M. Hoop, C. F. Walde, R. Riccò, F. Mushtaq, A. Terzopoulou, X.-Z. Chen, A. J. de Mello, C. J. Doonan, P. Falcaro, B. J. Nelson, J. Puigmartí-Luis and S. Panéa, Appl. Mater. Today, 2018, 11, 13–21 CrossRef.
- M. Zheng, S. Liu, X. Guan and Z. Xie, ACS Appl. Mater. Interfaces, 2015, 7, 22181–22187 CrossRef CAS PubMed.
- L. R. De Moura Ferraz, A. É. G. A. Tabosa, D. D. S. Da Silva Nascimento, A. S. Ferreira, V. de Albuquerque Wanderley Sales, J. Y. R. Silva, S. A. Júnior, L. A. Rolim, J. J. de Souza Pereira and P. J. Rolim-Neto, Sci. Rep., 2020, 10, 16815 CrossRef CAS PubMed.
- M. Maes, T. Vanhaecke, B. Cogliati, S. C. Yanguas, J. Willebrords, V. Rogiers and M. Vinken, Methods Mol. Biol., 2015, 1250, 349–361 CrossRef CAS PubMed.
- S. Jin, L. Weng, Z. Li, Z. Yang, L. Zhu, J. Shi, W. Tang, W. Ma, H. Zong and W. Jiang, J. Mater. Chem. B, 2020, 8, 4620–4626 RSC.
- H. Yang, Y. Zhang, L. Zeng, W. Yin, Y. Xu, J. Chen, S. Y. Liu, X. Zou, Z. He and Z. Dai, Small Methods, 2022, 6, 2101391 CrossRef CAS PubMed.
- X. Chen, Y. Huang, H. Chen, Z. Chen, J. Chen, H. Wang, D. Li and Z. Su, J. Nanobiotech., 2022, 20, 1–18 CrossRef PubMed.
- A. Tan, S. Gagné, I. A. Lévesque, S. Lachance, N. Boudreau and A. Lévesque, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 901, 79–84 CrossRef CAS PubMed.
- Y. Yuan, H. Wu, H.-F. Lu, Y. Zheng, J.-Y. Ying and Y.-G. Zhang, Chem. Commun., 2019, 55, 699–702 RSC.
- C. E. Torres, J. Cifuentes, S. C. Gómez, V. Quezada, K. A. Giraldo, P. R. Puentes, L. Rueda-Gensini, J. A. Serna, C. Muñoz-Camargo, L. H. Reyes, J. F. Osma and J. C. Cruz, Pharmaceutics, 2022, 14, 315 CrossRef CAS PubMed.
- X. Gu and A. F. Palmer, ACS Appl. Nano Mater., 2022, 5, 5670–5679 CrossRef CAS.
- D. Luo, C. Wang, Y. Tong, C. Liu, Y. Xiao, Z. Zhu, D. Liua and Y. Wang, RSC Adv., 2020, 10, 7360–7367 RSC.
- J. Hu, W. Wu, Y. Qin, C. Liu, P. Wei, J. Hu, P. H. Seeberger and J. Yin, Adv. Funct. Mater., 2020, 30, 1910084 CrossRef CAS.
- S. K. Bandyopadhyay, M. Azharuddin, A. K. Dasgupta, B. Ganguli, S. SenRoy, H. K. Patra and S. Deb, Front. Bioeng. Biotechnol., 2019, 7, 163 CrossRef PubMed.
- J. Lin, L. Huang, H. Ou, A. Chen, R. Xiang and Z. Liu, RSC Adv., 2021, 11, 21414–21425 RSC.
- E. D. Deeks, Drugs, 2018, 78, 1817–1828 CrossRef CAS PubMed.
- R. T. Gandhi, R. Bedimo, J. F. Hoy, R. J. Landovitz, D. M. Smith, E. F. Eaton, C. Lehmann, S. A. Springer, P. E. Sax, M. A. Thompson, C. A. Benson, S. P. Buchbinder, C. del Rio, J. J. Eron Jr., H. F. Günthard, J.-M. Molina, D. M. Jacobsen and M. S. Saag, JAMA, 2023, 329, 63–84 CrossRef CAS PubMed.
- J. Y. Feng, J. K. Ly, F. Myrick, D. Goodman, K. L. White, E. S. Svarovskaia, K. Borroto-Esoda and M. D. Miller, Retrovirology, 2009, 6, 44 CrossRef PubMed.
- F. Everson, A. Genis, T. Ogundipe, P. De Boever, N. Goswami, A. Lochner, D. Blackhurst and H. Strijdom, PLoS One, 2018, 13, e0208537 CrossRef PubMed.
- A. S. Nowacek, R. L. Miller, J. McMillan, G. Kanmogne, M. Kanmogne, R. L. Mosley, Z. Ma, S. Graham, M. Chaubal and J. Werling, Nanomedicine, 2009, 4, 903–917 CrossRef CAS PubMed.
- J. C. Kraft, L. A. McConnachie, J. Koehn, L. Kinman, J. Sun, A. C. Collier, C. Collins, D. D. Shen and R. J. Y. Ho, J. Controlled Release, 2018, 275, 229–241 CrossRef CAS PubMed.
- W. Li, F. Yu, Q. Wang, Q. Qi, S. Su, L. Xie, L. Lu and S. Jiang, AIDS, 2016, 30, 827–838 CrossRef CAS PubMed.
- M. Kovochich, M. D. Marsden and J. A. Zack, PLoS One, 2011, 6, e18270 CrossRef CAS PubMed.
- A. Bowen, E. E. Sweeney and R. Fernandes, Front. Immunol., 2020, 11, 789 CrossRef CAS PubMed.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- A. E. G. A. Tabosa, A. S. Ferreira, N. M. da Silva, D. D. S. S. Nascimento, L. R. M. Ferraz, J. Y. R. Silva, S. A. Junior, R. M. F. da Silva, L. A. Rolim and P. J. Rolim-Neto, Curr. HIV Res., 2020, 18, 396–404 CrossRef PubMed.
- C. L. Hobday, C. H. Woodall, M. J. Lennox, M. Frost, K. Kamenev, T. Düren, C. A. Morrison and S. A. Moggach, Nat. Commun., 2018, 9, 1429 CrossRef PubMed.
- Y. Hou, X.-J. Hu, H.-Y. Tong, Y.-B. Huang and R. Cao, Inorg. Chem. Commun., 2020, 114, 107825 CrossRef CAS.
- J. N. Weinstein, S. Yoshikami, P. Henkart, R. Blumenthal and W. A. Hagins, Science, 1977, 195, 489–492 CrossRef CAS PubMed.
- S. Zhang, J. Li, G. Lykotrafitis, G. Bao and S. Suresh, Adv. Mater., 2009, 21, 419–424 CrossRef CAS PubMed.
- D. W. Feng, W.-C. Chung, Z. W. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed.
- D. Pranantyo, E. Kang and M. B. Chan-Park, Biomater. Sci., 2021, 9, 1627–1638 RSC.
- I. J. Fuss, M. E. Kanof, P. D. Smith and H. Zola, Curr. Protoc. Immunol., 2009, 85, 7.1.1–7.1.7 Search PubMed.
- W. Strober, Curr. Protoc. Immunol., 2015, 111, A3.B.1–A3.B.3 Search PubMed.
- S. P. Perfetto, P. K. Chattopadhyay, L. Lamoreaux, R. Nguyen, D. Ambrozak, R. A. Koup and M. Roederer, Curr. Protoc. Cytom., 2010, 53, 9–34 Search PubMed.
- M. J. Stoddart, Mammalian Cell Viability: Methods and Protocols, Humana Press, Hatfield, 1st edn, 2011 Search PubMed.
- M. Zimmermann and N. Meyer, Methods Mol. Biol., 2011, 740, 57–73 CrossRef CAS PubMed.
- S. Dwivedi, Q. Saquib, A. A. Al-Khedhairy and J. Musarrat, Toxicology, 2012, 302, 77–87 CrossRef CAS PubMed.
- D. J. Webb and C. M. Brown, Methods Mol. Biol., 2013, 931, 29–59 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00272e |
| ‡ Authors with equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |