
Orally-administered nanomedicine systems targeting colon inflammation for the treatment of inflammatory bowel disease: latest advances
Shumeng
Hu
ab,
Runan
Zhao
 bc,
Yu
Xu
bd,
Zelin
Gu
a,
Beiwei
Zhu
*abd and
Jiangning
Hu
*bd
bc,
Yu
Xu
bd,
Zelin
Gu
a,
Beiwei
Zhu
*abd and
Jiangning
Hu
*bd
aCollege of Food Science and Engineering, Jilin Agricultural University, Changchun, 130118, P. R. China. E-mail: zhubeiwei@163.com
bState Key Laboratory of Marine Food Processing and Safety Control, National Engineering Research Center of Seafood, Dalian Polytechnic University, Dalian, 116034, P. R. China. E-mail: hujiangning2005@hotmail.com
cCollege of Biosystems Engineering and Food Science, Fuli Institute of Food Science, Zhejiang University, Hangzhou 310058, P. R. China
dSchool of Food Science and Technology, Collaborative Innovation Center of Seafood Deep Processing, Dalian Polytechnic University, Dalian, 116034, P. R. China
First published on 29th November 2023
Abstract
Inflammatory bowel disease (IBD) is a chronic and idiopathic condition that results in inflammation of the gastrointestinal tract, leading to conditions such as ulcerative colitis and Crohn's disease. Commonly used treatments for IBD include anti-inflammatory drugs, immunosuppressants, and antibiotics. Fecal microbiota transplantation is also being explored as a potential treatment method; however, these drugs may lead to systemic side effects. Oral administration is preferred for IBD treatment, but accurately locating the inflamed area in the colon is challenging due to multiple physiological barriers. Nanoparticle drug delivery systems possess unique physicochemical properties that enable precise delivery to the target site for IBD treatment, exploiting the increased permeability and retention effect of inflamed intestines. The first part of this review comprehensively introduces the pathophysiological environment of IBD, covering the gastrointestinal pH, various enzymes in the pathway, transport time, intestinal mucus, intestinal epithelium, intestinal immune cells, and intestinal microbiota. The second part focuses on the latest advances in the mechanism and strategies of targeted delivery using oral nanoparticle drug delivery systems for colitis-related fields. Finally, we present challenges and potential directions for future IBD treatment with the assistance of nanotechnology.
1 Introduction
IBD is a chronic and complex condition that causes inflammation in the gastrointestinal tract, leading to conditions such as ulcerative colitis (UC) and Crohn's disease (CD).1 In North America and Europe, more than 1.5 million and 2 million people, respectively, have been diagnosed with IBD since the turn of the century. The incidence of IBD is also rapidly increasing in Asia, including in China, India, and other underdeveloped regions. IBD is a debilitating disease that requires lifelong treatment due to the chronic development of inflammation and the significant damage caused by immune cell infiltration and organ destruction. This condition places a significant economic burden on individuals and the healthcare system. Consequently, IBD has become a global public health challenge that demands attention and resources to manage.2,31.1 Pathophysiology and pathogenesis of IBD
IBD is a condition that can lead to fever, abdominal pain, diarrhea, rectal bleeding, and weight loss, significantly impacting the patients' quality of life.4 UC symptoms are primarily characterized by continuous ulceration of the colon mucosa, abscesses, and inflammatory reactions that start in the rectum and gradually spread to the proximal colon. In some severe cases of colitis, the symptoms may also extend to the appendix.5 CD symptoms mainly affect the entire gastrointestinal tract, causing inflammation from the oral mucosa to the anus, and the inflammation can pass through multiple layers of the gastrointestinal tract, with the most severe site being the terminal ileum (Fig. 1).6 Our current understanding of complex diseases suggests that their etiology is often the result of multiple factors, including intrinsic triggering factors in the body (such as genetic susceptibility and immune regulatory dysfunction), external triggering factors from the environment (including daily diet, chemical exposure, and psychological stress), and microbial exposure.5,7 Recent research on the human microbiota has shown that ecological imbalance (changes in the regular composition of the microbiota) also plays a critical role in the development of IBD, adding complexity to the pathogenesis of IBD.8 While some reports attempt to uncover the pathological mechanisms of IBD, the exact causes remain unclear, and further exploration and research into the underlying mechanisms is necessary.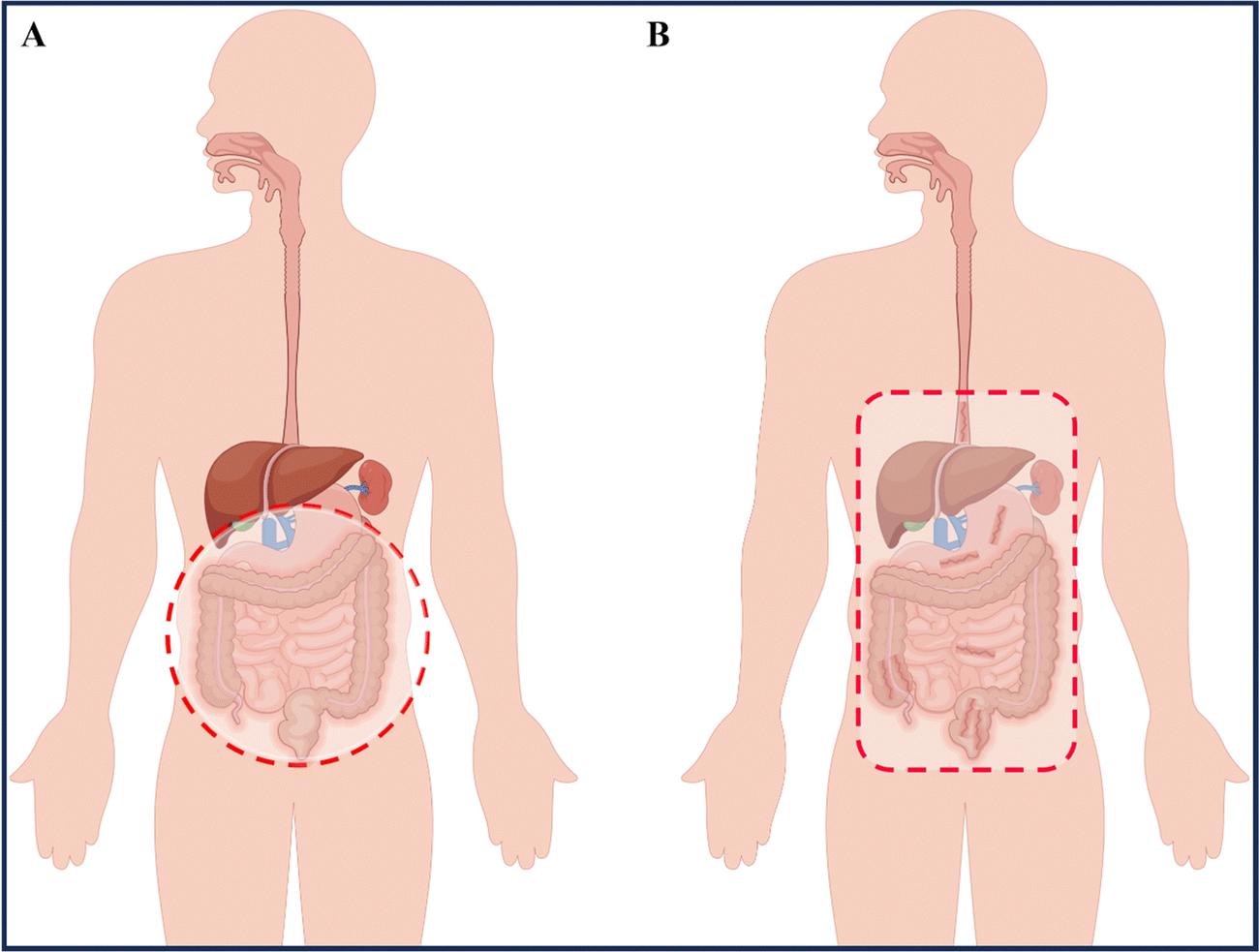 | ||
| Fig. 1 Location of ulcerative colitis (UC) and Crohn's disease (CD). (A) Inflammation of UC involves the entire colon and rectum. (B) Inflammation (dark red) of CD has a patchy pattern and predominantly involves the terminal ileum and colon. By Figdraw (ID: WTUPOa8caa). | ||
1.2 Current treatment regiments for IBD
At present, the primary methods for treating and relieving IBD involve reducing inflammation in the intestines and promoting healing of the intestinal mucosa.9 Drugs commonly used to regulate and treat IBD include anti-inflammatory drugs (such as amino salicylates and glucocorticoids), immunosuppressants, and antibiotics. Fecal microbiota transplantation is also being considered as a potential treatment.10 However, these drugs often have serious side effects. In the absence of targeted delivery, IBD drugs may be absorbed into healthy organs and tissues without any inflammatory effects, resulting in adverse effects.11 Therefore, the safety of drug action has emerged as a new concern, and the development of novel drug delivery systems that offer better therapeutic effects and minimal side effects has become imperative. Nanoparticles (NPs) are a novel type of biologically active carrier used to load hydrophobic drugs. By significantly reducing the required drug dosage, they enhance treatment efficiency while minimizing systemic side effects.12 Oral powders, tablets, intravenous injection solutions, and emulsions are currently widely used to enhance drug bioavailability.13 However, nanocarriers, which possess biocompatibility, biodegradability, site-specificity, and stability, can help overcome existing biological barriers and deliver drugs to specific sites in the gastrointestinal tract. This makes them a promising alternative to traditional drug delivery systems.14 Common nanomedicine systems for treating IBD by targeting inflammation in the intestinal tract include intravenous injection, rectal administration, and oral administration.15The administration of medication via injection can take several forms, including intravenous, subcutaneous, and intramuscular injection (Table 1).16 Compared to oral administration, injecting drugs has the advantage of higher bioavailability and faster delivery (Fig. 2A). Furthermore, it eliminates the need for drugs to pass through the stomach, removing the influence of the gastric environment on drug delivery design and creating more research opportunities for drug retention rates.17,18 However, injection administration requires professional personnel and specialized equipment, and patients often experience pain, which may lead to resistance. Additionally, injection administration can have significant systemic side effects due to its high efficiency compared to other administration routes.13,19 For instance, most biologics, including anti-tumor necrosis factor antibodies and some corticosteroids, are typically given via intravenous or subcutaneous injection. Unfortunately, the intravenous administration of anti-TNF-α biologics may suppress the body's entire immune system.19 Therefore, using nano delivery systems that target inflammatory sites and utilize various responsive mechanisms to achieve precise and multi-reactive degradation could improve IBD treatment efficacy while reducing systemic side effects. Injection administration is particularly useful for IBD treatment when the disease manifests outside the gastrointestinal tract, typically involving joints, skin, and eyes. However, oral, or rectal administration is preferred when the disease is limited to the gastrointestinal tract, guided by the anatomical location of the inflammation and the severity of the disease.
| Advantage | Disadvantage | |
|---|---|---|
| Injectable administration | 1. Rapid onset of action | 1. Invasive procedure |
| 2. High bioavailability | 2. Requires trained medical personnel | |
| 3. Can be used for drugs that cannot be taken orally | 3. Increased risk of infections or complications | |
| 4. Suitable for patients who cannot swallow | 4. Can be painful or uncomfortable for the patient | |
| Rectal administration | 1. Can be used for both local and systemic drug delivery | 1. Limited surface area for absorption |
| 2. Avoids first-pass metabolism | 2. Can cause irritation or discomfort | |
| 3. Can be used for patients who cannot swallow | 3. Not suitable for all drugs | |
| 4. Can be self-administered | 4. Compliance can be an issue | |
| Oral administration | 1. Convenient and non-invasive | 1. First-pass metabolism can reduce bioavailability |
| 2. High patient compliance | 2. Can be affected by food and drink intake | |
| 3. Generally safe and well-tolerated | 3. Slow onset of action for some drugs | |
| 4. Can be used for sustained drug release | 4. Difficult for patients who have difficulty swallowing | |
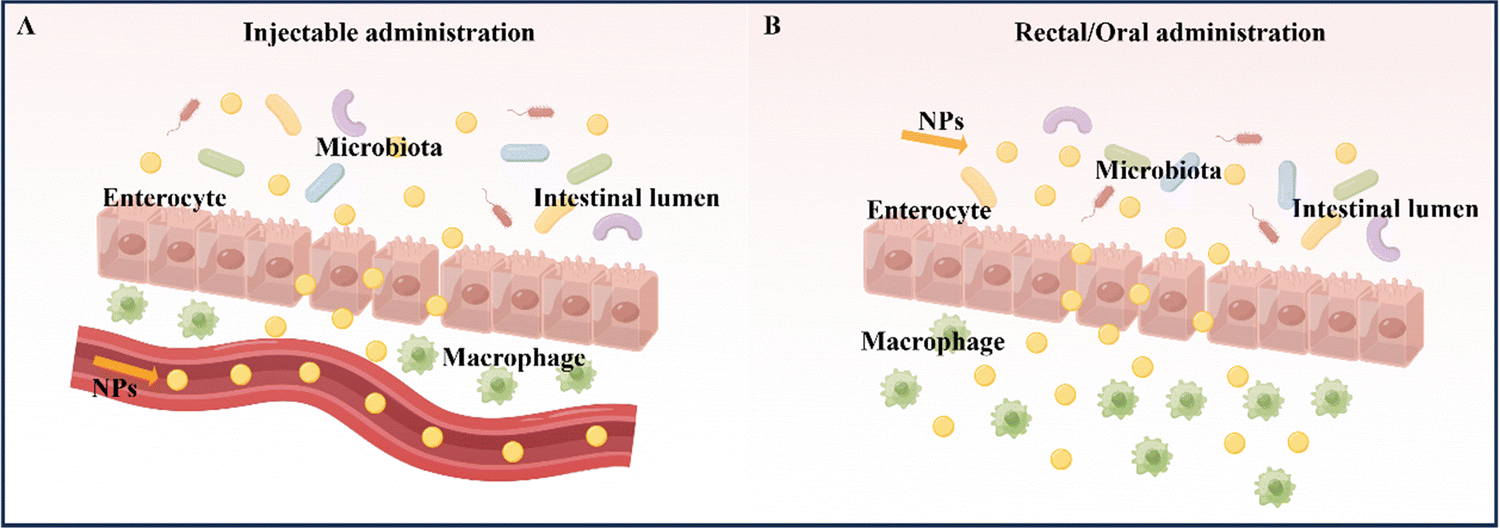 | ||
| Fig. 2 Schematic illustration of NPs for IBD treatment of injectable administration and rectal/oral administration. (A) Injectable administration. (B) Rectal/oral administration. By Figdraw (ID: RIPPA347a3 & ID: AWSAY17991). | ||
Rectal administration is an effective local drug delivery system for treating mild to moderate IBD, using enemas and suppositories to provide high drug concentrations to inflamed areas of the distal colon.20 The colon has a unique environment with slightly acidic conditions and a lower concentration of enzymes compared to other parts of the gastrointestinal tract (Fig. 2B).21 Importantly, rectal administration can bypass the liver and be absorbed systemically, reducing the liver's first-pass effect.22 As a result, rectal administration has been found to produce better response rates than oral administration or intravenous injection/infusion. This approach delivers higher concentrations of active drugs to affected tissues, while also resulting in lower systemic drug exposure.23 Despite its rapid targeting of the inflamed site in the intestine and its improved treatment efficacy, rectal drug formulations, such as suppositories, enemas, and foams, must remain in the colon for a certain period, which can be inconvenient for patients with diarrhea and fecal urgency. Large volumes and frequent administration via the rectal route can also cause discomfort and urgency in patients.24
Oral administration is considered the optimal route for treating IBD due to its high patient compliance, convenience in self-administration, good safety profile, and low production costs when compared to intravenous and rectal administration.25 The advantage of oral administration in IBD treatment lies in its ability to directly deliver therapeutic drugs to the intestinal mucosa (Fig. 2B).26 However, the gastrointestinal tract has unfavorable conditions, such as strong acidic gastric juice, abundant digestive enzymes, and diverse bacterial strains, that can cause drug formulations to be unstable and reduce the therapeutic effect of loaded drugs. Therefore, drug formulations should be designed to remain stable during their transit through the upper gastrointestinal tract and selectively release the loaded drug to inflamed intestinal tissues.
Prior research has summarized the mechanisms and strategies of various oral nanocarriers targeting IBD.13,27–30 Recently, Li et al. conducted a thorough and comprehensive analysis of the diverse development approaches utilized in recent years for nanocarrier systems targeting the colon. These methods encompassed lipid NPs, polymeric NPs, and metallic NPs, natural NPs, and plant-derived exosomes. Furthermore, the authors succinctly outlined the therapeutic effectiveness of colon-targeted nanocarrier systems in the treatment of IBD.31 However, there is a swift progression in the research of orally administered nanocarrier systems designed to target intestinal inflammation, marked by continuous advancements in the development of pertinent materials. Consequently, the initial section of this paper, leveraging a plethora of recent studies, extensively explores the understood pathophysiological landscape of IBD. This includes factors such as the pH levels of the gastrointestinal tract, various enzymes within the pathways, transit time, intestinal mucus, the intestinal epithelial barrier, immune cells in the intestine, and gut microbiota. The subsequent section provides a thorough and detailed scrutiny of the most recent developments in the mechanisms and strategies employed by orally administered nanocarrier systems in targeting intestinal inflammation, spanning the years 2021 to 2023. Lastly, considering the current safety considerations and limitations associated with orally administered nanocarrier systems, we outline the challenges and potential directions for utilizing nanotechnology in IBD treatment. The goal is to catalyze further advancements in the development of orally administered nanocarrier systems specifically designed for targeting intestinal inflammation in the context of IBD therapy.
2 Consideration of targeted colonic inflammation administration in the treatment of IBD
In recent years, there has been a growing body of literature indicating that oral nanomedicine systems are being developed to specifically target IBD based on its pathological and physiological characteristics.32,33 However, traditional colon-targeting systems used in IBD treatment, which rely on drug release triggered by factors such as pH, time, pressure, and microbiota, have been linked to inconsistent therapeutic efficacy among patients, largely due to variations in IBD pathophysiological changes and inter-individual variability in the gastrointestinal tract.29 Therefore, it is crucial to consider the pathophysiological characteristics of the inflammatory site to design drug delivery systems that can effectively target IBD inflammation (Fig. 3). Before introducing the various strategies of oral nanomedicine systems that target IBD, it is necessary to provide a brief overview of the pathological and physiological environment associated with the disease.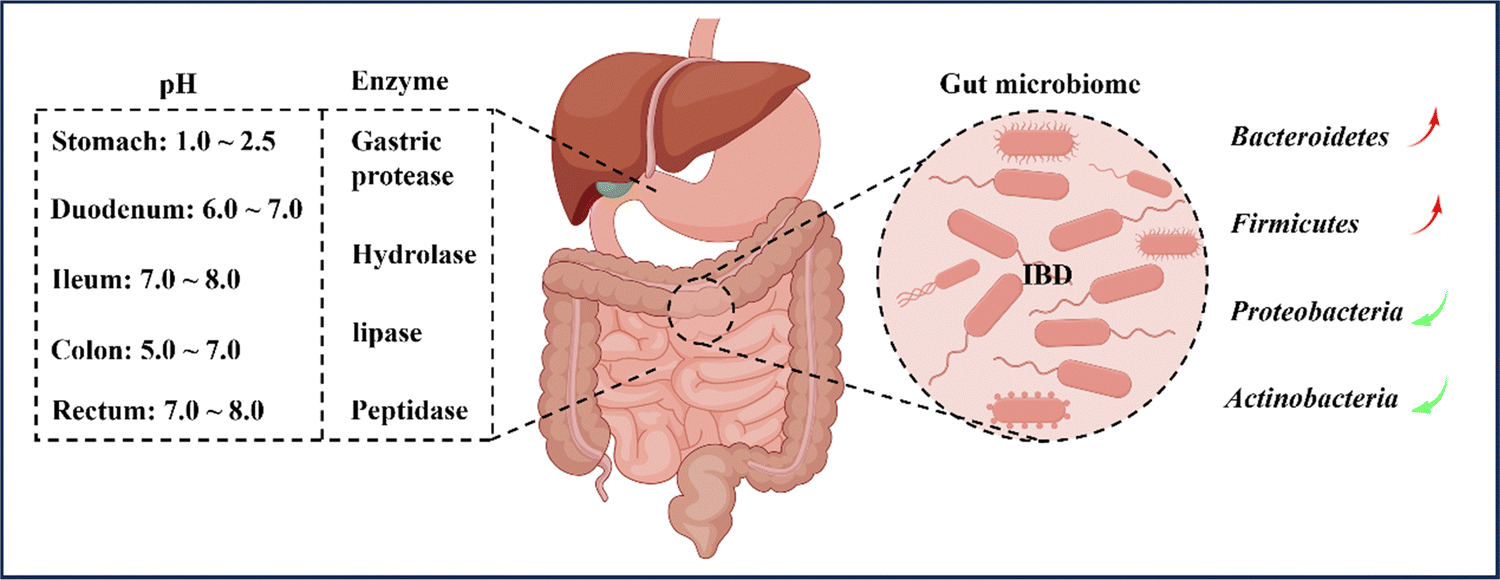 | ||
| Fig. 3 General physiological considerations for colonic oral administration. Colonic drug delivery relies on changes in gastrointestinal physiology, microbial changes, and digestive enzymes in IBD. Patients with IBD have significantly lower intestinal pH and abnormal changes in commensal populations in the gastrointestinal tract, thereby affecting the availability of drugs in the small intestine and colon. By Figdraw (ID: RYYUOad27d). | ||
2.1 pH
In general, the pH environment of the normal gastrointestinal tract can be divided into two parts. Firstly, the pH in the stomach ranges from 1.0 to 2.5, creating an acidic environment. After entering the duodenum, the pH rapidly increases to between 6.0 and 7.0 and gradually increases to between 7.0 and 8.0 at the end of the ileum. Upon entering the colon, the pH slightly decreases to between 5.0 and 7.0 due to microorganism fermentation. However, on the way to the rectum, the pH increases again to between 7.0 and 8.0, resulting in an alkaline environment.34,35 Nonetheless, the pH values of different parts of the intestine may vary among individuals, particularly the colon. Various factors, such as mucosal bicarbonate and lactate production, carbohydrates fermented by bacteria, short-chain fatty acids absorption in the mucosa, and intestinal transport, influence the luminal pH of the colon.7,36 It is reported that inflammation in the colon affects and disrupts the aforementioned factors, causing differences in colon pH between IBD patients and healthy individuals. Although fluctuations in colon pH have been observed, the overall trend of pH changes is towards increased acidity.35,37 Therefore, designing oral nanomedicine systems for IBD should make the most of the changes in gastrointestinal pH and the fluctuations in inflamed gastrointestinal pH to select suitable nanoparticle materials.2.2 Enzymes
When drugs are administered orally and enter the body, they are subject to the influence of various digestive enzymes during the passage through the oral and gastrointestinal tract. These enzymes, including salivary amylase in the oral cavity and gastric protease, hydrolase, lipase, and peptidase in the gastrointestinal tract, can all digest and degrade drugs, leading to a significant reduction in drug efficacy.38 The colon, in particular, harbors over 400 different types of aerobic and anaerobic microorganisms that contain numerous hydrolytic and reducing metabolic enzymes capable of breaking down monosaccharides and polysaccharides.8,39 As a result, polysaccharides like chitosan (CS) and pectin are often used as targeted drug delivery carriers for the colon, as they represent a common and effective strategy for overcoming gastrointestinal enzyme activity in the treatment of IBD.40,412.3 Transit time
The transit time of food through the gastrointestinal tract appears to be a key driver of changes in the gut microbiota. There are significant variations in gastrointestinal transit time both between and within individuals, which are linked to gut microbiota diversity, composition, and metabolism. Factors affecting gastrointestinal transit time include dietary structure, diet-microbe interactions, microbial metabolism, and gastrointestinal diseases.42 In patients with IBD, the gastrointestinal transit time is shorter than in healthy individuals, likely due to diarrhea, a common IBD symptom resulting in reduced transit time of the colon due to increased fluid content in the intestinal lumen.42,43 Hence, when designing targeted oral drug delivery systems for IBD, changes in gastrointestinal transit time should be considered as it can significantly affect drug retention at the site of inflammation. However, drug delivery systems designed solely based on transit time may be ineffective due to the wide variations in transit time among individuals.2.4 Mucus
Intestinal mucus is a complex biopolymer composite with both viscous and elastic properties, consisting predominantly of highly glycosylated mucin (MUC2) that covers the intestinal surface.44 This vital mucus layer is secreted by goblet cells and submucosal glands, serving as the first line of defense to restrict and isolate potential. Pathogens, digestive enzymes, and toxins from entering the gastrointestinal epithelium.45,46 The colon mucus layer comprises two distinct layers, with an inner firmly adherent layer closely attached to the intestinal epithelium that contains a high concentration of antibacterial molecules, creating a bacteria-free zone, and an outer layer that is loosely attached, allowing bacteria to penetrate through. Bacterial hydrolases partially degrade mucus, promoting mucus turnover. Overall, mucus facilitates nutrient passage while preventing the penetration of harmful substances.47 The thickness of the intestinal mucus layer is determined by its secretion rate and erosion rate.48 Since the colon hosts the most diverse microbiota, the colon mucus layer is typically thicker than other intestinal regions, providing adequate protection.49 However, in patients with IBD, the thickness of the colon mucus layer varies significantly from the normal state. In cases of active UC, the colon mucus layer becomes thinner than usual, particularly in inflamed areas. This is due to changes in glycosylation and increased sulfation degradation of mucin by the microbiota, leading to faster mucin degradation by bacteria with shorter oligosaccharides, resulting in mucus erosion. The degradation of the mucus layer exposes the gastrointestinal mucosa to higher bacterial levels, leading to a vicious cycle.50–52 In contrast, the colon mucus layer in patients with CD is thicker than normal due to goblet cell hypertrophy.51 Therefore, when developing oral colon-targeted drug delivery systems to treat IBD, it is important to consider changes in the thickness of the colon mucus layer, such as the potential of nanocarriers and drugs to stimulate the intestinal mucus layer.2.5 Epithelium barriers and tight junctions
The intestinal epithelium consists of a singular layer of columnar epithelial cells that are arranged in a specific order, with several specialized cells possessing unique properties and functions such as intestinal epithelial cells (IECs), goblet cells, intestinal microfold (M) cells, and Paneth cells.53–56 IECs play a critical role in absorbing various small molecules and have the capability of transporting immunoglobulin A produced by plasma B cells in the lamina propria to the intestinal lumen, which helps to maintain the overall stability of the intestinal epithelium.48,57 Furthermore, IECs play an active role in local immune responses. Extensive research has highlighted the involvement of IECs in the immunopathology of IBD. The latest developments in bacteriophage studies indicate that, in addition to their well-known antimicrobial capabilities, they also exhibit potential immunomodulatory properties that could have clinical applications.58 Consequently, the exploration of these immunomodulatory bacteriophage activities directed at IECs holds promise for pioneering novel approaches in the treatment of digestive disorders, specifically IBD. Goblet cells are responsible for secreting mucus to repair mucosa and safeguard the intestine.54 Goblet cells can generate mucin, primarily serving as a lubricant. Beyond this, mucin offers nonspecific protection, acting as a deterrent against the infiltration of harmful microorganisms. Utilizing glycosylation pathways, mucin engages with a variety of microbes in a bait-like fashion, thereby preserving the equilibrium of the intestinal microbiota. Emerging research indicates the potential significance of immunoreactive goblet cells in the development of IBD.59 Identifying antigens associated with goblet cells can enhance our comprehension of the mechanisms underlying IBD, presenting a new and potentially diagnostic tool. The secretions of Paneth cells contain lysozyme, phospholipase A2, and α-defensins, all of which have antibacterial functions.55 Impairment in the function of the intestinal mucosal barrier, particularly the dysfunction of Paneth cells, plays a significant role in initiating and advancing IBD. Recent research proposes that Paneth cells act as the primary site for the onset of IBD, influencing intestinal inflammatory responses through the release of antimicrobial peptides (AMPs).60 Moreover, the autophagic process in Paneth cells is instrumental in mitigating IBD by regulating several mechanisms linked to the condition, including endoplasmic reticulum stress (ERS), reactive oxygen species (ROS), and the composition of the intestinal microbiota.61 M cells are responsible for the uptake and transport of macromolecules and particles, and they capture foreign particles while processing antigens.56 They play a proactive role in transferring luminal antigens derived from food, viruses, and bacteria to Peyer's patches (PPs), consequently triggering a tailored immune response.27 Leveraging the pivotal role of M cells in antigen transport, directing antigens towards these cells has been recognized as a strategy to augment the effectiveness of oral vaccines.62Intestinal epithelial cells play a pivotal role as the primary protective barrier in the human body, safeguarding against external environmental factors. The compromised functionality of this intestinal barrier is a key contributor to the development of IBD. Up to now, the diagnosis of IBD has heavily leaned on endoscopic techniques, which are crucial in the comprehensive management of individuals with IBD. Nevertheless, prevalent clinical challenges linked to endoscopic procedures encompass issues such as endoscopic anesthesia and interventional operations.63 While information regarding the detection of the intestinal barrier is limited, the challenges associated with conventional endoscopic techniques have necessitated the exploration of novel strategies for mitigation or resolution. Research into organic fluorescent probes featuring aggregation-induced emission (AIE) characteristics offers a direct solution to this complex problem. Xu et al. have introduced an innovative AIE-active far-red fluorescent probe (PTZB-FR) based on a D-π-A structure for the in vivo detection of IBD. PTZB-FR boasts qualities favorable for in vivo imaging, including far-red emission, exceptional photostability, a substantial Stokes shift, and low cytotoxicity. Imaging experiments reveal notable distinctions between PTZB-FR and the control group in DSS-induced colitis mouse models, suggesting its potential as an effective diagnostic tool for IBD.64
Tight junctions, which are composed of adhesive molecules such as claudins, occludens, and junctional adhesion molecules-A, are the primary structures that link the single layer of columnar epithelial cells. As a vital component of the intestinal barrier, tight junctions play a crucial role in regulating the passage of water, ions, and nutrients.65,66 However, studies have demonstrated that overexpressed cytokines, such as TNF-a and IFN-γ, in inflamed tissues can impact the stability of tight junctions.65,67 Tight junctions between single layer columnar epithelial cells create a seal in the intercellular space, and the lack of tight junctions in IBD patients causes increased intestinal permeability.68 Therefore, considering changes in intestinal permeability in IBD patients, we can develop colon-targeted drug delivery systems that are more advantageous for transporting NPs through the intestinal epithelial barrier. Utilizing the enhanced permeability and retention effect, we can target inflamed sites for IBD treatment.69
2.6 Immune cells
The immune system of the intestines comprises the innate and adaptive systems and is separated from the intestinal lumen by the intestinal epithelium. Its primary function is to safeguard the intestine from pathogenic intrusion and maintain stability and tolerance towards ingested food and symbiotic microorganisms.70 The innate immune system is composed of various cells such as macrophages, monocytes, dendritic cells, granulocytes (neutrophils), and innate lymphoid cells, each having its own specific duties.71 Dendritic cells can search for external invasions via their pseudopodia penetrating the intestinal epithelium, whereas innate lymphoid cells play a significant role in controlling the chronic inflammatory response on the intestinal epithelial barrier.72 In individuals with active IBD, innate immune cells (including neutrophils, macrophages, dendritic cells, and natural killer cells) infiltrate the lamina propria significantly.71 The infiltration of macrophages and neutrophils is widely recognized as the hallmark of IBD pathophysiology.73,74 For IBD patients, the surface expression of the CD14 marker on macrophages increases significantly post-infiltration and produces pro-inflammatory cytokines such as TNF-α and IL-6. However, normal individuals have intestinal macrophages that secrete anti-inflammatory cytokines such as IL-10, as they lack the expression of the CD14 marker.75 Neutrophils respond to CXCL8, a chemokine ligand, when epithelial cells undergo inflammation, allowing them to reach the intestinal lumen and express their antibacterial ability through protein expression.76After the adaptive immune system is activated, T cells differentiate into effector T cells that promote inflammatory responses, and regulatory T cells that suppress inflammation.77 Dendritic cells typically express CD103 and maintain stability in the intestine.78 However, in active IBD, dendritic cells are significantly activated and upregulate the expression of TLR4 and IL-6 production, leading to a dominance of effector T cells over regulatory T cells, thereby promoting T cell-mediated colitis.77,79 Furthermore, B cells and effector B cells are highly infiltrated in the intestine. In active IBD, B cells upregulate the expression levels of IL-8 and TLR2, thereby promoting B cell-mediated colitis.80
2.7 Gut microbiome
According to reports, the range of microbial population in the colon is usually between 1011 and 1012 CFU per mL,81 including bacteria, fungi, and viruses,42 with their composition influenced by factors such as diet, inflammation, and host genotype.82 This article primarily focuses on the bacterial component of the gut. Previous studies have demonstrated that the microbiota in the intestines of IBD patients is significantly reduced in both quantity and diversity in comparison to healthy individuals.83,84 The human gut microbiota is primarily classified into four phyla: Bacteroidetes (67%), Firmicutes (28%), Proteobacteria (2%), and Actinobacteria (1%).85 In active IBD, there is a significant reduction in the number of Bacteroidetes and Firmicutes, particularly the Clostridium species.86 Conversely, Proteobacteria and Actinobacteria are significantly increased, mainly members of the Enterobacteriaceae family.87 Moreover, the microbial genes of IBD patients are relatively fewer than those of healthy individuals, consistent with the previous diversity comparison.88The intestinal microbiota serves not only as a protective barrier for the gut but also aids in the digestion and breakdown of indigestible food components, such as dietary fiber, into beneficial short-chain fatty acids (SCFAs) like butyrate and acetate.89 These SCFAs have immune-regulatory and anti-inflammatory properties.90 For instance, Clostridium and Bacteroides genera produce butyrate and acetate, which are involved in regulating adaptive immune responses by promoting the proliferation of Foxp3+IL-10 colon Treg cells, thereby regulating and treating IBD.91 Additionally, an ecological imbalance of the gut microbiota is characterized by a significant decrease in SCFA levels, leading to disturbed bile acid metabolism.92 Therefore, alterations in the composition and function of the gut microbiota in IBD patients suggest that the gut microbiota could play a crucial role in the pathogenesis and progression of IBD.
3 Mechanisms and strategies of oral nano-delivery systems for targeting IBD
In recent years, nanomedicine has rapidly evolved as a field with great potential for the treatment of IBD.14,33 Oral delivery has gained significant attention due to its convenience and ease of administration. By targeting the colon, nanocarriers for oral delivery have demonstrated the ability to effectively address the known pathophysiological changes present in inflamed gastrointestinal tracts. This includes increasing local drug concentrations, improving drug efficacy, reducing dosing frequency, and minimizing drug side effects.30,32 Additionally, these systems can directly deliver drugs to the specific site of colonic inflammation (Fig. 4).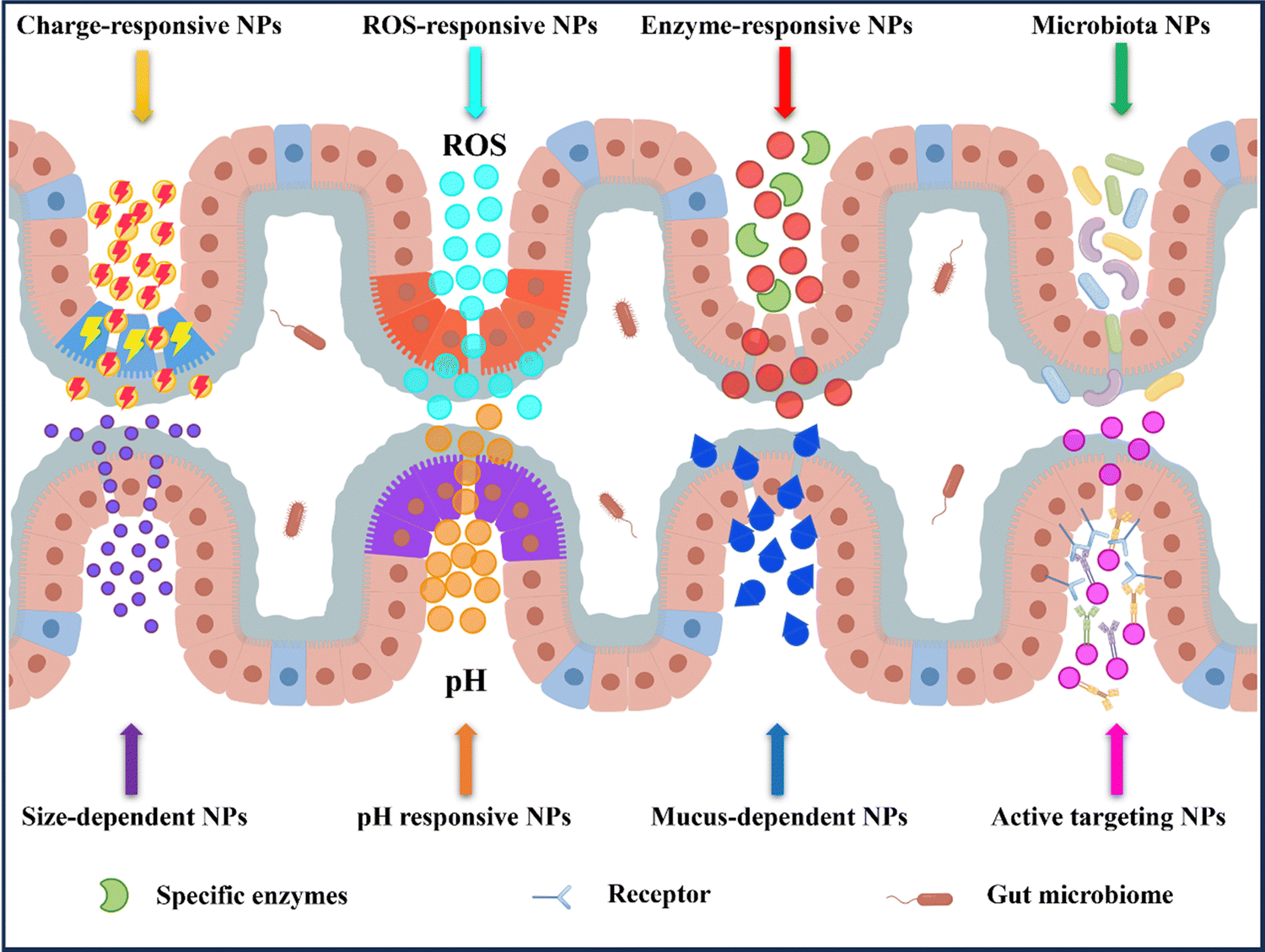 | ||
| Fig. 4 Nanoparticle-based oral drug delivery systems strategy in IBD therapy. After oral administration, the nanoparticles target the epithelial cells of the inflamed colon based on their specific pH, specific levels of enzymes, or ROS levels. Following oral administration, the nanoparticles actively target the inflamed colonic epithelium through ligand–receptor interactions, electrostatic interaction, and permeability. By Figdraw (ID: WYYYObb0b0). | ||
3.1 pH responsive NPs drug delivery systems
After being orally administered, drugs first pass through the stomach before reaching the inflamed intestine. However, the highly acidic environment in the stomach caused by the presence of stomach acid can lead to the breakdown of acid-sensitive drugs, resulting in a loss of bioactivity.93 Therefore, pH-responsive NPs drug delivery systems can be designed based on the significant difference in pH between the stomach and intestines. This type of drug delivery system can protect the loaded drug from the effects of stomach acid.94 Furthermore, the system must be capable of effectively dissolving or swelling in the neutral and alkaline environment of pH 6–8 after reaching the intestines, thereby allowing the loaded drug to be released and achieve targeted treatment of colonic inflammation.95 The most common pH-responsive mechanism is the alternate transformation of COOH/COO− groups in the materials used, which changes with pH. When the drug passes through the stomach, the carboxyl groups are protonated under low pH, effectively protecting the integrity of the loaded drug. When it reaches the intestine, the carboxylic acid easily undergoes deprotonation under the pH of the colon, causing electrostatic repulsion and swelling, thereby releasing the loaded drug.96Currently, acrylic acid esters and anionic polymers are commonly used to create pH-responsive NPs drug delivery systems. These materials are resistant to degradation under low pH conditions but dissolve or swell rapidly under high pH conditions. Nanoparticles made of such pH-sensitive materials can effectively protect loaded drugs under acidic conditions and release the loaded drugs after reaching the colon due to an increase in pH, thereby achieving the goal of treating inflammation.97,98 Methacrylic acid copolymers (Eudragit) are a popular pH-sensitive nanoparticle material, as they can be composed of different side chains to produce derivatives that are sensitive to different pH gradients. Eudragit has already been approved by the Food and Drug Administration (FDA) for oral formulations.99 Eudragit L100 dissolves at pH 6, while Eudragit S100 dissolves at pH 7. They can be combined in different ratios to control the pH at which the loaded drug dissolves and is released.100,101
As an illustration, Zhang et al. coated curcumin-loaded nanoparticles (CNs) with two pH-sensitive materials, Eudragit EPO and Eudragit L100, to create CNs@EPO@L100. This nano system exhibited pH-responsive drug release properties, increased in vitro anti-inflammatory efficacy, and greater accumulation at the site of colonic inflammation. Furthermore, orally administered CNs@EPO@L100 led to a significant improvement in the inflammatory symptoms of mice (Fig. 5A).102 In addition to using combinations of Eudragit derivatives, a single material can also be used as a coating to prepare pH-responsive nanoparticles. Feng et al. developed ES100/HA/CS nanoparticles (SK@SAC) loaded with shikonin (SK) as an oral drug delivery system for treating colitis in mice. The SK@SAC accumulated in RAW264.7 macrophage cells and demonstrated targeting ability for colitis by increasing the local drug concentration and reducing non-specific distribution after oral administration. Treatment with SK@SAC in mice with TNBS-induced IBD models showed significant therapeutic effects by regulating pro-inflammatory cytokines (TNF-a, IL-6, and IL-1β) and anti-inflammatory cytokines (IL-10 and TGF-β), and inhibiting the activity of COX-2 and iNOS (Fig. 5B).103 Lv et al. also utilized a combination of poly (lactic-co-glycolic acid) (PLGA), Eudragit S100 (ES100), CS, and hyaluronic acid (HA) to create a HA-CS/ES100/PLGA nanoparticle (MTX@hCEP) loaded with methotrexate (MTX). Under simulated gastrointestinal conditions, in vitro drug release experiments showed that MTX@hCEP displayed pH-sensitive drug release characteristics in the colon. The cellular uptake ability of hCEP nanoparticles in RAW 264.7 macrophages was notably increased. Moreover, MTX@hCEP was found to inhibit the proliferation of LPS-stimulated macrophages and the secretion of pro-inflammatory cytokines. In vivo imaging results not only confirmed the accumulation of MTX in the colons of colitis mice, but also demonstrated prolonged retention time in the colon. Additionally, MTX@hCEP alleviated inflammatory symptoms by reducing the activity of myeloperoxidase and pro-inflammatory cytokines, and promoting mucosal repair in vivo (Fig. 5C).104 In addition, Lee et al. developed pH-responsive nanoparticles for loading and controlled release of cyclosporine A (CSA) (CSA-PENPs) by combining biologically synthesized polyhydroxy butyrate (PHB) with Eudragit FS (EFS) and CSA. CSA-PENPs exhibited minimal release of CSA at pH 1.8, while the release amount gradually increased within 2 hours at pH 7.4. The results indicated that CSA-PENPs effectively delivered a sufficient amount of CSA to the inflamed site in the distal colon and demonstrated an enhanced therapeutic effect in a mouse model of DSS-induced colitis.105
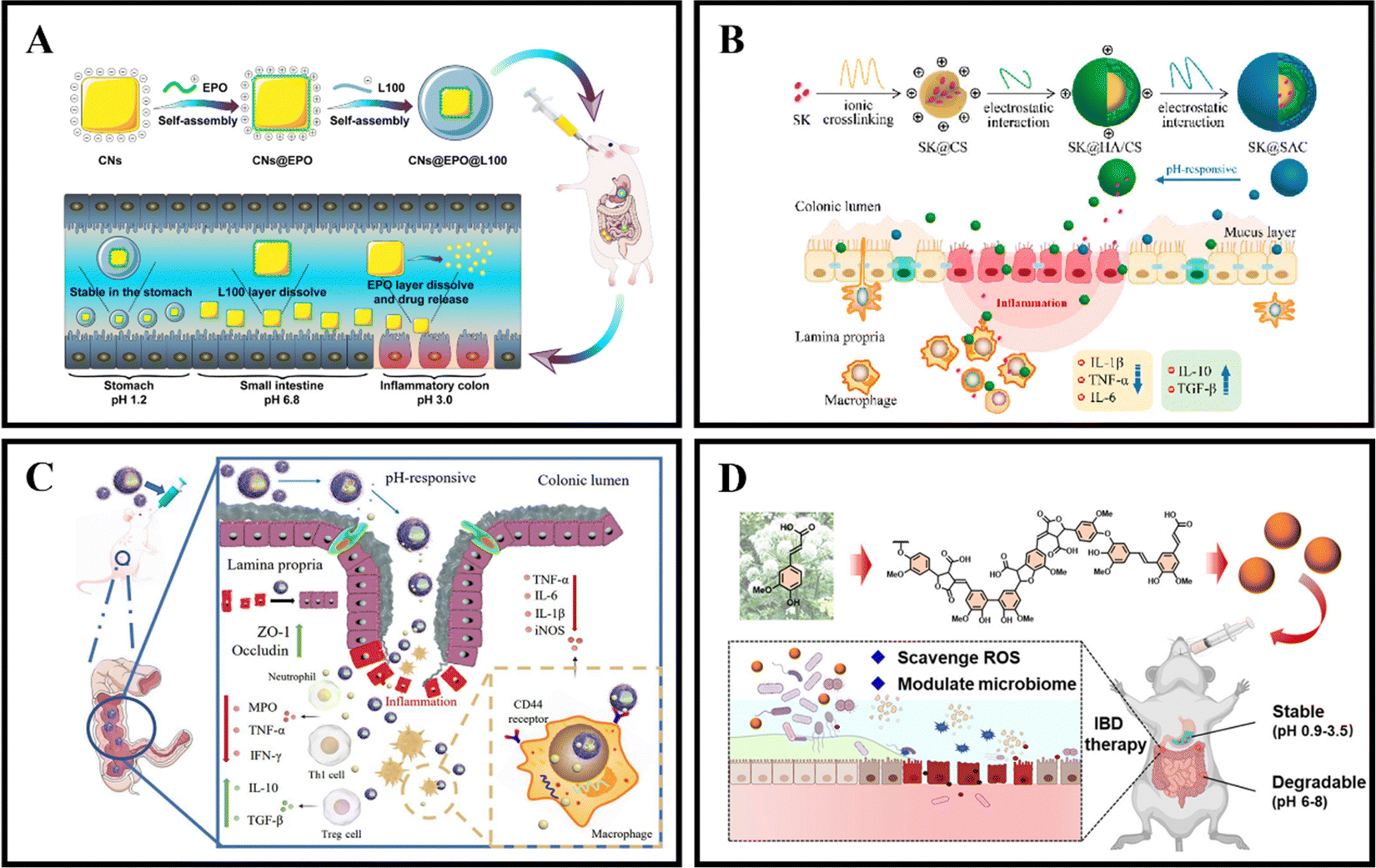 | ||
| Fig. 5 pH responsive NPs drug delivery systems. (A) Programmed pH-responsive core–shell nanoparticles for precisely targeted therapy of ulcerative colitis. Reproduced with permission.102 Copyright 2023, Royal Society of Chemistry Publishing Group. (B) XA pH-responsive and colitis-targeted nanoparticle loaded with shikonin for the oral treatment of inflammatory bowel disease in mice. Reproduced with permission.103 Copyright 2022, American Chemical Society Publishing Group. (C) Colon-specific delivery of methotrexate using hyaluronic acid modified pH-responsive nanocarrier for the therapy of colitis in mice. Reproduced with permission.104 Copyright 2023, Elsevier Publishing Group. (D) Synthetic lignin-derived therapeutic nano reagent as intestinal pH-sensitive drug carriers capable of bypassing the gastric acid environment for colitis treatment. Reproduced with permission.106 Copyright 2023, American Chemical Society Publishing Group. | ||
Besides synthetically produced pH-responsive nanoparticles, there are also pH-responsive nanoparticles obtained from natural drugs. For instance, Zhao et al. developed lignin-like nanoparticles derived from ferulic acid (FALNPs). These nanoparticles not only exhibited good antioxidant activity but also showed potential for the treatment and improvement of IBD. The pH-responsive and degradable nature of FALNPs enabled them to survive the harsh acidic environment of the stomach, cross physiological barriers, and specifically target the intestine. In vivo studies demonstrated that oral administration of FALNPs significantly reduced oxidative stress, regulated the gut microbiota, and alleviated pathological symptoms in mice with colitis (Fig. 5D).106
While targeting the intestinal pH environment through orally administered nanocarrier systems holds great promise as an efficient strategy for IBD treatment, research suggests that the colonic pH environment in IBD patients tends to be more acidic. Moreover, variations in subjective experiences and physiological conditions exist among individuals with IBD.107 Therefore, when designing pH-responsive nanocarrier systems, careful consideration of the pH fluctuations in the colonic environment due to IBD-induced inflammation is crucial. This approach is essential for the development of pH-responsive nanocarrier systems that are both effective and adaptable. Table 2 provides a concise summary of the oral delivery targeting strategies discussed in the third section of the paper.
| Drug | Nanoparticles | Targeting mechanism | Size and zeta | Efficacy | Ref. |
|---|---|---|---|---|---|
| Curcumin | Eudragit®EPO and Eudragit®L100 nanoparticles | pH | 571.8 nm | Enhance in vitro anti-inflammatory effects and significantly ameliorate the inflammatory symptoms in mice. | 102 |
| −18.1 mV | |||||
| Shikonin | Eudragit S100/hyaluronic acid/chitosan nanoparticles | pH | 190.3 ± 1.6 nm | Regulate of proinflammatory cytokines and anti-inflammatory cytokines and inhibited COX-2 and iNOS activity. | 103 |
| +22.2 ± 2.1 mV | |||||
| Methotrexate | Hyaluronic acid/chitosan/Eudragit®S100/poly(lactic-co-glycolic acid) nanoparticles | pH | 202.5 ± 3.8 nm | Alleviate inflammatory symptoms via decreasing the activities of myeloperoxidase and pro-inflammatory factors, promoting mucosal repair in vivo. | 104 |
| −18.7 ± 2.1 mV | |||||
| Cyclosporine A | Biosynthesized polyhydroxy butyrate combined with Eudragit FS nanoparticles | pH | 311.4 ± 9.1 nm | Can deliver enough CSA to inflamed tissues in the distal colon and exhibit potent anti-inflammatory effects in the DSS-induced murine colitis model. | 105 |
| −18.4 ± 0.06 mV | |||||
| Cyclosporine A | Ferulic acid-derived lignin nanoparticles | pH | 556 nm | Relieve pathological symptoms in a mouse model of acute colitis by reducing oxidative stress and regulating the gut microbiome. | 106 |
| −40 mV | |||||
| Pterostilbene | Folic acid-functionalized nanoparticles | ROS | 231.6 nm | Regulate dendritic cells, promote macrophage polarization, and regulate T cell infiltration. | 108 |
| −21.8 ± 1.76 mV | |||||
| Carbon monoxide | Mesoporous polydopamine nanoparticles | ROS | 331.1 nm | Reverse the pro-inflammatory microenvironment and restores gut barrier functions through multiple mechanisms, including scavenging oxidative stress, restoring immune homeostasis, and modulating the gut microbiota. | 109 |
| −45.6 mV | |||||
| Genistein | Dismutase-mimetic temporally conjugated β-cyclodextrin and 4-(hydroxymethyl)phenylboronic acid | ROS | 304 ± 60 nm | Increase the expression of estrogen receptor β, simultaneously reduced the expression of proinflammatory mediators (apoptosis-associated speck-like protein containing a CARD (ASC) and Caspase1-p20). | 110 |
| +6.63 ± 1.91 mV | |||||
| Luteolin | D-α-Tocopherol polyethylene glycol succinate-b-poly(β-thioester) copolymer | ROS | 426.3 nm | Regulate the inflammatory microenvironment by modulating the T helper (Th)1/Th2 and Th17/regulatory T cell (Treg) balance, thus resolving inflammation, and accelerating the healing of the intestinal mucosa | 111 |
| −16.7 mV | |||||
| Budesonide | Eudragit L/Eudragit S/Eudragit RS chitosan nanoparticles | Charge | 288 ± 7.5 nm | Release the drug throughout the GIT in a sustained fashion, and have long-lasting anti-inflammatory effects | 112 |
| +26.6 ± 1.1 mV | |||||
| Tripterine | Selenized polymer-lipid hybrid nanoparticles | Charge | 123.1 nm | Enable drug supersaturation in the gut and the sustained release of Tri to facilitate absorption, while selenium surface engineering reinforced the formulation performance and in vivo anti-inflammatory efficacy. | 113 |
| −29.70 mV | |||||
| Infliximab | Eudragit S100@folic acid-grafted amino clay nanoparticles | Charge | 20.1 ± 3.5 nm | Minimize premature drug release in gastric conditions and the upper intestine. | 114 |
| +24.9 ± 1.4 mV | |||||
| Cyclosporine A | HA functionalized PLGA-chitosan drug core | Charge | 396.65 nm | Improve colitis phenotype therapeutic efficacy in both animal models compared to free drug. Furthermore, ex vivo study of colon tissue biopsies from patients with colitis revealed that IT-NCs adhered preferentially to inflamed biopsies compared to normal. | 115 |
| −41.7 ± 1.39 mV | |||||
| Dexamethasone | Hydroxyethyl starch-curcumin nanoparticles | Enzyme | 40–60 nm | Can be effectively internalized by macrophages and show excellent cytocompatibility with macrophages since they are composed of food-derived compounds. | 116 |
| −28.0 mV | |||||
| Curcumin-cyclodextrin | Low molecular weight chitosan and unsaturated alginate resulting nanoparticles | Enzyme | 462.1 nm | Promote colonic epithelial barrier integrity and modulate production of inflammatory cytokines, reshape the gut microbiota in mice with dextran sodium sulfate (DSS)induced colitis. | 117 |
| −19 mV | |||||
| Curcumin | Palmitic acid-N-2-hydroxypropyl trimethyl ammonium chloride chitosan-cysteine nanoparticles | Mucus | 213.2 ± 4.78 nm | Can open tight junctions between cells for transepithelial transport while striking a balance between mucus interaction and diffusion through mucus. | 118 |
| −40 mV | |||||
| Resveratrol | Silk fibroin-based nanoparticles with the functionalization of pluronic F127 | Mucus | 168.1 ± 2.2 nm | Enhance the suppression of the secretion of proinflammatory cytokine TNF-α and reactive oxygen species (ROS) from RAW 264.7 macrophages under lipopolysaccharide stimulation in comparison. | 119 |
| +21.2 ± 0.5 mV | |||||
| Infliximab | PLGA-PEGNPs of varying particle sizes and polydispersity | Size | 131.74 ± 3.43 nm | Delivery of INF directly to the inflamed barrier by ∼600 nm NPs accelerated recovery of barrier integrity and reduced inflammatory cytokine secretion and cytotoxicity in comparison to treatment with INF alone. | 120 |
| 325.42 ± 5.22 nm | |||||
| 595.80 ± 90.4 nm | |||||
| −26.54 ± 3.47 mV | |||||
| −21.76 ± 1.57 mV | |||||
| −24.80 ± 2.43 mV | |||||
| Mannose | Mannose-modified liposomes and peroxisome proliferators-activated receptor γ | Size | 124.6 ± 10.45 nm | The targeting ability of MAN-LPs is obviously enhanced with the increasing of particle size, whereas the largest MAN-LPs particles achieve the best anti-inflammatory effect in cells, and a higher therapeutic efficiency in IBD mouse model. | 121 |
| 219.8 ± 15.68 nm | |||||
| 324.5 ± 22.75 nm | |||||
| NPs with ≈ 30, 50, and 70 nm of size | All nanoparticles were internalized by endothelial cells, LPS-stimulated macrophages, and fibroblasts | Size | 32.1 ± 1.2 nm | All NPs were cytocompatibility and internalized by the different cell types. However, NPs uptake was size-dependent, being the maximum uptake efficiency observed for the 30 nm NPs. | 122 |
| 50.7 ± 2.5 nm | |||||
| 69.6 ± 3.4 nm | |||||
| −13.5 ± 1.8 mV | |||||
| −18.9 ± 3.2 mV | |||||
| −24.2 ± 2.6 mV | |||||
| Escherichia coli Nissle 1917 | A bimolecular lipid coating is deposited on the surface of probiotics, Pt-Lipid | Microbiota | — | Protect the probiotics from strong digestive acids and enzymes and can significantly improve the colonization rate of the microorganisms in the intestinal tract. | 123 |
| Escherichia coli Nissle 1917 | Hyaluronic acid-poly(propylene sulfide) and EcN cells with a poly- norepinephrine layer | Microbiota | 2037 nm | Enhance prophylactic and therapeutic efficacy. Furthermore, the abundance and diversity of gut microbiota are increased after treatment with HPN-NE-EcN, contributing to the alleviation of IBDs. | 124 |
| −31.1 mV | |||||
| Escherichia coli Nissle 1917 and 5-ASA | Alginate-based coating | Microbiota | 250.0 ± 50.4 nm | Increase the microbiota richness and diversity, alleviating inflammation, and repairing intestinal barriers. | 125 |
| −29.2 ± 0.7 mV | |||||
| Escherichia coli Nissle 1917 (ECN-pE) | Chitosan/sodium alginate | Microbiota | −38.1 mV | Upregulate the expression level of tight junction associated proteins in colon tissue and protect colon epithelial cells from inflammation-induced apoptosis. Increase the density and richness of Lachnospiraceae_NK4A136 and Odoribacter. | 126 |
| Bifidobacterium longum | Artificial enzymes of single-atom catalyst | Microbiota | — | Can reduce ROS levels, inhibit proinflammatory cytokine production, restore the intestinal barrier functions, and increase the richness and diversity of the gut microbiota in murine models of IBD. | 127 |
| Dexamethasone | GAL-poly(lactic-co-glycolic acid) nanoparticles | Galactose | 118 nm | Have considerable hemocompatibility, biocompatibility and cellular uptake; macrophage uptake was inhibited by D-galactose confirming involvement of MGL2. | 128 |
| −9.5 mV | |||||
| NTs derived from tea leaves | Tea leaf-derived natural nanotherapeutics | Galactose | 140.0 nm | Reduce the production of reactive oxygen species, inhibit the expression of pro-inflammatory cytokines, and increase the amount of anti-inflammatory IL-10 secreted by macrophages. | 129 |
| −14.6 mV | |||||
| Mannose | Mannose-modified carbon nanotubes | Mannose | 10 nm | Significantly improve antigen delivery to macrophages and promote immune activity in vitro. Moreover, it effectively decreases the release rate of antigen, and dramatically enhance the in vivo humoral and cellular immune responses against OVA. | 130 |
| −29.1 ± 0.1 mV | |||||
| Ovalbumin | Poly(lactic-co-glycolic acid) microspheres and conjugated it with Mannose-modified chitosan | Mannose | 7.9 ± 0.5 μm | Act by activating the T cells to initiate an immune response by promoting the maturation of dendritic cells and improving their antigen presentation efficiency. | 131 |
| +30.8 ± 1.1 mV | |||||
| Tofacitinib citrate | Transferrin anchored poly(lactic-co-glycolic acid) nanocarriers | Transferrin | 208.0 nm | In vitro cell-based studies confirmed the cellular biocompatibility and considerable uptake of nanocarriers by colon and macrophage cells; the uptake was diminished by anti-CD71/TFR1 antibodies. | 132 |
| −8.64 mV | |||||
| Dexamethasone | Multilayers of chitosan, hyaluronic acid, and finally Eudragit S100 nanoparticles | CD44 | 179.7 ± 9.85 nm | Are ingested by macrophages via CD44 receptor-mediated endocytosis to regulate M1-to-M2 macrophage polarization and exert anti-inflammatory effects and show better colon-targeting properties than Dex-loaded chitosan. | 133 |
| +19 mV | |||||
| Curcumin | Are derived from a quaternate chitosan and surface functionalization with chondroitin sulfate | CD44 | 238.90 ± 4.51 nm | Are ingested by macrophages via CD44 receptor-mediated endocytosis to regulate M1-to-M2 macrophage polarization and exert anti-inflammatory effects and show better colon-targeting properties than Dex-loaded chitosan. | 134 |
| 41.93 ± 1.17 mV | |||||
| CD98 siRNA and berberine | Hyaluronic acid-modified chitosan-guanidine-CO2 NPs | CD98 | 219.4 ± 8.62 nm | Can target the colon, downregulate pro-inflammatory cytokines, and alleviate microbial dysbiosis in a mouse model of UC. | 135 |
| −17.4 ± 6.82 mV | |||||
| TNF-a-siRNA and F4/80 Ab (Fab'-bearing) | Poly(lactic acid) poly(ethylene glycol) block copolymer | F4/80 | 376 nm ± 19 nm | Can overcome physiological barriers and target the siRNA to inflamed areas of the colon. Increase the kinetics of uptake. | 136 |
| +2.56 mV | |||||
| Anti-MAdCAM-1 antibodies and MnO NPs | Poly(isobutylene-alt- maleic anhydride) | MAdCAM-1 | 41.1 nm ± 1.9 nm | Preferentially localize in the inflamed bowel, whereas untargeted nanoparticles were more rapidly washed out. | 137 |
| −44.7 ± 0.6 mV | |||||
| MAdCAM-1-Fc fusion protein and siRNA | Lipid nanoparticles | α4β7 | 40 nm | Exploit the conformational changes of an integrin to enhance cell-specific uptake is feasible and that it translates into therapeutic efficacy in experimental colitis. | 138 |
| −10 mV |
3.2 ROS-responsive NPs drug delivery systems
When in a healthy state, the body maintains a certain balance between the production rate of reactive oxygen species (ROS) and their clearance rate by antioxidants.139 ROS are made up of superoxide radicals, hydroxyl radicals, singlet oxygen, and hydrogen peroxide (H2O2), and are typically derived from white blood cells and macrophages at the inflamed site of the colon.140 ROS play a role in various physiological activities, including immune responses, cell metabolism, and signal transduction.141 Compared to the healthy intestine of normal individuals, the intestinal mucosa of IBD patients experiences oxidative stress due to the production of large amounts of ROS by neutrophils and macrophages.142 This results in a significant increase in ROS levels, leading to intestinal cell damage and mucosal destruction.111 UC is often associated with excessive ROS production, with the concentration of ROS in the intestinal mucosa of UC patients being 10-100 times higher than that in non-UC patients.32 By utilizing the pathological features of IBD, ROS-responsive NPs have tremendous potential for the relief and treatment of IBD.An effective method for treating IBD is the use of ROS-responsive nanoparticles administered orally. For example, Yan et al. developed PSB@NPs, which are ROS-responsive and folate-functionalized nanoparticles loaded with pterostilbene (PSB). These nanoparticles exhibit the ability to release PSB in response to ROS, effectively scavenge ROS, and protect intestinal cells against oxidative damage induced by H2O2. In a DSS-induced colitis model, PSB@NPs-FA demonstrated significant ROS scavenging ability and anti-inflammatory activity. Additionally, they exhibited both innate and adaptive immune responses, which regulated dendritic cells, promoted macrophage polarization, and regulated T cell infiltration. As a result, PSB@NPs-FA effectively alleviated and treated colonic injury in mice with colitis (Fig. 6A).108 Moreover, Zhang et al. have devised an innovative oral nanotherapeutic remedy named LBL-CO@MPDA, incorporating carbon monoxide (CO) for IBD treatment. The formulation involves loading a CO prodrug (Fe3(CO)12) onto polydopamine nanoparticles (CO@MPDA) using a layer-by-layer (LBL) self-assembly strategy, followed by encapsulation with a CS/alginate electrolyte layer. The colitis microenvironment's heightened oxidative stress may degrade the LBL shell, while excess ROS at the inflammatory site directly prompt the release of CO from CO@MPDA into the colonic cavity. Consequently, CO gas modulates macrophages to exert its pharmacological activity. Research findings suggest that CO activates heme oxygenase-1 (HO-1), inducing M2 polarization of macrophages through the Notch/Hes1/Stat3 signaling pathway, simultaneously suppressing inflammatory responses by downregulating the p38 MAPK and NF-κB (p50/p65) signaling pathways. In a murine model of UC, LBL-CO@MPDA effectively ameliorates the inflammatory microenvironment and restores intestinal barrier function through diverse mechanisms, including alleviating oxidative stress, reinstating immune homeostasis, and regulating the intestinal microbiota (Fig. 6B).109
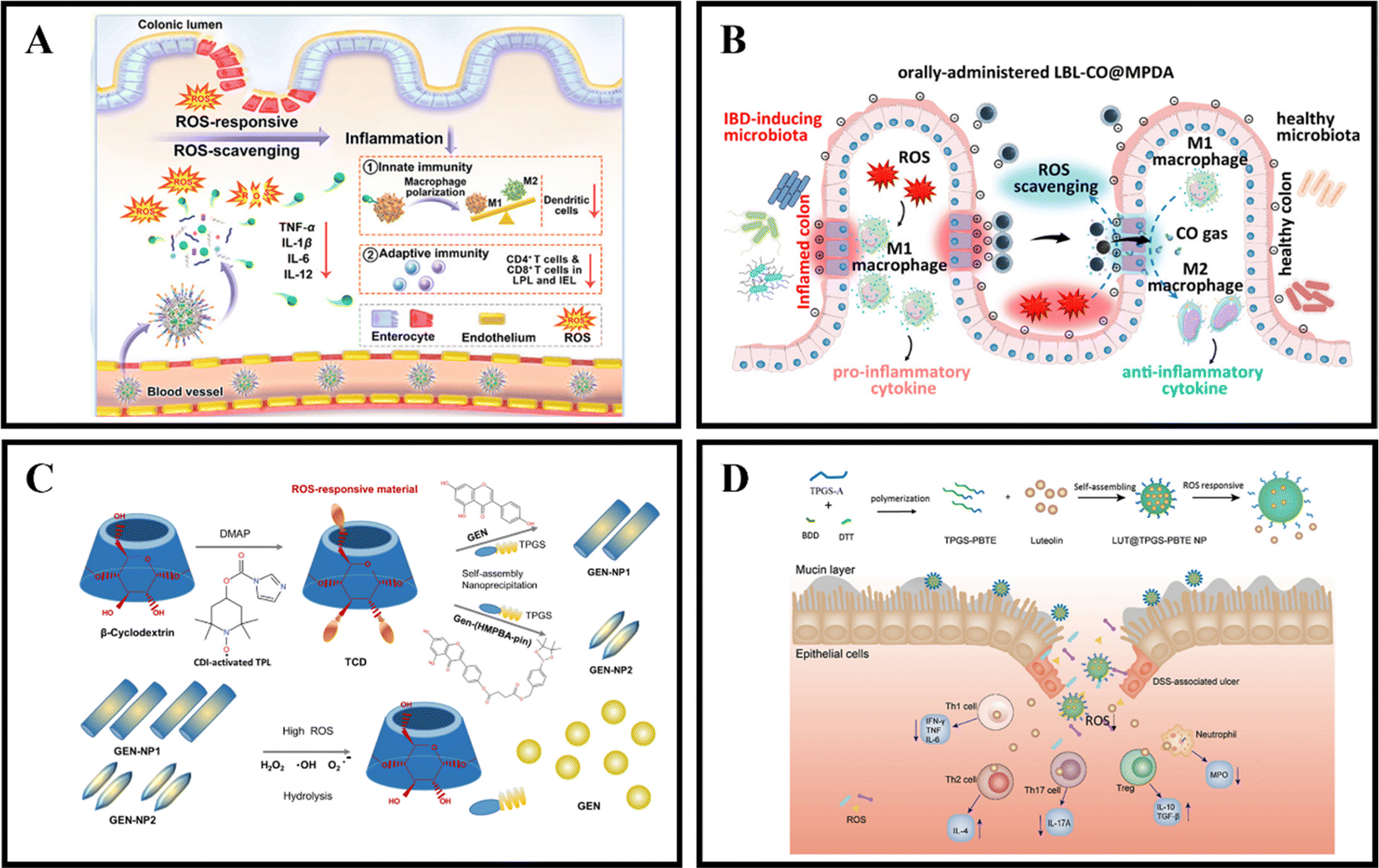 | ||
| Fig. 6 ROS-responsive NPs drug delivery systems. (A) Reactive oxygen species-responsive nanocarrier ameliorates murine colitis by intervening colonic innate and adaptive immune responses. Reproduced with permission. Reproduced with permission.108 Copyright 2023, Elsevier Publishing Group. (B) An orally-administered nanotherapeutics with carbon monoxide supplying for inflammatory bowel disease therapy by scavenging oxidative stress and restoring gut immune homeostasis. Reproduced with permission.109 Copyright 2023, American Chemical Society Publishing Group. (C) Genistein-derived ROS-responsive nanoparticles relieve colitis by regulating mucosal homeostasis. Reproduced with permission.110 Copyright 2021, American Chemical Society Publishing Group. | ||
Genistein (GEN), a bioactive compound primarily present in soy-based foods, exhibits the ability to modulate both cellular and humoral immunity, inhibit granule enzyme secretion, and maintain the internal balance of inflammation. Fan et al. devised a nanomaterial, GEN-NP 2, responsive to ROS. This material incorporates a time-conjugated β-cyclodextrin with superoxide dismutase mimetic properties and GEN modified with 4-(hydroxymethyl)phenylboronic acid pinacol ester. GEN-NP 2 effectively transports GEN to the inflammation site, shielding it from rapid metabolism and elimination in the gastrointestinal tract. In the presence of elevated ROS levels, GEN is specifically released and accumulates at the inflammation site. The findings suggest that GEN-NP 2 mechanistically enhances the expression of estrogen receptor β (ERβ), concurrently diminishes the expression of inflammatory mediators, mitigates inflammatory cell infiltration, fosters intestinal epithelial cell autophagy, suppresses the secretion of IL-1β and TNF-α, regulates the gut microbiota, and ultimately alleviates colitis (Fig. 6C).110 In a similar vein, Tan et al. employed D-α-tocopherol polyethylene glycol succinate-b-poly(β-thioester) copolymer (TPGS-PBTE) as the base material to craft NPs designed for the ROS-responsive release of luteolin (LUT), a natural flavonoid renowned for its exceptional anti-inflammatory and free radical scavenging properties. Termed LUT@TPGS-PBTE NPs, the resultant particles displayed size variations and exhibited responsiveness to ROS for drug release, attributed to the sulfur ether bond present on the polymer main chain. This feature facilitated ROS clearance and the selective accumulation of LUT in the inflamed colon. The findings suggest that LUT@TPGS-PBTE NPs ameliorated weight loss, colon shortening in a UC mouse model, and alleviated colon tissue damage by suppressing ROS and pro-inflammatory cytokines (such as TNF-α, IL-6). Moreover, an increase in glutathione levels and upregulation of anti-inflammatory factors (such as IL-10, IL-4) were observed. Crucially, LUT@TPGS-PBTE NPs modulated the balance of T helper (Th)1/Th2 and Th17/regulatory T cell (Treg), thereby regulating the inflammatory microenvironment, ultimately alleviating inflammation and expediting the healing of the intestinal mucosa (Fig. 6D).111
Nevertheless, nanocarrier systems intended for oral administration and designed to be responsive to ROS often concentrate solely on the interactive response with ROS. Unfortunately, in many cases, these drugs can inflict irreversible damage to the human body, especially at high doses. Additionally, there is a risk of premature drug release in ROS-responsive nanocarriers due to contact with ROS during the delivery process.31,143 Hence, when developing ROS-responsive oral nanocarrier systems, it is imperative to not only prioritize therapeutic effectiveness but also consider sustained release characteristics and the biocompatibility of the nanocarrier system. Striking a balance between efficacy and safety is paramount in the design process. As an illustration, Bai et al. investigated the treatment of IBD by encapsulating superoxide dismutase (SOD) within a zeolitic imidazolate framework-zinc (ZIF-zni) using biomimetic mineralization. The findings reveal that SOD@ZIF-zni effectively suppresses levels of ROS and pro-inflammatory cytokines, leading to the amelioration of colitis in a murine model of UC.144 This metal–organic framework (MOFs) material emerges as a promising solution to address the limitations associated with ROS responsiveness. By building upon the foundation of SOD@ZIF-zni, the design of a nano drug delivery system with ROS responsiveness has the potential to deliver both effective therapeutic outcomes and mitigate the issue of premature drug release in ROS-responsive nano drug delivery systems.
3.3 Charge-responsive NPs drug delivery systems
In cases of active IBD, the inflamed region of the intestine is drawn to areas of opposite charge through electrostatic interactions.145 Leveraging this mechanism, we can devise a NPs drug delivery system that responds to charges, enabling targeted delivery of medications to the inflamed region of the intestine and the alleviation of intestinal inflammation. Typically, the inflammatory area's mucosal components include negatively charged carbohydrates and colonic mucin.146,147 Consequently, positively charged NPs can interact electrostatically with the negatively charged inflamed mucosal area, increasing the drug's residence time and improving its efficacy.145 Research has found CS, a cationic natural polysaccharide, to possess good biodegradability, biocompatibility, and adhesion. It can attach to the intestinal mucosal surface of inflamed tissue by positively charged nanoparticle and negatively charged intestinal mucosa interactions, making it widely used in oral colon-targeted drug delivery systems.148 For example, Fatemeh et al. used the ionotropic gelation technique to load budesonide (BU) onto CS NPs (BCN), which were then pelletized via extrusion-spheronization and coated with a polymer solution consisting of two enteric-coating agents (Eudragit L and S) and a time-dependent polymer (Eudragit RS). Eudragits coatings, featuring pH-dependent and time-dependent properties, are designed to minimize the premature release and absorption of BCN in the upper gastrointestinal tract, ensuring optimal transportation of BCN to the site of colonic inflammation. Notably, BCN exhibits a sustained release pattern throughout the entire gastrointestinal tract, leading to a prolonged anti-inflammatory effect. Research indicates that encapsulating BCN within a well-designed colonic delivery coating system may result in a synergistic effect, potentially providing substantial and enduring anti-inflammatory benefits.112Furthermore, during inflammation of the intestine, the inflamed area rapidly and significantly produces transferrin and eosinophilic cationic protein, which attract NPs with negative charges.149,150 Thus, this offers a molecular target and anchoring site for drug carriers with negatively charged surfaces.151 Studies have shown that negatively charged liposomes accumulate more in the inflamed area than liposomes with positive or neutral charges, and NPs drug carriers with negative charges will preferentially adhere to the area of colitis.152 For instance, Ren et al. developed selenized polymer-lipid hybrid nanoparticles loaded with triptolide (Tri) and prepared Se@Tri-PLNs using a solvent diffusion in situ reduction technique. The zeta potential of Se@Tri-PLNs was −29.70 mV, which can effectively target the area of colitis. Compared to the corresponding unmodified group, Se@Tri-PLNs can significantly reduce the levels of inflammatory factors in vivo, alleviate DSS-induced colonic injury, and significantly improve the therapeutic effect on UC mice.113 In addition, Lee et al. utilized an in situ sol–gel method to prepare folate-grafted amino-clay (FA-AC). Drug-loaded nanocomposites were then formed by the electrostatic interaction between FA-AC and the model antibody Infliximab (IFX), and coated with ES100 to yield EFA-AC-IFXNPs. The positively charged FA-AC promoted the deposition of pH-sensitive polymer ES100 for surface coating, leading to an increase in particle size and reversal of surface charge. FA-AC exhibited desirable drug carrier properties, including low cytotoxicity, good target selectivity, and the ability to form nanocomposites with negatively charged macromolecules. Compared to orally administered IFX solution, EFA-AC-IFXNPs effectively alleviated UC in mice.114 Notably, Kotla et al. have introduced inflammation targeted nanocarriers (IT-NCs) for the delivery of CSA to inflamed sites. These nanocarriers, termed CSA/IT-NCs, possess a compact size, anionic surface charge, and mucin-adhesive properties, facilitating adhesion and penetration through the mucosal epithelium in inflamed regions. This capability was confirmed through experimentation in murine models of DSS- and TNBS-induced colitis. In contrast to free CSA, IT-NCs exhibited enhanced therapeutic efficacy in the treatment of colitis in both animal models. Moreover, ex vivo analyses using colon tissue biopsies from colitis patients revealed that IT-NCs preferentially adhered to inflamed colon tissues compared to normal tissue (Fig. 7).115
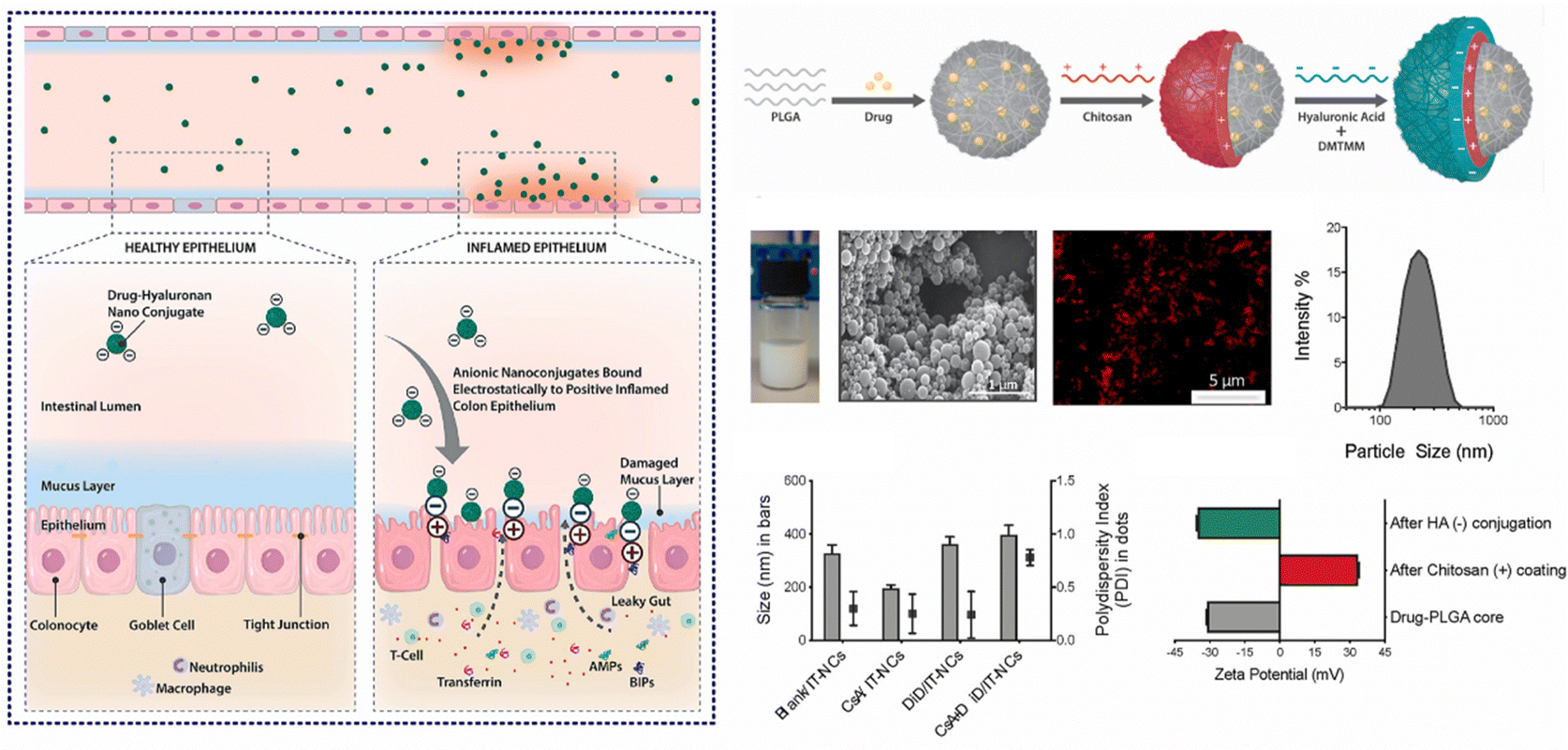 | ||
| Fig. 7 Charge-responsive NPs drug delivery systems. Inflammation-specific targeted carriers for local drug delivery to inflammatory bowel disease. Reproduced with permission.115 Copyright 2022, Elsevier Publishing Group. | ||
In summary, a wealth of research suggests that both positively and negatively charge-responsive nano-drug delivery systems can effectively target colonic inflammation sites and exhibit specific binding during the delivery process.153 However, studies have affirmed that positively charged nano-drug delivery systems engage in robust electrostatic interactions with negatively charged mucosal surfaces, impeding the diffusion of nano-drugs to inflammatory sites, and leading to a reduction in drug release rates.31 Moreover, it is crucial to acknowledge that charge-responsive nano-drug delivery systems may interact with other components in the gastrointestinal tract, such as bile acids and soluble proteins, potentially impacting therapeutic outcomes.154,155 Therefore, when designing charge-responsive nano-drug delivery systems, careful consideration should be given to selecting an appropriate outer shell to mitigate adverse effects from the external environment.
3.4 Enzyme-responsive NPs drug delivery systems
When orally administered drugs make their way to the intestines, they will encounter a range of digestive enzymes.38,39 For instance, α-amylase is a critical enzyme that plays a role in breaking down carbohydrates. This enzyme can break down starch molecules into smaller polysaccharides, such as maltose, glucose, and oligosaccharides.156 The specific way in which enzymes react depends on the enzymatic catalytic chemical reaction, which can cause the nanoparticle carrier to degrade, dissociate, or undergo morphological transformation, ultimately leading to the release of the drug.157,158 By designing enzyme-responsive nanoparticle drug delivery systems, we can target various digestive enzymes at the inflamed colon site in active IBD to achieve precise drug delivery.Xu et al. developed oral nanocarriers, referred to as DHC NPs, that simultaneously deliver an anti-inflammatory drug, dexamethasone (DEX), and a ROS scavenger. These carriers are formed through the self-assembly of hydroxyethyl starch (HES) and curcumin (CUR) conjugated polymers. DHC NPs exhibit good stability in the harsh gastric acid environment and can penetrate and retain in epithelial cells, then be internalized by macrophages in the inflamed colon. The overexpressed α-amylase in the inflamed colon degrades HES, and DHC NPs release the drug in an α-amylase-responsive manner, achieving therapeutic effects through both anti-inflammatory and ROS scavenging activities. In vivo experiments show that oral administration of DHC NPs significantly improves the therapeutic effect on the DSS-induced UC model in mice (Fig. 8A).116 Li et al. developed an enzyme-triggered controlled release system (CD-Cur-CANPs) using a curcumin-cyclodextrin (CD-Cur) polymer as the core and low molecular weight CS and unsaturated sodium alginate nanoparticles (CANPs) as the shell. In the presence of α-amylase, the enzyme-catalyzed degradation of β-CD causes chain scission within the CD, leading to degradation and release of Cur from CD-Cur-CANPs, achieving targeted drug delivery. Oral administration of CD-Cur-CANPs promotes colonic epithelial barrier integrity, regulates the production of inflammatory cytokines, and regulates the gut microbiota of DSS-induced UC model mice, as shown in in vivo experiments (Fig. 8B).117
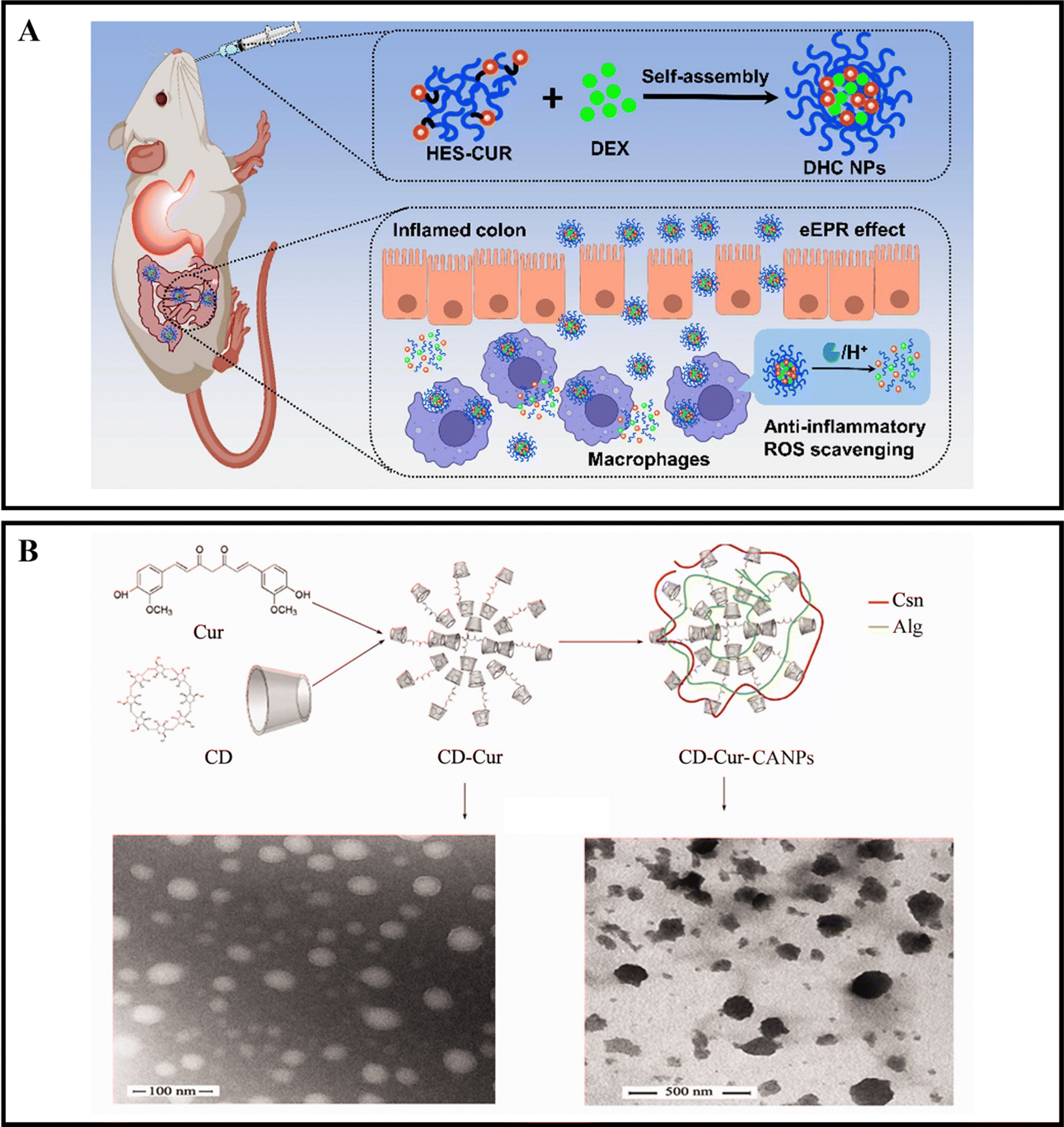 | ||
| Fig. 8 Enzyme-responsive NPs drug delivery systems. (A) Colon-targeted oral nanoparticles based on ROS-scavenging hydroxyethyl starch-curcumin conjugates for efficient inflammatory bowel disease therapy. Reproduced with permission.116 Copyright 2022, Elsevier Publishing Group. (B) An efficient enzyme-triggered controlled release system for colon-targeted oral delivery to combat dextran sodium sulfate (DSS)-induced colitis in mice. Reproduced with permission.117 Copyright 2021, Taylor & Francis Publishing Group. | ||
3.5 Mucus-dependent NPs drug delivery systems
The mucosal layer in the intestine presents a significant barrier for drugs to enter the biopolymer matrix, and it plays a crucial role in the oral delivery of NPs.45,159 To achieve effective therapeutic outcomes, orally administered NPs must traverse the upper gastrointestinal tract and the mucosal layer before accumulating in the corresponding inflamed tissue.36,160 Various strategies have been explored in previous studies to enhance the ability of NPs to penetrate and adhere to the intestinal mucosa, including alterations to their hydrophilicity, shape, size, charge, and rigidity.161–163 NPs with neutral and hydrophilic surface properties have demonstrated superior mucosal permeability, enabling them to overcome the mucosal barrier. However, the hydrophilic/neutral surface may also reduce their interaction with the cell membrane.164 It is important to note that, in addition to enhancing mucosal adhesion to extend drug retention time, researchers should consider the balance between the interaction with mucin and diffusion through it.165Xie et al. utilized a self-assembly method to create PA-N-2-HACC-Cys nanoparticles (NPs) that were loaded with CUR. The composition of the NPs included N-2-hydroxypropyltrimethylammonium chloride CS (N-2-HACC), hydrophobic palmitic acid (PA), and cysteine (Cys). These NPs exhibited strong mucosal adhesion, sustained release, and high permeability, indicating their ability to overcome mucosal and epithelial barriers, thus improving the hydrophilicity, stability, and bioavailability of orally administered drugs. Furthermore, these NPs were able to penetrate mucosal and epithelial barriers, facilitating cell uptake. The CUR@PA-N-2-HACC-Cys NPs could open tight junctions between cells, allowing for transport through the epithelium, while still maintaining a balance between interaction with mucin and diffusion through it. In vivo studies demonstrated that administering CUR@PA-N-2-HACC-Cys NPs orally significantly improved colitis in a mouse model of UC, outperforming free CUR and CUR@PA-N-2-HACC NPs and promoting mucosal epithelial repair.118 Xie et al. also developed silk-based nanoparticles that contained resveratrol (RSV) and were functionalized with Pluronic F127 (PF-127). The results showed that adding PF-127 enhanced the intestinal mucosal penetration of RSV NPs. In vitro studies revealed that, compared to other similar products, RSV NPs containing PF-127 exhibited improved anti-inflammatory and antioxidant activity by inhibiting pro-inflammatory cytokine TNF-α and ROS secretion in RAW 264.7 macrophages stimulated by lipopolysaccharide. RSV NPs containing PF-127 significantly enhanced the therapeutic effect of UC compared to blank silk NPs and RSV-loaded NPs (Fig. 9).119
 | ||
| Fig. 9 Mucus-dependent NPs drug delivery systems. Mucus-penetrating silk fibroin-based nanotherapeutics for efficient treatment of ulcerative colitis. Reproduced with permission.119 Copyright 2022, MDPI Publishing Group. | ||
3.6 Size-dependent NPs drug delivery systems
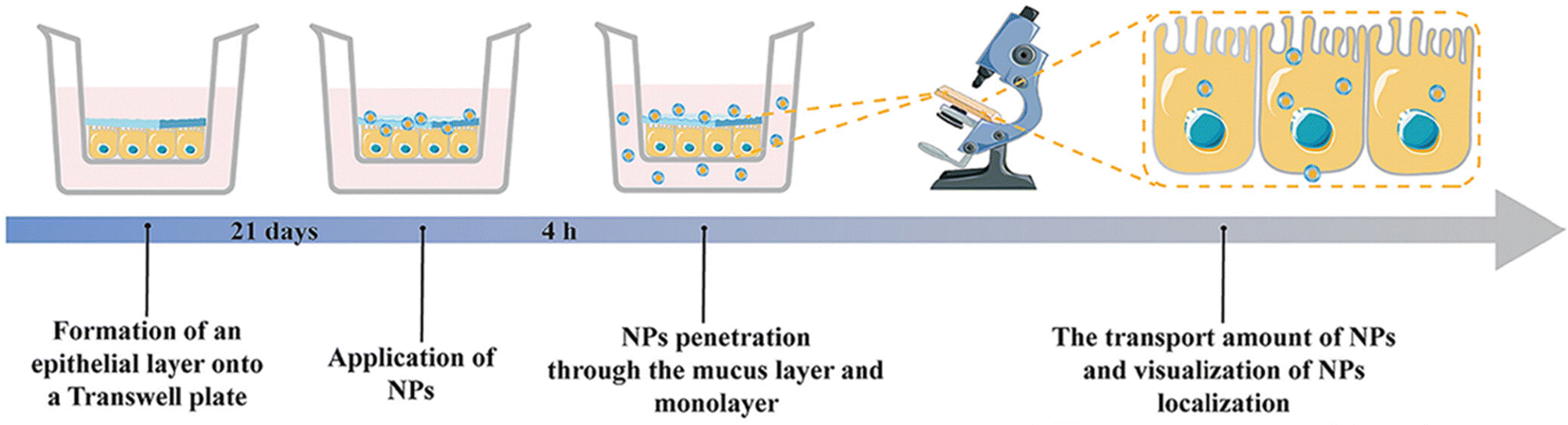
IBD is characterized by increased permeability in the intestinal epithelial cells and blood vessels of affected patients.68,166 This makes size-dependent NPs delivery systems a popular choice for targeted IBD treatment. Nanoparticles in the nanometer range can easily penetrate the tight junctions of the intestinal epithelial cells and reach the inflamed area, while also increasing permeability in the inflammatory site to prolong drug retention.167,168Lamprecht et al. were the first to report that 100 nm polystyrene particles exhibit stronger adhesion than 1 μm and 10 μm particles in the inflamed colon of TNBS-induced mice. Further studies have demonstrated that such particles can effectively improve the therapeutic effect on IBD.169 Mohan et al. recently explored the potential of INF nanoparticles for treating IBD, focusing on the size-dependent targeting effects of nanoparticles in regulating INF on the epithelial barrier. They created an in vitro model of healthy and inflamed intestinal epithelial barriers to observe the cell interactions of PLGA-PEGG NPs with various NPs sizes and polydispersities. INF-loaded NPs were formed through electrostatic interactions between INF and NPs, and their therapeutic effects were evaluated in the inflamed epithelial cell barrier model. The findings revealed that NPs interactions were notably intensified in the inflamed cell barrier model, with increased transport of 130–300 nm NPs and accumulation of larger NPs (∼600 nm) at the barrier. Delivering INF directly to the inflamed barrier through ∼600 nm NPs expedited barrier integrity recovery, lowered the secretion of inflammatory cytokines, and reduced cellular toxicity compared to using INF alone.120 Wang et al. developed mannosylated liposomes (MAN-LPs) targeting macrophages and utilized rosiglitazone (ROSI) as a peroxisome proliferator-activated receptor gamma (PPAR-γ) agonist. To investigate the size-dependent targeting effect of MAN-ROSI-LPs on macrophages, they prepared a series of liposomes of different sizes and compared them with unmodified polyethylene glycol (PEG)-ROSI-LPs. In a zebrafish model, MAN-LPs showed higher uptake by RAW 264.7 cells and better co-localization with macrophages compared to unmodified liposomes. Moreover, in an IBD mouse model, MAN-LPs could effectively accumulate in inflamed intestinal sites. Importantly, the targeting ability of MAN-LPs significantly increased with increasing particle size, and the largest MAN-LPs particles exhibited the best anti-inflammatory effect in cells and higher therapeutic efficiency in the IBD mouse model.121
However, the administration of size-dependent NPs delivery systems results in the passive accumulation of NPs at inflammatory sites. When NPs interact with tissues or cells in the inflamed colon, a significant number of local immune cells can affect the internalization of NPs, thereby influencing the results.170 To address this issue, Gimondi et al. developed a microfluidic device to synthesize homogeneous NPs with particle sizes of approximately 30, 50, and 70 nm. They subsequently investigated the levels and internalization mechanisms of these NPs when exposed to endothelial cells, macrophages, and fibroblasts. The results indicated that all NPs were compatible with cells and were internalized by different types of cells. However, NPs absorption was size-dependent, with the maximum absorption efficiency of 30 nm NPs. Furthermore, the nanoparticle size can lead to different interactions with cells. For instance, 30 nm NPs were internalized by endothelial cells and increased over time, while NPs internalization by LPS-stimulated macrophages and fibroblasts showed stable and decreasing trends, respectively. This evidence highlights the importance of size regulation in NPs design for interactions with specific cell types (Fig. 10).122
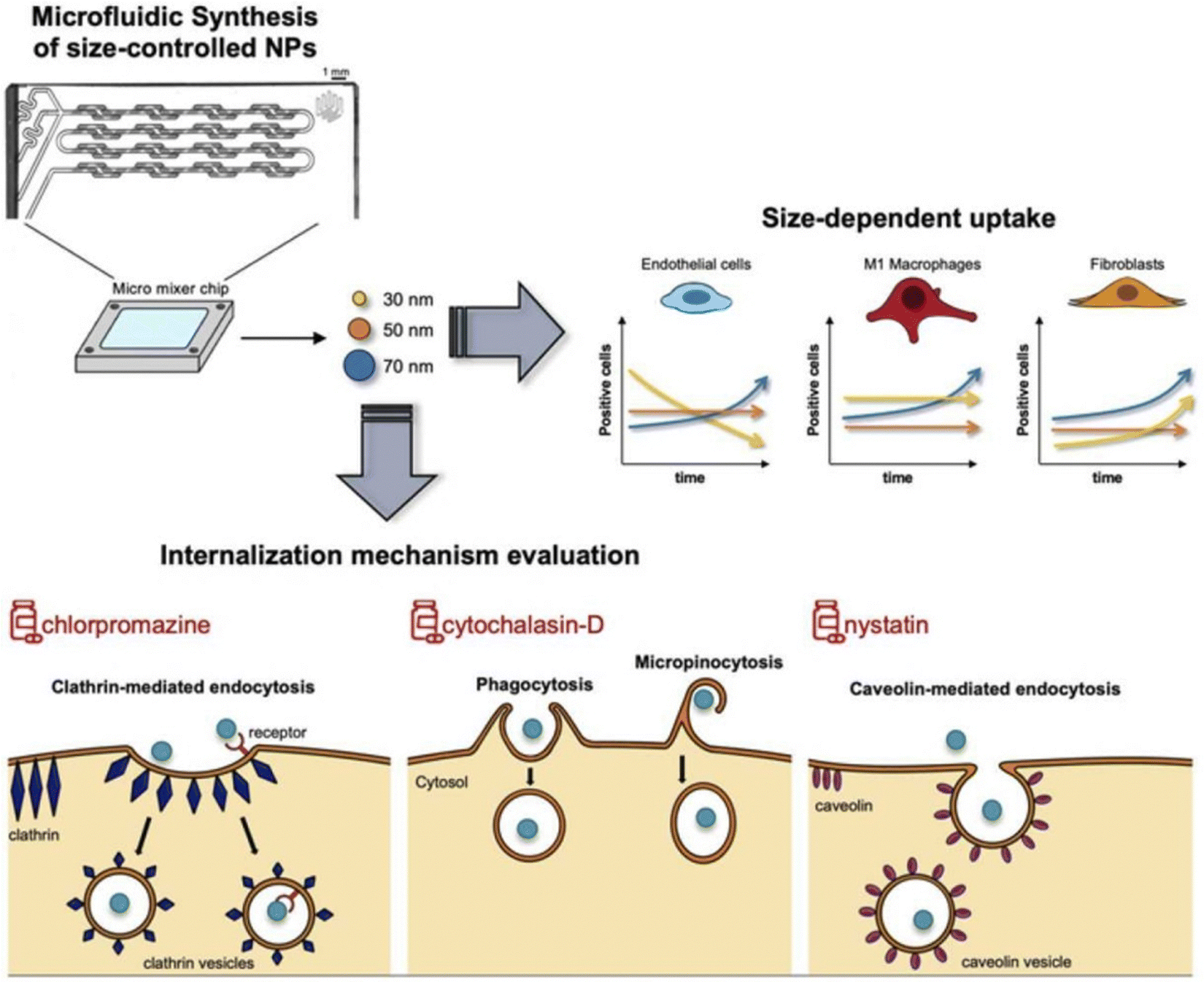 | ||
| Fig. 10 Size-dependent NPs drug delivery systems. On the size-dependent internalization of sub-hundred polymeric nanoparticles. Reproduced with permission.122 Copyright 2023, Elsevier Publishing Group. | ||
3.7 Microbiota-dependent NPs drug delivery systems
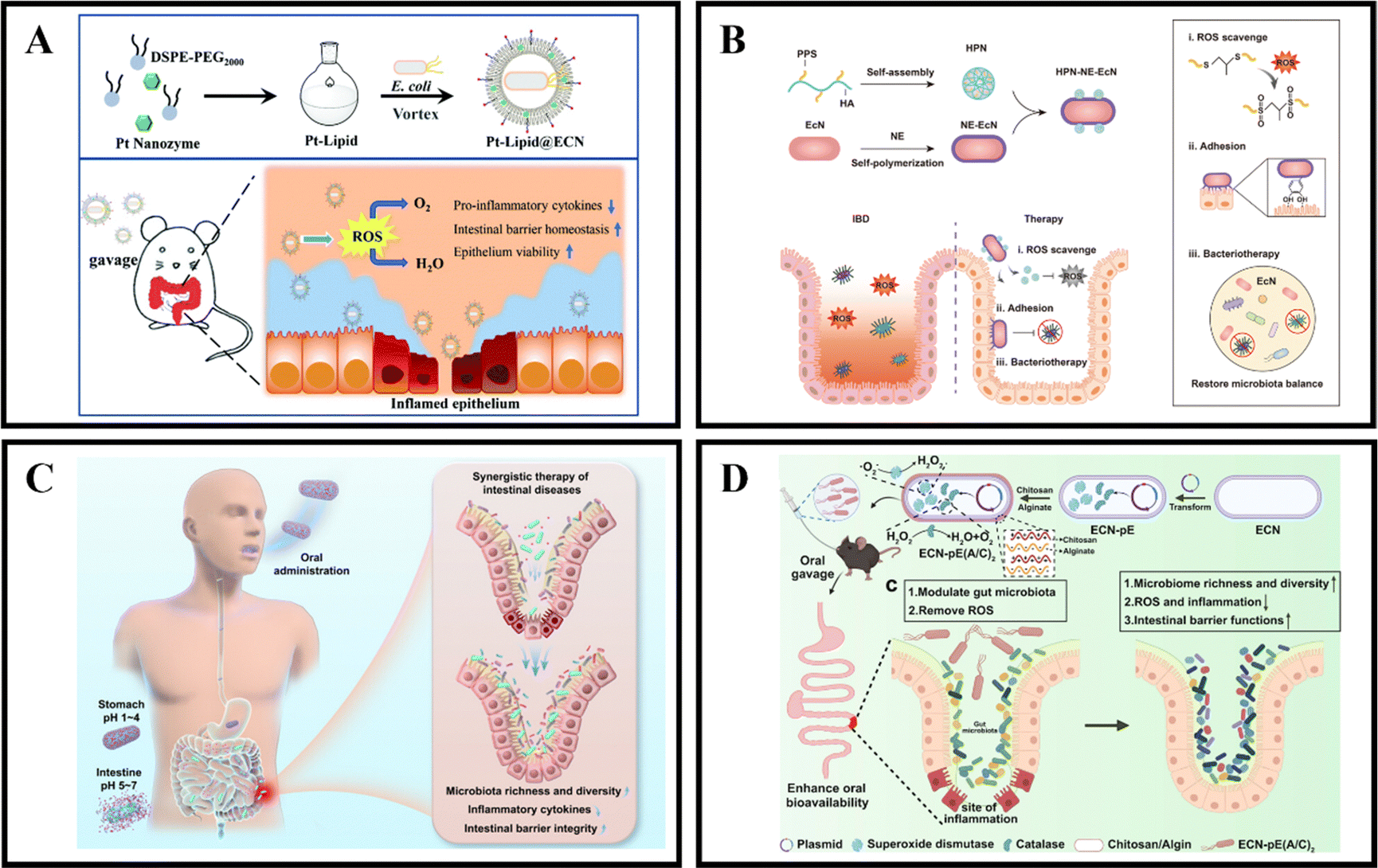
The gut microbiota is known to provide the host with essential vitamins and short-chain fatty acids, while also acting as a protective barrier against pathogen invasion and replication.171,172 Unfortunately, in cases of IBD, the colonic microbiota can become dysbiotic and lead to chronic inflammation, disrupted metabolism, and increased toxin production.173,174 However, it is possible to develop microbiota-dependent NPs delivery systems that can help alleviate and treat IBD by targeting changes in the colonic microbiota.175 Probiotics have been found to be beneficial in the treatment of gastrointestinal diseases like IBD, with preclinical and clinical studies showing significant therapeutic effects. Probiotics work by regulating intestinal microbiota homeostasis, inhibiting pathogen colonization, and reducing the production of inflammatory cytokines, which ultimately alleviates the severity of IBD and maintains its remission.176,177 Unfortunately, the therapeutic effect of probiotics is limited by their short transmission and retention time in the gastrointestinal tract, which results in insufficient regulation of the gut microbiota.175,178 Therefore, it is crucial to protect and enhance the retention of probiotics in the gastrointestinal tract after oral administration to improve their therapeutic effect.In recent years, the development of engineering microbes has been rapid and has become an efficient and significant therapeutic method.179 One of the genetically controllable probiotics with good safety is Escherichia coli Nissle 1917 (EcN),180 which has been used as a therapeutic approach for gastrointestinal diseases for over 100 years.181 Recently, Zhao et al. introduced a universal and simple method of preparing a Pt-Lipid@EcN by decorating a single probiotic with a nano-enzyme coating. The probiotic self-assembles with liposomes under cell-compatible conditions to deposit a bilayer lipid shield on the probiotic surface, providing effective protection against strong acid and various enzymes. The results demonstrate that the lipid-coated EcN can significantly increase the colonization rate of gut microbiota (Fig. 11A).123 Another approach, developed by Liu et al., is an HPN-NE-EcN system that involves coupling hyaluronic acid-polythiophene nanoparticles (HPN) to the modified EcN surface and then wrapping the EcN cells with a layer of norepinephrine (NE). The NE contains a catecholamine group rich in mussel adhesive protein that gives EcN strong mucosal adhesion and prolongs its retention time in the intestine without affecting its growth and proliferation. In addition, due to the physical and chemical conversion of PPS from hydrophobic to hydrophilic, the HPN nanoparticles can self-degrade after complete reaction with ROS, thus enhancing the safety of this method. The HPN-NE-EcN system has a synergistic effect in clearing ROS and regulating the stability of the microbial community in the colon, significantly improving the preventive and therapeutic effects of UC (Fig. 11B).124 Additionally, Peng et al. developed a collaborative approach by coating EcN bacteria with alginate (Alg), which responds to the gastrointestinal microenvironment, and incorporating the therapeutic drug 5-ASA (EcN/5-ASA@Alg). The polysaccharide structure of Alg, binding with Ca2+, forms a protective gel-like framework on the surface of EcN, shielding them from the harsh conditions of gastric fluids post-oral administration. Upon reaching the intestines, the Alg coating degrades under neutral pH, facilitating the release of both EcN and 5-ASA. Research findings suggest that EcN restored bioactivity in an IBD mouse model, effectively alleviating colitis in conjunction with ASA. This synergistic effect was achieved by promoting the richness and diversity of the microbiota, reducing the expression of inflammatory cytokines, and restoring intestinal barrier function. This synthetic strategy is versatile for correcting dysbiosis in the gut microbiota using a combination of probiotic strains and clinical drugs, presenting a universal approach for future collaborative therapies in intestinal diseases (Fig. 11C).125
 | ||
| Fig. 11 Microbiota-dependent NPs drug delivery systems. (A) Oral nanozyme-engineered probiotics for the treatment of ulcerative colitis. Reproduced with permission.123 Copyright 2022, Royal Society of Chemistry Publishing Group. (B) Mucoadhesive probiotic backpacks with ROS nanoscavengers enhance the bacteriotherapy for inflammatory bowel diseases. Reproduced with permission.124 Copyright 2022, Science Publishing Group. (C) Gastrointestinal microenvironment responsive nanoencapsulation of probiotics and drugs for synergistic therapy of intestinal diseases. Reproduced with permission.125 Copyright 2023, American Chemical Society Publishing Group. (D) Programmable probiotics modulate inflammation and gut microbiota for inflammatory bowel disease treatment after effective oral delivery. Reproduced with permission.126 Copyright 2022, Nature Publishing Group. | ||
Zhou et al. skillfully engineered ECN into a modified version, ECN-pE, with enhanced expression of CAT and superoxide SOD through genetic manipulation. To optimize ECN-pE's effectiveness in the gastrointestinal tract, they utilized the layer-by-layer self-assembly technique to envelop ECN-pE with CS and sodium alginate. In murine models of IBD induced by diverse chemical agents, the CS/sodium alginate-coated ECN-pE(C/A)2 effectively alleviated inflammatory responses and rejuvenated the colonic epithelial barrier. Unexpectedly, this engineered ECN-pE(C/A)2 showcased the capacity to influence the gut microbiota positively, elevating the presence of Lachnos-piraceae_NK4A136 and Odoribacter, pivotal microorganisms for preserving intestinal balance. Research findings suggest that ECN-pE(C/A)2 features self-secreting functional proteins and the capability to self-encapsulate within a biofilm, presenting a secure and efficient therapeutic approach for acute IBD (Fig. 11D).126 Similarly, Cao et al. designed a humanized enzymatic variant of Bifidobacterium longum (BL) for the modulation of intestinal inflammation (BL@B-SA50). BL not only promotes the host's well-being but also exhibits excellent colonizing capabilities in the colon. Single-atom catalytic artificial enzymes (SAzymes) emulate the functions of antioxidant enzymes such as SOD and CAT, effectively eliminating superoxide radicals (O2˙−) and H2O2. These SAzymes act as antioxidant biomolecules, efficiently neutralizing hydroxyl radicals (˙OH) to rapidly alleviate inflammation. Furthermore, they serve as a protective shield for BL, safeguarding it from oxidative damage at the inflammatory site, leading to a swift transition in barrier function and the intestinal microbiota towards a beneficial state. Research results indicate that BL@B-SA50 can reduce ROS levels, suppress the production of inflammatory cytokines, restore intestinal barrier function, and enhance the richness and diversity of the gut microbiota in mouse models of UC and CD. Most importantly, BL@B-SA50 exhibits remarkable therapeutic potential in Beagle dogs with colitis.127
3.8 Active targeting-dependent NPs drug delivery systems
To achieve greater precision in targeting the site of inflammation in colitis, researchers are exploring active targeted NPs delivery systems in addition to the responsive and dependent systems mentioned above. Among these systems, the ligand-receptor NPs delivery system designed to target the site of colitis can enhance the accumulation of drugs at the site while reducing their secondary distribution.182 This can lead to improved therapeutic outcomes with fewer systemic side effects.183 NPs surface ligands play a crucial role in this process by facilitating specific molecular binding with overexpressed molecules on the surface of target cells, triggering receptor-mediated endocytosis.184 For instance, sugar-based ligands such as galactose, mannose, glucose, N-acetylglucosamine, and fucose exhibit significant targeting affinity for the C-type lectin receptor expressed on macrophage surfaces.185,186The galactose receptor (GR) is a surface receptor found on macrophages and is a member of the galectin superfamily. It is widely distributed throughout various animal bodies.187 This receptor plays an important role in numerous physiological and pathological processes, such as cell adhesion, apoptosis, inflammation, and metastasis, by specifically recognizing, binding, and taking up D-galactose and N-acetyl galactosamine residues in endogenous and exogenous substances.188,189 In a study by Zeeshan et al., a ligand-based, sustained-release nanoparticle (Dexa-GAL-PLGA) containing dexamethasone was developed using the QBD method. This nanoparticle can be actively targeted to macrophage MGL-2 receptors and delivered as the sole carrier to inflamed intestines to prolong therapeutic effects. The results showed that macrophages at the inflamed site of the intestine promoted the uptake of PLGA nanocarriers with galactose binding, exhibiting prolonged adhesion and therapeutic efficacy while enhancing stability against dose variations in pH and enzymes in GIT. Furthermore, physical, and chemical characterization, in vitro, ex vivo, and preliminary studies supported this hypothesis and indicated the potential of GAL-PLGA nanoparticles as safer and more biocompatible carriers for active targeting of MGL-2 macrophage receptors in intestinal inflammation via oral administration.128 Zu et al. have isolated and characterized three types of extracellular vehicles (EVs), namely LNTs, MNTs, and BNTs, from small, medium, and large tea leaves. Each of these EVs shares similar characteristics with mammalian cell-derived EVs. The surface of these EVs contains galactose, which effectively promotes macrophage internalization. The results indicate that these EVs can decrease the levels of pro-inflammatory cytokines (TNF-α, IL-6, IL-12), increase the levels of anti-inflammatory cytokine IL-10, inhibit colon MPO activity, reduce MDA levels, and increase colon GSH levels. Furthermore, these EVs can upregulate the expression of tight junction-related proteins ZO-1 and MUC2. Oral administration of these EVs can effectively suppress inflammatory bowel responses, restore damaged colon barriers, enhance the diversity and overall abundance of intestinal flora, and prevent or alleviate IBD and colon cancer associated with colitis (Fig. 12A).129
 | ||
| Fig. 12 Active targeting-dependent NPs drug delivery systems. (A) ‘Green’ nanotherapeutics from tea leaves for orally targeted prevention and alleviation of colon diseases. Reproduced with permission.129 Copyright 2021, Elsevier Publishing Group. (B) Chondroitin sulfate-based nanocarriers for drug/gene delivery. Reproduced with permission.133 Copyright 2015, Elsevier Publishing Group. (C) Chondroitin sulfate functionalized palmitic acid and cysteine cografted-quaternized chitosan for CD44 and gut microbiota dual-targeted delivery of curcumin. Reproduced with permission.134 Copyright 2023, Elsevier Publishing Group. (D) An oral pH-activated “nano-bomb” carrier combined with berberine by regulating gene silencing and gut microbiota for site-specific treatment of ulcerative colitis. Reproduced with permission.135 Copyright 2022, Royal Society of Chemistry Publishing Group. | ||
The mannose receptor (MR), a member of the C-type lectin superfamily, is predominantly expressed on the surface of immune cells such as macrophages and DC cells. It is capable of binding and internalizing various pathogenic bacteria and toxic glycoproteins, presenting antigens, and maintaining internal environmental stability.190,191 The macrophage MR is the prototype of a multi-lectin receptor family that recognizes carbohydrates on the cell wall of pathogenic organisms.192 Hence, the use of mannose-modified drugs or delivery materials has emerged as a viable approach for targeted drug delivery through the MR.186,193 Feng et al. encapsulated ovalbumin (OVA) in PLGA MPs and conjugated them with CS modified with the MR to obtain MAN-R targeted nano-MPs (MAN-CS-OVA-PLGA-MPs). In vitro and in vivo experiments demonstrated that MAN-CS-OVA-PLGA-MPs have the potential to activate T cells, initiate immune responses, promote DC cell maturation, and enhance antigen presentation efficiency.130 The authors utilized the specific recognition between the MR on the surface of antigen-presenting cells (APCs) and mannose to synthesize mannose-modified carbon nanotubes (M-MWCNTs). In vitro experiments demonstrated that macrophages rapidly phagocytosed M-MWCNTs carrying a large amount of ovalbumin (OVA), effectively promoting macrophage proliferation and cytokine secretion (IL-1β, IL-6). Subsequent animal experiments showed that M-MWCNTs could induce DC cell maturation, increase antigen-specific antibody levels (IgG, IgG1, IgG2a, IgG2b), and cytokine levels (IFN-γ, IL-6).131 The MR is a promising glycan receptor for the treatment of IBD due to the abundance of immune cells such as macrophages in inflamed intestinal tissues.
The transferrin receptor (TfR) is a transmembrane glycoprotein that facilitates cellular iron uptake through receptor-mediated endocytosis, making it a crucial carrier protein. In a healthy gut, TfR expression is low in all cells.194 However, in active UC, TfR is heavily upregulated on the surface of colon epithelial cells and macrophages.195,196 This upregulation can be exploited for ligand-receptor active targeting and to combat activated inflammatory pathways in colon macrophages and epithelial cells.194In vitro and in vivo binding studies were conducted to assess the targeting capability of anti-TfR antibody-loaded nanoliposomes to colitis regions. Results revealed that anti-TfR immunoliposomes significantly increased accumulation in the mucosa of DNBS-induced colitis rats compared to non-specific immunoliposomes.149 Zeeshan et al. developed PLGA nanocarriers loaded with citric acid Tofacitinib (Tofa-P/TfR NCs) targeting transferrin receptor (TFR-1/CD 71) overexpressed on colon epithelial cells and macrophages using the QBD method. They investigated the nanocarrier-mucin interaction to determine its mucosal adhesion potential. Tofa-P/TfR NCs exhibited stronger mucin binding due to the presence of TfR content adhered to negatively charged mucin compared to Tofa-PLGA NCs. In vitro cell studies confirmed that this nanocarrier was effectively taken up by colon epithelial cells and macrophages. In the DSS-induced UC mouse model experiment, it demonstrated promising colon inflammation site-targeting potential based on tissue histopathological score, vascular integrity, and antioxidant levels.132 Therefore, targeting TfR through drug delivery systems may be a promising active targeting therapy for IBD.
CD44 is a transmembrane glycoprotein that is highly expressed on epithelial cells and macrophages in inflamed colon tissue, making it an effective target for drug delivery in IBD treatment.197 HA is a natural anionic polysaccharide that plays a critical role in biological functions, such as cell migration and adhesion, through its interaction with CD44.198 Furthermore, HA is used as an active targeting ligand to modify nanocarriers, improving cellular uptake of drugs or nanocarriers via CD44-mediated endocytosis.199,200 Zhang et al. employed CS and HA as carrier materials to produce CD NPs and HCD NPs utilizing ion cross-linking and electrostatic adsorption methods, and then prepared ECHCD MPs by coating NPs with ES100. ECHCD MPs effectively prevented premature release of Dex in the stomach and delivered the drug specifically to the colon. Additionally, ECHCD MPs exhibited stronger mucosal adhesion, rendering them better for targeting the colon in colitis treatment. HCD NPs released from ECHCD MPs could be internalized by macrophages through CD44 receptor-mediated endocytosis, regulating M1 to M2 macrophage polarization.201 Therefore, HA NPs have the potential to be an effective strategy for targeting the inflamed area in IBD treatment. Besides HA, CD44 is also a specific receptor for chondroitin sulfate (Chs), which can be expressed in blood cells, epithelial cells, endothelial cells, and cartilage (Fig. 12B).133 Xie et al. incorporated CUR into resynthesized chondroitin sulfate nanoparticles based on N-2-hydroxypropyltrimethylammonium chloride (HACC) (CUR@Chs-PNC NPs) and aimed to enhance the oral absorption of CUR through CD44-mediated endocytosis after oral administration. CUR@Chs-PNC NPs exhibited good sustained release in simulated gastrointestinal environments and demonstrated adequate drug release in simulated colon environments. Chs functionalization endowed the NPs with a significant CD44-targeting ability, thereby increasing the concentration of CUR in the plasma of SD rats. The in vivo biodistribution of CUR@Chs-PNC NPs showed that the NPs prolonged the intestinal retention time, thereby promoting the interaction between CUR and GM. Importantly, in a mouse model of SSD-induced colitis, CUR@Chs-PNC NPs reduced the disease activity index, improved oxidative stress and inflammation, promoted the production of short-chain fatty acids (SCFAs), regulated immune cells, and maintained intestinal microbiota homeostasis (Fig. 12C).134
CD98 is a type II transmembrane protein heterodimer that regulates integrin signaling through its interaction with β1 integrin.202,203 It is in the cytoplasmic domain and has been found to be significantly upregulated in intestinal epithelial cells during active IBD.204 Overexpression of CD98 increases intestinal barrier permeability, pro-inflammatory cytokine production, and intestinal epithelial layer ulceration.205 Small interfering RNA (siRNA) has been shown to alleviate CD98-mediated epithelial barrier dysfunction, and the natural product berberine (BBR) can improve dysbiosis.206,207 Yang et al. have developed a pH-sensitive “nano-bomb” (HA-CG-CO2@NPs) that can deliver siCD98 and is HA-modified. Under lysosomal pH conditions, the CS-guanidine-CO2 (CG-CO2) in the HA-CG-CO2@NPs carrier can release CO2 and produce a “nano-bomb effect” to trigger siCD98 lysosomal escape. HA-CG-CO2@NPs can effectively bind to colon epithelial cells and macrophages through CD44-mediated endocytosis. By downregulating CD98 expression, HAsiCD98@NPs can alleviate inflammation. Oral administration of HA-siCD98@NPs has been shown to alleviate UC symptoms (Fig. 12D).135
F4/80 serves as a macrophage-specific surface antigen utilized as a marker in diverse organs and tissues.208 There are indications that F4/80 significantly influences the antigen-specific regulation of regulatory T cell (Treg) generation, playing a pivotal role in inducing peripheral tolerance.209 Importantly, monoclonal antibodies or antibody fragments directed against F4/80 find widespread application in macrophage-targeted delivery systems. For instance, Hamed et al. showcased the potential of F4/80 antibody Fab' fragments in targeted therapy for IBD. They incorporated TNF-α-siRNA into nanoparticles composed of a poly(lactic-co-glycolic acid)–polyethylene glycol (PLA–PEG) copolymer. Covalently grafting the Fab' of F4/80 onto the nanoparticle surface, they ultimately encapsulated the particles with a chitosan/alginate shell. Research results suggest that Fab'-loaded nanoparticles significantly improve uptake kinetics and targeting efficiency upon macrophage entry. Moreover, in vivo assessments of inflammatory markers reveal that Fab'-covered nanoparticles effectively alleviate symptoms of mouse colitis compared to those without Fab' coverage. Hence, the development of a nanodrug delivery system centered around F4/80 emerges as a promising and efficient strategy for treating IBD.136
MAdCAM-1 stands out as a pivotal adhesion molecule, emblematic of IBD inflammation and extensively investigated as a therapeutic target for both UC and CD.210 In the initial phases of IBD, MAdCAM-1 predominantly expresses on the high endothelial venules of intestinal lymphoid tissues, forming bonds with the α4β7 integrin found on homing T cells in the gut. This interaction subsequently fuels chronic inflammation within the corresponding intestinal regions.211 Notably, MAdCAM-1 demonstrates heightened expression in non-responsive patients, whereas its expression diminishes post anti-TNF-α treatment. Thus, MAdCAM-1 is intricately linked to the onset and progression of IBD, contributing to treatment responsiveness. For instance, Marta et al. conjugated anti-MAdCAM-1 antibodies with manganese oxide nanoparticles (MnO NPs) coated with poly (isobutylene-alt-maleic anhydride) (PMA). This led to the development of MnO-MdC. Research results highlight the excellent stability and biocompatibility of MnO-MdC. Furthermore, upon administration in a murine model of IBD, MnO-MdC selectively accumulates at the inflamed sites in the intestines. This orally administered targeted delivery system precisely mirrors the expression pattern of MAdCAM-1 and effectively identifies lesions in IBD.137 It is noteworthy that, despite the recognition of the α4β7 integrin as a crucial protein facilitating leukocyte homing to the intestine during intestinal inflammation, only 70% of intestinal T cells and 35% of CD4+ T cells express this integrin in healthy individuals. This observation implies that exclusively targeting the α4β7 integrin lacks specificity.212 Moreover, the functionality of integrins is contingent upon their conformational state, which undergoes changes in response to stimulation, substantially enhancing their affinity for ligands. The α4β7 integrin has the capability to bind to VCAM-1 and MAdCAM-1, facilitating homing to surrounding tissues but not concurrently to intestinal tissues. The affinity of the integrin α4β7 for VCAM-1 or MAdCAM-1 is contingent on specific stimuli, signal transduction, and conformational alterations.213 For example, Niels et al. engineered a fusion of the integrin binding domains D1 and D2 from the natural ligand MAdCAM-1 onto IgG-Fc, specifically for the α4β7 integrin. Subsequently, they selectively silenced genes in T cells in an inflammation-dependent manner. This approach resulted in improved therapeutic outcomes in IL-10KO mice with active colitis, achieved by employing siIFN-γ to knock out the targeted gene. The research findings underscore the feasibility of conformation-sensitive targets, especially in the context of IBD. The correlation between conformational states and protein functionality is a recurring phenomenon, and the pursuit of proteins based on specific conformations introduces innovative possibilities for lipid nanoparticles targeting strategies.138
4 Conclusion and prospect
IBD is a chronic inflammation of the gastrointestinal tract that affects people worldwide and is becoming more prevalent. Recent advances in nanomedicine have shown significant benefits for IBD treatment, such as enhanced drug bioavailability and reduced systemic side effects compared to conventional tablets. Combining different artificial or biological materials to create nanoparticles allows for multiple advantageous properties. Among these, orally-administered nanomedicine systems that specifically target inflammation in the intestinal tract are currently a highly researched area. The size, shape, and surface chemistry of nanoparticles affect their ability to perform and target the inflamed site, which ultimately influences their overall effectiveness in targeting IBD.The first part of this review provides a comprehensive overview of the pathophysiological environment of IBD, including the pH of the gastrointestinal tract, various enzymes in the pathway, transit time, intestinal mucus, intestinal epithelium, intestinal immune cells, and intestinal microbiota. The second part focuses on the latest advances, from 2021 to 2023, in the mechanisms and strategies of orally-administered nanomedicine systems targeting inflammation in the intestinal tract. In summary, nanomedicine systems targeting inflammation in the intestinal tract, when administered orally, hold great potential to offer a new treatment option that could overcome current limitations in IBD treatment and open a new era of IBD therapy.
In most studies, researchers typically use a single inducing agent to create an IBD model. However, the pathology of IBD is incredibly complex, and neither DSS- nor TNBS-induced pathological models can represent the full spectrum of IBD's pathology. Moreover, the IBD pathology observed in mice or rats does not fully replicate the human IBD pathology. For instance, there are significant variations between the intestinal microbiome, pH value, transit time, and mucosal barrier of mice and humans. Thus, it is essential to consider clinical applications involving the participation of human pathological and physiological tissues and volunteers to address these gaps. Additionally, it is crucial to evaluate more animal species to screen for animals with IBD pathology that closely resembles that of humans.
In addition to the concerns, drug safety is of utmost importance. Although most attention is paid to the adverse reactions of nanocarriers, there is still no practical data or conclusion. Generally, nanotoxicology studies are necessary and crucial in animal models and the human gastrointestinal tract. Furthermore, considering commercial aspects, both the manufacturing cost and the safety of nanocarrier products for the human body need to be carefully evaluated. As a result, more and more researchers are turning towards studying and developing nanocarriers derived from natural animal and plant sources, which could potentially pave the way for the future development of nano delivery systems for oral targeting of intestinal inflammatory sites.
Conflicts of interest
There is no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation (32172208); Liaoning BaiQianWan Talents Program, and Central Funds Guiding the Local Science and Technology Development (2020JH6/10500002).References
- S. Massironi, C. Vigano, A. Palermo, L. Pirola, G. Mulinacci, M. Allocca, L. Peyrin-Biroulet and S. Danese, Lancet Gastroenterol. Hepatol., 2023, 8, 579–590 CrossRef PubMed.
- S. C. Ng, H. Y. Shi, N. Hamidi, F. E. Underwood, W. Tang, E. I. Benchimol, R. Panaccione, S. Ghosh, J. C. Y. Wu, F. K. L. Chan, J. J. Y. Sung and G. G. Kaplan, Lancet, 2017, 390, 2769–2778 CrossRef.
- S. C. Ng, J. W. Y. Mak, P. Pal and R. Banerjee, Lancet Gastroenterol. Hepatol., 2020, 5, 1089–1100 CrossRef PubMed.
- P. Sudhakar, J. Wellens, B. Verstockt, M. Ferrante, J. Sabino and S. Vermeire, Gut, 2023, 72, 192–204 CrossRef CAS PubMed.
- A. J. Watson and A. R. Hart, Gastroenterology, 2011, 141, 768–770 CrossRef.
- C. G. Gonzalez, R. H. Mills, Q. Zhu, C. Sauceda, R. Knight, P. S. Dulai and D. J. Gonzalez, Microbiome, 2022, 10, 133 CrossRef CAS.
- T. E. Adolph, M. Meyer, J. Schwarzler, L. Mayr, F. Grabherr and H. Tilg, Nat. Rev. Gastroenterol. Hepatol., 2022, 19, 753–767 CrossRef.
- A. Vich Vila, S. Hu, S. Andreu-Sánchez, V. Collij, B. H. Jansen, H. E. Augustijn, L. A. Bolte, R. Ruigrok, G. Abu-Ali, C. Giallourakis, J. Schneider, J. Parkinson, A. Al-Garawi, A. Zhernakova, R. Gacesa, J. Fu and R. K. Weersma, Gut, 2023, 72, 1472–1485 CrossRef.
- S. D'Alessio, F. Ungaro, D. Noviello, S. Lovisa, L. Peyrin-Biroulet and S. Danese, Nat. Rev. Gastroenterol. Hepatol., 2022, 19, 169–184 CrossRef PubMed.
- H. Luo, G. Cao, C. Luo, D. Tan, C. T. Vong, Y. Xu, S. Wang, H. Lu, Y. Wang and W. Jing, Pharmacol. Res., 2022, 178, 106146 CrossRef CAS PubMed.
- S. Singh, Nat. Rev. Gastroenterol. Hepatol., 2023, 20, 411–412 CrossRef.
- W. Wang, Y. Gao, M. Zhang, Y. Li and B. Z. Tang, ACS Nano, 2023, 17, 7394–7405 CrossRef CAS.
- M. Zu, Y. Ma, B. Cannup, D. Xie, Y. Jung, J. Zhang, C. Yang, F. Gao, D. Merlin and B. Xiao, Adv. Drug Delivery Rev., 2021, 176, 113887 CrossRef CAS PubMed.
- W. Sun, Y. Chen, L. Wang, Z. Wang, S. Liu, M. Zhang, Y. Liu, Q. Li and H. Zhang, Biomaterials, 2023, 295, 122039 CrossRef CAS PubMed.
- P. Holko, P. Kawalec and M. Mossakowska, Eur. J. Gastroenterol. Hepatol., 2018, 30, 174–180 CrossRef PubMed.
- D. R. Owens, M. K. Jones, T. M. Hayes, L. G. Heding, K. G. Alberti, P. D. Home and J. M. Burrin, Lancet, 1981, 2, 118–122 CrossRef CAS.
- Y. Cong, S. Wu, J. Han, J. Chen, H. Liu, Q. Sun, Y. Wu and Y. Fang, J. Pharm. Biomed. Anal., 2016, 129, 405–409 CrossRef CAS PubMed.
- B. J. Molloy, A. King, L. A. Gethings, R. S. Plumb, R. J. Mortishire-Smith and I. D. Wilson, Xenobiotica, 2023, 1–30, DOI:10.1080/00498254.2023.2179952.
- C. Mowat, A. Cole, A. Windsor, T. Ahmad, I. Arnott, R. Driscoll, S. Mitton, T. Orchard, M. Rutter, L. Younge, C. Lees, G. T. Ho, J. Satsangi and S. Bloom, Gut, 2011, 60, 571–607 CrossRef PubMed.
- T. J. Purohit, S. M. Hanning and Z. Wu, Pharm. Dev. Technol., 2018, 23, 942–952 CrossRef CAS PubMed.
- R. D. Horniblow, G. O. Latunde-Dada, S. E. Harding, M. Schneider, F. M. Almutairi, M. Sahni, A. Bhatti, C. Ludwig, I. T. Norton, T. H. Iqbal and C. Tselepis, Mol. Nutr. Food Res., 2016, 60, 2098–2108 CrossRef CAS PubMed.
- K. Butler, J. Yi, M. Wasson, J. Klauschie, D. Ryan, J. Hentz, J. Cornella, P. Magtibay and R. Kho, Am. J. Obstet. Gynecol., 2017, 216, 491 e491–491 e496 CrossRef.
- S. E. Berends, A. S. Strik, M. Lowenberg, G. R. D'Haens and R. A. A. Mathot, Clin. Pharmacokinet., 2019, 58, 15–37 CrossRef CAS.
- S. Hua, Front. Pharmacol., 2019, 10, 1196 CrossRef CAS.
- Q. Huang, Y. Yang, Y. Zhu, Q. Chen, T. Zhao, Z. Xiao, M. Wang, X. Song, Y. Jiang, Y. Yang, J. Zhang, Y. Xiao, Y. Nan, W. Wu and K. Ai, Small, 2023, e2207350, DOI:10.1002/smll.202207350.
- J. Lou, H. Duan, Q. Qin, Z. Teng, F. Gan, X. Zhou and X. Zhou, Pharmaceutics, 2023, 15, 484 CrossRef CAS PubMed.
- H. Peng, J. Wang, J. Chen, Y. Peng, X. Wang, Y. Chen, D. L. Kaplan and Q. Wang, Expert Opin. Drug Delivery, 2023, 20, 1349–1369 CrossRef CAS.
- Z. Liu, H. Liu, J. Cheng, H. Wang, Y. Yang, J. Ye and Y. Liu, Chin. Chem. Lett., 2024, 35, 109074 CrossRef.
- S. Zhang, R. Langer and G. Traverso, Nano Today, 2017, 16, 82–96 CrossRef CAS PubMed.
- C. Yang and D. Merlin, Int. J. Nanomed., 2019, 14, 8875–8889 CrossRef CAS.
- D.-f Li, M.-f Yang, H.-m Xu, M.-z Zhu, Y. Zhang, C.-m Tian, Y.-q Nie, J.-y Wang, Y.-j Liang, J. Yao and L.-S. Wang, J. Mater. Chem. B, 2022, 10, 5853–5872 RSC.
- W. Liu, Z. Dong, K. Liu, Y. Lu, W. Wu, J. Qi and Z. Chen, Int. J. Pharm., 2021, 600, 120461 CrossRef CAS.
- H. S. Sardou, P. R. Vosough, M. Abbaspour, A. Akhgari, T. Sathyapalan and A. Sahebkar, Inflammopharmacology, 2023, 31, 1095–1105 CrossRef CAS PubMed.
- H. C. Vadlamudi, Y. P. Raju, B. R. Yasmeen and J. Vulava, Curr. Drug Delivery, 2012, 9, 556–565 CrossRef CAS.
- Y. Sun, Y. Koyama and S. Shimada, Biochem. Biophys. Res. Commun., 2022, 620, 129–134 CrossRef CAS.
- S. G. Nugent, D. Kumar, D. S. Rampton and D. F. Evans, Gut, 2001, 48, 571–577 CrossRef CAS PubMed.
- R. Caprilli, P. Vernia, O. Colaneri and A. Torsoli, Gut, 1976, 17, 763–769 CrossRef CAS PubMed.
- A. Bak, M. Ashford and D. J. Brayden, Adv. Drug Delivery Rev., 2018, 136–137, 2–27 CrossRef CAS.
- M. Goldberg and I. Gomez-Orellana, Nat. Rev. Drug Discovery, 2003, 2, 289–295 CrossRef CAS PubMed.
- T. Kou, M. Faisal, J. Song and A. Blennow, Crit. Rev. Food Sci. Nutr., 2022, 1–15, DOI:10.1080/10408398.2022.2104209.
- R. C. Cheung, T. B. Ng, J. H. Wong and W. Y. Chan, Mar. Drugs, 2015, 13, 5156–5186 CrossRef CAS.
- N. Prochazkova, G. Falony, L. O. Dragsted, T. R. Licht, J. Raes and H. M. Roager, Gut, 2023, 72, 180–191 CrossRef CAS PubMed.
- H. Pan, X. Chen, P. Wang, J. Peng, J. Li and K. Ding, Food Funct., 2023, 14, 1627–1635 RSC.
- Q. Zhao and C. L. Maynard, Gut Microbes, 2022, 14, 2041342 CrossRef.
- A. Zhao, H. Qin, M. Sun, M. Tang, J. Mei, K. Ma and X. Fu, Sci. Adv., 2021, 7, eabb2213 CrossRef CAS PubMed.
- D. Xi, L. Hofmann, T. Alter, R. Einspanier, S. Bereswill, M. M. Heimesaat, G. Golz and S. Sharbati, Gut Pathog., 2021, 13, 42 CrossRef CAS PubMed.
- A. Schutte, A. Ermund, C. Becker-Pauly, M. E. Johansson, A. M. Rodriguez-Pineiro, F. Backhed, S. Muller, D. Lottaz, J. S. Bond and G. C. Hansson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12396–12401 CrossRef CAS.
- T. T. Roh, Y. Chen, S. Rudolph, M. Gee and D. L. Kaplan, Trends Biotechnol., 2021, 39, 274–285 CrossRef CAS.
- X. Li, H. Sun, B. Li, X. Zhang, J. Cui, J. Yun, Y. Yang, L. Zhang, Q. Meng, S. Wu, J. Duan, H. Yang, J. Wu, Z. Sun, Y. Zou and R. Chen, Adv. Sci., 2019, 6, 1900972 CrossRef CAS PubMed.
- V. Strugala, P. W. Dettmar and J. P. Pearson, Int. J. Clin. Pract., 2008, 62, 762–769 CrossRef CAS PubMed.
- X. Liu, F. Zhao, H. Liu, Y. Xie, D. Zhao and C. Li, Sci. Rep., 2021, 11, 9073 CrossRef CAS.
- T. Zuo, X. J. Lu, Y. Zhang, C. P. Cheung, S. Lam, F. Zhang, W. Tang, J. Y. L. Ching, R. Zhao, P. K. S. Chan, J. J. Y. Sung, J. Yu, F. K. L. Chan, Q. Cao, J. Q. Sheng and S. C. Ng, Gut, 2019, 68, 1169–1179 CrossRef CAS PubMed.
- G. Collin, J. P. Foy, N. Aznar, N. Rama, A. Wierinckx, P. Saintigny, A. Puisieux and S. Ansieau, Cancers, 2023, 15, 973 CrossRef CAS PubMed.
- J. Hindson, Nat. Rev. Gastroenterol. Hepatol., 2023, 20, 202 CrossRef PubMed.
- F. M. Yavitt, B. E. Kirkpatrick, M. R. Blatchley, K. F. Speckl, E. Mohagheghian, R. Moldovan, N. Wang, P. J. Dempsey and K. S. Anseth, Sci. Adv., 2023, 9, eadd5668 CrossRef.
- U. V. Chembazhi, W. S. Tung, H. Hwang, Y. Wang, A. Lalwani, K. L. Nguyen, S. Bangru, D. Yee, K. Chin, J. Yang, A. Kalsotra and W. Mei, Nucleic Acids Res., 2023, 51, 2397–2414 CrossRef CAS.
- X. Song, X. He, X. Li and Y. Qian, Cell. Mol. Immunol., 2016, 13, 418–431 CrossRef CAS PubMed.
- A. Górski, E. Jończyk-Matysiak, M. Łusiak-Szelachowska, R. Międzybrodzki, B. Weber-Dąbrowska and J. Borysowski, Cell. Mol. Life Sci., 2018, 75, 589–595 CrossRef PubMed.
- B. Ardesjo, G. M. Portela-Gomes, F. Rorsman, E. Gerdin, L. Loof, L. Grimelius, O. Kampe and O. Ekwall, Inflammatory Bowel Dis., 2008, 14, 652–661 CrossRef.
- A. J. Ouellette, Curr. Opin. Gastroenterol., 2010, 26, 547–553 CrossRef PubMed.
- S. L. Wang, B. Z. Shao, S. B. Zhao, J. Fang, L. Gu, C. Y. Miao, Z. S. Li and Y. Bai, Front. Immunol., 2018, 9, 693 CrossRef.
- N. Gicheva, M. S. Macauley, B. M. Arlian, J. C. Paulson and N. Kawasaki, Biochem. Biophys. Res. Commun., 2016, 479, 1–4 CrossRef CAS PubMed.
- R. Kiesslich, M. Goetz, M. Vieth, P. R. Galle and M. F. Neurath, Nat. Clin. Pract. Oncol., 2007, 4, 480–490 CrossRef PubMed.
- W. Xu, L. He, Q. Xia, C. Jia, L. Geng, M. Yang, Z. Xu, P. Chen, Y. Cheng, J. Zhao, H. Wang, H. Chen, Y. Zhang, S. Gong and R. Liu, J. Mater. Chem. B, 2018, 6, 809–815 RSC.
- X. Wang, S. Lin, L. Wang, Z. Cao, M. Zhang, Y. Zhang, R. Liu and J. Liu, Sci. Adv., 2023, 9, eade5079 CAS.
- Z. Cao, R. Liu, C. Wang, S. Lin, L. Wang and Y. Pang, ACS Nano, 2023, 17, 2279–2293 CrossRef CAS.
- X. Zong, W. Hu, D. Song, Z. Li, H. Du, Z. Lu and Y. Wang, Biochem. Pharmacol., 2016, 104, 74–82 CrossRef CAS PubMed.
- F. Weiss, C. Czichos, L. Knobe, L. Voges, C. Bojarski, G. Michel, M. Fromm and S. M. Krug, Cells, 2022, 11, 1541 CrossRef CAS PubMed.
- J. Park, J. Choi, J. E. Lee, H. Choi and H. J. Im, Small Methods, 2022, 6, e2201091 CrossRef PubMed.
- R. Del Sordo, V. Lougaris, G. Bassotti, A. Armuzzi and V. Villanacci, Clin. Immunol., 2022, 234, 108916 CrossRef PubMed.
- A. Saez, B. Herrero-Fernandez, R. Gomez-Bris, H. Sanchez-Martinez and J. M. Gonzalez-Granado, Int. J. Mol. Sci., 2023, 24, 1526 CrossRef CAS PubMed.
- E. Vivier, S. A. van de Pavert, M. D. Cooper and G. T. Belz, Nat. Immunol., 2016, 17, 790–794 CrossRef CAS PubMed.
- T. Sato, T. Nakai, N. Tamura, S. Okamoto, K. Matsuoka, A. Sakuraba, T. Fukushima, T. Uede and T. Hibi, Gut, 2005, 54, 1254–1262 CrossRef CAS PubMed.
- V. E. Diaz-Ochoa, D. Lam, C. S. Lee, S. Klaus, J. Behnsen, J. Z. Liu, N. Chim, S. P. Nuccio, S. G. Rathi, J. R. Mastroianni, R. A. Edwards, C. M. Jacobo, M. Cerasi, A. Battistoni, A. J. Ouellette, C. W. Goulding, W. J. Chazin, E. P. Skaar and M. Raffatellu, Cell Host Microbe, 2016, 19, 814–825 CrossRef CAS PubMed.
- M. Z. Cader and A. Kaser, Gut, 2013, 62, 1653–1664 CrossRef CAS PubMed.
- Y. Zhu, S. Yang, N. Zhao, C. Liu, F. Zhang, Y. Guo and H. Liu, Biomed. Pharmacother., 2021, 138, 111427 CrossRef CAS PubMed.
- L. Calvo-Barreiro, L. Zhang, S. A. Abdel-Rahman, S. P. Naik and M. Gabr, Int. J. Mol. Sci., 2023, 24, 1806 CrossRef CAS PubMed.
- H. Y. Fung, M. Teryek, A. D. Lemenze and T. Bergsbaken, Sci. Immunol., 2022, 7, eabl9925 CrossRef CAS PubMed.
- T. Ogino and K. Takeda, Front. Immunol., 2023, 14, 1138971 CrossRef CAS PubMed.
- A. M. Noronha, Y. Liang, J. T. Hetzel, H. Hasturk, A. Kantarci, A. Stucchi, Y. Zhang, B. S. Nikolajczyk, F. A. Farraye and L. M. Ganley-Leal, J. Leukocyte Biol., 2009, 86, 1007–1016 CrossRef CAS PubMed.
- S. K. Lathrop, S. M. Bloom, S. M. Rao, K. Nutsch, C. W. Lio, N. Santacruz, D. A. Peterson, T. S. Stappenbeck and C. S. Hsieh, Nature, 2011, 478, 250–254 CrossRef CAS PubMed.
- D. Rios Garza, D. Gonze, H. Zafeiropoulos, B. Liu and K. Faust, Cell Syst., 2023, 14, 109–121 CrossRef CAS PubMed.
- P. He, L. Yu, F. Tian, H. Zhang, W. Chen and Q. Zhai, Adv. Nutr., 2022, 13, 1628–1651 CrossRef PubMed.
- Y. Wang, H. Zhu, X. Wang, Y. Yu and J. Xie, Foods, 2021, 10, 1288 CrossRef CAS PubMed.
- X. Wang, T. Tsai, F. Deng, X. Wei, J. Chai, J. Knapp, J. Apple, C. V. Maxwell, J. A. Lee, Y. Li and J. Zhao, Microbiome, 2019, 7, 109 CrossRef.
- M. Zhang, B. Liu, Y. Zhang, H. Wei, Y. Lei and L. Zhao, J. Clin. Microbiol., 2007, 45, 496–500 CrossRef CAS PubMed.
- R. Kotlowski, C. N. Bernstein, S. Sepehri and D. O. Krause, Gut, 2007, 56, 669–675 CrossRef CAS.
- K. Ray, Nat. Rev. Gastroenterol. Hepatol., 2016, 13, 377 CrossRef CAS PubMed.
- S. Sanna, N. R. van Zuydam, A. Mahajan, A. Kurilshikov, A. Vich Vila, U. Vosa, Z. Mujagic, A. A. M. Masclee, D. Jonkers, M. Oosting, L. A. B. Joosten, M. G. Netea, L. Franke, A. Zhernakova, J. Fu, C. Wijmenga and M. I. McCarthy, Nat. Genet., 2019, 51, 600–605 CrossRef CAS PubMed.
- S. D. de Ferranti, J. Steinberger, R. Ameduri, A. Baker, H. Gooding, A. S. Kelly, M. Mietus-Snyder, M. M. Mitsnefes, A. L. Peterson, J. St-Pierre, E. M. Urbina, J. P. Zachariah and A. N. Zaidi, Circulation, 2019, 139, e603–e634 CrossRef.
- E. M. Shevach, Immunity, 2009, 30, 636–645 CrossRef CAS.
- A. Zarrinpar, A. Chaix, Z. Z. Xu, M. W. Chang, C. A. Marotz, A. Saghatelian, R. Knight and S. Panda, Nat. Commun., 2018, 9, 2872 CrossRef.
- M. O. Mohammed, K. S. Hussain and N. Q. Haj, Molecules, 2017, 22, 1629 CrossRef PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- Q. Jin, Y. Deng, X. Chen and J. Ji, ACS Nano, 2019, 13, 954–977 CAS.
- B. Ginovska-Pangovska, A. Dutta, M. L. Reback, J. C. Linehan and W. J. Shaw, Acc. Chem. Res., 2014, 47, 2621–2630 CrossRef CAS.
- I. Saleem, A. G. A. Coombes and M. A. Chambers, Pharmaceutics, 2019, 11, 270 CrossRef CAS PubMed.
- Q. Wu, L. Wang, H. Yu, J. Wang and Z. Chen, Chem. Rev., 2011, 111, 7855–7875 CrossRef CAS PubMed.
- D. Han and A. J. Steckl, ACS Appl. Mater. Interfaces, 2017, 9, 42653–42660 CrossRef CAS PubMed.
- B. Xu, W. Zhang, Y. Chen, Y. Xu, B. Wang and L. Zong, Int. J. Biol. Macromol., 2018, 113, 534–542 CrossRef CAS.
- G. Wang, Y. Yang, D. Yi, L. Yuan, P. H. Yin, X. Ke, W. Jun-Jie and M. F. Tao, J. Liposome Res., 2022, 32, 250–264 CrossRef CAS PubMed.
- G. Zhang, W. Han, P. Zhao, Z. Wang, M. Li, X. Sui, Y. Liu, B. Tian, Z. He and Q. Fu, Nanoscale, 2023, 15, 1937–1946 RSC.
- J. Feng, Y. Wang, Y. Lv, S. Fang, M. Ren, M. Yao, M. Lan, Y. Zhao and F. Gao, Mol. Pharmaceutics, 2022, 19, 4157–4170 CrossRef CAS PubMed.
- Y. Lv, M. Ren, M. Yao, J. Zou, S. Fang, Y. Wang, M. Lan, Y. Zhao and F. Gao, Int. J. Pharm., 2023, 635, 122741 CrossRef CAS PubMed.
- J. Lee, A. Saparbayeva, S. P. Hlaing, D. Kwak, H. Kim, J. Kim, E. H. Lee and J. W. Yoo, Pharmaceutics, 2022, 14, 2811 CrossRef CAS.
- C. Zhao, J. Yang, M. Chen, W. Chen, X. Yang, H. Ye, L. Wang, Y. Wang, J. Shi, F. Yue and X. Ma, ACS Nano, 2023, 17, 811–824 CrossRef CAS.
- H. Y. Kim, J. H. Cheon, S. H. Lee, J. Y. Min, S. Y. Back, J. G. Song, D. H. Kim, S. J. Lim and H. K. Han, J. Nanobiotechnol., 2020, 18, 17 CrossRef CAS PubMed.
- X. Yan, L. Meng, X. Zhang, Z. Deng, B. Gao, Y. Zhang, M. Yang, Y. Ma, Y. Zhang, K. Tu, M. Zhang and Q. Xu, Mol. Ther., 2023, 31, 1383–1401 CrossRef CAS PubMed.
- X. Zhang, Z. Yuan, J. Wu, Y. He, G. Lu, D. Zhang, Y. Zhao, R. Wu, Y. Lv, K. Cai and S. He, ACS Nano, 2023, 17, 21116–21133 CrossRef.
- W. Fan, S. Zhang, Y. Wu, T. Lu, J. Liu, X. Cao, S. Liu, L. Yan, X. Shi, G. Liu, C. Huang and S. Song, ACS Appl. Mater. Interfaces, 2021, 13, 40249–40266 CrossRef CAS.
- C. Tan, H. Fan, J. Ding, C. Han, Y. Guan, F. Zhu, H. Wu, Y. Liu, W. Zhang, X. Hou, S. Tan and Q. Tang, Mater. Today Bio, 2022, 14, 100246 CrossRef CAS.
- F. Soltani, H. Kamali, A. Akhgari, M. Ghasemzadeh Rahbardar, H. Afrasiabi Garekani, A. Nokhodchi and F. Sadeghi, Pharmaceutics, 2022, 15, 69 CrossRef.
- Y. Ren, C. Qi, S. Ruan, G. Cao, Z. Ma and X. Zhang, Pharmaceutics, 2023, 15, 821 CrossRef CAS PubMed.
- S. H. Lee, J. G. Song and H. K. Han, Acta Pharm. Sin. B, 2022, 12, 4249–4261 CrossRef CAS PubMed.
- N. G. Kotla, R. Singh, B. V. Baby, S. Rasala, J. Rasool, S. O. Hynes, D. Martin, L. J. Egan, P. K. Vemula, V. R. Jala, Y. Rochev and A. Pandit, Biomaterials, 2022, 281, 121364 CrossRef CAS.
- C. Xu, S. Chen, C. Chen, Y. Ming, J. Du, J. Mu, F. Luo, D. Huang, N. Wang, Z. Lin and Z. Weng, Int. J. Pharm., 2022, 623, 121884 CrossRef CAS.
- S. Li, M. Jin, Y. Wu, S. Jung, D. Li, N. He and M. S. Lee, Drug Delivery, 2021, 28, 1120–1131 CrossRef CAS.
- Y. Xie, Z. Jin, D. Ma, T. H. Yin and K. Zhao, Bioeng. Transl. Med., 2023, 8, e10510 CrossRef CAS.
- D. Xie, X. Zhou, B. Xiao, L. Duan and Z. Zhu, Biomolecules, 2022, 12, 1263 CrossRef CAS PubMed.
- L. J. Mohan, J. S. Daly, B. M. Ryan and Z. Ramtoola, Eur. J. Pharm. Sci., 2023, 183, 106379 CrossRef CAS PubMed.
- E. Wang, R. Han, M. Wu, Y. He, Y. Cheng, J. Lu and Y. Zheng, Chin. Chem. Lett., 2023, 35 DOI:10.1016/j.cclet.2023.108361.
- S. Gimondi, J. Vieira de Castro, R. L. Reis, H. Ferreira and N. M. Neves, Colloids Surf., B, 2023, 225, 113245 CrossRef CAS PubMed.
- H. Zhao, Y. Du, L. Liu, Y. Du, K. Cui, P. Yu, L. Li, Y. Zhu, W. Jiang, Z. Li, H. Tang and W. Ma, J. Mater. Chem. B, 2022, 10, 4002–4011 RSC.
- J. Liu, Y. Wang, W. J. Heelan, Y. Chen, Z. Li and Q. Hu, Sci. Adv., 2022, 8, eabp8798 CrossRef CAS.
- P. Peng, T. Feng, X. Yang, C. Nie, L. Yu, R. Ding, Q. Zhou, X. Jiang and P. Li, ACS Nano, 2023, 17, 14718–14730 CrossRef CAS.
- J. Zhou, M. Li, Q. Chen, X. Li, L. Chen, Z. Dong, W. Zhu, Y. Yang, Z. Liu and Q. Chen, Nat. Commun., 2022, 13, 3432 CrossRef CAS.
- F. Cao, L. Jin, Y. Gao, Y. Ding, H. Wen, Z. Qian, C. Zhang, L. Hong, H. Yang, J. Zhang, Z. Tong, W. Wang, X. Chen and Z. Mao, Nat. Nanotechnol., 2023, 18, 617–627 CrossRef CAS.
- M. Zeeshan, H. Ali, Q. U. Ain, M. Mukhtar, R. Gul, A. Sarwar and S. Khan, Mater. Sci. Eng., C, 2021, 126, 112183 CrossRef CAS.
- M. Zu, D. Xie, B. S. B. Canup, N. Chen, Y. Wang, R. Sun, Z. Zhang, Y. Fu, F. Dai and B. Xiao, Biomaterials, 2021, 279, 121178 CrossRef CAS PubMed.
- H. Feng, X. Yang, L. Zhang, Q. Liu, Y. Feng, D. Wu, Y. Liu and J. Yang, Polymers, 2021, 13, 2208 CrossRef CAS.
- H. Feng, Y. Feng, L. Lin, D. Wu, Q. Liu, H. Li, X. Zhang, S. Li, F. Tang, Z. Liu and L. Zhang, Int. J. Mol. Sci., 2022, 23, 4239 CrossRef CAS.
- M. Zeeshan, Q. U. Ain, A. Sunny, F. Raza, M. Mohsin, S. Khan, B. Weigmann and H. Ali, Biomater. Sci., 2023, 11, 1373–1397 RSC.
- L. Zhao, M. Liu, J. Wang and G. Zhai, Carbohydr. Polym., 2015, 133, 391–399 CrossRef CAS PubMed.
- Y. Xie, W. Xu, Z. Jin and K. Zhao, Mater. Today Bio, 2023, 20, 100617 CrossRef CAS PubMed.
- M. Yang, C. Yang, Y. Zhang, X. Yan, Y. Ma, Y. Zhang, Y. Cao, Q. Xu, K. Tu and M. Zhang, Biomater. Sci., 2022, 10, 1053–1067 RSC.
- H. Laroui, E. Viennois, B. Xiao, B. S. Canup, D. Geem, T. L. Denning and D. Merlin, J. Controlled Release, 2014, 186, 41–53 CrossRef CAS PubMed.
- M. Truffi, M. Colombo, J. Peñaranda-Avila, L. Sorrentino, F. Colombo, M. Monieri, V. Collico, P. Zerbi, E. Longhi, R. Allevi, D. Prosperi and F. Corsi, Nanomedicine, 2017, 12, 1547–1560 CrossRef CAS.
- N. Dammes, M. Goldsmith, S. Ramishetti, J. L. J. Dearling, N. Veiga, A. B. Packard and D. Peer, Nat. Nanotechnol., 2021, 16, 1030–1038 CrossRef CAS.
- B. Yang, Y. Chen and J. Shi, Chem. Rev., 2019, 119, 4881–4985 CrossRef CAS PubMed.
- J. Liu, B. Jia, Z. Li and W. Li, Front. Bioeng. Biotechnol., 2023, 11, 1115603 CrossRef PubMed.
- H. Sies, V. V. Belousov, N. S. Chandel, M. J. Davies, D. P. Jones, G. E. Mann, M. P. Murphy, M. Yamamoto and C. Winterbourn, Nat. Rev. Mol. Cell Biol., 2022, 23, 499–515 CrossRef CAS.
- L. Coppo, P. Mishra, N. Siefert, A. Holmgren, S. Paabo and H. Zeberg, Sci. Adv., 2022, 8, eabm1148 CrossRef CAS PubMed.
- S. Zhang, F. Zheng, K. Liu, S. Liu, T. Xiao, Y. Zhu and L. Xu, Int. J. Mol. Sci., 2022, 23, 12624 CrossRef CAS.
- S. Bai, X. Shao, Y. Tao, S. Wang, H. Han and Q. Li, J. Mater. Chem. B, 2022, 10, 5174–5181 RSC.
- K. P. Lim and L. P. Tan, Acta Biomater., 2009, 5, 84–92 CrossRef CAS.
- J. Youshia and A. Lamprecht, Expert Opin. Drug Delivery, 2016, 13, 281–294 CrossRef CAS.
- C. P. Ivory and K. Chadee, Eur. J. Immunol., 2007, 37, 385–394 CrossRef CAS PubMed.
- Y. Chen, Y. Liu, Q. Dong, C. Xu, S. Deng, Y. Kang, M. Fan and L. Li, Int. J. Biol. Macromol., 2023, 235, 123716 CrossRef CAS PubMed.
- E. Harel, A. Rubinstein, A. Nissan, E. Khazanov, M. Nadler Milbauer, Y. Barenholz and B. Tirosh, PLoS One, 2011, 6, e24202 CrossRef CAS PubMed.
- A. Loktionov, World J. Gastroenterol., 2019, 25, 3503–3526 CrossRef CAS PubMed.
- A. Soumia, M. Adel, S. Amina, B. Bouhadjar, D. Amal, Z. Farouk, B. Abdelkader and S. Mohamed, Int. J. Biol. Macromol., 2020, 145, 466–475 CrossRef CAS PubMed.
- T. T. Jubeh, Y. Barenholz and A. Rubinstein, Pharm. Res., 2004, 21, 447–453 CrossRef CAS PubMed.
- A. Beloqui, R. Coco, M. Alhouayek, M. Solinís, A. Rodríguez-Gascón, G. G. Muccioli and V. Préat, Int. J. Pharm., 2013, 454, 775–783 CrossRef CAS.
- Q. Liu, F. Chen, L. Hou, L. Shen, X. Zhang, D. Wang and L. Huang, ACS Nano, 2018, 12, 7812–7825 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- N. Mandal, M. Bhattacharjee, A. Chattopadhyay and D. Bandyopadhyay, Biosens. Bioelectron., 2019, 124–125, 75–81 CrossRef CAS PubMed.
- K. R. Torgeson, M. W. Clarkson, D. Granata, K. Lindorff-Larsen, R. Page and W. Peti, Sci. Adv., 2022, 8, eabo5546 CrossRef CAS PubMed.
- E. Mahootchi, S. Cannon Homaei, R. Kleppe, I. Winge, T. A. Hegvik, R. Megias-Perez, C. Totland, F. Mogavero, A. Baumann, J. C. Glennon, H. Miletic, P. Kursula and J. Haavik, Sci. Adv., 2020, 6, eabb3713 CrossRef CAS PubMed.
- R. Limage, E. Tako, N. Kolba, Z. Guo, A. Garcia-Rodriguez, C. N. H. Marques and G. J. Mahler, Small, 2020, 16, e2000601 CrossRef PubMed.
- M. Boegh, S. G. Baldursdottir, A. Mullertz and H. M. Nielsen, Eur. J. Pharm. Biopharm., 2014, 87, 227–235 CrossRef CAS PubMed.
- C. Bao, B. Liu, B. Li, J. Chai, L. Zhang, L. Jiao, D. Li, Z. Yu, F. Ren, X. Shi and Y. Li, Nano Lett., 2020, 20, 1352–1361 CrossRef CAS PubMed.
- Y. Huang, B. S. B. Canup, S. Gou, N. Chen, F. Dai, B. Xiao and C. Li, J. Mater. Chem. B, 2021, 9, 1604–1615 RSC.
- X. Y. Bai, Y. Yan, L. Wang, L. G. Zhao and K. Wang, Drug Delivery, 2016, 23, 1926–1932 CAS.
- C. T. Nordgard and K. I. Draget, Adv. Drug Delivery Rev., 2018, 124, 175–183 CrossRef PubMed.
- X. Zhang, H. Cheng, W. Dong, M. Zhang, Q. Liu, X. Wang, J. Guan, H. Wu and S. Mao, J. Controlled Release, 2018, 272, 29–38 CrossRef CAS PubMed.
- C. Zhang, H. Chen, Q. He, Y. Luo, A. He, A. Tao and J. Yan, Cell. Mol. Gastroenterol. Hepatol., 2021, 11, 683–696 CrossRef CAS PubMed.
- J. Chang, R. W. Leong, V. C. Wasinger, M. Ip, M. Yang and T. G. Phan, Gastroenterology, 2017, 153, 723–731 CrossRef PubMed.
- I. Hossen, W. Hua, L. Ting, A. Mehmood, S. Jingyi, X. Duoxia, C. Yanping, W. Hongqing, G. Zhipeng, Z. Kaiqi, Y. Fang and X. Junsong, Crit. Rev. Food Sci. Nutr., 2020, 60, 1321–1345 CrossRef CAS.
- A. Lamprecht, U. Schafer and C. M. Lehr, Pharm. Res., 2001, 18, 788–793 CrossRef CAS.
- M. Colaco, T. Roquito, J. P. Costa, M. T. Cruz and O. Borges, Pharmaceutics, 2023, 15, 623 CrossRef CAS.
- A. Brauer-Nikonow and M. Zimmermann, Cell Host Microbe, 2022, 30, 1063–1066 CrossRef CAS.
- J. Cong, P. Zhou and R. Zhang, Nutrients, 2022, 14, 1977 CrossRef CAS PubMed.
- L. Drago, C. Valentina and P. Fabio, Dig. Liver Dis., 2019, 51, 1209–1213 CrossRef PubMed.
- Y. C. Pai, Y. H. Li, J. R. Turner and L. C. Yu, J. Crohn’s Colitis., 2023, 17, 1471–1488 CrossRef.
- S. Crunkhorn, Nat. Rev. Drug Discovery, 2021, 20, 588 Search PubMed.
- K. Ray, Nat. Rev. Gastroenterol. Hepatol., 2021, 18, 594 Search PubMed.
- B. M. Scott, C. Gutierrez-Vazquez, L. M. Sanmarco, J. A. da Silva Pereira, Z. Li, A. Plasencia, P. Hewson, L. M. Cox, M. O'Brien, S. K. Chen, P. M. Moraes-Vieira, B. S. W. Chang, S. G. Peisajovich and F. J. Quintana, Nat. Med., 2021, 27, 1212–1222 CrossRef CAS PubMed.
- K. L. Glassner, B. P. Abraham and E. M. M. Quigley, J. Allergy Clin. Immunol., 2020, 145, 16–27 CrossRef CAS PubMed.
- R. Tsoi, Z. Dai and L. You, Biotechnol. Adv., 2019, 37, 107372 CrossRef CAS PubMed.
- S. Ramadevi, R. Shelin and M. Shanmugaraja, Curr. Microbiol., 2023, 80, 150 CrossRef CAS PubMed.
- J. P. Lynch, L. Goers and C. F. Lesser, Trends Pharmacol. Sci., 2022, 43, 772–786 CrossRef CAS PubMed.
- Y. Kim, H. J. Jung, Y. Lee, S. Koo, R. Thangam, W. Y. Jang, S. Y. Kim, S. Park, S. Lee, G. Bae, K. D. Patel, Q. Wei, K. B. Lee, R. Paulmurugan, W. K. Jeong, T. Hyeon, D. Kim and H. Kang, J. Am. Chem. Soc., 2022, 144, 5769–5783 CrossRef CAS PubMed.
- X. Song, R. Li, H. Deng, Y. Li, Y. Cui, H. Zhang, W. Dai, B. He, Y. Zheng, X. Wang and Q. Zhang, Biomaterials, 2018, 180, 78–90 CrossRef CAS PubMed.
- T. Tanaka, Y. Zhou, T. Ozawa, R. Okizono, A. Banba, T. Yamamura, E. Oga, A. Muraguchi and H. Sakurai, J. Biol. Chem., 2018, 293, 2288–2301 CrossRef CAS PubMed.
- Y. Cheng, T. R. Hall, X. Xu, I. Yung, D. Souza, J. Zheng, F. Schiele, M. Hoffmann, M. L. Mbow, J. P. Garnett and J. Li, EBioMedicine, 2022, 75, 103758 CrossRef CAS.
- F. Chen, G. Huang and H. Huang, Future Med. Chem., 2020, 12, 161–171 CrossRef CAS PubMed.
- N. Porebska, M. Pozniak, A. Matynia, D. Zukowska, M. Zakrzewska, J. Otlewski and L. Opalinski, Cytokine Growth Factor Rev., 2021, 60, 89–106 CrossRef CAS.
- D. Zukowska, A. Gedaj, N. Porebska, M. Pozniak, M. Krzyscik, A. Czyrek, D. Krowarsch, M. Zakrzewska, J. Otlewski and L. Opalinski, Cell. Mol. Life Sci., 2023, 80, 113 CrossRef CAS PubMed.
- X. Liu, Y. Zhang, W. Li, J. Yin, B. Zhang, J. Wang and S. Wang, Food Res. Int., 2022, 162, 112072 CrossRef CAS PubMed.
- A. Ara, K. A. Ahmed and J. Xiang, Cell. Mol. Immunol., 2018, 15, 986–988 CrossRef CAS PubMed.
- R. D. Cummings, Curr. Opin. Struct. Biol., 2022, 75, 102394 CrossRef CAS PubMed.
- M. Erreni, F. D'Autilia, R. Avigni, E. Bolli, S. M. Arnouk, K. Movahedi, P. Debie, A. Anselmo, R. Parente, C. Vincke, F. W. B. van Leeuwen, P. Allavena, C. Garlanda, A. Mantovani, A. Doni, S. Hernot and J. A. Van Ginderachter, Theranostics, 2023, 13, 355–373 CrossRef CAS PubMed.
- J. Hu, P. Wei, P. H. Seeberger and J. Yin, Chem. – Asian J., 2018, 13, 3448–3459 CrossRef CAS PubMed.
- H. Kawabata, Free Radical Biol. Med., 2019, 133, 46–54 CrossRef CAS.
- G. Gu, X. Lv, G. Liu, R. Zeng, S. Li, L. Chen, Z. Liang, H. Wang, F. Lu, L. Zhan and X. Lv, Front. Pharmacol., 2021, 12, 734040 CrossRef CAS PubMed.
- L. M. Larcher, T. Wang and R. N. Veedu, Molecules, 2019, 24, 2489 CrossRef CAS.
- S. Muller, F. Sindikubwabo, T. Caneque, A. Lafon, A. Versini, B. Lombard, D. Loew, T. D. Wu, C. Ginestier, E. Charafe-Jauffret, A. Durand, C. Vallot, S. Baulande, N. Servant and R. Rodriguez, Nat. Chem., 2020, 12, 929–938 CrossRef CAS PubMed.
- M. Mohamadzadeh, H. DeGrendele, H. Arizpe, P. Estess and M. Siegelman, J. Clin. Invest., 1998, 101, 97–108 CrossRef CAS.
- E. Chiesa, A. Greco, F. Riva, R. Dorati, B. Conti, T. Modena and I. Genta, Pharmaceuticals, 2022, 15, 103 CrossRef CAS PubMed.
- J. M. Rios de la Rosa, A. Tirella, A. Gennari, I. J. Stratford and N. Tirelli, Adv. Healthcare Mater., 2017, 6, 1601012 CrossRef PubMed.
- Y. Zhang, R. Ma, C. You, X. Leng, D. Wang, S. Deng, B. He, Z. Guo, Z. Guan, H. Lei, J. Yu, Q. Zhou, J. Xing and Y. Dong, Carbohydr. Polym., 2023, 313, 120884 CrossRef CAS PubMed.
- Y. Lee, P. Wiriyasermkul, C. Jin, L. Quan, R. Ohgaki, S. Okuda, T. Kusakizako, T. Nishizawa, K. Oda, R. Ishitani, T. Yokoyama, T. Nakane, M. Shirouzu, H. Endou, S. Nagamori, Y. Kanai and O. Nureki, Nat. Struct. Mol. Biol., 2019, 26, 510–517 CrossRef CAS PubMed.
- R. Zent, C. A. Fenczik, D. A. Calderwood, S. Liu, M. Dellos and M. H. Ginsberg, J. Biol. Chem., 2000, 275, 5059–5064 CrossRef CAS PubMed.
- Y. Ma, W. Gao, Y. Zhang, M. Yang, X. Yan, Y. Zhang, G. Li, C. Liu, C. Xu and M. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 6358–6369 CrossRef CAS PubMed.
- B. Xiao, H. Laroui, E. Viennois, S. Ayyadurai, M. A. Charania, Y. Zhang, Z. Zhang, M. T. Baker, B. Zhang, A. T. Gewirtz and D. Merlin, Gastroenterology, 2014, 146, 1289–1300 CrossRef CAS.
- H. Laroui, D. Geem, B. Xiao, E. Viennois, P. Rakhya, T. Denning and D. Merlin, Mol. Ther., 2014, 22, 69–80 CrossRef CAS PubMed.
- C. Wu, Y. Zhao, Y. Zhang, Y. Yang, W. Su, Y. Yang, L. Sun, F. Zhang, J. Yu, Y. Wang, P. Guo, B. Zhu and S. Wu, J. Adv. Res., 2022, 37, 197–208 CrossRef CAS PubMed.
- Y. Yang, M. Chen, J. Zhou, J. Lv, S. Song, L. Fu, J. Chen, M. Yang and C. Mei, J. Am. Soc. Nephrol., 2018, 29, 2310–2325 CrossRef CAS PubMed.
- H. H. Lin, D. E. Faunce, M. Stacey, A. Terajewicz, T. Nakamura, J. Zhang-Hoover, M. Kerley, M. L. Mucenski, S. Gordon and J. Stein-Streilein, J. Exp. Med., 2005, 201, 1615–1625 CrossRef CAS.
- E. Liaskou, M. Karikoski, G. M. Reynolds, P. F. Lalor, C. J. Weston, N. Pullen, M. Salmi, S. Jalkanen and D. H. Adams, Hepatology, 2011, 53, 661–672 CrossRef CAS.
- H. Cheroutre and M. Kronenberg, Springer Semin. Immunopathol., 2005, 27, 147–165 CrossRef.
- J. Meenan, J. Spaans, T. A. Grool, S. T. Pals, G. N. Tytgat and S. J. van Deventer, Gut, 1997, 40, 241–246 CrossRef CAS PubMed.
- H. Sun, J. Liu, Y. Zheng, Y. Pan, K. Zhang and J. Chen, Dev. Cell, 2014, 30, 61–70 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |