
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced reduction of CO to C2 products on MoS2 nanoribbons by edge engineering†
Jiangke
Tao
a,
Di
Liu
 a,
Zhichao
Yu
a and
Hui
Pan
*ab
a,
Zhichao
Yu
a and
Hui
Pan
*ab
aInstitute of Applied Physics and Materials Engineering, University of Macau, Macao SAR, China. E-mail: huipan@um.edu.mo
bDepartment of Physics and Chemistry, Faculty of Science and Technology, University of Macau, Macao SAR, China
First published on 30th October 2024
Abstract
Two-dimensional (2D) transition metal dichalcogenides (TMDs) nanomaterials, such as MoS2 and WSe2, are promising for CO2 reduction to CO with high selectivity. However, the further reduction of CO to high-value C2 products is difficult on these 2D TMDs. To solve the challenging issues, we present an edge-engineering strategy by tuning the edge composition of 2D MoS2 nanoribbons through incorporation based on density-functional theory (DFT) calculations. A group of elements (Nb, Ta, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Bi, Sn, Sb) are searched to obtain the optimal dopants for high reduction activity. We find that the d-band center and electronegativity of the dopants influence the adsorption strength and configuration of CO, respectively. Compared to Mo ions, dopants with lower d-band centers can reduce the limiting potential and enhance the C–C coupling in CO reduction reaction. The targeted C2 products can be achieved on Fe, Mn, Ta, and Cu-incorporated MoS2 zigzag edges. Among these, the Cu-incorporated one exhibits the best selectivity for C2 products (CH3CH2OH) due to the lower d-band center energy of Cu ions relative to the Fermi level, which promotes the CO hydrogenation, the C–C coupling, and the product desorption. Our findings may provide guidance for the design of electrocatalysts for selective C2 production.
Introduction
Electrochemical reduction of carbon dioxide (CO2) to value-added chemicals is one of the important solutions to alleviate energy problems and promote low-carbon development.1–5 Using renewable energy sources, such as wind and solar energy, to power the electrochemical reduction reaction of CO2 (e-CO2RR) is an attractive direction. In the past decades, CO2 reduction has been extensively investigated.6–10 Due to the inert nature of the CO2 molecule, suitable catalysts have been widely explored to reduce the reaction barrier and improve selectivity. For example, metallic copper (Cu)-based materials have been demonstrated to be effective for the reduction of CO2 to various products, such as CO, HCOOH, CH3OH, CH4, and C2H4.11,12 Generally, the C2 products are more valuable and easier to collect. However, the direct reduction of CO2 to C2 products shows poor efficiency and selectivity because of the multiple-electron transfer and high energy barrier. Considering reducing CO2 to CO with a Faraday efficiency of ∼100% is available,13,14 it is natural to have a two-step reaction for the CO2 reduction to C2 products with improved performance, that is, CO2 → CO → C2 products, because the electrochemical CO reduction reaction (e-CORR) simplifies the reaction network and makes it easier to achieve C2 products. Similarly, catalysts play a critical role in producing high-valuable chemicals in the e-CORR too. Many Cu-based materials have also been reported as effective catalysts for the CO reduction to C2 products.15–21 However, the selectivity on C2 products and corresponding Faraday efficiency are still too low to be possible for practical application, which has encouraged us to find new catalysts.Two-dimensional (2D) materials have attracted great attention since they exhibit distinguished physical and chemical properties.22–27 Among these, transition metal dichalcogenides (TMDs) such as MoS2, WS2, and MoSe2 are recognized as promising catalysts for various reactions because of their high specific surface area and abundant active sites.28–32 Notably, MoS2 stands out as an electrocatalyst for electrochemical CO2 reduction (e-CO2RR) due to its remarkable catalytic performance.33–35 The unique monolayer structure of MoS2, characterized by short diffusion path lengths and terminated edges, serves as active centers that facilitate multiple-electron transfer reactions in the CO2 reduction pathway.36 Furthermore, the metallic edges of MoS2 enhance the charge transfer and migration.37 In addition to its superior catalytic properties, MoS2 is cost-effective, nontoxic, highly earth-abundant, and easy to synthesize, making it a more sustainable choice compared to other TMDs. Experimental results demonstrated that MoS2 showed superior CO2 reduction performance for selectively converting CO2 to CO at 65 mA cm−2 under −0.764 V.33 Moreover, studies have shown that N-incorporated MoS2 nanosheets had high CO faradaic efficiency and low onset overpotential of 130 mV in the CO2 reduction.34 Additionally, Nb-incorporated MoS2 nanoribbons exhibited improved electrocatalytic performance for the CO2 reduction to CO by modifying the binding energies of intermediates on the MoS2 edge.35 Theoretical studies also suggested that replacing the transition metal on the edge of MoS2 with other elements could enhance the CO2 reduction,38,39 predominantly yielding C1 products.
The major obstacle to obtaining C2 products lies in the strenuous C–C coupling, which is usually restrained by the repulsive dipole–dipole interaction between adjacent carbonaceous intermediates on equivalent adsorption sites in the e-CORR. It is believed that the uneven adsorption sites may endow the adjacent CO intermediates with distinct charge densities, thus decreasing their dipole–dipole repulsion, and hence benefit for triggering C–C coupling to form the multi-carbon products.40,41 Given that the Mo-terminated edge of MoS2 exhibits excellent carbon dioxide reduction capability and has unsaturated bonds, which facilitate the incorporation of other transition-metal elements, we selected the MoS2 nanoribbon with Mo-terminated edge as the substrate. We report a strategy for improving the catalytic selectivity toward C2 products by alternately replacing Mo ions on the Mo-terminated edge of MoS2 to create uneven adsorption sites between the two of them. We take the zigzag metallic edge of MoS2 as the model, and 13 metal elements are considered. By calculating their adsorption behavior and energy pathways, we find that alternate replacement can greatly enhance the selectivity of C2 products on the MoS2 edge in the e-CORR. We show that the Cu-incorporated edge gives out the best performance with a limiting potential of −0.71 V for selectively producing CH3CH2OH among all candidates.
Computational details
All calculations were carried out using the Vienna ab initio simulation package (VASP) with the projector augmented wave potential and the Perdew–Burke–Ernzerhof (PBE) energy functional.42–47 The cut-off energy for the plane-wave basis set was 500 eV and the convergence threshold was 10−5 eV per atom by considering the accuracy and reliability.46,48,49 A 1 × 3 × 1 Monkhorst–pack k-point mesh was used for the structural optimization.50A supercell with a size of 15.08 Å × 12.75 Å × 26.05 Å was used as the starting structure. A nanoribbon was constructed with the lattice parameter of 12.75 Å along the zigzag edge and a width of 10.91 Å. In addition, the adsorption of CO on the Mo-terminated zigzag edge is much stronger than that on the S-terminated zigzag edge.51 So, only the Mo-terminated zigzag edge is investigated. There are two Mo-terminated zigzag edges of MoS2 (Fig. S1a & b†). One of the edge structures undergoes deformation after structural relaxation (Fig. S1a†), which impacts the catalytic process. Therefore, we selected the stable edge structure (Fig. S1b†). The vacuum was set to 15.08 Å and 26.05 Å along the x and z directions, respectively, to avoid the interlayer interaction between two neighboring cells. The Mo ions were alternately replaced by other elements in our model, denoted at MoS2-2M (Fig. 1a). The ions in the dashed box were fixed to study the catalytic reaction on the zigzag Mo-edge of MoS2 (Fig. 1a). Spin-polarization was included in the calculations.
 | ||
| Fig. 1 (a) The schematic structure of MoS2 nanoribbon with alternate replacement. The dashed box shows the fixed atoms. (b) 13 metal elements considered. | ||
In this DFT study, 13 elements—V to Zn, Nb, Sn, Sb, Ta, and Bi—were selected as dopants (Fig. 1b). The common fourth-period transition metals (from V to Zn) exhibit increasing electronegativity and d-orbital filling as the atomic number rises. Although V, Ta, and Nb are all group V elements, they display distinct metallic properties and electronegativity due to differences in atomic radius. These trends facilitate a systematic investigation of their synergistic interactions with Mo ions, highlighting the influence of varying electron configurations on catalysis. Bi, Sn, and Sb are well-known for their use in CO2 reduction, making them ideal candidates for exploring how they enhance Mo-based catalysts in producing C2 products.52–56
The replacement energy for one dopant is calculated by
| EF = (EMoS2+2M − EMoS2 − 2EM + 2EMo)/2 | (1) |
 , where EM−bulk is the energy of bulk metal.
, where EM−bulk is the energy of bulk metal.  , where EMo−bulk is the energy of Mo crystal. n is the number of metal atoms in the crystal.
, where EMo−bulk is the energy of Mo crystal. n is the number of metal atoms in the crystal.
As an indicator for each elemental step of e-CORR, the Gibbs free reaction energy change (ΔG) is calculated by
| ΔG = ΔE + ΔEZPE − TΔS + ΔH | (2) |
| H = U + kBT | (3) |
| Gad(*CO) = E(MoS2 + CO) − E(MoS2) − E(CO) + ΔEZPE − TΔS + ΔH | (4) |
 | (5) |
The adsorption Gibbs free energies of hydroxide ion and H2O were calculated in the same way.
The d-band center was calculated using the following equation58
 | (6) |
Results and discussion
We proposed a “five-step” screening strategy for the C2 production on the MoS2-2M edge via the e-CORR process (Fig. 2a). Firstly, the modified edge should possess high thermodynamic stability and feasibility (EF < 0, where EF is the replacement energy). Secondly, the CO should be more favorable to adsorb on the two active sites (Mo and dopant site) in e-CORR (step 2 and step 3). Thirdly, the final products on these candidates should be the C2 products and the limiting potential should not be too high to prevent possible HER (step 4). If the limiting potential is very high, the negative voltage on the electrode should increase the hydrogen adsorption ability. Fourthly, a screening principle is proposed as below:| ULe + ΔG(*H) ≫ ΔG(*CO) |
 | ||
| Fig. 2 (a) Catalyst-screening strategy for e-CORR toward C2 products, (b) the replacement energy of dopant on the zigzag edge of MoS2, and (c) the adsorption Gibbs free energies of CO on Mo and dopant sites. (d) The adsorption process of CO on the incorporated edge: the first row shows the adsorption process of CO on the Sn, Cr, Mn, Fe, Co, Ni, Cu, and Zn-incorporated edges and the second row shows that on the V, Nb, and Ta-incorporated edges. | ||
Theoretically, if ULe + ΔG(*H) is greater than ΔG(*CO), the adsorption capacity of proton on the active site cannot compete with that of CO. However, due to the limitation of theoretical calculation, when the two values are very close, it increases the possibility of the side reaction, hydrogen evolution reaction (HER), occurring in practical applications. Therefore, we set the difference between the two to be 0.2 eV, allowing for adjustability. Finally, the desorption energy of products at the edge of the catalyst should be lower than 0.75 eV for releasing the products easily and fast, and benefiting the continuous reaction (step 5). A barrier of 0.75 eV is generally regarded as the threshold for efficient kinetics, corresponding to a turnover frequency (TOF) of 1 s−1.60 Therefore, the value is considered as the threshold for the product release rate in this context.
The replacement energies of Bi and Sb on the edges are positive (Fig. 2b), indicating their difficulty to be realized in experiments. The replacement energies for other dopants are negative and considered further for the following steps in e-CORR. After the first step screening, we calculated the Gibbs free energy for the adsorption of CO on the Mo and dopant sites, respectively (Fig. 2c). Clearly, CO is more favorable to adsorb on the Mo site compared with the dopant site because of lower adsorption energy. Therefore, CO will first occupy the Mo sites and then adsorb to the dopant sites. However, CO can adopt two adsorption configurations on the doped edge. On the Sn, Cr, Mn, Fe, Co, Ni, Cu, and Zn-incorporated edges, CO binds to the active site solely through the C atom, while CO is adsorbed via dual sites involving both the C and O atoms on the V, Nb, and Ta-incorporated edges. Consequently, there are two distinct CO adsorption processes (Fig. 2d).
As CO initially adsorbs on the Mo atom at the edge, we then calculated the competitive adsorptions on the site (Fig. 3a). CO is more favorable to adsorb on Mo than hydrogen and water for all considered modified edges. On the Co and Zn-incorporated edges, the hydroxide ions are more easily to adsorb on the Mo site than CO, which are not considered. For the Ni and Cr-incorporated edges, the Gibbs free energies for the adsorption of hydroxide and CO are nearly the same. As the negative voltage applied on the catalyst and the free hydroxide ions repel each other during the e-CORR process in the experiment, Ni and Cr are still considered.
 | ||
| Fig. 3 Adsorption energy differences between CO and: (a) hydrogen, hydroxide ions, and H2O at Mo sites; (b) hydrogen, hydroxide ions, and H2O at dopant sites. | ||
Similarly, we analyzed the competitive reactions on the dopant (Fig. 3b). The Gibbs free energy changes for the adsorption of hydrogen are smaller than that of CO in all candidates, indicating that CO can adsorb on the dopant site more easily than hydrogen. CO is also more favorable to adsorb on the dopant site than hydroxide ion, except for the Nb and Ta-incorporated edges. The Gibbs free energies for the adsorption of hydroxide ions are approximately equal to those of CO on Nb and Ta-incorporated edges, which are considered further due to the repulsion of negative voltage to hydroxide ions. Finally, the dopant sites at the Sn-incorporated edge are more likely to be occupied by H2O, resulting in the elimination of MoS2-2Sn. The adsorption configuration of the 2nd CO shows that CO is difficult to adsorb on the Sn site (Fig. S2†). Therefore, eight elements (Nb, Ta, V, Cr, Mn, Fe, Ni, and Cu) are screened out for e-CORR.
We calculated the CO reduction pathway for each incorporated edge. Considering that the carbonaceous intermediates adsorbed on each pair of adjacent sites may undergo protonation or C–C coupling, we computed the Gibbs free energy changes associated with these processes at each intermediate step. The step with the largest free energy reduction was selected, and this calculation was repeated for the subsequent steps until the reaction was completed. Configurations that cannot be stable are excluded from the pathway. The calculations show that the product is CH3OH on the Ni- and V-incorporated edges in the e-CORR process (Table 1 & Fig. S3†). On the Nb-incorporated edge, the catalytic energy barrier for the C2 production is higher than that for the C1 production (Fig. S4†). The dominant product on the Nb-incorporated edge is CH3OH (Table 1). As for the Cr-incorporated edge, both C1 and C2 products are possible (Fig. S5†). The reaction path to CH3OH is energetically easier to occur than C2 products. However, the limiting potential of CORR on the Cr-incorporated edge is relatively high, which may limit the reaction, leading to competitive HER (Table 1). Similar situations also happen on the Fe and Mn-incorporated edges. The catalytic products of CO on both Fe and Mn-incorporated edges are CH3CH2OH (Table 1). However, the limiting potential of catalytic reactions are both relatively high and may cause competitive HER (Table 1, Fig. S6 & S7†). Finally, there are only Ta and Cu-incorporated edges possible toward C2 products.
| Dopants | Products | U L (V) |
|
|---|---|---|---|
| V | CH3OH | −0.72 | −1.57 |
| Ni | CH3OH | −0.64 | −1.19 |
| Nb | CH3OH | −0.89 | −1.77 |
| Cr | CH3OH | −1.2 | −1.16 |
| Fe | CH3CH2OH | −1.32 | −1.35 |
| Mn | CH3CH2OH | −1.01 | −1.08 |
| Ta | CH3CHO/CH2CH2 | 0/−0.17 | −2.16 |
| Cu | CH3CH2OH | −0.71 | −1.17 |
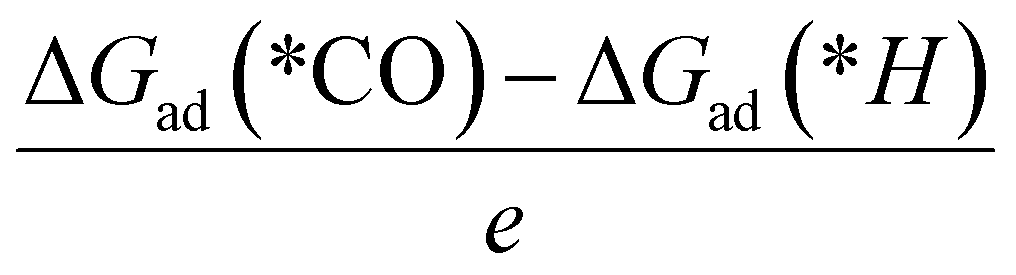
The catalytic products on the Ta-incorporated edge are CH3CHO and CH2CH2 (Fig. 4a). The limiting potential of the catalytic path to C2 products is about −0.17 V for CH2CH2 (Table 1). The catalytic pathway to CH3CHO is energetically spontaneous. However, the desorption energies of these two products are very high, that is, −2.92 eV for CH3CHO and −2.16 V for CH2CH2 (Fig. 4a), respectively, which are difficult to desorb from the Ta-incorporated edge. Consequently, based on our fifth-step screening strategy, the Ta-incorporated edge is eliminated.
 | ||
| Fig. 4 The reaction pathways of CO reduction on: (a) Ta-incorporated and (b) Cu-incorporated edges at 0 V. | ||
The Cu-incorporated edge is the best one among all candidates for the CO reduction to C2 products. The products on the Cu-incorporated edge are CH3OH and CH3CH2OH (Fig. 4b). Energetically, the path to the C2 product is easier to proceed. The limiting potential of the catalytic pathway on the Cu-incorporated edge is −0.71 V and the desorption energy of CH3CH2OH is 0.64 eV, indicating that the negative voltage shall not cause competitive HER during e-CORR and the products are easy to desorb from the edge.
Because the CO is easier to adsorb on the Mo site (Fig. 2c), the 1st CO adsorbs on the Mo site and the 2nd CO is also more likely to adsorb on the other Mo site after the 1st CO adsorption (Fig. 4b). Then the Cu site shall be occupied by CO because the adsorption energy of CO is lower than those of proton, hydroxide and H2O (Fig. 3b). The first protonation of *CO occurs on the *CO adsorbed on Cu site. After the first protonation, the C–C coupling between *CHO on the Cu site and *CO on the Mo site energetically prefers to happen than the protonation of *CHO. After the C–C coupling, the *OCHCO is protonated step by step and the CH3CH2OH is obtained finally.
According to the Bader charge analysis, we find the adsorption configuration of CO is dependent on the electronegativity of dopants (Fig. 5a). The Bader charge calculation results for different incorporated edges with CO adsorption are presented in the ESI (Fig. S8).† Statistical comparisons reveal that the O ions connected to the Nb, Ta, and V sites gain more electrons than the O ions of CO adsorbed on the Mo site of the other five incorporated edges (Cr, Mn, Fe, Ni, Cu) (Fig. 5a & S8†). Additionally, the number of electrons given by the Nb, Ta, or V atoms after CO adsorption is greater than that of Cr, Mn, Fe, Ni, or Cu atoms. A clear linear relationship exists between the electrons gained by O atoms and those given by the dopants (Fig. 5a). The Bader charge of the dopant reflects their electronegativity. Nb, Ta, and V are less electronegative than Cr, Mn, Fe, Ni, and Cu, making them more prone to losing electrons and bonding with the O atom of CO, and resulting in a two-site adsorption configuration of C and O atoms. In contrast, Cr, Mn, Fe, Ni, and Cu, being more electronegative, are less likely to lose electrons and bond with O atoms, leading to a single-site adsorption configuration.
 | ||
| Fig. 5 (a) The Bader charge of the dopants and the O atoms in CO adsorbed on the Mo sites of different incorporated edges, (b) the Gibbs free energy changes for the adsorption of CO (ΔG(*CO-M)) on the dopant sites and the integrated COHP (ICOHP) of the dopants and C atoms on the Cr, Mn, Fe, Ni, and Cu-incorporated edges, (c) the Gibbs free energy changes for the hydrogenation of C atoms in CO adsorbed on dopant sites, as well as for the adsorption of CO on those sites at the Cr, Mn, Fe, Ni, and Cu-incorporated edges, and (d) the relationship between the ICOHP between the dopant and C of adsorbed CO, and the d-band center of the dopant. | ||
On the Cr, Mn, and Fe-incorporated edges, the limiting potential for CO reduction is very high due to the significant energy barrier for hydrogenation (Fig. S5–S7†). Therefore, we calculated the Gibbs free energy changes for the CO hydrogenation on Mo sites and dopant sites for Cr, Mn, Fe, Ni, and Cu-incorporated edges (Table 2), as CO on these five incorporated edges exhibits single-site adsorption. For the Cr, Mn, and Fe-incorporated edges, the hydrogenation of CO on both Mo and dopant sites is relatively challenging, whether involving O or C atoms (Table 2). In contrast, on the Ni and Cu-incorporated edges, the C atom of CO adsorbed on the dopant site is more readily hydrogenated, with a Gibbs free energy change of 0.64 eV and 0.71 eV, respectively (Table 2). We analyzed the crystal orbital Hamiltonian population (COHP) between the dopant and the C atom of adsorbed CO (Fig. S9†). The integrated COHP (ICOHP) between dopant and C on the Ni and Cu-incorporated edges is higher than those on Cr, Mn, and Fe-incorporated edges (Fig. 5b), suggesting that the adsorption strength of CO on Ni and Cu atoms is lower. The reduced CO adsorption strength facilitates the hydrogenation of the C atom in CO (Fig. 5c). Conversely, Fe, Cr, and Mn exhibit relatively strong adsorption strengths for CO, making hydrogenation more difficult. The bonding strength between the dopant and CO (ICOHP) is determined by the d-band center of the dopant (Fig. 5d).
| ΔGMo(CO–COH) | ΔGMo(CO–CHO) | ΔGM(CO–COH) | ΔGM(CO–CHO) | |
|---|---|---|---|---|
| Cr | 1.2 | 1.34 | 1.2 | 2.15 |
| Mn | 1.0 | 1.34 | 1.07 | 1.31 |
| Fe | 0.98 | 1.04 | 1.98 | 1.78 |
| Ni | 1.02 | 0.87 | 3.57 | 0.64 |
| Cu | 1.02 | 1.24 | 1.83 | 0.71 |
To study the effect of dopant on C–C coupling, we calculated the Gibbs free energy change for the adjacent CO coupling on the Cr, Mn, Fe, Ni, and Cu-incorporated edges (Fig. 6a). The results indicate that stronger adsorption of CO on the dopant site correlates with a higher energy required the adjacent CO–CO coupling, making the *OCCO structure more unstable. This instability arises because the Mo site has a strong adsorption capacity for CO, and the d-band centers of Mo atoms on the incorporated edges are near the Fermi level (Fig. 6d). If the d-band center of the dopant is close to the Fermi level and exhibits strong CO adsorption, the force of CO adsorption on adjacent sites can easily disrupt the C–C bond. Among the selected dopants, the d-band center of Cu is the lowest (Fig. 6b and c), resulting in the weakest CO adsorption. Consequently, the Gibbs free energy change for adjacent CO coupling is minimized, making the *OCCO structure relatively stable. In our calculated reaction pathway, C–C coupling does not occur through the combination of adjacent CO molecules. The C–C coupling paths vary across different incorporated edges due to the hydrogenation occurring at different sites, leading to distinct lowest energy paths. However, we can observe the influence of the dopant's d-band center on C–C coupling through the energy changes during the formation of *OCCO. For Nb, Ta, and V-incorporated edges, when multiple CO molecules are adsorbed on the active sites, the adsorption configurations differ from those of the edges incorporated with the other five elements (Cr, Mn, Fe, Ni, and Cu). In this case, two CO molecules are adsorbed on the Mo atom, while the dopant site is occupied by oxygen atoms (Fig. 5a, top). The adsorption configurations of *OCCO on these edges also differ from those on the other five incorporated edges (Fig. S10†). Here, *OCCO is adsorbed above the Mo site, with the O atom connected to the dopant site. The adsorption energy of CO at the dopant site has a minimal impact on the stability of *OCCO (Fig. S10†). However, on the V and Nb-incorporated edges, the hydrogenation of O atoms is challenging due to the strong adsorption of the O atoms on dopant (Fig. S11a–c†). In the case of Ta incorporation, the strong adsorption of O atoms on Ta atoms causes the O atoms to disconnect from the carbonaceous intermediate after hydrogenation (Fig. S11d†).
 | ||
| Fig. 6 (a) The Gibbs free energy changes for the coupling of CO adsorbed on the neighbor asymmetric active sites of the Cr, Mn, Fe, Ni and Cu-incorporated edges, and the adsorption of CO on the dopant sites, (b) the Gibbs free energy changes for the adsorption of *CO (ΔGad(*CO)) on the dopant sites and the d-band center of the dopant atoms, (c) partial density of states (PDOS) for the d orbitals of the dopants on the incorporated edges, where the first one shows the PDOS of Mo atoms on the pure edge of MoS2. (d) Partial density of states (PDOS) for d orbitals of the Mo atoms on the incorporated edges, where the first one shows the PDOS of Mo atoms on the pure edge of MoS2. | ||
Additionally, the lower d-band center of Cu facilitates the release of the final product which weakens its CO adsorption, making the final product easier to desorb according to the Sabatier criterion.61 At the Cu-incorporated edge, the desorption energy of the final product, CH3CH2OH, is 0.64 eV, significantly lower than that on the Ta-incorporated edge (2.16 eV).
Conclusion
In summary, we proposed a strategy for efficiently generating C2 products on MoS2 nanoribbons by alternately replacing Mo atoms on the zigzag edges with other transition metals. By comparing the CO adsorption configurations and reduction pathways on different incorporated edges, we found that the d-band center of the dopant determines the adsorption strength of CO on the dopant site. Stronger CO adsorption on the dopant site makes hydrogenation more difficult, resulting in a higher limiting potential for CO reduction. Additionally, strong CO adsorption reduces the stability of the C2 intermediate, hindering C–C coupling. However, when the electronegativity of the dopant is relatively low, the CO adsorption configuration changes: CO binds to the dopant through the O atom and to the Mo atom through the C atom, allowing adjacent CO molecules to be co-adsorbed on the Mo atom, facilitating C–C coupling. But strong adsorption of the O atom on the dopant makes the hydrogenation of the O atom difficult. The d-band center also influences the release of final product from the catalyst edge. After screening, the Cu-incorporated edge emerges as the most effective catalyst for the reduction of CO to C2 products. The d-band center of the Cu atom is the lowest among all candidates, resulting in a low energy barrier for CO hydrogenation. Additionally, due to the weak CO adsorption on the Cu site, the formation of the first C2 intermediate (*OCHCO) is relatively easy, and the desorption energy of the final product (CH3CH2OH) is also low (0.64 eV). Our work demonstrates that the catalytic pathway for electrochemical CO reduction to targeted C2 products on two-dimensional materials can be achieved through edge engineering, guiding the design of novel electrocatalysts with high selectivity and efficiency for the electrochemical reduction of CO to valuable chemicals.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There is no conflicts of interest to declare.Acknowledgements
This work was supported by the Science and Technology Development Fund (FDCT) from Macau SAR (0050/2023/RIB2, 0023/2023/AFJ, 006/2022/ALC, and 0111/2022/A2), Multi-Year Research Grants (MYRG-GRG2023-00010-IAPME and MYRG2022-00026-IAPME) from Research & Development Office at University of Macau, and Shenzhen-Hong Kong-Macao Science and Technology Research Programme (Type C) (SGDX20210823103803017) from Shenzhen.References
- J. Feng, J. Ni and H. Pan, Synergistic carbon and hydrogen reactions in the electrochemical reduction of CO2 to liquid fuels, J. Mater. Chem. A, 2021, 9, 10546–10561 RSC.
- S. Chu, Y. Cui and N. Liu, The path towards sustainable energy, Nat. Mater., 2016, 16, 16–22 CrossRef.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364, 6482 CrossRef.
- F. Li, H. Ai, C. Shi, K. H. Lo and H. Pan, Single transition metal atom catalysts on Ti2CN2 for efficient CO2 reduction reaction, Int. J. Hydrogen Energy, 2021, 46, 12886–12896 CrossRef CAS.
- Y. Li, L. Yu, Y. Yu, K. Maliutina, Q. Wu, C. He and L. Fan, Understanding CO2 electrochemical reduction kinetics of mixed-conducting cathodes by the electrical conductivity relaxation method, Int. J. Hydrogen Energy, 2021, 46, 9646–9652 CrossRef CAS.
- Y. Meng, K. Li, D. Xiao, Y. Yuan, Y. Wang and Z. Wu, High selective and efficient Fe2–N6 sites for CO2 electroreduction: A theoretical investigation, Int. J. Hydrogen Energy, 2020, 45, 14311–14319 CrossRef CAS.
- Z. Feng, Y. Tang, Y. Ma, Y. Li, Y. Dai, W. Chen, G. Su, Z. Song and X. Dai, Theoretical computation of the electrocatalytic performance of CO2 reduction and hydrogen evolution reactions on graphdiyne monolayer supported precise number of copper atoms, Int. J. Hydrogen Energy, 2021, 46, 5378–5389 CrossRef CAS.
- T. He, C. Tang, A. R. Puente Santiago, R. Luque, H. Pan and A. Du, Tuning CO binding strength via engineering the copper/borophene interface for highly efficient conversion of CO into ethanol, J. Mater. Chem. A, 2021, 9, 13192–13199 RSC.
- M. de Jesus Gálvez-Vázquez, P. Moreno-García, H. Xu, Y. Hou, H. Hu, I. Z. Montiel, A. V. Rudnev, S. Alinejad, V. Grozovski and B. J. Wiley, et al., Environment Matters: CO2RR Electrocatalyst Performance Testing in a Gas-Fed Zero-Gap Electrolyzer, ACS Catal., 2020, 10, 13096–13108 CrossRef.
- R. M. Aran-Ais, D. Gao and B. Roldan Cuenya, Structure- and Electrolyte-Sensitivity in CO2 Electroreduction, Acc. Chem. Res., 2018, 51, 2906–2917 CrossRef CAS.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. de Arquer, A. Kiani, J. P. Edwards, P. De Luna and O. S. Bushuyev, et al., CO electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang and H. Xie, et al., Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons, Nat. Chem., 2018, 10, 974–980 CrossRef CAS.
- N. Zhang, X. Zhang, Y. Kang, C. Ye, R. Jin, H. Yan, R. Lin, J. Yang, Q. Xu and Y. Wang, et al., A Supported Pd(2) Dual-Atom Site Catalyst for Efficient Electrochemical CO2 Reduction, Angew. Chem., Int. Ed., 2021, 60, 13388–13393 CrossRef CAS.
- J. S. Derrick, M. Loipersberger, R. Chatterjee, D. A. Iovan, P. T. Smith, K. Chakarawet, J. Yano, J. R. Long, M. Head-Gordon and C. J. Chang, Metal-Ligand Cooperativity via Exchange Coupling Promotes Iron-Catalyzed Electrochemical CO2 Reduction at Low Overpotentials, J. Am. Chem. Soc., 2020, 142, 20489–20501 CrossRef CAS.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, Mechanistic Understanding of CO2 Reduction Reaction (CO2RR) Toward Multicarbon Products by Heterogeneous Copper-Based Catalysts, ACS Catal., 2019, 10, 1754–1768 CrossRef.
- Z. Li, Y. Yang, Z. Yin, X. Wei, H. Peng, K. Lyu, F. Wei, L. Xiao, G. Wang and H. D. Abruña, et al., Interface-Enhanced Catalytic Selectivity on the C2 Products of CO2 Electroreduction, ACS Catal., 2021, 11, 2473–2482 CrossRef CAS.
- S. Y. Lee, S. Y. Chae, H. Jung, C. W. Lee, D. L. T. Nguyen, H.-S. Oh, B. K. Min and Y. J. Hwang, Controlling the C2+ product selectivity of electrochemical CO2 reduction on an electrospray Cu catalyst, J. Mater. Chem. A, 2020, 8, 6210–6218 RSC.
- C. Kim, K. M. Cho, K. Park, J. Y. Kim, G. T. Yun, F. M. Toma, I. Gereige and H. T. Jung, Cu/Cu2O Interconnected Porous Aerogel Catalyst for Highly Productive Electrosynthesis of Ethanol from CO2, Adv. Funct. Mater., 2021, 31, 2102142 CrossRef CAS.
- S. Chen, H. Yuan, S. I. Morozov, L. Ge, L. Li, L. Xu and W. A. Goddard III, Design of a Graphene Nitrene Two-Dimensional Catalyst Heterostructure Providing a Well-Defined Site Accommodating One to Three Metals, with Application to CO2 Reduction Electrocatalysis for the Two-Metal Case, J. Phys. Chem. Lett., 2020, 11(7), 2541–2549 CrossRef CAS PubMed.
- Y. Ying, X. Luo, J. Qiao and H. Huang, “More is Different:” Synergistic Effect and Structural Engineering in Double-Atom Catalysts, Adv. Funct. Mater., 2020, 31, 2007423 CrossRef.
- C. Chen, X. Sun, L. Lu, D. Yang, J. Ma, Q. Zhu, Q. Qian and B. Han, Efficient electroreduction of CO2 to C2 products over B-doped oxide-derived copper, Green Chem., 2018, 20, 4579–4583 RSC.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Fundamentals and Challenges of Electrochemical CO2 Reduction Using Two-Dimensional Materials, Chem, 2017, 3, 560–587 CAS.
- J. H. Liu, L. M. Yang and E. Ganz, Efficient electrocatalytic reduction of carbon dioxide by metal-doped beta(12)-borophene monolayers, RSC Adv., 2019, 9(47), 27710–27719 RSC.
- J.-H. Liu, L.-M. Yang and E. Ganz, Efficient and Selective Electroreduction of CO2 by Single-Atom Catalyst Two-Dimensional TM–Pc Monolayers, ACS Sustainable Chem. Eng., 2018, 6(11), 15494–15502 CrossRef CAS.
- J.-H. Liu, L.-M. Yang and E. Ganz, Electrocatalytic reduction of CO2 by two-dimensional transition metal porphyrin sheets, J. Mater. Chem. A, 2019, 7(19), 11944–11952 RSC.
- J.-H. Liu, L.-M. Yang and E. Ganz, Electrochemical reduction of CO2 by single atom catalyst TM–TCNQ monolayers, J. Mater. Chem. A, 2019, 7(8), 3805–3814 RSC.
- J. H. Liu, L. M. Yang and E. Ganz, Two-Dimensional Organometallic TM3–C12S12 Monolayers for Electrocatalytic Reduction of CO2, Energy Environ. Mater., 2019, 2(3), 193–200 CrossRef CAS.
- D. Wang, B. Su, Y. Jiang, L. Li, B. K. Ng, Z. Wu and F. Liu, Polytype 1T/2H MoS2 heterostructures for efficient photoelectrocatalytic hydrogen evolution, Chem. Eng. J., 2017, 330, 102–108 CrossRef CAS.
- W. Tu, Y. Li, L. Kuai, Y. Zhou, Q. Xu, H. Li, X. Wang, M. Xiao and Z. Zou, Construction of unique two-dimensional MoS2-TiO2 hybrid nanojunctions: MoS2 as a promising cost-effective cocatalyst toward improved photocatalytic reduction of CO2 to methanol, Nanoscale, 2017, 9(26), 9065–9070 RSC.
- L. S. Byskov, J. K. Norskov, B. S. Clausen and H. Topsoe, Edge termination of MoS and CoMoS catalyst particles, Catal. Lett., 2000, 64(2–4), 95–99 CrossRef CAS.
- L. Sun, H. Xu, Z. Cheng, D. Zheng, Q. Zhou, S. Yang and J. Lin, A heterostructured WS2/WSe2 catalyst by heterojunction engineering towards boosting hydrogen evolution reaction, Chem. Eng. J., 2022, 443, 136348 CrossRef CAS.
- Y. Huang, H. Lu, H. Gu, J. Fu, S. Mo, C. Wei, Y. E. Miao and T. Liu, A CNT@MoSe2 hybrid catalyst for efficient and stable hydrogen evolution, Nanoscale, 2015, 7(44), 18595–18602 RSC.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu and R. Haasch, et al., Robust carbon dioxide reduction on molybdenum disulfide edges, Nat. Commun., 2014, 5, 4470 CrossRef CAS PubMed.
- K. Lv, W. Suo, M. Shao, Y. Zhu, X. Wang, J. Feng, M. Fang and Y. Zhu, Nitrogen doped MoS2 and nitrogen doped carbon dots composite catalyst for electroreduction CO2 to CO with high Faradaic efficiency, Nano Energy, 2019, 63, 103834 CrossRef CAS.
- P. Abbasi, M. Asadi, C. Liu, S. Sharifi-Asl, B. Sayahpour, A. Behranginia, P. Zapol, R. Shahbazian-Yassar, L. A. Curtiss and A. Salehi-Khojin, Tailoring the Edge Structure of Molybdenum Disulfide toward Electrocatalytic Reduction of Carbon Dioxide, ACS Nano, 2017, 11, 453–460 CrossRef CAS PubMed.
- S. Singh, A. Modak, K. K. Pant, A. Sinhamahapatra and P. Biswas, MoS2–Nanosheets-Based Catalysts for Photocatalytic CO2 Reduction: A Review, ACS Appl. Nano Mater., 2021, 4(9), 8644–8667 CrossRef CAS.
- R. Lv, H. Terrones, A. L. Elías, N. Perea-López, H. R. Gutiérrez, E. Cruz-Silva, L. P. Rajukumar, M. S. Dresselhaus and M. Terrones, Two-dimensional transition metal dichalcogenides: Clusters, ribbons, sheets and more, Nano Today, 2015, 10(5), 559–592 CrossRef CAS.
- W. Shao, X. Li, J. Zhu, X. Zu, L. Liang, J. Hu, Y. Pan, J. Zhu, W. Yan and Y. Sun, et al., How doped MoS2 breaks transition-metal scaling relations for CO2 electrochemical reduction, ACS Catal., 2016, 6, 4428–4437 CrossRef.
- J. Xu, X. Li, W. Liu, Y. Sun, Z. Ju, T. Yao, C. Wang, H. Ju, J. Zhu, S. Wei and Y. Xie, Carbon dioxide electroreduction into syngas boosted by a partially delocalized charge in molybdenum sulfide selenide alloy monolayers, Angew. Chem., Int. Ed., 2017, 56, 9121–9125 CrossRef CAS PubMed.
- W. Shao, X. Li, J. Zhu, X. Zu, L. Liang, J. Hu, Y. Pan, J. Zhu, W. Yan, Y. Sun and Y. Xie, Metaln+-Metalδ+ pair sites steer C-C coupling for selective CO2 photoreduction to C2 hydrocarbons, Nano Res., 2021, 15, 1882–1891 CrossRef.
- X. Wang, Z. Wang, T. T. Zhuang, C. T. Dinh, J. Li, D. H. Nam, F. Li, C. W. Huang, C. S. Tan and Z. Chen, et al., Efficient upgrading of CO to C3 fuel using asymmetric C-C coupling active sites, Nat. Commun., 2019, 10, 5186 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blochl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- D. Lal, T. Konnur, A. M. Verma, M. Shaneeth and A. Govind Rajan, Unraveling Low Overpotential Pathways for Electrochemical CO2 Reduction to CH4 on Pure and Doped MoS2 Edges, Ind. Eng. Chem. Res., 2023, 62, 21191–21207 CrossRef CAS.
- L. F. Xie, X. Liu, F. Y. Huang, J. S. Liang, J. Y. Liu, T. Y. Wang, L. M. Yang, R. G. Cao and Q. Li, Regulating Pd-catalysis for electrocatalytic CO reduction to formate via intermetallic PdBi nanosheets, Chin. J. Catal., 2022, 43(7), 1680–1686 CrossRef CAS.
- J. Yang, X. Wang, Y. Qu, X. Wang, H. Huo, Q. Fan, J. Wang, L. M. Yang and Y. Wu, Bi-Based Metal-Organic Framework Derived Leafy Bismuth Nanosheets for Carbon Dioxide Electroreduction, Adv. Energy Mater., 2020, 10(36), 2001709 CrossRef CAS.
- T. Wang, J. Chen, X. Ren, J. Zhang, J. Ding, Y. Liu, K. H. Lim, J. Wang, X. Li and H. Yang, et al., Halogen-Incorporated Sn Catalysts for Selective Electrochemical CO2 Reduction to Formate, Angew. Chem., Int. Ed., 2023, 62(10), e202211174 CrossRef CAS PubMed.
- J. Li, J. Li, X. Liu, J. Chen, P. Tian, S. Dai, M. Zhu and Y.-F. Han, Probing the role of surface hydroxyls for Bi, Sn and In catalysts during CO2 Reduction, Appl. Catal., B, 2021, 298, 120581 CrossRef CAS.
- H. Li, T.-W. Jiang, X. Qin, J. Chen, X.-Y. Ma, K. Jiang, X.-G. Zhang and W.-B. Cai, Selective Reduction of CO2 to CO on an Sb-Modified Cu Electrode: Spontaneous Fabrication and Physical Insight, ACS Catal., 2021, 11(12), 6846–6856 CrossRef CAS.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- B. J. K. N. Hammer and J. K. Nørskov, Electronic factors determining the reactivity of metal surfaces, Surf. Sci., 1995, 343(3), 211–220 CrossRef CAS.
- J. C. Liu, X. L. Ma, Y. Li, Y. G. Wang, H. Xiao and J. Li, Heterogeneous Fe3 single-cluster catalyst for ammonia synthesis via an associative mechanism, Nat. Commun., 2018, 9(1), 1610 CrossRef PubMed.
- J. K. Nørskov, F. Studt, F. Abild-Pedersen, T. Bligaard, Fundamental Concepts in Heterogeneous Catalysis, John Wiley & Sons, 2014 Search PubMed.
- P. Sabatier, La catalyse en chimie organique, C. Béranger, 1920 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05552g |
| This journal is © The Royal Society of Chemistry 2024 |