
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimodal effects on lipid droplets induced in cancer and non-cancer cells by chemotherapy drugs as revealed with a green-emitting BODIPY fluorescent probe†
Artūras
Polita
 *a,
Rokas
Žvirblis
b,
Jelena
Dodonova-Vaitkūnienė
c,
Arun Prabha
Shivabalan
a,
Karolina
Maleckaitė
d and
Gintaras
Valinčius
a
*a,
Rokas
Žvirblis
b,
Jelena
Dodonova-Vaitkūnienė
c,
Arun Prabha
Shivabalan
a,
Karolina
Maleckaitė
d and
Gintaras
Valinčius
a
aInstitute of Biochemistry, Life Sciences Center, Vilnius University, Saulėtekio av. 7, Vilnius, LT-10257, Lithuania. E-mail: arturas.polita@gmc.vu.lt
bLife Sciences Center, Institute of Biotechnology, Vilnius University, Saulėtekio av. 7, Vilnius, LT-10257, Lithuania
cInstitute of Chemistry, Faculty of Chemistry and Geosciences, Vilnius University, Naugarduko st. 24, Vilnius, LT-03225, Lithuania
dCenter of Physical Sciences and Technology, Saulėtekio av. 3, Vilnius, LT-10257, Lithuania
First published on 20th February 2024
Abstract
Lipid droplets (LDs) are cytoplasmic lipid-rich organelles with important roles in lipid storage and metabolism, cell signaling and membrane biosynthesis. Additionally, multiple diseases, such as obesity, fatty liver, cardiovascular diseases and cancer, are related to the metabolic disorders of LDs. In various cancer cells, LD accumulation is associated with resistance to cell death, reduced effectiveness of chemotherapeutic drugs, and increased proliferation and aggressiveness. In this work, we present a new viscosity-sensitive, green-emitting BODIPY probe capable of distinguishing between ordered and disordered lipid phases and selectively internalising into LDs of live cells. Through the use of fluorescence lifetime imaging microscopy (FLIM), we demonstrate that LDs in live cancer (A549) and non-cancer (HEK 293T) cells have vastly different microviscosities. Additionally, we quantify the microviscosity changes in LDs under the influence of DNA-damaging chemotherapy drugs doxorubicin and etoposide. Finally, we show that doxorubicin and etoposide have different effects on the microviscosities of LDs in chemotherapy-resistant A549 cancer cells.
Introduction
Lipid droplets (LDs) are dynamic lipid-rich organelles1 composed of a neutral lipid hydrophobic core surrounded by a single layer of phospholipids decorated with various proteins (Fig. 1A).2 Importantly, the composition of LDs and their size, localization and number change rapidly in response to cellular states and nutrient availability.3,4 Not only do LDs function as lipid storage compartments,5 but also play important roles in cell signalling and inflammation,6,7 lipid metabolism8 and membrane biosynthesis.9 The metabolic disorders of LDs are related to multiple diseases, such as obesity,10 fatty liver,11 cardiovascular diseases,12 and cancer.13 Accumulation of LDs has been observed in cancer cells and cancerous tissues such as breast, prostate, and colorectal cancers, as well as hepatocellular and renal carcinomas.14 In addition, LD accumulation also affects anti-cancer drug efficiency, alters their cellular distribution, and impairs drug-induced apoptosis and immunogenic cell death, causing chemotherapy resistance.14,15 Moreover, LDs have been identified in all the processes involved in cancer development, including proliferation, evading growth suppressors, activating invasion and metastasis, evasion of immune destruction and resisting death.1,15 On top of this, increased storage of lipids in LDs is also beneficial for cancer cell survival,16,17 as additional lipids serve as energy sources to meet the metabolic needs of proliferating cancer cells. Conventional chemotherapeutic drugs have been shown to alter the fatty acid distribution in LDs, producing highly saturated LDs and unsaturated plasma membrane lipids.18 Changes in the fatty acid distribution increase the amount of LDs and decrease the fluidity of the plasma membrane, potentially triggering the invasion and metastasis of cancer cells.19 Importantly, LDs themselves may be appropriate targets for cancer chemotherapy,1,20 and the elevated levels of LDs are used as a diagnostic biomarker for cancer.14,21 Consequently, the ability to image the compositional changes of LDs may be particularly useful for the development of cancer diagnostic tools and studying the effects of chemotherapeutic agents on LDs.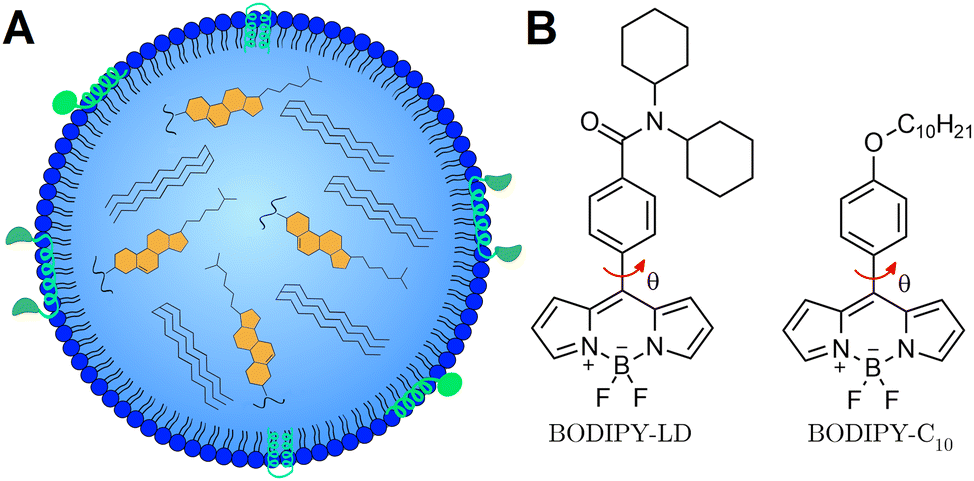 | ||
| Fig. 1 (A) Simplified structure of the LD. (B) Structure of one of the most widely used molecular rotors – BODIPY-C10, and its variant for the detection of LDs – BODIPY-LD. The red arrows indicate the intramolecular rotation which causes BODIPY to display viscosity sensitivity. | ||
Viscosity measurements provide a convenient way to observe the compositional changes that take place in LDs. The efficiency of lipid packaging and the order of lipids have a great influence on the viscosity values of lipid structures.22 For example, cholesterol-rich lipid systems form highly viscous liquid-ordered (Lo) phases, whereas highly unsaturated lipids produce non-viscous liquid disordered (Ld) phases.23 Moreover, the viscosity of the Ld phase is directly affected by the amount of cholesterol present in the bilayer.24 The viscosity differences arising due to lipid packaging in the Lo and Ld phases allow for lipid order determination using fluorescent viscosity-sensitive probes.25–27 LDs, depending on the cellular state, undergoing phase transitions from an ordered state to a disordered state have varying levels of cholesteryl esters and occasionally may feature ordered nanodomains in the centre.28
Viscosity-sensitive dyes, called molecular rotors, can be used to quantitatively image the microviscosity changes in LDs.29,30 The term microviscosity here refers to the lipid packaging and molecular mobility of the probe's local environment.31 In the excited state, molecular rotors undergo intramolecular rotation, which results in the molecular rotor entering the dark state.32 In low viscosity or non-crowded environments, the intramolecular rotation is not restricted, and the non-radiative decay dominates, leading to a decrease in the fluorescence quantum yield and lifetime.33 Using the fluorescence lifetime to quantify the microviscosity is particularly advantageous, since the lifetime is independent of the probe's concentration, excitation setup, and optical properties of the medium or the microscope. Molecular rotors can be combined with fluorescence lifetime imaging microscopy (FLIM) to create spatial microviscosity maps of different lipid structures in live cells.34–36 Currently, BODIPY-C10 (Fig. 1B) is one of the most widely used molecular rotors for viscosity measurements.37,38 Previously, BODIPY-C10 and its derivatives were used to quantify viscosity in model lipid bilayers,39 plasma membranes,40,41 mitochondria,42 and lysosomes.43 Due to its lack of specificity, BODIPY-C10 internalizes into various cellular membranes and the cytoplasm,44 making LDs less detectable and data analysis more difficult.
Although fluorescence lifetime-based viscosity probes for LDs do exist (TPE-Cy and NLV-1),45,46 to our knowledge, both these probes considerably stain the cytoplasm (and membrane-bound organelles in the case of TPE-Cy),45 thus making LDs harder to identify. Despite the fact that both TPE-Cy and NLV-1 feature multiple rotating groups for increased viscosity sensitivity, their fluorescence lifetimes do not rise rapidly with increasing solvent viscosity, resulting in more challenging detection of minor microviscosity changes in live specimens.45,46 The aim of this research is to explore the possibility of discriminating cancer and non-cancer cells based on the microviscosity of LDs. In this work, we introduce BODIPY-LD, a BODIPY-C10 derivative probe with high specificity for LDs and fluorescence lifetime-based ability to measure the microviscosity (Fig. 1B). By comparing fluorescence lifetimes of BODIPY-LD in solvents of different polarities and viscosities, we show that BODIPY-LD is affected mainly by the viscosity of the solvent. We explore the ability of BODIPY-LD to distinguish Lo and Ld phases in large unilamellar vesicles (LUVs) – one of the most frequently used model lipid systems. Furthermore, by using BODIPY-LD in combination with FLIM, we image LDs in human lung cancer cells (A549) and immortalised human embryonic kidney cells (HEK 293T). In addition, we investigate how DNA-damaging chemotherapeutic agents47,48 – etoposide (ETP) and doxorubicin (DOX) – affect the microviscosities of LDs. Although both DOX and ETP cause DNA damage by poisoning enzyme topoisomerase II and converting it into a DNA-damaging agent,47,48 DOX is additionally known for its ability to intercalate into DNA and generate free radicals, which can damage cellular membranes, DNA and proteins.48 Numerous anticancer drugs have been reported to be sequestered in the hydrophobic cores of LDs.1,16 However, studies investigating the physical changes that occur in LDs during chemotherapeutic treatment are still lacking. Our results demonstrate that the microviscosities of LDs in human lung cancer cells (A549) vary significantly between different cells in the same culture, but are similar within individual cells. In contrast, the microviscosities of LDs in human immortalised kidney cells (HEK 293T) are similar between different cells. We found that both DOX and ETP induce an increased number of LDs in both A549 and HEK 293T cells. Furthermore, DOX and ETP treatments of A549 cells induce the formation of both highly viscous and highly non-viscous LD populations within individual cells. In contrast, DOX treatment of HEK 293T cells produces LDs with varying microviscosities in individual cells, whereas ETP treatment induces the formation of LDs with uniform microviscosities within each cell. Finally, we show that highly viscous LDs are absent in DOX- and ETP-resistant A549 cells.
Results and discussion
Probe design
The BODIPY-LD design was based on one of the most applicable and successful rotors to date – BODIPY-C10. In order to retain the same mechanism of the viscosity sensitivity of BODIPY-C10, we decided to keep the rotation of the phenyl group versus the BODIPY unhindered and modified the para-position of the phenyl group. Our goal was to create a probe with high specificity for LDs with minimal cytoplasm staining. As a result, we introduced two cyclohexyl rings to increase the size and hydrophobicity of the probe, making the probe too hydrophobic for cytoplasm staining (Fig. 1B). The non-charged nature of the probe should also favour its accumulation in neutral, lipid-rich environments. The synthesis of BODIPY-LD is described in the ESI.†Absorption, steady-state and time-resolved fluorescence spectroscopies
In order to investigate the spectroscopic properties of BODIPY-LD, we performed absorption, steady-state, and time-resolved fluorescence experiments in solvents of varying polarity, from non-polar [cyclohexane] to very polar [DMSO] (Fig. 2). Similar to BODIPY-C10,37 BODIPY-LD's absorption spectra include a low intensity band at 300–400 nm and the main band in the 450–525 nm region with maximum positioned at 500–505 nm (Fig. 2A). The fluorescence spectra of BODIPY-LD feature a slight red shift as the polarity of the solvent increases, from the fluorescence peak maximum at 518 nm in cyclohexane to 524 nm in DMSO (Fig. 2A). Importantly, no aggregation of the dye was observed in polar solvents such as methanol and DMSO. To explore the effects of solvent polarity on the fluorescence lifetimes of BODIPY-LD, we performed time-resolved fluorescence measurements in cyclohexane, toluene, chloroform, dichloromethane, DMSO and methanol (Fig. 2B and Fig. S1, ESI†). In all solvents tested, the fluorescence decay curves of BODIPY-LD were monoexponential, with fluorescence lifetimes ranging from 226 ps in chloroform to 153 ps in DMSO. In contrast, the fluorescence lifetimes of BODIPY-C10 range from 300 to 800 ps in the solvents of 0.5 cP viscosity.40 We found no correlation between the polarity of the solvent and the fluorescence lifetimes of BODIPY-LD since the fluorescence decay curves differ marginally (Fig. 2B).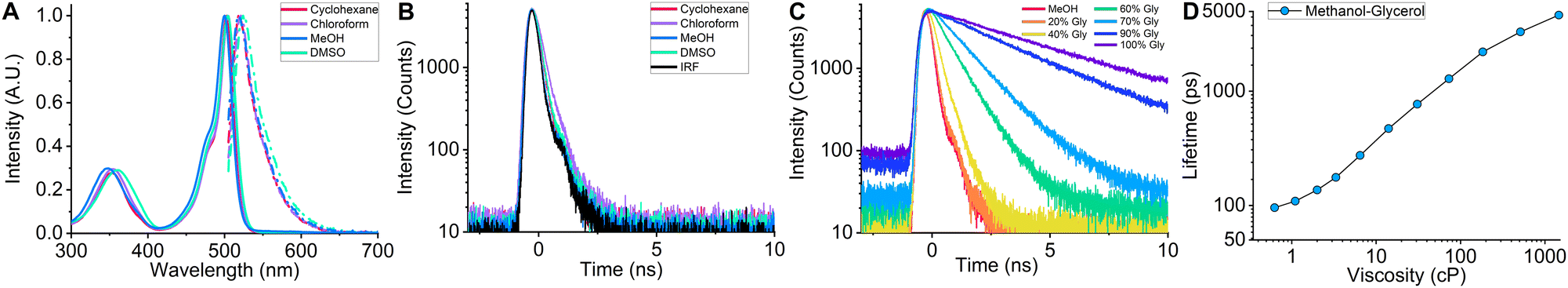 | ||
| Fig. 2 (A) Fluorescence (dotted-dashed lines) and absorption (solid lines) spectra of BODIPY-LD obtained in cyclohexane, chloroform, methanol and DMSO. (B) Time-resolved fluorescence decay curves of BODIPY-LD in cyclohexane, chloroform, methanol and DMSO. (C) Time-resolved fluorescence decay curves of BODIPY-LD obtained in methanol–glycerol mixtures of varying viscosities. (D) Fluorescence lifetimes of BODIPY-LD in methanol–glycerol mixtures. | ||
Next, we measured time-resolved and steady-state fluorescence of BODIPY-LD in methanol–glycerol mixtures that cover a viscosity range from 0.6 to 1457 cP to evaluate the probe's viscosity sensitivity and construct a fluorescence lifetime-viscosity calibration curve (Fig. 2C and Fig. S2, ESI†). The calibration curve can be utilized to convert the fluorescence lifetimes of BODIPY-LD in LDs into microviscosity values. The fluorescence lifetimes of BODIPY-LD range from 92 ps in methanol to 4639 ps in pure glycerol (Fig. 2D). The fluorescence decays were monoexponential for compositions ranging from methanol to 40% methanol-60% glycerol. At higher glycerol concentrations, the fluorescence decays become biexponential with low amplitude short lifetime components (Fig. S2, ESI†). Due to low fluorescence lifetime values in non-viscous solvents, BODIPY-LD has a significantly expanded dynamic viscosity range, which can be calculated from the lifetime ratio at high and low viscosities, compared to BODIPY-C10. The dynamic range of BODIPY-LD in methanol–glycerol mixtures is 50.4, as determined from the ratio of fluorescence lifetimes in glycerol (4639 ps) and methanol (92 ps), compared to 11.2 for BODIPY-C10 in the same mixture.37 These results demonstrate that the fluorescence lifetimes of BODIPY-LD are mainly affected by solvent viscosity and are minimally influenced by polarity.
Given that LDs contain varying amounts of cholesteryl esters and occasionally may feature ordered nanodomains,28 we aimed to explore the ability of BODIPY-LD to integrate into highly viscous Lo phases, as well as probe's sensitivity to cholesterol and capability to distinguish Lo and Ld lipid phases. We carried out steady-state and time resolved fluorescence measurements on LUVs with Lo (DOPC:DPPC:Chol 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5) and Ld (DOPC, POPC, DOPC:Chol) lipid phases (Fig. 3). While BODIPY-LD does not exhibit solvatochromism in the Ld phases, the steady-state fluorescence spectra display a slight red shift in the Lo phases, with peak maximum shifting from 521 (Ld) to 526 nm (Lo). (Fig. 3A). Notably, BODIPY-LD successfully integrates into both Lo and Ld LUVs and displays similar fluorescence intensity values in both phases (Fig. S3, ESI†). The ability of BODIPY-LD to partition into highly ordered lipid phases is highly important because LDs occasionally feature highly ordered lipid nanodomains28 and undergo phase transitions from amorphous to the liquid crystalline lipid phases.49 The time-resolved fluorescence decays of BODIPY-LD in Lo LUVs are remarkably longer compared to the Ld LUVs (Fig. 3B), with biexponential intensity-weighted fluorescence lifetimes being 1.24, 1.3 and 3.6 ns for DOPC, POPC and DOPC:DPPC:Chol LUVs, respectively. We also examined how the cholesterol concentration in Ld LUVs affects the fluorescence lifetimes of BODIPY-LD. Cholesterol is especially known for its ability to induce order and condense Ld lipid bilayers.50 Addition of cholesterol to DOPC LUVs (Ld) resulted in significantly longer fluorescence decays, with intensity weighted fluorescence lifetimes increasing from 1.24 ns for pure DOPC LUVs to 1.63 and 2.13 ns for DOPC/Chol 75/25 and DOPC/Chol 50/50 LUVs, respectively (Fig. 3B). We hypothesize that the biexponential nature of fluorescence decays in LUVs is likely caused by the different positions that the dye can occupy in the lipid bilayer. Most importantly, the ability to determine the lipid order and distinguish Lo and Ld phases based on the fluorescence lifetime of BODIPY-LD may be particularly useful for estimating the cholesterol levels in LDs, studying the biophysical changes induced in LDs upon binding of proteins, imaging ordering and disordering effects induced by small molecules, detecting the lipid phase transitions and observing the lipid exchange processes between LDs and organelles.
:5:5) and Ld (DOPC, POPC, DOPC:Chol) lipid phases (Fig. 3). While BODIPY-LD does not exhibit solvatochromism in the Ld phases, the steady-state fluorescence spectra display a slight red shift in the Lo phases, with peak maximum shifting from 521 (Ld) to 526 nm (Lo). (Fig. 3A). Notably, BODIPY-LD successfully integrates into both Lo and Ld LUVs and displays similar fluorescence intensity values in both phases (Fig. S3, ESI†). The ability of BODIPY-LD to partition into highly ordered lipid phases is highly important because LDs occasionally feature highly ordered lipid nanodomains28 and undergo phase transitions from amorphous to the liquid crystalline lipid phases.49 The time-resolved fluorescence decays of BODIPY-LD in Lo LUVs are remarkably longer compared to the Ld LUVs (Fig. 3B), with biexponential intensity-weighted fluorescence lifetimes being 1.24, 1.3 and 3.6 ns for DOPC, POPC and DOPC:DPPC:Chol LUVs, respectively. We also examined how the cholesterol concentration in Ld LUVs affects the fluorescence lifetimes of BODIPY-LD. Cholesterol is especially known for its ability to induce order and condense Ld lipid bilayers.50 Addition of cholesterol to DOPC LUVs (Ld) resulted in significantly longer fluorescence decays, with intensity weighted fluorescence lifetimes increasing from 1.24 ns for pure DOPC LUVs to 1.63 and 2.13 ns for DOPC/Chol 75/25 and DOPC/Chol 50/50 LUVs, respectively (Fig. 3B). We hypothesize that the biexponential nature of fluorescence decays in LUVs is likely caused by the different positions that the dye can occupy in the lipid bilayer. Most importantly, the ability to determine the lipid order and distinguish Lo and Ld phases based on the fluorescence lifetime of BODIPY-LD may be particularly useful for estimating the cholesterol levels in LDs, studying the biophysical changes induced in LDs upon binding of proteins, imaging ordering and disordering effects induced by small molecules, detecting the lipid phase transitions and observing the lipid exchange processes between LDs and organelles.
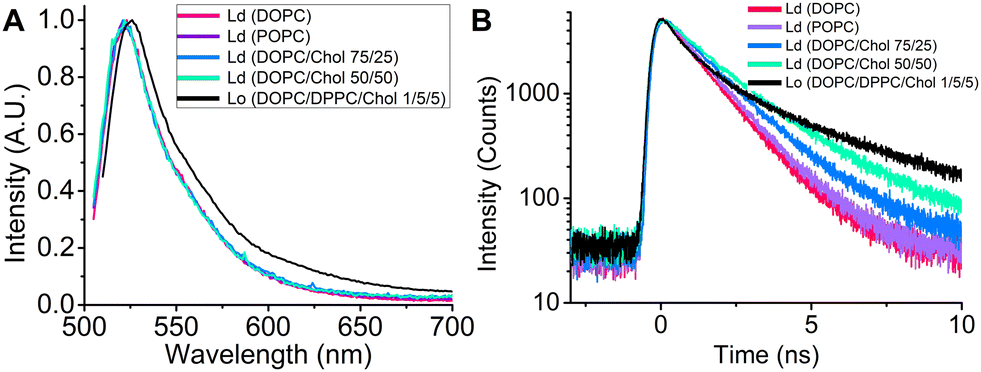 | ||
| Fig. 3 (A) Steady-state fluorescence spectra of BODIPY-LD in Ld and Lo LUVs. (B) Time-resolved fluorescence spectra of BODIPY-LD in Ld and Lo LUVs. | ||
FLIM of LDs in human epithelial lung cancer (A549) and immortalised embryonic kidney (HEK 293T) cells
Next, we used FLIM to image LDs in human lung cancer (A549) and immortalised embryonic kidney (HEK 293T) cells (Fig. 4 and 5). BODIPY-LD very effectively stained both highly viscous and non-viscous LDs (1 μM concentration, 5 min), with negligible cytoplasm staining. Fluorescence intensities in the LDs were about 102 times higher than those in the cytoplasm. Curiously, BODIPY-LD did not stain the plasma membrane, allowing for an easy identification of peripheral LDs (Fig. 4 and 5). We hypothesize that poor plasma membrane staining is due to the highly lipophilic nature of the dye and a preference for the neutral lipid environments. The plasma membrane, on the other hand, features highly polar lipid headgroups, which give rise to higher dielectric constants in the lipid bilayer.51 Notably, we did not observe any dye-induced morphology changes in the cells even when the concentration of BODIPY-LD was increased to 5 μM (Fig. S4, ESI†). Furthermore, we have confirmed that the spherical, slowly moving in the cytoplasm, vesicles (Fig. S5, ESI†) were indeed LDs by co-staining the cells with Nile Red – a well-known marker of LDs (Fig. S6, ESI†).52 Additionally, we recorded the fluorescence spectra of LDs stained with BODIPY-LD to ensure that no aggregation of the dye occurs in the LDs and the fluorescence lifetimes are measured accurately (Fig. S7, ESI†).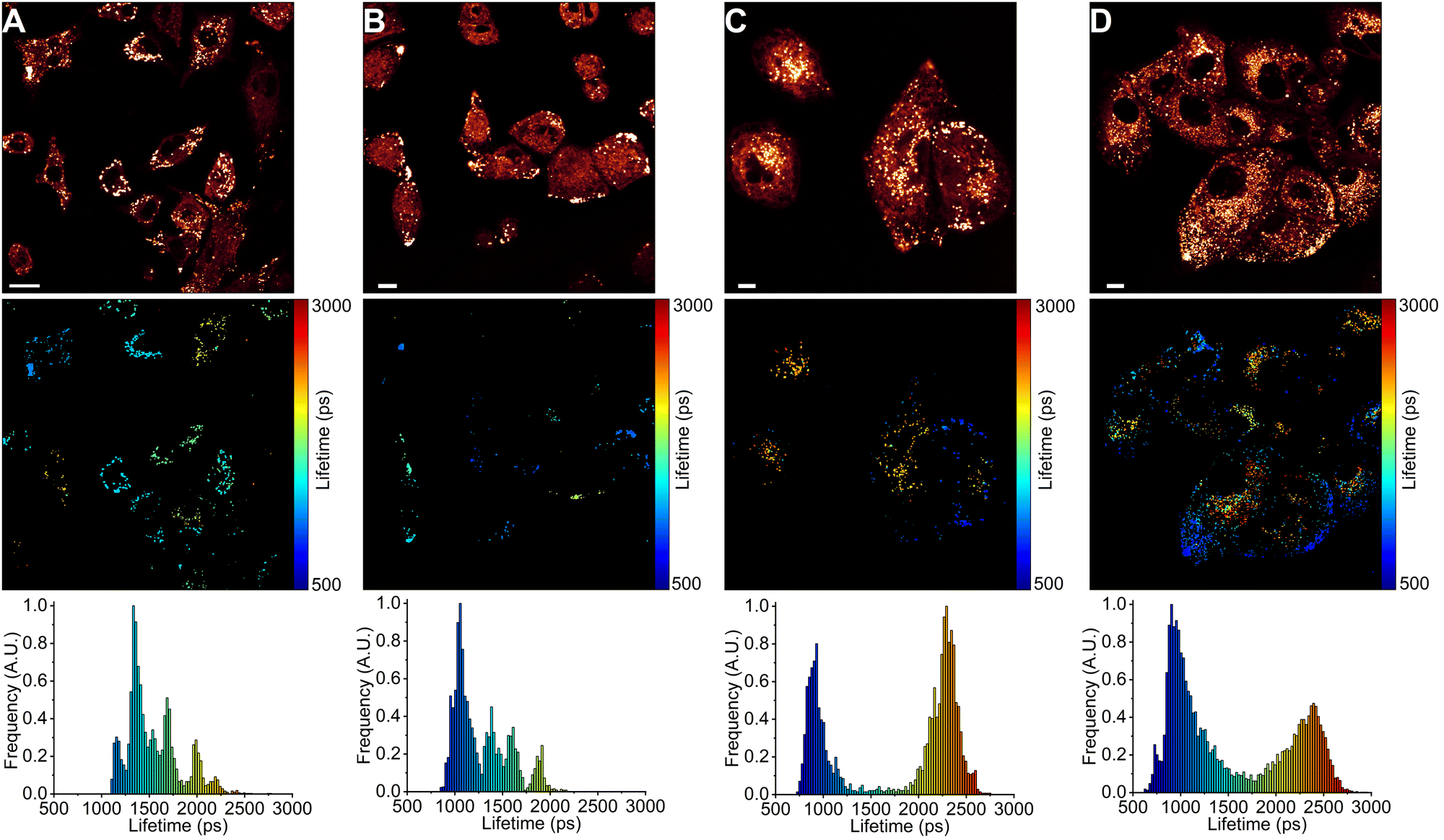 | ||
| Fig. 4 FLIM images of BODIPY-LD in LDs of A549 cells. (A) A549 cells treated with DOX for 6 h. (B) A549 cells treated with DOX for 24 h. (C) A549 cells treated with DOX for 48 h. (D) The top panel shows images of fluorescence intensity. FLIM images are shown in the middle panel. The corresponding lifetime histograms are shown in the bottom panel. Scale bars are 10 μm. | ||
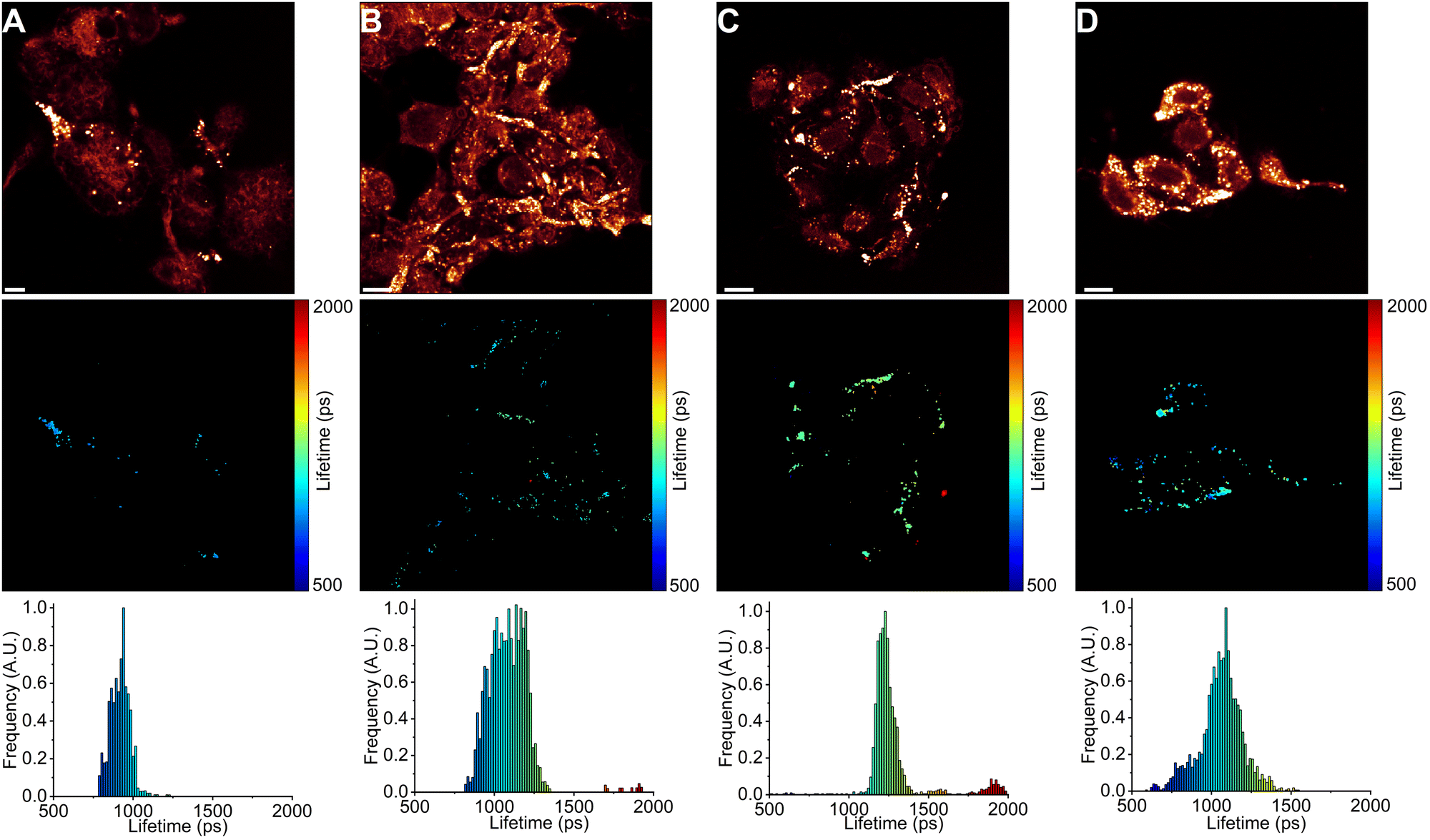 | ||
| Fig. 5 FLIM images of BODIPY-LD in LDs of HEK 293T cells. (A) HEK 293T cells treated with DOX for 6 h. (B) HEK 293T cells treated with DOX for 24 h. (C) HEK 293T cells treated with DOX for 48 h. (D) The top panel shows images of fluorescence intensity. FLIM images are shown in the middle panel. The corresponding lifetime histograms are shown in the bottom panel. Scale bars are 10 μm. | ||
Interestingly, in lung cancer cells (A549), we observed that the microviscosities of LDs vary significantly between individual cells within the same culture, with the intensity-weighted fluorescence lifetimes of BODIPY-LD ranging from 1100 to 2500 ps, corresponding to the viscosities of 60 to 270 cP in methanol–glycerol calibration mixtures (Fig. 4A and Fig. S8A, ESI†). Furthermore, the microviscosities of LDs in individual A549 cells were very similar (Fig. 4A and Fig. S8A, ESI†). In contrast, LDs in human embryonic kidney cells (HEK 293T) possessed similar microviscosities in different cells, with the intensity-weighted fluorescence lifetimes of BODIPY-LD ranging from 700 to 1000 ps, corresponding to the viscosities of 25 and 50 cP in methanol–glycerol calibration mixtures (Fig. 5A and Fig. S9A, ESI†). In addition, the number of LDs in A549 cells far surpasses the number of LDs in HEK 293T (Fig. 4A and 5A). Most importantly, the large differences in fluorescence lifetimes between the cancerous and non-cancerous cells can be used for the detection of malignant cells. We hypothesize that the fluorescence lifetime differences, arising due to different viscosities of LDs, are representative of different cellular or metabolic states3,53 in malignant cells and fluorescent environmental probes for LDs may be useful for the studies of cancer progression.
Next, we treated A549 and HEK 293T cells with half maximal inhibitory concentrations (IC50) of anti-cancerous DNA-damaging drugs DOX (18 nM for A549 and 20 μM for HEK 293T)54,55 and ETP (107 μM for A549 and 22 μM for HEK 293T).55,56 The IC50 value represents the concentration of a drug capable of causing 50% cell death. We imaged LDs after 6, 24 and 48 hours of drug treatment. In both A549 and HEK 293T cells, DOX treatment gradually increased the number of LDs (Fig. 4 and 5). Similarly, ETP treatment of A549 and HEK 293T cells also resulted in a gradual increase in the number of LDs (Fig. S8 and S9, ESI†). Furthermore, after 24 and 48 h, DOX treatment of A549 cells slowly induced the formation of highly viscous and highly non-viscous LD populations within individual cells, with fluorescence lifetimes of BODIPY-LD ranging from 750 to 2600 ps, corresponding to the microviscosities of 30 and 300 cP (Fig. 4C and D). In addition, LDs with viscosities of about 90 cP (corresponding to BODIPY-LD fluorescence lifetimes of about 1500 ps) almost completely disappeared after 24 h of DOX treatment (Fig. 4C and D). Correspondingly, treatment of A549 cells with ETP also resulted in the disappearance of LDs with 90 cP viscosities after 24 h (Fig. S8, ESI†). Instead, highly viscous and highly non-viscous LD populations were formed, with viscosities ranging from 30 to 300 cP (Fig. S8, ESI†).
In contrast, the microviscosities of LDs in DOX-treated HEK 293T cells increased after 6 h from 25–50 cP to 25–70 cP (Fig. 5B) and a further 24 h treatment with DOX resulted in the formation of LDs with 60–70 cP viscosities (Fig. 5C). Additionally, some LDs had viscosities of 100 and 160 cP, corresponding to the fluorescence lifetimes of BODIPY-LD of 1500 ps and 2000 ps (Fig. 5C). Unlike in A549 cells, 48 h of DOX treatment in HEK 293T cells did not further increase the microviscosities of LDs; instead, heterogenous LD populations were present in all cells, with viscosities ranging from 25 to 70 cP (Fig. 5D). Finally, treatment of HEK 293T cells with ETP resulted in the formation of LDs with viscosities of 45–70 cP after 6 h (Fig. S9B, ESI†). Further 24 and 48 h treatment of HEK 293T cells with ETP produced LDs with viscosities ranging from 50 to 160 cP (Fig. S9C and D, ESI†). Curiously, after 24 h of ETP treatment, LDs in individual cells had more or less the same viscosities (Fig. S9C, ESI†).
We hypothesize that the increase in the number of LDs in cells undergoing DOX or ETP treatment is a result of cellular stress response.17,57 Furthermore, cellular stress can induce the formation of highly hydrophobic LDs, which are likely enriched with cholesterol esters.57 The increase in LD viscosity in A549 and HEK 293T cells upon DOX and ETP treatment may be due to the increased amount of cholesterol esters.57 DOX and 5-fluorouracil have also been reported to alter the fatty acid distribution in LDs and induce the formation of highly saturated LDs in hepatocellular carcinoma and colorectal cancer cells.18 We suspect that the presence of saturated fatty acids, as well as cholesteryl esters, promote the formation of highly ordered lipid phases58 and account for the formation of viscous LDs after DOX and ETP treatments in A549 cells. However, we also observed non-viscous LDs in A549 cells upon DOX and ETP treatments. In addition, while DOX and ETP exerted similar effects on LDs in A549 cells, ETP treatment in HEK 293T cells produced moderately and highly viscous LDs, with the absence of non-viscous LDs, while DOX produced non-viscous and moderately viscous LDs.
FLIM of A549 and HEK 293T cells that are resistant to DOX and ETP treatment
Finally, we have quantified the microviscosities of LDs in A549 and HEK 293T cells that are resistant to chemotherapeutic drug treatment (Fig. 6). To perform this, we treated A549 and HEK 293T cells for 48 h with twice the IC50 concentrations of DOX (36 nM for A549 and 40 μM for HEK 293T) and ETP (214 μM for A549 and 44 μM for HEK 293T). Surprisingly, we found that DOX and ETP-resistant A549 and HEK 293T cells have LDs with very dissimilar microviscosities over their respective 48 h drug treatment (Fig. 6). With 2IC50 treatment of A549 cells with DOX, we no longer observed LDs with microviscosities of 30 and 300 cP; instead, LDs ranging from 60 to 100 cP were present (Fig. 6A). The 2IC50 treatment of A549 cells with ETP also resulted in the absence of LDs with microviscosities of 30 and 300 cP, but a very homogeneous population of LDs with microviscosities of about 50–65 cP was formed in each cell (Fig. 6B). In contrast, the 2IC50 treatment of HEK 293T cells with DOX induced the formation of LDs with moderate viscosities (25–90 cP), similar to IC50 DOX treatment (Fig. 6C). Of note, some LDs in HEK 293T cells treated with 2IC50 DOX concentrations displayed very low viscosities of about 10 cP (corresponding to the 300 ps fluorescence lifetime of BODIPY-LD). Similar to the IC50 treatment of HEK 293T cells with ETP, the 2IC50 treatment of HEK 293T cells with ETP resulted in the formation of LDs with homogeneous microviscosities in individual cells, with some cells having LD populations of 50, 90, 160 and 300 cP (Fig. 6D).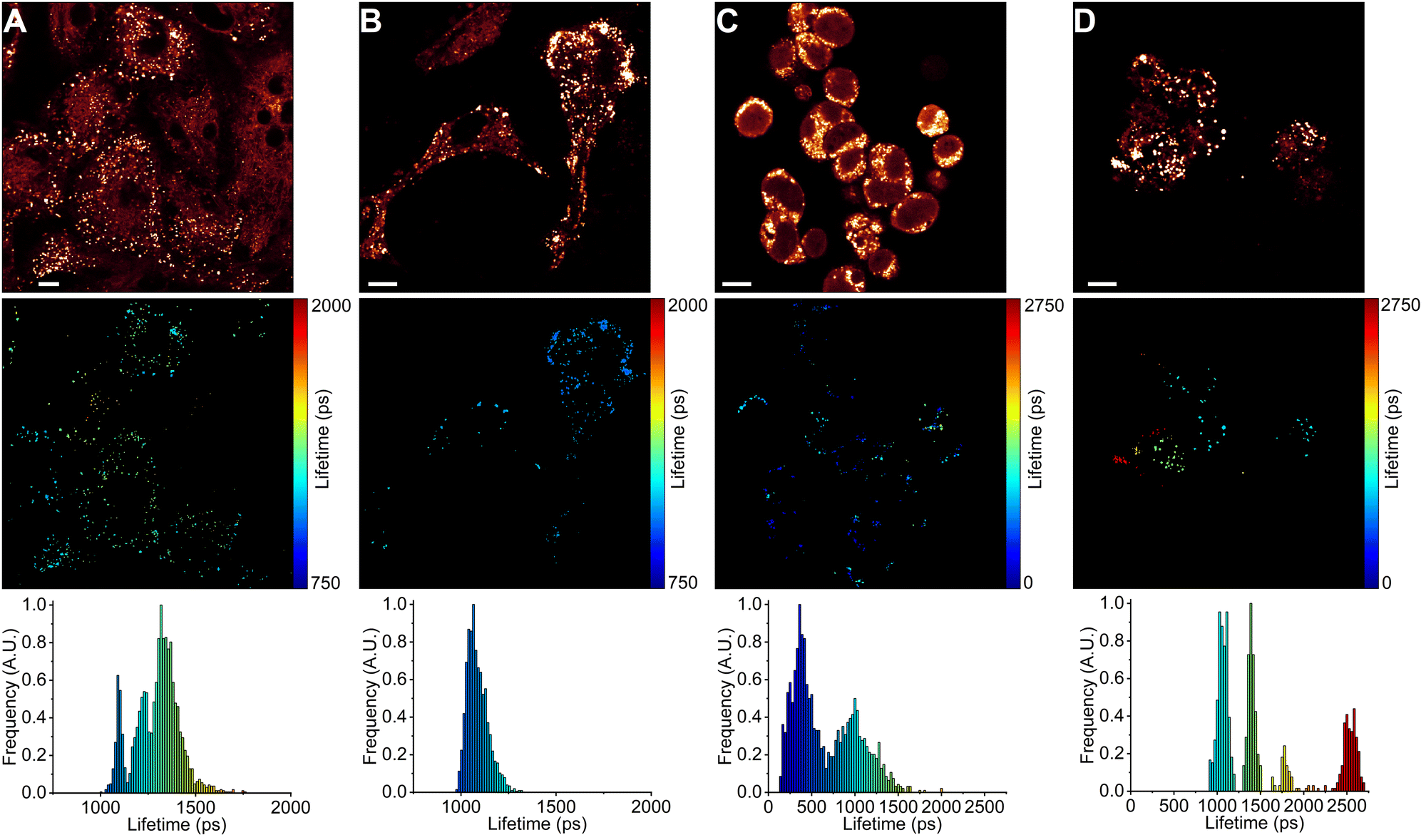 | ||
| Fig. 6 FLIM images of BODIPY-LD in LDs of A549 cells treated with DOX (2IC50) for 48 h. (A) A549 cells treated with ETP (2IC50) for 48 h. (B) HEK 293T cells treated with DOX (2IC50) for 48 h. (C) HEK 293T cells treated with ETP (2IC50) for 48 h. (D) The top panel shows images of fluorescence intensity. FLIM images are shown in the middle panel. The corresponding lifetime histograms are shown in the bottom panel. Scale bars are 10 μm. | ||
To summarize, DOX and ETP-resistant A549 cells possessed completely different LD populations compared to their corresponding IC50 treatments. The IC50 treatments of A549 cells with DOX and ETP resulted in very similar LD populations with high and low microviscosities in each cell. In contrast, DOX- and ETP-resistant A549 cells did not exhibit similar microviscosities of LDs. Instead, DOX-resistant cells possessed LDs with heterogenous (60–100 cP) microviscosities, whereas ETP-resistant cells featured homogeneous LDs of 50–65 cP. On the other hand, DOX- and ETP-resistant HEK 293T cells possessed LDs with microviscosities similar to their respective 48 h IC50 treatments. However, while ETP-resistant A549 cells had LDs with homogeneous microviscosities, LDs in ETP-resistant HEK 293T cells showed highly variable microviscosities. Importantly, it appears that the microviscosities of LDs, and their lipid compositions in chemotherapy-resistant cells differ depending on drug treatment and cell type.
Conclusions
In conclusion, we have synthesised and explored the photophysical properties of a new BODIPY-based molecular rotor – BODIPY-LD. We showed that fluorescence decays of BODIPY-LD are sensitive to viscosity over a large range, from 0.5 to 1457 cP, and are minimally influenced by solvent polarity. Importantly, time-resolved fluorescence measurements on LUVs with Lo and Ld lipid phases demonstrate that BODIPY-LD possesses vastly different fluorescence lifetimes in lipid-ordered and lipid-disordered environments, allowing this probe to successfully distinguish lipid phase transitions and estimate the cholesterol concentrations in Ld phases. We found that BODIPY-LD preferentially partitions into the LDs of live cells without staining the cytoplasm or plasma membrane. Our results demonstrate that LDs in cancerous A549 and non-cancerous HEK 293T cells highly differ in their microviscosity, making the dye applicable as a diagnostic biomarker for cancer. Furthermore, the chemotherapeutic drugs DOX and ETP induce different microviscosity changes in LDs in cancerous and non-cancerous cells. Finally, we demonstrate that microviscosities of LDs in chemotherapy-resistant cells differ depending on the drug treatment and cell type.Experimental section
Dyes, reagents, and solvents
The synthetic details of BODIPY-LD, with mass and NMR spectra, are presented in the ESI.† NMR spectra were recorded on a Bruker Ascend 400 spectrometer (400 MHz for 1H, 100 MHz for 13C, 128.4 MHz for 11B, 376.5 MHz for 19F). NMR spectra were referenced to residual solvent peaks. HRMS spectra were recorded on a quadrupole, time-of-flight mass spectrometer (microTOF-Q II, Bruker Daltonik GmbH, Germany). Column chromatography was performed using silica gel 60 (0.040–0.063 mm) (Merck). Thin layer chromatography (TLC) was performed using TLC-aluminium sheets with silica gel (Merck 60 F254). Visualisation was accomplished by UV light. Melting points were determined in open capillaries with a digital melting point IA9100 series apparatus (Thermo Fisher) and were not corrected. Reagents and solvents for the organic synthesis of the BODIPY compounds were purchased directly from commercial suppliers; solvents were purified by known procedures. Stock solutions of 1 mM BODIPY-LD were prepared in methanol or DMSO and diluted for further experiments in solvents or their mixtures. All solvents used were spectroscopic grade obtained from Sigma Aldrich. DOPC, POPC, DPPC, and cholesterol were obtained from Avanti Polar Lipids. DOX and ETP were obtained from Sigma Aldrich, and 10 mM stock solutions of DOX and ETP were prepared in DMSO and diluted for subsequent experiments.Formation of LUVs
LUVs were prepared by mixing lipids in appropriate ratios; afterwards, BODIPY-LD was added to the mixture with a dye to lipid ratio of 1:800, and chloroform was evaporated under a nitrogen stream for at least 2 hours. After evaporation, PBS buffer was added on top of the film. The total concentration of lipids in the PBS solution was 1 mM. In the case of DOPC/DPPC/Chol (1/5/5) LUVs, PBS buffer was heated to 60 °C. After resuspension, mixtures were placed in an ultrasonic bath (1 h) and extruded through 100 nm polycarbonate membranes with a syringe extruder (Avanti Polar Lipids). Extrusions were performed above the melting temperature of the lipids.
Absorption, steady-state, and time-resolved fluorescence spectroscopies
Absorption spectra were measured using a Jasco V-670 spectrophotometer. Fluorescence spectra were recorded with an Edinburgh-F900 (Edinburgh Instruments) fluorimeter using an Fianum white laser, together with bandpass filters (Thorlabs), emitting at 488 nm as an excitation source. Fluorescence decays were measured using time-correlated single-photon counting (TCSPC). Fluorescence decays had 5000 counts at the peak of the decay with 20 ns time window being used with 4096 channels in the time domain. 10 mm quartz cuvettes were used for absorption and fluorescence measurements with a BODIPY-LD concentration up to 2 μM. Fluorescence decays of BODIPY-LD in solvent mixtures and pure solvents were taken at 20 °C.Imaging of live cells
Cell imaging experiments were performed using the human lung cancer A549 and immortalized human embryonic kidney HEK 293T cell lines (ATCC). The cells were cultured in Dulbecco‘s modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 100 IU mL−1 penicillin and 100 μg mL−1 streptomycin (Thermo Fisher). The cells were incubated at 37 °C with 5% CO2. Before imaging, cells were seeded into Ibidi μ-Dish (Ibidi) at a seeding density of 10000 cells mL−1 and allowed to grow for 24 hours. For cell imaging, 1 μM of BODIPY-LD solution (in DMSO) was added to the culture medium for 5 min at 37 °C. FLIM imaging was performed using a Leica SP8 microscope with a 63× objective (HC PL APO oil immersion, N.A. – 1.4, Leica).
Fluorescence lifetime imaging microscopy (FLIM)
FLIM was performed with a Leica SP8 microscope using a 63× objective (HC PL APO oil immersion, N.A. – 1.4, Leica). The fluorescence decay signal was measured over the 505–550 nm range using a 488 nm filter-supported white light laser excitation line. The FLIM images were produced in a 512 × 512 pixel resolution with 128 channels in the time domain, and pixels were binned to have at least 1000 counts at the peak of the decay curve for reliable biexponential fitting. Only lipid droplet-containing pixels in FLIM images were fitted. Lipid droplets were identified based on their 100-fold greater fluorescence counts per pixel relative to the cytoplasm. The instrument response function (IRF) was measured by recording the laser reflection signal off a glass coverslip.Data analysis
FLIM images were analysed with FLIMFIT software (v4.6.1, Imperial College London).59 The biexponential fluorescence decay model with intensity-weighted mean lifetimes was applied for both FLIM and fluorimeter time-correlated single photon counting (TCSPC) measurements:where ai and τi are the amplitudes of the individual components. The goodness-of-fit parameter (χ2) was 1.3 or less for both FLIM images and fluorimeter TCSPC decays. Fluorimeter TCSPC decays of BODIPY-LD were fitted using the Edinburgh-F900 software package F900.
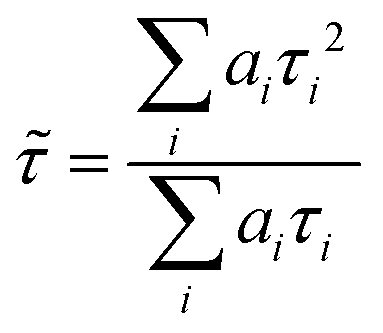
Conflicts of interest
The authors declare no conflicts of interest.Notes and references
- A. L. S. Cruz, E. D. A. Barreto, N. P. B. Fazolini, J. P. B. Viola and P. T. Bozza, Cell Death Dis., 2020, 11, 105 CrossRef PubMed.
- J. A. Olzmann and P. Carvalho, Nat. Rev. Mol. Cell Biol., 2019, 20, 137–155 CrossRef CAS PubMed.
- A. R. Thiam and M. Beller, J. Cell Sci., 2017, 130, 315–324 CAS.
- A. Herms, M. Bosch, B. J. N. Reddy, N. L. Schieber, A. Fajardo, C. Ruperez, A. Fernandez-Vidal, C. Ferguson, C. Rentero, F. Tebar, C. Enrich, R. G. Parton, S. P. Gross and A. Pol, Nat. Commun., 2015, 6, 7176 CrossRef PubMed.
- C.-W. Wang, Biochim. Biophys. Acta, 2016, 1861, 793–805 CrossRef CAS PubMed.
- C. Cheng, F. Geng, X. Cheng and D. Guo, Cancer Commun., 2018, 38, 1–14 CrossRef CAS PubMed.
- J. E. Norman, H. H. Aung, D. W. Wilson and J. C. Rutledge, Food Funct., 2018, 9, 6245–6256 RSC.
- T. C. Walther and R. V. Farese, Annu. Rev. Biochem., 2012, 81, 687–714 CrossRef CAS PubMed.
- C. F. Kurat, H. Wolinski, J. Petschnigg, S. Kaluarachchi, B. Andrews, K. Natter and S. Kohlwein, Mol. Cell, 2009, 33, 53–63 CrossRef CAS PubMed.
- S. Xu, X. Zhang and P. Liu, Biochim. Biophys. Acta - Mol. Basis Dis., 2018, 1864, 1968–1983 CrossRef CAS PubMed.
- N. L. Gluchowski, M. Becuwe, T. C. Walther and R. V. Farese, Nat. Rev. Gastroenterol. Hepatol., 2017, 14, 343–355 CrossRef CAS PubMed.
- H. Wang, M. Lei, R.-C. Hsia and C. Sztalryd, Methods Cell Biol., 2013, 116, 129–149 CAS.
- H. Abramczyk, J. Surmacki, M. Kopeć, A. K. Olejnik, K. Lubecka-Pietruszewska and K. Fabianowska-Majewska, Analyst, 2015, 140, 2224–2235 RSC.
- Z. Li, H. Liu and X. Luo, Am. J. Cancer Res., 2020, 10, 4112–4122 CAS.
- C. Jin and P. Yuan, Oncol. Lett., 2020, 20, 2091–2104 CrossRef CAS PubMed.
- Y. Jin, Y. Tan, J. Wu and Z. Ren, Cell Death Discovery, 2023, 9, 254 CrossRef CAS PubMed.
- A. Zadoorian, X. Du and H. Yang, Nat. Rev. Endocrinol., 2023, 19, 443–459 CrossRef CAS PubMed.
- A. Mehdizadeh, M. Bonyadi, M. Darabi, R. Rahbarghazi, S. Montazersaheb, K. Velaei, M. Shaaker and M.-H. Somi, Bioimpacts, 2017, 7, 31–39 CrossRef CAS PubMed.
- V. Ajdzanovic, M. Mojic, D. Maksimovic-Ivanic, M. Bulatovic, S. Mijatovic, V. Milosevic and I. Spasojevic, J. Membr. Biol., 2013, 246, 307–314 CrossRef CAS PubMed.
- Y. Wang, H. Pan, D. Chen, D. Guo and X. Wang, J. Funct. Foods, 2021, 83, 104570 CrossRef CAS.
- F. Geng and D. Guo, Int. Med. Rev., 2017, 3, 10 Search PubMed.
- Y. Wu, M. Štefl, A. Olzyńska, M. Hof, G. Yahioglu, P. Yip, D. R. Casey, O. Ces, J. Humpolíčková and M. K. Kuimova, Phys. Chem. Chem. Phys., 2013, 15, 14986–14993 RSC.
- J. Zidar, F. Merzel, M. Hodošček, K. Rebolj, K. Sepčić, P. Maček and D. Janežič, J. Phys. Chem. B, 2009, 113, 15795–15802 CrossRef CAS PubMed.
- O. G. Mouritsen and M. J. Zuckermann, Lipids, 2004, 39, 1101–1113 CrossRef CAS PubMed.
- M. R. Dent, I. López-Duarte, C. J. Dickson, N. D. Geoghegan, J. M. Cooper, I. R. Gould, R. Krams, J. A. Bull, N. J. Brooks and M. K. Kuimova, Phys. Chem. Chem. Phys., 2015, 17, 18393–18402 RSC.
- M. R. Dent, I. López-Duarte, C. J. Dickson, P. Chairatana, H. L. Anderson, I. R. Gould, D. Wylie, A. Vyšniauskas, N. J. Brooks and M. K. Kuimova, Chem. Commun., 2016, 52, 13269–13272 RSC.
- M. Páez-Pérez, I. López-Duarte, A. Vyšniauskas, N. J. Brooks and M. K. Kuimova, Chem. Sci., 2021, 12, 2604–2613 RSC.
- J. Mahamid, D. Tegunov, A. Maiser, J. Arnold, H. Leonhardt, J. M. Plitzko and W. Baumeister, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16866–16871 CrossRef CAS PubMed.
- H. Chen, J. Zhao, J. Lin, B. Dong, H. Li, B. Geng and M. Yan, RSC Adv., 2021, 11, 8250–8254 RSC.
- C. W. Song, U. Tamima, Y. J. Reo, M. Dai, S. Sarkar and K. H. Ahn, Dyes Pigm., 2019, 171, 107718 CrossRef CAS.
- M. R. Bittermann, M. Grzelka, S. Woutersen, A. M. Brouwer and D. Bonn, J. Phys. Chem. Lett., 2021, 12, 3182–3186 CrossRef CAS PubMed.
- M. K. Kuimova, Phys. Chem. Chem. Phys., 2012, 14, 12671–12686 RSC.
- S.-C. Lee, J. Heo, H. C. Woo, J.-A. Lee, Y. H. Seo, C.-L. Lee, S. Kim and O.-P. Kwon, Chem. – Eur. J., 2018, 24, 13706–13718 CrossRef CAS PubMed.
- M. Kubánková, I. López-Duarte, D. Kiryushko and M. K. Kuimova, Soft Matter, 2018, 14, 9466–9474 RSC.
- P. S. Sherin, I. López-Duarte, M. R. Dent, M. Kubánková, A. Vyšniauskas, J. A. Bull, E. S. Reshetnikova, A. S. Klymchenko, Y. P. Tsentalovich and M. K. Kuimova, Chem. Sci., 2017, 8, 3523–3528 RSC.
- T. Mukherjee, R. J. Martinez-Sanchez, K. T. Fam, S. Bou, L. Richert, D. Garnier, Y. Mély, S. Kanvah, A. S. Klymchenko and M. Collot, Mater. Chem. Front., 2021, 5, 2459–2469 RSC.
- A. Polita, S. Toliautas, R. Žvirblis and A. Vyšniauskas, Phys. Chem. Chem. Phys., 2020, 22, 8296–8303 RSC.
- A. Vyšniauskas, M. Qurashi, N. Gallop, M. Balaz, H. L. Anderson and M. K. Kuimova, Chem. Sci., 2015, 6, 5773–5778 RSC.
- A. Polita, M. Stancikaitė, R. Žvirblis, K. Maleckaitė, J. Dodonova-Vaitkūnienė, S. Tumkevičius, A. P. Shivabalana and G. Valinčius, RSC Adv., 2023, 13, 19257–19264 RSC.
- I. López-Duarte, T. T. Vu, M. A. Izquierdo, J. A. Bull and M. K. Kuimova, Chem. Commun., 2014, 50, 5282–5284 RSC.
- J. T. Mika, A. J. Thompson, M. R. Dent, N. J. Brooks, J. Michiels, J. Hofkens and M. K. Kuimova, Biophys. J., 2016, 111, 1528–1540 CrossRef CAS PubMed.
- Z. Yang, Y. He, J.-H. Lee, N. Park, M. Suh, W.-S. Chae, J. Cao, X. Peng, H. Jung, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 9181–9185 CrossRef CAS PubMed.
- L. Wang, Y. Xiao, W. Tian and L. Deng, J. Am. Chem. Soc., 2013, 135, 2903–2906 CrossRef CAS PubMed.
- J. A. Levitt, M. K. Kuimova, G. Yahioglu, P.-H. Chung, K. Suhling and D. Phillips, J. Phys. Chem. C, 2009, 113, 11634–11642 CrossRef CAS.
- S. Chen, Y. Hong, Y. Zeng, Q. Sun, Y. Liu, E. Zhao, G. Bai, J. Qu, J. Hao and B. Z. Tang, Chem. – Eur. J., 2015, 21, 4315–4320 CrossRef CAS PubMed.
- R. Guo, J. Yin, Y. Ma, G. Li, Q. Wang and W. Lin, Sens. Actuators, B, 2018, 271, 321–328 CrossRef CAS.
- V. Singh, P. Johansson, E. Ekedahl, Y.-L. Lin, O. Hammarsten and F. Westerlund, Biochem. Biophys. Res. Commun., 2022, 594, 57–62 CrossRef CAS PubMed.
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandez-Boussard, H. McLeod, T. E. Klein and R. B. Altmana, Pharmacogenet. Genomics, 2011, 21, 440–446 CrossRef CAS PubMed.
- I. A. Windham and S. Cohen, J. Cell Biol., 2022, 221, e202210008 CrossRef CAS PubMed.
- T. Róg, M. Pasenkiewicz-Gierula, I. Vattulainen and M. Karttunen, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 97–121 CrossRef PubMed.
- G. Gramse, A. Dols-Perez, M. A. Edwards, L. Fumagalli and G. Gomila, Biophys. J., 2013, 104, 1257–1262 CrossRef CAS PubMed.
- S. I. Suarez, C. C. Warner, H. Brown-Harding, A. M. Thooft, B. VanVeller and J. C. Lukesh III, Org. Biomol. Chem., 2020, 18, 495–499 RSC.
- T. C. Walther, J. Chung and R. V. Farese, Annu. Rev. Cell Dev. Biol., 2017, 33, 491–510 CrossRef CAS PubMed.
- R. Punia, K. Raina, R. Agarwal and R. P. Singh, PLoS One, 2017, 12, e0182870 CrossRef PubMed.
- V. Chaudhary, S. Das, A. Nayak, S. K. Guchhait and C. N. Kundu, RSC Adv., 2015, 5, 26051–26060 RSC.
- F. M. Siswanto, A. Tamura, R. Sakuma and S. Imaoka, Mol. Pharmacol., 2022, 101, 257–273 CrossRef CAS PubMed.
- A. E. Ventura, S. Pokorna, N. Huhn, T. C. B. Santos, M. Prieto, A. H. Futerman and L. C. Silva, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2023, 1868, 159347 CrossRef CAS PubMed.
- L. J. Pike, J. Lipid Res., 2003, 44, 655–667 CrossRef CAS PubMed.
- L. S. C. Warren, A. Margineanu, D. Alibhai, D. J. Kelly, C. Talbot, Y. Alexandrov, I. Munro, M. Katan, C. Dunsby and P. M. W. French, PLoS One, 2013, 8, e70687 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb02979d |
| This journal is © The Royal Society of Chemistry 2024 |