
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radiolabeled multi-layered coated gold nanoparticles as potential biocompatible PET/SPECT tracers†
Cristina M.
Uritu‡
 a,
Cristina M.
Al-Matarneh‡
b,
Denisse I.
Bostiog‡
b,
Adina
Coroaba‡
b,
Vlad
Ghizdovat
c,
Silviu I.
Filipiuc
a,
Natalia
Simionescu
b,
Cipriana
Stefanescu
*c,
Wael
Jalloul
c,
Valentin
Nastasa
*d,
Bogdan I.
Tamba
*a,
Stelian S.
Maier
be and
Mariana
Pinteala
*b
a,
Cristina M.
Al-Matarneh‡
b,
Denisse I.
Bostiog‡
b,
Adina
Coroaba‡
b,
Vlad
Ghizdovat
c,
Silviu I.
Filipiuc
a,
Natalia
Simionescu
b,
Cipriana
Stefanescu
*c,
Wael
Jalloul
c,
Valentin
Nastasa
*d,
Bogdan I.
Tamba
*a,
Stelian S.
Maier
be and
Mariana
Pinteala
*b
aAdvanced Center for Research and Development in Experimental Medicine “Prof. Ostin C. Mungiu”, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania. E-mail: bogdan.tamba@umfiasi.ro; Tel: +4072 929 0877
bCentre of Advanced Research in Bionanoconjugates and Biopolymers, “Petru Poni” Institute of Macromolecular Chemistry, Iasi, Romania. E-mail: pinteala@icmpp.ro; Tel: +40333 288 0221
cDepartment of Biophysics and Medical Physics, Nuclear medicine, Faculty of Medicine, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania. E-mail: cipriana.stefanescu@umfiasi.ro; Tel: +4072 285 2158
dFaculty of Veterinary Medicine, “Ion Ionescu de la Brad” Iasi University of Life Science, Iasi, Romania. E-mail: vnastasa67@gmail.com; Tel: +4075 426 8911
ePolymers Research Center, “Gheorghe Asachi” Technical University of Iasi, Romania
First published on 15th March 2024
Abstract
The demand for tailored, disease-adapted, and easily accessible radiopharmaceuticals is one of the most persistent challenges in nuclear imaging precision medicine. The aim of this work was to develop two multimodal radiotracers applicable for both SPECT and PET techniques, which consist of a gold nanoparticle core, a shell involved in radioisotope entrapment, peripherally placed targeting molecules, and biocompatibilizing polymeric sequences. Shell decoration with glucosamine units located in sterically hindered molecular environments is expected to result in nanoparticle accumulation in high-glucose-consuming areas. Gold cores were synthesized using the Turkevich method, followed by citrate substitution with linear PEG α,ω-functionalized with thiol and amine groups. The free amine groups facilitated the binding of branched polyethyleneimine through an epoxy ring-opening reaction by using PEG α,ω-diglycidyl ether as a linker. Afterwards, the glucose–PEG–epoxy prepolymer has been grafted onto the surface of AuPEG–PEI conjugates. Finally, the AuPEG–PEI–GA conjugates were radiolabeled with 99mTc or 68Ga. Instant thin-layer chromatography was used to evaluate the radiolabeling yield. The biocompatibility of non-labeled and 99mTc or 68Ga labeled nanoparticles was assessed on normal fibroblasts. The 99mTc complexes remained stable for over 22 hours, while the 68Ga containing ones revealed a slight decrease in stability after 1 hour.
1. Introduction
Because of the consistently escalating incidence of cancer, there is an ever-increasing need for the development of effective treatment options and, critically, the possibility of early detection by using accessible and low-cost methods. In this respect, nuclear medicine can provide a rapid and precise diagnosis, as long as appropriate radiotracers for the target tissue are available. The nuclear medicine procedures, known as positron emission tomography (PET) and single photon emission computerized tomography (SPECT), rely on the distribution of radiotracers (gamma and positron-emitters) throughout the body and subsequently imaged using a gamma camera. These radiotracers facilitate the quantification of biological processes and enable the acquisition of comprehensive whole-body images that demonstrate the locations of radiotracer accumulation. The benefits of PET imaging with 18F-FDG (2-deoxy-2-[fluorine-18]fluoro-D-glucose) as a radiotracer for the purpose of identifying areas of abnormal glucose metabolism,1 including various forms of tumours, are an indisputable aspect. However, the exorbitant expense associated with this diagnostic procedure significantly restricts patients’ ability to benefit from this investigation method. Carbon nanotubes, gold nanoparticles/nanorods, quantum dots, and magnetic nanoparticles are just a few of the many nanoscale materials being studied for imaging applications.Gold nanoparticles (AuNPs) have various biological applications2–4 owing to their unique surface chemical,5 electrical,6 and optical properties.7 Contrast agents based on gold nanoparticles8 have been developed for various applications, including computed tomography (CT),9 magnetic resonance imaging (MRI),10 single-photon emission computed tomography (SPECT),11 fluorescence imaging,12 and others, with the aim of amplifying the sensitivity and precision of these imaging modalities. For instance, high-quality CT imaging of the blood pool/liver was achieved by the use of polyethylene glycol (PEG) stabilized 30 nm AuNPs.13,14 Targeted tumor SPECT imaging, on the other hand, involved the application of gold nanoparticles (AuNPs) with a diameter of 5 nm, which have been doped with 199Au and coated with peptide molecules.11
It is currently postulated that gold nanoparticles function as a form of radiosensitizer in the context of radiotherapy.15 The reason for this phenomenon is attributed to the powerful photoelectric absorption and secondary electron effects of gamma or X-ray radiation, which can accelerate the occurrence of DNA strand breaks. The toxicity of gold nanoparticles is influenced by various factors such as size, shape, surface charge, and surface coating.16 However, it is worth noting that the overall toxicity dose of these nanoparticles falls within the safe limits, allowing their use in radiation therapy for cancer.
Experimental research conducted in vivo has indicated that the administration of gold nanoparticles with a diameter of 1.9 nm can considerably increase the effectiveness of radiotherapy.17 Subsequent in vitro investigations demonstrated that PEGylated gold nanoparticles with sizes of 4.6 and 6.1 nm could potentially decrease the cell survival rates of both EMT-6 and CT26 cells.18 According to Kong et al.,19 the radiation sensitivity of 10 nm gold nanoparticles coated with cysteamine and glucose was significantly enhanced by high-energy rays, with the most sensitive energy level being 200 keV.
Previous studies have revealed that gold nanoparticles (NPs) with a diameter of 50 nm exhibit greater susceptibility to radiation in comparison to their counterparts with sizes of 14 and 74 nm. The observed phenomenon can be attributed to the enhanced endocytosis and cellular uptake of 50 nm uncoated gold particles, as reported in the literature.20
Therapeutic gold nanoparticles that measure less than 50 nm in diameter can easily pass through cell membranes, while particles that measure less than 20 nm have the ability to traverse the endothelium of blood vessels.21 The stability of gold nanoparticles can be enhanced through surface coatings, such as PEG-SH, as reported in the literature.22 Hence, it is desirable to clarify the radiosensitization effects that are dependent on the size of PEG-coated gold nanoparticles ranging from 5 to 60 nm. This gives a clear indication of how well the radiation and drug delivery are working.23
It is imperative to employ a suitable platform that facilitates the conjugation, encapsulation, stability, and modification of functional groups on the surface of NPs.24 Branched polyethyleneimine (PEI), which has abundant surface amine groups and high water solubility, has been employed as a potent carrier to coat nanoparticles or encapsulate medicines. The incorporation of a targeting ligand onto the surface of PEGylated PEI can yield a multifunctional nanoplatform capable of targeted tumour CT/MR imaging.25
The present study represents the first steps in creating a multimodal radiotracer capable of efficiently incorporating both SPECT and PET radionuclides. In this regard, a hybrid nanoparticle was designed, consisting of a metal core, a radioisotope entrapment shell, a recognition molecule, and a polymeric sequence that confers biocompatibility. According to the literature,26–29 it is anticipated that the grafting of glucosamine (GA) units on the surface of a polymer will lead to its accumulation in regions of high glucose consumption. This is attributed to the steric hindrance caused by the complex architectures, which renders GA a vector molecule for these specific tissue domains.
The employment of 99mTc and 68Ga radionuclides represents a highly accessible method for radiolabeling a wide range of molecules. These radionuclides are synthesized in the laboratory through 99Mo/99mTc and 68Ge/68Ga generators, respectively, eliminating the requirement for a cyclotron. The use of sodium pertechnetate (99mTcO4−Na+) presents numerous advantages, such as its favorable half-life of approximately 6 hours and suitable energy (140 keV), which leads to minimized radiation exposure to the patient. Finally, due to its ability to exist in multiple oxidation states, 99mTc exhibits a wide range of chemical properties.30,31 This fact provides the possibility of creating a broad spectrum of complexes with distinct properties, which is a notable benefit in the advancement of radiopharmaceuticals. The present study suggests that utilizing 99mTc as a radionuclide for the purpose of labeling nanoparticles that exhibit glucose on their surface may serve as a promising substitute for 18F-FDG and PET. On the other hand, 68Ga is a radionuclide with a relatively short half-life of 68 minutes, making it appropriate for PET imaging as a result of its β+ decay (with a positron energy of 1.90 MeV, as per the manufacturer's specifications). Currently, PET stands out as one of the most sophisticated imaging techniques that can monitor biochemical, physiological, and pharmacological mechanisms with remarkable precision, boasting an exceptional sensitivity range of 10−11–10−12 mol L−1. The 68Ga radionuclide has garnered growing attention among researchers in the field owing to its numerous advantages. Numerous articles in the literature discuss the radiolabeling of peptides, proteins, and other biomolecules for use in various fields such as oncology, neurology, and cardiology.32–34 Typically, DOTA/NOTA chelators have been commonly utilized for the purpose of radionuclide complexation.35 These chelators are characterized by their polydentate macrocyclic structures, which enable them to effectively accommodate di- and tri-valent metal ions through coordinative binding. Conjugation of macrocyclic structures capable of radionuclide chelation with the polymeric shell of nanoparticles would result in complex structures that require supplementary synthesis and purification procedures, thereby causing a significant increase in the production costs of a radiopharmaceutical compound. To address this issue, an initial hypothesis was formulated suggesting that the polymer configuration, such as hyperbranched polyethyleneimine (PEI) selected for the purpose of coating the surface of gold nanoparticles, could represent an ideal molecular environment for the coordinative attachment of 99mTc and 68Ga. The experimental confirmation of this hypothesis is detailed in the Results section of this study.
2. Materials and methods
2.1. Materials
Gold(III) chloride trihydrate (HAuCl4·3H2O; ≥99.9%, Sigma-Aldrich, St. Louis, MO, USA), tri-sodium citrate dihydrate (C6H5Na3O7·2H2O; ≥99%, Carl Roth, Karlsryhe, Germany), poly(ethylene glycol)diglycidyl ether (Mn = 500, Sigma-Aldrich, St. Louis, MO, USA), poly(ethylene glycol) (Mn = 2000, Sigma-Aldrich, St. Louis, MO, USA), thiol PEG amine (HS-PEG2000-NH2·HCl, Mw = 2000, JenKem Technology, Xi Xiaokou Road, Beijing), polyethyleneimine solution (Mw = 2000, 50 wt% in H2O; Sigma-Aldrich, St. Louis, MO, USA), (D)-(+)-glucosamine hydrochloride (Sigma Aldrich), sodium borohydride (NaBH4, Sigma-Aldrich), stannous chloride (SnCl2, Sigma-Aldrich), and deionized water were used in this study. All reagents and solvents were used without further purification.2.2. Instrumental analyses
Absorption measurements (UV-Vis) were performed using a Lambda 35 device (PerkinElmer, USA). The absorption spectra were recorded in the 300–700 nm range for identical sample volumes (3 mL) with the following parameters: slit width 1 nm, scan speed 480 nm min−1, and data interval 1 nm. The spectra of the samples were recorded at room temperature using 1 cm path-length quartz cuvettes.Fourier transform infrared (FTIR) spectra were obtained in transmission mode using a Bruker Vertex instrument (Bruker Physik GmbH, Ettlingen, Germany), model 70. The samples were prepared by depositing the nanoparticle suspension on KBr pellets which were then subjected to a drying process (using a UV lamp) before recording the spectra. The spectra ranged from 4000 to 400 cm−1 with a resolution of 2 cm−1.
The hydrodynamic diameter and zeta potential of the nanoparticles were determined by DLS using a Delsa Nano C Submicron Particle Size Analyzer (Beckman Coulter, Brea, CA, USA) with the corresponding module (flow cell module).
Mass spectrometry analysis was performed using an Agilent 6500 Series accurate-mass quadrupole time-of-flight (Q-TOF) LC/MS instrument. The solutions were introduced into the electrospray ion source (ESI) via a syringe pump at a flow rate of 0.01 mL min−1. After optimization of the Q/TOF MS parameters, they were set as follows: electrospray ionization (positive ion mode), drying gas (N2) flow rate 8 L min−1; drying gas temperature 325 °C; nebulizer pressure 40 psig, capillary voltage 4000 V; and fragmentation voltage 175 V; the full-scan mass spectra of the investigated compounds were acquired in the m/z range of 50–3000. The mass scale was calibrated using the standard calibration procedure and compounds provided by the manufacturer. Data were collected and processed using Mass Hunter Workstation Software Data Acquisition for 6200/6500 Series, version B.07.00 (Agilent Technologies, Inc., Santa Clara, CA, USA).
The morphology of the samples was investigated with a scanning electron microscope type Verios G4 UC (Thermo Fisher Scientific, Waltham, MA, USA) working in STEM mode at 30 kV, with a STEM 3+ detector (bright-field mode). The samples were prepared on 24 carbon-coated copper grids with a 300-mesh size. The size of the nanoparticles was determined using a transmission electron microscope (HT7700 TEM, Hitachi, Tokyo, Japan). The elemental composition of the samples was determined through the EDAX analysis using a Verios G4 UC field-emission scanning electron microscope (Thermo Scientific, Czech Republic) equipped with an Octane Elect Super SDD detector operating at 20 kV with a spot size of 6.4 nA. The samples were deposited on aluminum supports and subsequently dried prior to being coated with a 6 nm layer of platinum to prevent charge buildup during the electron beam exposure. The coating process was carried out using a Leica EM ACE200 Sputter coater. The sizes of the nanoparticles under study were determined by analyzing the STEM/TEM images through the use of ImageJ software. The resulting values were then presented as the mean core diameters in nanometers, together with the standard deviation of the obtained values.
The X-ray photoelectron spectroscopy (XPS) results were obtained using an Axis NOVA equipment (Kratos Analytical, Manchester, United Kingdom), with AlK (1486.6 eV) as the X-ray source, operating at 20 mA current, 15 kV voltage (300 W), and 10−8–10−9 Torr pressure. For each element of interest, a high-resolution spectrum was obtained by averaging five scans recorded with a pass energy of 20 eV and a step size of 0.1 eV. The binding energy of the C 1s peak has been established as the reference value for all binding energies, normalized at 284.6 eV. The ESCApe program was used to fit the XPS data using a Gaussian–Lorentzian mixed function.
2.3. Preparation of gold nanoparticle-based nanoconjugates
Gold nanoparticles (AuNPs) were chemically synthesized (Scheme 1) by the modified Turkevich method.36 Briefly, gold(III) chloride trihydrate (12.5 mg, 0.0313 mmol) was added over 100 mL of H2O mQ, into a 250 mL flask, then was placed in a preheated oil bath at 110 °C, under vigorous stirring (700 rpm). Once the gold salt is dissolved and after the temperature of the solution remains constant (∼80–85 °C, about 30 minutes), a solution of 50 mg of sodium citrate (0.17 mmol) dissolved in 50 mL of H2O mQ was warmed up to the same temperature and added over. The reaction between the auric chloride and citrate begins within the first minute. Afterward, the solution is allowed to gradually reach room temperature while undergoing magnetic agitation and being covered in aluminum foil for a duration of roughly 12 hours. A nanodispersion of a distinct ruby-red color was obtained. It was then subjected to purification through centrifugation at 8000g for a duration of 8 minutes. The purified precipitate was then dispersed in mQ water. | ||
| Scheme 1 Synthesis of polymer-coated gold nanoparticles. | ||
In the next step, the production of AuPEG–NH2 is initiated by subjecting the previously obtained AuNPs to a reaction with α,ω heterobifunctional PEG (Mw 2000), containing both thiol and amine moieties. The AuNP nanodispersion was subjected to stirring at ambient temperature for 24 hours, in the presence of a bifunctional PEG solution at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 atomic ratio (Au:PEG). For the next step, in order to obtain AuPEG–epoxy, we maintained the aforementioned conditions (24 hours reaction time, room temperature, 1:200 atomic ratio between components, reported to gold) and reacted AuNP–PEG with PEG diglycidyl ether of a Mw of 500. The previously mentioned step was deemed essential to acquire a suitable terminal reacting group (epoxy) for subsequent reaction steps, which involved the grafting of the nanoconjugate with polyethyleneimine (PEI). AuPEG–PEI was synthesized by reacting the epoxy terminal group of AuPEG–epoxy with the primary amine of PEI (Mw: 2000). The two solutions, AuPEG–epoxy and PEI (in a 1:1 ratio of PEG diglycidyl ether to PEI), were combined and stirred vigorously for 72 hours at room temperature, in an aluminum-covered flask. The procedure for the reaction involving the epoxy ring and primary amine groups was conducted in a manner consistent with a previously reported protocol.37,38
:200 atomic ratio (Au:PEG). For the next step, in order to obtain AuPEG–epoxy, we maintained the aforementioned conditions (24 hours reaction time, room temperature, 1:200 atomic ratio between components, reported to gold) and reacted AuNP–PEG with PEG diglycidyl ether of a Mw of 500. The previously mentioned step was deemed essential to acquire a suitable terminal reacting group (epoxy) for subsequent reaction steps, which involved the grafting of the nanoconjugate with polyethyleneimine (PEI). AuPEG–PEI was synthesized by reacting the epoxy terminal group of AuPEG–epoxy with the primary amine of PEI (Mw: 2000). The two solutions, AuPEG–epoxy and PEI (in a 1:1 ratio of PEG diglycidyl ether to PEI), were combined and stirred vigorously for 72 hours at room temperature, in an aluminum-covered flask. The procedure for the reaction involving the epoxy ring and primary amine groups was conducted in a manner consistent with a previously reported protocol.37,38
In a separate synthesis, the PEG diglycidyl ether (Mw 500) was subjected to a reaction with glucosamine (GA) in a 1:1 molar ratio. The resulting mixture was stirred in an aqueous medium for 72 hours, at ambient temperature. Lyophilization was employed to extract water from the system, resulting in the formation of the product hereafter referred as GA–PEG–epoxy.
The GA–PEG–epoxy subsequently reacted with the polymeric shell of AuPEG–PEI via oxirane ring opening reaction and primary amine substitution. The molar ratio of GA–PEG–epoxy to PEI was adjusted to 1:1. The system was maintained in the dark for 72 h, at room temperature, under vigorous stirring.
Following each synthesis step, the nanoparticles were purified through centrifugation at 8000g, for 8 minutes, and then dispersed in deionized water. Each synthesis stage was monitored and the composition, structure, and morphology of the reaction products were verified through a range of analytical techniques, including UV-Vis, FTIR, XPS, DLS, zeta potential, and STEM/TEM analyses. In the case of GA–PEG–epoxy, mass spectrometry was also employed.
2.4. Radiolabeling of gold nanoparticle-based nanoconjugates
Before starting the experiment, fresh stock solutions of AuPEG–PEI (1 mg mL−1), AuPEG–PEI–GA (1 mg mL−1), NaBH4 (6.25 and 12.5 mg mL−1), and SnCl2 (1 and 2 mg mL−1) dissolved in ultrapure water were prepared. Each sample was prepared in a total volume of 1750 μL, comprising the following: 125 μL of gold nanoparticle dispersion, 500 μL of ultrapure water, 125 μL of reducing agent, and 1000 μL of Na+99mTcO4−. The radioactive solution of Na+99mTcO4− was diluted so that samples with radioactivity of 185 MBq were obtained. A total of 8 samples of radiolabeled compounds were prepared. The first step of the radiolabeling process consisted of the reduction of sodium pertechnetate with each of the reducing agents, by vortexing the above-mentioned samples for 30 minutes. Then, a volume of 125 μL of nanoparticle suspension was added to each product of the reduction reaction. The samples were mixed for about 2 minutes with a vortex and left to incubate for one hour at room temperature, before determining the radiolabeling yield by instant thin layer chromatography (iTLC). Taking into account the nanoparticle concentration (1 mg mL−1) related to the 99mTc activity (185 MBq/5mCi), the specific activity (As) of 1480 GBq g−1 was calculated.
Reducing agents. 99mTcO4− is a convenient precursor for the preparation of a large variety of radiopharmaceuticals because Tc is able to adopt several oxidation states.40 The oxidation state and coligands dictate the specificity of radiopharmaceuticals, which are based on coordination linkages. To produce the desired coordination complexes, TcO4− can be treated with a wide range of reducing agents in the presence of a coordinating ligand. Among the reducing agents, stannous chloride (SnCl2) was the most used to obtain complexes of Tc(V) and Tc(I), while boron-hydrides were considered more suitable for organometallic Tc(I) complexes.41 The selection of reducing agents is highly limited by the fact that transition metals compete with 99mTc for ligands.
Sodium borohydride (NaBH4) is proficient in alkaline media, whereas SnCl2 is commonly used in acidic environments. Using SnCl2, high yields of technetium-labeled compounds could be achieved, without the requirement to eliminate free pertechnetate. The reducing agents considered for the present study, NaBH4 and SnCl2, were taken in a large excess compared to pertechnetate.42–45 The working concentrations in the radiolabeling solutions were 0.45 and 0.9 mg mL−1 for NaBH4, and 0.07 and 0.14 mg mL−1 for SnCl2.
The instant thin layer chromatography (iTLC) method was used to determine the radiolabeling yield of each nanoparticle suspension. As a stationary phase, an iTLC glass microfiber chromatography paper impregnated with silica gel (ITLC-SG paper, Agilent) was used. Twenty μL of samples were deposited on the chromatographic strips, dried in the atmosphere, and then placed in the chromatographic chamber by immersing the spotted end of the strip into the solvent used as the mobile phase to carry out the compound migration. The mobile phase has been chosen in both cases so that the nanoparticles migrate very little above the starting line, and the solutions containing the radionuclides migrate completely in the upper half of the chromatographic paper. Therefore, the mobile phase for radiolabeling using Na+99mTcO4− was sodium acetate 1 M, while a mixture of acetone:glacial acetic acid in a ratio of 3:1 (v/v) was considered when 68GaCl3 was the subject. iTLC was performed vertically in sealed beakers with a mobile phase in the bottom. After the complete migration of the solvent, the strips were dried under a jet of hot air and cut into two equal pieces, and then the activity of each strip was measured. As indicated in the literature data,47,48 due to the polyethyleneimine grafted onto the surface of the gold nanoparticles, they migrated very little, with the spot of the gold particles subjected to TLC analysis being visualized above the starting line. Therefore, the activity measured in the upper half (RF, μCi) of the strip represents the unbound radionuclides, Na+99mTcO4− or 68GaCl3, while the one measured in the bottom half (RB, μCi) of the strip represents the radiolabeled nanoparticles. The RF and RB values of the radioactivity were taken into account by subtracting the activity of the environment. Accordingly, the radiolabeling yield was determined using the following calculation formula: η = RB × 100/(RB + RF). The radioactivity of samples was measured with a VDC-603 dose calibrator (Comecer, Joure, Netherlands), by successively introducing each half of each TLC paper strip, protected in a glass test tube, into the analysis chamber.
2.5. In vitro biocompatibility assessment on normal fibroblasts
Biocompatibility of nanoparticles was assessed on human gingival fibroblasts (HGFs, CLS Cell Lines Service GmbH, Eppelheim, Germany) using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, WI USA), according to the manufacturer's instructions. Cells were seeded into 96-well tissue culture-treated opaque white plates (50000 cells per mL) and allowed to adhere overnight in DMEM with low glucose, containing GlutaMAX™ supplement and pyruvate, 5% fetal bovine serum (FBS, both from Gibco, Thermo Fisher Scientific, Waltham, MA USA) and 1% antibiotic–antimycotic (Lonza, Basel, Switzerland). Cells were incubated with nanoparticles (0, 1, 5, 10, and 25 μg mL−1) for 24 h and then luminescence readings were taken using a FLUOstar® Omega microplate reader (BMG LABTECH, Ortenberg, Germany). The experiments were carried out in triplicate, and the viability of treated cells was expressed as a percentage of the viability of control cells (0 μg mL−1). Data were represented as means ± standard deviations and statistically analyzed by the independent two-tailed (Student's) t-test, considering p < 0.05 to be statistically significant.
2.6. In vivo experiments
The animals subjected to PET-MRI scanning were injected with 20 MBq kg−1 bw of each 99mTc based radiotracer or free 68GaCl3 for the control group, equivalent to about 6 MBq per animal. The MRI sequence (T1 GRE) precedes the PET scan; it has the role of locating the anatomical structures where the radiotracer accumulates and this took place for approximately 15 min for the chosen sequence.
The animals subjected to SPECT examination have received 60 MBq kg−1 bw of each 68Ga based radiotracer or free Na+99mTcO4−, in the case of control group, corresponding to about 18 MBq per animal. The radiolabeling protocol was conducted taking into account the option where the maximum yield was obtained, using SnCl2 as the reducing agent, at a concentration of 2 mg mL−1. As indicated in the section detailing the radiolabeling protocol, the production of 99mTc radiolabeled complexes in high yields and purity requires the reduction 99mTc(VII) in 99mTcO4− to a lower oxidation state. After demonstrating the efficiency, stability and nontoxicity of stannous salts, these reducing agents have been introduced for commercial use. Moreover, stannous chloride is considered the most appropriate reducing agent for 99mTc-based pharmaceuticals.41
SPECT standard parameters were used to achieve anterior and posterior planar scans, including a 256 × 256 matrix and a 20% window centered around the 140-keV photopeak, using a low-energy, high-resolution parallel collimator. The scans were performed for 7 hours (taking into account the 99mTc half-life of 6 hours) after the injection of the radiotracer. The animals’ whole-body scans were accomplished in the supine position. Furthermore, tomographic images were obtained for more fields of view as well as precise localisation. The uptake quantification was studied through measuring the total counts and pixels (given by the software of the Gamma Camera), by drawing a region of interest (ROI) in every uptake localisation. Each ROI uptake was reported to an equal ROI in a background area (the right leg of the animal). The (total counts/pixels) uptake/(total counts/pixels) background ratio was used in order to generate uptake graphics for 7 hours following the injection of the radiopharmaceuticals.
3. Results and discussion
3.1. Synthesis and the structural, morphological and optical properties of AuNP-based nanoconjugates
The structure of intermediate compounds was confirmed by mass spectrometry, where m/z values indicate their molecular weight and the oxirane ring opening with primary amines was established with the help of FTIR spectroscopy. The particle size and morphology were investigated using UV-Vis, DLS, and STEM/TEM techniques. The elemental composition was determined by XPS, and the concentration of gold nanoparticles was determined by UV-Vis.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, primary and secondary amines I, and C–H bending. The last region, ranging from 2800 to 3600 cm−1, is the characteristic of O–H, N–H, and C–H stretching vibrations.37,52
O stretching, primary and secondary amines I, and C–H bending. The last region, ranging from 2800 to 3600 cm−1, is the characteristic of O–H, N–H, and C–H stretching vibrations.37,52
The FTIR spectrum of the AuPEG–NH2, as illustrated in Fig. S3 (ESI†), exhibits the characteristic bands for primary aliphatic amines at 3440 cm−1, and at around 1620 cm−1 which is overlapped with the S–C–C– bond. The band at around 1150 cm−1 is attributed to the C–N, C–S and C–O–C bonds. In the domain of C–H and –CH2 stretching, bands could be observed at around 2850 cm−1, which can be attributed to the methylene groups of PEG chains and residual S–H moieties. Moreover, the AuPEG–NH2 spectrum displays a band at 1400 cm−1, which is consistent with the band seen in AuNPs and is ascribed to the presence of residual COO− from the AuNPs.37 The Au–S vibration occurs at 557 cm−1, and aligns well within the 500–600 cm−1 range.53 The band corresponding to C–O–C moieties of the AuPEG–NH2, at around 1100 cm−1, becomes larger and is shifted to a smaller wavenumber in Au–PEG–epoxy conjugates (Fig. S3, ESI†) due to the conjugation with the epoxy precursor. Also, the FTIR spectrum of AuPEG–epoxy conjugates (Fig. S3, ESI†) reveals the strong symmetric stretching vibration at 947 cm−1 and 846 cm−1, as well as asymmetric stretching vibrations at 1352 and 1251 cm−1 corresponding to the oxirane group.54 The FTIR spectrum corresponding to the AuPEG–PEI intermediate compound confirms its expected structure with the presence of primary and secondary amine absorption bands at 1639 cm−1 and 1571 cm−1, respectively. The presence of PEI moieties in the conjugate is responsible for the appearance of characteristic bands for primary aliphatic amines at 3430 cm−1. Additionally, a weak band associated with secondary amines is observed as a shoulder at 3240 cm−1. The other existing bands are in agreement with the proposed structure. Finally, the FTIR spectrum of the AuPEG–PEI–GA compound (Fig. S3, ESI†), bearing glucosamine grafted onto the PEI surface of gold nanoparticles, exhibits only slight modifications in the intensity and shape of the characteristic bands, with minor shifting owed to the secondary amines.37
The C 1s high-resolution spectrum of AuNPs (Fig. S5, ESI†) exhibits four discrete binding energies at 284.6, 286, 287.9, and 289.3 eV which are attributed to adventitious carbons (C–C or C–H), the hydroxyl (C–OH alch) and/or the α-carbons (CH2), the coordinated carboxylates (COO–Au), and free carboxyl moieties (COOH or COO−), respectively.37,58 In contrast, the high-resolution spectrum of AuPEG–NH2's C 1s, (Fig. S5, ESI†) displays six distinct peaks positioned at 284.6, 286.2, 287.4, 288.2, 288.9, and 289.3 eV. The observed peaks have been ascribed to adventitious carbons (C–C or C–H), the hydroxyl (C–OH) and/or the α-carbons (CH2), the coordinated carboxylates (COO–Au), carbon–nitrogen bonds originating from the PEG sidechain (C–N), carbon–sulfur bonds arising from the heterobifunctional PEG (C–S), and free carboxyl moieties (COOH or COO−), respectively.37,58
The high-resolution spectra of Au 4f indicate that the thiol bond has been attached to the gold nanoparticle's surface. Fig. S5 (ESI†) depicts the existence of Au(0) and Au(1) in the case of AuNPs. On the other hand, for the AuPEG–NH2 conjugate (Fig. S5, ESI†), three separate peaks were observed which correspond to Au(0), Au(1), and Au–S bonds.37,58 The appearance of surface Au(1) species in both instances can be attributed to the coordination of Au–carboxylates, as reported in the literature.58–60
| A = εextBC | (1) |
Changes in the structure and the mean hydrodynamic diameters of the gold nanoparticles were observed through the use of the DLS technique (as depicted in Fig. S8, ESI†). The DLS histograms demonstrated homogeneous formations, exhibiting a distinct tendency towards an enhanced hydrodynamic diameter as the length of the sidechain conjugated to gold nanoparticles increased. The hydrodynamic diameters of AuNPs and their four generations of conjugates, namely AuPEG–NH2, AuPEG–epoxy, AuPEG–PEI, and AuPEG–PEI–GA, were observed to be around 143, 324, 367, 581, and 707 nm, respectively. However, the polydispersity index was found to be fairly high, ranging from 0.8 to 0.9, despite the unimodal curves. The zeta potential values of AuNPs, AuPEG–NH2, AuPEG–epoxy, AuPEG–PEI, and AuPEG–PEI–GA conjugates were observed to be −33.9, 19.27, −0.79, 38.29, and 2.69 mV, respectively (Fig. S8, ESI†), indicating the unique surface modifications of each conjugate. It is noteworthy that the hydrodynamic diameter values of AuNPs were considerably high, while the zeta potential values were low, as a result of the implementation of the Turkevich method,36 corresponding to the citrate coating of AuNPs. The colloidal stability of AuPEG–PEI and AuPEG–NH2 was found to be superior, as anticipated. Additionally, the zeta potential values indicated that the surface charge of AuPEG–PEI–GA nanosystems was nearly neutral, measuring approximately 3 mV. It is noteworthy that negative nanosystems exhibit a moderate level of systemic exposure, whereas positive nanosystems are immediately eliminated. On the other hand, neutral nanosystems show a considerable degree of systemic absorption upon intravenous administration and exhibit a low clearance rate when administered intraperitoneally. The reduced plasma clearance is observed to be a reflection of tumor uptake in this particular context.69 The aforementioned statement implies that neutral nanosystems, specifically AuPEG–PEI–GA nanosystems, have the potential to be efficient in cancer diagnosis and tumor uptake.
The STEM method was employed in order to clarify the morphology and dimensions of the resultant nanoparticles. Fig. 1 displays STEM images of the AuNP sequences, demonstrating their organization into spherical nano-sized particles. The calculated diameter distribution for each sample under investigation is also presented. The AuNPs and their conjugates exhibited a spherical morphology with average diameters of 11.98 ± 1.85, 22.75 ± 3.237, 23.77 ± 3.32, 23.81 ± 3.74 and 26.59 ± 3.57 nm for AuNPs, AuPEG–NH2, AuPEG–epoxy, AuPEG–PEI, and AuPEG–PEI–GA, respectively. The ImageJ software was used to determine the average core diameters of the particles (D, nm). The STEM images exhibit a consistent trend similar to the DLS analysis, wherein homogeneous assemblies are observed to have an evident increasing diameter with an increase in the length of the sidechain that is linked to the gold nanoparticles. Despite this, the polymer coating's impact on the particle size is minimal according to DLS and STEM measurements, while its effect on aggregation behavior is significantly greater. It is essential to note that when utilizing difunctional reactants, to prevent the participation of the second function in the reaction (which could result in the coupling of two NPs), two parameters should be considered: (a) ensuring an optimal ratio between the reactants and (b) maintaining a low concentration of the difunctional compound. According to our findings, the zeta potential values of AuNPs, AuPEG–NH2, AuPEG–epoxy, AuPEG–PEI, and AuPEG–PEI–GA conjugates were observed to be −33.9, 19.27, −0.79, 38.29, and 2.69 mV, respectively (Fig. S8, ESI†), varying from negative to positive values as the surface of the NPs was modified, leading to the hypothesis that the NPs were gradually functionalized. The non-PEGylated AuNPs showed a zeta potential of around −34 mV, indicating the good stability of the particle in dispersion. PEGylation changed the sign of the NP charge and brought it to about +20 mV. Subsequent functionalization with PEI increased the positive charge of NPs, practically doubling it. The presence of PEG–glucosamine in the third shell of NPs largely neutralized their charge, in agreement with the literature data.70 This fact enhances the ability of nanoparticles to evade protein adsorption, increasing their half-life in the bloodstream by a factor of several times as a result of blocking their association.71 Moreover, a weakly positively charged surface is preferred in the case of specific binding by receptor-mediated endocytosis.72 On the other hand, neutral nanosystems show a considerable degree of systemic absorption upon intravenous administration and exhibit a low clearance rate when administered intraperitoneally. The reduced plasma clearance is observed to be a reflection of tumor uptake in this particular context.69 The aforementioned statement implies that neutral nanosystems, including AuPEG–PEI–GA, have the potential to be efficient in cancer diagnosis and tumor uptake.
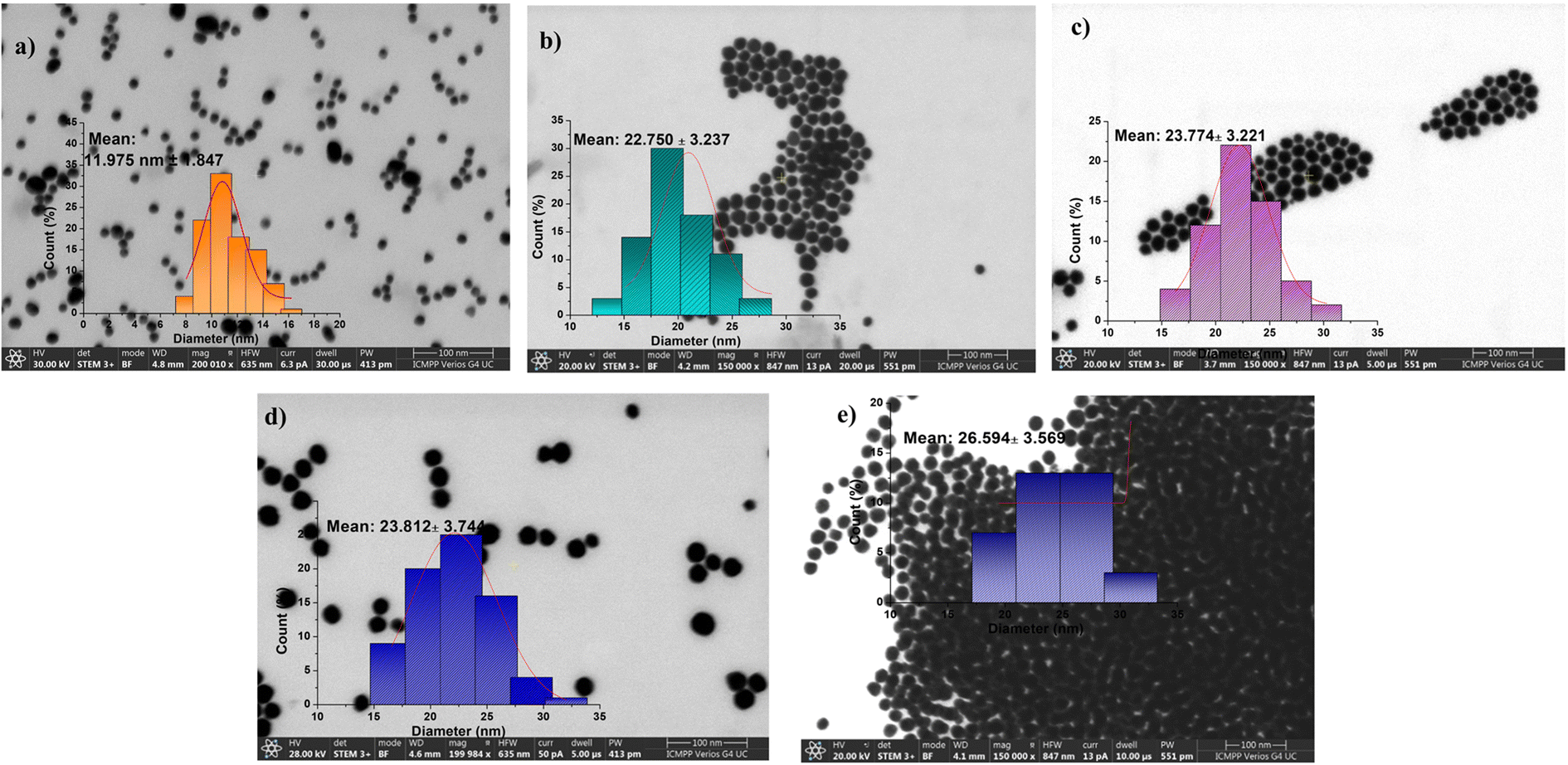 | ||
| Fig. 1 STEM images of (a) AuNPs, (b) AuPEG–NH2, (c) AuPEG–epoxy, (d) AuPEG–PEI, and (e) AuPEG–PEI–GA. The polymer coating slightly influences the particle size but shows a more significant effect on aggregation behavior. The insets show STEM histograms of particle sizes measured from about 1000 particles per image. | ||
Following the findings of the STEM analysis, the DLS and zeta potential measurements (ξ) indicate that both AuNPs and gold nanoparticle-based nanoconjugates exhibit a spherical morphology with sizes varying between 11 and 30 nm (as depicted in Fig. 1). Additionally, the zeta potential values (as illustrated in Fig. S8, ESI†) are indicative of the shell's characteristics, with positively charged measurements observed in the case of AuPEG–NH2, AuPEG–PEI, and AuPEG–PEI–GA conjugates.
3.2. In vitro biological assays – the non-toxic effect of AuPEG–PEI and AuPEG–PEI–GA nanoconjugates on normal fibroblasts
Fig. 2 shows that AuNPs, AuPEG–PEI nanoparticles and AuPEG–PEI–GA nanoparticles are biocompatible at the tested concentrations (>70% cell viability). A significant statistical difference in cell viability was observed between the lowest and highest concentrations of AuPEG–PEI (p = 0.001) and AuPEG–PEI–GA (p = 0.005) nanoparticles.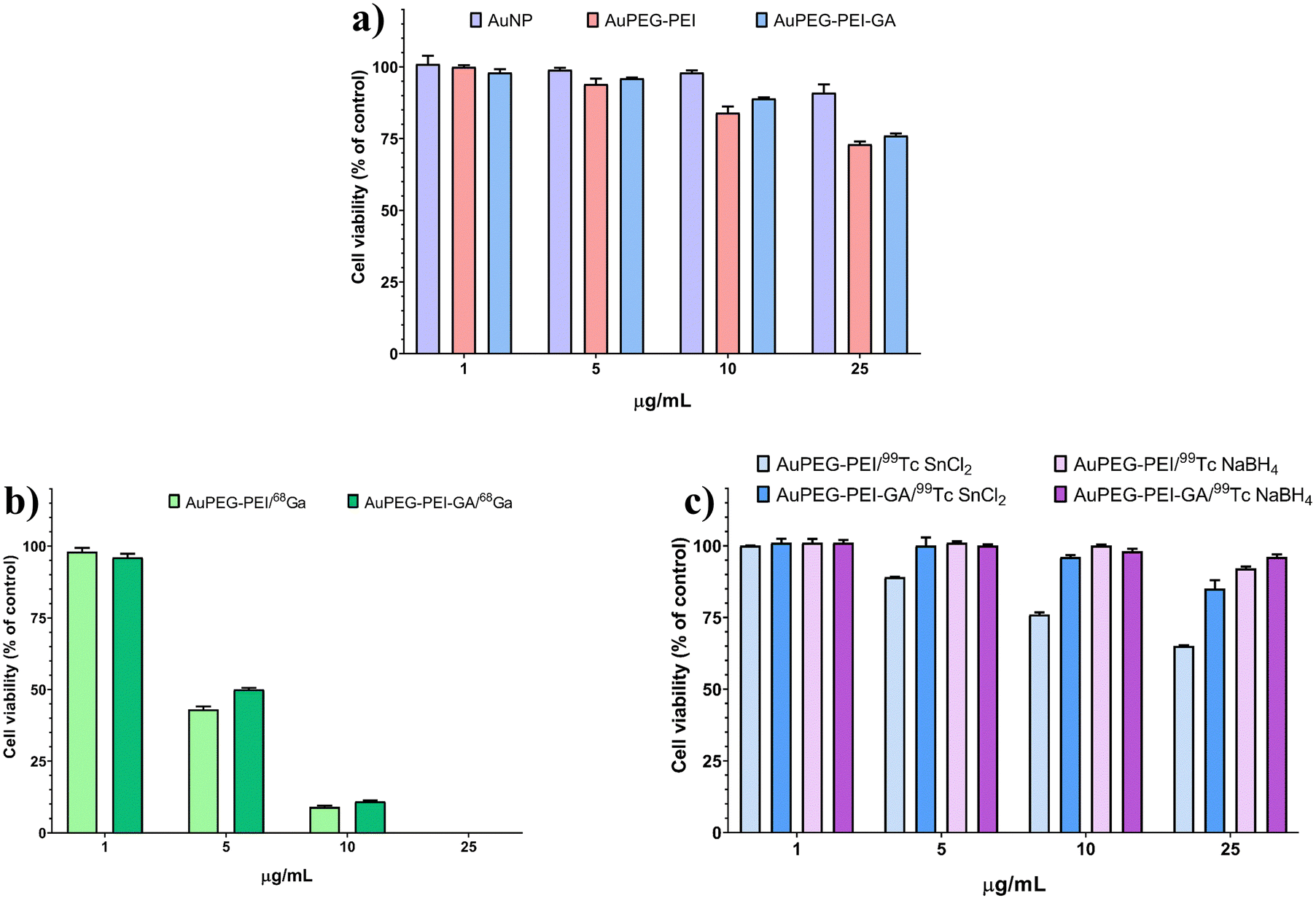 | ||
| Fig. 2 Cytotoxicity assessment of non-radiolabeled and radiolabeled nanoparticles on normal fibroblasts at 24 h incubation of (a) AuNPs, AuPEG–PEI nanoparticles and AuPEG–PEI–GA nanoparticles; (b) AuPEG–PEI and AuPEG–PEI–GA labeled with 68Ga; and (c) AuPEG–PEI and AuPEG–PEI–GA labeled with 99mTc. | ||
3.3. Radiolabeling of AuNP-based nanoconjugates
The radiolabeling was performed using two radioisotopes, 99mTc and 68Ga, respectively, to highlight the potential of the gold nanoparticle conjugates (AuPEG–PEI and AuPEG–PEI–GA) to be applied in both SPECT and PET nuclear medicine techniques.In order to achieve the highest radiolabeling yield, the procedure with 99mTc was conducted with the variation of the Na+99mTcO4− activity (185 MBq/5 mCi), along with the nature and concentrations of NaBH4 or SnCl2 reducing agents. The complex resulting between [TcO4−] and the highly positive charged PEI was confirmed by the decrease of zeta potential values (ξ) measured at 6 h after the radiolabeling process. The values decreased from +38 to −38 mV for AuPEG–PEI/99mTc and from +3 to −60 mV for AuPEG–PEI–GA/99mTc, as depicted in Fig. S9 (ESI†).
The difference observed in the ξ-potential values (the inset of Fig. S9, ESI†) before and after radiolabeling of both types of nanoconjugates (AuPEG–PEI and AuPEG–PEI–GA) is notably significant, measuring approximately 60 mV. However, it was observed that this difference decreased gradually over time. After 22 hours, the zeta potential measurements of the two radiolabeled conjugates were recorded to be +12 and +0.6 mV, respectively. These findings suggest that the aforementioned conjugates may be more appropriate to be used in cancer detection and tumor uptake.69 Nevertheless, these two values are much more logical and justified and, although they appear to favor the agglomeration of nanoparticles, this phenomenon has not been observed experimentally. The variations in zeta potential values observed over time for the two examined conjugates subsequent to exposure to the sodium pertechnetate solution were ascribed to the occurrence of intra- and intermolecular rearrangement phenomena. These phenomena resulted in the redistribution of electrical charges within the polymer coating that decorates the surface of the nanoparticles. During the first stage, the zeta potential values exhibit a highly negative trend, which can be attributed to the dominant effect of the ion [TcO4−] present on the surface of the polymer coating of the nanoparticles (the inset of Fig. S9, ESI†). This effect is observed when it is assumed that 99mTc has not yet been trapped in a stable coordination network. It is noteworthy to mention that the aforementioned rearrangements occur concomitantly with the presence of reducing agents, namely 0.07/0.14 mg mL−1 SnCl2 and 0.45/0.9 mg mL−1 NaBH4, which exert a significant impact on the yield of radiolabeling. This study revealed that the stability of the systems and the resulting radiolabeling yields exhibit a decline over time, which is dependent upon the concentration and nature of the reducing agent. It was found that the optimal stability was achieved with SnCl2 at a concentration of 0.14 mg mL−1, as evidenced by the superior performance of AuPEG–PEI–GA (as depicted further in Fig. 4(b)).
The mechanism of radiolabeling of AuPEG–PEI and AuPEG–PEI–GA conjugates with 99mTc has not been extensively studied. However, based on their structural resemblance to those previously reported by Costa et al.,73 it is hypothesized that the labeling occurred through the [99mTc = O]+ complex, which is commonly involved in such reactions. In this complex, Tc is in an oxidation state of +5, having been reduced from +7 by the reducing agent. The hyperbranched PEI chains exhibit a specific spatial configuration that enables the polymer to function as a tetradentate ligand, thereby facilitating the formation of stable complexes with [99mTc = O]+.
The hydrodynamic diameter of radiolabeled nanoparticles, determined using the DLS technique, showed an increase in their size, suggesting the formation of clusters with average diameters larger than 1000 nm as a result of 99mTc complexation, as shown in Fig. S9 (ESI†).
Nevertheless, the morphological and dimensional investigation by TEM did not provide evidence in favor of the clustering hypothesis. Although regions of particle agglomeration were observed in the analyzed fields, it is not possible to definitively conclude the formation of clusters due to the presence of numerous well-defined individual particles, as depicted in Fig. 3. Additionally, it has been observed that the mean sizes of nanoconjugates labeled with 99mTc are smaller in comparison to those that are not labeled. This phenomenon may be attributed to the constriction of the polymeric shell during the complexation process with the 99mTc radioisotope.
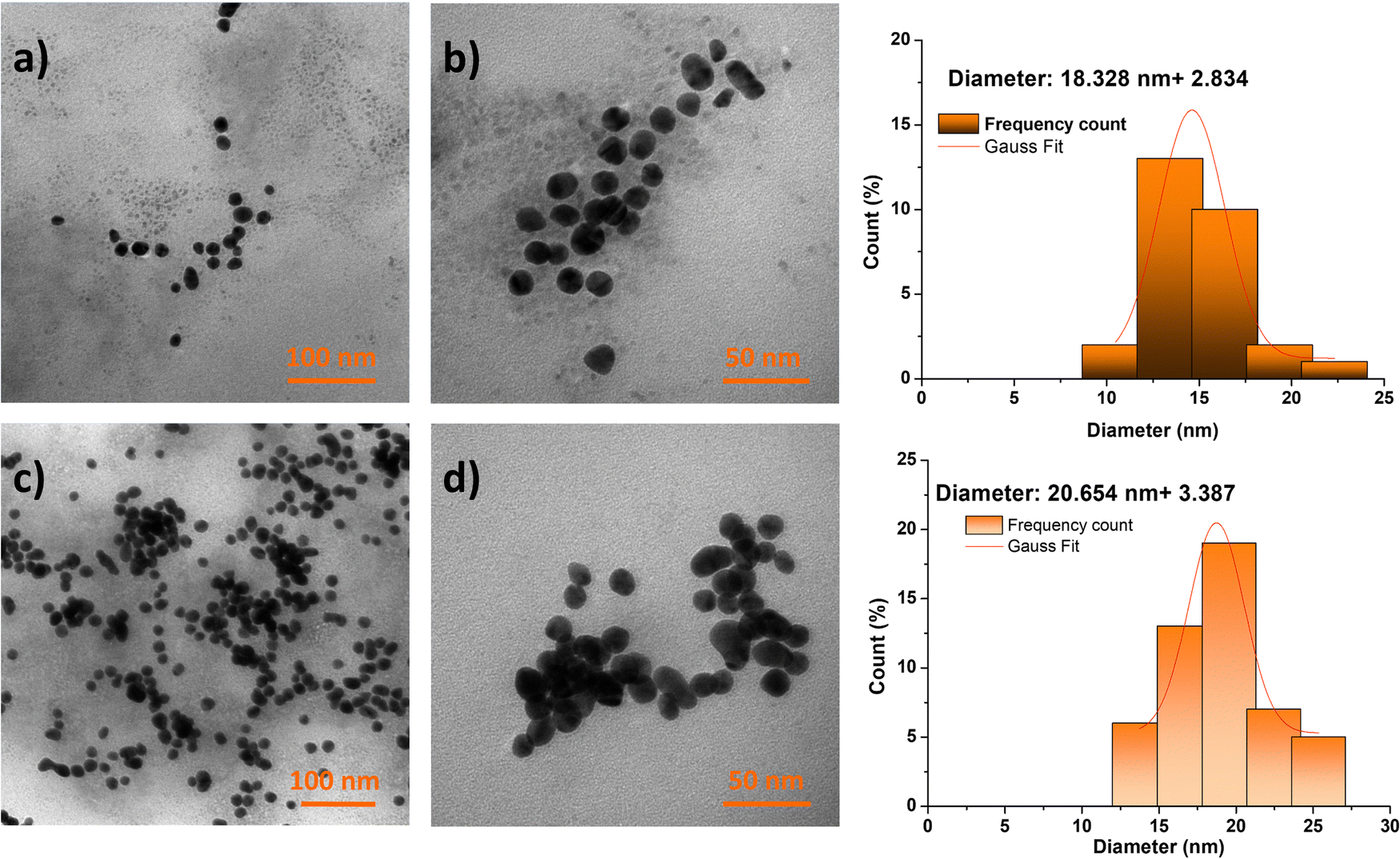 | ||
| Fig. 3 TEM images of AuPEG–PEI/99mTc (a) and (b) and AuPEG–PEI–GA/99mTc (c) and (d) after 6 hours from the starting moment of the radiolabeling process. | ||
The radiolabeling efficiency of AuPEG–PEI and AuPEG–PEI–GA was observed to reach values as high as 99%, primarily within the initial 6-hour period, with minor variations based on the type and concentration of the reducing agent employed. The data presented in Fig. 4(a)–(c) indicate that the stability of the system is significantly impacted by the presence of the reducing agent after a duration of 6 hours. The radiolabeling efficiency of the AuPEG–PEI–GA–99mTc complex was found to be 91% after 22 hours of incubation with SnCl2 (0.14 mg mL−1).
 | ||
| Fig. 4 The radiolabeling yield of (a) AuPEG–PEI and (b) AuPEG–PEI–GA nanoparticles with 99mTc, depending on the type and concentration of reducing agents, performed with sodium pertechnetate of 185 MBq (5 mCi) radioactivity, which corresponds to an N/99mTc ratio of approximately 100/1. (c) The influence of reducing agent type and its concentration on the stability of complexes between AuPEG–PEI–GA nanoparticles and 99mTc. The bar chart highlights the optimal conditions for AuPEG–PEI–GA radiolabeling. (d) Radiolabeling yields of AuPEG–PEI and AuPEG–PEI–GA with 68Ga depending on the ratio between nitrogen and gallium (N/Ga). | ||
 | ||
| Fig. 5 STEM images of AuPEG–PEI/68Ga (a), (c) and (e) and AuPEG–PEI–GA/68Ga (b), (d), and (f) at the selected N/Ga ratios: 50/1 (upper line), 100/1 (middle line) and 150/1 (bottom line). The histograms indicate the dimensional distribution by measuring about 1000 particles from several STEM images. | ||
This study highlights the significance of the variance in zeta values (the inset of Fig. S10, ESI†), indicating that the N/Ga ratios used in the experiment yield zeta potential values that are suitable for imaging and cancer uptake through parenteral administration of the conjugates discussed in the research.69 The zeta potential values of radiolabeled nanoparticles exhibit a positive trend owing to the existence of the cationic 68Ga3+ radioisotope. The stability of the investigated systems, namely AuPEG–PEI–68Ga and AuPEG–PEI–GA–68Ga, was maintained throughout the 68-minute half-life of 68Ga. The radiolabeling yields of both AuPEG–PEI and AuPEG–PEI–GA achieved values of up to 100%. However, a slight decrease in yield was observed after one hour, with values ranging between 73% and 99%. The relationship between the yield and the analyzed concentrations is evident, as illustrated in Fig. 4(c). The optimal yields for both analyzed compounds were achieved at the highest concentrations examined.
Fig. S11 and S12 (ESI†) depict the FTIR spectra of gallium and technetium-labeled compounds, AuPEG–PEI and AuPEG–PEI–GA. The technetium-labeled compounds were subjected to reducing agents SnCl2 and NaBH4, respectively. Differences are observed among the four primary characteristic regions of the spectra. The initial spectral region (400–750 cm−1), which is indicative of metallic bonds, exhibits a distinct shape in each case. The subsequent region (750–1300 cm−1) is characterized by a broader shape and less well-defined profile. The third spectral region (1300–1800 cm−1) displays broader vibration bands, fewer in number, and less well-defined due to the emergence of new N–metal bonds. The spectral region spanning from 2800 to 3600 cm−1 is recognized for its characteristic O–H, N–H, and C–H stretching vibrations. Within this region, variations are observed in the 2800–3000 cm−1 region, which can be attributed to dissimilarities in the bond for C–H stretching vibrations. This phenomenon may be explained by the existence of a metal ion in close proximity to the bond, which induces a distinct vibration of the bond in its vicinity.
Comparison between three different concentrations of gold nanoparticles and 68Ga3+ complex (corresponding to the N/Ga ratios of 50/1, 100/1, and 150/1) was analyzed by the FTIR technique and the results are presented in Fig. S13 (ESI†) for the sample AuPEG–PEI and Fig. S14 (ESI†) for the sample AuPEG–PEI–GA. The findings indicate that the radiolabeling yields of AuPEG–PEI and AuPEG–PEI–GA with 68Ga are concentration-dependent after 1 hour. Analysis of the FTIR spectra reveals that the most prominent changes in the shape and wavenumber shifting occur at a N/Ga ratio of 100/1. The fine structure of the initial samples is preserved in all spectra.
Fig. S15 (ESI†) illustrates the EDAX spectra of the gallium and technetium-labeled conjugates (AuPEG–PEI and AuPEG–PEI–GA), alongside the corresponding quantitative results presented in Fig. S16 and S17 (ESI†). These findings provide the percentage of each atomic species encountered in the achieved radiolabeled products, thereby enabling the calculation of the ratio between non-labeled nanoconjugates and radionuclides, expressed by the ratio between N from PEI and 68Ga (N/Ga) and 99mTc (N/Tc), respectively. It is noteworthy to indicate that the FTIR and EADX spectra were obtained subsequent to the full radioactive decay of 99mTc and 68Ga radionuclides, which were converted into their stable isotopes 99Tc/99Ru and 68Zn, respectively.74
3.4. In vitro biological assay – cytotoxicity assessment of radiolabeled AuPEG–PEI and AuPEG–PEI–GA nanoparticles on normal fibroblasts
The results depicted in Fig. 2(b) indicate that the radiolabeling of AuPEG–PEI and AuPEG–PEI–GA nanoparticles with 68Ga (AuPEG–PEI/68Ga and AuPEG–PEI–GA/68Ga, respectively) resulted in significant cytotoxicity towards normal fibroblasts. This effect was observed even at low concentrations of 5 μg mL−1 (p = 0.001 vs. 1 μg mL−1). Hence, it is imperative to take into account an ideal nanoparticle concentration of 1 μg mL−1, as it has been observed to have no impact on cell viability. On the other hand, it can be observed from Fig. 2(c) that the impact of 99mTc labeling on cell viability was noticeable solely at elevated concentrations (specifically, for the AuPEG–PEI/99mTc compound that employed SnCl2 as the reducing agent at a concentration of 0.14 mg mL−1), which exhibited cytotoxicity at 25 μg mL−1 with a statistical significance of p < 0.001. A reduction in cellular viability was noted in the non-toxic range (>70% cell viability) as the concentration of nanoparticles increased. Statistical analysis revealed significant differences between the lowest and highest concentrations for AuPEG–PEI–GA/99mTc SnCl2 (p = 0.042), AuPEG–PEI/99mTc NaBH4 (p = 0.015), and AuPEG–PEI–GA/99mTc NaBH4 (p = 0.019).3.5. In vivo biodistribution of radiolabeled AuPEG–PEI and AuPEG–PEI–GA nanoparticles
The results obtained in the radiolabeling experiments can be best highlighted in vivo in an animal model. In this regard, the in vivo biodistribution studies represent a first attempt, which demonstrates on the one hand the stability of the radiolabeled complexes in the biological environment, and also provides a means to track their path from administration to elimination, as long as the complex is stable and the radionuclide continues to exhibit activity. Fig. 6 and 7 illustrate the in vivo biodistribution of AuPEG–PEI and AuPEG–PEI–GA systems, radiolabeled with 68Ga and 99mTc, according to the experimental procedure described in the Materials and methods section. | ||
| Fig. 6 Tissue uptake of the 99mTc radiolabeled nanoparticles investigated as compared to the sodium pertechnetate uptake. T = thyroid, S = stomach, B = bladder, L = liver. | ||
 | ||
| Fig. 7 Tissue uptake of 68Ga radiolabeled nanoparticles in comparison with free 68GaCl3 highlighted by fused PET-MRI images. | ||
Fig. 6 clearly illustrates that both systems exhibited favorable characteristics when used as radiotracers. The stability of the two new radiotracer complex until 7 hours was tested and demonstrated. Physiological thyroid uptake through the NIS symporter75 was present in the case of 99mTc, but absent in the case of AuPEG–PEI and AuPEG–PEI–GA systems.
Analyzing Fig. 7, it can be seen that the free radionuclide (68Ga) has a relatively non-specific biodistribution, with visible accumulation in several tissues such as the heart, brain, liver, stomach, and intestine. In the case of AuPEG–PEI/68Ga, the radiotracer uptake is quite weak in most tissues, being more visible in the bladder and kidneys, therefore with a predisposition to rapid elimination via the renal route. The presence of GA in AuPEG–PEI–GA/68Ga provides an interesting biodistribution of the radiotracer, obviously different compared to the compound without GA, which shows a much better uptake in the nervous system (in the brain and spinal cord), being also present in the kidneys and bladder. The preferential fixation in nervous tissues confirms the hypothesis according to which such a compound could be useful for imaging visualization of large glucose-consuming tissues. This finding was confirmed only for radiolabeling with 68Ga (PET), but not in the case of 99mTc (SPECT), which reflects the fact that radiolabeling occurs through different mechanisms for the two radionuclides, possibly leading to conformational changes that result in different biodistributions.
However, the major difference between the biodistribution of the free radionuclide as compared to those of radiolabeled compounds indicates with certainty the stability of the complex in the biological environment at least in the tested time interval.
The radiolabeling process and the stability of the radiolabeled compounds were evaluated and clearly demonstrated, in accordance with the data already verified in other experiments39 through in vivo imaging on an animal model (Fig. 6 and 7). The radiolabeling protocol39,46 was developed starting from the bibliographic sources already mentioned, being, therefore, already verified. The main aspect targeted by our studies refers to the versatility of the nanoparticulate complex to bind the radioisotope (both SPECT and PET) and the vector molecule, an original aspect that, to date, according to the data in the literature, has not been studied in this way, and which could bring an enormous benefit to the development of new SPECT and PET radiotracers.
4. Conclusions
The present study reports on the reproducible synthesis of two hybrid nanoparticle conjugates, namely AuPEG–PEI and AuPEG–PEI–GA. These nanoparticles are composed of a metallic gold core and coated with a positively charged polymer. The physical–chemical characterization results provided evidence for the successful synthesis of these products.The noteworthy capabilities of Au nanoparticles for various biomedical applications, along with the previously reported results of our research team, led us towards their deeper exploitation, in an attempt to produce a versatile product for diagnostics or presumably theranostics. On the other hand, the polymeric coating of these metallic nanoparticles, made of PEG and PEI, has been the subject of extensive research and proved its ability in the design of agents for gene or drug transport and delivery. All these assets render the new nanoparticulate products excellent candidates as radiotracers for nuclear medicine in SPECT and PET imaging since they displayed a great radiolabeling feature with any of the tested radionuclides, namely 99mTc or 68Ga. Both conjugates exhibited good radiolabeling yields, reaching up to 100% at certain ratios between nanoparticles and radionuclides, expressed as N/99mTc or N/68Ga ratios. Furthermore, it has been established that the radiolabeled compounds had satisfactory stability, for the 99mTc complexes, maintaining their stability for over 22 hours. However, complexes containing 68Ga indicated a slight decrease in stability after 1 hour, with yield values ranging between 73 and 99%. The in vitro biocompatibility evaluation conducted on normal fibroblasts demonstrated promising potential for using these agents as diagnostic tools in subsequent in vivo evaluations.
The radiopharmaceutical market continues to be a constantly untapped niche due to the undeniable benefits of nuclear medicine imaging techniques, which might theoretically explore a considerably broader range of disorders if appropriate radiotracers would be available. The present study approaches this field from the perspective of identifying a compound that can serve as a radiotracer for both PET and SPECT depending on the radionuclide used, much more accessible and versatile, suitable especially in the examination of malignancies. The process of attaching glucosamine (GA) onto the surface of nanoparticles is expected to enhance their accumulation in tissues with high glucose consumption. This has significant potential for use as a diagnostic tool for tumors and as a basis for creating accessible radiopharmaceuticals with structural–functional hybrid properties. The main aspect targeted by our studies, additionally strengthened by in vivo testing, refers to the versatility of the nanoparticulate complex to bind the radioisotope (both SPECT and PET) and the vector molecule, an original aspect that, to date, according to the data in the literature, has not been studied in this way, and which could bring an enormous benefit in the development of new SPECT and PET radiotracers.
Author contributions
Cristina M. Uritu: investigation, formal analysis, and writing – original draft. Cristina M. Al-Matarneh: investigation, formal analysis, and writing – original draft. Denisse I. Bostiog: investigation, formal analysis, and writing – original draft. Adina Coroaba: investigation, formal analysis, and writing – original draft. Vlad Ghizdovat: investigation and formal analysis. Silviu I. Filipiuc: investigation and formal analysis. Bogdan I. Tamba: methodology, validation, and formal analysis. Natalia Simionescu: investigation, formal analysis, and writing – original draft. Cipriana Stefanescu: methodology, validation, formal analysis, and writing – original draft. Wael Jalloul: investigation, formal analysis, and writing. Valentin Nastasa: methodology, validation, formal analysis, and writing – original draft. Stelian S. Maier: conceptualization and writing – original draft. Mariana Pinteala: conceptualization, methodology, validation, formal analysis, resources, writing – original draft, visualization, supervision, project administration, and funding acquisition.Conflicts of interest
The authors have declared no conflicts of interest.Acknowledgements
This work was supported by a grant of the Romanian Ministry of Education and Research, CNCS – UEFISCDI, project number PN-III-P4-ID-PCE-2020-1523 (TM-Vector), within PNCDI III. We acknowledge the support provided by the European Union's Horizon Europe research and innovation program under grant agreement no. 101086667, project BioMat4CAST (BioMat4CAST—“Petru Poni“ Institute of Macromolecular Chemistry Multi-Scale In Silico Laboratory for Complex and Smart Biomaterials). In vivo studies were supported by “Grigore T. Popa” University of Medicine and Pharmacy Iasi, grant number 4716/25.02.2021.References
- A. Almuhaideb, N. Papathanasiou and J. Bomanji, Ann. Saudi Med., 2011, 31, 3–13 CrossRef PubMed.
- X. Luan, K. Rahme, Z. Cong, L. Wang, Y. Zou, Y. He, H. Yang, J. D. Holmes, C. M. O’Driscoll and J. Guo, Eur. J. Pharm. Biopharm., 2019, 137, 56–67 CrossRef CAS PubMed.
- Q. Chen, C. Fang, F. Xia, Q. Wang, F. Li and D. Ling, Acta Pharm. Sin. B, 2024, 14(3), 1132–1149 CrossRef CAS.
- R. Han, Y. Xiao, Q. Bai and C. H. J. Choi, Acta Pharm. Sin. B, 2023, 13, 1847–1865 CrossRef CAS PubMed.
- X. Zhang, Cell Biochem. Biophys., 2015, 72, 771–775 CrossRef CAS PubMed.
- S.-J. Park, T. A. Taton and C. A. Mirkin, Science, 2002, 295, 1503–1506 CrossRef CAS.
- X. Huang and M. A. El-Sayed, J. Adv. Res., 2010, 1, 13–28 CrossRef.
- D. Luo, X. Wang, C. Burda and J. P. Basilion, Cancers, 2021, 13, 1825 CrossRef CAS PubMed.
- L. E. Cole, R. D. Ross, J. M. Tilley, T. Vargo-Gogola and R. K. Roeder, Nanomedicine, 2015, 10, 321–341 CrossRef CAS.
- K. C. Kwon, E. Jo, Y.-W. Kwon, B. Lee, J. H. Ryu, E. J. Lee, K. Kim and J. Lee, Adv. Mater., 2017, 29, 1701146 CrossRef.
- Y. Zhao, B. Pang, H. Luehmann, L. Detering, X. Yang, D. Sultan, S. Harpstrite, V. Sharma, C. S. Cutler, Y. Xia and Y. Liu, Adv. Healthcare Mater., 2016, 5, 928–935 CrossRef CAS PubMed.
- H. He, C. Xie and J. Ren, Anal. Chem., 2008, 80, 5951–5957 CrossRef CAS PubMed.
- J. F. Hainfeld, D. N. Slatkin, T. M. Focella and H. M. Smilowitz, Br. J. Radiol., 2006, 79, 248–253 CrossRef CAS PubMed.
- D. Kim, S. Park, J. H. Lee, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2007, 129, 7661–7665 CrossRef CAS PubMed.
- A. J. Mieszawska, W. J. M. Mulder, Z. A. Fayad and D. P. Cormode, Mol. Pharmaceutics, 2013, 10, 831–847 CrossRef CAS PubMed.
- J. Guo, K. Rahme, Y. He, L.-L. Li, J. Holmes and C. O’Driscoll, Int. J. Nanomed., 2017, 12, 6131–6152 CrossRef CAS PubMed.
- K. T. Butterworth, J. A. Coulter, S. Jain, J. Forker, S. J. McMahon, G. Schettino, K. M. Prise, F. J. Currell and D. G. Hirst, Nanotechnology, 2010, 21, 295101 CrossRef CAS.
- W. N. Rahman, N. Bishara, T. Ackerly, C. Fa He, P. Jackson, C. Wong, R. Davidson and M. Geso, Nanomedicine, 2009, 5, 136–142 CrossRef CAS PubMed.
- T. Kong, J. Zeng, X. Wang, X. Yang, J. Yang, S. McQuarrie, A. McEwan, W. Roa, J. Chen and J. Z. Xing, Small, 2008, 4, 1537–1543 CrossRef CAS PubMed.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS.
- J. H. Kim, J. H. Kim, K. W. Kim, M. H. Kim and Y. S. Yu, Nanotechnology, 2009, 20, 505101 CrossRef.
- B. D. Chithrani and W. C. W. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS PubMed.
- X. D. Zhang, D. Wu, X. Shen, J. Chen, Y. M. Sun, P. X. Liu and X. J. Liang, Biomaterials, 2012, 33, 6408–6419 CrossRef CAS.
- B. Zhou, R. Wang, F. Chen, L. Zhao, P. Wang, X. Li, I. Bányai, Q. Ouyang, X. Shi and M. Shen, ACS Appl. Mater. Interfaces, 2018, 10, 6146–6154 CrossRef CAS.
- B. Zhou, Z. Xiong, P. Wang, C. Peng, M. Shen, S. Mignani, J.-P. Majoral and X. Shi, Drug Delivery, 2018, 25, 178–186 CrossRef CAS PubMed.
- C. C. Barron, P. J. Bilan, T. Tsakiridis and E. Tsiani, Metabolism, 2016, 65, 124–139 CrossRef CAS PubMed.
- S. A. Torres-Pérez, C. E. Torres-Pérez, M. Pedraza-Escalona, S. M. Pérez-Tapia and E. Ramón-Gallegos, Front. Oncol., 2020, 10, 605037 CrossRef PubMed.
- E. Shin and J. S. Koo, Front. Cell Dev. Biol., 2021, 9, 728759 CrossRef PubMed.
- M. Pliszka and L. Szablewski, Cancers, 2021, 13, 4184 CrossRef CAS PubMed.
- R. J. Kowalsky, Technetium Radiopharmaceutical Chemistry, The University of New Mexico Health Sciences Center College of Pharmacy, Albuquerque, New Mexico, 2006 Search PubMed.
- O. U. Akakuru, Z. Zhang, M. Z. Iqbal, C. Zhu, Y. Zhang and A. Wu, Acta Pharm. Sin. B, 2022, 12, 2640–2657 CrossRef CAS.
- T. Olafsen, C.-w Cheung, P. J. Yazaki, L. Li, G. Sundaresan, S. S. Gambhir, M. A. Sherman, L. E. Williams, J. E. Shively, A. A. Raubitschek and A. M. Wu, Protein Eng., Des. Sel., 2004, 17, 21–27 CrossRef CAS.
- G. Sugiura, H. Kühn, M. Sauter, U. Haberkorn and W. Mier, Molecules, 2014, 19, 2135–2165 CrossRef.
- J. Wang and L. Maurer, Curr. Top. Med. Chem., 2005, 5, 1053–1075 CrossRef CAS PubMed.
- R. Li, H. He, X. Li, X. Zheng, Z. Li, H. Zhang, J. Ye, W. Zhang, C. Yu, G. Feng and W. Fan, Eur. J. Nucl. Med. Mol. Imaging, 2023, 50, 2100–2113 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- R. V. Lupusoru, D. A. Pricop, C. M. Uritu, A. Arvinte, A. Coroaba, I. Esanu, M. F. Zaltariov, M. Silion, C. Stefanescu and M. Pinteala, Sci. Rep., 2020, 10, 6591 CrossRef CAS.
- V. Harabagiu, M. Pinteala, C. Cotzur, M. N. Holerca and M. Ropot, J. Macromol. Sci., Part A: Pure Appl. Chem., 1995, 32, 1641–1648 CrossRef.
- B. I. Tamba, A. Dondas, M. Leon, A. N. Neagu, G. Dodi, C. Stefanescu and A. Tijani, Eur. J. Pharm. Sci., 2015, 71, 46–55 CrossRef CAS PubMed.
- K. Schwochau, Angew. Chem., Int. Ed. Engl., 1994, 33, 2258–2267 CrossRef.
- H. Spies and H.-J. Pietzsch, Technetium-99m pharmaceuticals: Preparation and quality control in nuclear medicine, Springer, 2007, pp.59–66 Search PubMed.
- N. Arulsudar, N. Subramanian, P. Mishra, R. Sharma and R. Murthy, J. Drug Targeting, 2003, 11, 187–196 CAS.
- P. Richards and J. Steigman, Chemistry of technetium as it is applied to radiopharmaceuticals, Brookhaven National Lab., 1973 Search PubMed.
- J. Steigman, G. Meinken and P. Richards, Int. J. Appl. Radiat. Isot., 1975, 26, 601–609 CrossRef CAS.
- M. Sadeghzadeh, G. Charkhlooiea and F. J. Daha, Iran. J. Pharm. Res., 2013, 12, 273 CAS.
- D. Mueller, W. A. Breeman, I. Klette, M. Gottschaldt, A. Odparlik, M. Baehre, I. Tworowska and M. K. Schultz, Nat. Protoc., 2016, 11, 1057–1066 CrossRef CAS.
- P. S. Choi, J. Y. Lee, S. D. Yang and J. H. Park, J. Mater. Chem. B, 2021, 9, 8237–8245 RSC.
- A. Aghanejad, A. R. Jalilian, K. Ardaneh, F. Bolourinovin, H. Yousefnia and A. B. Samani, Asia Ocean J. Nucl. Med. Biol., 2015, 3, 99 Search PubMed.
- C. M. Uritu, M. Calin, S. S. Maier, C. Cojocaru, A. Nicolescu, D. Peptanariu, C. A. Constantinescu, D. Stan, M. Barboiu and M. Pinteala, J. Mater. Chem. B, 2015, 3, 8250–8267 RSC.
- A.-R. Petrovici, M. Silion, N. Simionescu, R. Kallala, M. Pinteala and S. S. Maier, Int. J. Mol. Sci., 2022, 23, 5944 CrossRef CAS PubMed.
- A. I. Dascalu, R. Ardeleanu, A. Neamtu, S. S. Maier, C. M. Uritu, A. Nicolescu, M. Silion, D. Peptanariu, M. Calin and M. Pinteala, J. Mater. Chem. B, 2017, 5, 7164–7174 RSC.
- A. Durdureanu-Angheluta, C. M. Uritu, A. Coroaba, B. Minea, F. Doroftei, M. Calin, S. S. Maier, M. Pinteala, M. Simionescu and B. C. Simionescu, J. Biomed. Nanotechnol., 2014, 10, 131–142 CrossRef CAS PubMed.
- E. Kuzmann, E. Csapó, S. Stichleutner, V. K. Garg, A. C. De Oliveira, S. W. Da Silva, L. H. Sing, S. S. Pati, E. M. Guimaraes, A. Lengyel, I. Dékány and K. Lázár, Colloids Surf., A, 2016, 504, 260–266 CrossRef CAS.
- D. Anderson and A. L. Smith, Analysis of silicones, Wiley-Interscience, New York, 1974, p. 247 Search PubMed.
- I.-A. Turin-Moleavin, A. Fifere, A.-L. Lungoci, I. Rosca, A. Coroaba, D. Peptanariu, V. Nastasa, S.-A. Pasca, A.-C. Bostanaru, M. Mares and M. Pinteala, Nanomaterials, 2019, 9, 1565 CrossRef CAS PubMed.
- I.-E. Bordianu, G. David, B. Simionescu, M. Aflori, C. Ursu, A. Coroaba, G. Hitruc, C. Cotofana and M. Olaru, J. Mater. Chem. B, 2015, 3, 723–727 RSC.
- A. Durdureanu-Angheluta, M.-E. Ignat, S. S. Maier, L. Pricop, A. Coroaba, A. Fifere, M. Pinteala and A. Chiriac, Appl. Surf. Sci., 2014, 292, 898–905 CrossRef CAS.
- J.-W. Park and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2014, 136, 1907–1921 CrossRef CAS.
- Z. Huo, C. Tsung, W. Huang, X. Zhang and P. Yang, Nano Lett., 2008, 8, 2041–2044 CrossRef CAS PubMed.
- Y. Mikhlin, M. Likhatski, A. Karacharov, V. Zaikovski and A. Krylov, Phys. Chem. Chem. Phys., 2009, 11, 5445–5454 RSC.
- D. E. Newbury and N. W. M. Ritchie, Scanning, 2013, 35, 141–168 CrossRef CAS PubMed.
- D. Son, S. Cho, J. Nam, H. Lee and M. Kim, Polymers, 2020, 12, 1053 CrossRef CAS PubMed.
- K. S. Oh, R. S. Kim, J. Lee, D. Kim, S. H. Cho and S. H. Yuk, J. Appl. Polym. Sci., 2008, 108, 3239–3244 CrossRef CAS.
- J. Manson, D. Kumar, B. J. Meenan and D. Dixon, Gold Bull., 2011, 44, 99–105 CrossRef CAS.
- S. Thambiraj, S. Hema and D. R. Shankaran, Mater. Today: Proc., 2018, 5, 16763–16773 CAS.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Maragò and M. A. Iatì, J. Phys.: Condens. Matter, 2017, 29, 203002 CrossRef PubMed.
- J. Tang, K. Gao, Q. Ou, X. Fu, S.-Q. Man, J. Guo and Y. Liu, Spectrochim. Acta, Part A, 2018, 191, 513–520 CrossRef CAS PubMed.
- D.-K. Kim, Y. J. Hwang, C. Yoon, H.-O. Yoon, K. S. Chang, G. Lee, S. Lee and G.-R. Yi, Phys. Chem. Chem. Phys., 2015, 17, 20786–20794 RSC.
- R. R. Arvizo, O. R. Miranda, D. F. Moyano, C. A. Walden, K. Giri, R. Bhattacharya, J. D. Robertson, V. M. Rotello, J. M. Reid and P. Mukherjee, PLoS One, 2011, 6, e24374 CrossRef CAS PubMed.
- R. Machado Cruz, M. J. Santos-Martinez and L. Tajber, Eur. J. Pharm. Biopharm., 2019, 144, 57–67 CrossRef CAS PubMed.
- L. Shi, J. Zhang, M. Zhao, S. Tang, X. Cheng, W. Zhang, W. Li, X. Liu, H. Peng and Q. Wang, Nanoscale, 2021, 13, 10748–10764 RSC.
- S. Honary and F. Zahir, Trop. J. Pharm. Res., 2013, 12, 255–264 Search PubMed.
- B. Costa, D. Ilem-Özdemir and R. Santos-Oliveira, J. Coord. Chem., 2019, 72, 1759–1784 CrossRef CAS.
- B. Salbu and E. Holm, Encyclopedia of Analytical Science, Elsevier, 2nd edn, 2005, pp. 24–32 Search PubMed.
- P. R. Franken, J. Guglielmi, C. Vanhove, M. Koulibaly, M. Defrise, J. Darcourt and T. Pourcher, Thyroid, 2010, 20, 519–526 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb02654j |
| ‡ First Authors with equal contributions. |
| This journal is © The Royal Society of Chemistry 2024 |