
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurfactant templated biogenic nanoporous silica thermal insulation composite†
Long
Zhu
 ,
Taotao
Meng
,
Saurabh
Khuje
and
Shenqiang
Ren
*
,
Taotao
Meng
,
Saurabh
Khuje
and
Shenqiang
Ren
*
Department of Materials Science and Engineering, University of Maryland, College Park, MD 20742, USA. E-mail: sren@umd.edu
First published on 25th September 2024
Abstract
Carbon-sequestration biogenic insulation materials offer a promising solution for sustainable and energy-efficient buildings. However, minimizing energy-intensive manufacturing processes and ensuring a uniform microstructure are essential to achieve widespread application. In this work, we introduce cetyltrimethylammonium bromide (CTAB) surfactant as both synthesis and dispersion template for nanoporous silica to create biogenic straw-based thermal insulation composites. As the amount of nanoporous silica increases to 72 wt%, the elastic insulation composite dried using the solvent exchange method exhibits a density of 0.058 g cm−3, a thermal conductivity of 30.3 mW m−1 K−1 and a flexural modulus of 3.96 MPa, while demonstrating materials circularity and fire retardance. The use of CTAB surfactant effectively captures nanoporous silica, preventing its loss during manufacturing and enhancing the homogeneity and dispersion of the silica within the composite. In addition, the results show that the ethanol solvent exchange drying at ambient conditions provides the optimum thermal insulation performance with less energy consumption compared to freeze-drying.
1. Introduction
Biogenic straw has demonstrated significant potential for reducing carbon emissions in the building sector due to its renewable nature and carbon sequestration capabilities.1 However, its widespread application is limited by intrinsic disadvantages such as high thermal conductivity, flammability, and water absorption.2–5 These limitations undermine its effectiveness as an insulation material and compromise its safety in green building applications.6–8 To address these challenges, silica aerogel has been recognized as a promising additive to enhance the performance of biogenic insulation composites.9–12 Silica aerogel is renowned for its exceptional thermal insulation properties, low density, and high porosity.13,14 However, integrating nanoporous silica into biogenic materials presents several challenges:15–19 (1) loss of silica during the manufacturing process, such as filtration during molding, which is inherently difficult to avoid in water-based processes; (2) achieving homogeneous mixing of nanoporous silica within the straw network and preventing silica aggregation. Coating nanoporous silica on cellulose can partially address this issue, as reported in the literature.20,21 However, the problem of silica loss persists, as some free-standing silica remains in the water and is inevitably lost during water removal; (3) addressing the shrinkage issue during the water removal process in water-based manufacturing,15 which often results in dense structures and deteriorates thermal insulation performance. Although supercritical drying and freeze-drying can resolve this issue, these processes are energy-intensive.15Here we report cetyltrimethylammonium bromide (CTAB) as a dual-functioning surfactant – serving as both synthesis and dispersant template for nanoporous silica aerogel – for manufacturing biogenic straw and nanoporous silica insulation composite. The use of CTAB effectively mitigates silica loss by encapsulating it within its micelles, ensuring uniform dispersion throughout the composite. Furthermore, the study systematically investigates the impact of different drying methods – water based drying in an oven (60 °C), in air (20 °C) and freeze-drying (−84 °C), and ethanol solvent exchange (20 °C) based drying – on thermal insulation, mechanical strength, and fire retardance of biogenic composite material. The findings reveal that the solvent exchange method offers optimal performance with reduced energy consumption. Composites dried using this method exhibit lightweight characteristics, excellent elasticity under compression, and enhanced fire retardance properties.
2. Results and discussion
The CTAB surfactant was used as the synthesis template for nanoporous silica and its dispersing template for manufacturing biogenic insulation composites (Fig. 1a). Using a CTAB template, nanoporous silica can be synthesized with controlled pore structure and improved dispersion properties (Fig. 1b–d), on which it significantly improves the uniformity of nanoporous silica distribution in the composite. The nanoporous silica powder tends to aggregate, especially when dispersed in water, while the nanoporous silica is encapsulated within the CTAB micelles, thereby eliminating loss during manufacturing. The concentration of CTAB significantly influences the dispersion of nanoporous silica. Higher CTAB concentrations result in larger micelles, which can disperse greater amounts of silica. | ||
| Fig. 1 (a) Schematic illustration of the nanoporous silica dispersion by CTAB micelles. TEM images of the sintered nanoporous silica (b); dispersed nanoporous silica with a low concentration of CTAB (c), and dispersed nanoporous silica with a high concentration of CTAB (d). (e) Picture of a prepared composite supported by tree leaves. (f) FTIR spectroscopy of pristine straw, NaOH-treated straw, silica aerogel, and silica aerogel/straw composite. | ||
The as-prepared composite insulation panel is lightweight, as shown in Fig. 1e. Fig. 1f shows the Fourier-transform infrared spectroscopy (FTIR) spectrum for pristine straw, NaOH-treated straw, nanoporous silica, and the silica/straw composite. The absorption band between 3100 and 3500 cm−1 corresponds to O–H stretching vibrations.22 Peaks at 2918 cm−1 and 2851 cm−1 are attributed to the C–H stretching vibrations of aliphatic groups in lignin.23 The distinct band at 1733 cm−1 in the pristine straw indicates the presence of hemicellulose acetyl groups, as well as uronic ester or ester linkages of the carboxylic groups of ferulic and p-coumaric acids in hemicellulose/lignin.24,25 Bands at 1461 and 1420 cm−1 are characteristic absorptions of the C–O–C groups in hemicellulose and lignin. The absence of these bands in NaOH-treated straw, which are assigned to hemicellulose carbonyl stretching conjugated with aromatic rings, signifies the removal of lignin/hemicellulose. The absorption at 1032 cm−1 corresponds to C–O–C skeletal vibrations, characteristic of cellulose. Additionally, the peak at 960 cm−1 is associated with Si–O in-plane stretching vibrations of silanol Si–OH groups.26 The band at 802 cm−1 is due to Si–O stretching. The cellulose network and silica are crosslinked through hydrogen bonding between the silanol groups on the surface of silica and the hydroxyl end groups of cellulose. Fig. S1† presents the X-ray diffraction (XRD) spectra of pristine straw, NaOH-treated straw, nanoporous silica, and the silica/straw composite. The XRD spectra for both pristine and NaOH-treated straw exhibit two characteristic peaks at 16.5° and 22.3°, corresponding to the (101) and (002) crystalline planes of cellulose.27 The broad diffraction peak around 21.6° indicates amorphous silica.28
The concentration of CTAB plays a key role in dispersing nanoporous silica, which could significantly influence thermal insulation performance of its composites. When its concentration is low, the CTAB micelle is small in size (Fig. 2a). The size of the micelles increases with higher CTAB concentrations (Fig. 2b).29 Further increases in CTAB concentration result in negligible increases in micelle size, but the number of CTAB molecules per micelle increases (Fig. 2c). The transmission electron microscopy image (TEM, Fig. 2d) shows a large CTAB micelle with densely dispersed nanoporous silica. The effect of CTAB concentration on the porosity and density of the composites is detailed in Fig. 2e. As shown, when the CTAB concentration increases from 2.5 mM to 10 mM, the porosity of the composite increases from 87% to 93%, as more nanoporous silica is dispersed in larger micelles. However, further increasing the CTAB concentration to 40 mM decreases the porosity to 90.8%. The density of the composite follows an opposite trend, first decreasing from 0.078 g cm−3 to 0.06 g cm−3, then increasing to 0.08 g cm−3 within this CTAB concentration range. The thermal conductivity of the composite closely follows the changes in density as the CTAB concentration changes from 2.5 mM to 40 mM (Fig. 2f). It first decreases from 33 mW m−1 K−1 to 30.3 mW m−1 K−1 at a CTAB concentration of 10 mM, then increases to 34.1 mW m−1 K−1 at a CTAB concentration of 40 mM.
 | ||
| Fig. 2 (a–c) Schematic illustration of the nanoporous silica dispersion by CTAB micelles with increasing CTAB concentrations. (d) TEM image of the dispersed sintered nanoporous silica with a high concentration of CTAB. (e) Change in porosity and density of the composite with increasing CTAB concentration. (f) Thermal conductivity of the composite with increasing CTAB concentration. | ||
The scanning electron microscopy (SEM) images (Fig. 3a–c) of the composite with silica weight percentages of 0%, 20%, and 65% reveal that the nanoporous silica coats the straw fibers, as shown by EDX O and Si mapping in Fig. S2,† and fills the gaps in the composite, thereby enhancing its thermal insulation properties. The density of the composite decreases continuously with the increase in silica weight percentage (Fig. 3d). Drying the composite at 60 °C and 20 °C in air results in a relatively dense material (Fig. S3a and b†), with the density decreasing from 0.3 g cm−3 to 0.19 g cm−3. In contrast, composites dried using water-based freeze drying and ethanol solvent exchange methods exhibit more porous structures, as shown in Fig. S3c and d,† and have much lower densities, decreasing from approximately 0.15 g cm−3 to 0.06 g cm−3 as the silica weight percentage increases from 0 wt% to 72 wt%. The porosity of the composite follows the opposite trend of density, increasing as the weight percentage of silica increases for all samples (Fig. 3e). Samples dried using the ethanol solvent exchange method exhibit the highest porosity, rising from 84.2% to 94.5% as the silica weight percentage increases from 0 wt% to 72 wt%. Freeze-dried samples show slightly lower porosity. The samples dried at 60 °C have the lowest porosity due to significant shrinkage during the drying process, with porosity increasing from 80.5% to 90% as the silica weight percentage increases from 0 wt% to 72 wt%. We also characterized the specific surface area and pore size distribution of the composite with 20 wt% silica using Brunauer–Emmett–Teller (BET) analysis, as shown in Fig. S4.† The N2 adsorption/desorption isotherms of the composite display a type IV isotherm with an H3-type hysteresis loop (Fig. S4a†), indicating the presence of disordered pore structures with ill-defined pores ranging from narrow mesopores to macropores.30 The specific surface area of the composite, determined by BET analysis, is around 34 m2 g−1. From Fig. S4b,† the pore size distribution based on the adsorption branch of the Barrett–Joyner–Halenda (BJH) method is mainly concentrated between 20 and 40 nm.
 | ||
| Fig. 3 SEM images of the composite with silica weight percentages of 0 wt% (a), 20 wt% (b), and 65 wt% (c). Porosity (d) and density (e) of the composite with different drying methods at various silica weight percentages. Thermal conductivity (f) and flexural modulus (g) of the composite with different drying methods at various silica weight percentages. | ||
Fig. 3f illustrates the impact of silica weight percentage on the thermal conductivity of composites dried using different processes. With the increase of the silica weight percentage from 0% to 72%, the thermal conductivity of the composite decreases continuously. The fast drying at 60 °C results in a high thermal conductivity, which decreases from 45.6 mW m−1 K−1 to 37.8 mW m−1 K−1 as the silica weight percentage increases from 0% to 72%. This high thermal conductivity is due to the high density of the composite resulting from significant shrinkage during the fast drying process. Compared with the different drying methods, water-based drying at 60 °C, 20 °C, and freeze drying, and ethanol solvent exchange drying, the latter two methods, freeze drying and solvent exchange, significantly decrease the thermal conductivity. Both freeze drying and solvent exchange involve freezing the samples first in a refrigerator, which largely preserves the porous structure of the composite, resulting in a very lightweight composite (Fig. 1e). The prepared composite also exhibited excellent machinability, as demonstrated in Video S1.† The solvent exchange process results in the lowest thermal conductivity, decreasing from 36.1 mW m−1 K−1 to 30.3 mW m−1 K−1 as the silica weight percentage increases from 0% to 72%, due to the low thermal conductivity of nanoporous silica. We also conducted three point bending test to investigate the flexural modulus of the composites. A typical force–displacement curve and corresponding pictures captured during testing are shown in Fig. S5.† The flexural modulus of the composites follows a trend similar to that of thermal conductivity (Fig. 3g). The highest flexural modulus of the composite is achieved using the drying method at 60 °C, with the flexural modulus decreasing from 162 MPa to 87 MPa. The lowest flexural modulus of the composite is achieved using the solvent exchange method, with the flexural modulus decreasing from 10.7 MPa to 3.96 MPa as the silica weight percentage increases from 0% to 72%. The highly porous composite behaves like an elastic sponge under compression with 50% strain, fully recovering to original state after the compression is released (Fig. 4a–c), indicating ultrahigh compressibility and good recoverability. SEM images of the composite in its initial state, compressed state, and after unloading are shown in Fig. 4d–f, respectively. The large pores in the initial state are squeezed during compression but recover after the compression force is removed (Video S2†), verifying its structure stability. We conducted the compression tests under different strains of 10% to 50% for the composite with different silica weight percentages: 0 wt% (Fig. 4g), 20 wt% (Fig. 4h), and 65 wt% (Fig. 4i). As the compressive strain increases, the compressive stress also increases. Additionally, higher silica weight percentages result in higher compressive stress. We summarized the compressive stress and compressive modulus in Fig. 4j. With increasing compressive strain, both compressive stress and compressive modulus increase. Specifically, at 50% strain, the compressive stress of the composite increases from 33.8 kPa to 112.5 kPa as the silica weight percentage increases from 0 wt% to 65 wt%. The compressive modulus increases from 1.7 MPa to 7.4 MPa in this range. We also conducted cyclic tests with 200 cycles at different strains of 20%, 30%, 40%, and 50%. The corresponding stress and strain curves are shown in Fig. S6a–c† and 4k respectively. The maximum compressive stresses of the composite at different strains (20%, 30%, 40%, and 50%) under 200 cyclic tests are shown in Fig. 4l. The maximum compressive stress exhibits a small decrease: a 12.3% drop from 16.1 kPa to 14.1 kPa at 20% strain, a 15.4% drop from 25.7 kPa to 21.7 kPa at 30% strain, a 12.5% drop from 32.2 kPa to 28.2 kPa at 40% strain, and an 18% drop from 39.0 to 31.8 kPa at 50% strain, likely due to structural degradation during these high cyclic tests. This indicates that the composite demonstrates robust mechanical performance, consistently withstanding large deformations without collapsing at high compressive strains. Achieving the circular economy is critical for reducing energy consumption and fostering sustainable development.31,32 By promoting the reuse, repair, refurbishment, and recycling of materials, the circular economy minimizes the need for new resource extraction and manufacturing, which are typically energy-intensive and environmentally unfriendly (Fig. 5a).33 In this context, it would be desirable that the obtained insulation composite can be recycled and reused effectively. We summarized the thermal conductivity of the composite after recycling and drying using different methods. The thermal conductivity of the composite showed an increase of 1.5 mW m−1 K−1 to 3 mW m−1 K−1 after recycling, demonstrating its recyclability (Fig. 5b). This indicates that the composite maintains its functional properties even after undergoing recycling processes, further supporting the principles of the circular economy. The hydrophobicity of the composite is crucial for its application as a thermal insulation material in buildings. The wax-coated composite exhibits hydrophobic properties, with a water-contact angle of 131° (the inset of Fig. 5c). Its water absorption capacity is significantly reduced from 4.88 g g−1 to 0.85 g g−1 compared to the uncoated composite. We further investigated the thermal conductivity of the composite under different humidity levels ranging from 30% to 80%. The wax-coated composite demonstrated stability, with a 2.5% change of thermal conductivity at 80% humidity. In contrast, the uncoated composite showed a 23% change of thermal conductivity under the same conditions compared to its performance at 30% humidity. Fire retardance is another critical factor for green building applications. The pure straw sample ignites quickly and produces large flames (Fig. 5e, Video S3†), while silica/straw composite with 65 wt% silica ignites very slowly and shows good fire retardance, with negligible flame (Fig. 5f, Video S4†). The mass loss of the samples during fire tests is summarized in Fig. 5g. The pure straw sample exhibits poor fire retardance, with a mass loss of 97% after 8 minutes of burning. However, the composite with 72 wt% silica shows significantly improved fire resistance, with a mass loss of only 15.1% after 8 minutes of burning.
 | ||
| Fig. 4 Schematic illustration of the composite in initial (a); compressed (b) and recovered (c) states. SEM images of the composite sample in initial (d); compressed (e) and recovered (f) states. (Insets): Corresponding pictures of the sample during testing. Compressive stress–strain curves of the composite with the silica weight percentage of 0 wt% (g); 20 wt% (h) and 65 wt% (i) under different strains. (j) Compressive stress and modulus of the composite under different strains for the composites with different silica weight percentages. (k) Compressive stress–strain curves from the cyclic test at 40% strain. (l) Maximum compressive stress of the composite at different strains. | ||
 | ||
| Fig. 5 (a) Schematic illustration of the circular economy. (b) Thermal conductivity of the fresh and recycled composite dried using four different drying methods. (c) Water absorption capacity of the uncoated and wax-coated composite. (Inset): Contact angle of the wax-coated composite. (d) Thermal conductivity change (ΔTC) of the uncoated and wax-coated composite under different humidity levels. Pictures of pure straw (e) and silica/straw composite with 65 wt% silica (f) during the fire test. (g) Mass loss of the composite with different silica weight percentages during the fire test. | ||
3. Conclusions
In summary, integrating nanoporous silica within biogenic insulation composites by utilizing surfactant addresses critical manufacturing challenges, such as the uniform dispersion and porosity/density control of the composite. The resulting composite, dried using the solvent exchange method, benefits from a substantial improvement in thermal insulation, with a thermal conductivity of 30.3 mW m−1 K−1 at a silica weight percentage of 72% and enhances energy efficiency in buildings. Additionally, nanoporous silica imparts fire resistance to the composite, significantly mitigating the flammability and moisture condensation associated with biogenic based materials. The ethanol solvent exchange drying not only enhances material mechanical and thermal insulation performance but also aligns with sustainable manufacturing practices by minimizing energy use. This research contributes valuable insights into advancing eco-friendly building materials with improved functionality and performance metrics.4. Experimental methods
4.1. Materials
Wheat straw was sourced from Dumor Straw. Cetyltrimethylammonium bromide (CTAB), mesitylene, ammonium hydroxide, tetraethyl orthosilicate (TEOS), and sodium hydroxide (NaOH) were obtained from Sigma-Aldrich.4.2. NaOH treatment on wheat straw
First, 30 g of NaOH was dissolved in 500 mL of deionized water. Then, 90 g of natural wheat straw was blended with 1 L of deionized water using a blender for 5 minutes. The resulting slurry was added to the NaOH solution and heated at 160 °C for 2 hours. Afterward, the treated straw slurry was filtered through a nylon mesh and rinsed with deionized water until it reached a neutral pH. Finally, the treated straw was dried in an oven at 60 °C for two days.4.3. Nanoporous silica preparation and dispersion in CTAB
Initially, 12.5 g of CTAB was dissolved in 1 L of deionized water at 40 °C and stirred for 1.5 hours to ensure complete dissolution. Next, 1 mL of ammonium hydroxide and 10 mL of mesitylene were added to the solution, which was then stirred for an additional 2.5 hours. Following this, 10 mL of TEOS was slowly added, and the mixture was kept at 60 °C in an oven for gelation. The dried powder was then sintered in an oven at 600 °C for 1 hour to remove any organic residue, and the nanoporous silica was thus obtained.To disperse the nanoporous silica in the CTAB solution, different concentrations of CTAB solutions were prepared. Specifically, varying amounts of CTAB were dissolved in deionized water while stirring at 300 rpm at 40 °C for 1 hour. Subsequently, different amounts of nanoporous silica were added to the CTAB solution while stirring at 200 rpm.
4.4. Prepare nanoporous silica/straw composite
Different amounts of dried treated straw were first blended for 2 minutes. The blended straw was then added to the previously obtained nanoporous silica dispersion and mixed using mechanical stirring for 5 minutes. The resulting nanoporous silica/straw slurry was poured into a mold with a stainless-steel mesh at the bottom to filter out water. For the CTAB-template-dispersed nanoporous silica, almost all the silica was retained during filtration. However, for the slurry without CTAB, a large amount of nanoporous silica leached out during filtration. The mold was then removed, and the filtered samples were subjected to different drying methods. For oven drying at 60 °C, the filtered composite was clamped between metal perforated sheets to avoid warping during drying and dried overnight in an oven at 60 °C. For air drying at 20 °C, the filtered composite was clamped between metal perforated sheets and placed in a well-ventilated area for one week. For freeze drying, the sample was first frozen in a freezer for 3 hours, then placed in a freeze dryer (FreeZone Triad Cascade Benchtop Freeze Dry System, Labconco, Kansas City, MO, USA). For solvent exchange, the samples were first frozen in a freezer for 3 hours, then placed in ethanol for solvent exchange, with the ethanol being replaced twice during the process. For recycling the composite samples, the tested samples were blended with water, remolded, and dried using the same process as for fresh samples. For wax coating, the composite panel was coated with a thin layer of Minwax® Paste Finishing Wax, and the coating was then dried in an oven overnight at 60 °C.4.5. Characterization
Transmission electron microscopy (TEM, JEOL JEM 2100 LaB6) was used to investigate the morphology of the synthesized nanoporous silica. Scanning electron microscopy (Hitachi SU-70 FEG SEM) was employed to examine the microstructural morphology of the composite. Fourier-transform infrared spectroscopy (FTIR-Agilent Cary 560) was used to study the chemical bonding in the composite. The bulk density (ρm) of the composites was calculated by dividing the mass (m) by the volume (V) of the composite, as shown in eqn (1). | (1) |
The porosity of the composite samples was determined using eqn (2), where ρm represents the bulk density and ρs indicates the skeletal density, which was determined using the pycnometer system (Ultrapyc 3000, Anton Paar). The equation for porosity calculation is as follows:
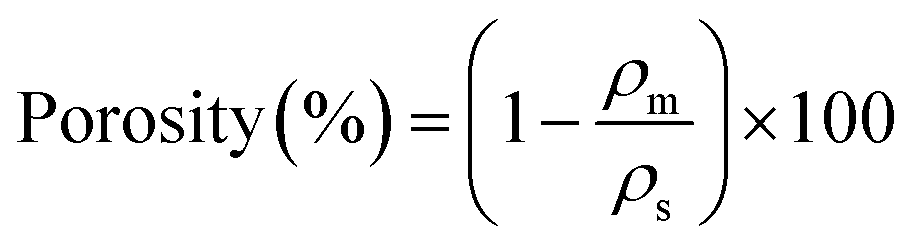 | (2) |
At least five samples were tested to obtain an average value for each data point. Brunauer–Emmett–Teller (BET) analysis is performed using a Micromeritics ASAP 2020 Porosimeter (Micromeritics Instrument Co., USA). The specific surface area and pore size distributions are evaluated using the low-temperature nitrogen adsorption–desorption isotherm measurement method. The composite sample is degassed at 140 °C for 7 hours before analysis. The Thermtest Heat Flow Meter 100 series (HFM-100) was used to measure the thermal conductivity of the composite according to the ASTM C518 standard. Prior to measurement, calibration was performed using NIST SRM 1450e reference material with a thermal conductivity of 32.5 mW m−1 K−1. For testing under different humidity levels, the samples were first exposed to each humidity environment for 5 hours before measuring thermal conductivity. For the fire test, the composite was exposed to the flame generated by a propane torch lighter, with both the lighter and the sample's positions fixed to ensure consistency during each test. The mass of the sample was recorded as burning progressed. The flexural modulus was evaluated through a three-point bending test using an Instron 3367 universal testing machine (Instron Co. Ltd, USA) at a testing speed of 5 mm min−1, while compression tests were conducted with the same machine at a testing speed of 3 mm min−1.
The water absorption capacity of the composite was evaluated according to ASTM D570. Before testing, samples were conditioned in an oven at 50 °C for 24 hours, and their weight was measured immediately after removal from the oven. The conditioned samples were then immersed in distilled water for 24 hours. After removal, their weight was measured immediately after wiping off the surface water with a dry cloth. The water absorption capacity was calculated using the formula:
 | (3) |
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Author contributions
Long Zhu: conceptualization, data curation, methodology, investigation, validation, formal analysis, writing – original draft. Taotao Meng: investigation, methodology. Saurabh Khuje: investigation. Shenqiang Ren: conceptualization, funding acquisition, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the funding support on this work by the United States Department of Energy (DOE) Advanced Research Projects Agency-Energy (ARPA-E) award DE-AR0001771.References
- B. Ismail, N. Belayachi and D. Hoxha, Constr. Build. Mater., 2022, 317, 126049 CrossRef CAS.
- S. Zou, H. Li, L. Liu, S. Wang, X. Zhang and G. Zhang, J. Build. Eng., 2021, 43, 103172 CrossRef.
- C. Rojas, M. Cea, A. Iriarte, G. Valdés, R. Navia and J. P. Cárdenas-R, Sustainable Mater. Technol., 2019, 20, e00102 CrossRef CAS.
- R. Ahmadi, B. Souri and M. Ebrahimi, J. Cleaner Prod., 2020, 254, 120043 CrossRef CAS.
- A. Sarkar, A. Islam, L. Zhu and S. Ren, Appl. Mater. Today, 2024, 36, 102080 CrossRef.
- L. Liu, S. Zou, H. Li, L. Deng, C. Bai, X. Zhang, S. Wang and N. Li, Energy Build., 2019, 201, 19–36 CrossRef.
- Y. Li, N. Zhu and J. Chen, J. Mater. Sci., 2023, 58, 2361–2380 CrossRef CAS.
- P. S. Jadhav, A. Sarkar, L. Zhu and S. Ren, J. Appl. Polym. Sci., 2024, 141, e55137 CrossRef CAS.
- A. Lamy-Mendes, A. D. R. Pontinha, P. Alves, P. Santos and L. Durães, Constr. Build. Mater., 2021, 286, 122815 CrossRef CAS.
- Y. Liu, H. Wu, Y. Zhang, J. Yang and F. He, Energy Build., 2020, 228, 110452 CrossRef.
- G. Wei, Y. Liu, X. Zhang, F. Yu and X. Du, Int. J. Heat Mass Transfer, 2011, 54, 2355–2366 CrossRef CAS.
- L. Zhu, Y. Huang, J. Armstrong and S. Ren, ACS Appl. Eng. Mater., 2024, 2, 1959–1965 CrossRef CAS.
- C.-Y. Zhu, H.-B. Xu, X.-P. Zhao, L. Gong and Z.-Y. Li, Energy Storage Sav., 2022, 1, 217–240 CrossRef CAS.
- M. A. Hasan, R. Sangashetty, A. C. M. Esther, S. B. Patil, B. N. Sherikar and A. Dey, J. Inst. Eng. (India): Ser. D, 2017, 98, 297–304 CAS.
- T. Linhares, M. T. P. de Amorim and L. Durães, J. Mater. Chem. A, 2019, 7, 22768–22802 RSC.
- J. C. Wong, H. Kaymak, P. Tingaut, S. Brunner and M. M. Koebel, Microporous Mesoporous Mater., 2015, 217, 150–158 CrossRef CAS.
- X. Yang, Y. Sun, D. Shi and J. Liu, Mater. Sci. Eng., A, 2011, 528, 4830–4836 CrossRef.
- Z. Mazrouei-Sebdani, M. Naeimirad, S. Peterek, H. Begum, S. Galmarini, F. Pursche, E. Baskin, S. Zhao, T. Gries and W. J. Malfait, Mater. Des., 2022, 223, 111228 CrossRef CAS.
- A. Sarkar, P. K. Singh, L. Zhu, D. Faghihi and S. Ren, ACS Appl. Eng. Mater., 2024, 2, 136–142 CrossRef CAS.
- J. Cai, S. Liu, J. Feng, S. Kimura, M. Wada, S. Kuga and L. Zhang, Angew. Chem., 2012, 9, 2118–2121 CrossRef.
- Y. Chen, S. Sepahvand, F. Gauvin, K. Schollbach, H. Brouwers and Q. Yu, Constr. Build. Mater., 2021, 293, 123289 CrossRef CAS.
- S.-k. Du, X. Su, W. Yang, Y. Wang, M. Kuang, L. Ma, D. Fang and D. Zhou, Carbohydr. Polym., 2016, 140, 279–286 CrossRef CAS PubMed.
- Q. Wang, J. Hu, F. Shen, Z. Mei, G. Yang, Y. Zhang, Y. Hu, J. Zhang and S. Deng, Bioresour. Technol., 2016, 199, 245–257 CrossRef CAS PubMed.
- F. Neudecker, M. Jakob, S. C. Bodner, J. Keckes, H. Buerstmayr and W. Gindl-Altmutter, ACS Sustainable Chem. Eng., 2023, 11, 7596–7604 CrossRef CAS.
- S. H. Ghaffar and M. Fan, Biomass Bioenergy, 2015, 77, 123–134 CrossRef CAS.
- R. Al-Oweini and H. El-Rassy, J. Mol. Struct., 2009, 919, 140–145 CrossRef CAS.
- A. Kaushik, M. Singh and G. Verma, Carbohydr. Polym., 2010, 82, 337–345 CrossRef CAS.
- M. Kellermeier, F. Glaab, R. Klein, E. Melero-Garcia, W. Kunz and J. M. García-Ruiz, Nanoscale, 2013, 5, 7054–7065 RSC.
- N. C. Das, H. Cao, H. Kaiser, G. T. Warren, J. R. Gladden and P. E. Sokol, Langmuir, 2012, 28, 11962–11968 CrossRef CAS PubMed.
- F. He, X. He, W. Yang, X. Zhang and L. Zhou, J. Non-Cryst. Solids, 2018, 488, 36–43 CrossRef CAS.
- H. Corvellec, A. F. Stowell and N. Johansson, J. Ind. Ecol., 2022, 26, 421–432 CrossRef.
- M. Geissdoerfer, P. Savaget, N. M. Bocken and E. J. Hultink, J. Cleaner Prod., 2017, 143, 757–768 CrossRef.
- L. Keßler, S. A. Matlin and K. Kümmerer, Curr. Opin. Green Sustainable Chem., 2021, 32, 100535 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05605a |
| This journal is © The Royal Society of Chemistry 2024 |