
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of Au(III) with amino acids: a vade mecum for medicinal chemistry and nanotechnology†
Edoardo Jun
Mattioli
 ,
Beatrice
Cipriani
,
Francesco
Zerbetto
,
Tainah Dorina
Marforio
* and
Matteo
Calvaresi
*
,
Beatrice
Cipriani
,
Francesco
Zerbetto
,
Tainah Dorina
Marforio
* and
Matteo
Calvaresi
*
Dipartimento di Chimica ‘‘G. Ciamician’’, Alma Mater Studiorum – Universita di Bologna, via F. Selmi 2, 40126 Bologna, Italy. E-mail: tainah.marforio2@unibo.it; matteo.calvaresi3@unibo.it
First published on 16th April 2024
Abstract
Au(III) is highly reactive. At odds with its reduced counterpart, Au(I), it is hardly present in structural databases. And yet, it is the starting reactant to form gold nanoclusters (AuNCs) and the constitutive component of a new class of drugs. Its reactivity is a world apart from that of the iso-electronic Pt(II) species. Rather than DNA, it targets proteins. Its interaction with amino acid residues is manifold. It can strongly interact with the residue backbones, amino acid side chains and protein ends, it can form appropriate complexes whose stabilization energy reaches up to more than 40 kcal mol−1, it can affect the pKa of amino acid residues, and it can promote charge transfer from the residues to the amount that it is reduced. Here, quantum chemical calculations provide quantitative information on all the processes where Au(III) can be involved. A myriad of structural arrangements are examined in order to determine the strongest interactions and quantify the amount of charge transfer between protonated and deprotonated residues and Au(III). The calculated interaction energies of the amino acid side chains with Au(III) quantitatively reproduce the experimental tendency of Au(III) to interact with selenocysteine, cysteine and histidine and negatively charged amino acids such as Glu and Asp. Also, aromatic residues such as tyrosine and tryptophan strongly interact with Au(III). In proteins, basic pH plays a role in the deprotonation of cysteine, lysine and tyrosine and strongly increases the binding affinity of Au(III) toward these amino acids. The amino acid residues in the protein can also trigger the reduction of Au(III) ions. Sulfur-containing amino acids (cysteine and methionine) and selenocysteine provide almost one electron to Au(III) upon binding. Tyrosine also shows a considerable tendency to act as a reductant. Other amino acids, commonly identified in Au–protein adducts, such as Ser, Trp, Thr, Gln, Glu, Asn, Asp, Lys, Arg and His, possess a notable reducing power toward Au(III). These results and their discussion form a vade mecum that can find application in medicinal chemistry and nanotech applications of Au(III).
1. Introduction
Amino acids, the basic building blocks of proteins, interact with Au(III)1,2 to determine the pharmacological profile of Au(III) drugs,3–9 and trigger the protein-assisted synthesis of gold nanoclusters (AuNCs).10–14Au(III) compounds are isoelectronic with Pt(II) complexes. In medicinal chemistry, it was initially suggested that the mechanism of pharmacological activity of Au(III) compounds would be similar to that of Pt(II)-compounds, i.e., interaction with DNA. It then appeared that the modes of action of Au(III) compounds are multifaceted and distinct from those of Pt species.3–9,15–22 On the one hand, there is evidence suggesting that rather than nucleic acids some specific protein targets primarily mediate the biological effects of Au(III) compounds.3–9,15–24 On the other hand, the cytotoxic activity of some Au(III) compounds exploit metalation and inactivation of a few key intracellular proteins that are effective cancer targets.3–9,15–23,25
Proteins further offer the opportunity to synthesize AuNCs directly in their pockets through a series of sequential steps of gold deposition, reduction, and aggregation.10–14 A variety of functional groups, such as sulfhydryl, hydroxyl, carboxyl and amine groups, are suitable for accumulating and reducing Au(III) ions.10–14 The bulky protein can also protect sterically the resulting AuNCs.10–14 Many proteins (for instance, bovine and serum albumin, lysozyme, transferrin, trypsin, pepsin, insulin and others) were used to synthesize protein–AuNC hybrids.10–14 External reducing agents (i.e., NaBH4) or some of the protein amino acid residues trigger the reduction of the Au(III) ions leading to the synthesis of the AuNCs.10–14 During the AuNC formation process, amino acid residues play a three-fold role of being anchoring sites for gold ions, reducing agents, and stabilizers.10–14 AuNCs are among the most promising theranostic agents for applications in nanomedicine.10,26–33
Interaction of Au(III) with proteins may also be useful for (i) developing innovative photodynamic and photoactivated therapies;34,35 (ii) radiotherapeutic purposes, since Au possesses two β-emitting isotopes, 198Au and 199Au,9 and the proteins can be used as carriers;36 (iii) catalysis and metal-mediated transformations in biological systems.37
The increasing interest in interactions between Au(III) and amino acids/peptides/proteins spurred a thorough investigation of the chemical mechanism governing their binding.38–46 Comparison of the structural characteristics of known Au–protein adducts contained in the Protein Data Bank (PDB)2,47 revealed that cysteine is the most preferred residue for Au binding, followed by histidine.2 Side chains of other amino acids, such as Lys, Arg, Gln, Met, Glu, Asp, Asn, Thr, and Ser, as well as the terminal residues of proteins, interact with Au.2 Sulfur, nitrogen, and oxygen lone pairs commonly bind Au ions.2
Au ions can interact with more than one side chains (i.e. His/Cys, His/Gln, Cys/Cys, Cys/Asp, or Cys/Asn dyads) to complete their coordination spheres or substitute ligands already present in the initial Au complex.2 The binding of Au ions to proteins generally does not significantly affect the overall structure of the protein.2
Amino acids can be the starting point to determine the mechanisms that control peptide/protein interactions with Au(III) ions.48 Computational chemistry represents a tool to understand and design gold–bio interfaces.49–52 Systematic studies of the interaction of amino acids with the Au(111) surface53,54 or with the Au(I) ion55,56 were already carried out, but surprisingly this analysis is lacking for Au(III). Here, using quantum chemical calculations, we investigated the interactions between twenty-one proteinogenic amino acids (including selenocysteine) and Au(III) ions.
2. Results and discussion
All the possible interaction sites between Au(III) and a protein/peptide were considered: (i) the protein/peptide backbone, (ii) the amino acid (AA) side chains, and (iii) N-terminal and C-terminal amino acids (Scheme 1). To mimic the usual interactions between Au(III) and an amino acid inserted into the peptide/protein sequence, the amino acids were capped at the N- and C-termini with acetyl (ACE) and n-methyl amide (NME) groups (ACE-AA-NME), while the N-terminal and C-terminal amino acids were capped only on one side. | ||
| Scheme 1 (A) Amino acids (in black) in a peptide/protein chain. (B) N-terminal and (C) C-terminal amino acids. On the left is the real system. On the right is the model system used in the calculations, capped with ACE/NME residues. Structures 1 to 4 represent the possible interaction sites of an Au(III) ion in a peptide/protein. 1. Peptide/protein backbone. 2. Amino acid side chains. 3. N-terminal and 4. C-terminal amino acids. | ||
2.1 Interactions of Au(III) with the peptide/protein backbone
Structural, spectroscopic and computational investigations of the interactions between Au(III) ions and model peptides indicated that Au(III) can(a) deprotonate and bind the amide nitrogen of a peptide bond and/or the terminal amino nitrogen atom of these peptides,57,58
(b) be involved in the binding of the terminal carboxylate groups of the peptides,59–63
In the interaction of Au(III) with the –NH3+ and –COO− terminals, the binding of the Au(III) ion may induce deamination and decarboxylation of the amino acids.64,65
The interactions of Au(III) with the backbone and the N, C terminal tails usually take place cooperatively. To dissect and separate the individual contributions, we considered them singularly and calculated the interaction of the Au(III) ion with (i) the amino nitrogen at the N-terminal (Fig. 1A), (ii) the amide nitrogen (Fig. 1B) or the (iii) the carbonyl (Fig. 1C) of the peptide/protein backbone, and (iv) the carboxylate oxygen atoms at the C-terminal (Fig. 1D).
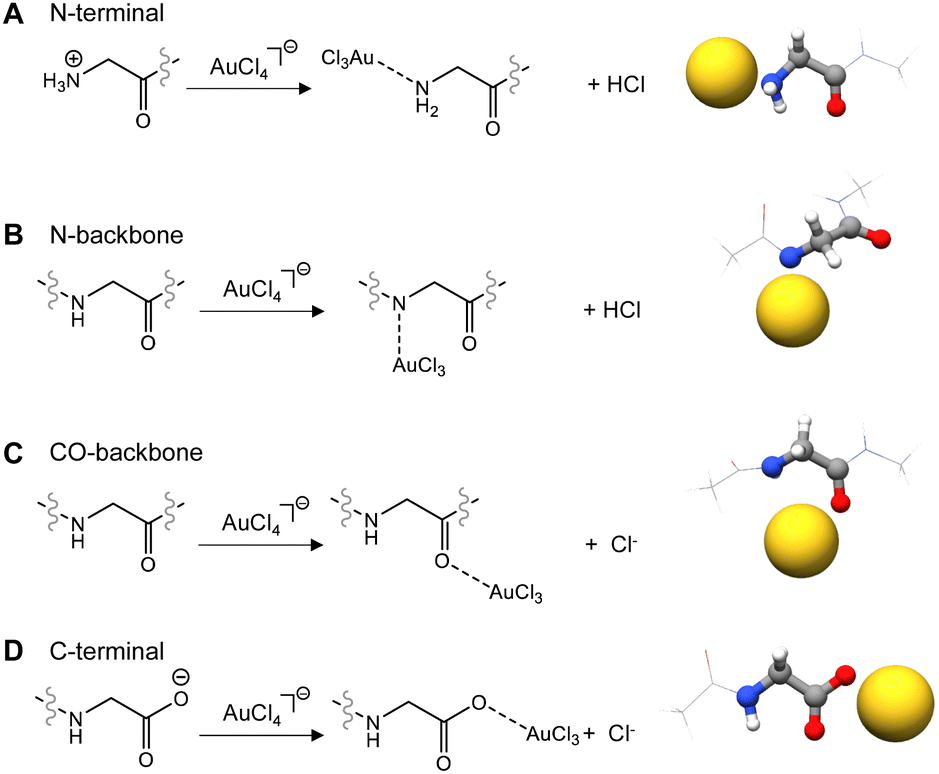 | ||
Fig. 1 Interaction of Au(III) with the (A) N-terminal amino acid, (B) N–H peptide/protein backbone, (C) C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peptide/protein backbone, and (D) C-terminal amino acid. Chlorine atoms are removed for clarity. O peptide/protein backbone, and (D) C-terminal amino acid. Chlorine atoms are removed for clarity. | ||
When the Au(III) ion interacts with the peptide bond, perhaps counterintuitively, the binding with the amide nitrogen (ΔEbinding = −13.1 kcal mol−1) is strongly favoured with respect to binding with the carbonyl group (ΔEbinding = −5.1 kcal mol−1).
The ability of Au(III) to induce amide deprotonation upon binding, already experimentally demonstrated,57,58 explains this behaviour, making the lone pair of the amide available for the interaction with the Au(III) ion. Under physiological conditions, when the Au(III) ion interacts with the terminal amino acids, the binding with the carboxylate oxygen atoms of the C-terminal residue (ΔEbinding = −17.1 kcal mol−1) is preferred with respect to the interaction with the N-terminal amino nitrogen (ΔEbinding = −11.1 kcal mol−1), which becomes deprotonated upon Au(III) binding. When a neutral N-terminal group is taken into account, for example in solutions where the pH is above 10, the ΔEbinding between Au(III) and the lone pair of the amine nitrogen becomes strongly favoured, with a ΔEbinding value of −36.5 kcal mol−1.
However, since peptide bonds are usually engaged in the formation of secondary structures, the side chains of amino acids are important hot-spots to bind Au(III).
2.2 Interactions of Au(III) with amino acid side chains at a glance
We determined the interaction energies of the amino acid side chains with Au(III), ranking their propensity to interact with the Au(III) ion (Fig. 2). A systematic search was carried out to analyse all possible binding sites of Au(III). We considered the amino acid side chains in their physiological protonation state. | ||
| Fig. 2 Ranking of the interaction of Au(III) with amino acid side chains in their physiological protonation state. For comparison, the interaction energies of Au(III) with the N-terminal (blue line), N-backbone (green line), C-terminal (red line), and O-backbone (orange line) are shown. The side chains of SEL, CYS, HIS, GLU, TRP, ASP, MET, TYR, and LYS can successfully compete with the backbone to host Au(III). | ||
The rank quantitatively reproduces the experimental tendency of Au(III) to interact with selenocysteine, cysteine and histidine and negatively charged amino acids such as Glu and Asp, which is observed by the analysis of the interactions of the known Au–protein adducts in the PDB.2
From the calculation, we can also classify the tendency of different amino acids to reduce Au(III), or alternatively to be oxidized by Au(III), considering the charge transfer from the amino acid to the Au(III) ion upon binding (Fig. 3).
 | ||
| Fig. 3 Ranking of the reduction potential of amino acid side chains toward Au(III). For comparison, the reduction potential of the N-terminal (blue line), N-backbone (green line), C-terminal (red line), and O-backbone (orange line) toward Au(III) is shown. The side chains of SEL, CYS, TYR, MET, SER, TRP, THR, and GLN show a tendency to reduce Au(III) better than the backbone. | ||
The amount of charge transfer from the various moieties to the Au(III) provides a proxy for the tendency to reduce Au(III). Structural data of protein/gold complexes suggest that the reduction of Au(III) into Au(I) determines the mechanism of action of many biologically active Au(III) compounds, viz. the oxidation of catalytic residues such as selenocysteine or cysteine.2 Following the binding of Au(III) ions to specific amino acid residues in a protein pocket, the reduction of Au(III) is also a crucial step for the formation of protein-protected AuNCs.10
Amino acids containing sulfur (cysteine and methionine) and selenium (selenocysteine) provide almost one electron to Au(III) upon binding. Tyrosine also shows a considerable tendency to act as a reductant. Other amino acids, commonly identified in Au–protein adducts, such as Ser, Trp, Thr, Gln, Glu, Asn, Asp, Lys, Arg and His, possess a notable reducing power toward Au(III).
These results indicate that, when Au(III) is bound in a protein by a dyad/triad of reducing amino acids, it can spontaneously be reduced to Au(I), without the need for an external reductant. Such a behaviour is observed in many drugs and AuNCs,2,10 see for instance the interaction of the drug Auoxo6 with the model protein bovine pancreatic ribonuclease66 or the binding of Au ions in Apo-ferritin (Fig. 4).67
 | ||
| Fig. 4 (A) Interaction of the drug Auoxo6 with the model protein bovine pancreatic ribonuclease (PDB:4MXF).66 The Au atom is simultaneously coordinated with imidazoles of His12 and His119. (B) Linear coordination structure of the Au atom with Cys 48 and Cys54 in apo-ferritin (PDB:5GU3) before the formation of AuNCs.67 In both cases, the coordination geometries are nearly linear, strongly indicating that gold is in the +1 oxidation state. | ||
Below, we analyse the results in detail, grouping the amino acids in families, following their chemical characteristics.
Selenocysteine (Sec) is considered to be the 21st proteinogenic amino acid. Sec is a cysteine analogue with selenium instead of sulfur. There are about 25 selenoproteins, i.e., proteins that contain Sec, known in humans. They play biological functions essential for preserving redox equilibrium.
The biological activity of many Au(III) drugs are usually mediated via Au–S or Au–Se coordination via ligand replacement with protein thiols/selenols.2 By directly inhibiting the active sites, cysteine (or selenocysteine) in proteins, Au(III) compounds can cause cytotoxicity.2 For example, thioredoxin reductase (TrxR) enzymes possesses a Gly–Cys–Sec–Gly C-terminal active site motif, which is the target of several Au(III) drugs,73 explaining their anti-tumor proliferative activities.
The most important difference between Cys and Sec is the basicity of selenolate, that is 3–4 pKa units lower than that of thiolate, due to the weaker bond to hydrogen and the larger size and increased polarizability of Se.74 Thus, at neutral pH, Cys (pKa = 8.25) is mostly in the protonated state whereas selenocysteine is virtually entirely ionized to a selenolate (pKa = 5.43).74 This behaviour determines the difference in the binding affinity of the Au(III) ion to Sec and Cys (Fig. 5). Even if the binding of Au(III) is able to deprotonate the Cys, the interaction energy of Au(III) with the negatively charged Se (ΔEbinding = −44.7 kcal mol−1) is obviously higher than that with Cys (ΔEbinding = −27.6 kcal mol−1).
 | ||
| Fig. 5 Interaction of Au(III) with (A) selenocysteine (Sec), (B) cysteine (Cys), and (C) methionine (Met). Chlorine atoms are removed for clarity. | ||
However, even in a physiological environment, the thiol may exist as a thiolate.75 Electropositive local environments tend to lower the pKa of Cys by stabilizing the thiolate.75 α-Helices exhibit a dipole moment with a more positive charge towards the N-terminal end that stabilizes the thiolate form of the Cys residues located in this region.75 Also, the presence of specific residues near Cys can modify its pKa, for example proximity of Cys to His usually determine the deprotonation of the SH group to form a thiolate–imidazolium ion pair.76 In this case, when Au(III) interacts with the thiolate, the interaction energy between Au(III) and Cys is comparable (ΔEbinding = −45.4 kcal mol−1) to that observed with Sec.
Also, Met strongly interacts with Au(III) (ΔEbinding = −17.4 kcal mol−1), even if it remains in its neutral form upon interaction with gold because of the lack of acid hydrogens on the sulfur atom, demonstrating the natural affinity of the sulfur atom toward gold.
Histidine was analysed individually because it is difficult to classify this amino acid residue in a single family because it is an aromatic amino acid, potentially charged in a physiological environment, it is polar and characterized by acid–base properties (a characteristic relevant to the catalytic mechanism of many enzymes). The unique chemical properties of His are due to its imidazole ring. At physiological values (the pKa of the imidazole group in proteins is 6.04), the two nitrogens of the imidazole ring can bind or release a proton to form the acid or the base form of His. In its neutral form, two tautomers of His exist (N1–H or Nδ and N3–H or Nε tautomers), depending on which nitrogen the proton of the imidazole ring resides.
Interactions with the Au(III) indicate that histidine in its neutral form interacts using preferentially the lone pairs of the histidine imidazole nitrogens (Fig. 6), with the Nδ tautomer slightly preferred (ΔEbinding = −25.5 kcal mol−1) to the Nε tautomer (ΔEbinding = −23.1 kcal mol−1). Due to the resonance effect, also the C4 position of the His ring represents a potential Au(III) binding site, even if these interactions are considerably smaller (ΔEbinding = −10.3 kcal mol−1), when compared to the imidazole nitrogens.
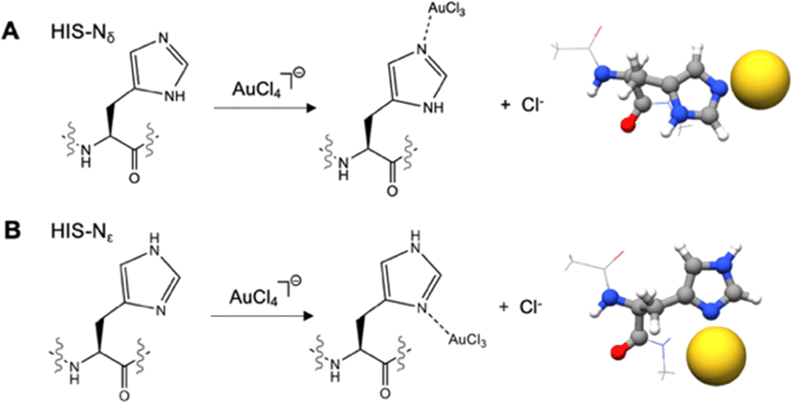 | ||
| Fig. 6 Interaction of Au(III) with a (A) histidine (His)-Nδ tautomer and (B) histidine (His)-Nε tautomer. Chlorine atoms are removed for clarity. | ||
The pKa value of histidine is very close to the physiological pH value, which makes it very common to find protonated histidines in proteins. However, also when the histidine is in its protonated form, deprotonation easily occurs upon Au(III) binding, releasing the lone pairs of the two nitrogens, that can bind Au(III) with ΔEbinding values of −20.3 kcal mol−1 for Nε and −16.3 kcal mol−1 for Nδ.
Aromatic amino acids such as tryptophan, tyrosine and phenylalanine may interact with gold ions with (i) their aromatic π-system,2 (ii) with the lone pairs of the heteroatoms (when present),2 or (iii) with the nucleophilic carbon atoms, composing the aromatic ring.2 Analysis of the protein data bank and quantum chemical calculations evidenced Au⋯π interactions involving Au(I) and aromatic amino acids (Phe, Tyr, and Trp).84 These Au⋯π interactions (alternatively defined as regium-π bond) are not observed in the interactions between Au(III) ions and aromatic residues. The softer nature of the Au(I) ion, compared to Au(III), is at the basis of this different behaviour. The interaction of Au(III) with Trp and Tyr is structurally interesting, because even if free lone pairs are available due the presence of O/N heteroatoms in the amino acid side chains, Au(III) interacts preferentially with nucleophilic carbon atoms of the aromatic system. Trp and indole are π-excessive aromatic heterocycles that are able to strongly interact with electrophiles, such as Au(III).85 In the case of Trp, Au(III) shows the strongest interaction with the C3 position of the indole ring (ΔEbinding = −20.9 kcal mol−1), similarly to the well-known gold-catalyzed functionalization reactions of C2-substituted indoles (Fig. 7).85 Alternative interaction sites can also be identified in the Trp ring. Au(III) can deprotonate the N–H bond in Trp. In this case, two competitive Au(III) interaction sites can be identified, i.e., C3 with a ΔEbinding value of −19.9 kcal mol−1 and N1 with a ΔEbinding value of −16.4 kcal mol−1.
 | ||
| Fig. 7 Interaction of Au(III) with (A) tryptophan (Trp), (B) tyrosine (Tyr), and (C) phenylalanine (Phe). Chlorine atoms are removed for clarity. | ||
In Tyr, the preferred interaction site is not the phenolic oxygen atom (ΔEbinding = −3.6 kcal mol−1), but the ortho (ΔEbinding = −14.9 kcal mol−1) and para (ΔEbinding = −13.3 kcal mol−1) positions of the phenyl ring. It is the well-known ability of cations to trigger the deprotonation of Tyr.86 Au(III) induces deprotonation of the hydroxyl group of the Tyr residue, but delocalization of the lone pair makes the softer “ortho” and “para” carbons favourite over the hard oxygen-site.
Experimentally, the binding/reduction capability of Tyr can be greatly improved by adjusting the pH above the pKa of Tyr (∼10).10 The calculations, repeated on a deprotonated Tyr, showed a huge increase in the binding energy (ΔEbinding = −41.4 kcal mol−1). This value is comparable to those of Sec and Cys, demonstrating the importance of this residue in the binding/reduction of Au(III), especially at pH > 10, when in deprotonated form.
Considering the aromatic system of Phe, Au(III) interacts more strongly with the backbone of the amino acid that with its side chain (ΔEbinding = −1.5 kcal mol−1), where the para position is favoured on the ortho (ΔEbinding = −1.3 kcal mol−1).
In this case, the strongly positive Au(III) clearly prefers the interaction with negatively charged Glu and Asp residues, than with the basic Lys and Arg (Fig. 8). Glu (ΔEbinding = −21.9 kcal mol−1) and Asp (ΔEbinding = −18.9 kcal mol−1) residues interact with the positive Au(III) ion giving electrostatic interactions with their negatively charged carboxylates. Lys (ΔEbinding = −13.2 kcal mol−1) and Arg (ΔEbinding = −8.2 kcal mol−1) instead use the lone pair of their basic nitrogens, after deprotonation induced by Au(III) binding, to coordinate the Au(III) ion.
 | ||
| Fig. 8 Interaction of Au(III) with (A) aspartate (Asp), (B) glutamate (Glu), (C) lysine (Lys), and (D) arginine (Arg). Chlorine atoms are removed for clarity. | ||
The interactions with Asp, Glu, and Lys strongly resemble, energetically and structurally, the interactions with the C-terminal and N-terminal residues of a peptide/protein, but they are numerically more common and consequently more important. Even in this case, if basic pH solutions are considered, there is the possibility to deprotonate the side chains of Lys (pKa = 10.79) and Arg (pKa = 12.48), strongly improving their binding with Au(III) (ΔEbinding = −23.2 kcal mol−1 for Lys ΔEbinding = −24.9 kcal mol−1 for Arg).
The interactions with Asp, Glu, and Lys strongly resemble, energetically and structurally, the interactions with the C-terminal and N-terminal residues of a peptide/protein, but they are numerically more common and consequently more important.
In analogy with the peptide bond, Au(III) interacts with Asn (ΔEbinding = −8.0 kcal mol−1) and Gln (ΔEbinding = −7.8 kcal mol−1) through their amide nitrogens, deprotonated upon binding (Fig. 9). The interaction energy of the Au(III) with the side chains of Asn and Gln is lower than the interaction with the peptide backbone because of the different nature of the amide group that is primary in the side chain of Asn and Gln and secondary, and thus more electron-rich, in the peptide backbone.
 | ||
| Fig. 9 Interaction of Au(III) with (A) asparagine (Asn), (B) glutamine (Gln), (C) serine (Ser), and (D) threonine (Thr). Chlorine atoms are removed for clarity. | ||
Also, the interaction of Au(III) with the hydroxyl groups of Ser (ΔEbinding = −4.4 kcal mol−1) and Thr (ΔEbinding = −2.8 kcal mol−1) may induce deprotonation, but small binding energies are observed in this case.
Apart the direct interaction of the Au(III) ion with the side chain of polar amino acids, it was recently demonstrated that amino acids improve their properties as H-bond donors and become less efficient H-bond acceptors upon coordination with metal ions.93
3. Methodology
All structures were fully optimized by density functional theory (DFT) calculations employing the PBE functional94 adding the atom-pair wise dispersion correction with Becke–Johnson damping (D3).95 Frequency calculations were carried out at the same level of theory to check the nature of critical points of the potential energy surface, finding only real frequencies for the minima. In such structures, single point calculations were performed at the MP296 level of theory. In the manuscript, we provided the MP2 energies obtained using optimized DFT geometries. The atoms belonging to the amino acids (i.e., N, C, O, H, S, and Se) and chlorine atoms were treated with the Karlsruhe basis set def2TZVP, and the Los Alamos electron core potential LanL2DZ was used to describe the metal center Au.97 This computational procedure has been reported to accurately treat the interaction of amino acids with metals such as silver and gold,56 reproducing the experimental findings.98 To take into account the presence of the water media on the molecular structures and energetics, we used the integral equation formalism polarizable continuum model (IEFPCM) approach,99 employing water as a solvent. All calculations were carried out using the Gaussian16 software.100 Cartesian coordinates of all the molecular structures can be found in the ESI.†The interaction of the Au(III) species, here investigated in the tetrachloroaurate ion [AuCl4]− form, with the amino acid (AA) leads to the formation of the [AA·AuCl3] complex. If the AA contains a deprotonable site, the formation of [AA·AuCl3] generates a hydrochloric acid molecule. Being in water media, HCl dissociates. As previously reported, the dissociation of HCl is observed computationally only considering a minimum of four water molecules.101 Therefore, four molecules of H2O were explicitly added to reliably reproduce the behavior of the acid in water solution.
Overall, the equilibrium considered throughout this work for a deprotonable AA is described in eqn (1):
| [AuCl4]− + AA + 4H2O → [AA·AuCl3] + [3H2O·H3O−·Cl−] | (1) |
| ΔEbinding = {E[AA·AuCl3] + E[3H2O·H3O−·Cl−]} − E[AuCl4]− + E(AA) + E(4H2O) | (2) |
| [AuCl4]− + AA + 4H2O → [AA·AuCl3] + [4H2O·Cl−] | (3) |
| ΔEbinding = {E[AA·AuCl3] + E[4H2O·Cl−]} − E[AuCl4]− + E(AA) + E(4H2O) | (4) |
4. Conclusions
In order to comprehend and design protein–gold interactions, broad principles that govern the binding between amino acids and Au(III) can be obtained by analysing their binding energies and tendency to oxidation.Experimentally, it was suggested that:
(i) cysteine and, even more, selenocysteine residues are the primary targets of the interaction of Au(III);
(ii) histidine and, in general, nitrogen-containing residues are key groups to complex/reduce Au (III) ions;
(iii) oxygen-containing residues have a fundamental role in the complex formation/reduction of Au(III);
(iv) aromatic residues are critical for the formation and stabilization of the Au nanoclusters in proteins;
(v) a basic pH (10–12) is usually needed to favor the formation of Au nanoclusters in proteins;
(vi) the reduction of Au(III) can be activated by external reducing agents (i.e., NaBH4) or directly by the protein amino acid residues.
The calculations provided in this paper address the different findings in a comprehensive manner:
(i) selenocysteine and cysteine are the two amino acids that undergo higher interaction with Au(III) due to the natural affinity of sulfur/selenium atoms to gold. In particular, selenocysteine is usually favored over cysteine because at physiological pH selenocysteine is virtually entirely ionized to a selenolate, while cysteine is mostly in the thiol state;
(ii) histidine possesses a free nitrogen lone pair, characterized by high coordination ability to bind Au(III). In cases when the lone pair of the nitrogen is unavailable because of protonation, N–H deprotonation easily occurs upon Au(III) binding, releasing the lone pair for the Au(III) coordination;
(iii) amino acid residues such as aspartate and glutamate hold a negative charge enabling them to electrostatically interact with the positive Au(III), while residues such as serine and threonine have an hydroxyl group in which the free lone pair of oxygen enables them to coordinate with the Au(III) ion;
(iv) aromatic residues such as tyrosine and tryptophan strongly interact with Au(III) with nucleophilic carbon atoms belonging to their aromatic system, resulting in soft–soft interactions;
(v) a basic pH (10–12) determines the deprotonation of cysteine, lysine and tyrosine in proteins, strongly increasing the binding affinity of Au(III) toward these amino acids;
(vi) many residues are able to donate, upon binding, an electron to Au(III), triggering its reduction without the need for external reducing agents.
The present data and discussion provide an essential platform to (i) fully comprehend the forces at play in the interaction between proteins and Au(III) and (ii) improve our capacity to engineer such complexes; and (iii) predict in advance the structure and strength of Au(III) adsorption on amino acids. They can be applied to (i) decipher the driving factors in the interaction of peptides/proteins with Au(III) drugs and understand their pharmacological mechanism and (ii) design protein pockets to synthesize AuNCs with desired properties. The complexation of Au(III) with amino acids is generally favourable thermodynamically; however, kinetic aspects should be clarified in future works, since processes such as ligand exchange, side chain deprotonation, and solvation and desolvation of the gold ion and the coordination sphere are crucial steps in the dynamics of the complexation process.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. J. M. was supported by Fondazione Umberto Veronesi.Notes and references
- B. Glišić, U. Rychlewska and M. I. Djuran, Dalton Trans., 2012, 41, 6887–6901 RSC.
- A. Giorgio and A. Merlino, Coord. Chem. Rev., 2020, 407, 213175 CrossRef CAS.
- M. D. Đurović, Ž. D. Bugarčić and R. van Eldik, Coord. Chem. Rev., 2017, 338, 186–206 CrossRef.
- R. T. Mertens, S. Gukathasan, A. S. Arojojoye, C. Olelewe and S. G. Awuah, Chem. Rev., 2023, 123, 6612–6667 CrossRef CAS PubMed.
- S. Radisavljević and B. Petrović, Front. Chem., 2020, 8, 379 CrossRef PubMed.
- D. Van Der Westhuizen, D. I. Bezuidenhout and O. Q. Munro, Dalton Trans., 2021, 50, 17413–17437 RSC.
- M. A. Malik, A. A. Hashmi, A. S. Al-Bogami and M. Y. Wani, J. Mater. Chem. B, 2024, 12, 552–576 RSC.
- T. Lazarević, A. Rilak and Ž. D. Bugarčić, Eur. J. Med. Chem., 2017, 142, 8–31 CrossRef PubMed.
- P. I. Da Silva Maia, V. M. Deflon and U. Abram, Future Med. Chem., 2014, 6, 1515–1536 CrossRef PubMed.
- N. El-Sayed and M. Schneider, J. Mater. Chem. B, 2020, 8, 8952–8971 RSC.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- Y. Xu, J. Sherwood, Y. Qin, D. Crowley, M. Bonizzoni and Y. Bao, Nanoscale, 2014, 6, 1515–1524 RSC.
- M. Di Giosia, F. Zerbetto and M. Calvaresi, Acc. Mater. Res., 2021, 2, 594–605 CrossRef CAS.
- R. Antoine, D. Maysinger, L. Sancey and V. Bonačić-Koutecký, Commun. Chem., 2022, 5, 1–5 CrossRef PubMed.
- L. Messori, G. Marcon and P. Orioli, Bioinorg. Chem. Appl., 2003, 1, 177–187 CrossRef CAS PubMed.
- C. Gabbiani, M. A. Cinellu, L. Maiore, L. Massai, F. Scaletti and L. Messori, Inorg. Chim. Acta, 2012, 393, 115–124 CrossRef CAS.
- K. C. Tong, D. Hu, P. K. Wan, C. N. Lok and C. M. Che, Front. Chem., 2020, 8, 1–11 CrossRef PubMed.
- L. Kou, S. Wei and P. Kou, Front. Chem., 2021, 9, 1–13 Search PubMed.
- C. I. Yeo, K. K. Ooi and E. R. T. Tiekink, Molecules, 2018, 23, 14–23 CrossRef PubMed.
- T. Gamberi, A. Pratesi, L. Messori and L. Massai, Coord. Chem. Rev., 2021, 438, 213905 CrossRef CAS.
- D. Hu, Y. Liu, Y. T. Lai, K. C. Tong, Y. M. Fung, C. N. Lok and C. M. Che, Angew. Chem., Int. Ed., 2016, 55, 1387–1391 CrossRef CAS PubMed.
- K. C. Tong, C. N. Lok, P. K. Wan, D. Hu, Y. M. E. Fung, X. Y. Chang, S. Huang, H. Jiang and C. M. Che, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1321–1329 CrossRef CAS PubMed.
- L. Massai, D. Cirri, E. Michelucci, G. Bartoli, A. Guerri, M. A. Cinellu, F. Cocco, C. Gabbiani and L. Messori, Biometals, 2016, 29, 863–872 CrossRef CAS PubMed.
- M. N. Wenzel, S. M. Meier-Menches, T. L. Williams, E. Rämisch, G. Barone and A. Casini, Chem. Commun., 2018, 54, 611–614 RSC.
- M. G. Milutinović, N. N. Milivojević, N. M. Đorđević, D. D. Nikodijević, S. R. Radisavljević, A. S. Đeković Kesić and S. D. Marković, J. Pharm. Sci., 2022, 111, 3215–3223 CrossRef PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- G. Bergamaschi, P. Metrangolo and V. Dichiarante, Photochem. Photobiol. Sci., 2022, 21, 787–801 CrossRef CAS PubMed.
- S. M. van de Looij, E. R. Hebels, M. Viola, M. Hembury, S. Oliveira and T. Vermonden, Bioconjugate Chem., 2022, 33, 4–23 CrossRef CAS PubMed.
- X. Ge, M. Zhang, F. Yin, Q. Sun, F. Mo, X. Huang, Y. Zheng, G. Wu, Y. Zhang and Y. Shen, J. Mater. Chem. B, 2024, 12, 1446–1466 RSC.
- S. Zhang, X. Zhang and Z. Su, J. Mater. Chem. B, 2020, 8, 4176–4194 RSC.
- E. Porret, X. Le Guével and J. L. Coll, J. Mater. Chem. B, 2020, 8, 2216–2232 RSC.
- H. Chen, Y. Jiang, T. Xu, J. Xu, J. Yu, Z. Chu, Y. Jiang, Y. Song, H. Wang and H. Qian, J. Mater. Chem. B, 2022, 10, 4789–4799 RSC.
- J. Dai, J. Lei, T. Zhang, J. You, D. Qin, Y. Wu, Y. Liu and Y. Zheng, J. Mater. Chem. B, 2024, 12, 1775–1781 RSC.
- J. Jiang, B. Cao, Y. Chen, H. Luo, J. Xue, X. Xiong and T. Zou, Angew. Chem., Int. Ed., 2022, 61, e2022011 Search PubMed.
- Y. Luo, B. Cao, M. Zhong, M. Liu, X. Xiong and T. Zou, Angew. Chem., Int. Ed., 2022, 61, e2022126 Search PubMed.
- J. Zhang, R. Fang, Y. Li, J. Jin, F. Yang and J. Chen, Mol. Pharm., 2023, 20, 3632–3644 CrossRef CAS PubMed.
- S. R. Thomas and A. Casini, Curr. Opin. Chem. Biol., 2020, 55, 103–110 CrossRef CAS PubMed.
- C. Martín-Santos, E. Michelucci, T. Marzo, L. Messori, P. Szumlas, P. J. Bednarski, R. Mas-Ballesté, C. Navarro-Ranninger, S. Cabrera and J. Alemán, J. Inorg. Biochem., 2015, 153, 339–345 CrossRef PubMed.
- A. Djeković, B. Petrović, Ž. D. Bugarčić, R. Puchta and R. Van Eldik, Dalton Trans., 2012, 41, 3633–3641 RSC.
- M. D. Durović, Ž. D. Bugarčić, F. W. Heinemann and R. Van Eldik, Dalton Trans., 2014, 43, 3911–3921 RSC.
- C. Gabbiani, L. Massai, F. Scaletti, E. Michelucci, L. Maiore, M. A. Cinellu and L. Messori, J. Biol. Inorg. Chem., 2012, 17, 1293–1302 CrossRef CAS PubMed.
- L. Messori, M. A. Cinellu and A. Merlino, ACS Med. Chem. Lett., 2014, 5, 1110–1113 CrossRef CAS PubMed.
- G. Ferraro, A. Giorgio, A. M. Mansour and A. Merlino, Dalton Trans., 2019, 48, 14027–14035 RSC.
- L. Massai, C. Zoppi, D. Cirri, A. Pratesi and L. Messori, Front. Chem., 2020, 8, 1–14 CrossRef PubMed.
- A. Pratesi, D. Cirri, D. Fregona, G. Ferraro, A. Giorgio, A. Merlino and L. Messori, Inorg. Chem., 2019, 58, 10616–10619 CrossRef CAS PubMed.
- Y. N. Tan, J. Y. Lee and D. I. C. Wang, J. Am. Chem. Soc., 2010, 132, 5677–5686 CrossRef CAS PubMed.
- D. Loreto, G. Ferraro and A. Merlino, Int. J. Biol. Macromol., 2020, 163, 970–976 CrossRef CAS PubMed.
- Y. Shimazaki, M. Takani and O. Yamauchi, Dalton Trans., 2009, 7854–7869 RSC.
- P. Pyykkö, Chem. Soc. Rev., 2008, 37, 1967–1997 RSC.
- P. Charchar, A. J. Christofferson, N. Todorova and I. Yarovsky, Small, 2016, 12, 2395–2418 CrossRef CAS PubMed.
- I. Tolbatov, A. Marrone, C. Coletti and N. Re, Molecules, 2021, 26, 7600 CrossRef CAS PubMed.
- I. Tolbatov and A. Marrone, J. Organomet. Chem., 2022, 965–966 Search PubMed.
- M. Hoefling, F. Iori, S. Corni and K. E. Gottschalk, ChemPhysChem, 2010, 11, 1763–1767 CrossRef CAS PubMed.
- Z. Futera, Phys. Chem. Chem. Phys., 2021, 23, 10257–10266 RSC.
- I. Tolbatov, C. Coletti, A. Marrone and N. Re, Int. J. Mol. Sci., 2019, 20, 1–15 Search PubMed.
- A. A. Buglak and A. I. Kononov, RSC Adv., 2020, 10, 34149–34160 RSC.
- M. Wienken, B. Lippert, E. Zangrando and L. Randaccio, Inorg. Chem., 2002, 31, 1983–1985 CrossRef.
- S. L. Best, T. K. Chattopadhyay, M. I. Djuran, R. A. Palmer, P. J. Sadler, I. Sóvágó and K. Varnagy, J. Chem. Soc., Dalton Trans., 1997, 15, 2587–2596 RSC.
- T. Kolev, B. B. Koleva, S. Y. Zareva and M. Spiteller, Inorg. Chim. Acta, 2006, 359, 4367–4376 CrossRef CAS.
- B. D. Glišić, S. Rajković, M. D. Živković and M. I. Djuran, Bioorg. Chem., 2010, 38, 144–148 CrossRef PubMed.
- B. B. Koleva, S. Zareva, T. Kolev and M. Spiteller, J. Coord. Chem., 2008, 61, 3534–3548 CrossRef CAS.
- B. B. Ivanova, J. Coord. Chem., 2005, 58, 587–593 CrossRef CAS.
- B. B. Koleva, T. Kolev, S. Y. Zareva and M. Spiteller, J. Mol. Struct., 2007, 831, 165–173 CrossRef CAS.
- J. Zou, Z. Guo, J. A. Parkinson, Y. Chen and P. J. Sadler, Chem. Commun., 1999, 1359–1360 RSC.
- A. Chipman, A. Gouranourimi, K. Farshadfar, A. Olding, B. F. Yates and A. Ariafard, Chem. – Eur. J., 2018, 24, 8361–8368 CrossRef CAS PubMed.
- L. Messori, F. Scaletti, L. Massai, M. A. Cinellu, I. Russo Krauss, G. Di Martino, A. Vergara, L. Paduano and A. Merlino, Metallomics, 2014, 6, 233–236 CrossRef CAS PubMed.
- B. Maity, S. Abe and T. Ueno, Nat. Commun., 2017, 8, 14820 CrossRef CAS PubMed.
- B. A. Al-Maythalony, M. I. M. Wazeer and A. A. Isab, Inorg. Chim. Acta, 2010, 363, 3244–3253 CrossRef CAS.
- A. J. Canumalla, N. Al-Zamil, M. Phillips, A. A. Isab and C. F. Shaw, J. Inorg. Biochem., 2001, 85, 67–76 CrossRef CAS PubMed.
- P. M. Yangyuoru, J. W. Webb and C. F. Shaw, J. Inorg. Biochem., 2008, 102, 584–593 CrossRef CAS PubMed.
- E. Bordignon, L. Cattalini, G. Natile and A. Scatturin, J. Chem. Soc., Chem. Commun., 1973, 22, 878–879 RSC.
- G. Natile, E. Bordignon and L. Cattalini, Inorg. Chem., 2002, 15, 246–248 CrossRef.
- A. Bindoli, M. P. Rigobello, G. Scutari, C. Gabbiani, A. Casini and L. Messori, Coord. Chem. Rev., 2009, 253, 1692–1707 CrossRef CAS.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed.
- L. B. Poole, Free Radical Biol. Med., 2015, 80, 148–157 CrossRef CAS PubMed.
- M. Zaffagnini, S. Fermani, M. Calvaresi, R. Orrù, L. Iommarini, F. Sparla, G. Falini, A. Bottoni and P. Trost, Antioxid Redox Signal, 2016, 24, 502–517 CrossRef CAS PubMed.
- R. J. Sundberg, Chem. Rev., 1974, 74, 471–517 CrossRef CAS.
- P. Chakrabarti, Protein Eng., Design Selection, 1990, 4, 57–63 CrossRef CAS PubMed.
- J. A. Cuadrado, W. Zhang, W. Hang and V. Majidi, J. Environ. Monit., 2000, 2, 355–359 RSC.
- B. A. Al-Maythalony, A. A. Isab, M. I. M. Wazeer and A. Ibdah, Inorg. Chim. Acta, 2010, 363, 3200–3207 CrossRef CAS.
- S. K. Burley and G. A. Petsko, Science, 1985, 229, 23–28 CrossRef CAS PubMed.
- E. A. Meyer, R. K. Castellanod and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- M. Cordes, B. Giese, A. Bauzá, M. De Las Nieves Pina and A. Frontera, J. Phys. Chem. Lett., 2020, 11, 8259–8263 CrossRef PubMed.
- M. De Las Nieves Pina, A. Frontera and A. Bauzá, J. Phys. Chem. Lett., 2020, 11, 8259–8263 CrossRef PubMed.
- V. Pirovano, Eur. J. Org. Chem., 2018, 1925–1945 CrossRef CAS.
- P. Dupuis, T. C. Corcoran and M. A. El-Sayed, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 3662–3664 CrossRef CAS PubMed.
- F. B. Sheinerman, R. Norel and B. Honig, Curr. Opin. Struct. Biol., 2000, 10, 153–159 CrossRef CAS PubMed.
- J. Gao, P. Müller, M. Wang, S. Eckhardt, M. Lauz, K. M. Fromm and B. Giese, Angew. Chem., Int. Ed., 2011, 50, 1926–1930 CrossRef CAS PubMed.
- A. Keramidas, A. J. Moorhouse, P. R. Schofield and P. H. Barry, Prog. Biophys. Mol. Biol., 2004, 86, 161–204 CrossRef CAS PubMed.
- S. Kumar and R. Nussinov, ChemBioChem, 2002, 3, 604–617 CrossRef CAS PubMed.
- M. S. Lawrence, K. J. Phillips and D. R. Liu, J. Am. Chem. Soc., 2007, 129, 10110–10112 CrossRef CAS PubMed.
- T. Dudev and C. Lim, Chem. Rev., 2003, 130, 773–788 CrossRef PubMed.
- S. S. Zrilić, J. M. Živković and S. D. Zarić, J. Inorg. Biochem., 2023, 242, 112151 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 3865–3868 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- T. S. Sych, A. A. Buglak, Z. V. Reveguk, V. A. Pomogaev, R. R. Ramazanov and A. I. Kononov, J. Phys. Chem. C, 2018, 122(45), 26275–262280 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. C. 01 Search PubMed.
- K. R. Leopold, Annu. Rev. Phys. Chem., 2011, 62, 327–349 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00204k |
| This journal is © The Royal Society of Chemistry 2024 |