
Structure–activity relationship of drug conjugated polymeric materials against uropathogenic bacteria colonization under in vitro and in vivo settings†
Sourav
Sarkar
 a,
Parikshit
Moitra
bc and
Santanu
Bhattacharya
*acde
a,
Parikshit
Moitra
bc and
Santanu
Bhattacharya
*acde
aSchool of Applied & Interdisciplinary Sciences, Indian Association for the Cultivation of Science, Kolkata 700032, India. E-mail: sb23in@yahoo.com; sb@iisc.ac.in
bDepartment of Nuclear Engineering, The Pennsylvania State University, University Park, Pennsylvania 16802, USA
cTechnical Research Centre, Indian Association for the Cultivation of Science, Kolkata 700032, India
dDepartment of Organic Chemistry, Indian Institute of Science, Bangalore 560012, India
eDepartment of Chemistry, Indian Institute of Science Education and Research Tirupati, Srinivasapuram, Yerpedu Mandal, Tirupati District, Andhra Pradesh 517619, India
First published on 21st November 2023
Abstract
The human world has been plagued with different kinds of bacterial infections from time immemorial. The increased development of resistance towards commercial antibiotics has made these bacterial infections an even more critical challenge. Bacteria have modified their mode of interactions with different types of commercial drugs by bringing changes to the receptor proteins or by other resisting mechanisms like drug efflux. Various chemical approaches have been made to date to fight against these smart adapting species. Towards this, we hypothesize chemically modifying the commercial antibacterial drugs in order to deceive the bacteria and destroy the bacterial biomass. In this study, different molecular weight polyethyleneimines are taken and conjugated with some well-known commercial drugs like penicillin and chloramphenicol to explore their antibacterial properties against some of the laboratory and uro-pathogenic strains of Gram-positive and Gram-negative bacteria. A detailed structure–activity relationship of these polymeric prodrug-like materials has been evaluated to determine the optimum formulation. The standardized system not only shows significant ∼90% bacterial killing in liquid broth culture, but also demonstrates promising bacterial inhibition towards biofilm formation for the pathogenic strains on inanimate surfaces like urinary catheters and on an in vivo mouse skin abrasion model. The reported bioactive polymeric materials can be successfully used for widespread therapeutic applications with promising medical relevance.
1. Introduction
Most bacteria are harmless and some of them have even been found to have some beneficiary roles in our day to day to life. However, about one percent of the total bacterial population are accountable for causing a large number of bacterial infections with varying intensities and complications.1–3 These bacterial infections are still considered to be a major cause of annual human death. The use of several chemotherapeutic drugs for the treatment of different cancers, other diseases and the rise in number of immunocompromised individuals have paved a way for bacterial infections which were otherwise a rare event in the past. The abundant use of antibiotics, and the long exposure to low concentrations of antibiotics results in the development of antibiotic resistant bacteria which are difficult to eradicate compared to their normal naturally occurring counterparts.4–9 These types of infections are becoming more prevalent in hospital setups because of the prolonged exposure to drugs, and as a result a large number of patients are developing a number of bacterial infections whilst being under treatment for other diseases.10–15 A major hospital-borne infection is the acquirement of urinary tract infections (UTI) which generally affects a greater number of females compared to males. The use of urinary catheters is common for individuals admitted to healthcare facilities and these catheters are becoming the most potent site of bacterial colonization and forming a biofilm mass which is a kind of defensive barrier for the bacteria making it tougher to kill thereby causing serious health hazards.16–21 UTIs still pose a potential threat with almost 10 million cases per year in India alone. The incidence of the infections starts with the invasion of the body's initial line of defense barrier before colonizing the urethra and the following migration to bladders, thereby causing serious complications. The bacteria mostly responsible for causing UTIs is the Gram-negative uropathogenic bacteria Escherichia coli.22,23 Several other bacterial species even some fungi are also responsible for causing this infection in individuals which involves Klebsiella pneumoniae, Staphylococcus saprophyticus, Enterococcus faecalis, Proteus vulgaris, Proteus mirabilis, Pseudomonas aeruginosa, Staphylococcus aureus and Candida spp. etc.The scientific community has been coming up with new improved strategies to combat these hard to kill bacteria and creating more resistant biofilms. There have been several studies which include antimicrobial peptides and new age antibiotics like daptomycin, gramicidin b and bacitracin. But these antibiotics are usually employed as a last resort when all other drugs have failed mainly because of their heightened toxicity. These treatments also incur huge costs, along with limited supply. All these factors along with some severe pharmacological and toxicological issues lead to a lack of development in this field.24–26 One way to find a solution to these problems is exploring different polymers and utilizing their antibacterial potentials.27–31 The polymers which were studied extensively for evaluating their antimicrobial properties include polyurethane, pyridinium polymers, alkoxysilanes, polyionenes and polyethyleneimine.32–35 These compounds' low propensity to develop resistance and their quick bactericidal impact are just two of their many benefits. In the present study, we have also used a well-known polycationic polymer polyethyleneimine with multiple primary, secondary and tertiary amines.36–40 Although different polymeric systems have already been explored for their bactericidal efficacy, there are some limitations that often curtail their application inluding their poor stability in biological systems, hydrophobicity, enhanced cytotoxicity toward the host organism, rapid degradation, etc.41 On the other hand, encapsulation of the hydrophilic antibacterial drugs to the polymeric micelles, gels or polymeric vesicles often leads to premature leakage thus reducing their potential as a potent antibacterial strategy.42,43 All these shortcomings could be overcome by chemically conjugating the known antibacterial drugs to the polymeric backbone. This chemical modification is also beneficial to evade the common issues or limitations arising out of the use of the drugs alone, which includes systemic toxicity, a short half-life and greater susceptibility to developing resistance.
PI molecules have widespread importance in biology research studies ranging from anti-cancer to anti-bacterial studies. It is often considered to be a gold standard for the transfection of a gene in several anti-cancer therapeutic approaches.44–47 We tried to explore this well-established polymeric PI system and made further modifications to enhance its ability in antibacterial studies. The ease of functionalizing the amines of a PI backbone enables us to conjugate a few of the well-known anti-bacterial drugs like penicillin and chloramphenicol (Scheme 1). Antibiotics can be conjugated to polymers to make use of particular polymer characteristics. These polymeric antibiotic systems differ in antibiotic class type, polymer composition, bond lability, and antibacterial activity. They also exhibit regulated and prolonged drug release.48 The two drugs employed in this study follow separate pathways to showcase their antibacterial potency. The mechanism of action of penicillin G drug relies on the inhibition of the biosynthesis of peptidoglycan in the bacterial cell wall thereby making the cell walls vulnerable for osmotic lysis. The chloramphenicol, on other hand, targets the protein synthesis mechanism of the bacteria by binding to the 50S subunit of the 70S ribosome and preventing the action of the peptidyl transferase enzyme. The attachment of drug to the PI backbones provided us with a dual mode effectivity against the bacteria, because the PI with free amines will be positively charged in aqueous media enhancing its interaction with the negatively charged bacterial peptidoglycan thus destabilizing the cell wall and causing osmotic lysis and killing of bacteria.49,50 On the other hand, some of the free amines of PI bound with drugs like penicillin G and chloramphenicol will provide an effective way for this drug to interact with the bacteria and show its bacterial inhibition properties. This will have a greater advantage because drugs like penicillin are losing their foot in the bacterial community because of the increase in number of penicillin resistant bacteria.51–53 Previously, few reports of conjugation of the penicillin drug to other polymeric backbones appeared leading to an increase in their antibacterial efficacy to evade the microbial resistance. However, these studies were found to be concentrated on Gram-positive bacteria only.54,55 Herein, we employed our conjugated polymeric system against some potent Gram-positive as well as Gram-negative uropathogens isolated from UTI patient's urine samples. The outcomes were satisfactory in highlighting how the penicillin conjugated PI system, as opposed to the drug alone, helped to revive the antibacterial capability. Now, to see whether the molecular weight of PI plays a role in the antibacterial property both the drugs were conjugated to four PIs of different molecular weights ranging from 800 to 750![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.
000.
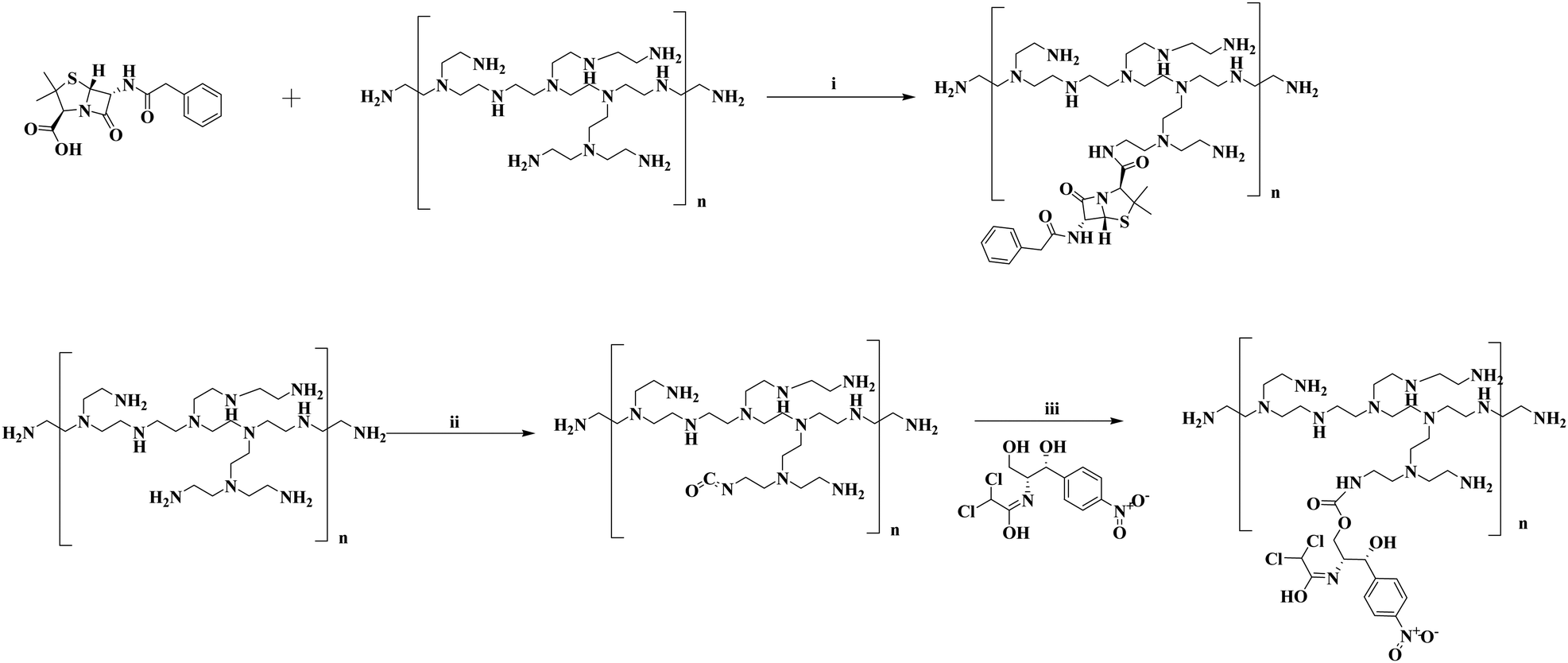 | ||
| Scheme 1 Synthesis of penicillin G and chloramphenicol conjugated PI molecules of different molecular weights. Reagents and conditions: (i) EDC, HOBt, cat. DMAP, Et3N, DMF, rt, 12 h; (ii) Triphosgene, DIPEA, DMF, 170 °C, 3 h; (iii) rt, 12 h. | ||
The antibacterial study was initially monitored on liquid broth cultures of respective bacteria. But we wanted to see the efficacy of the system not only for liquid culture techniques but also for bacterial biomass inside a biofilm. The bacteria when found to have a suitable adherent surface inside the body or in inanimate objects like urinary catheters they tend to produce different extracellular elements like DNA, proteins, polysaccharides which along with pili and flagella forms an extra cellular scaffold known as the biofilm thereby earning protection from antimicrobial drugs, stresses and even host immune systems.56–61 Since we tried to explore the anti-bacterial properties against some potential uropathogenic bacteria isolated from patient's urine samples, the catheter and its role in the spread of the disease came into play. Although urinary catheters are an important part of the urology medicine setup, the spread of the disease because of the indwelling catheters is still a major concern that needs to be addressed as soon as possible. Although several modifications in the inner and outer surfaces of the catheters are already under consideration to stop the spread of catheter associated UTIs, the problem is still far from being resolved.62–65 Here we also tried to coat the catheter with the synthesized material and investigate whether the system can significantly bring down the bacterial population of the biofilm on coated surfaces. The bacteria after being treated with the synthesized drug conjugated polymers were also tested for their ability to form biofilms on mouse skin. It was seen that the polymeric drug conjugates were able to significantly reduce the bacterial biomass on both the surfaces of catheters and even on the mouse skin infection model.
2. Experimental
2.1 Materials
All the chemical and biological reagents used in this study were of the highest commercial grade and were procured from good reliable sources. The different molecular weight branched polyethyleneimines used for the study (average Mw 800, 2000, 25000, 750000) were purchased from Sigma Aldrich. The anti-bacterial drugs penicillin-G sodium salts and cloramphenicol were obtained from Sisco Research Laboratories Pvt. Ltd. The biological reagents like the bacterial growth media Luria Bertani broth, Nutrient agar, and Hi-chrome UTI agar was all purchased from HIMEDIA. The glass wares were purchased from Sarada Glass and Chemicals. 1H-NMR (nuclear magnetic resonance) spectra were recorded on a FT-NMR Bruker DPX 400 MHz NMR spectrometer. Fourier transform infrared spectroscopy (FTIR) was carried out using a PerkinElmer spectrum BX FTIR spectrometer with the ATR method. NMR and FT-IR spectra were obtained for all synthesized compounds along with only penicillin and chloramphenicol. Dynamic light scattering (DLS) and zeta potential measurements were taken for each compound at room temperature in a Malvern Zetasizer Nano ZS (Malvern Instrument Inc., MA). FESEM analysis was carried out using a JEOL JSM 6700F at an operating voltage of 5 kV. The catheter used in the study is an uncoated rubber latex foley catheter procured from a local pharmacy store.
2.2 Methods
000, 750000) were used along with penicillin G sodium salt (100 mg, 0.31 mM). The two reagents were dissolved in an appropriate volume of dimethyl formamide (DMF). Then triethylamine (98 μl, 0.702 mM), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) (108 mg, 0.562 mM), 1-hydroxy benzotriazole (HOBt) (58 mg, 0.422 mM), and 4-dimethylaminopyridine (DMAP) (7 mg, 0.056 mM) were added and stirred overnight at room temperature over a magnetic stirrer. The next day the reaction was stopped and ethyl acetate was added to the reaction mixture to precipitate out the compound (Scheme 1). Finally, the solution was put in 50 ml falcon tubes and centrifuged to obtain the precipitate, followed by washing again with ethyl acetate, and the samples were then dried in a vacuum desiccator to remove the traces of solvent and subjected to characterization by NMR and FT-IR.
| %Drug conjugation= 4/(API − ADrug) × 100 |
For the zone of inhibition studies the sterile nutrient agar was prepared by autoclaving the media at 121 °C at 15 lb pressure for 15 min. Then cooling the agar to around 55–60 °C they were poured into sterile 100 mm Petri plates and kept undisturbed to let them solidify. Then on the day of the experiment, the first the bacterium inoculum was equally distributed on the agar surface using the spread plate technique followed by punching holes on the surface with the back side of sterile autoclaved tips. The wells were then filled with the desired materials whose antibacterial properties need to be investigated. The plates were incubated overnight under stationary conditions at 37 °C in a BOD incubator for observation of the inhibition zone.
:1 ratio to coat the catheter surface taking into account the lubricating and hydrophilic properties of PVP.68,69 The ensured the adherence of the synthesized polymers to the catheter surface and made them more hydrophilic smoother surfaces, making it difficult for the bacterial adhesion and consequent biofilm formation.70 The PVP also increased the stability of the coated polymers in aqueous media, enhancing the overall antibacterial potency.71 Only PVP coated catheters and non-coated catheter surfaces were used as controls. The dip coating was carried out for 30 min, followed by drying in a vacuum desiccator overnight to remove any traces of solvent. The coated catheter pieces were then subjected to SEM analysis to monitor their surface morphology.
For the biofilm assay studies, the dip coated catheter pieces were dried following a similar protocol as mentioned above, and then they were employed for the biofilm formation on their surfaces. The biofilm was formed on their surface following a similar biofilm formation technique as mentioned previously, just in this case in the 12 well sterile Petri plates each well was introduced with a single piece of catheter coated with the synthesized polymers. After every 48 hours the bacterial media was replaced with fresh media and the incubation was continued for 7 days. Then the catheters were rinsed with 1× PBS buffers thrice, the catheter pieces were taken in a 300 μl PBS buffer containing autoclaved microcentrifuge tubes and sonicated in an ultrasonication bath for 15 min to dislodge the adhered bacterial biomass from the biofilm. The catheter pieces were removed from the tube and the dislodged bacterial population was centrifuged down in a REMI C24 plus centrifugation machine at 5000 rpm. The bacteria were then stained for live/dead cell quantification by Syto-9 (Invitrogen™ SYTO™ 9 green fluorescent nucleic acid stain) and propidium iodide, PI (Sigma-Aldrich) in a 1:1 staining mixture. The respective concentrations of the two dyes were 5 μM for Syto-9 and 30 μM for propidium iodide prepared in 1× PBS buffer. The bacterial pellet thus obtained from centrifugation was dissolved in 50 μl 1× PBS buffer containing the dyes and incubated in the dark for 20 minutes, then 15 μl from that mixture was introduced onto a clean sterile glass slide and covered with a coverslip. The slides were then kept at 4 °C for 30 minutes for the bacteria to settle down and then the fluorescent images were obtained at 40× magnification in the EVOS M5000 imaging system to monitor the living vs. dead bacterial population in each sample.
Another fraction of the skin homogenized supernatant was subjected to a live/dead cell staining procedure by SYTO-9 and PI. Following the incubation with dyes in the dark for 20 minutes, an inoculum of the solution was made into a clean sterile glass slide and covered with a coverslip. The slides were then kept at 4 °C for 30 minutes and fluorescent images of the bacterial biomass was obtained using an EVOS M5000 imaging system.
For the SEM analysis two mice groups were considered, one receiving an untreated bacterial population as the control and another group receiving HPIC incubated bacterial cells. A fraction of the isolated skin from the mouse dorsal surface was similarly obtained following a previously mentioned protocol. The skin portion was isolated from the respective group of mice and was subsequently washed with 0.9% NaCl thrice, and the skin tissues were fixed in 4% paraformaldehyde overnight. Then the tissues were dehydrated by dipping in gradually increasing ethanol concentrated solution (30%, 50%, 70%, 90% and 100%). The skin tissues were then fixed on silicon wafers using conductive tapes and gold coated for final observation in FESEM (ZEISS Gemini 500).
To check whether the synthesized material has any kind of toxicity towards the RBC cells a hemolysis assay was performed for the compound HPIC on fresh drawn mouse blood. The blood was obtained from the heart after sacrificing the mouse and kept in sodium–citrate buffer containing vials to prevent coagulation. Then the RBC was diluted 25× and taken in respective marked tubes with the desired concentrations of materials, 0.1% Triton-X (positive control) and 1× PBS (negative control). The RBC were then incubated at 37 °C for 1.5 hours. The tubes were centrifuged at 2000 RPM for 5 min, the supernatant was taken out and put in a 96 well plate to record the absorbance at 576 nm to monitor the release of haemoglobin.72 All the samples were carried out in triplet and the experiment was repeated twice.
| % Haemolysis = [(O.D. of treated sample − O.D. of negative control)/(O.D. of positive control − O.D. of negative control)] × 100 |
3. Result and discussion
3.1 Characterisation of the synthesised material
The polymeric backbone of PI of different molecular weights were considered for our study and they were named accordingly as VP, HP, MP and LP for their respective 750000, 25000, 2000, and 800 molecular weights. The two drugs for our study involve penicillin G (P) and chloramphenicol (C). On the basis of the drugs, the synthesized materials were given the nomenclature VPIP, HPIP, MPIP, and LPIP for penicillin conjugated series and similarly VPIC, HPIC, MPIC and LPIC for chloramphenicol conjugated series. The amount of drug grafted in each material of the series was measured from the NMR spectra as mentioned in the material and methods section. It was found that the percentage grafting for both the drugs was quite different as compared to each other and also with different molecular weight PI backbones. The percentage grafting of penicillin G with VP, HP, MP and LP are approximately 41%, 45%, 9% and 41% respectively. Whereas the percentage grafting of chloramphenicol in VP, HP, MP and LP were 8%, 12%, 29% and 9% respectively.
In this study we have employed the PI molecules of different molecular weight but out of the four PI polymers VP and MP were linear polyethyleneimine, whereas the HP and LP were the branched polyethyleneimine of respective molecular weights. The grafting of the drugs to PI molecules was random especially with the MP polymeric system, which did not follow any obvious trend. The two higher molecular weight PI molecules (VP and HP) were found to follow a pattern in conjugation of drugs in accordance with their branching nature and among them, the one (HP) with the branched nature having more primary amines provided better grafting sites for conjugating the two antibacterial drugs. For the two types of drugs conjugation, penicillin G was grafted using EDC coupling methods which resulted in better grafting percentages, whereas in the case of chloramphenicol the PI molecules were first modified by triphosgene reaction to form an isocyanate intermediate which was followed by grafting of the drug, which may have resulted in less drug grafting percentage.
For further characterization the FTIR spectrum of all the samples were recorded. The presence of the characteristic peak corresponding to the carbonyl groups in all the compounds further ensures the presence and attachment of both the drugs to the respective PI backbone (Fig. S6, ESI†). The zeta potential was also measured for all synthesized molecules because the presence of cationic charges over the molecules will be essential for interacting with the negatively charged bacterial cell wall (Fig. S7, ESI†). The zeta measurements of all the materials showed varying positive charge intensities with LP being the least positive among the group (having an average charge distribution of 11.8 for LPIC and 12.8 for LPIP respectively). VP and HP molecules showed maximum positive charge intensities followed by MP conjugated polymers. The average charge densities for VPIC and VPIP were 22.9 and 21.65 whereas that of HPIC and HPIP were 20.8 and 23.5, respectively. The MP series on the other hand showed the average charge distribution around 12.45 for MPIC and 17.05 for MPIP. The dynamic light scattering (DLS) technique was also employed to determine the average size distribution of these materials when dispersed in aqueous solution (Fig. S7a and b, ESI†). The materials were found to form smaller aggregates with average hydrodynamic diameters of 326 nm, 239 nm, 246 nm and 300 nm for VPIC, HPIC, MPIC and LPIC respectively. Whereas the average sizes range from 458 nm, 198 nm, 342 nm and 375 nm for VPIP, HPIP, MPIP and LPIP respectively.
3.2. Anti-bacterial study with the synthesized material
The series of molecules after being synthesized and characterized now needs to be evaluated for their anti-bacterial properties. To evaluate that, a normal E. coli strain was initially used for this study. The bacterial inoculum was incubated overnight with the desired concentration of the polymeric materials. All the concentrations mentioned in this article are represented based on the percent conjugated drug with PI, since we will be comparing with the same concentration of free drug for their bacterial growth inhibition. In the case of both drugs, the higher molecular weight PI namely VPIP, HPIP showed about 50% antibacterial property whereas VPIC, HPIC showed better anti-bacterial efficacy of almost 100% at high concentrations when compared to MP and LP conjugated drugs respectively (Fig. 1a and b). Among the employed PI backbones for drug conjugation and subsequent studies, the VP and MP were linear polymers while the HP and LP were branched polymers. As we already know in the case of branched polymers the presence of all three types of amines (primary, secondary and tertiary) makes it a highly branched structure with higher cationic charge density.73,74 So, when the effectiveness of the conjugated polymers was considered, the two higher molecular weight PI molecules (VP and HP) were found to be better in their antibacterial property compared to others (MP and LP). The higher molecular weight PI molecules having higher cationic charge densities were found to be more efficient than the low molecular weight counterparts.75–77 Among these two polymers the one (HP) with the branched nature having more primary amines provided better grafting sites for conjugating the two antibacterial drugs. Thus, VP despite having a higher molecular weight were less effective as compared to the branched HP polymers. So, from the initial studies we found HPIP to be more efficient in inhibiting bacterial growth compared to VPIP, MPIP and LPIP respectively. In the case of chloramphenicol conjugated polymer series, the observation was similar but, in this case, also among the best two PI formulations HPIC was marginally better than VPIC.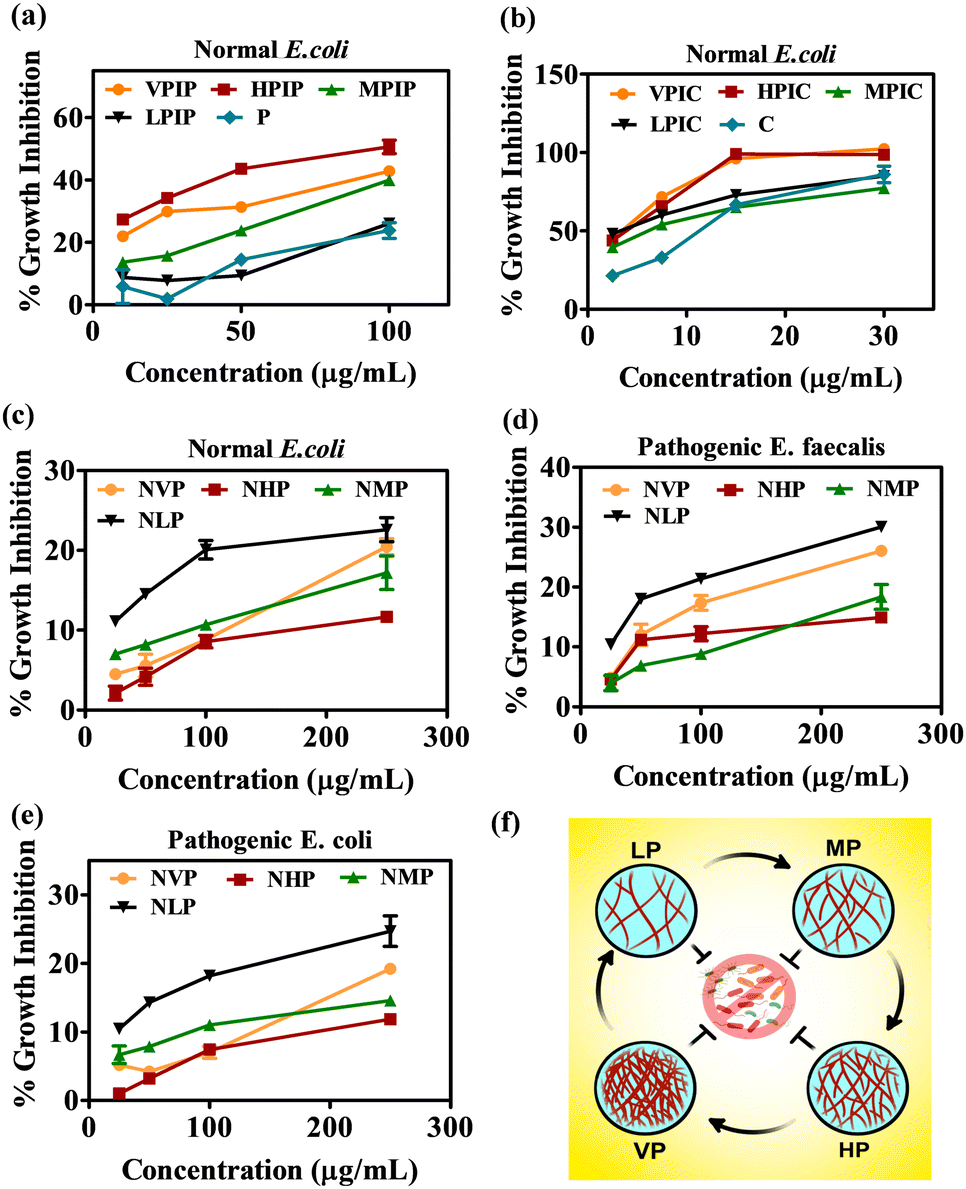 | ||
| Fig. 1 The anti-bacterial screening of the synthesized molecules on normal E. coli strain (a) and (b). The evaluation of the anti-bacterial property of only PI backbones on normal E. coli (c), pathogenic E. faecalis (d) and pathogenic E. coli (e). The scheme to describe the four molecular weight PI molecules considered in the current antibacterial study (f). | ||
Since we will be working mainly on uro-pathogenic strains of E. coli and E. faecalis isolated and identified on Hichrome UTI agar (Fig. S8, ESI†), now before moving ahead we tested the anti-bacterial property of only the polymeric PI backbones of all four molecular weight against the pathogenic and normal bacterial strains. The result in all three bacterial strains showed a similar pattern where the LP molecule were found to have an intrinsic toxicity (about 25–30%) towards the bacteria higher than all other PI backbones (Fig. 1c–e). Although having greater toxicity itself when conjugated with drugs LP had the least killing effect on the bacteria as evident from Fig. 1a and b. From this data we were able to decide to move ahead with VP and HP conjugated penicillin G and chloramphenicol moieties because the MP and LP conjugated drug showed the least antibacterial effectivity. The initial screening of the VPIP, HPIP and VPIC, HPIC was then carried out in two pathogenic strains of bacteria and was compared with free drug of the same concentration (Fig. 2). The results indicated that the synthesized materials showed better bacterial inhibition compared to the drug alone and the drug conjugated polymer HP series were found to be better than the VP series in the case of both the drugs. Chloramphenicol conjugated PI molecules showed almost more than 80% bactericidal potential compared to about 40% killing by the penicillin tagged PI molecules. The chloramphenicol drug itself being a broad-spectrum antibiotic compared to the narrow spectrum penicillin G molecules, also showed good antibacterial effect in this liquid broth study. Similarly, the chloramphenicol conjugated PI molecules VPIC and HPIC were found to have superior bactericidal properties when compared to penicillin G conjugated VPIP and HPIP polymers respectively.
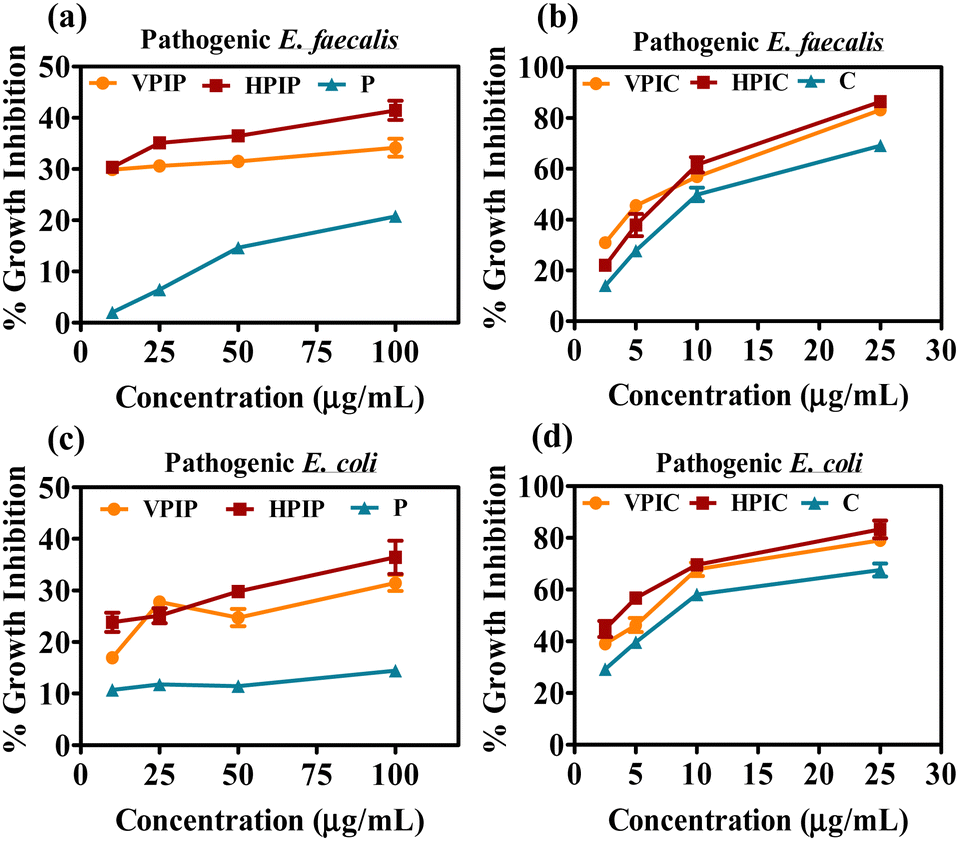 | ||
| Fig. 2 The screening of penicillin G (a) and (c) and chloramphenicol (b) and (d) conjugated VP and HP polymers on clinically isolated E. coli and E. faecalis after overnight incubation. | ||
The cationic PI molecules contributed to this added antibacterial potency and established the usefulness of the polymeric structure in antibacterial strategies. Among the two most potent PI compounds VPIC and HPIC, the latter was found to have a slight advantage with the minimum inhibitory concentration (MIC50) of 5 μg ml−1 for E. coli and around 10 μg ml−1 for E. faecalis respectively.
Before moving ahead, the modification of HP and VP polymers before and after the conjugation of two antibacterial drugs was validated using Gel Permeation Chromatography (GPC). The GPC curves showed that the modification results in an increase in molecular weight, thus having lower retention time than the backbone itself. The pattern was similar for both penicillin as well as chloramphenicol conjugation in the HP and VP polymer backbone (Fig. S9, ESI†).
3.3. Anti-bacterial property of materials on bacterial growth kinetics and biofilms
The anti-bacterial property of the synthesized materials was initially estimated after overnight incubation, now we wanted to see the interference of the materials with the bacterial growth cycle.78,79 The bacterial population as observed from the O.D values at different time intervals ranging from 1 h to 18 hours showing that among the two molecular weight PI, the HP conjugated drugs showed slightly improved anti-bacterial properties compared to VP conjugated drugs (Fig. 3).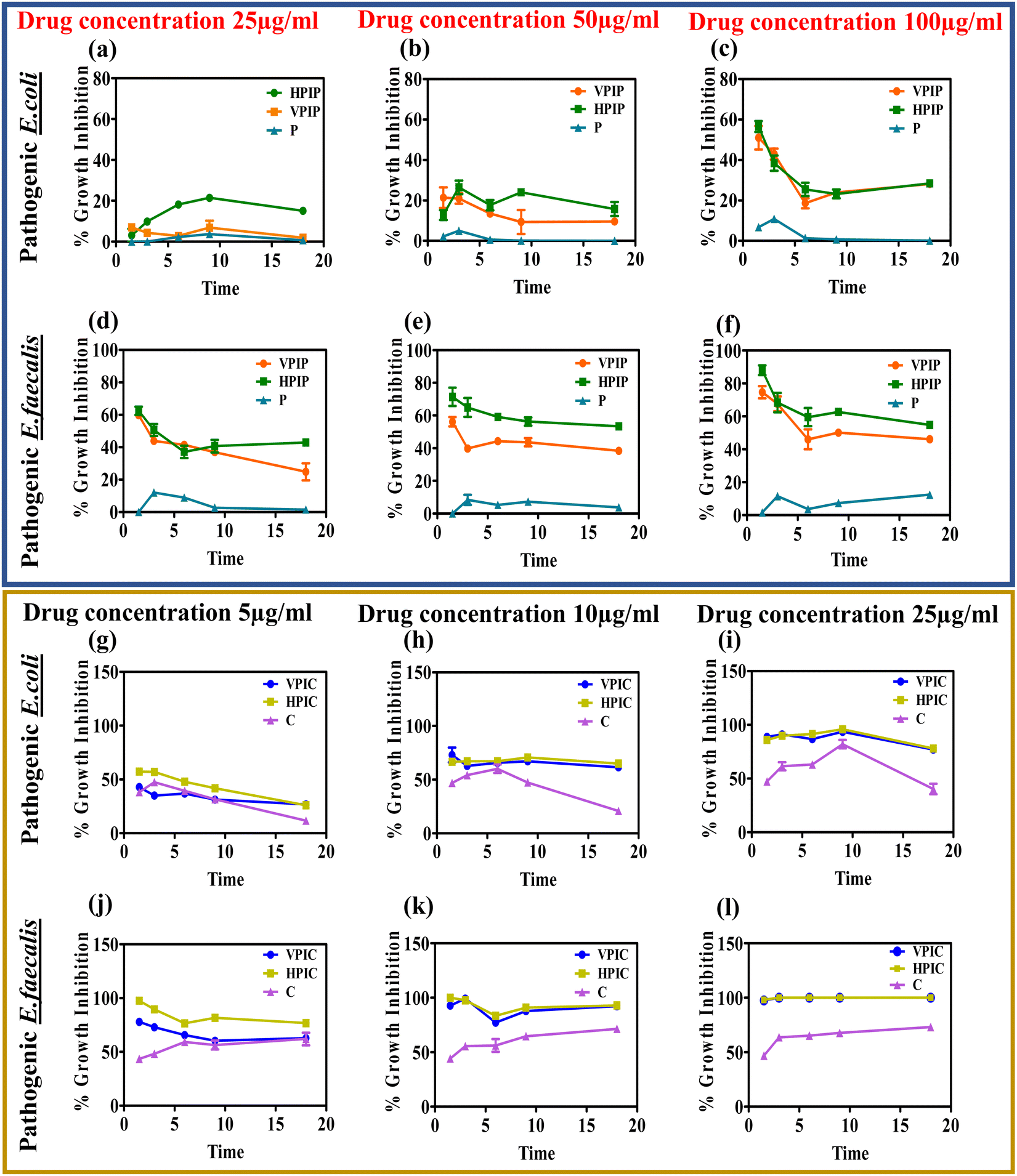 | ||
| Fig. 3 The effect of the synthesized PI conjugated penicillin G (a)–(f) and chloramphenicol (g)–(l) on the growth kinetics of the bacterial population at different time intervals and different concentrations of respective conjugated drug on pathogenic strains of E. coli (a)–(c) and (g)–(i) and E. faecalis (d)–(f) and (j)–(l). | ||
In the case of both penicillin G and chloramphenicol conjugated PI molecules the materials showed optimum activity around 9 hours with a slight dip in the effectivity at 18 hours in some cases while in other cases the inhibitory effect persisted at a similar rate until the 18 hour time point. This study actually helps us to better understand the anti-bacterial property of our synthesized materials that were capable of inhibiting the bacteria growth in a time dependent manner. Although in some concentrations of the respective drugs we could see that after 9 hours the bacteria tries to regain its pathogenicity a bit but in all those cases the polymeric systems were far more superior in tackling them compared to the drug alone. It was also observed that the PI conjugated penicillin and chloramphenicol when taken according to the concentration of the conjugated drugs, penicillin conjugated PI were most effective at 50 and 100 μg concentrations whereas in the case of chloramphenicol it was around 10 and 25 μg respectively. The materials having a positive charge as we already know from our zeta potential studies will interact with the negatively charged bacterial wall thereby causing instability in the peptidoglycan cell wall, diffusing through the wall to adsorb onto the cytoplasmic membrane and resulting in death of bacteria by disrupting it.35,80 In addition to their electrostatic contact with the bacterial membrane, the drugs' subsequent attachment to the PI backbone may give them a chance to engage in interactions with different bacterial metabolic pathway machineries and subsequently demonstrate their antibacterial potency.81
The anti-bacterial efficacy of these materials was also tested on bacterial biofilms and evaluated by crystal violet staining. The bacteria were allowed to form biofilms on 12-well Petri-plates in the presence of the desired concentrations of VPIP, HPIP and VPIC, HPIC. After incubation and crystal violet staining of each well, the crystal violet absorbance data as well the stained biofilm images further established that the HPIC and HPIP formulations were better at handling the bacterial growth compared to the control samples (Fig. S10, ESI†). This result was consistent for both the pathogenic bacteria E. coli and E faecalis.
3.4. Elucidating the individual effect of the polymer and drug in the drug conjugated PI molecules and their effect on bacterial biofilm inhibition on urinary catheters
Since we were achieving an enhanced antibacterial property of the drug conjugated PI molecules when compared to drug alone and only the polymer backbone, it was evident that the conjugated material provided an added advantage. But to elucidate the individual contribution of the polymer and the respective drugs in showing the enhanced bactericidal effect, an experimental strategy was deployed. In this study, we have measured the zeta potential charge distribution of the two potent drug conjugated PI molecules HPIP and HPIC respectively. The cationic charge densities thus obtained were then neutralized by using a biocompatible and least bactericidal anionic backbone, calf thymus DNA (ct-DNA). In the case of HPIP and HPIC, the observed positive zeta potentials were neutralized or brought down to approximate charge densities around zero by adding a particular concentration of DNA for each of the compounds. Based on this observation, we found that for neutralization of 25 μg ml−1 HPIC, approximately 70 μg ml−1 ct-DNA is needed whereas 12 μg ml−1 DNA is needed for 100 μg ml−1 concentration of HPIP. As per these data, a control experiment was performed where an equal primary inoculum of bacteria (pathogenic E. coli) was added into labelled tubes. The tubes had a blank control, along with an only HP polymer control (200 μg ml−1) and an only DNA control (70 μg ml−1). The two drugs penicillin G (100 μg ml−1) and chloramphenicol (25 μg ml−1) were also taken as the only drug controls. Now two of the tubes were inoculated with HPIC (25 μg ml−1) and HPIP (100 μg ml−1) each, of which one from each group were then neutralized through the addition of respective DNA concentration as obtained from the above-mentioned zeta potential studies. The bacterial tubes with respective treatments were then incubated under shaking conditions at 37 °C overnight and the bacterial growth was observed by measuring the O.D at 600 nm. This strategy ensures that once the positive charge densities of the PI backbone are neutralized by the drug conjugated polymers, HPIC and HPIP, will lose its ability to adhere to the bacterial surface through electrostatic interaction and thus the attached drug molecules will also be rendered ineffective. The obtained result was also conclusive of this fact because it shows that only HPIC and HPIP were far more superior in inhibiting the bacterial growth when compared to their DNA neutralized counterparts. We further did a spread plate zone of inhibition study with respective wells containing HPIC, HPIC + DNA, only DNA and only HP. Here also a clean and distinct zone of inhibition was found only in the case of HPIC, but no such zone was seen for its neutralized counterpart (Fig. S11, ESI†).The synthesized material HPIC and HPIP, proven to be better in inhibiting the bacterial population from the previous studies was chosen for further investigation of its capability in reducing the bacterial biomass in a biofilm. The urinary catheter, one of the most potent sites for bacterial colonization and biofilm formation was employed in in vitro bacterial culture setup after being coated with respective synthesized materials along with PVP lubricant. The catheters after being coated were subjected to SEM analysis to monitor the coating of the materials and to find the differences in their surface topology. The non-coated control rubber latex catheters appeared as the most rough or rugged surfaces, while the PVP lubricant coated catheters have shown a smoother topology. Due to the presence of the drug-conjugated PI molecules, the surface properties of the catheter coated with HPIC and HPIP were slightly different from those of a catheter coated simply with PVP (Fig. 4a). The live dead cell staining of the bacterial biofilm on the catheter surface showed a significant reduction in the total bacteria biomass in HPIC and HPIP coated catheter surfaces compared to the non-coated and only PVP coated catheter control samples. Among the two coated specimens the HPIC was found to be better than HPIP in inhibiting the bacterial growth in a biofilm marked by the presence of less green fluorescent signal of SYTO-9 a marker of living cells and a greater number of red stained bacterial cells indicating the dead bacterial cells being stained with PI82–84 (Fig. 4). The only PVP coated catheters also showed some bactericidal property because of its hydrophilicity reducing bacterial adhesion to surfaces when compared to the non-coated catheter control, but the PVP-HPIC and PVP-HPIP were much ahead in inhibiting the bacterial biofilm formation.
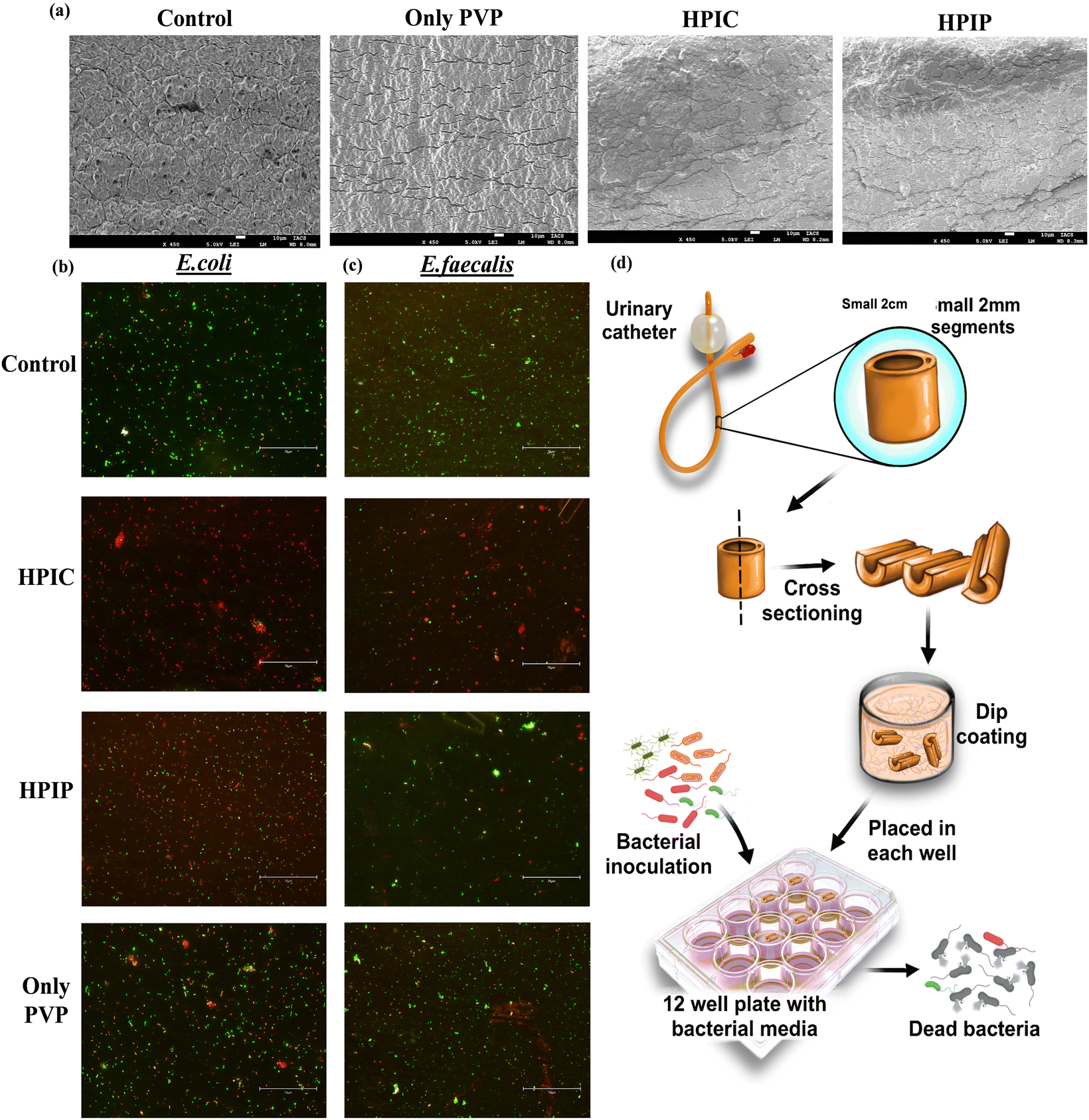 | ||
| Fig. 4 The coatings on the catheter surfaces were monitored by SEM analysis (a). Live/dead cell staining of the bacterial biofilm of E. coli (b) and E. faecalis (c) on coated urinary catheters to monitor the anti-biofilm properties of HPIC and HPIP respectively. The scheme (d) describes the processing of catheters into small 2 cm cross-sections followed by dip coating of polymeric materials and evaluation of its antibacterial property. | ||
The repeated experiments done in this setup with very similar observation actually helped us to confer that among the two drug conjugated materials the chloramphenicol conjugated with 25000 MW PI was found to be the best formulation to inhibit the bacterial growth in both broth culture as well as in biofilms. These studies also prove that our synthesized materials when used as a protective coating to coat the urinary catheters were very efficient in bringing down the bacterial biomass, and that of the pathogenic bacterial strain from UTI patients. These uropathogenic bacteria obtained from the patients urine samples have an intrinsic nature of forming more resistant biofilms on catheter surfaces, but the coating of such catheters with synthesized modified polymers have actually proven to be detrimental for their propagation and growth.
3.5. Biofilm inhibition on a mouse skin infection model
The previous experiments helped us to determine the best formulation among the synthesized materials with better bacterial killing properties. Thus, HPIC was explored in an in vivo mouse skin infection model to ensure its ability of inhibiting the bacterial biofilm formation even on mouse skin. To ensure that the material (HPIC) itself doesn’t have any toxicity, a hemolysis assay was carried out on fresh blood drawn from mice with different concentrations of HPIC (concentrations represented with respect to the conjugated drug). The HPIC molecule did not have any significant toxicity toward the red blood cells as observed from the release of hemoglobin monitored spectrophotometrically (Fig. S12, ESI†). The half minimal hemolytic concentration (HC50) for the material was found to be around 75 μg ml−1 (concentration given in terms of the conjugated chloramphenicol drug), which is much higher than what was employed in the in vivo study. The claim was further validated by performing a cytotoxicity assay of the materials by MTT at similar concentrations on the McCoy cell line (Fig. S13, ESI†) where the IC50 values were found to be around 100 μg ml−1. In both experimental setups the materials did not show any significant toxicity even at concentrations higher than what has been employed in the current study. The selectivity index (SI) of HPIC when calculated taking into account the inhibitory concentration of HPIC against normal mouse fibroblast (McCoy) cells and the MIC50 values against E. coli and E. faecalis, was found to be more than 20 and 10 respectively, further proving that this system indeed has significant potential for widespread applications in in vivo systems.When the mice skin was processed post experimentation and the number of bacteria that were able to colonize the skin and initiate biofilm formation was evaluated by live/dead cell staining showed that the skin of the mice group receiving bacteria pre-incubated with HPIC were not able to colonize the skin and cause a biofilm infection on the skin surface, compared to the ‘Control’ and ‘Drug only’ mice groups.85–87 The presence of very few green and yellow stained bacterial cells in the HPIC groups indicates a marked reduction in the number of bacterial populations (Fig. 5d–f). The CFU assay done with the bacteria isolated from the mouse skins of respective groups also validated the previous result where the number of CFU's were considerably very high in the case of ‘Control’ and ‘Drug only’ mice when compared with the treated (HPIC) one (Fig. 5b and c). These data shows that the synthesized drug conjugated polymers were able to decrease the bacterial biomass to such a significant level that the samples treated with HPIC were not able to form any kind of biofilm colonization on the abrasion skin surfaces of the mice.
 | ||
| Fig. 5 A schematic to describe the overall plan and dosages administered to mice groups for this antibacterial study (a). A CFU assay was carried out with bacteria isolated from respective mice skin (b), with the representative image of a mice skin infection model in the inset. The number of CFU's were counted and represented in a graph format (c). The live dead staining of the bacterial biomass isolated from each mice group was shown using fluorescence microscopy (d)–(f). Scanning electron microscopy of the skin of the control mice with only bacterial inoculation (g) and HPIC mice with treated bacterium inoculation (h) after 48 hours incubation. | ||
To further corroborate the above findings SEM analysis of the mouse skin after being colonized with the HPIC treated bacteria was compared with the mice skin which receives only bacteria as a control. The SEM images clearly showed a dense bacterial biofilm formation on the skin surface in the case of the control group which is quite a characteristic feature of the catheter colonizing uropathogenic bacteria whereas the mouse skin abrasions receiving the treated samples of the same bacterial strain were unable to colonize and infect (Fig. 5g and h). This observation further validated the anti-bacterial potency of HPIC in bringing down the biofilm forming capabilities of pathogenic bacteria.
4. Conclusions
Bacterial infections especially UTI infections are occurring in huge numbers every year all over the world, which poses a serious threat to mankind. Furthermore, an increasing number of bacteria are developing resistance toward available commercial drugs and the eradication of bacterial infection has become more difficult. However, the scientific community is constantly making efforts by introducing new systems and modifying the existing systems to combat them. In this study, we have also employed a similar setup where a polymeric cationic backbone of polyethyleneimine has been selected and modified by percentage grafting of two well-known commercial drugs penicillin G and chloramphenicol, thereby devising a modified polymeric material to combat clinical isolates of E. coli and E. faecalis. Our synthesized system, especially the chloramphenicol conjugated to 25000 MW PI was found to be highly effective in reducing the bacterial population to a greater extent in not only liquid bacterial culture setup but was highly efficient in bringing down the bacterial biomass in bacterial biofilm. The ability of the materials to inhibit the bacteria from colonizing a surface and forming biofilms makes it an important compound to be explored as an antibacterial commercial drug alternative, because the biofilms being one of the defense mechanism of bacteria are even tougher for the commercial drugs to eradicate. The dual mode of action of these synthesized materials lies in its unique structural integrity. The cationic polymeric backbone interacts with the negatively charged bacterial cell wall and thereby brings an instability in the cell wall, and as a result the conjugated drugs attached to the PI backbone can easily interact with the bacteria and target their respective metabolic pathways showing its promising bactericidal properties against potent pathogens. The well explored antibiotics like penicillin G and chloramphenicol which are slowly losing their ground because of the increased incidence of resistance among the microbes, are involved in hospital acquired infections like UTIs. However modifications of these drugs by simple grafting in polymeric PI systems helps them to regain the drug activity by simply modifying the mode of interactions of these drugs with bacterial membranes. This system has potential to be a better anti-bacterial system against some tough to kill bacterial strains.
Abbreviations
| PI | Polyethyleneimine |
| VPI | Very high molecular weight PI-75000 |
| HPI | High molecular weight PI-25000 |
| MPI | Middle molecular weight PI-2000 |
| LPI | Low molecular weight PI-800 |
| Penicillin G (P) | Penicillin conjugated with different molecular weights PI as VPIP, HPIP, MPIP and LPIP respectively, |
| Chloramphenicol (C) | Chloramphenicol conjugated different PI as VPIC,HPIC,MPIC and LPIC respectively. |
Author contributions
S. B. originated and conceptualized the study. P. M. helped S. S. with the chemical synthesis and characterization of the compounds. S. S. performed all the biologicals experiments both in vitro and in vivo. S. S. prepared the manuscript and P. M. and S. B. revised it. All authors have contributed to data interpretations and writing.Conflicts of interest
There are no competing financial or emotional interests among the authors.Acknowledgements
S. S. acknowledges the DST Inspire fellowship (IF160191) and S. B. thanks the J. C. Bose fellowship towards the successful completion of the project. P. M. and S. B. thanks Department of Science and Technology, GoI for the funding provided through Technical Research Centre at IACS. We would also like to thank the IACS central facility for the use of SEM and other instruments. The clinical isolates were generously provided to us by Calcutta Medical College.References
- S. Doron and S. Gorbach, Int. Encyclopedia Public Health, 2008, 273 CrossRef.
- H. C. Neu, Rev. Infect. Dis., 1985, 7, S778–S782 CrossRef.
- G. G. Grassi, J. Antimicrob. Chemother., 1988, 21, 1–7 CrossRef.
- M. Frieri, K. Kumar and A. Boutin, J. Infection Public Health, 2017, 10, 369–378 CrossRef.
- J. M. Munita and C. A. Arias, Microbiol. Spectrum, 2016, 4(4.2), 15 Search PubMed.
- A. MacGowan and E. Macnaughton, Medicine, 2013, 41, 642–648 CrossRef.
- K. Bush, P. Courvalin, G. Dantas, J. Davies, B. Eisenstein, P. Huovinen, G. A. Jacoby, R. Kishony, B. N. Kreiswirth and E. Kutter, Nat. Rev. Microbiol., 2011, 9, 894–896 CrossRef CAS.
- R. Capita and C. Alonso-Calleja, Crit. Rev. Food Sci. Nutr., 2013, 53, 11–48 CrossRef CAS PubMed.
- M. Webber and L. Piddock, J. Antimicrob. Chemother., 2003, 51, 9–11 CrossRef CAS.
- B. A. Rogers, Z. Aminzadeh, Y. Hayashi and D. L. Paterson, Clin. Infect. Dis., 2011, 53, 49–56 CrossRef.
- M. R. Rameshkumar and N. Arunagirinathan, Adv. HIV AIDS Control, 2018, 83, 341–344 Search PubMed.
- S. Morris and E. Cerceo, Antibiotics, 2020, 9, 196 CrossRef CAS.
- E. Tacconelli, G. De Angelis, M. A. Cataldo, E. Mantengoli, T. Spanu, A. Pan, G. Corti, A. Radice, L. Stolzuoli and S. Antinori, Antimicrob. Agents Chemother., 2009, 53, 4264–4269 CrossRef CAS.
- N. Hassoun-Kheir, Y. Stabholz, J.-U. Kreft, R. De La Cruz, J. L. Romalde, J. Nesme, S. J. Sørensen, B. F. Smets, D. Graham and M. Paul, Sci. Total Environ., 2020, 743, 140804 CrossRef CAS PubMed.
- D. Hocquet, A. Muller and X. Bertrand, J. Hosp. Infect., 2016, 93, 395–402 CrossRef CAS PubMed.
- A. L. Flores-Mireles, J. N. Walker, M. Caparon and S. J. Hultgren, Nat. Rev. Microbiol., 2015, 13, 269–284 CrossRef CAS.
- W. E. Stamm and S. R. Norrby, J. Infect. Dis., 2001, 183, S1–S4 CrossRef.
- E. J. Dielubanza and A. J. Schaeffer, Medical clinics, 2011, 95, 27–41 Search PubMed.
- S. Salvatore, S. Salvatore, E. Cattoni, G. Siesto, M. Serati, P. Sorice and M. Torella, Eur. J. Obstet. Gynecol. Reprod. Biol., 2011, 156, 131–136 CrossRef.
- F. Ostadhossein, P. Moitra, E. Altun, D. Dutta, D. Sar, I. Tripathi, S.-H. Hsiao, V. Kravchuk, S. Nie and D. Pan, Commun. Biol., 2021, 4, 1–16 CrossRef.
- J. R. Johnson, M. A. Kuskowski and T. J. Wilt, Ann. Intern. Med., 2006, 144, 116–126 CrossRef.
- K. E. Sivick and H. L. Mobley, Infect. Immun., 2010, 78, 568–585 CrossRef CAS.
- G. R. Nielubowicz and H. L. Mobley, Nat. Rev. Urol., 2010, 7, 430–441 CrossRef CAS PubMed.
- F. Rabanal and Y. Cajal, New Weapons to Control Bacterial Growth, 2016, 433–451 Search PubMed.
- N. Ganesan, B. Mishra, L. Felix and E. Mylonakis, Microbiol. Mol. Biol. Rev., 2023, e00037–00022 Search PubMed.
- A. K. Marr, W. J. Gooderham and R. E. Hancock, Curr. Opin. Pharmacol., 2006, 6, 468–472 CrossRef CAS.
- S. Ghosh, R. Mukherjee, S. Mukherjee, S. Barman and J. Haldar, ACS Appl. Mater. Interfaces, 2022, 14, 34527–34537 CrossRef CAS PubMed.
- S. Dhingra, S.-P. Su, Y.-H. Chan and S. Saha, Biomater. Sci., 2023, 11, 4308–4326 RSC.
- J. Peng, K. Li, Y. Du, Y. Feng, L. Wu and G. Liu, J. Mater. Chem. B, 2023, 11, 8020–8032 RSC.
- N. Wang, K.-K. Yu, K. Li and X.-Q. Yu, J. Mater. Chem. B, 2023, 11, 1232–1239 RSC.
- S. Bochani, A. Zarepour, A. Kalantari, F. Haghi, M. Shahbazi, A. Zarrabi, S. Taheri and A. Maleki, J. Mater. Chem. B, 2023, 11, 8056–8068 RSC.
- K.-S. Huang, C.-H. Yang, S.-L. Huang, C.-Y. Chen, Y.-Y. Lu and Y.-S. Lin, Int. J. Mol. Sci., 2016, 17, 1578 CrossRef.
- Z. Si, W. Zheng, D. Prananty, J. Li, C. H. Koh, E.-T. Kang, K. Pethe and M. B. Chan-Park, Chem. Sci., 2022, 13, 345–364 RSC.
- G. Li, J. Shen and Y. Zhu, J. Appl. Polym. Sci., 1998, 67, 1761–1768 CrossRef CAS.
- I. Cakmak, Z. Ulukanli, M. Tuzcu, S. Karabuga and K. Genctav, Eur. Polym. J., 2004, 40, 2373–2379 CrossRef CAS.
- M. Azevedo, P. Ramalho, A. Silva, R. Teixeira-Santos, C. Pina-Vaz and A. Rodrigues, J. Med. Microbiol., 2014, 63, 1167–1173 CrossRef CAS.
- I. Yudovin-Farber, J. Golenser, N. Beyth, E. I. Weiss and A. J. Domb, J. Nanomater., 2010, 2010, 1–11 CrossRef.
- I. M. Helander, H.-L. Alakomi, K. Latva-Kala and P. Koski, Microbiology, 1997, 143, 3193–3199 CrossRef CAS.
- X. Wang, D. Niu, C. Hu and P. Li, Curr. Pharm. Des., 2015, 21, 6140–6156 CrossRef CAS PubMed.
- D. Xu, Q. Wang, T. Yang, J. Cao, Q. Lin, Z. Yuan and L. Li, Int. J. Environ. Res. Public Health, 2016, 13, 334 CrossRef.
- M. Rofeal, F. Abdelmalek and A. Steinbüchel, Int. J. Mol. Sci., 2022, 23, 4101 CrossRef CAS.
- X. Ding, A. Wang, W. Tong and F. J. Xu, Small, 2019, 15, 1900999 CrossRef PubMed.
- S. Bhattacharya and S. Haldar, Langmuir, 1995, 11, 4748–4757 CrossRef CAS.
- P. Moitra, S. K. Misra, K. Kumar, P. Kondaiah, P. Tran, W. Duan and S. Bhattacharya, ACS Biomater. Sci. Eng., 2021, 7, 2508–2519 CrossRef CAS PubMed.
- A. Bajaj, P. Kondaiah and S. Bhattacharya, Bioconjugate Chem., 2008, 19, 1640–1651 CrossRef CAS.
- S. K. Misra, S. Naz, P. Kondaiah and S. Bhattacharya, Biomaterials, 2014, 35, 1334–1346 CrossRef CAS PubMed.
- M. Kamra, P. Moitra, D. Ponnalagu, A. A. Karande and S. Bhattacharya, ACS Appl. Mater. Interfaces, 2019, 11, 37442–37460 CrossRef CAS PubMed.
- N. D. Stebbins, M. A. Ouimet and K. E. Uhrich, Adv. Drug Delivery Rev., 2014, 78, 77–87 CrossRef CAS.
- P. Vicennati, A. Giuliano, G. Ortaggi and A. Masotti, Curr. Med. Chem., 2008, 15, 2826–2839 CrossRef CAS.
- K. Lewis and A. M. Klibanov, Trends Biotechnol., 2005, 23, 343–348 CrossRef CAS.
- J. A. Kelly, O. Dideberg, P. Charlier, J.-P. Wery, M. Libert, P. Moews, J. Knox, C. Duez, C. Fraipont and B. Joris, Science, 1986, 231, 1429–1431 CrossRef CAS PubMed.
- J.-M. Ghuysen, Int. J. Antimicrob. Agents, 1997, 8, 45–60 CrossRef CAS PubMed.
- A. Zapun, C. Contreras-Martel and T. Vernet, FEMS Microbiol. Rev., 2008, 32, 361–385 CrossRef CAS PubMed.
- N. Aumsuwan, S. Heinhorst and M. W. Urban, Biomacromolecules, 2007, 8, 3525–3530 CrossRef CAS PubMed.
- H. Yang and S. T. Lopina, J. Biomater. Sci., Polym. Ed., 2003, 14, 1043–1056 CrossRef CAS PubMed.
- H. Wu, C. Moser, H.-Z. Wang, N. Høiby and Z.-J. Song, Int. J. Oral Sci., 2015, 7, 1–7 CrossRef CAS PubMed.
- M. Jamal, W. Ahmad, S. Andleeb, F. Jalil, M. Imran, M. A. Nawaz, T. Hussain, M. Ali, M. Rafiq and M. A. Kamil, J. Chin. Medi. Association, 2018, 81, 7–11 CrossRef.
- D. Dufour, V. Leung and C. M. Lévesque, Endodontic Topics, 2010, 22, 2–16 CrossRef.
- C. B. Whitchurch, T. Tolker-Nielsen, P. C. Ragas and J. S. Mattick, Science, 2002, 295, 1487 CrossRef CAS PubMed.
- L. Hall-Stoodley, J. W. Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95–108 CrossRef CAS PubMed.
- N. Høiby, O. Ciofu, H. K. Johansen, Z. J. Song, C. Moser, P. Ø. Jensen, S. Molin, M. Givskov, T. Tolker-Nielsen and T. Bjarnsholt, Int. J. Oral Sci., 2011, 3, 55–65 CrossRef.
- C. Zhou, Y. Wu, K. R. V. Thappeta, J. T. L. Subramanian, D. Pranantyo, E.-T. Kang, H. Duan, K. Kline and M. B. Chan-Park, ACS Appl. Mater. Interfaces, 2017, 9, 36269–36280 CrossRef CAS PubMed.
- D. M. Siddiq and R. O. Darouiche, Nat. Rev. Urol., 2012, 9, 305–314 CrossRef CAS.
- J. R. Johnson, P. L. Roberts, R. J. Olsen, K. A. Moyer and W. E. Stamm, J. Infect. Dis., 1990, 162, 1145–1150 CrossRef CAS PubMed.
- M. A. Yassin, T. A. Elkhooly, S. M. Elsherbiny, F. M. Reicha and A. A. Shokeir, Heliyon, 2019, 5, e02986 CrossRef.
- A. Bajaj, S. K. Misra, P. Kondaiah and S. Bhattacharya, Biochim. Biophys. Acta Biomembr., 2008, 1778, 1222–1236 CrossRef CAS.
- K. Kumar, G. Vulugundam, P. Kondaiah and S. Bhattacharya, J. Mater. Chem. B, 2015, 3, 2318–2330 RSC.
- D. Mahata, S. M. Mandal, A. Basak and G. B. Nando, RSC Adv., 2015, 5, 69215–69219 RSC.
- K. Kristinsson, B. Jansen, U. Treitz, F. Schumacher-Perdreau, G. Peters and G. Pulverer, J. Biomater. Appl., 1991, 5, 173–184 CrossRef CAS.
- N. Cerca, G. B. Pier, M. Vilanova, R. Oliveira and J. Azeredo, Res. Microbio., 2005, 156, 506–514 CrossRef CAS.
- D. Lubasova, H. Niu, X. Zhao and T. Lin, RSC Adv., 2015, 5, 54481–54487 RSC.
- A. Pascual, J. P. Tan, A. Yuen, J. M. Chan, D. J. Coady, D. Mecerreyes, J. L. Hedrick, Y. Y. Yang and H. Sardon, Biomacromolecules, 2015, 16, 1169–1178 CrossRef CAS PubMed.
- C. N. Lungu, M. V. Diudea, M. V. Putz and I. P. Grudziński, Int. J. Mol. Sci., 2016, 17, 555 CrossRef.
- A. Prud’homme and F. Nabki, Sensors, 2020, 20, 1824 CrossRef PubMed.
- K. A. Gibney, I. Sovadinova, A. I. Lopez, M. Urban, Z. Ridgway, G. A. Caputo and K. Kuroda, Macromol. Biosci., 2012, 12, 1279–1289 CrossRef CAS.
- J. Pachla, R. J. Kopiasz, G. Marek, W. Tomaszewski, A. Głogowska, K. Drężek, S. Kowalczyk, R. Podgórski, B. Butruk-Raszeja and T. Ciach, Biomacromolecules, 2023, 24, 2237–2249 CrossRef CAS.
- A. K. Tiwari, M. K. Gupta, G. Pandey, R. J. Narayan and P. C. Pandey, J. Mater. Res., 2020, 35, 2405–2415 CrossRef CAS.
- R. Dutta, B. P. Nenavathu, M. K. Gangishetty and A. Reddy, Colloids Surf., B, 2012, 94, 143–150 CrossRef CAS PubMed.
- K. R. Raghupathi, R. T. Koodali and A. C. Manna, Langmuir, 2011, 27, 4020–4028 CrossRef CAS PubMed.
- C. Wiegand, M. Bauer, U.-C. Hipler and D. Fischer, Int. J. Pharm., 2013, 456, 165–174 CrossRef CAS.
- E. Zamani, S. Chatterjee, T. Changa, C. Immethun, A. Sarella, R. Saha and S. K. Dishari, Sci. Rep., 2019, 9, 20411 CrossRef CAS.
- L. Boulos, M. Prevost, B. Barbeau, J. Coallier and R. Desjardins, J. Microbiol. Methods, 1999, 37, 77–86 CrossRef CAS PubMed.
- M. Berney, F. Hammes, F. Bosshard, H.-U. Weilenmann and T. Egli, Appl. Environ. Microbiol., 2007, 73, 3283–3290 CrossRef CAS PubMed.
- M. Rosenberg, N. F. Azevedo and A. Ivask, Sci. Rep., 2019, 9, 1–12 CrossRef CAS.
- J. Hoque and J. Haldar, ACS Appl. Mater. Interfaces, 2017, 9, 15975–15985 CrossRef CAS PubMed.
- T. Dai, G. P. Tegos, T. Zhiyentayev, E. Mylonakis and M. R. Hamblin, Lasers in Surgery and Medicine: The Official Journal of the American Society for Laser Medicine and Surgery, 2010, 42, 38–44 CrossRef PubMed.
- T. Dai, A. Gupta, Y.-Y. Huang, M. E. Sherwood, C. K. Murray, M. S. Vrahas, T. Kielian and M. R. Hamblin, Photomedicine Laser Surgery, 2013, 31, 531–538 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S11. See DOI: https://doi.org/10.1039/d3tb01841e |
| This journal is © The Royal Society of Chemistry 2024 |