
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Delithiation-induced secondary phase formation in Li-rich cathode materials
Yin-Ying
Ting
 abd,
Ben
Breitung
c,
Simon
Schweidler
c,
Junbo
Wang
c,
Michael
Eikerling
ad,
Piotr M.
Kowalski
ab,
Olivier
Guillon
e and
Payam
Kaghazchi
*ef
abd,
Ben
Breitung
c,
Simon
Schweidler
c,
Junbo
Wang
c,
Michael
Eikerling
ad,
Piotr M.
Kowalski
ab,
Olivier
Guillon
e and
Payam
Kaghazchi
*ef
aInstitute of Energy Technologies (IET-3), Forschungszentrum Jülich, Wilhelm-Johnen-Straße, 52428 Jülich, Germany
bJülich Aachen Research Alliance, JARA Energy & Center for Simulation and Data Science (CSD), 52428 Jülich, Germany
cInstitute of Nanotechnology, Karlsruhe Institute of Technology, Kaiserstraße 12, 76131 Karlsruhe, Germany
dChair of Theory and Computation of Energy Materials, Faculty of Georesources and Materials Engineering, RWTH Aachen University, 52062 Aachen, Germany
eInstitute of Energy Materials and Devices: Materials Synthesis and Processing (IMD-2), Forschungszentrum Jülich, Wilhelm-Johnen-Straße, 52428 Jülich, Germany
fMESA+ Institute for Nanotechnology, University of Twente, Enschede, 7500 AE, The Netherlands
First published on 14th November 2024
Abstract
Li-rich layered oxides can potentially provide high capacity, thereby enhancing energy density as cathode materials in Li-ion batteries. However, one of the main drawbacks is their low cycling stability. It has been proposed that the structural stability of a solid solution compound might be enhanced by exploiting the high-entropy concept. Here, we studied two Li-rich layered oxide cathode materials with multiple cations in their transition metal sites, categorized as medium or high entropy: Li(Li0.2Co0.18Ni0.18Mn0.44)O2 and Li(Li0.2Co0.18Ni0.18Mn0.18Ti0.26)O2. The synthesized materials, however, experienced a large capacity loss during the first charge/discharge cycle. We performed first-principles calculations to understand the mechanism behind the capacity fading and discovered significant structural changes in both systems. Specifically, we observed extensive Li/Ni interchange, migration of transition metal ions to Li sites, and formation of secondary phases. For the Ti-containing material, which shows a larger capacity fade than the other system, we even observed the formation of a spinel phase. The computed enthalpies of secondary phase formation reactions exhibit large negative values. However, the estimated (maximum) configurational entropy contributions to the free energies of these reactions are much smaller and therefore not determining factors. This study provides crucial insights into degradation mechanisms in Li-rich high-entropy systems, aiding the future design and development of advanced cathode materials for next-generation lithium-ion batteries.
1 Introduction
Lithium-ion batteries (LIBs) are important energy storage systems due to their relatively high energy density and long-term cycle stability. However, their performance still needs improvement for future mobile applications. As cathode electrodes play a key role in determining the energy density and stability of LIBs, they have been extensively studied.1,2 Nevertheless, many fundamental questions regarding the mechanism of the charge/discharge (delithiation/lithiation) process remain to be addressed to aid in the design of novel cathode materials.The high-entropy (HE) concept is a relatively recent development in the field of materials science. A HE solid solution compound involves a combination of multiple different elements, typically five or more, in a single phase lattice structure.3 The high configurational entropy resulting from a large number of possible combinations is assumed to play a crucial role in stabilizing the material's structure.4,5 The application of HE concept to various ceramics shows promise for developing new materials in battery research. For example, high-entropy fluorides (HEFs) serve as high-capacity conversion cathodes for LIBs,6 while high-entropy Prussian White has been demonstrated to enhance structural stability upon electrochemical cycling when used as cathode material in sodium-ion batteries.7 Another emerging category of cathode materials gaining recognition due to their distinct properties is high-entropy oxides (HEOs).8–13 Numerous theoretical and experimental analyses have revealed superior electrochemical properties of HEOs, including high capacity, cycle stability, and relatively high rate capability.3,4,9,14–16 For example, a cation-disordered high-entropy cathode with five different transition metal (TM) species can achieve 590 mA h g−1 at 50 mA g−1, demonstrating better cycle stability compared to cathodes with only four different TM species.15 A considerable compositional adaptability enables a wide exploration of element combinations to uncover optimized compositions with diverse elements decreasing the dependence on critical elements. Thus, HEOs can be potentially promising alternatives to traditional cathode materials.
Additionally, Li-rich layered oxides (LLOs) have been identified as another interesting cathode material.17,18 They can potentially provide high capacity (over 250 mA h g−1), making them attractive candidates for high-performance battery applications.19–21 Unlike traditional cathodes in lithium batteries, which predominantly utilize the redox activity of transition metals for electrochemical reactions, LLOs can offer a mixed redox potential of both TMs and oxygen.22–24 By this synergistic approach, they are able to achieve an exceptionally high capacity, surpassing what conventional cathode materials can provide. However, the structural instability of these materials exhibits non-negligible voltage decay over electrochemical cycling, presenting an obstacle to their practical application.25–28
In this work, we investigate synergy effects between the high-entropy and Li-rich layered concepts. Structural stability, redox mechanism, and phase stability of two materials were analyzed using a combination of computational and experimental techniques.
2 Method
2.1 Computational method
We employed spin-polarized density functional theory (DFT) calculations using the Quantum-ESPRESSO simulation package29,30 to compute the atomistic structure and magnetic properties of LLOs, namely Li(Li0.2Co0.18Ni0.18Mn0.44)O2 (MELLO) and Li(Li0.2Co0.18Ni0.18Mn0.18Ti0.26)O2 (HELLO). Ultrasoft pseudopotentials31 and the PBEsol exchange–correlation functional32 were utilized for the optimization of geometries and unit cells. The application of this functional for the prediction of structural parameters of solids has been validated in previous studies.33,34 A comparison with the PBE exchange–correlation functional35 will be discussed. For the calculation of strongly correlated electrons, the Hubbard-U correction was applied, with U values of 6.0, 3.4, and 3.9 eV for Ni, Co, and Mn respectively, following the ref. 36. We also calculated the electronic structure of the delithiated MELLO system employing the HSE06 hybrid functional.37 We used a plane-wave cutoff energy of 50 Ry and a 2 × 2 × 2 k-point mesh, except for the hybrid functional calculation, for which a k-point mesh of 1 × 1 × 1 supplemented by a q-point mesh of 1 × 1 × 1 was used. An energy and force convergence criterion of 1.36 × 10−3 eV and 2.57 × 10−2 eV Å−1, respectively, were applied. The plane-wave cutoff energy and k-point mesh were selected to ensure convergence of all reported values. To ensure we obtain the magnetic ground state, we imposed a constraint on the total magnetic moment, which was estimated from the most likely oxidation state of each TM. We calculated all possible total magnetization values based on these oxidation states and then performed calculations for each spin configuration. After reaching electronic and geometric convergence (including cell optimization), we relaxed the constraint and re-ran the calculations to see if the system would converge into a different state. Finally, we selected the configuration with the lowest total energy, ensuring that this is the most stable magnetic state.To mimic the disordered atomic arrangement of the LLOs, the special quasirandom structure (SQS) approach38 was applied. The SQSs were generated using the mcsqs code as part of the Alloy Theoretic Automated Toolkit (ATAT) package.39 The SQS method statistically mimics a truly disordered state by providing an “average” atomic configuration that represents the fully disordered structure. As an average disordered configuration, the SQS is well suited for the computation of thermodynamic parameters of the material. This method is particularly suited for high-entropy systems, where considering all potential configurations is impractical. The determined structures by SQS method have even been used to quantify the phase stability, electronic structure, and voltage curves in other studies.16,40–42 We modeled the MELLO system using 2 × 6 × 2 supercells comprising 192 atoms per cell with a space group of C2/m and 4 × 4 × 1 supercells containing 192 atoms per cell with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. This was to determine the most stable structure for this compound. Although the C2/m and Rm structures do not have exactly the same TM configuration due to the different number of TM sites in these structures, we ensured that the short-range correlations (pair correlations) between TM atoms are very similar, allowing for a fair comparison of their relative stabilities. For the HELLO system, a 4 × 4 × 1 supercell was used, having 192 atoms per cell and a space group of Rm. To obtain the atomistic structure of LLO systems with Li/Ni interchanges and the low Li concentration of x = 0.25, we performed total Coulomb Energy (CE) calculations utilizing the ‘supercell’ code.43 We selected five configurations with the lowest Coulomb energies and calculated the total energies using DFT. Our results confirm that the configuration with the lowest Coulomb energy is the most stable. Subsequently, we applied the DFT+U method with the PBEsol functional and HSE06 hybrid functional calculations to these configurations.
m. This was to determine the most stable structure for this compound. Although the C2/m and Rm structures do not have exactly the same TM configuration due to the different number of TM sites in these structures, we ensured that the short-range correlations (pair correlations) between TM atoms are very similar, allowing for a fair comparison of their relative stabilities. For the HELLO system, a 4 × 4 × 1 supercell was used, having 192 atoms per cell and a space group of Rm. To obtain the atomistic structure of LLO systems with Li/Ni interchanges and the low Li concentration of x = 0.25, we performed total Coulomb Energy (CE) calculations utilizing the ‘supercell’ code.43 We selected five configurations with the lowest Coulomb energies and calculated the total energies using DFT. Our results confirm that the configuration with the lowest Coulomb energy is the most stable. Subsequently, we applied the DFT+U method with the PBEsol functional and HSE06 hybrid functional calculations to these configurations.
2.2 Materials synthesis
The medium-entropy oxide Li(Li0.2Co0.18Ni0.18Mn0.44)O2 and the high-entropy oxide Li(Li0.2Co0.18Ni0.18Mn0.18Ti0.26)O2 were synthesized employing a modified Pechini method. Co(CH3COO)2·4H2O, Ni(CH3COO)2·4H2O, Mn(CH3COO)2·4H2O, titanium butoxide (C16H36O4Ti), and excess (50%) LiNO3 (all Sigma-Aldrich) were utilized as initial materials. Stoichiometric quantities of these materials were dissolved and dispersed in ethanol with citric acid and ethylene glycol (molar ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) under stirring to form a precursor. Following 4 hours of reaction with stirring and overnight drying, the mixtures were heated to 400 °C for 2 h, during which they swelled up and burned spontaneously, forming foamy masses as the temperature increased. The resulting black powder was pressed into pellets and calcined at 950 °C for 12 h in air. The final products were obtained by natural cooling to ambient temperature and kept inside an Ar-filled glovebox (O2 < 0.1 ppm, H2O < 0.1 ppm).
:4) under stirring to form a precursor. Following 4 hours of reaction with stirring and overnight drying, the mixtures were heated to 400 °C for 2 h, during which they swelled up and burned spontaneously, forming foamy masses as the temperature increased. The resulting black powder was pressed into pellets and calcined at 950 °C for 12 h in air. The final products were obtained by natural cooling to ambient temperature and kept inside an Ar-filled glovebox (O2 < 0.1 ppm, H2O < 0.1 ppm).
2.3 Electrode preparation
Cathodes were prepared by mixing oxide, Super C65 black carbon, and Solef 5130 polyvinylidene fluoride (PVDF) in a ratio of 7:2:1, uniformly dispersed in N-methyl-2-pyrrolidone (NMP). The slurry was then cast onto an Al current collector by doctor blading (200 μm slit size) and dried under vacuum at 120 °C for 12 h. The mass of the loaded active material on the dried foil was 2 to 3 mg cm−2. Electrode discs of diameter 13 mm were punched from the cathode sheet and used as cathode electrodes for CR2032 coin cell assembly. The cells were assembled in an Ar-filled glovebox, using glass microfiber filter paper (GF/C, Whatman, Freiburg, Germany) as the separator, 1 M LiPF6 in a 3:7 weight mixture of ethylene carbonate:ethyl methyl carbonate (LP57, BASF SE, Ludwigshafen, Germany) as the electrolyte, and Li metal foil (Gelon LIB Co., Ltd) as the counter electrode.
2.4 Cycling
Galvanostatic cycling tests were conducted using a multichannel battery test system (CT2001A, Wuhan LAND Electronic Co., Ltd). The voltage range was set to 2.0–4.8 V vs. Li+/Li, and the specific capacity was calculated based on the mass of the active material (1C = 250 mA g−1).2.5 X-ray diffraction
XRD patterns of powder samples were collected using a Bruker D8 Advance diffractometer with CuKα (λ = 1.5406 Å) radiation source.3 Results and discussion
In this work, we studied two distinct LLO compounds, namely Li(Li0.2Co0.18Ni0.18Mn0.44)O2 and Li(Li0.2Co0.18Ni0.18Mn0.18Ti0.26)O2. These compounds were specifically chosen to represent different levels of configurational entropy, attributed to their varying concentrations of excess Li and TMs. The structures comprise consecutive layers of …–Li–O–TM–O–Li–… along the c-axis, with an excess of Li ions that are mixed with TMs in the TM sites: (Li/Co/Ni/Mn)TM and (Li/Co/Ni/Mn/Ti)TM. The selection of Ti was based on previous research demonstrating its ability to stabilize the disordered state.41,42,44In an ideal solid solution with random elemental distribution in lattice sites, the molar configurational entropy is

The term ‘high-entropy’ in the context of materials like MELLO and HELLO primarily reflects the elevated configurational entropy arising from the incorporation of different types of elements at specific site types, rather than indicating entropy stabilization. This concept aligns with the broader definition of high-entropy materials, which are distinct from entropy-stabilized oxides (ESOs).4
Single-phase structures were observed in both synthesized MELLO and HELLO at elevated temperatures through XRD analysis, as shown in Fig. 1. The reflected peaks from the MELLO sample (Fig. 1) can be indexed to either the rhombohedral structure with the Rm space group or the monoclinic structure with the C2/m space group. A minor peak appearing in the range of 20° to 23° was identified as the characteristic peak of C2/m symmetry. To ascertain the most stable crystal structure for our simulation and analysis, we evaluated the total energy of both possible structures employing DFT-PBE, -PBEsol, -PBE + U, and -PBEsol + U (Fig. 2). The total energy differences are normalized to the chemical formula of MELLO, Li(Li0.2Co0.18Ni0.18Mn0.44)O2, which consists of 4 atoms. All the functionals indicated that the monoclinic structure has a lower energy of around 2.1 meV per atom compared to the rhombohedral one. The minimal total energy difference of 8.3 meV (0.8 kJ mol−1) at 0 K between the two considered structures suggests a possibility for phase co-existence or transformation at room temperature. Hereafter, we focused on the most stable structure, according to DFT, namely the C2/m structure. The XRD pattern for the HELLO system (Fig. 1) indicates a rhombohedral structure with the Rm space group.
 | ||
| Fig. 1 XRD patterns and voltage–capacity curves for (a) MELLO and (b) HELLO. The black arrow in (a) MELLO highlights the characteristic peaks of C2/m symmetry. The XRD pattern of (b) HELLO closely matches the Rm space group. | ||
 | ||
| Fig. 2 Computed energy difference between the rhombohedral (Rm) and monoclinic (C2/m) structures for the MELLO system using DFT-PBE, -PBEsol, -PBE + U, and -PBEsol + U. | ||
Based on the Gibbs free energy equation (ΔG = ΔH − TΔS), the absolute value of the entropy term (−TΔS) increases with temperature. If the enthalpy of mixing (ΔHmix) is low, the entropy term can play a role in stabilizing the (disordered) single-phase structure. However, the enthalpy of formation for likely secondary phases might be so large that ΔHmix and −TΔS terms become negligible.
Our electrochemical test (Fig. 1) shows a large capacity fading after the first cycle, being larger in the case of HELLO compared to MELLO. This result most likely shows that these two compounds do not represent ESO materials. To understand the reason behind large capacity fading in these two systems after the first cycle, we applied DFT calculation to study structural change, defect formation, redox mechanism, and second phase formation.
Table 1 presents the computed lattice parameters (a, b, and c) for MELLO and HELLO. It is found that the PBEsol functional can reproduce the experimental a, b, and c values within 0.77%. The Ni/Li exchange (NiLi + LiNi), a common anti-site defect in the Ni-containing layered oxide materials, has been well documented in the previous experimental works.46,47 In the present study, our calculations show that HELLO has a higher tendency to form NiLi + LiNi as shown in Fig. 3. The observed increase in energy gain for higher Ni/Li ratio in the presence of Ti cation can be attributed to its larger ionic radius (0.605 Å) relative to the other transition metals such as Mn3+ (0.53 Å), Ni3+ (0.56 Å), and Co4+ (0.545 Å). This observation correlates with a larger O–TM–O intralayer separation in HELLO (2.18 Å) relative to MELLO (2.13 Å) for a Ni/Li concentration of 22%, as depicted in Fig. 3, which provides more space for Li+ ions (0.76 Å) within the lattice structure of HELLO. For further study, we selected the same NiLi + LiNi concentration of c = 22.22% for MELLO and HELLO, to focus on the influence of elemental composition on other parameters such as oxidation and migration of ions.
| a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | Vol. (Å3) | ||
|---|---|---|---|---|---|---|---|---|
| MELLO | Exp. | 4.937 | 8.547 | 5.026 | 90.000 | 109.260 | 90.000 | 200.210 |
| PBEsol | 4.962 | 8.608 | 5.011 | 90.096 | 108.882 | 90.005 | 202.117 | |
| PBE | 5.015 | 8.680 | 5.065 | 90.130 | 109.144 | 89.919 | 208.265 | |
| HELLO | Exp. | 2.875 | 2.875 | 14.326 | 90.000 | 90.000 | 120.000 | 102.560 |
| PBEsol | 2.853 | 2.858 | 14.295 | 89.916 | 89.956 | 119.893 | 101.036 | |
| PBE | 2.886 | 2.866 | 14.861 | 90.739 | 89.021 | 120.071 | 106.371 | |
 | ||
| Fig. 3 Visualization of O–TM–O intralayer separation in MELLO (2.13 Å) and HELLO (2.18 Å) for a Ni/Li concentration of 22%. Li/Ni interchanges are represented by black arrows as an example. The middle figure shows the Ni/Li interchange energy (with respect to the initial energies without interchange) for MELLO (left) and HELLO (right). The Ni/Li interchange concentration (expressed as a percentage) is calculated with respect to Ni concentration. | ||
To understand the stability of MELLO and HELLO during electrochemical cycling, we analyzed their delithiated states by modeling Li0.25(Co0.18Ni0.18Mn0.44)O2 and Li0.25(Co0.18Ni0.18Mn0.18Ti0.26)O2, respectively. The low lithium concentration of 0.25 was chosen to study the extreme delithiation scenario. Following the geometry and unit cell optimization (using DFT-PBEsol) of the delithiated MELLO and HELLO, we found the formation of oxygen dimers, as shown in Fig. 4. The presence of charged oxygen dimers (O2x−) has been reported in several previous studies on Li-rich layered oxide systems.22,48 Given that the optimized structures might be confined to a local minimum, we further modeled additional oxygen dimers by manually adjusting the positions of isolated oxygen atoms in a sequential manner, followed by structural optimization to determine whether these manually induced dimers were energetically favorable or not. Detailed analysis, as illustrated in Fig. 4, reveals that HELLO can have up to 10.4%, while MELLO up to 6.3% oxygen dimers. This leads to a larger delithiation-induced change in the oxidation state of oxygen in HELLO compared to MELLO (see Table 2).
 | ||
| Fig. 4 (Left) Graph depicting the energy gain from O2 dimerization as a function of O2 concentration, expressed as a percentage of the total oxygen concentration within each compound for MELLO and HELLO systems. (Right) Visual snapshots illustrating the O2 dimers formed in MELLO (top two images) and HELLO (bottom two images). | ||
| MELLO | HELLO | |||
|---|---|---|---|---|
| x = 1.2 | x = 0.25 | x = 1.2 | x = 0.25 | |
| Ni | +3.00 | +4.00 | +3.00 | +4.00 |
| Co | +3.00 | +4.00 | +3.00 | +4.00 |
| Mn | +4.00 | +4.93 | +4.00 | +4.97 |
| Ti | — | — | +4.00 | +4.00 |
| O | −2.00 | −1.91 | −2.00 | −1.83 |
We observed partial migration of Ti to Li-layers and Mn to tetrahedral sites (between TM- and Li-layers) in the delithiated systems, which could potentially react with the oxygen dimers forming secondary phases. The migration of cations from TM sites to lithium sites during the delithiation process has been reported both experimentally and theoretically in several multicomponent Li-rich layered oxide systems.49–53 In the HELLO system, the extraction of Li from Li-layers, followed by extraction from TM-layers, provides free space for TM migration. As presented in Fig. 5, HELLO systems exhibit significant Ti ion migration after delithiation. One reason could be the low energy penalty for Ti to distort/migrate due to its empty d-orbital42 and relatively large ionic size. In the extreme delithiated state of x = 0.25, we found 2% Mn migration from octahedral to tetrahedral sites.
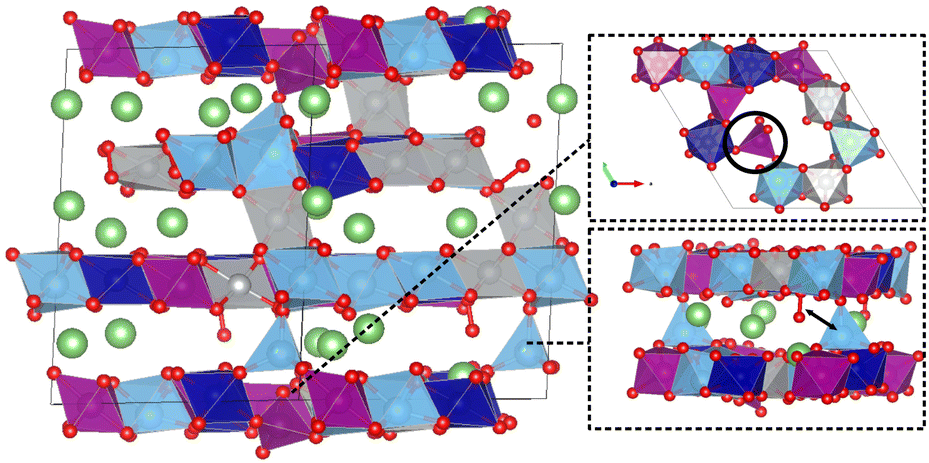 | ||
| Fig. 5 Schematic representation of cation migration during the delithiation process in the HELLO system. The upper-right figure shows the migration of Mn7+ to the tetrahedral site at low Li content, denoted by the black circle. The lower-right figure demonstrates the migration of Ti from the TM-layer to the Li layer, adjacent to an oxygen dimer. The migrated Ti atom proceeds to react with the oxygen dimer, culminating in the formation of a secondary phase as marked by the black arrow. The Li, O, Ti, Co, Ni, and Mn elements are represented by green, red, light blue, dark blue, gray, and purple spheres/polyhedra, respectively. | ||
The concomitant occurrence of TM migration and oxygen dimer formation suggests a correlation where each phenomenon influences the other. We suggest that oxygen dimers reduce the activation barrier for TM migration by disrupting the TMO6 octahedral structure, which facilitates TM mobility. Meanwhile, the migration of TM ions appears to have a significant impact on the oxygen redox process and subsequent dimerization. Gent et al.49 suggests that TM migration triggers a substantial reorganization of electronic states, leading to an elevation of the energy levels of oxygen's projected density of states (pDOS). This reorganization enhances the reactivity involved in oxygen redox activities and fosters the dimerization of oxygen. Hence, TM migration and oxygen dimerization emerge as interlinked processes, each facilitating the other, and together influencing the electrochemical properties and stability of the material.
The oxidation states were determined by examining the atomic magnetic moment and by analyzing the 3d occupation matrices for each atom. It is found that during the initial stage of delithiation when predominantly extraction of Li ions from the Li layers occurs, the charge neutrality is preserved by the oxidation of Ni3+ to Ni4+ and Co3+ to Co4+. Further delithiation involves the extraction of Li ions from TM sites and TM migration, as well as the oxidation of Mn from +4 to +5 states. The depletion of Li in the TM layer, combined with vacancies created by TM migration, leads to unstable electrons on isolated oxygen atoms facilitating the formation of oxygen dimers. Oxidation states of +5 and +7 were found for Mn, with +5 in a distorted octahedral site and +7 in the tetrahedral site (see Fig. 5). This finding is in line with other studies,54,55 which showed that MnO4 tetrahedra can only be realized when the Li+ content is sufficiently low. These migrations underscore the critical impact on the stability and capacity retention of MELLO and HELLO, particularly in the latter case where more MnO4 tetrahedra form. Mn typically exhibits electrochemical activities with oxidation/reduction states of Mn2+ ↔ Mn3+ ↔ Mn4+, providing a maximum of two electron transfers. Our calculation shows the oxidation of Mn from +4 to +5/+7 occurring for x = 1.2 → 0.25, which may be considered unrealistic. To address this, we reintroduced Li ions into the structures, adjusting the Li concentration from x = 0.25 to x = 0.83. We maintained (i) migrated Ti and interchanged Ni (with Li) in the Li-layers, and (ii) the formed O2 dimers. The concentration of x = 0.83 was chosen to achieve a more reasonable oxidation state for Mn, which gives a capacity of ∼115 mA h g−1 for both systems. Interestingly, this value lies between the observed second cycle capacities of 150 mA h g−1 and 100 mA h g−1 for MELLO and HELLO, respectively (see Fig. 1), underscoring the critical role of Mn in determining the reversible capacity of these compounds. The reintroduction of Li atoms did not affect the thermodynamic favorability of oxygen dimerization after DFT geometry relaxation. The previous experimental and theoretical works by other groups suggest both (i) a reversible redox reaction of oxygen in the form of single ion and/or charged dimer56 as well as (ii) an irreversible release of oxygen.18,57 Increasing the Li concentration from 0.25 to 0.83 leads to the reduction of highly oxidized Mn cations (with charges larger than 4+ oxidation). This shows the importance of oxygen redox reaction, in particular in the form of O2 dimer, in determining the capacity. The limited formation of oxidized oxygen in the form of dimer and single anion leads to the higher Mn oxidation states for x = 0.25. This is because of the specific composition of the studied compounds in this work. The computed charge states of ions for x = 0.83 indicate (Table 4) oxidation of Ni, Co, and O compared to the pristine case of x = 1.2. This shows that these elements are active during charge/discharge processes. Mn is slightly reduced in both HELLO and MELLO systems with x = 0.83 compared to that with x = 1.2 which can be attributed to the participation of oxygen in the redox mechanism.
The irreversible high first-cycle capacity (Fig. 1) in both systems can be due to the tendency of TMs to migrate from TM to Li sites and/or irreversibly O2 formation. The migration of Ti cations to Li sites and increased O2 dimer formation in HELLO contributes further to its capacity fading, making this fading more pronounced compared to MELLO. Migration of Ni, Mn, and Ti facilitated by O2 formation might indicate a layered-to-spinel phase transition, which has been proposed for Li-rich systems.58,59 This might explain why voltage and capacity decrease after the first cycle, which is in line with voltage fade primarily ascribed to the layered-to-spinel phase transition in the lattice structure of Li-rich electrodes.59,60
Afterwards, we investigated the formation of a few possible secondary oxide phases that might form besides the primary initial active phase during delithiation (charging) in both MELLO and HELLO systems. This was achieved by calculating the reaction enthalpies at 0 K for a series of probable reactions under the assumption that changes in zero-point energy and volume are negligible. A lithium concentration of x = 0.83 was chosen to avoid the formation of unusual Mn oxidation states higher than +4. The thermodynamically favored formation of 6.3% and 10.4% O2 dimers, respectively, in MELLO and HELLO (see Fig. 4) was kept as well. The O2 dimers or unstable oxidized oxygen ions from which the dimers form can react with the unstable TMs, in particular those that migrated from TM sites to Li sites. This can lead to the formation of the secondary phase comprising O and migrating TM atoms. The configuration of the primary phase after the reaction is constructed using SQS, in which the atomic distribution is selected based on pair correlations calculated from the configuration before the reaction, but with the migrated TM and oxygen atoms removed. This method captures the structural changes due to migration while still representing the system after secondary phase formation. These reactions and the corresponding changes of the primary phase in oxidation states before and after the secondary phase formation are detailed in Tables 3 and 4. Reactions that are thermodynamically favorable, as indicated by negative enthalpy values, are predicted to occur spontaneously. Our calculations indicate that the formation of MnO2 in MELLO and TiO2 in HELLO is thermodynamically favored. As shown in Table 4, the oxidation states of Ni and Mn increase in the primary phase following secondary phase formation, whereas those of Co and O decrease. This alteration in oxidation states can result in a reduced activity of TM ions, especially for Ni and Mn, which generally provide a more reversible redox reaction than O. Consequently, secondary phase formation may limit the capacity of the materials. Notably, the decrease in TM activity is more significant in the HELLO system.
| Reaction | ΔH (meV p.f.u.) |
|---|---|
| MELLO | |
| Li0.83Co0.18Mn0.44Ni0.18O2 → 0.91Li0.91Co0.20Mn0.38Ni0.20O2 + 0.09MnO2 | −171.0 |
| Li0.83Co0.18Mn0.44Ni0.18O2 → 0.875Li0.95Co0.21Mn0.36Ni0.21O2 + 0.125MnO2 | 140.0 |
| Li0.83Co0.18Mn0.44Ni0.18O2 → 0.91Li0.74Co0.20Mn0.38Ni0.20O2 + 0.09LiMnO2 | 199.6 |
|
|
| HELLO | |
| Li0.83Co0.18Mn0.18Ni0.18Ti0.26O2 → 0.87Li0.95Co0.21Mn0.21Ni0.21Ti0.15O2 + 0.13TiO2 | −69.6 |
| Li0.83Co0.18Mn0.18Ni0.18Ti0.26O2 → 0.92Li0.82Co0.20Mn0.20Ni0.20Ti0.20O2 + 0.08LiTiO2 | 251.3 |
| Li0.83Co0.18Mn0.18Ni0.18Ti0.26O2 → 0.87Li0.91Co0.21Mn0.21Ni0.21Ti0.15O2 + 0.09TiO2 + 0.04LiTiO2 | 35.8 |
| MELLO | HELLO | |||
|---|---|---|---|---|
| Before | After | Before | After | |
| Li | +1.00 | +1.00 | +1.00 | +1.00 |
| Ni | +3.78 | +4.00 | +3.72 | +4.00 |
| Co | +3.91 | +3.40 | +3.77 | +3.50 |
| Mn | +3.64 | +4.00 | +3.72 | +4.00 |
| Ti | — | — | +4.00 | +4.00 |
| O | −1.91 | −1.94 | −1.94 | −1.95 |
The enthalpy changes in the secondary phase formation reactions are more significant than the entropy changes. We assume that the vibrational and electronic entropy contributions are negligible due to the structural similarity of the primary phase before and after the secondary phase reaction. The configurational entropy changes for secondary phase formation reactions at room temperature, TΔSconf, for the thermodynamically favorable reactions listed in Table 3, are −1.1 meV and −0.07 meV p.f.u. for MELLO and HELLO, respectively. Notably, the negative entropy values indicate that the entropic contribution even slightly favors the formation of the secondary phase.
The formation of secondary phases is closely linked to TM vacancies, which result from the extraction of lithium and the migration of TMs, as well as the process of oxygen dimerization. Consequently, secondary phases can only form during the cycling process when Li is removed from the TM sites. A greater amount of secondary phase formation is expected in HELLO compared to MELLO due to its higher oxidation of O in the form of O2 dimers. The last reaction in Table 3, namely the formation of LiTiO2 together with TiO2, with a small positive enthalpy of 35.8 meV per formula unit, may still be kinetically driven at room temperature for the HELLO system. This is because the energy associated with molecular motion at this temperature is approximately 26 meV, and computational uncertainties must also be considered. Therefore, a larger amount of inactive secondary phases is expected to form in HELLO compared to the MELLO system.
To further investigate the effect of Ni/Li anti-site defects, we performed calculations with a higher Ni/Li interchange concentration of c = 44.44% for HELLO at lithium concentrations of 0.25 and 0.83. We again observed TM migration and dimer formation under these conditions. Furthermore, our calculations revealed that with Ni/Li interchange c = 44.44%, the secondary phase reaction becomes even more favorable, with an enthalpy of −75.8 meV p.f.u., indicating an increased instability in HELLO. These results support the mechanism proposed in this study.
Fig. 6 presents the proposed mechanism underlying the capacity fading observed in both MELLO and HELLO systems. During cycling, the formation of irreversible oxygen dimers, spinel phases, and/or secondary phases may occur. These phenomena, particularly during the first cycle, lead to a significant capacity decrease in both MELLO and HELLO. The higher concentration of migrated TM cations, O2 formation, and secondary phase formation, as well as the layered-to-spinel phase transition in the form of LiTiO2, might explain the larger capacity fading in the HELLO system.
 | ||
| Fig. 6 The proposed sequential stages of degradation during charging (delithiation). In the pristine stage, we found Li/Ni interchange. During the charging process, the lithium ions are extracted from the Li layers and the TM layers, leading to the oxygen dimerization and TM migration. Subsequently, oxygen dimers react with the migrated TM ions, leading to the formation of the secondary TMO2 phase. Oxygen atoms are denoted by red circles, while lithium atoms are represented by green circles. The multi-colored circles represent a mixture of various cationic species present in the system. | ||
4 Conclusions
In this study, we synthesized and characterized two multi-doped Li-rich layered oxide (LLO) systems to explore their structural and electrochemical properties. We observed a preferential Ni/Li interchange in both LLOs, with a higher tendency in the presence of larger ionic radius cations, namely Ti. Furthermore, we found simultaneous oxygen dimer formation and TM migration during delithiation. The formation of dimers is attributed to the removal of Li at TM layers as well as to the TM vacancies created by the TM migration, which results in isolated unstable oxygen anions. This phenomenon is particularly observed under conditions of extreme delithiation. Partial oxygen dimerization and TM migration contribute to the observed irreversible capacity loss, correlating with the concentration of formed oxygen dimers.We further investigated the tendency for secondary phase formation. Our findings indicate a thermodynamically favorable mechanism for this process. The secondary phase formation can be viewed as a reaction product between unfavorably oxidized oxygen anions and TM cations that are not stable at TM sites during cycling. Our calculations show that the formation of secondary phases, such as MnO2 and TiO2, decreases the overall activity of TM cations in the primary phase. Due to this and the likely lower electronic and ionic conductivity of secondary phases, their formation can reduce the capacity of these materials. In particular, we found a formation of a spinel LiTiO2 phase in the system with the largest capacity fading. We examined the implications of the high-entropy effect on material stability. Our analysis indicates that entropy does not play a role in stabilizing these materials throughout the cycling process. Notably, HELLO, despite its higher theoretical configurational entropy, exhibits even poorer stability. This observation suggests that a simple increase in the configurational entropy by integrating a large number of various elements does not necessarily improve cyclability. Furthermore, our investigation suggests that the increased disorder inherent in high-entropy systems may intensify certain undesirable phenomena during cycling, such as oxygen dimerization and TM migration. These effects can lead to phase transitions, structural decomposition, and secondary phase formation. This indicates that while the high-entropy approach contributes to the stabilization of disordered phases, it can also potentially worsen certain destabilizing processes during electrochemical cycling. Thus, when evaluating the benefits of “high-entropy” materials, it is crucial to balance the gains in entropy-driven stability against the potential for increased disorder-related instabilities such as oxygen dimerization and TM migration.
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y.-Y. T., P. M. K. and P. K. acknowledge the Jülich Aachen Research Alliance-Center for Simulation and Data Science for providing the computational resources (JARA-CSD, project no. cjiek61). B. B. and S. S. acknowledge financial support from the KIT via the project Auto.MAP and the Helmholtz Program “Materials Systems Engineering” under program no. 43.31.01.Notes and references
- D. Deng, Energy Sci. Eng., 2015, 3, 385–418 CrossRef.
- Y. Liang, C.-Z. Zhao, H. Yuan, Y. Chen, W. Zhang, J.-Q. Huang, D. Yu, Y. Liu, M.-M. Titirici, Y.-L. Chueh, H. Yu and Q. Zhang, InfoMat, 2019, 1, 6–32 CrossRef CAS.
- Y. Ma, Y. Ma, Q. Wang, S. Schweidler, M. Botros, T. Fu, H. Hahn, T. Brezesinski and B. Breitung, Energy Environ. Sci., 2021, 14, 2883–2905 RSC.
- C. M. Rost, E. Sachet, T. Borman, A. Moballegh, E. C. Dickey, D. Hou, J. L. Jones, S. Curtarolo and J.-P. Maria, Nat. Commun., 2015, 6, 8485 CrossRef CAS PubMed.
- A. Sarkar, B. Breitung and H. Hahn, Scr. Mater., 2020, 187, 43–48 CrossRef CAS.
- Y. Cui, P. A. Sukkurji, K. Wang, R. Azmi, A. M. Nunn, H. Hahn, B. Breitung, Y.-Y. Ting, P. M. Kowalski, P. Kaghazchi, Q. Wang, S. Schweidler and M. Botros, J. Energy Chem., 2022, 72, 342–351 CrossRef CAS.
- Y. He, S. L. Dreyer, Y.-Y. Ting, Y. Ma, Y. Hu, D. Goonetilleke, Y. Tang, T. Diemant, B. Zhou, P. M. Kowalski, M. Fichtner, H. Hahn, J. Aghassi-Hagmann, T. Brezesinski, B. Breitung and Y. Ma, Angew. Chem., 2023, e202315371 Search PubMed.
- Q. Wang, A. Sarkar, D. Wang, L. Velasco, R. Azmi, S. S. Bhattacharya, T. Bergfeldt, A. Düvel, P. Heitjans, T. Brezesinski, H. Hahn and B. Breitung, Energy Environ. Sci., 2019, 12, 2433–2442 RSC.
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, Adv. Mater., 2019, 31, 1806236 CrossRef PubMed.
- A. Sarkar, R. Djenadic, N. J. Usharani, K. P. Sanghvi, V. S. K. Chakravadhanula, A. S. Gandhi, H. Hahn and S. S. Bhattacharya, J. Eur. Ceram. Soc., 2017, 37, 747–754 CrossRef CAS.
- H. Xiang, Y. Xing, F.-z. Dai, H. Wang, L. Su, L. Miao, G. Zhang, Y. Wang, X. Qi, L. Yao, H. Wang, B. Zhao, J. Li and Y. Zhou, J. Adv. Ceram., 2021, 10, 385–441 CrossRef CAS.
- R. Wang, X. Li, L. Liu, J. Lee, D.-H. Seo, S.-H. Bo, A. Urban and G. Ceder, Electrochem. Commun., 2015, 60, 70–73 CrossRef CAS.
- D. Bérardan, S. Franger, A. K. Meena and N. Dragoe, J. Mater. Chem. A, 2016, 4, 9536–9541 RSC.
- J. Lee, D.-H. Seo, M. Balasubramanian, N. Twu, X. Li and G. Ceder, Energy Environ. Sci., 2015, 8, 3255–3265 RSC.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kübel, T. Brezesinski, S. S. Bhattacharya, H. Hahn and B. Breitung, Nat. Commun., 2018, 9, 3400 CrossRef PubMed.
- Z. Lun, B. Ouyang, D.-H. Kwon, Y. Ha, E. E. Foley, T.-Y. Huang, Z. Cai, H. Kim, M. Balasubramanian, Y. Sun, J. Huang, Y. Tian, H. Kim, B. D. McCloskey, W. Yang, R. J. Clément, H. Ji and G. Ceder, Nat. Mater., 2021, 20, 214–221 CrossRef CAS PubMed.
- C. S. Johnson, J.-S. Kim, C. Lefief, N. Li, J. T. Vaughey and M. M. Thackeray, Electrochem. Commun., 2004, 6, 1085–1091 CrossRef CAS.
- Y. Xie, Y. Jin and L. Xiang, Particuology, 2022, 61, 1–10 CrossRef CAS.
- Z. Lu, D. D. MacNeil and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A191 CrossRef CAS.
- W. Zuo, M. Luo, X. Liu, J. Wu, H. Liu, J. Li, M. Winter, R. Fu, W. Yang and Y. Yang, Energy Environ. Sci., 2020, 13, 4450–4497 RSC.
- H.-Y. Jang, D. Eum, J. Cho, J. Lim, Y. Lee, J.-H. Song, H. Park, B. Kim, D.-H. Kim, S.-P. Cho, S. Jo, J. H. Heo, S. Lee, J. Lim and K. Kang, Nat. Commun., 2024, 15, 1288 CrossRef CAS PubMed.
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M.-L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J.-M. Tarascon, Nat. Mater., 2013, 12, 827–835 CrossRef CAS PubMed.
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y.-S. Liu, K. Edström, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684–691 CrossRef CAS PubMed.
- G. Assat and J.-M. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- H. Konishi, T. Hirano, D. Takamatsu, A. Gunji, X. Feng and S. Furutsuki, J. Power Sources, 2015, 298, 144–149 CrossRef CAS.
- S. H. Lee, J.-S. Moon, M.-S. Lee, T.-H. Yu, H. Kim and B. M. Park, J. Power Sources, 2015, 281, 77–84 CrossRef CAS.
- G. Assat, D. Foix, C. Delacourt, A. Iadecola, R. Dedryvère and J.-M. Tarascon, Nat. Commun., 2017, 8, 2219 CrossRef PubMed.
- G. Assat, S. L. Glazier, C. Delacourt and J.-M. Tarascon, Nat. Energy, 2019, 4, 647–656 CrossRef CAS.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. Otero-de-la Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys. Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- P. M. Kowalski, Z. He and O. Cheong, Front. Energy Res., 2021, 9, 653542 CrossRef.
- Y.-Y. Ting and P. M. Kowalski, Electrochim. Acta, 2023, 443, 141912 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Jain, G. Hautier, C. J. Moore, S. Ping Ong, C. C. Fischer, T. Mueller, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2011, 50, 2295–2310 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- A. Zunger, S.-H. Wei, L. G. Ferreira and J. E. Bernard, Phys. Rev. Lett., 1990, 65, 353–356 CrossRef CAS PubMed.
- A. van de Walle, P. Tiwary, M. de Jong, D. L. Olmsted, M. Asta, A. Dick, D. Shin, Y. Wang, L. Q. Chen and Z. K. Liu, Calphad, 2013, 42, 13–18 CrossRef CAS.
- A. Abdellahi, A. Urban, S. Dacek and G. Ceder, Chem. Mater., 2016, 28, 3659–3665 CrossRef CAS.
- A. Urban, I. Matts, A. Abdellahi and G. Ceder, Adv. Energy Mater., 2016, 6, 1600488 CrossRef.
- A. Urban, A. Abdellahi, S. Dacek, N. Artrith and G. Ceder, Phys. Rev. Lett., 2017, 119, 176402 CrossRef PubMed.
- K. Okhotnikov, T. Charpentier and S. Cadars, J. Cheminf., 2016, 8, 17 Search PubMed.
- W. D. Richards, S. T. Dacek, D. A. Kitchaev and G. Ceder, Adv. Energy Mater., 2018, 8, 1701533 CrossRef.
- B. S. Murty, J.-W. Yeh, S. Ranganathan and P. P. Bhattacharjee, High-Entropy Alloys, Elsevier, 2019 Search PubMed.
- J. Xu, F. Lin, M. M. Doeff and W. Tong, J. Mater. Chem. A, 2017, 5, 874–901 RSC.
- J. Zheng, Y. Ye, T. Liu, Y. Xiao, C. Wang, F. Wang and F. Pan, Acc. Chem. Res., 2019, 52, 2201–2209 CrossRef CAS PubMed.
- E. McCalla, M. T. Sougrati, G. Rousse, E. J. Berg, A. Abakumov, N. Recham, K. Ramesha, M. Sathiya, R. Dominko, G. Van Tendeloo, P. Novák and J.-M. Tarascon, J. Am. Chem. Soc., 2015, 137, 4804–4814 CrossRef CAS PubMed.
- W. E. Gent, K. Lim, Y. Liang, Q. Li, T. Barnes, S.-J. Ahn, K. H. Stone, M. McIntire, J. Hong, J. H. Song, Y. Li, A. Mehta, S. Ermon, T. Tyliszczak, D. Kilcoyne, D. Vine, J.-H. Park, S.-K. Doo, M. F. Toney, W. Yang, D. Prendergast and W. C. Chueh, Nat. Commun., 2017, 8, 2091 CrossRef PubMed.
- D. Qian, B. Xu, M. Chi and Y. S. Meng, Phys. Chem. Chem. Phys., 2014, 16, 14665–14668 RSC.
- Q. Li, D. Ning, D. Wong, K. An, Y. Tang, D. Zhou, G. Schuck, Z. Chen, N. Zhang and X. Liu, Nat. Commun., 2022, 13, 1123 CrossRef CAS PubMed.
- B. Silván, E. Gonzalo, L. Djuandhi, N. Sharma, F. Fauth and D. Saurel, J. Mater. Chem. A, 2018, 6, 15132–15146 RSC.
- J. Huang, B. Ouyang, Y. Zhang, L. Yin, D.-H. Kwon, Z. Cai, Z. Lun, G. Zeng, M. Balasubramanian and G. Ceder, Nat. Mater., 2023, 22, 353–361 CrossRef CAS PubMed.
- M. D. Radin, J. Vinckeviciute, R. Seshadri and A. Van der Ven, Nat. Energy, 2019, 4, 639–646 CrossRef CAS.
- Z. Zhang, S. Zhao, B. Wang and H. Yu, Cell Rep. Phys. Sci., 2020, 1, 100061 CrossRef.
- K. Kawai, X.-M. Shi, N. Takenaka, J. Jang, B. M. d. Boisse, A. Tsuchimoto, D. Asakura, J. Kikkawa, M. Nakayama, M. Okubo and A. Yamada, Energy Environ. Sci., 2022, 15, 2591–2600 RSC.
- H. Zhang, H. Liu, L. F. J. Piper, M. S. Whittingham and G. Zhou, Chem. Rev., 2022, 122, 5641–5681 CrossRef CAS PubMed.
- T. Zhao, N. Zhou, X. Zhang, Q. Xue, Y. Wang, M. Yang, L. Li and R. Chen, ACS Omega, 2017, 2, 5601–5610 CrossRef CAS PubMed.
- W. Hua, S. Wang, M. Knapp, S. J. Leake, A. Senyshyn, C. Richter, M. Yavuz, J. R. Binder, C. P. Grey, H. Ehrenberg, S. Indris and B. Schwarz, Nat. Commun., 2019, 10, 5365 CrossRef PubMed.
- W. Liu, P. Oh, X. Liu, S. Myeong, W. Cho and J. Cho, Adv. Energy Mater., 2015, 5, 1500274 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |