
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomic tuning of 3D printed carbon surface chemistry for electrocatalytic nitrite oxidation and reduction to ammonia†
Wanli
Gao
a,
Jan
Michalička
b and
Martin
Pumera
 *acd
*acd
aFuture Energy and Innovation Laboratory, Central European Institute of Technology, Brno University of Technology, Purkyňova 123, 61200 Brno, Czech Republic. E-mail: martin.pumera@ceitec.vutbr.cz
bCentral European Institute of Technology, Brno University of Technology, Purkyňova 123, 61200 Brno, Czech Republic
cFaculty of Electrical Engineering and Computer Science, VSB – Technical University of Ostrava, 17. Listopadu 2172/15, 70800, Ostrava, Czech Republic
dDepartment of Medical Research, China Medical University Hospital, China Medical University, No. 91 Hsueh-Shih Road, Taichung 40402, Taiwan
First published on 11th November 2024
Abstract
Nitrite contamination in agricultural and industrial wastewater presents a critical impact on environmental sustainability, demanding efficient strategies for monitoring and remediation. This study addresses this challenge by developing cost-effective electrocatalysts for both nitrite detection and conversion to value-added ammonia. 3D printed carbon materials are explored as bifunctional platforms for the electrochemical nitrite oxidation reaction (NO2OR) and nitrite reduction reaction (NO2RR). Benefiting from the inherent Ti-dominated metallic impurities and intrinsic surface features of carbon nanotubes, 3D printed carbon electrodes exhibit electrocatalytic activity for both reactions. To enhance this activity, we further introduce an effective fabrication methodology that combines 3D printing of carbon substrates with precise surface modification using atomic layer deposition (ALD) of TiO2. The resulting TiO2-coated carbon electrode demonstrates significantly improved electrocatalytic properties. For NO2OR, it exhibits a peak current density of 0.75 mA cm−2 at 1.53 V vs. RHE, while for NO2RR, it achieves a yield rate of 630.5 µg h−1 cm−2 with a faradaic efficiency of 81.9% at −1.06 V vs. RHE. This enhancement in electrocatalytic activity is primarily attributed to the formation of abundant interfaces between the conductive carbon and ALD-coated TiO2. The developed methodology not only enables precise modification of 3D printed carbon surface chemistry but also presents a scalable method for electrocatalyst production.
1. Introduction
Ammonia (NH3) is a cornerstone of global agriculture and economy.1 It is increasingly recognized as a potential hydrogen-rich and carbon-free energy carrier, which makes it valuable in the pursuit of sustainable energy storage and transport. Traditionally, NH3 has been produced via the Haber–Bosch (HB) process, which requires stringent conditions of high temperatures (>400 °C) and pressures (>100 bar) to facilitate the reaction between nitrogen and hydrogen (N2 + 3H2 → 2NH3). The HB process relies heavily on carbon-intensive steam methane reforming for H2 production, consuming approximately 1% of the global energy supply derived from fossil fuels. As a result, this process generates a substantial amount of carbon dioxide, accounting for 1–2% of global carbon emissions.2 This significant environmental impact has motivated growing scientific efforts to search for sustainable alternatives to the HB process.Electrochemical NH3 synthesis offers a promising pathway for NH3 production due to its compatibility with intermittent renewable electricity sources.3–6 Among various electrochemical approaches, the electrochemical nitrite reduction reaction (NO2RR) emerges as a compelling alternative to the HB process for decentralized NH3 production.7–9 This primarily stems from the high water solubility of nitrite and the relatively low dissociation energy of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (204 kJ mol−1) compared to the direct cleavage of N
O bond (204 kJ mol−1) compared to the direct cleavage of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N (941 kJ mol−1).10,11 Furthermore, due to excessive emissions and the lack of effective treatment methods, nitrite is prevalent in agricultural and industrial wastewater, which presents a critical challenge to environmental sustainability.12,13 In this context, electrochemical nitrite oxidation reaction (NO2OR)14–17 and conversion to value-added NH3 play crucial roles in both monitoring ecosystem health and mitigating environmental degradation.18 The practical implementation of these electrocatalytic processes is closely linked to the development of efficient electrocatalysts. Recent research on NO2OR and NO2RR has mainly focused on metal-based catalysts,19–21 while carbon materials, despite their proven efficacy in other electrochemical processes (e.g., water splitting) and their widespread use as conductive substrates for electrocatalysts,22–24 remain largely unexplored as direct electrocatalysts for these two reactions. Additionally, scalable and cost-efficient fabrication methods for these carbon electrocatalysts are another important consideration. Although emerging research has begun to explore advanced fabrication techniques, there remains a significant lack of effective methodologies for precisely tuning the surface chemistry of these carbon electrocatalysts to achieve desired functionalities.
N (941 kJ mol−1).10,11 Furthermore, due to excessive emissions and the lack of effective treatment methods, nitrite is prevalent in agricultural and industrial wastewater, which presents a critical challenge to environmental sustainability.12,13 In this context, electrochemical nitrite oxidation reaction (NO2OR)14–17 and conversion to value-added NH3 play crucial roles in both monitoring ecosystem health and mitigating environmental degradation.18 The practical implementation of these electrocatalytic processes is closely linked to the development of efficient electrocatalysts. Recent research on NO2OR and NO2RR has mainly focused on metal-based catalysts,19–21 while carbon materials, despite their proven efficacy in other electrochemical processes (e.g., water splitting) and their widespread use as conductive substrates for electrocatalysts,22–24 remain largely unexplored as direct electrocatalysts for these two reactions. Additionally, scalable and cost-efficient fabrication methods for these carbon electrocatalysts are another important consideration. Although emerging research has begun to explore advanced fabrication techniques, there remains a significant lack of effective methodologies for precisely tuning the surface chemistry of these carbon electrocatalysts to achieve desired functionalities.
3D printing is a promising technique for fabricating self-supported electrocatalysts with on-demand structure and high efficiency, opening up possibilities for scalable production of electrocatalysts.25–27 Particularly, fused deposition modeling (FDM) 3D printing of 1-dimensional (1D) carbon filaments inherently containing Ti-dominated impurities has attracted growing interest in the fabrication of electrocatalytic electrodes.28 Although such 3D-printed 1D electrodes show promising electrocatalytic performance,29 their activity is closely related to the exposure of Ti-dominated impurities on the electrode surface.30,31 However, these impurities account for only about 1 wt%,24 with an even smaller fraction exposed on the electrode surface, thus greatly restricting their broader applications. To overcome this limitation, precise surface engineering of 3D-printed 1D carbon frameworks is essential. Atomic layer deposition (ALD) stands out as an effective and compatible technique for modifying the surface chemistry of 1D electrodes. Unlike conventional methods such as chemical vapor deposition and sputtering, ALD enables the deposition of conformal thin films on high-surface-area, tortuous networks through sequential, self-limiting surface reactions.32,33 This unique capability allows for the creation of uniform functional layers even on complex 3D-printed architectures.24 Furthermore, ALD offers unparalleled control over coating thickness and uniformity at the Ångström scale, a level of precision that preserves the initial porous electrode architecture while introducing desired surface functionalities. This combination of conformality and atomic-level control makes ALD an ideal technique for enhancing the electrocatalytic properties of 3D-printed 1D carbon frameworks, potentially unlocking their full potential in electrochemical NO2OR and NO2RR.
This work explores the potential of 3D-printed 1D carbon materials as direct electrocatalysts for both nitrite detection and conversion. To expand their applications, an advanced electrocatalyst fabrication methodology is developed, which starts with the 3D printing of 1D carbon substrates and culminates in atomic-scale tuning of surface chemistry using TiO2. Precise tuning of TiO2 deposition via ALD is used to engineer the surface structure of 1D carbon, creating abundant interfaces between the conductive carbon nanotubes (CNTs) and ALD-coated TiO2. These CNT/TiO2 interfaces play a pivotal role in enhancing electrocatalytic reactions. The developed methodology enables effective modification of the 3D-printed carbon surface chemistry, thereby expanding the potential of 3D-printed carbon materials for electrocatalytic applications.
2. Experimental section
2.1. Fabrication of carbon frameworks by 3D printing and post-printing activation
Carbon frameworks, with a thickness of 0.45 mm and a diameter of 9 mm, were designed using Autodesk Fusion 360 software. To facilitate connection to the working electrode, a connecting strip was integrated into each carbon framework. The design was then sliced via PrusaSlicer software and exported as a G-code file. Subsequently, the carbon frameworks were 3D printed based on the G-code file using a Prusa i3 MK3 printer (Prusa Research, Czech Republic) and commercially available conductive carbon filament (BlackMagic, Graphene Supermarket, USA). The nozzle and the bed temperature of the printer were set to 230 °C and 60 °C, respectively. The as-printed carbon frameworks were immersed in a 1 M NaOH solution for 3 h to partially remove the surface polylactic acid (PLA) through the chemical saponification reaction, followed by rinsing with deionized water and drying in the air.2.2. Atomic layer deposition of titanium dioxide (TiO2)
The TiO2 was deposited on 3D printed carbon frameworks using an oxygen plasma atomic layer deposition system (ALD, Ultratech/CambridgeNanoTech Fiji 200). Tetrakis(dimethylamido)titanium(IV), 99%, TDMAT, Strem Chemicals, was used as Ti precursor, and oxygen at a rate of 30 sccm was used as the reactant gas under a plasma power of 300 W. Argon served as the carrier gas, flowing at a rate of 60 sccm for Ti precursor and 200 sccm for O2 plasma during the entire deposition process. Prior to deposition, the ALD chamber was heated to 150 °C and stabilized for 60 min, while the stainless steel cylinder containing the Ti precursor was heated and maintained at 75 °C. The relatively low temperature of 150 °C is used during the ALD process to prevent excessive pyrolysis loss of PLA in the 3D-printed carbon frameworks, which is crucial for preserving the mechanical integrity of the 3D-printed structure.34 To achieve varying thicknesses of TiO2 deposits on the carbon frameworks, ALD was performed for 100, 300, and 500 cycles. The resulting samples were denoted as 100-, 300-, and 500-TiO2, respectively. Each ALD cycle consisted of 0.1 s precursor pulse, 30 s purging, 20 s oxygen plasma exposure, and another 5 s purging.2.3. Characterizations
Scanning electron microscopy (SEM) was performed by Verios 460L (Thermo Fisher Scientific, USA) with an electron beam accelerating voltage of 20 kV. The nano-characterization was conducted via transmission electron microscopy (TEM) performed by Thermo Fisher Scientific TITAN Themis 60-300 microscope (Thermo Fisher Scientific, USA) utilizing a high-angle annular dark-field detector for scanning TEM (STEM-HAADF), a Super-X energy-dispersive X-ray (EDX) spectrometer, and a Gatan GIF Quantum ERS electron energy loss spectroscopy (EELS) spectrometer. For STEM-HAADF high-resolution imaging and STEM-EDX mapping, the microscope was operated at an accelerating voltage of 300 kV. To minimize beam damage to the nanoclusters' structure and chemistry during STEM-EELS analysis, the accelerating voltage was reduced to 60 kV. Velox software version 2.12 and GMS software version 3.2 were used to acquire and process STEM-EDX data and STEM-EELS data, respectively. STEM-EDX elemental mapping was performed using net intensities, with the maximum likelihood fit method applied for deconvoluting overlapping peaks and an empirical model for spectrum background subtraction. STEM-EELS elemental mapping acquisition was performed in a dual EELS mode and the maps were created from C–K, Ti–L and O–K edge signal intensities subtracted after Fourier–Ratio plural scattering removal and model-based background removal.X-ray photoelectron spectroscopy (XPS) was performed by an AXIS Supra instrument (Kratos Analytical, UK) using monochromatized AlKα excitation (1486.7 eV) with a pass energy of 100 eV and 20 eV for acquisition of the survey and high-resolution spectra, respectively. The element content analysis was carried out by inductively coupled plasma optical emission spectrometry (ICP-OES, Arcos MV, SPECTRO Analytical Instruments, Kleve, Germany). A detailed procedure for ICP-OES measurements is described in the ESI.† X-ray diffraction (XRD) measurements were performed using Rigaku SmartLab 3 kW diffractometer with a CuKα source (λ = 1.54184 Å), operated at 40 kV and 30 mA. For Raman spectroscopy, a WITec alpha 300R Confocal Raman Microscope was used at a laser excitation of 532 nm and power of 5 mW. Differential scanning calorimetry (DSC) measurements were conducted using the DSC600 Thermal Analysis System (HITACHI) under a nitrogen atmosphere. Further details on the DSC procedures and the calculation of the PLA crystallinity degree are described in the ESI.†
2.4. Electrochemical nitrite oxidation reaction (NO2OR)
The NO2OR activity of different carbon frameworks was probed by cyclic voltammetry (CV) on an AutoLab workstation (PGSTAT204, Metrohm) using three-electrode configuration in an electrolyte containing 0.5 M Na2SO4 and varying concentration of NaNO2. CV tests were performed using a commercial Ag/AgCl electrode (filled with KCl gel electrolyte) and a platinum wire as the reference and counter electrode, respectively. Each experiment was repeated three times to ensure reliability. To study the carbon defects on the electrochemical NO2OR, the commercial pyrolytic graphite electrode (0.071 cm2) from BAS, Inc. (Japan) with basal (PGEb) and edge orientation (PGEe) were used as received. The electrochemically active surface area (ECSA) of PGEb and PGEe was determined using the electrochemical double-layer capacitance, derived from CV measurements in 0.5 M Na2SO4 at varying scan rates, within a non-faradaic potential window. Electrode potentials were converted to the reversible hydrogen electrode (RHE) scale using E (vs. RHE) = E (vs. Ag/AgCl) + 0.059 V × pH + 0.199 V.2.5. Electrochemical nitrite reduction reaction (NO2RR)
The NO2RR experiments were performed on an AutoLab workstation (PGSTAT204, Metrohm), using an H-type glass cell divided by a frit in ambient environment. A commercial Ag/AgCl electrode (filled with KCl gel electrolyte) served as the reference electrode, and a platinum wire acted as the counter electrode. A solution containing 0.1 M NaNO2/0.5 M Na2SO4 was evenly distributed in the anodic and cathodic compartments (20 mL each) of the H-type cell for electrocatalytic assessments. The electrocatalytic tests were carried out at specific potentials for 1 hour with magnetic stirring at 200 rpm to evaluate ammonia yield and Faraday efficiency (FE) of different electrodes. Linear sweep voltammetry (LSV) tests were carried out at a scan rate of 5 mV s−1 to preliminarily assess the NO2RR performance of different electrodes. To ensure reliability, each electrolysis test was repeated three times.2.6. Determination of ammonia
The NH3 concentration after each electrolysis was determined using an ultraviolet-visible (UV-vis) spectrophotometer (JASCO V-750). Firstly, a certain volume of solution was extracted from the electrolytic cell and diluted to a suitable detection range of up to 600 µL. Subsequently, a solution composed of 3 M NaOH, 10 wt% salicylic acid, and 10 wt% sodium citrate (600 µL) was introduced into the diluted solution, followed by the sequential addition of 0.20 M NaClO (300 µL) and 2.0 wt% sodium nitroferricyanide (60 µL). The mixed solution was rested for 2 hours before proceeding with UV-vis spectrophotometry measurements. The concentration of the resulting indophenol blue product was quantified by the absorbance at a wavelength of 655 nm according to a calibration curve, which was established in our prior work by measuring the absorbance of a range of standard NH4Cl solutions in 0.5 M Na2SO4.242.7. Determination of ammonia by 1H-NMR (isotopic labelling experiment)
The isotopic labeling experiment was performed using an electrolyte containing 0.5 M Na2SO4 and 0.1 M K15NO2. After the electrolysis at −1.06 V vs. RHE for 1 h, the catholyte was collected. 50 µL of concentrated H2SO4 was added to 1 mL catholyte to adjust the pH to be acid for quantification by 1H-NMR (700 MHz, Bruker, TXI probe). Then, 20 µL maleic acid (20 mg mL−1) was added as the internal standard. After that, 500 µL of this solution was placed in an NMR tube with addition of 50 µL deuterium oxide (D2O) for NMR detection.3. Results and discussion
3.1. Characterization of 3D printed 1D carbon framework
1D carbon framework was prepared via 3D printing using commercially available 1D carbon filament. After post-printing activation in NaOH to partially eliminate the surface PLA, the CNTs in 1D carbon framework are sufficiently exposed, resulting in high porous structure formed by the presence of entangled CNTs and residual PLA (Fig. 1a and b). The ICP-OES analysis reveals that Ti is the predominant impurity, with a substantial content of 1.07 wt%, which is approximately 7 times higher than the Fe impurities (Fig. 1c). This finding suggests a significant presence of inherent Ti within 3D printed 1D carbon framework, which can potentially affect the carbon framework's electrocatalytic properties.24,31 Therefore, it is necessary to understand the distribution of Ti impurities as well as their chemical states. The distribution of metal impurities was investigated by EDX elemental mapping in STEM. Unlike Fe impurities, which are scarcely observed, Ti-based impurities are readily detectable and found to exist both in the hollow interior (Fig. 1d and e) and on the surface of the CNTs (Fig. S1†). XPS was used to investigate the chemical state of metal impurities and the surface chemistry of the 1D carbon framework. The XPS wide spectrum clearly shows an obvious signal attributed to Ti impurities, alongside the expected C and O peaks (Fig. 1f, upper panel). This confirms the extensive presence of Ti impurities on the surface of the 1D carbon framework. Furthermore, unlike the barely detectable Fe signal (Fig. S2†), the high-resolution Ti 2p spectrum exhibits a well-defined pair of peaks at binding energies of 465.1 eV and 459.4 eV (Fig. 1f, lower panel). This distinct doublet is characteristic of stoichiometric TiO2 with Ti in the 4+ oxidation state, which is further supported by the 5.7 eV peak separation between the Ti 2p3/2 and Ti 2p1/2 peaks.31,35 However, although TiO2 has demonstrated electrocatalytic activity for various electrochemical reactions,24,28,31 its content and distribution within 1D carbon frameworks are currently not well controlled during industrial fabrications of 1D carbon filaments. This lack of controllability over TiO2-dominated impurities results in 3D-printed 1D carbon electrodes with unpredictable and inconsistent electrocatalytic properties, which can vary significantly across different industrial fabrication processes. Therefore, to achieve a uniform surface chemistry for reliable electrocatalytic performance, an atomically precise TiO2 layer is further deposited onto the surface of the 1D carbon framework using ALD technique. | ||
| Fig. 1 (a and b) SEM images and (c) ICP-OES element analyses of 1D carbon framework, excluding elements with content below 0.1 wt%. (d) STEM-HAADF image of a carbon nanotube from 1D carbon framework and corresponding STEM-EDX maps of C, O, and Ti. (e) An EDX spectrum integrated from the whole area of the map in (d); the Cu signal is from the TEM grid. (f) XPS full survey of 1D carbon framework (upper panel), as well as its high-resolution Ti 2p core level spectrum (lower panel). | ||
3.2. Characterization of TiO2-coated 1D carbon framework by ALD
The surface morphology of TiO2-coated 1D carbon framework was firstly studied by SEM. As shown in Fig. 2a–f, the TiO2-coated 1D carbon frameworks maintain their porous structure. Additionally, a distinct increase in the diameter of CNTs is observed with an increase in the number of ALD cycles, implying the possible deposition of TiO2 on 1D carbon frameworks. This observation is supported by ICP-OES analysis, particularly exemplified by a 0.15 wt% increase in Ti content after 500 ALD cycles (Fig. 2g). XPS was employed to investigate the chemical composition of the ALD-coated TiO2 and the resulting surface environment change of the 1D carbon framework. The survey spectra of the TiO2-coated electrodes, depicted in Fig. 2h, exhibit three major peaks corresponding to O 1s, Ti 2p, and C 1s, like the 1D carbon framework. As the number of ALD cycles increases, the TiO2-coated electrodes display a progressive intensification in the O 1s and Ti 2p signals, accompanied by a diminishing C 1s signal. This observation aligns well with the development of an ALD-coated TiO2 shell enveloping the CNT core. The Ti 2p high-resolution spectra of the TiO2-coated electrodes resemble that of the 1D carbon framework (Fig. 2i), exhibiting a spin–orbit splitting of approximately 5.8 eV between the Ti 2p3/2 and Ti 2p1/2 peaks. This indicates the formation of solely TiO2 during the ALD process.36,37 The increasing presence of TiO2 on the 1D carbon framework, as the number of ALD cycles increases, is further evidenced by the growing signal of the lattice O of ALD-coated TiO2 at 530.7 eV (Fig. 2j). Additionally, Raman spectrum of the 1D carbon framework clearly exhibits a D band at 1340 cm−1 (Fig. 2k), demonstrating the inherent existence of carbon defects within the 1D carbon framework. The degree of defects can be quantified by the intensity ratio of the D to G band (ID/IG). Compared to the 1D carbon substrate, the ID/IG ratio remains relatively constant in the TiO2-coated electrodes. It suggests that the ALD process performed in this study does not introduce significant defects onto the CNT surface. Besides, the TiO2 peak (ITiO2) at approximately 150 cm−1 is also visible in the TiO2-coated electrodes, and its ratio to the G band (ITiO2/IG) increases with the number of ALD cycles.32 These observations confirm the effective coating of artificial TiO2 on 1D carbon framework without generating additional carbon defects. Furthermore, the XRD pattern of the TiO2-coated electrodes does not exhibit any significant diffraction peaks of TiO2 (Fig. 2l), suggesting either the formation of amorphous TiO2 or the presence of extremely small nanosized TiO2 crystals that are below the detection limit of XRD.34 It should be noted that PLA diffraction peaks emerge after the ALD process, which implies the PLA crystallization (Fig. S3†). Such crystallization reduces the amorphous regions of PLA substrate, consequently limiting the space available for accommodating CNTs and promoting the formation of a strengthened CNT conductive network.38 This enables more CNTs to contribute to the formation of an electron transfer pathway. To further validate this description, a conductivity test was performed on different TiO2-coated 1D carbon frameworks. As illustrated in Fig. S4,† the 1D carbon substrate exhibits the lowest slope value in the I–V curve, while the highest slope value is observed after 100 ALD cycles of TiO2 coating. This confirms the formation of a strengthened CNT conductive network due to increased crystalline content in the 1D carbon framework following the ALD process. However, further increasing the ALD coating through additional cycles results in reduced electron transfer capability, as evidenced by the smaller I–V slopes in 300-TiO2 and 500-TiO2 electrodes. This decrease in conductivity is attributed to the thicker semiconducting TiO2 layer, which begins to significantly impede electron flow. The change in electron transfer capability resulting from different ALD cycles is closely related to the ECSA, which plays an important role in electrocatalytic reactions (Section 3.5).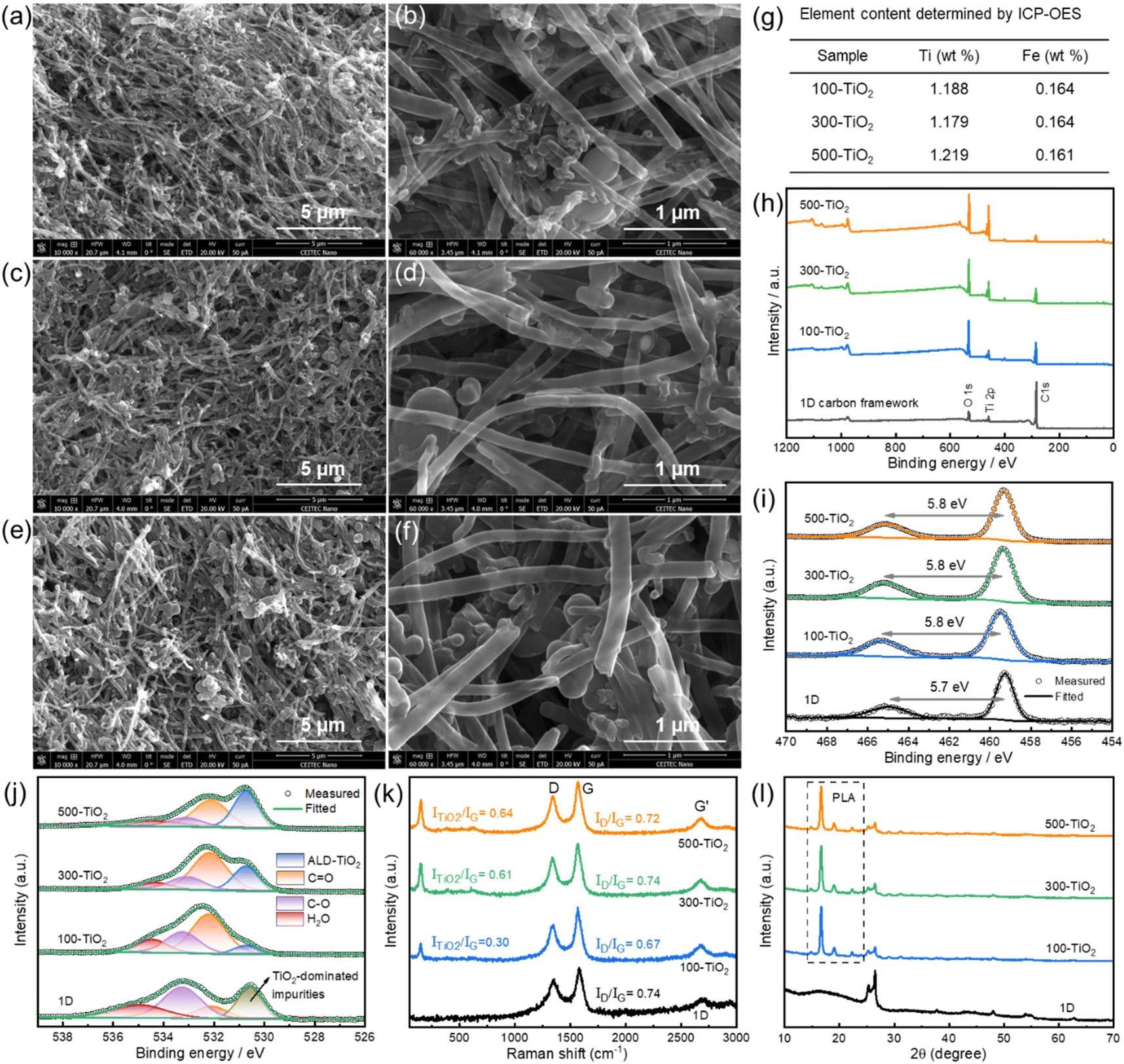 | ||
| Fig. 2 SEM images of (a and b) 100-TiO2, (c and d) 300-TiO2, and (e and f) 500-TiO2. (g) ICP-OES element analyses, excluding elements with weight content below 0.1 wt%. (h) XPS full survey and high-resolution spectra of (i) Ti 2p and (j) O 1s core levels for different electrodes. (k) Raman and (l) XRD spectra of 1D carbon framework before and after ALD of TiO2. For comparison, the pertinent results of the 1D carbon substrate are also displayed in panels (h) to (l). | ||
Nanoscale structural analysis of TiO2-coated 1D carbon frameworks was conducted under STEM-HAADF conditions allowing imaging with a high atomic number contrast. As depicted in Fig. S5,† the CNT surface becomes decorated by TiO2 nanostructures after 500 ALD cycles, which is in significant contrast to the pristine smooth surface of the 1D carbon substrate. Meanwhile, the performed highly localized STEM-EELS clearly reveals the scattered TiO2 nanoclusters on the CNT surface after 100 ALD cycles (Fig. 3a and d and S6†). As the number of ALD cycles increases to 500, the TiO2 nanoclusters progressively grow, overlap each other, and form an almost continuous layer encapsulating the CNT surface (Fig. 3b and S7†). This process leads to the formation of core–shell structured TiO2 electrodes, consisting of a 3D printed conductive CNT core and an ALD-coated electrocatalytic TiO2 shell. Such core–shell structures are clearly visible after 500 cycles, with an ultrathin TiO2 layer ranging from 1 to 3 nm in thickness, as observed in EELS maps (Fig. 3c). These results, in conjunction with SEM, ICP-OES, XPS, and Raman analyses, demonstrate the effectiveness of ALD-coated TiO2 in precisely tuning the surface morphology and chemistry of 3D printed 1D carbon frameworks.
 | ||
| Fig. 3 STEM-ELLS analyses of (a) 100-TiO2, and (b and c) 500-TiO2 electrode. (d) Schematic illustrations for ALD of TiO2 on 3D printed 1D carbon framework. | ||
3.3. Electrocatalytic performance for NO2OR
The 1D carbon framework and 100-TiO2 were used as the working electrodes for CV measurements to assess their electrocatalytic activities towards NO2OR. As shown in Fig. 4a, no electrochemical oxidation peaks are observed from either electrode in the absence of nitrite. However, upon the introduction of nitrite, the 100-TiO2 electrode exhibits nearly a twofold higher peak current density of 0.75 mA cm−2 at a lower peak potential of 1.53 V vs. RHE compared to the 1D carbon electrode (Fig. 4a and b). This indicates the effectiveness of ALD-coated TiO2 nanoclusters in enhancing the electrocatalytic activity on the surface of the 1D carbon electrodes through a two-electron transfer process: NO2− + H2O → NO3− + 2H+ + 2e−.14,39Fig. 4c shows the effect of scan rates on the electrochemical NO2OR activity of 100-TiO2 in 1 mM NaNO2/0.5 M Na2SO4 mixed electrolyte. With the increase of the scan rate from 5 to 100 mV s−1, a positively shifted anodic peak potential is observed from 1.46 V to 1.55 V. Concomitantly, there exists a linear relationship between anodic peak current and the square root of scan rate (v in mV s−1): Ip (mA) = 0.0961 v1/2 + 0.0564 (Fig. 4d). This is indicative of a diffusion-controlled process of nitrite oxidation on the surface of 100-TiO2 electrodes.14,16 Furthermore, the anodic peak current also progressively increases with the increase of nitrite concentration from 0.05 mM to 5 mM (Fig. 4e). Two linear regression regions bounded by the concentration (C in mM) of 0.8 mM are identified (Fig. 4f), which can be expressed as Ip (mA) = 0.7529C + 0.0340 (C ≤ 0.8 mM), and Ip (mA) = 0.5901C + 0.2050 (C ≥ 0.8 mM). The observed change in slope at higher concentrations can be attributed to the transition from an electron transfer controlled regime to a diffusion controlled regime. At low NO2− concentrations, the reaction rate is primarily determined by the kinetics of electron transfer at the electrode surface. As the NO2− concentration increases, mass transport limitations begin to play a more significant role. The reaction becomes increasingly limited by the rate at which NO2− ions can diffuse to the electrode surface. This transition from electron transfer to diffusion control leads to a less steep slope in the current density vs. concentration plot at higher concentrations. | ||
| Fig. 4 (a) CV curves of 1D and 100-TiO2 electrodes at 50 mV s−1 in the absence and presence of 1 mM NaNO2 in 0.5 M Na2SO4 electrolyte. (b) The peak current density and related potential in the presence of 1 mM NaNO2 from CV curves in (a). (c) CV curves of 100-TiO2 electrode in 1 mM NaNO2/0.5 M Na2SO4 mixed electrolyte at different scan rates, as well as (d) the corresponding anodic peak currents as a function of the square root of scan rate. (e) CV curves of 100-TiO2 electrode in 0.5 M Na2SO4 with varying NaNO2 concentrations from 0.05 mM to 5 mM at 50 mV s−1. (f) Linear relationship between the anodic peak current density and nitrite concentration. | ||
3.4. Electrocatalytic performance for NO2RR
The electrochemical NO2RR performance is initially evaluated using LSV. As illustrated in Fig. 5a, both 1D carbon substrate and 100-TiO2 electrode display enhanced current densities in the NaNO2-containing electrolyte compared to the electrolyte without NaNO2, implying their electrocatalytic activity for NO2RR. Notably, the 100-TiO2 electrode exhibits a much higher current density enhancement relative to the 1D carbon electrode. At −1.06 V, for example, the 100-TiO2 electrode presents a current density enhancement of 4.06 mA cm−2 in the NaNO2-containing electrolyte, which is 2.2-fold higher than that of the 1D carbon electrode. This suggests a more efficient NO2RR process occurring on the surface of the 100-TiO2 electrode. To further investigate the impact of ALD-coated TiO2 on the electrochemical NO2RR for NH3 production, electrolysis experiments were performed. It should be noted that nitrogen-containing contaminants from both the external environment and the electrochemical system itself could potentially interfere with post-electrolysis ammonia analysis. To address this concern, control experiments were conducted using a 0.5 M Na2SO4 solution without NaNO2 (Fig. 5b and S8†). These control experiments reveal negligible NH3 production compared to the solution containing NaNO2. This strongly indicates that the NH3 formed during electrolysis primarily results from the electrochemical NO2RR process, rather than from potential contaminants. Such phenomenon is further consolidated by the 15N isotope labelling experiments. As shown in Fig. 5c, typical double peaks of 15NH4+ with Na15NO2 as reactants and triple peaks of 14NH4+ with Na14NO2 as reactants are detected.40 In contrast, no signal is detected when only a blank solution is used during electrolysis. These results conclusively exhibit that the NH3 produced during electrolysis is derived from the NO2− electroreduction. After confirming the reliability of electrochemical NO2RR for NH3 production, chronoamperometry measurements were further performed at different selective potentials (Fig. S9†). The 100-TiO2 electrodes outperform the 1D carbon substrates in NH3 production across all potentials (Fig. 5d and e). For example, 100-TiO2 electrodes deliver an NH3 yield rate of 630.5 µg h−1 cm−2 with a NH3 FE of 81.9% at −1.06 V, which is significantly higher than 204.4 µg h−1 cm−2 and 48.1% for the 1D carbon substrate at the same potential. The durability of the 100-TiO2 electrode was assessed by seven consecutive cycling tests at −1.06 V. Both current density and charge consumption remain nearly constant across all cycles (Fig. S10†). Meanwhile, the electrode maintains roughly constant NH3 yield rates and FE throughout the cycling tests (Fig. 5f). Additionally, SEM (Fig. S11†) and XPS analyses (Fig. S12†) after the cycling test show similar results to those before the test, further confirming the durability of 100-TiO2 electrode during NO2RR process.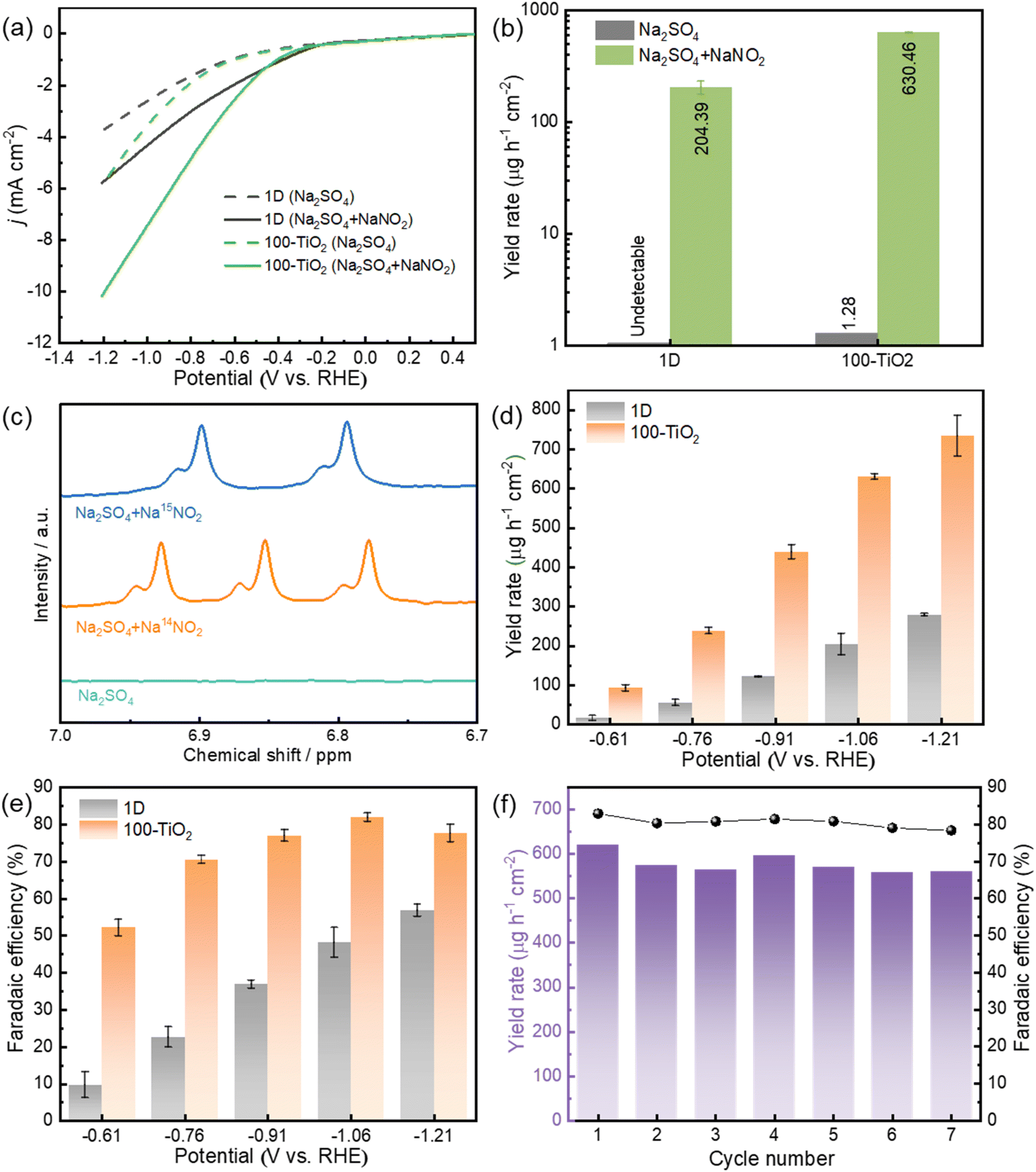 | ||
| Fig. 5 (a) LSV curves of 1D and 100-TiO2 in 0.5 M Na2SO4 with and without 0.1 M NaNO2. (b) NH3 yield of 1D and 100-TiO2 after NO2RR in 0.5 M Na2SO4 with and without 0.1 M NaNO2 at −1.06 V vs. RHE. (c) NMR spectra of the cathodic solutions after electrolysis on 100-TiO2 at −1.06 V vs. RHE using different electrolytes. (d) NH3 yield and (e) FE of 1D and 100-TiO2 at different potentials. (f) Stability test of 100-TiO2 at −1.06 V vs. RHE with refreshed electrolyte for each cycle. | ||
3.5. Electrocatalytic active sites for NO2OR and NO2RR
Carbon defects within 1D carbon substrate have been demonstrated as potential electrocatalytic active sites.24,41,42 Particularly, when the ALD-coated material does not fully cover the carbon substrate, carbon defects may remain exposed and accessible to the electrolyte, potentially participating in electrocatalytic reactions. Therefore, elucidating the influence of defective CNTs on the electrocatalytic activity of ALD-coated 1D carbon electrodes is of fundamental importance. To this end, control experiments were conducted on both PGEb and PGEe. Fig. 6a presents well-defined nitrite oxidation peaks on both PGEb and PGEe, confirming the electrocatalytic activity of both basal and edge carbon planes for nitrite oxidation. Notably, PGEe exhibits a negatively shifted peak potential with higher peak current density compared to PGEb, indicating its higher activity over PGEb for nitrite oxidation. Furthermore, the ALD-coated TiO2 layer plays a crucial role in modulating the electrocatalytic performance of the 1D carbon framework (Fig. 6b and S13†). It is observed that 100 ALD cycles of TiO2 on 1D carbon framework significantly enhance the electrocatalytic activity for nitrite oxidation. This enhancement is evidenced by a negatively shifted anodic peak from 1.62 V to 1.53 V, accompanied by an increase in peak current density from 0.39 mA cm−2 to 0.75 mA cm−2. However, further increasing the TiO2 coating to 300 and 500 ALD cycles leads to a progressive decrease in activity, represented by positively shifted anodic peaks and diminishing peak current densities. This phenomenon is attributed to the morphological evolution of the TiO2 layer with increasing ALD cycles. The 100-TiO2 electrode exhibits a non-continuous distribution of TiO2 nanoclusters (Fig. 3a) due to the inefficient nucleation at early stages during the ALD process.43 This non-continuous distribution creates abundant conductive CNT/semiconductive TiO2 interfaces, which enhance electrocatalytic reactions by facilitating electron transfer between TiO2 and CNT (Fig. S4†), and providing increased ECSA (Fig. S14†) for electrocatalytic reactions. As the number of ALD cycles increases to 300 and 500, the TiO2 layer becomes denser and more continuous (Fig. 3b and c), resulted from the coalescence of TiO2 nanoclusters and progressive gap-filling between initial nucleation sites. This evolution reduces the CNT/TiO2 interfaces, leading to a decrease in electron transfer capability and in ECSA for electrocatalytic reactions. | ||
| Fig. 6 (a) CV curves of graphite with different plane orientations at 50 mV s−1 in the absence and presence of 1 mM NaNO2 in 0.5 M Na2SO4 electrolyte. (b) The peak current density and related potential in the presence of 1 mM NaNO2 from CV curves of different electrodes. (c) LSV curves of graphite with different plane orientations in 0.5 M Na2SO4 with and without 0.1 M NaNO2. (d) NH3 yield and FE of graphite with different plane orientations at −1.06 V vs. RHE. (e) LSV curves of 100-TiO2, 300-TiO2, and 500-TiO2 at 5 mV s−1 in 0.5 M Na2SO4 with and without 0.1 M NaNO2. (f) NH3 yield and FE of 1D carbon framework with TiO2 coating by varying ALD cycles at −1.06 V vs. RHE. | ||
In parallel, we investigated the origin of electrocatalytic active sites for NO2RR. The LSV profile of PGEe (Fig. 6c) exhibits a significantly higher current density relative to PGEb in the presence of nitrite, with an onset potential around −0.66 V. This suggests more efficient NO2RR on the surface of PGEe over PGEb. At −1.06 V, PGEe exhibits a current density enhancement of 10.96 mA cm−2 in nitrite-containing electrolyte compared to nitrite-free electrolyte, which is 3.4-fold higher than that for PGEb. Electrolysis experiments conducted at −1.06 V (Fig. 6d and S15†) demonstrate that PGEe produces a higher NH3 yield than PGEb (231.6 vs. 163.7 µg h−1 cm−2), but with comparable low FE (10.5% vs. 11.4%). This suggests that PGEb and PGEe possess similar intrinsic activity for NO2RR to NH3. The enhanced electrocatalytic NH3 yield on PGEe is believed to be associated with its higher ECSA (Fig. S16†).
Furthermore, we investigated the effect of ALD-coated TiO2 layers on the 1D carbon framework for NO2RR performance. Fig. 6e illustrates that, compared to 100-TiO2, the LSV profiles of 300- and 500-TiO2 exhibit progressively decreased current densities in nitrite-containing electrolyte, while showing comparable current densities in nitrite-free electrolyte. This suggests that the morphology of ALD-coated TiO2 plays a crucial role in NO2RR. To quantitatively analyze this effect, electrolysis experiments were further conducted (Fig. S17†). In contrast to 1D carbon framework, 100-TiO2 with its abundant carbon/TiO2 interfaces significantly enhances the NH3 yield rate from 204.4 to 630.5 µg h−1 cm−2, accompanied by a substantial increase in FE from 48.1% to 81.9% (Fig. 6f). Similar to the trend observed in NO2OR, further increasing the TiO2 coating leads to the decreased NO2RR performance due to a reduction in carbon/TiO2 interfaces by the formation of a denser and more continuous TiO2 layer encapsulating the CNT surface. Specifically, 300-TiO2 exhibits an NH3 yield of 555.1 µg h−1 cm−2 with an FE of 74.8%, which remains approximately constant for 500-TiO2. These findings highlight the critical role of optimizing the morphology of ALD-coated TiO2 and preservation of carbon/TiO2 interfaces for optimum electrocatalytic activity.
4. Conclusion
This study addresses the growing need for efficient and cost-effective electrocatalysts by demonstrating the efficacy of 3D printed 1D carbon materials for electrochemical NO2OR and NO2RR. The findings reveal that entangled carbon nanotubes, inherently containing TiO2-dominated impurities and carbon defects, exhibit electrocatalytic activity for both reactions. Control experiments confirm that the intrinsic surface of carbon nanotubes is electrocatalytically active for NO2OR and NO2RR, while carbon defects further enhance this activity by providing a higher ECSA. To enhance the electrocatalytic performance of the 3D printed 1D carbon framework, ALD of TiO2 has been explored as a surface modification technique. Results demonstrate that the surface morphology and chemistry of the TiO2-coated 1D carbon electrode are intimately related to the layer of the ALD-coated TiO2, which can be precisely controlled by adjusting the number of ALD cycles. Remarkably, the 100-TiO2 electrode, characterized by non-continuous TiO2 deposition on the 1D carbon framework, facilitates the formation of abundant carbon/TiO2 interfaces. These interfaces play a crucial role in electrocatalytic reactions. The methodology presented in this study, encompassing 3D printing of 1D carbon substrates and subsequent atomic-scale tuning of surface chemistry, offers a scalable fabrication process for electrocatalysts, which opens up new possibilities for tailoring materials at the nanoscale for specific electrochemical applications.Data availability
Data will be made available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the ERDF/ESF project TECHSCALE (No. CZ.02.01.01/00/22_008/0004587). This research was co-funded by the European Union under the REFRESH-Research Excellence For Region Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Programme Just Transition. Material fabrication and characterizations were carried out with the support of CzechNanoLab Research Infrastructure (ID LM2023051, MEYS CR). The authors gratefully thank Dr Martin Loula for IPC-OES measurements and analyses, and Dr Richard Slávik for DSC measurements and analyses.References
- S. Z. Andersen, V. Colic, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Norskov and I. Chorkendorff, A rigorous electrochemical ammonia synthesis protocol with quantitative isotope measurements, Nature, 2019, 570, 504–508, DOI:10.1038/s41586-019-1260-x.
- H. Iriawan, S. Z. Andersen, X. Zhang, B. M. Comer, J. Barrio, P. Chen, A. J. Medford, I. E. L. Stephens, I. Chorkendorff and Y. Shao-Horn, Methods for nitrogen activation by reduction and oxidation, Nat. Rev. Methods Primers, 2021, 1, 56, DOI:10.1038/s43586-021-00053-y.
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Electrocatalytic nitrate reduction for sustainable ammonia production, Joule, 2021, 5, 290–294, DOI:10.1016/j.joule.2020.12.025.
- K. Kim, A. Zagalskaya, J. L. Ng, J. Hong, V. Alexandrov, T. A. Pham and X. Su, Coupling nitrate capture with ammonia production through bifunctional redox-electrodes, Nat. Commun., 2023, 14, 823, DOI:10.1038/s41467-023-36318-1.
- Z. Xu, L. Wan, Y. Liao, M. Pang, Q. Xu, P. Wang and B. Wang, Continuous ammonia electrosynthesis using physically interlocked bipolar membrane at 1000 mA cm−2, Nat. Commun., 2023, 14, 1619, DOI:10.1038/s41467-023-37273-7.
- C. Feng, W. Zhou, H. Wu, Q. Huo, J. Shao, X. Li, H. Yang, Q. Hu and C. He, Tuning work function difference of copper/cobalt oxides heterointerfaces enables efficient electrochemical nitrate reduction, Appl. Catal., B, 2024, 341, 123280, DOI:10.1016/j.apcatb.2023.123280.
- J. Lim, C.-Y. Liu, J. Park, Y.-H. Liu, T. P. Senftle, S. W. Lee and M. C. Hatzell, Structure sensitivity of Pd facets for enhanced electrochemical nitrate reduction to ammonia, ACS Catal., 2021, 11, 7568–7577, DOI:10.1021/acscatal.1c01413.
- Y. Zhang, Y. Wang, L. Han, S. Wang, T. Cui, Y. Yan, M. Xu, H. Duan, Y. Kuang and X. Sun, Nitrite electroreduction to ammonia promoted by molecular carbon dioxide with near-unity faradaic efficiency, Angew. Chem., Int. Ed., 2023, 62, e202213711, DOI:10.1002/anie.202213711.
- J. V. Perales-Rondon, D. Rojas, W. Gao and M. Pumera, Copper 3D-printed electrodes for ammonia electrosynthesis via nitrate reduction, ACS Sustainable Chem. Eng., 2023, 11, 6923–6931, DOI:10.1021/acssuschemeng.2c06851.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper–molecular solid catalyst, Nat. Energy, 2020, 5, 605–613, DOI:10.1038/s41560-020-0654-1.
- K. Fan, W. Xie, J. Li, Y. Sun, P. Xu, Y. Tang, Z. Li and M. Shao, Active hydrogen boosts electrochemical nitrate reduction to ammonia, Nat. Commun., 2022, 13, 7958, DOI:10.1038/s41467-022-35664-w.
- S. Liu, L. Cui, S. Yin, H. Ren, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Heterointerface-triggered electronic structure reformation: Pd/CuO nano-olives motivate nitrite electroreduction to ammonia, Appl. Catal., B, 2022, 319, 121876, DOI:10.1016/j.apcatb.2022.121876.
- J. Yuan, H. Yin, X. Jin, D. Zhao, Y. Liu, A. Du, X. Liu and A. P. O'Mullane, A practical FeP nanoarrays electrocatalyst for efficient catalytic reduction of nitrite ions in wastewater to ammonia, Appl. Catal., B, 2023, 325, 122353, DOI:10.1016/j.apcatb.2022.122353.
- W. Zhu, Y. Zhang, J. Gong, Y. Ma, J. Sun, T. Li and J. Wang, Surface engineering of carbon fiber paper toward exceptionally high-performance and stable electrochemical nitrite sensing, ACS Sens., 2019, 4, 2980–2987, DOI:10.1021/acssensors.9b01474.
- J. F. S. Pereira, R. G. Rocha, S. V. F. Castro, A. F. João, P. H. S. Borges, D. P. Rocha, A. de Siervo, E. M. Richter, E. Nossol, R. V. Gelamo and R. A. A. Muñoz, Reactive oxygen plasma treatment of 3D-printed carbon electrodes towards high-performance electrochemical sensors, Sens. Actuators, B, 2021, 347, 130651, DOI:10.1016/j.snb.2021.130651.
- H. Wang, X. Wang and J. Cheng, Bionic enzyme-assisted ion-selective amperometric biosensor based on 3D porous conductive matrix for point-of-care nitrite testing, ACS Nano, 2022, 16, 14849–14859, DOI:10.1021/acsnano.2c05752.
- B. R. Kozub, N. V. Rees and R. G. Compton, Electrochemical determination of nitrite at a bare glassy carbon electrode; why chemically modify electrodes?, Sens. Actuators, B, 2010, 143, 539–546, DOI:10.1016/j.snb.2009.09.065.
- Z. Cai, D. Zhao, X. Fan, L. Zhang, J. Liang, Z. Li, J. Li, Y. Luo, D. Zheng, Y. Wang, T. Li, H. Yan, B. Ying, S. Sun, A. A. Alshehri, H. Yan, J. Xu, Q. Kong and X. Sun, Rational construction of heterostructured Cu3P@TiO2 nanoarray for high-efficiency electrochemical nitrite reduction to ammonia, Small, 2023, 19, 2300620, DOI:10.1002/smll.202300620.
- F. Wang, H. Zhao, G. Zhang, H. Zhang, X. Han and K. Chu, Electroreduction of nitrite to ammonia over Ni1Ru single-atom alloys, Adv. Funct. Mater., 2024, 34, 2308072, DOI:10.1002/adfm.202308072.
- H. Zhao, J. Xiang, Z. Sun, S. Shang and K. Chu, Electroreduction of nitrite to ammonia over a cobalt single-atom catalyst, ACS Sustainable Chem. Eng., 2024, 12, 2783–2789, DOI:10.1021/acssuschemeng.3c07388.
- Z. Cai, C. Ma, D. Zhao, X. Fan, R. Li, L. Zhang, J. Li, X. He, Y. Luo, D. Zheng, Y. Wang, B. Ying, S. Sun, J. Xu, Q. Lu and X. Sun, Ni doping enabled improvement in electrocatalytic nitrite-to-ammonia conversion over TiO2 nanoribbon, Mater. Today Energy, 2023, 31, 101220, DOI:10.1016/j.mtener.2022.101220.
- Y. Cheng, J. Zhang and S. P. Jiang, Are metal-free pristine carbon nanotubes electrocatalytically active?, Chem. Commun., 2015, 51, 13764–13767, 10.1039/c5cc02218e.
- L. Cheng, T. Ma, B. Zhang, L. Huang, W. Guo, F. Hu, H. Zhu, Z. Wang, T. Zheng, D.-T. Yang, C.-K. Siu, Q. Liu, Y. Ren, C. Xia, B. Z. Tang and R. Ye, Steering the topological defects in amorphous laser-induced graphene for direct nitrate-to-ammonia electroreduction, ACS Catal., 2022, 12, 11639–11650, DOI:10.1021/acscatal.2c03219.
- W. Gao, J. V. Perales-Rondon, J. Michalička and M. Pumera, Ultrathin manganese oxides enhance the electrocatalytic properties of 3D printed carbon catalysts for electrochemical nitrate reduction to ammonia, Appl. Catal., B, 2023, 330, 122632, DOI:10.1016/j.apcatb.2023.122632.
- E. M. Richter, D. P. Rocha, R. M. Cardoso, E. M. Keefe, C. W. Foster, R. A. A. Munoz and C. E. Banks, Complete additively manufactured (3D-printed) electrochemical sensing platform, Anal. Chem., 2019, 91, 12844–12851, DOI:10.1021/acs.analchem.9b02573.
- F. Xie, X. Cui, X. Zhi, D. Yao, B. Johannessen, T. Lin, J. Tang, T. B. F. Woodfield, L. Gu and S.-Z. Qiao, A general approach to 3D-printed single-atom catalysts, Nat. Synth., 2023, 2, 129–139, DOI:10.1038/s44160-022-00193-3.
- W. Gao and M. Pumera, 3D Printed nanocarbon frameworks for Li-ion battery cathodes, Adv. Funct. Mater., 2021, 31, 2007285, DOI:10.1002/adfm.202007285.
- C. W. Foster, M. P. Down, Y. Zhang, X. Ji, S. J. Rowley-Neale, G. C. Smith, P. J. Kelly and C. E. Banks, 3D printed graphene based energy storage devices, Sci. Rep., 2017, 7, 42233, DOI:10.1038/srep42233.
- F. Novotny, V. Urbanova, J. Plutnar and M. Pumera, Preserving fine structure details and dramatically enhancing electron transfer rates in graphene 3D-printed electrodes via thermal annealing: toward nitroaromatic explosives sensing, ACS Appl. Mater. Interfaces, 2019, 11, 35371–35375, DOI:10.1021/acsami.9b06683.
- M. Pumera, A. Ambrosi and E. L. K. Chng, Impurities in graphenes and carbon nanotubes and their influence on the redox properties, Chem. Sci., 2012, 3, 3347–3355, 10.1039/c2sc21374e.
- M. P. Browne, V. Urbanova, J. Plutnar, F. Novotný and M. Pumera, Inherent impurities in 3D-printed electrodes are responsible for catalysis towards water splitting, J. Mater. Chem. A, 2020, 8, 1120–1126, 10.1039/c9ta11949c.
- P. Kaushik, M. Eliáš, J. Michalička, D. Hegemann, Z. Pytlíček, D. Nečas and L. Zajíčková, Atomic layer deposition of titanium dioxide on multi-walled carbon nanotubes for ammonia gas sensing, Surf. Coat. Technol., 2019, 370, 235–243, DOI:10.1016/j.surfcoat.2019.04.031.
- J. Han, Y. Kim, D. H. K. Jackson, H. Chang, H. W. Kim, J. Lee, J.-R. Kim, Y. Noh, W. B. Kim, K.-Y. Lee and H. J. Kim, Enhanced catalytic performance and changed reaction chemistry for electrochemical glycerol oxidation by atomic-layer-deposited Pt-nanoparticle catalysts, Appl. Catal., B, 2020, 273, 119037, DOI:10.1016/j.apcatb.2020.119037.
- W. Gao, J. Michalicka and M. Pumera, Hierarchical atomic layer deposited V2O5 on 3D printed nanocarbon electrodes for high-performance aqueous zinc-ion batteries, Small, 2022, 18, 2105572, DOI:10.1002/smll.202105572.
- A. C. Bronneberg, C. Höhn and R. van de Krol, Probing the interfacial chemistry of ultrathin ALD-grown TiO2 films: an in-line XPS study, J. Phys. Chem. C, 2017, 121, 5531–5538, DOI:10.1021/acs.jpcc.6b09468.
- M. E. Dufond, M. W. Diouf, C. Badie, C. Laffon, P. Parent, D. Ferry, D. Grosso, J. C. S. Kools, S. D. Elliott and L. Santinacci, Quantifying the extent of ligand incorporation and the effect on properties of TiO2 thin films grown by atomic layer deposition using an alkoxide or an alkylamide, Chem. Mater., 2020, 32, 1393–1407, DOI:10.1021/acs.chemmater.9b03621.
- M. P. Browne, J. Plutnar, A. M. Pourrahimi, Z. Sofer and M. Pumera, Atomic layer deposition as a general method turns any 3D-printed electrode into a desired catalyst: case study in photoelectrochemisty, Adv. Energy Mater., 2019, 9, 1900994, DOI:10.1002/aenm.201900994.
- W. Gao, C. Iffelsberger and M. Pumera, Dual polymer engineering enables high-performance 3D printed Zn-organic battery cathodes, Appl. Mater. Today, 2022, 28, 101515, DOI:10.1016/j.apmt.2022.101515.
- G. Li, Y. Xia, Y. Tian, Y. Wu, J. Liu, Q. He and D. Chen, Review-recent developments on graphene-based electrochemical sensors toward nitrite, J. Electrochem. Soc., 2019, 166, B881–B895, DOI:10.1149/2.0171912jes.
- Z. Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F. Y. Chen, J. Y. T. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst, Nat. Commun., 2021, 12, 2870, DOI:10.1038/s41467-021-23115-x.
- C. E. Banks, T. J. Davies, G. G. Wildgoose and R. G. Compton, Electrocatalysis at graphite and carbon nanotube modified electrodes: edge-plane sites and tube ends are the reactive sites, Chem. Commun., 2005, 829–841, 10.1039/b413177k.
- M. Velický, P. S. Toth, C. R. Woods, K. S. Novoselov and R. A. W. Dryfe, Electrochemistry of the basal plane versus edge plane of graphite revisited, J. Phys. Chem. C, 2019, 123, 11677–11685, DOI:10.1021/acs.jpcc.9b01010.
- C. Marichy, M. Bechelany and N. Pinna, Atomic layer deposition of nanostructured materials for energy and environmental applications, Adv. Mater., 2012, 24, 1017–1032, DOI:10.1002/adma.201104129.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06800a |
| This journal is © The Royal Society of Chemistry 2024 |